

Abstract
Abnormal tau protein impairs mitochondrial function, including transport, dynamics, and bioenergetics. Mitochondria interact with the endoplasmic reticulum (ER) via mitochondria‐associated ER membranes (MAMs), which coordinate and modulate many cellular functions, including mitochondrial cholesterol metabolism. Here, we show that abnormal tau loosens the association between the ER and mitochondria in vivo and in vitro. Especially, ER‐mitochondria interactions via vesicle‐associated membrane protein‐associated protein (VAPB)—protein tyrosine phosphatase‐interacting protein 51 (PTPIP51) are decreased in the presence of abnormal tau. Disruption of MAMs in cells with abnormal tau alters the levels of mitochondrial cholesterol and pregnenolone, indicating that conversion of cholesterol into pregnenolone is impaired. Opposite effects are observed in the absence of tau. Besides, targeted metabolomics reveals overall alterations in cholesterol‐related metabolites by tau. The inhibition of GSK3β decreases abnormal tau hyperphosphorylation and increases VAPB–PTPIP51 interactions, restoring mitochondrial cholesterol and pregnenolone levels. This study is the first to highlight a link between tau‐induced impairments in the ER‐mitochondria interaction and cholesterol metabolism.
Keywords: cholesterol, endoplasmic reticulum, GSK3β, mitochondria, tau protein
Subject Categories: Membranes & Trafficking, Metabolism, Molecular Biology of Disease
Endoplasmic reticulum (ER)—mitochondria coupling regulates many cellular functions, including mitochondrial cholesterol metabolism. This study shows that abnormal tau protein disrupts ER—mitochondria contacts, impairing cholesterol metabolism and pregnenolone synthesis within mitochondria.
Introduction
Tauopathies comprise a group of familial and sporadic neurodegenerative diseases, including frontotemporal lobar degeneration (FTLD) and Alzheimer's disease (AD; Ballatore et al, 2007). They are characterized by an aberrant intracellular accumulation of neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein (Spillantini & Goedert, 1998). In familial cases of FTLD, mutations such as P301L have been identified in the tau (MAPT) gene (Hutton et al, 1998; Poorkaj et al, 1998; Spillantini et al, 1998). These render tau more susceptible to hyperphosphorylation, favor dissociation from microtubules, and further convey a greater propensity in promoting NFT aggregation (von Bergen et al, 2001), similarly to what is observed in AD where no MAPT mutations have been identified (Götz & Ittner, 2008).
Although compelling evidence has related abnormal tau to neurodegeneration and clinical deficits, the mechanisms underlying tau‐induced neuronal dysfunction and loss are still not entirely understood. Nonetheless, numerous studies highlight the dysfunction of mitochondria in tauopathies, and malfunctioning mitochondria are already present at early disease stages, possibly occurring even before the onset of cognitive impairments (Schmitt et al, 2012; Wu et al, 2019). In this regard, we recently reviewed the impact of abnormal tau on mitochondria. Remarkably, aberrantly hyperphosphorylated tau appears to compromise almost every aspect of mitochondrial function (Szabo et al, 2020). In particular, previous findings of our groups demonstrated that abnormal tau disturbs mitochondrial bioenergetics and dynamics both in vitro and in vivo (David et al, 2005; Rhein et al, 2009; DuBoff et al, 2012; Schulz et al, 2012; Grimm et al, 2015). More recently, we reported that abnormal tau also impairs mitochondrial neurosteroidogenesis (Grimm et al, 2019). In fact, steroids can be synthesized de novo in the brain and are hence termed neurosteroids. For that, cholesterol is imported into mitochondria and subsequently converted into pregnenolone, the precursor of all neurosteroids (Porcu et al, 2016). Pregnenolone and the downstream neuroactive steroids play important functions in the nervous system, including in neuroprotection, neuroplasticity, and memory processes (Lin et al, 2022). We showed previously that pregnenolone synthesis is decreased in cells bearing the P301Ltau mutation, but the underlying causes remained unknown (Grimm et al, 2019).
Strikingly, all the abovementioned alterations observed in mitochondrial functions are processes that are also regulated at the interface between mitochondria and the endoplasmic reticulum (ER). Indeed, mitochondria are closely connected to the ER via mitochondria‐associated ER membranes (MAMs), allowing them to communicate physically and biochemically (Csordás et al, 2006). Accordingly, MAM cross talk is involved in numerous fundamental metabolic processes (Hayashi et al, 2009), including cholesterol metabolism, calcium homeostasis, phospholipid metabolism, the transfer of lipids between these two organelles, and the regulation of mitochondrial bioenergetics and dynamics (Rusinol et al, 1994; Vance, 2003; Csordás et al, 2010; Volgyi et al, 2015). Besides, multiple tethering protein complexes have been revealed to be enriched at MAMs in order to maintain stable contact sites (Wilson & Metzakopian, 2021).
Over the last years, potential disturbances of MAMs have drawn great attention, particularly in connection to neurodegenerative diseases (Paillusson et al, 2016). For instance, several amyloid‐β (Aβ)‐related AD models are showing a structurally and functionally altered ER‐mitochondria coupling (Area‐Gomez & Schon, 2016; Fernandes et al, 2021). In contrast, the impact of tau on the ER‐mitochondria axis has so far received limited attention.
Therefore, in the present study, we investigated the impact of disease‐associated tau on MAMs in vitro and in vivo, with a specific focus on cholesterol homeostasis. We report that abnormal tau (mutation and depletion of tau) modifies the mitochondrial network morphology, disrupts the ER‐mitochondria interaction, and impairs the intracellular cholesterol homeostasis with a defective cholesterol import into mitochondria. Moreover, we demonstrate that both an artificial MAM tethering (by knocking down mitofusin 2 [MFN2]) and glycogen synthase kinase‐3β (GSK3β) inhibition increase the ER‐mitochondria association and thereby rescue the abnormal tau‐induced defects in intramitochondrial cholesterol metabolism.
Results
P301L‐tau modifies mitochondrial network morphology and disrupts the ER‐mitochondria association in vivo and in vitro
By crossing P301L‐tau transgenic pR5 mice with the mitochondrial reporter strain mitoCFP, we first studied the impact of P301L‐tau on mitochondrial network morphology and the ER‐mitochondria association in neurons specifically in mitoCFP+/P301L+ mice, using mitoCFP+/P301L− mice as a control (see Fig 1A and Materials and Methods). P301L+ mice show a pronounced pathology in several brain areas, including the amygdala and hippocampus. In the amygdala, we observed a significant increase in mitochondrial length, while in the hippocampus we observed an additional increase in two other indicators of mitochondrial elongation, the form factor and area‐weighted form factor in mitoCFP+/P301L+ mice compared with mitoCFP+/P301L− mice (Figs 1B–E and EV1A–E).
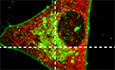
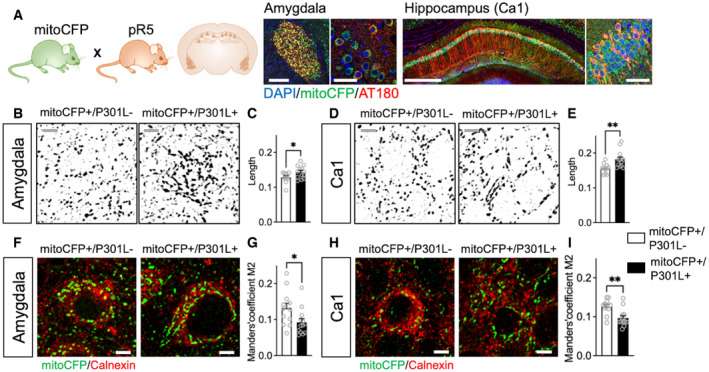
Figure 1. Abnormal tau disturbs mitochondrial network morphology and disrupts the ER‐mitochondria association in pR5 mice.
-
AMitoCFP mice, expressing Cyan Fluorescent Protein (CFP) in neuronal mitochondria, crossed with P301L‐tau mutant pR5 mice present with abnormal tau hyperphosphorylation (AT180 staining in red) in neurons of the amygdala and the hippocampus (CA1). P301L‐tau can be found in the same neurons as those expressing mitoCFP (in green). Nuclei are labeled with DAPI (in blue). Scale bars amygdala left panel (×10): 500 μm, right panel (×63): 50 μm; Scale bars hippocampus: left panel (×10): 500 μm, right panel (×63): 50 μm.
-
B–E(B, D) Representative microscopy images of the mitochondrial network morphology in neurons of the amygdala (B) and the CA1 region of the hippocampus (D) in mitoCFP+/P301L− mice and mitoCFP+/P301L+ mice. MitoCFP signal is displayed in gray on the images. Cropped images, see Fig EV1A for whole images. Gray scale bars: 5 μm. (C, E) Mitochondrial length in neurons of the amygdala (C) and in Ca1 (E) of mitoCFP+/P301L− and mitoCFP+/P301L+ mice.
-
F–I(F, H) Representative microscopy images of the endoplasmic reticulum stained with calnexin (in red) and mitochondria (mitoCFP) in neurons of the amygdala (F) and Ca1 (H) in mitoCFP+/P301L− and mitoCFP+/P301L+ mice. Cropped images, see Fig EV1F for whole images. White scale bars: 5 μm. (G, I) Colocalization analysis between calnexin and mitoCFP in neurons of the amygdala (G) and Ca1 (I) of mitoCFP+/P301L− and mitoCFP+/P301L+ mice. The Manders' overlap coefficients M2 is represented on the graphs, as it indicates the proportion of overlaps between the mitoCFP signal (channel 2) and calnexin signal (channel 1) relative to the total pixel intensity of channel 2 (i.e., only in neurons, as mitoCFP is expressed only in these cells).
Data information: Values represent the mean ± SEM of n = 11 MitoCFP+/P301L− mice and n = 13 MitoCFP+/P301L+ mice. Each gray open circle represents the mean of 6 technical replicates (data from 6 images) per animal. *P < 0.05, **P < 0.01, Student unpaired t‐test.
Source data are available online for this figure.
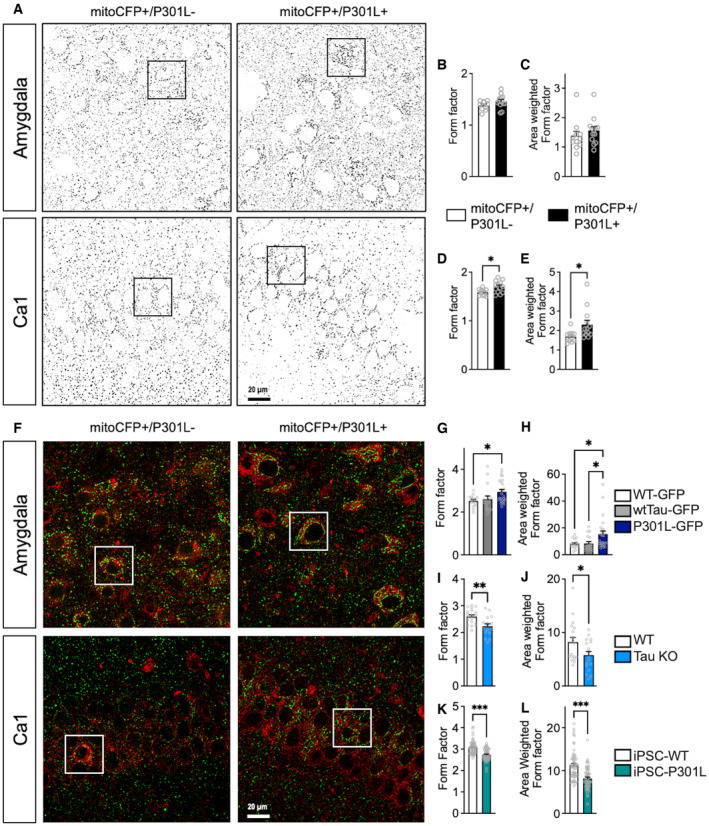
Figure EV1. Tau affects mitochondrial morphology and ER‐mitochondria association.
-
ARepresentative microscopy images of the mitochondrial network morphology in neurons of the amygdala (top) and the Ca1 region of the hippocampus (bottom) in mitoCFP+/P301L− mice and mitoCFP+/P301L+ mice. MitoCFP signal is displayed in gray on the images. Squares represent the cropped area presented in Fig 1B and D.
-
B–EComplementary metrics of mitochondrial network morphology: form factor (mitochondrial elongation) (B, D), and area‐weighted form factor, a variant of form factor with a bias toward larger mitochondria (C, E) in neurons of the amygdala (B, C) and in Ca1 (D, E) of mitoCFP+/P301L− and mitoCFP+/P301L+ mice. Values represent the mean ± SEM of n = 11 MitoCFP+/P301L− mice and n = 13 MitoCFP+/P301L+ mice. Each gray open circle represents the mean of 6 technical replicates (data from 6 images) per animal. *P < 0.05, Student unpaired t‐test.
-
FRepresentative microscopy images of the endoplasmic reticulum stained with calnexin (in red) and mitochondria (mitoCFP) in neurons of the amygdala (top) and Ca1 (bottom) in mitoCFP+/P301L− and mitoCFP+/P301L+ mice. Squares represent the cropped area presented in Fig 1F and H.
-
G–LComplementary metrics of mitochondrial network morphology: form factor (mitochondrial elongation) (G, I, K) and area‐weighted form factor (H, J, L) in WT‐GFP, wtTau‐GFP and P301L‐GFP expressing SH‐SY5Y cells (G, H), WT and Tau KO SH‐SY5Y cells (I, J), and iPSC‐WT and iPSC‐P301L (K, L). On average 1,000–2,500 mitochondrial organelles were analyzed per group (n = 20–45 images per group, 3 experiments).
Data information: (G, H) *P < 0.05; One‐way ANOVA + Tukey's post hoc test. (I–L) *P < 0.05, **P < 0.01, ***P < 0.001; Student unpaired t‐test.
To evaluate the ER‐mitochondria association in neurons, we performed a colocalization analysis between the mitoCFP signal (mitochondria) and the ER marker calnexin in both brain areas (Figs 1F–I and EV1F). We observed a decreased colocalization in mitoCFP+/P301L+ mice compared with mitoCFP+/P301L− mice in both brain areas, indicating a decreased proximity between the ER and mitochondria in animals expressing abnormal tau (Fig 1G and I).
We next confirmed these data in vitro. A significant increase in mitochondrial length, form factor, and area‐weighted form factor was observed in SH‐SY5Y cells transfected with green fluorescent protein (GFP)‐tagged P301L‐tau when compared to GFP‐only transfected cells (wild‐type WT‐GFP; Figs 2A and B, and EV1G and H, Movies [Link], [Link]). No difference was observed between the WT‐GFP cells and wild‐type tau‐GFP (wtTau‐GFP)‐transfected cells (Figs 2A and B, and EV1G and H). Nonetheless, both wtTau‐GFP and P301L‐GFP‐transfected cells presented a perinuclear clustering of mitochondria (Fig 2A and C), suggesting that tau overexpression affects mitochondrial distribution and the P301L mutation affects in addition mitochondrial morphology. To visualize and quantify the ER‐mitochondria interaction, we next performed a proximity ligation assay (PLA) between proteins of the outer mitochondrial membrane and the ER membrane at the MAM interface (Fig 2D–I; Tubbs & Rieusset, 2016). Three different pairs of antibodies targeting MAM tethers were used: IP3R (inositol triphosphate receptor, ER membrane) together with VDAC (voltage‐dependent anion channel, mitochondrial membrane), IP3R with GRP75 (glucose‐regulated protein 75, mitochondrial membrane), and VAPB (vesicle‐associated membrane protein‐associated protein, ER membrane) with PTPIP51 (protein tyrosine phosphatase‐interacting protein 51, mitochondrial membrane; Fig 2D–F). A significant decrease in the number of contact points between the ER and mitochondria was detected for the three pairs in P301L cells, with the greatest difference observed for the VAPB–PTPIP51 pair when compared to WT cells (Fig 2G–I). Interestingly, a decrease in the number of ER‐mitochondria contacts was also observed in wtTau compared with WT cells, but only with the VAPB–PTPIP51 pair (Fig 2I).
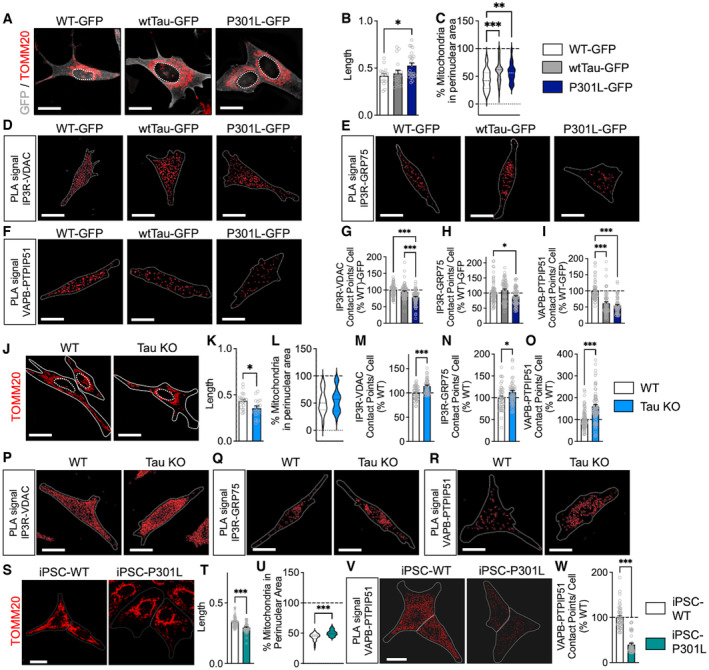
Figure 2. Effects of tau on mitochondrial network morphology and the ER‐mitochondria association.
-
ARepresentative microscopy images (z‐projections) of the mitochondrial network stained with TOMM20 in SH‐SY5Y cells stably expressing a green fluorescent protein (GFP)‐tagged wild‐type form of tau protein (wtTau‐GFP), a GFP‐tagged P301L mutant form of tau protein (P301L‐GFP) or cell expression the GFP only (control cells = WT‐GFP). GFP signal is displayed in gray on the images, TOMM20 signal is in red, and the nuclei are delimited by white dotted lines. See Movies [Link], [Link] for the three‐dimensional reconstruction of mitochondrial network of the WT‐GFP, wtTau‐GFP and P301L‐GFP expressing cells.
-
B, CMitochondrial length (B) and percentage of mitochondria in the perinuclear area (C) in WT‐GFP, wtTau‐GFP, and P301L‐GFP expressing cells. On average 1,000–2,500 mitochondrial organelles were analyzed per group (n = 20–25 images per group, 3 independent experiments).
-
D–FVisualization of the IP3R–VDAC (D), IP3R–GRP75 (E), and VAPB–PTPIP51 (F) interactions by in situ proximity ligation assay (PLA) WT‐GFP, wtTau‐GFP, and P301L‐GFP expressing cells. Interactions between the two targeted proteins are depicted in red (63X magnification) and cell surfaces are delimited by a white line.
-
G–IQuantitative analysis of the PLA signal represented as number of contact points between IP3RVDAC (G), IP3R–GRP75 (H), and VAPB–PTPIP51 (I) per cell in percentage of the WT‐GFP cells. In total, 60–75 cells were analyzed per group (5 independent experiments).
-
JRepresentative microscopy images (z‐projections) of the mitochondrial network stained with TOMM20 in wild‐type (WT) SH‐SY5Y cells and tau knockout (Tau KO) SH‐SY5Y cells. TOMM20 signal is in red on the images, cell surfaces are delimited by white lines, and nuclei are delimited by white dotted lines. See Movies EV4 and EV5 for the three‐dimensional reconstruction of mitochondrial network of the WT and Tau KO cells.
-
K, LMitochondrial length (K) and percentage of mitochondria in the perinuclear area (L) in WT cells and Tau KO cells. On average 1,000–2,500 mitochondrial organelles were analyzed per group (n = 20–25 images per group, 3 independent experiments).
-
M–OQuantitative analysis of the PLA signal represented as number of contact points between IP3R–VDAC (M), IP3R–GRP75 (N), and VAPB–PTPIP51 (O) per cell in percentage of the WT cells. In total, 60–75 cells were analyzed per group (3 independent experiments).
-
P–RVisualization of the IP3R–VDAC (P), IP3R–GRP75 (Q), and VAPB–PTPIP51 (R) interactions by in situ PLA in WT and Tau KO cells. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line.
-
SRepresentative microscopy images (z‐projections) of the mitochondrial network stained with TOMM20 in patient‐derived induced pluripotent stem cells (iPSC) bearing a P301L tau mutation (iPSC‐P301L) and the corresponding isogenic wild‐type cells (iPSC‐WT). TOMM20 signal is in red on the images, cell surfaces are delimited by white lines, and nuclei are delimited by white dotted lines. See Movies EV6 and EV7 for the three‐dimensional reconstruction of mitochondrial network of the iPSC‐WT and iPSC‐P301L.
-
T, UMitochondrial length (T) and percentage of mitochondria in the perinuclear area (U) in iPSC‐WT cells and iPSC‐P301L. On average 1,000–2,500 mitochondrial organelles were analyzed per group (n = 51 cells per group, 3 independent experiments).
-
VVisualization of the VAPB–PTPIP51 interactions by in situ PLA in iPSC‐WT cells and iPSC‐P301L. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line.
-
WQuantitative analysis of the PLA signal represented as number of contact points between VAPB–PTPIP51 per cell in percentage of the iPSC‐WT cells. In total, 45 cells were analyzed per group (3 independent experiments).
Data information: Data are presented as mean ± SEM. White scale bars: 20 μm. B, C, G–I *P < 0.05, **P < 0.01, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test. K–O, T–U, W *P < 0.05, ***P < 0.001; Student unpaired t‐test. GRP75: glucose‐regulated protein 75; IP3R: inositol trisphosphate receptor; PTPIP51: protein tyrosine phosphatase‐interacting protein 51; TOMM20: translocase of the outer mitochondrial membrane complex subunit 20; VAPB: vesicle‐associated membrane protein‐associated protein B; VDAC: voltage‐dependent anion channel.
Source data are available online for this figure.
To assess whether tau itself influences mitochondrial network morphology and ER‐mitochondria association, we next compared CRISPR Cas9‐generated tau knockout SH‐SY5Y cells (Tau KO) with WT cells expressing endogenous tau. Strikingly, a decrease in mitochondrial length, form factor, and area‐weighted form factor was detected in Tau KO cells when compared to WT cells (Figs 2J and K, and EV1I and J, Movies EV4 and EV5), but no difference was observed in the mitochondrial distribution (Fig 2L). Also, different from what we had observed in tau‐transfected cells, a significant increase in the number of ER‐mitochondria contact points was detected in Tau KO with the three pairs of antibodies, and again, the greatest difference was observed with the VAPB–PTPIP51 pair (Fig 2M–R).
To validate our key data in cells expressing endogenous levels of P301L‐tau, we used human induced pluripotent stem cells (iPSCs) derived from a patient bearing the P301L‐tau mutation (iPSC‐P301L) as well as CRISPR/Cas9‐corrected wild‐type isogenic control cells (iPSC‐WT; Fig 2S–W). Intriguingly, a decrease in mitochondrial length, form factor, and area‐weighted form factor was detected in iPSC‐P301L when compared to iPSC‐WT cells (Figs 2S and T, and EV1K and L; Movies EV6 and EV7). As observed in P301L‐transfected SH‐SY5Y, iPSC‐P301L presented a perinuclear clustering of mitochondria compared with iPSC‐WT (Fig 2S and U). Besides, a significant decrease in the number of ER‐mitochondria contact points was detected in iPSC‐P301L with the VAPB–PTPIP51 antibody pair (Fig 2V and W).
Taken together, these data demonstrate, on the one hand, that mitochondrial elongation is increased in P301L‐transfected cells, paralleled with a perinuclear mitochondrial clustering and a decrease in the ER‐mitochondria association. On the other hand, mitochondrial elongations are decreased in the absence of tau, while the ER‐mitochondria association is increased. Data obtained in iPSCs suggest that it is the P301L overexpression in SH‐SY5Y cells (mimicking abnormal tau accumulation in tauopathy) that would be responsible for mitochondrial elongation, while the P301L mutation per se (in iPSC‐P301L) induces mitochondrial fragmentation and disruption of the ER‐mitochondria association.
P301L‐tau affects intracellular cholesterol metabolism
To better understand the effects of P301L‐tau on the ER‐mitochondria association, we next assessed the expression level of genes involved in mitochondrial dynamics, ER‐mitochondria tethering, as well as calcium and cholesterol homeostasis at the MAMs (Fig 3A). Strikingly, opposing effects were again observed between P301L cells and Tau KO cells regarding the expression of most of the genes investigated by us, with some genes being downregulated in P301L cells while upregulated in Tau KO cells, and vice versa, when compared to WT cells (Fig 3B; Table EV1).
Figure 3. Abnormal tau impairs intracellular cholesterol metabolism.
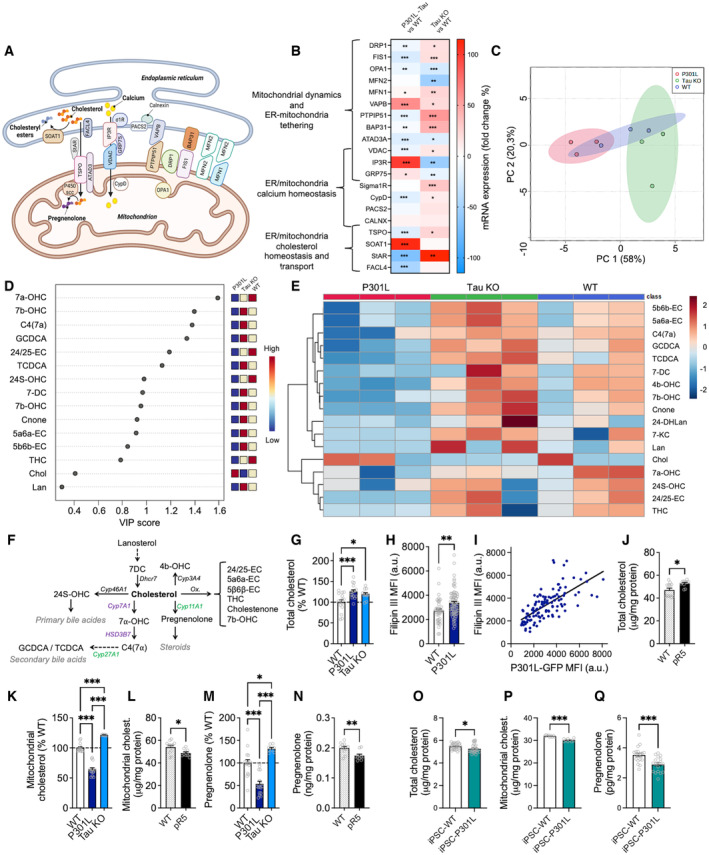
-
ASchematic representation of the mitochondria‐associated endoplasmic reticulum (ER) membranes (MAMs) showing the proteins of interest in our study. Two important MAM functions are depicted: the transfer of calcium from the ER to mitochondria via IP3R and VDAC, as well as cholesterol transfer from the ER to mitochondria for conversion into pregnenolone.
-
BStudy of the impact of tau on the mRNA level of MAM proteins using quantitative real‐time PCR analysis of gene expression. The heat map depicts the fold changes in mRNA expression in P301L‐transfected SH‐SY5Y cells and in tau knockout (Tau KO) SH‐SY5Y cells as percentage of the WT cells. Red colors represent an upregulation of gene expression, while blue colors represent a downregulation. The corresponding values can be found in Table EV1. *P < 0.05, **P < 0.01, ***P < 0.001; Student unpaired t‐test P301L versus WT, or Tau KO versus WT, n = 6–12 replicates/group (3 independent experiments).
-
CPrincipal component analysis of cholesterol‐related metabolites in wild‐type (WT, in blue), Tau KO (in green), and P301L‐expressing SH‐SY5Y cells (in red): scores plot between the selected principal component 1 and 2. Each colored dot represents one sample (3 independent cell cultures per group).
-
DImportant features identified by partial least squares‐discriminant analysis (PLS‐DA). The colored boxes on the right indicate the relative concentrations of the corresponding metabolite in WT, Tau KO, and P301L cells. Dots represent the variable importance in projection (VIP) score for each metabolite identified by PLS‐DA.
-
EClustering result of cholesterol metabolites shown as heat map (distance measure using Euclidean, and clustering algorithm using ward.D).
-
FSimplified representation of cholesterol metabolic pathways highlighted in the present study. Enzymes written in violet are located in the ER, while those in green are located in mitochondria.
-
GQuantification of total cholesterol levels in cell lysates from WT, P301L, and Tau KO cells.
-
HMean fluorescence intensity (MFI) of the Filipin III (cholesterol) staining in WT and P301L cells. Data are presented as mean ± SEM of n = 70–90 cells per group (3 independent experiments).
-
ICorrelation between Filipin III fluorescence intensity (in ordinate) and GFP fluorescence intensity (in abscissa) in GFP‐tagged P301L cells. Each dot represents one P301L cell. Simple linear regression, Pearson R squared = 0.459, P < 0.01.
-
JQuantification of total cholesterol levels in brain preparations from wild‐type (WT) and pR5 (P301L) mice.
-
K–NMitochondrial cholesterol levels (K, L) and pregnenolone levels (M, N) in WT, P301L and Tau KO cells (K, M) and in brain preparations from WT and pR5 mice (L, N).
-
O–QQuantification of total cholesterol levels (O), mitochondrial cholesterol levels (P) and pregnenolone levels (Q) in patient‐derived induced pluripotent stem cells (iPSC) bearing a P301L tau mutation (iPSC‐P301L) and the corresponding isogenic wild‐type cells (iPSC‐WT). (C–E) Graphs were generated on Metaboanalyst.ca.
Data information: (G, K, M) Data are presented as mean ± SEM of n = 12–18 replicates per group (4 independent experiments) in percentage of the WT cells. *P < 0.05, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test. (J, L, N) Data are presented as mean ± SEM of n = 6 WT and n = 5 pR5 mice with 2 technical replicates per animal. *P < 0.05, **P < 0.01, Student unpaired t‐test. (O–Q) Data are presented as mean ± SEM of n = 27 replicates/group (O), n = 9 replicates/group (P) and n = 18 replicates/group (Q). *P < 0.05, ***P < 0.001, Student unpaired t‐test. Abbreviations (A, B): ATAD3, ATPase family AAA domain‐containing protein 3; BAP31, b‐cell receptor‐associated protein 31; CALNX, calnexin; CypD, cyclophilin D; DRP1, dynamin‐related protein 1; ER, endoplasmic reticulum; FACL4, fatty‐acid‐coenzyme A ligase, Long‐Chain 4; FIS1, fission protein 1; GRP75, glucose‐regulated protein 75; IP3R, inositol trisphosphate receptor; MFN1, mitofusin 1; MFN2, mitofusin 2; OPA1, optic atrophy 1; P450scc, cholesterol side‐chain cleavage cytochrome P450; PACS2, phosphofurin acidic cluster sorting protein 2; PTPIP51, protein tyrosine phosphatase‐interacting protein 51; SOAT1, sterol o‐acyltransferase 1; StAR, steroidogenic acute regulator protein; TSPO, translocator protein; VAPB, vesicle‐associated membrane protein‐associated protein B; VDAC, voltage‐dependent anion channel; σ1R, sigma 1 receptor. Abbreviations (D–F): 24/25‐EC: 24,25‐Epoxycholesterol; 24‐DHLan: 24,25‐Dihydro‐lanosterol; 24S‐OHC: 24S‐Hydroxy‐cholesterol; 4b‐OHC: 4‐beta‐Hydroxy‐cholesterol; 5a6a‐EC: 5‐alpha,6‐alpha‐Epoxycholesterol; 5b6b‐EC: 5‐beta,6‐beta‐Epoxycholesterol; 7DC: 7‐Dehydrocholesterol; 7a‐OHC: 7‐alpha‐Hydroxy‐cholesterol; 7b‐OHC: 7‐beta‐Hydroxy‐cholesterol; 7KC: 7‐Ketocholesterol; C4(7a): 7‐alpha‐Hydroxy‐cholestenone; Chol.: Cholesterol; Cnone: Cholestenone; Cyp11A1: Cytochrome P450 Family 11 Subfamily A Member 1; Cyp27A1: Cytochrome P450 Family 27 Subfamily A Member 1; Cyp3A4: Cytochrome P450 Family 3 Subfamily A Member 4; Cyp7A1: Cytochrome P450 Family 7 Subfamily A Member 1; Dhcr7: 7‐dehydrocholesterol reductase; GCDCA: Glycochenodeoxycholic acid; HSD3B7: 3 beta‐hydroxysteroid dehydrogenase type 7; Lan: Lanosterol; TCDCA: Taurochenodeoxycholic acid; THC: 5‐alpha,6‐beta‐Dihydroxycholestanol.
Source data are available online for this figure.
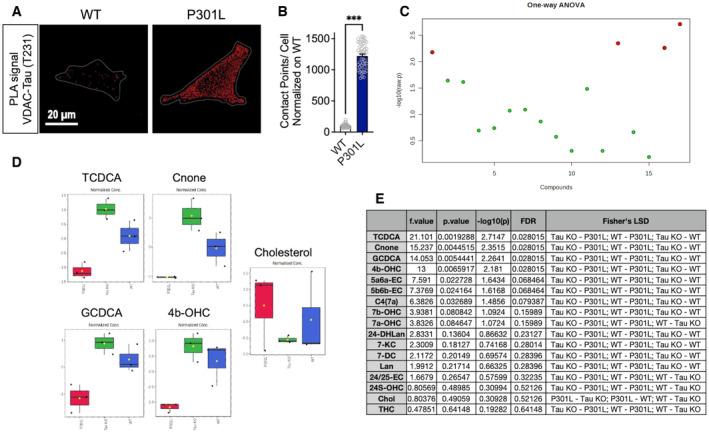
Given that genes involved in ER‐mitochondria tethering were either up or downregulated in P301L cells, the disruption of the ER‐mitochondria association observed in these cells could not be solely explained by changes in mRNA levels of related genes, and probably involved other mechanisms. Therefore, guided by a previous study showing that phosphorylated tau can interact with VDAC in human and mouse brains (Manczak & Reddy, 2012), we performed a PLA between VDAC and phospho‐tau (T231). We observed numerous contact points in the P301L cells compared with WT cells (Fig EV2A and B), suggesting that phospho‐tau interacts directly with VDAC, which may disturb the ER‐mitochondria associations.
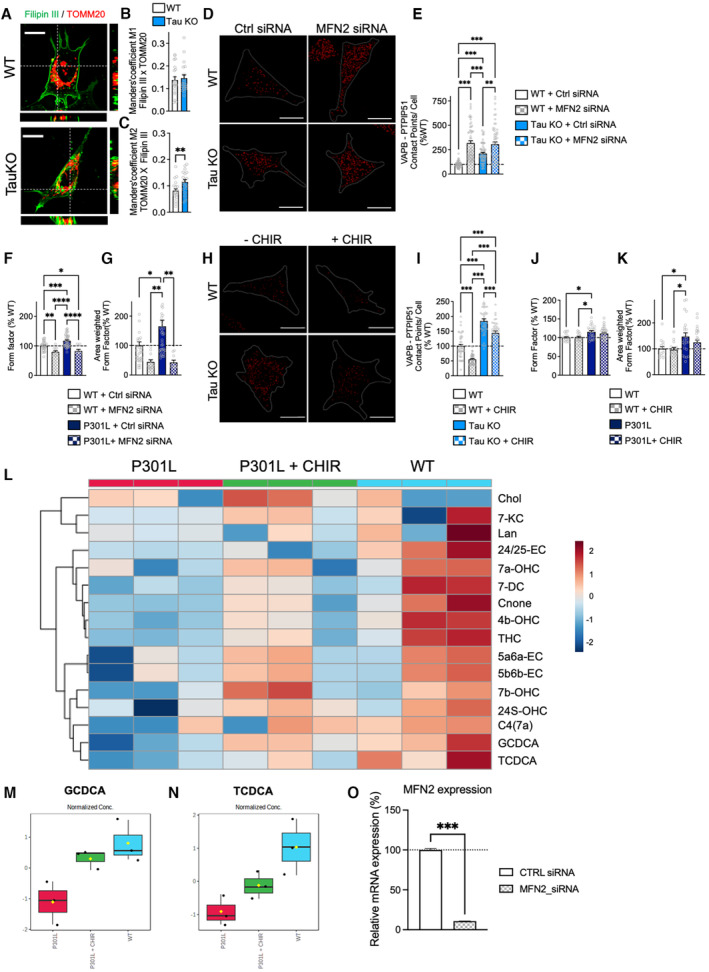
Figure EV3. Supplementary data obtained in Tau KO cells.
-
ARepresentative microscopy images (z‐projections) of WT and Tau KO cells co‐stained with Filipin III (cholesterol) in green and TOMM20 (mitochondria) in red with xy‐ and xz‐axis orthogonal views framing the corresponding image. Scale bar: 10 μm.
-
B, CManders' coefficients M1 (B) representing the proportion of Filipin III overlapping with TOMM20, and M2 (C) representing the proportion of TOMM20 overlapping with Filipin III in WT versus Tau KO cells. Data are presented as mean ± SEM (n = 20–23 images per group, 2 independent experiments). **P < 0.01, Student unpaired t‐test.
-
DVisualization of the VAPB–PTPIP51 interactions by in situ proximity ligation assays (PLAs) in wild‐type (WT) cells and Tau KO cells transfected with the control (Ctrl) siRNA, as well as in MFN2 siRNA‐transfected cells. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line. Scale bars: 20 μm.
-
EQuantitative analysis of the PLA signal represented as number of contact points between VAPBPTPIP51 per cell in percentage of the WT cells. In total, 60 cells were analyzed per group (4 independent experiments). Data are presented as mean ± SEM. **P < 0.01, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test.
-
F, GComplementary metrics of mitochondrial network morphology: form factor (mitochondrial elongation) (F) and area‐weighted form factor (G) in WT and P301L transfected with MFN2 siRNA or control (Ctrl) siRNA. On average 500–1,500 mitochondrial organelles were analyzed per group (n = 15–20 images per group, 2 independent experiments). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; One‐way ANOVA + Tukey's post hoc test.
-
HVisualization of the VAPB–PTPIP51 interactions by in situ proximity ligation assay (PLA) in untreated wild‐type (WT) cells and Tau KO cells as well as in CHIR99021 (CHIR)‐treated cells. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line. Scale bar: 20 μm.
-
IQuantitative analysis of the PLA signal represented as number of contact points between VAPB–PTPIP51 per cell in percentage of the WT cells. In total, 45 cells were analyzed per group (3 independent experiments). Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test.
-
J, KComplementary metrics of mitochondrial network morphology: form factor (mitochondrial elongation) (J) and area‐weighted form factor (K) in WT and P301L untreated or treated with CHIR99021 in percentage of the WT cells. On average 500–1,500 mitochondrial organelles were analyzed per group (n = 15–20 images per group, 2 independent experiments). Data are presented as mean ± SEM. *P < 0.05; One‐way ANOVA + Tukey's post hoc test.
-
LClustering result of cholesterol metabolites shown as heat map (distance measure using Euclidean, and clustering algorithm using ward.D).
-
M, NCorresponding graphs of GCDCA (glycochenodeoxycholic acid) and TCDCA: (taurochenodeoxycholic acid) levels in WT cells, P301L cells, and P301L cells + CHIR. N = 3 independent cell culture/group. Data are presented as boxplot, the central band represents the median, the box represents the 10–90 percentile, and the whiskers represent the minimum to maximum. (M) P = 0.018483, One‐way ANOVA + Fisher's LSD: P301L + CHIR vs P301L; WT vs P301L; WT vs P301L + CHIR. (N) P = 0.022415, One‐way ANOVA + Fisher's LSD: P301L + CHIR vs P301L; WT vs P301L; WT vs P301L + CHIR. (L–N) Graphs were generated on Metaboanalyst.ca.
-
OExpression level of MFN2 (mRNA expression) in SH‐SY5Y cells transfected with the control (Ctrl) siRNA, as well as in MFN2 siRNA‐transfected cells. Data represent the mean and SEM, normalized to 100% of the Ctrl siRNA condition (n = 4 replicates per group).
Data information: 24/25‐EC, 24,25‐Epoxycholesterol; 24S‐OHC, 24S‐Hydroxy‐cholesterol; 4b‐OHC, 4‐beta‐Hydroxy‐cholesterol; 5a6a‐EC, 5‐alpha,6‐alpha‐Epoxycholesterol; 5b6b‐EC, 5‐beta,6‐beta‐Epoxycholesterol; 7DC, 7‐Dehydrocholesterol; 7a‐OHC, 7‐alpha‐Hydroxy‐cholesterol; 7b‐OHC, 7‐beta‐Hydroxy‐cholesterol; 7KC, 7‐Ketocholesterol; C4(7a), 7‐alpha‐Hydroxy‐cholestenone; Chol., Cholesterol; Cnone, Cholestenone; GCDCA, Glycochenodeoxycholic acid; Lan, Lanosterol; TCDCA, Taurochenodeoxycholic acid; THC, 5‐alpha,6‐beta‐Dihydroxycholestanol; TOMM20, translocase of the outer mitochondrial membrane complex subunit 20; VDAC, voltage‐dependent anion channel.
Among the genes that were the most dysregulated in P301L cells were those involved in cholesterol metabolism and transport at the MAMs (Fig 3B). Previous studies had shown an accumulation of cholesterol in neurons affected by tau (Distl et al, 2001; Glöckner & Ohm, 2014). Interestingly, an important MAM function is the regulation of cholesterol homeostasis (Martin et al, 2014). We therefore assessed the impact of abnormal tau and tau knockout on cholesterol metabolism. To do so, we used a targeted metabolomics approach to compare the profile of cholesterol‐related metabolites in WT, P301L, and tau KO cells. Among 55 metabolites assessed, 17 were above the limit of detection of the assay and were analyzed in three independent cell cultures per group (Dataset EV1). Principal component analysis (PCA) revealed distinct alterations in cholesterol‐related metabolite levels in P301L and Tau KO cells versus WT cells (Fig 3C). Especially, a statistically significant difference was detected between the three cell lines regarding the levels of the oxysterols 4‐beta‐Hydroxy‐cholesterol (4b‐OHC) and cholestenone, and the secondary bile acids glycochenodeoxycholic acid (GCDCA) and taurochenodeoxycholic acid (TCDCA; Fig EV2C–E). The partial least squares‐discriminant analysis (PLS‐DA) reveals the main metabolites driving the differences between groups (Fig 3D). Overall, cholesterol‐related metabolites were decreased in P301L cells and increased in Tau KO cells when compared to WT cells (Fig 3E). Interestingly, the cholesterol level seemed to be increased in P301L cells and decreased in Tau KO cells, although this difference was not statistically significant (Fig EV2D and E) due to the low replicate number (N = 3/group). We therefore aimed to validate these data with a higher sample number using a (fluorescence‐based) cholesterol quantitation assay. In line with previous findings (Glöckner & Ohm, 2014), we observed a significant increase in the total cholesterol amount in P301L cells compared with WT cells (Fig 3G). Of note, total cholesterol was also increased in Tau KO cells. This may be because the cholesterol‐related metabolites were increased in these cells and the cholesterol quantitation assay used by us cannot discriminate between free cholesterol and other oxysterol metabolites. Microscopic analysis of Filipin III staining (cholesterol dye) revealed an increased fluorescence intensity in P301L cells compared with WT cells (Fig 3H), indicating cholesterol accumulation in the presence of abnormal tau. Moreover, Filipin III fluorescence intensity positively correlated with P301L‐tau expression (GFP‐tagged P301L‐tau, Fig 3I), arguing for an impact of abnormal tau on cellular cholesterol homeostasis. In line with this finding, total cholesterol levels were increased in brain lysates of pR5 mice compared with nontransgenic littermate controls (Fig 3J).
Targeted metabolomics data revealed that the secondary bile acids GCDCA and TCDCA were decreased in P301L cells and increased in Tau KO cells when compared to WT cells (Figs 3E and EV2D). Interestingly, the conversion of cholesterol into secondary bile acids requires enzymatic steps taking place first in the ER and then in mitochondria (Fig 3F; Monte et al, 2009). Alterations in secondary bile acid levels might therefore be linked to defects in the cholesterol transfer from the ER to mitochondria. Indeed, MAMs seem to be involved in cholesterol transport to mitochondria (Martin et al, 2014). When assessing mitochondrial cholesterol levels, we observed a significant decrease in P301L cells and brain lysates from pR5 mice (Fig 3K and L) and an increase in Tau KO cells compared with WT cells (Fig 3K). In the brain, cholesterol is the precursor for the biogenesis of neurosteroids, with the first step of steroidogenesis being the conversion of cholesterol to pregnenolone in mitochondria (Fig 3F). Therefore, we measured the amount of pregnenolone (as a readout of intramitochondrial cholesterol metabolism) that can be easily quantified by enzyme immunoassay. We observed a significant decrease in pregnenolone levels in P301L cells compared with WT cells (Fig 3M). Again, these data were recapitulated in our in vivo model (Fig 3N). On the contrary, pregnenolone levels were increased in Tau KO cells when compared to WT cells (Fig 3M).
To validate our findings, key experiments were performed in iPSC‐P301L versus iPSC‐WT (Fig 3O–Q). Total cholesterol, mitochondrial cholesterol, and pregnenolone were all decreased in iPSC‐P301L when compared to iPSC‐WT. These data confirm that P301L‐tau impairs cholesterol transport to mitochondria and its conversion to pregnenolone. Cholesterol accumulation in P301L SH‐SY5Y cells could rather be due to P301L overexpression, which mimics abnormal tau accumulation in tauopathy. Besides, data obtained in Tau KO cells suggest that tau may per se play a role in cholesterol homeostasis and transfer to mitochondria.
P301L‐tau impairs intracellular cholesterol distribution
To confirm our data on the altered mitochondrial cholesterol levels in P301L cells, we performed a deep colocalization analysis between Filipin III staining and mitochondrial staining (TOMM20: translocase of the outer mitochondrial membrane complex subunit 20) using the Manders' overlap coefficients M1 and M2 (see also Materials and Methods). A significant decrease in the overlap between cholesterol staining and TOMM20 was observed in SH‐SY5Y P301L cells (Fig 4A–C), confirming decreased cholesterol in mitochondria. Conversely, an increased overlap between Filipin III and TOMM20 was observed in Tau KO cells compared with WT cells (Fig EV3A–C).
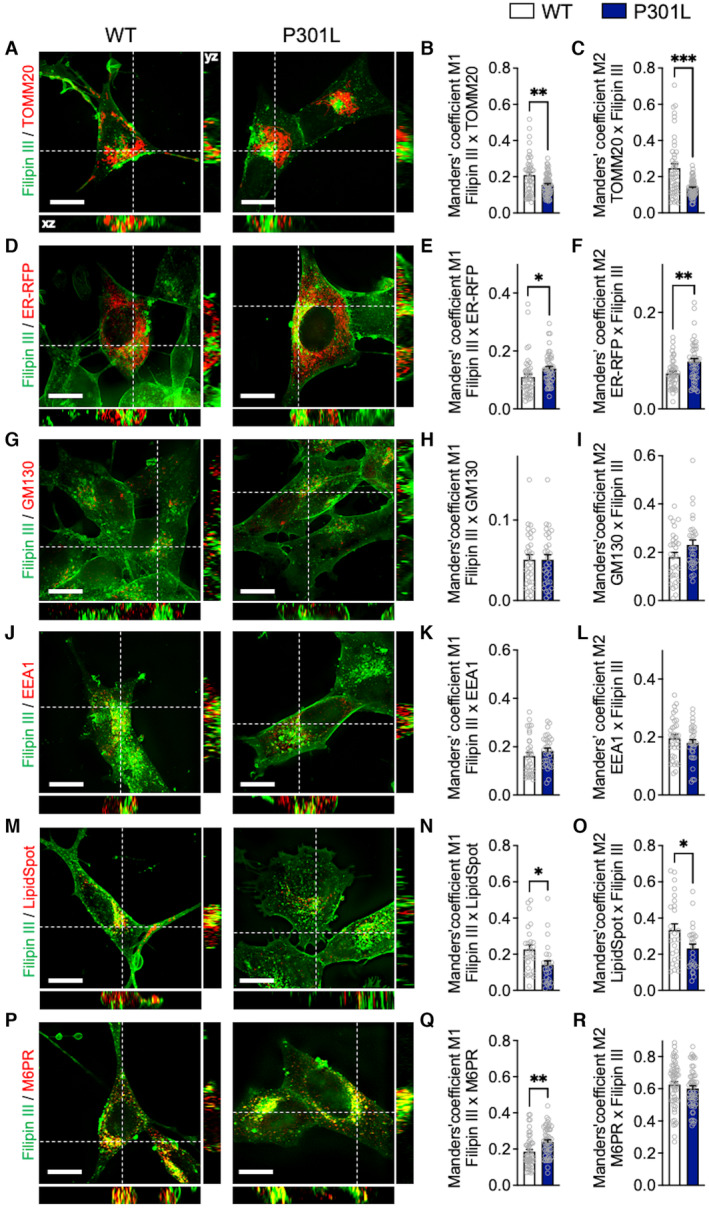
Figure 4. Abnormal tau impairs intracellular cholesterol distribution.
-
A–R(A, D, G, J, M, P) Representative microscopy images (z‐projections) of co‐staining with Filipin III (cholesterol) in green and (A) TOMM20 (mitochondria), (D) ER‐RFP (endoplasmic reticulum), (G) GM130 (Golgi apparatus), (J) EEA1 (endosomes), (M) LipidSpot (lipid droplets), or (P) M6PR (lysosomes) in red with xy‐ and xz‐axis orthogonal views framing the corresponding image. (B–C, E–F, H–I, K–L, N–O, Q–R) Manders' coefficients M1 representing the proportion of Filipin III overlapping with (B) TOMM20, (E) ER‐RFP, (H) GM130, (K) EEA1, (N) LipidSpot or (Q) M6PR, and M2 representing the proportion of (C) TOMM20, (F) ER‐RFP, (I) GM130, (L) EEA1, (O) LipidSpot or (R) M6PR overlapping with Filipin III in wild‐type (WT) versus P301L cells.
Data information: Data are presented as mean ± SEM (n = 40–90 images per group, 3–4 independent experiments). *P < 0.05, **P < 0.01, ***P < 0.001; Student unpaired t‐test. White scale bars: 10 μm. EEA1, early endosome antigen 1; ER‐RFP, endoplasmic reticulum red fluorescent protein; GM130, Golgi matrix protein 130; M6DR, mannose‐6‐phosphate receptor; TOMM20, translocase of the outer mitochondrial membrane complex subunit 20.
Source data are available online for this figure.
Figure EV2. Tau binds to VDAC and affects intracellular cholesterol metabolism.
-
AVisualization of the VDAC–phospho‐Tau (T231) interactions by in situ proximity ligation assay (PLA) in wild‐type (WT) and P301L cells. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line.
-
BQuantitative analysis of the PLA signal represented as number of contact points between VDAC–phospho‐Tau per cell. In total, 45–50 cells were analyzed per group (3 independent experiments). Data are presented as mean ± SEM. ***P < 0.001; Student unpaired t‐test.
-
CImportant features of cholesterol‐related metabolites selected by ANOVA plot with P‐value threshold 0.05. Red dots represent feature for which a statistically significant difference was detected between WT, Tau KO, and P301L cells. One‐way ANOVA + Fisher's least significant difference (Fisher's LSD).
-
DCorresponding graphs of features for which a statistically significant difference was detected between WT, Tau KO, and P301L cells. Of note, no statistical difference was detected regarding cholesterol (probably due to high variation within samples), but the corresponding graph is shown for information. n = 3 independent cell culture/group. Data are presented as boxplot, the central band represents the median, the box represents the 10–90 percentile, and the whiskers represent the minimum to maximum. One‐way ANOVA + Fisher's least significant difference (Fisher's LSD).
-
ETable showing the details of the features depicted in (C).
Data information: 24/25‐EC, 24,25‐Epoxycholesterol; 24‐DHLan, 24,25‐Dihydro‐lanosterol; 24S‐OHC, 24S‐Hydroxy‐cholesterol; 4b‐OHC, 4‐beta‐Hydroxy‐cholesterol; 5a6a‐EC, 5‐alpha,6‐alpha‐Epoxycholesterol; 5b6b‐EC, 5‐beta,6‐beta‐Epoxycholesterol; 7DC, 7‐Dehydrocholesterol; 7a‐OHC, 7‐alpha‐Hydroxy‐cholesterol; 7b‐OHC, 7‐beta‐Hydroxy‐cholesterol; 7KC, 7‐Ketocholesterol; C4(7a), 7‐alpha‐Hydroxy‐cholestenone; Chol., Cholesterol; Cnone, Cholestenone; GCDCA, Glycochenodeoxycholic acid; Lan, Lanosterol; TCDCA, Taurochenodeoxycholic acid; THC, 5‐alpha,6‐beta‐Dihydroxycholestanol; VDAC, voltage‐dependent anion channel.
Because the ER‐mitochondria interaction, a coupling that mediates cholesterol transfer to mitochondria, is disrupted in P301L cells, we next analyzed the colocalization between the Filipin III staining and the ER (using an ER‐red fluorescent protein as ER marker). Strikingly, P301L cells presented a higher overlapping coefficient than WT cells, indicating a higher cholesterol amount in the ER compared with WT cells (Fig 4D–F).
Using the same approach as above, we also verified whether cholesterol was accumulating in other cellular compartments. No differences were detected between P301L and WT cells regarding the colocalization between the cholesterol staining and Golgi apparatus (GM130: Golgi matrix protein 130) or the endosomes (EEA1: Early Endosome Antigen 1; Fig 4G–L). A lower overlapping coefficient was measured between the cholesterol staining and the lipid droplet staining (LipidSpot) in P301L cells, suggesting that cholesterol is not accumulating in lipid droplets (Fig 4M–O). Finally, the overlapping coefficient measured between cholesterol staining and the lysosome staining (M6PR: mannose‐6‐phosphate receptor) was increased only in the Filipin III channel (coefficient M1) but not in the M6PR channel (coefficient M2; Fig 4P–R). This suggests that a higher proportion of cholesterol locates to lysosomes in P301L cells when compared to WT cells.
Taken together, these data indicate that cholesterol accumulates in abnormal tau‐bearing neurons. This accumulation appears to result from reduced mitochondrial cholesterol in the presence of abnormal tau, which leads to a decrease in pregnenolone synthesis and accumulation of cholesterol in the ER and lysosomes. We propose that this defect in cholesterol transport to mitochondria is due, at least in part, to the P301L‐induced disruption of the ER‐mitochondria association.
An increase in the ER‐mitochondria association restores mitochondrial cholesterol transfer in disease‐associated tau‐bearing cells
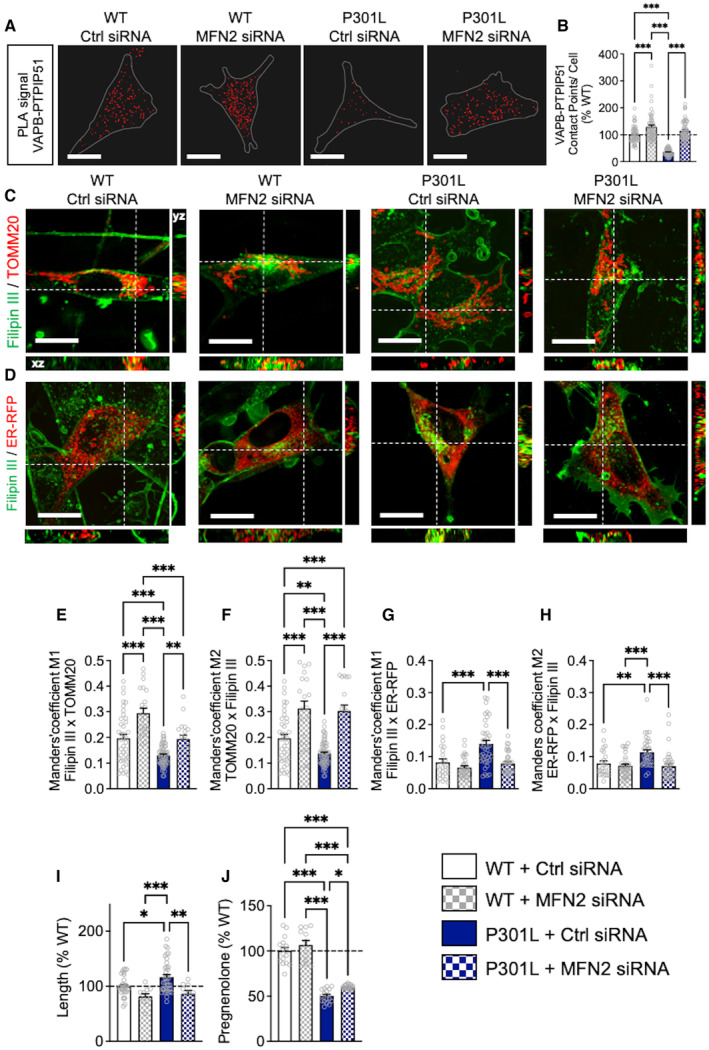
To verify our latest assumption, we asked whether an artificial increase in the ER‐mitochondria association would influence the transfer of cholesterol from the ER to mitochondria in P301L cells. To do so, we used a siRNA‐mediated knockdown (KD) of mitofusin 2 (MFN2; Filadi et al, 2015). The number of contact points between the MAM tethers VAPB and PTPIP51 was significantly increased in MFN2 siRNA‐transfected WT and P301L cells compared with P301L and WT cells transfected with the control (Ctrl) siRNA (Fig 5A and B). Of note, a similar increase in VAPB–PTPIP51 contacts was observed in MFN2 KD Tau KO cells (Fig EV3D and E).
Figure 5. Increase of ER‐mitochondria association counterbalances the effects of abnormal tau on mitochondrial cholesterol transfer.
-
AVisualization of the VAPB–PTPIP51 interactions by in situ proximity ligation assays (PLAs) in wild‐type (WT) cells and P301L cells transfected with the control (Ctrl) siRNA, as well as in MFN2 siRNA‐transfected cells. Interactions between the two targeted proteins are depicted in red (63× magnification) and cell surfaces are delimited by a white line. White scale bars: 20 μm.
-
BQuantitative analysis of the PLA signal represented as number of contact points between VAPB–PTPIP51 per cell in percentage of the WT cells. In total, 60 cells were analyzed per group (4 independent experiments).
-
C, DRepresentative microscopy images (z‐projections) of co‐staining with Filipin III in green and (C) TOMM20 (mitochondria) or (D) ER‐RFP (endoplasmic reticulum) in red with xy‐ and xz‐axis orthogonal views framing the corresponding image. White scale bars: 10 μm.
-
E–HManders' coefficient M1 representing the proportion of Filipin III overlapping with (E) TOMM20 or (G) ER‐RFP, and M2 representing the proportion of (F) TOMM20 or (H) ER‐RFP, overlapping with Filipin III. In total, 20–60 cells were analyzed per group (4 independent experiments).
-
IMitochondrial length in percentage of the WT cells. On average 500–1,500 mitochondrial organelles were analyzed per group (n = 15–20 images per group, 2 independent experiments);
-
JQuantification of pregnenolone levels in Ctrl siRNA‐transfected WT cells and P301L cells as well as in MFN2 siRNA‐transfected WT and P301L cells. Data are presented as mean ± SEM of n = 12–16 replicates per group (4 independent experiments) in percentage of the WT cells.
Data information: (B, E–J) Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test. ER‐RFP, endoplasmic reticulum red fluorescent protein; PTPIP51, protein tyrosine phosphatase‐interacting protein 51; TOMM20, translocase of the outer mitochondrial membrane complex subunit 20; VAPB, vesicle‐associated membrane protein‐associated protein B.
Source data are available online for this figure.
Regarding the overlap between the Filipin III staining and TOMM20, we observed a significant increase in MFN2 KD cells (WT and P301L) compared with Ctrl siRNA‐transfected cells (Fig 5C, E and F). The colocalization between the Filipin III staining and ER‐RFP was significantly decreased only in MFN2 KD P301L cells (Fig 5D, G and H). Given that MFN2 is involved in mitochondrial fusion, we also assessed the impact of the MFN2 KD on mitochondrial network morphology. As expected, a significant decrease in mitochondrial length, form factor, and area‐weighted form factor was measured in P301L cells + MFN2 siRNA versus P301L cells + Ctrl siRNA (Figs 5I and EV3F and G), indicating a decrease in mitochondrial elongation. Only the form factor was significantly decreased in MFN2 KD WT cells compared with Ctrl siRNA‐transfected cells (Fig EV3F and G). In addition, we observed a slight but significant increase in pregnenolone in P301L cells + MFN2 siRNA (Fig 5J), indicating that the increase of ER‐mitochondria association via MFN2 KD can partially restore pregnenolone synthesis in P301L cells. No significant difference between MFN2 siRNA and Ctrl siRNA‐transfected WT cells was observed regarding pregnenolone level.
GSK3β inhibition restores ER‐mitochondria interactions and intramitochondrial cholesterol metabolism in P301L‐tau expressing cells
Glycogen synthase kinase‐3β (GSK3β) is one of the key enzymes involved in abnormal tau hyperphosphorylation (Grimm, 2021). GSK3β activation has also been shown to disrupt the binding of VAPB to PTPIP51, reducing the ER‐mitochondria coupling in a model of amyotrophic lateral sclerosis and frontotemporal dementia (ALS/FTD; Stoica et al, 2014). Therefore, we next assessed whether GSK3β inhibition would increase the ER‐mitochondria interaction and restore the transfer of cholesterol from the ER to mitochondria that is disrupted by abnormal tau.
GSK3β inhibition (using CHIR99021) significantly decreased tau phosphorylation at serine 202/threonine 205 (AT8 epitope) and threonine 231 (AT180 epitope) in P301L cells (Fig 6A–D). In parallel, CHIR99021 treatment normalized the number of contacts between VAPB and PTPIP51 in P301L cells to those observed in the WT cells (Fig 6E and F). Intriguingly, the number of VAPB–PTPIP51 contacts was decreased in CHIR99021‐treated WT cells when compared to untreated cells (Fig 6E and F). The same observation was made in Tau KO cells, which showed a decreased VAPB–PTPIP51 association after CHIR99021 treatment (Fig EV3H and I). This suggests that distinct pathways may influence the ER‐mitochondria association when abnormal tau is overexpressed in cells, or when tau is expressed at endogenous levels, or completely ablated.
Figure 6. GSK3β inhibition restores ER‐mitochondria interactions and intramitochondrial cholesterol metabolism disrupted by abnormal tau.
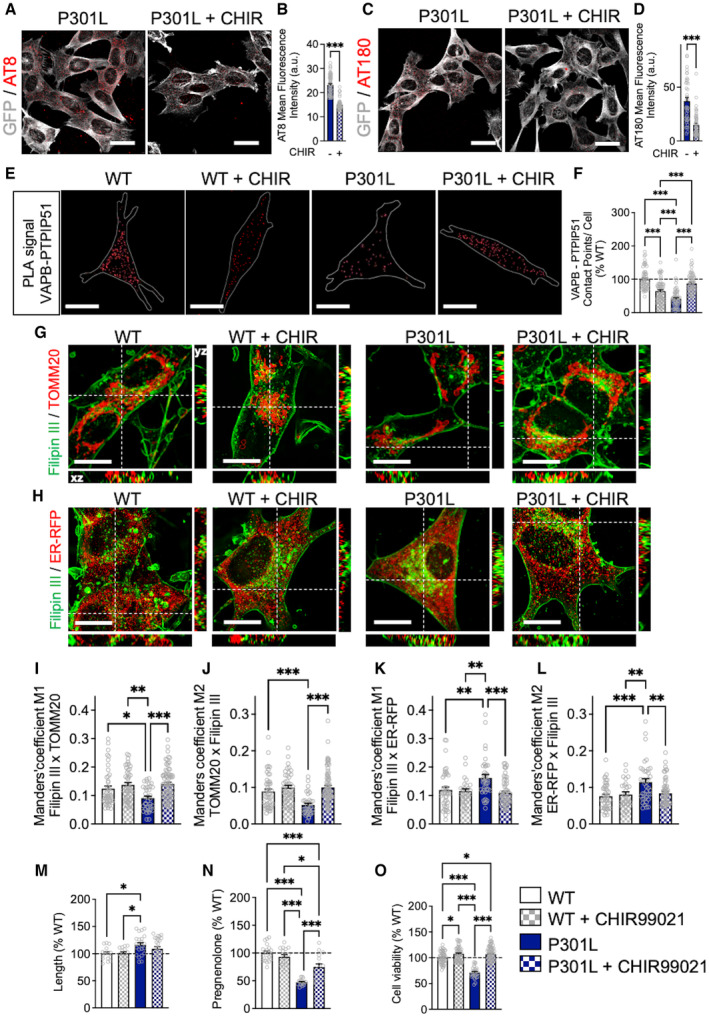
-
A–D(A, C) Representative microscopy images (z‐projections) of tau phosphorylation using (A) AT8 and (C) AT180 antibodies (in red) in GFP‐tagged P301L cells (GFP signal in gray) treated for 24 h with CHIR99021 (CHIR; right) and untreated cells (left). White scale bars: 25 μm. (B, D) Quantification of (B) AT8 and (D) AT180 staining fluorescence intensity. Data are presented as mean ± SEM of n = 50–60 cells per group (4 independent experiments). ***P < 0.001; Student unpaired t‐test.
-
EVisualization of the VAPB–PTPIP51 interactions by in situ proximity ligation assay (PLA) in untreated wild‐type (WT) cells and P301L cells as well as in CHIR‐treated cells. Interactions between the two targeted proteins are depicted in red (63X magnification) and cell surfaces are delimited by a white line. White scale bars: 20 μm.
-
FQuantitative analysis of the PLA signal represented as number of contact points between VAPB–PTPIP51 per cell in percentage of the WT cells. In total, 60 cells were analyzed per group (4 independent experiments).
-
G, HRepresentative microscopy images (z‐projections) of co‐staining with Filipin III in green and (G) TOMM20 (mitochondria) or (H) ER‐RFP (endoplasmic reticulum) in red with xy‐ and xz‐axis orthogonal views framing the corresponding image. White scale bars: 10 μm.
-
I–LManders' coefficients M1 representing the proportion of Filipin III overlapping with (I) TOMM20 or (K) ER‐RFP, and M2 representing the proportion of (J) TOMM20 or (L) ER‐RFP, overlapping with Filipin III in untreated WT cells and P301L cells as well as in CHIR‐treated cells. In total, 35–75 cells were analyzed per group (4 independent experiments).
-
MMitochondrial length in percentage of the WT cells. On average 500–1,500 mitochondrial organelles were analyzed per group (n = 15–20 images per group, 2 independent experiments).
-
NQuantification of pregnenolone levels in WT cells and P301L cells as well as in CHIR‐treated cells. Data are presented as mean ± SEM of n = 16–20 replicates per group (4 independent experiments) in percentage of the WT cells.
-
OAssessment of cell viability in WT cells and P301L cells as well as in CHIR‐treated cells.
Data information: Data are presented as mean ± SEM of n = 50 replicates per group (5 independent experiments) in percentage of the WT cells. (F, I–O) * P < 0.05, ** P < 0.01, ***P < 0.001; One‐way ANOVA + Tukey's post hoc test. ER‐RFP, endoplasmic reticulum red fluorescent protein; PTPIP51, protein tyrosine phosphatase‐interacting protein 51; TOMM20, translocase of the outer mitochondrial membrane complex subunit 20; VAPB, vesicle‐associated membrane protein‐associated protein B.
Source data are available online for this figure.
Similar to the effect of MFN2 KD, GSK3β inhibition also normalized cholesterol levels in mitochondria and the ER in P301L cells to those found in WT cells (Fig 6G–L). No effects of CHIR99021 were observed in WT cells with regard to cholesterol localization in the ER and mitochondria (Fig 6G–L).
Regarding mitochondrial network morphology, we only observed a trend to a decrease in mitochondrial length, form factor, and area‐weighted form factor between CHIR‐treated P301L cells and vehicle‐treated P301L (Figs 6M and EV3J and K). Of note, we also did not detect a significant difference between WT cells and CHIR‐treated P301L cells on parameters of the mitochondrial network morphology, indicating that GSK3β inhibition slightly alleviated the impact of abnormal tau on mitochondrial dynamics. GSK3β inhibition showed no effect on mitochondrial morphology in WT cells (Figs 6M and EV3J and K).
Nevertheless, treatment with CHIR99021 significantly increased pregnenolone levels in P301L cells compared with vehicle‐treated P301L cells (Fig 6N). Again, no effect was observed in CHIR99021‐treated WT cells. Targeted metabolomics revealed an overall increase in cholesterol‐related metabolites after treatment of P301L cells with CHIR99021 (Fig EV3L), in particular a significant increase in the level of the secondary bile acids GCDCA and TCDCA (Fig EV3M and N). This suggests that GSK3β inhibition can, at least partially, restore cholesterol metabolism in the P301L cells.
Finally, we asked whether CHIR99021 would improve the viability of P301L cells. Indeed, P301L cells present a decreased cell viability (−30%) compared with WT cells (Fig 6O). CHIR99021 increased the viability of P301L cells (+35%) and, to a lower extent, of WT cells (+7%).
Together, these data revealed that GSK3β inhibition decreased abnormal tau phosphorylation and increased ER‐mitochondria interaction in P301L cells, thereby restoring the transfer of cholesterol between both organelles and thus pregnenolone synthesis. The increase of P301L cell viability, observed after GSK3β inhibition, might therefore be due, in part, to a reduction in tau phosphorylation or an increase of pregnenolone that has neuroprotective properties, or a combination of both mechanisms.
Discussion
Our previous studies had shown that P301L‐tau impairs mitochondria bioenergetics, dynamics, and mitophagy in vitro and in vivo. Specifically, P301L‐tau inhibits mitochondrial complex I activity, decreases mitochondrial respiration, and reduces adenosine triphosphate (ATP) levels and the mitochondrial membrane potential, while the production of reactive oxygen species (ROS) is increased (David et al, 2005; Rhein et al, 2009; Schulz et al, 2012; Grimm et al, 2015). P301L‐tau also inhibits mitophagy by sequestering the key mitophagy protein Parkin in the cytosol (Cummins et al, 2018). Moreover, disease‐associated tau increases actin stabilization, leading to dynamin‐related protein 1 (DRP1) mislocalization, which promotes excessive mitochondrial elongation (DuBoff et al, 2012), and further clusters mitochondria around the nucleus (Schulz et al, 2012). In the present study, we showed that neuronal mitochondria are indeed more elongated in the amygdala and hippocampus (Ca1) of pR5 mice compared with WT littermates. This observation was also demonstrated in vitro, with P301L‐overexpressing cells showing an increase in mitochondrial length and a perinuclear clustering of mitochondria. On the contrary, Tau KO cells and iPSC‐P301L showed a more fragmented mitochondrial network, suggesting that the absence of tau or the presence of pathological tau at endogenous levels differently impacts mitochondrial shape. Indeed, P301L‐tau overexpression rather mimics the accumulation of pathological tau, which may lead to the disruption of the physical association of mitochondria and the mitochondrial fusion protein dynamin‐related protein 1 (DRP1), leading to mitochondrial elongation, as previously described (DuBoff et al, 2012).
Because abnormal tau impacts mitochondrial dynamics by disturbing mitochondrial shaping proteins (DuBoff et al, 2012; Li et al, 2016), and as these proteins are also involved in the ER‐mitochondria coupling, we decided to investigate the impact of disease‐associated tau on the MAMs. Indeed, increasing evidence shows the involvement of MAMs in different human pathologies, including neurodegenerative disorders (Pinton, 2018). To date, Aβ has received the bulk of attention regarding MAMs dysfunction in AD (Area‐Gomez et al, 2018), while studies focusing on tau are still scarce. In the present study, we demonstrate that abnormal tau interferes with ER‐mitochondria associations, leading to a decreased transfer of cholesterol from the ER to mitochondria, and a decrease in pregnenolone production.
Regarding the effects of abnormal tau on the MAMs, our findings differ from previous studies, which may be due to differences in the experimental model and the way how the ER‐mitochondria contact points were quantified. For example, by using electron microscopy, Perreault et al (2009) showed an increase in the ER‐mitochondria association in the spinal cord of JNPL3 mice expressing the same (P301L) mutation but under the control of the mouse prion promoter, that is expressed in glia and neurons, with the mice developing a very pronounced early motor phenotype. In contrast, we used a colocalization analysis that focused on the hippocampus and amygdala which are two brain regions heavily affected in pR5 mice that express the P301L mutation under the control of the murine Thy1 promoter and develop more subtle age‐related cognitive impairments. Likewise, Cieri et al (2018) showed that truncated tau (2N4RΔC20) increases short ER‐mitochondria interactions coupled with calcium dyshomeostasis. They used a split‐GFP‐based sensor (SPLICS) expressed in Hela cells to assess the ER‐mitochondria interactions in the short (8–10 nm) and long (40–50 nm) range, while we applied a PLA approach (detecting protein–protein interaction at distances < 40 nm) in SH‐SY5Y cells. Apart from differences in the technical approach, these findings suggest also that the effects of tau on the MAMs may differ depending on the tissue that is being investigated (hippocampus/amygdala versus spinal cord).
In support of our findings, by using PLAs with a total of three different pairs of proteins involved in the ER‐mitochondria interaction (IP3R–VDAC, IP3R–GRP75, and VAPB–PTPIP51), we revealed that the ER‐mitochondria association is strongly decreased in P301L‐expressing cells compared with control cells, at least via these markers. Of note, alterations in mitochondrial distribution and ER‐mitochondria contacts were also observed in wtTau cells, albeit to a lesser extent. As observed in previous studies, the effects of elevated tau levels are exacerbated in the presence of FTD‐associated tau mutations (Cummins et al, 2018; Evans et al, 2021). That tau has a direct impact on MAMs is supported by our finding, as we observed the exact opposite effect in Tau KO cells. Besides, data obtained in patient‐derived iPSCs bearing the P301L‐tau mutation, hence expressing endogenous levels of tau, suggested that the disruption of the ER‐mitochondria contacts is due to the P301L mutation and not to P301L‐tau overexpression. Finally, the artificial tethering of the MAMs under MFN2 KD conditions completely restored the number of ER‐mitochondria contact points in P301L cells to a level found in control cells, indicating that it is indeed possible to modulate the impact of tau on the MAMs.
Because cholesterol is enriched at the ER‐mitochondria contact sites, MAMs have been suggested to be important hubs for cholesterol synthesis and metabolism (Hayashi et al, 2009). Besides, the transfer of cholesterol from the ER to mitochondria is important for the synthesis of cholesterol‐derived metabolites, including steroids and secondary bile acids (Martin et al, 2014). We therefore assessed whether tau had an impact on cholesterol homeostasis and transport from the ER to mitochondria. Targeted metabolomics data obtained in Tau KO cells revealed that the absence of tau deeply affects cholesterol levels and metabolism, suggesting that tau per se influences cellular cholesterol homeostasis. In P301L cells, cholesterol seems to accumulate, and cholesterol‐related metabolites were overall reduced. Especially, the secondary bile acids GCDCA and TCDCA, whose synthesis requires enzymatic steps first in the ER and then in mitochondria, were significantly decreased in P301L cells compared with WT cells. Accordingly, levels of pregnenolone levels, whose synthesis requires the transfer of cholesterol within mitochondria, were also decreased in P301L cells and pR5 mice. These new data support those obtained in a previous study in which we reported that P301L‐tau affects pregnenolone level by impacting its synthesis (Grimm et al, 2019). Indeed, the downstream conversion of pregnenolone was blocked by the addition of trilostane and abiraterone, which inhibits the conversion of pregnenolone into progesterone or dehydroepiandrosterone, respectively. Therefore, compared with the wild‐type cells, the decrease in pregnenolone level measured in P301L cells was due to a decrease in pregnenolone synthesis and not a rapid conversion to other steroids. In line, lower neurosteroid levels were observed in the brains of demented patients (postmortem analysis). Namely, reduced levels of pregnenolone, as well as other neurosteroids, were found in the hippocampus, amygdala, and frontal cortex, which were negatively correlated with the occurrence of NFTs (Weill‐Engerer et al, 2002; Schumacher et al, 2003). Interestingly, impairments in neuronal cholesterol homeostasis have been demonstrated in different tauopathies, as evidenced by an accumulation of cholesterol in neurons bearing tau aggregates (Distl et al, 2001, 2003; Glöckner & Ohm, 2014). Of note, this effect was independent of the apolipoprotein E (APOE) gene, which encodes a cholesterol transporter protein, and is the strongest genetic risk factor for AD (Glöckner & Ohm, 2014). Interestingly, NFTs similar to those seen in AD are also observed in Niemann–Pick type C (NPC) disease, an inherited neurovisceral storage disorder characterized by the impairment of intracellular transport of cholesterol, and its trapping in the lysosomal compartment (Distl et al, 2003). Our study shows an accumulation of cholesterol in pR5 brains and P301L cells, as well as an accumulation of cholesterol in lysosomes and in the ER, while mitochondrial cholesterol levels were decreased compared with control cells. In fact, recent findings have connected cholesterol metabolism to tau pathology. Van der Kant and colleagues have shown that different drugs inhibiting cholesterol synthesis reduced tau phosphorylation in iPSC‐derived AD neurons (van der Kant et al, 2019). A link between membrane cholesterol levels and tau seeding has also been established. On the one hand, high levels of cholesterol at the plasma membrane seem to act as a barrier to tau entry and aggregation into cells, whereas reduced or missorted cholesterol (e.g., lysosome accumulation) seem to increase tau entering the cytosol and permit seeded aggregation (Tuck et al 2022).
Mitochondria‐associated ER membranes appear to be involved in steroidogenesis (Hayashi et al, 2009; Martin et al, 2014). Indeed, the number of ER‐mitochondria contact points was increased in response to hormone (human choriogonadotropin, hCG) treatment in mouse tumor Leydig cells, in parallel with an increase in progesterone levels (Issop et al, 2015). The steroidogenic acute regulatory protein (StAR) was shown to interact with VDAC at the MAMs to foster cholesterol into the adrenal and gonadal mitochondria to initiate steroidogenesis (Prasad et al, 2015). Interestingly, tau was also shown to interact with VDAC in the brains of AD patients and AD transgenic mice, causing mitochondrial dysfunction (Manczak & Reddy, 2012). Given that we also observed an interaction between abnormal tau and VDAC in our cellular model, we hypothesize that tau disturbs the binding of StAR to VDAC, leading to decreased steroidogenesis.
We tested whether artificially increasing the ER‐mitochondria interaction (using MFN2 siRNA) in P301L‐tau expressing cells could restore cholesterol transfer and steroidogenesis to the level of the control cells (Fig 4). By doing this, we could decrease the cholesterol level in the ER and increase it in mitochondria, but this was only paralleled with a slight improvement of pregnenolone synthesis in mutant tau cells. Besides, no increase in pregnenolone levels was observed in MFN2 siRNA‐transfected WT cells. These data indicate that increasing ER‐mitochondria contacts improves cholesterol transport in the presence of abnormal tau but the increase is not sufficient to restore pregnenolone levels. Of note, MFN2 is also involved in mitochondrial fusion and knocking down MFN2 leads to mitochondrial fragmentation. We previously showed that MFN2 KD disturbs the circadian variations of mitochondrial fusion/fission and circadian variations of pregnenolone synthesis, without affecting pregnenolone level per se (Witzig et al, 2020). These data suggested that it is the dynamic process of mitochondrial fusion that plays a role in mitochondrial steroidogenesis. In line, others have shown that MFN2 KD impairs mitochondrial fusion and steroid synthesis (with progesterone P4 level used as readout of steroid synthesis) upon cAMP stimulation, but not under basal (unstimulated) condition (Duarte et al, 2012).
Based on these observations, we decided to target both MAM tethering and tau hyperphosphorylation by pharmacologically inhibiting GSK3β. Of note, this enzyme is not only involved in abnormal tau phosphorylation but has also been linked to MAM function (Stoica et al, 2014; Gomez et al, 2015). Indeed, Stoica et al (2014) have shown that GSK3β regulates the VAPB–PTPIP51 interaction in a model of ALS/FTD. In their study, TDP‐43, a protein involved in the pathophysiology of this disease, activated GSK3β, which disrupted the VAPB–PTPIP51 interaction.
Intriguingly, in our study, GSK3β inhibition induced a reduction of the VAPB–PTPIP51 contact points in WT and Tau KO cells. Considering that GSK3β regulates numerous targets other than tau or the VAPB–PTPIP51 interaction via phosphorylation, CHIR‐mediated GSK3β inhibition may thus affect also several other cellular processes. For instance, GSK3β lies downstream of various major signal transduction pathways, including insulin, Wnt, Hedgehog, and Notch signaling and hence modulates the function of proteins involved in glycogen metabolism, transcriptional regulation, cellular transport, cell cycle, pluripotency, and cell fate (Kaidanovich‐Beilin & Woodgett, 2011; Woodgett, 2013). Regarding the canonical Wnt/β‐catenin signaling pathway, GSK3β as a central component controls the phosphorylation of β‐catenin and consequently enhances its proteasome‐dependent degradation. Accordingly, GSK3β inhibition might consecutively impact processes that are essential for cell proliferation, polarity, division, and movement (Patel & Woodgett, 2017; Law & Zheng, 2022). Moreover, a growing body of evidence indicates the participation of GSK3β in regulating apoptosis. In general, it is perceived that an increased GSK3β activity promotes apoptosis in a wide range of conditions, while GSK3β inhibition attenuates or prevents apoptosis (Jope & Johnson, 2004; Manji et al, 2009). Intriguingly, GSK3β has been linked to neuronal survival and it has been shown that an enhanced GSK3β activity is associated with neuronal apoptosis. Conversely, the inactivation of GSK3β stimulates neuroprotection, and hence, GSK3β activity appears to correlate inversely with neuronal viability (Kaytor & Orr, 2002). Interestingly, the inhibition of GSK3β was demonstrated to support various aspects related to mitochondrial function, thereby positively ameliorating mitochondrial impairments and thus activity (Yang et al, 2017). Several studies have shown that inhibition of GSK3β (using LiCl in a millimolar concentration range) reduced mitochondrial fusion (Chou et al, 2012; Wu et al, 2013; Huang et al, 2015). In our study, GSK3β inhibition did not induce significant changes in mitochondrial morphology. Here, we used the GSK3β inhibitor CHIR99021 at 100 nM, which might explain why we do not see a clear effect of mitochondrial morphology. Our data also showed that GSK3β inhibition increases the level of cholesterol metabolites and restores the level of cholesterol in the ER in P301L cells. However, pregnenolone level was not fully restored in P301L cells. To our knowledge, it is still unknown whether and how GSK3β influences cholesterol homeostasis and steroidogenesis. Therefore, this important point deserves to be investigated in future studies.
It was recently shown that tau accumulation also activates GSK3β, leading to a vicious cycle between tau hyperphosphorylation and GSK3β activation (Zhou et al, 2022). This might explain why we find an increased ER‐mitochondria association, an increased cholesterol transfer from the ER to mitochondria, and an increase in pregnenolone synthesis only in GSK3β‐inhibited P301L cells but not WT cells. P301L‐tau is overexpressed in our P301L SH‐SY5Y cells, which would activate GSK3β, leading to a decrease in the ER‐mitochondria association, which is restored after GSK3β inhibition. In healthy conditions (WT cells), GSK3β inactivation might act on other pathways, like those mentioned above, leading to a decreased ER‐mitochondria association. This assumption needs to be investigated in more detail in further studies. Overall, these findings emphasize the therapeutic potential of GSK3β inhibition in tauopathies.
Based on the recent findings showing that tau accumulation activates GSK3β (Zhou et al, 2022) and factoring in the data presented here, we propose a model in which abnormal tau activates GSK3β, leading to the disruption of VAPB–PTPIP51 association and the loosening of the ER‐mitochondria associations (Fig 7). As a direct consequence, the cholesterol transfer between the two organelles is decreased and the mitochondrial cholesterol metabolism (including pregnenolone and secondary bile acid synthesis) is reduced. GSK3β inhibition reduces tau phosphorylation and increases the number of ER‐mitochondria contact points (VAPB–PTPIP51 interaction), paralleled by an increased cholesterol transfer from the ER to mitochondria, increased pregnenolone synthesis, and an improvement of cell viability (Figs 6 and 7). Other aspects of MAM's function, such as the effect on calcium homeostasis, require attention in future experiments as GSK3β seems also to play a role in the calcium exchange between the ER and mitochondria (Gomez et al, 2015). Importantly, pregnenolone is one of the most abundant neurosteroids in the brain, as well as the precursor of all neurosteroids (Lin et al, 2022). It has been shown to play a role in important brain functions, ranging from neurotransmission, inflammation, and myelinization to neuroplasticity, learning, and memory (Lin et al, 2022; Ramírez et al, 2022). Therefore, alterations in pregnenolone levels caused by abnormal tau may be involved more generally in neuronal and phenotypic impairments observed in AD‐related tauopathy.
Figure 7. Abnormal tau protein loosens endoplasmic reticulum (ER)–mitochondria associations and impairs cholesterol transfer to mitochondria.
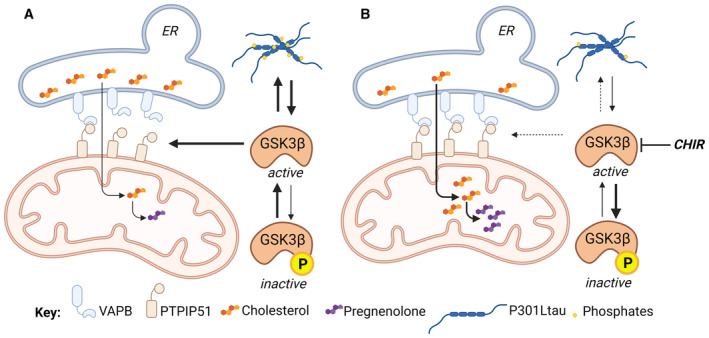
-
AP301L‐tau activates GSK3β, which is involved in abnormal tau hyperphosphorylation. GSK3β activation disrupts the binding of VAPB to PTPIP51, reducing ER‐mitochondria associations and cholesterol transfer between the two organelles. The decrease of mitochondrial cholesterol levels then leads to a reduction of pregnenolone synthesis.
-
BInhibition of GSK3β with CHIR decreases abnormal tau hyperphosphorylation and restores the binding of VAPB to PTPIP51 in the presence of mutant tau. The increase of ER‐mitochondria associations allows the transfer of cholesterol between the two organelles and its conversion to pregnenolone.
CHIR, CHIR99021 (GSK3β inhibitor); ER, endoplasmic reticulum; GSK3β, glycogen synthase kinase 3β; PTPIP51, protein tyrosine phosphatase‐interacting protein 51; VAPB, vesicle‐associated membrane protein‐associated protein B. Created with BioRender.com
In summary, our findings describe a novel pathogenic mechanism for disease‐associated tau, by which tau impairs steroidogenesis via disruption of ER‐mitochondria associations. Given that MAMs have been presented as a therapeutic target in neurodegenerative disorders, our findings extend this notion to tauopathies.
Limitations of the study
There are noteworthy limitations of the study. First, we focused on P301L‐tau, mainly because mice bearing this mutation present NFTs similar to those observed in the brain of AD patients. Future work will focus on assessing whether the features we observed are also present for other tau mutations. Additionally, although SH‐SY5Y cells are human neuron‐like cells, studies on advanced cellular models like human iPSCs‐derived neurons will allow for a more relevant translation to the human brain physiology. It would also be pertinent to confirm our findings in brain samples from patients with tauopathies. Besides, neurons are not the only cell‐type synthetizing neurosteroids, as glial cells also have this ability. Because tau was recently shown to accumulate in hilar astrocytes of the dentate gyrus of AD patients (Richetin et al, 2020), this cell type deserves future attention regarding MAM function, cholesterol homeostasis and steroidogenesis. The effects of tau on cholesterol metabolism also deserve to be studied in more depth. Indeed, as already mentioned, a study highlighted cholesterol metabolism as a druggable axis reducing aberrant tau phosphorylation (van der Kant et al, 2019). The interrelationship between cholesterol and tau still needs to be unraveled. Finally, more studies must be done to understand the role of GSK3β in the ER‐mitochondria interaction in healthy and pathological (presence of abnormal tau) conditions.
Materials and Methods
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| Human: wild‐type (WT), wild‐type tau (wtTau) and P301L‐Tau‐transfected SH‐SY5Y neuroblastoma cells | The Götz Laboratory | |
| Human: Tau knockout SH‐SY5Y neuroblastoma cells and corresponding wild‐type cells | The Paganetti Laboratory | Sola et al (2020) |
| Human: Induced pluripotent stem cells (iPSC) from a patient bearing a P301Ltau mutation and the corresponding CRISPR/Cas9‐edited isogenic wild‐type control iPSCs | The Karch Laboratory | F0510.2, F0510.2Δ2 (Karch et al, 2019) |
| Experimental models: organisms/strains | ||
| Mouse: pR5 (P301L) | The Götz Laboratory | Götz et al (2001) |
| Mouse: mitoCFP | Jackson Laboratories, USA | B6.Cg‐Tg(Thy1‐CFP/COX8A)C1Lich/J (Misgeld et al, 2007) |
| Mouse: mitoCFP+/P301L+ (mitoCFP x pR5) | The Götz Laboratory | |
| Antibodies | ||
| Anti‐AT180 | Thermofisher | MN1040 |
| Anti‐AT8 | Thermofisher | MN1020 |
| Anti‐calnexin | Abcam | ab22595 |
| Anti‐EEA1 Alexa Fluor® 647 | Abcam | ab196186 |
| Anti‐GFP | Abcam | ab13970 |
| Anti‐GM130 Alexa Fluor® 647 | Abcam | ab195303 |
| Anti‐GRP75 | Santa Cruz Biotechnology | sc‐133137 |
| Anti‐IP3R1 | Merck Millipore | # 07‐1213 |
| Anti‐M6PR (cation independent) Alexa Fluor® 647 | Abcam | ab198324 |
| Anti‐PTPIP51 | Proteintech | 16850215 |
| Anti‐Tau (phospho T231) | Abcam | ab151559 |
| Anti‐TOMM20 Alexa fluor® 555 | Abcam | ab221292 |
| Anti‐β‐tubulin, class III, Alexa Fluor® 488 mouse | BD Pharmingen™ | # 560381 |
| Anti‐VAPB | Proteintech | 16827845 |
| Anti‐VDAC1 | Abcam | ab14734 |
| Donkey anti‐rabbit Alexa fluor® 647 | Abcam | ab150075 |
| Goat anti‐rabbit Alexa fluor® 488 | Thermofisher | A11034 |
| Goat anti‐mouse Alexa fluor® 568 | Thermofisher | A11031 |
| Goat anti‐mouse Alexa fluor® 488 | Thermofisher | A11029 |
| Chemicals, peptides, and recombinant proteins | ||
| CHIR 99021 | MedChemExpress | HY‐10182 |
| DAPI | Merck | 10236276001 |
| Formaldehyde | Sigma‐Aldrich | F8775 |
| Glycine | Sigma‐Aldrich | G8898 |
| LipidSpot™ 610 Lipid Droplet Stain | Biotium | BIO70069‐T |
| Rat tail Collagen I coated German glass coverslips | Neuvitro Corporation | GG‐18‐15‐Collagen |
| Triton X‐100 | Sigma‐Aldrich | T8787 |
| Xfect transfection reagent (Standard) | Takara | 631317 |
| Xfect transfection reagent (RNA) | Takara | 631450 |
| NaCl | Merk | 1.06404.1000 |
| Na2HPO4 | Fluka | 71629 |
| KH2PO4 | Sigma‐Aldrich | P‐5655 |
| KCl | Sigma‐Aldrich | P9541 |
| Glucose | Sigma‐Aldrich | G‐5146 |
| Saccharose | Eurobio | 18363 |
| Mannitol | Sigma‐Aldrich | 114125 |
| Hepes | GibcoBRL | 11344‐033 |
| EDTA | Sigma‐Aldrich | E6758 |
| BSA | Sigma‐Aldrich | A9647 |
| DTT | Calbiochem | 233155 |
| MTT | Sigma‐Aldrich | M2128 |
| DMSO | Sigma‐Aldrich | 276855 |
| PFA | Sigma‐Aldrich | P6148 |
| Chemicals, enzymes, and other reagents | ||
| DMEM | Invitrogen | D6429 |
| Fetal bovine serum (FBS) | Corning | 35‐079‐CV |
| Horse serum (HS) | Bioconcept | 2‐05F00‐1 |
| Penicillin/Streptomycin | Bioconcept | 4‐01F00‐5 |
| GlutaMax | Thermo Fisher Scientific | 35050087 |
| Blasticidin | InvivoGen | Ant‐bl‐1 |
| Opti‐MEM™ Reduced Serum Medium | Thermo Fisher Scientific | 31985070 |
| 5x Roche complete protease inhibitor | Sigma‐Aldrich | 11697498001 |
| T150 flasks | TPP | 90151 |
| PBS | Dominique Dutscher | X0520‐500 |
| Collagen I, Rat Tail, 100 mg | Corning | 354236 |
| Cellartis DEF‐CS 500 Culture System | Takara Bio | Y30010 |
| Cellartis DEF‐CS 500 COAT‐1 | Takara Bio | Y30012 |
| Cellartis DEF‐CS additives GF‐1, GF‐2, GF‐3 | Takara Bio | Y30016 |
| TripLE™ Select Enzyme | Thermo Fisher Scientific | 12605010 |
| Vectashield H‐1000 mounting medium | Vector | H‐1000 |
| Critical commercial assays | Thermo Fisher Scientific | |
| Amplex® Red Cholesterol Assay Kit | Invitrogen | A12216 |
| Pregnenolone EIA | DRG | EIA‐4170 |
| Duolink® In Situ Red Starter Kit Mouse/Rabbit (proximity ligation assay) | Sigma‐Aldrich | DUO92101 |
| Duolink® In Situ Detection Reagents Far Red | Sigma‐Aldrich | DUO92013 |
| Cholesterol Assay Kit (Cell‐Based) | Abcam | ab133116 |
| Xfect transfection reagent (Standard) | Takara | 631317 |
| Xfect transfection reagent (RNA) | Takara | 631450 |
| RNeasy Mini Kit (250) | Qiagen | 74106 |
| GoScript™ Reverse Transcription Mix, Oligo(dT) | Promega | A2791 |
| GoTaq® Probe qPCR Master Mix | Promega | A6102 |
| MycoAlert® PLUS Mycoplasma Detection Kit | Lonza | LT07‐701 |
| Oligonucleotides | ||
| AllStars Negative Control siRNA | Qiagen | Cat. #SI03650318/Prod. #1027417 |
| Hs_MFN2_5 FlexiTube siRNA | Qiagen | SI04188835 |
| Hs_MFN2_8 FlexiTube siRNA | Qiagen | SI04375406 |
| Primers and Probes for RT–qPCR (see corresponding table) | Microsynth AG | Custom‐made |
| Recombinant DNA | ||
| ER‐RFP | Addgene | 62236 |
| Software and algorithms | ||
| Prism version 9.2.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji (Fiji is just ImageJ) | NIH | https://imagej.net/software/fiji/ |
| JACoP (Just Another Colocalization Plugin) | NIH | https://imagej.nih.gov/ij/plugins/track/jacop.html |
| Huygens Deconvolution Software | Scientific Volume Imaging | https://svi.nl/Huygens-Software |
| MetaboAnalyst 5.0 | MetaboAnalyst | https://www.metaboanalyst.ca/home.xhtml |
| BioRender | BioRender.com | |
Methods and Protocols
Mice
All animal experiments were conducted with mice under the guidelines of the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes and approved by the University of Queensland Animal Ethics Committee (QBI/412/14/NHMRC; QBI/554/17). Male and female transgenic mice and wild‐type littermates were housed in standard conditions (mice were maintained on a 12 h light/dark cycle), in group cages (housed 3–5 mice per cage) with ad libitum access to food and water.
pR5 mice express 2N4R tau with the P301L mutation under the control of the mThy.1.2 promoter (Götz et al, 2001). Neurofibrillary tangles (NFT) formation is initiated in the amygdala and also found in the hippocampus (Götz & Ittner, 2008; Fig 1A).
MitoCFP mice express Cyan Fluorescent Protein (CFP) under the control of the mouse Thy1 promoter. CFP is specifically localized to the mitochondria by a human cytochrome c oxidase, subunit 8A (ubiquitous), targeting signal fused to the N terminus (Misgeld et al, 2007). MitoCFP mice were crossed with pR5 mice, and the genotype of MitoCFP+/P301L+ animals was verified. These mice present abnormal tau hyperphosphorylation in the same neurons as those expressing the mitoCFP, especially in the amygdala and the hippocampus by 12–14 months of age (Fig 1A). Therefore, this unique mouse model allows studying the impact of P301L‐mutant tau on mitochondrial network morphology and the ER‐mitochondria association in neurons specifically.
The mice were sacrificed between 12.5 and 14 months of age, when tau pathology is well present in the Ca1 region of the hippocampus and the amygdala (Fig 1A). The mice were anesthetized and transcardially perfused with PBS. Brains were either snap frozen in liquid nitrogen for biochemical studies or immersion fixed in 4% paraformaldehyde for immunostaining. The number of animals used for brain immunostaining was N = 12 MitoCFP+/P301L− and N = 13 MitoCFP+/P301L+. The number of animals used for biochemical analysis was N = 6 wild‐type (WT) and N = 5 pR5 (P301L+).
SH‐SY5Y cell culture
Human SH‐SY5Y cells were grown in DMEM containing 10% heat‐inactivated fetal bovine serum (FBS), 5% heat‐inactivated horse serum, 1% penicillin/streptomycin, and 1% GlutaMax. The cells were kept in culture in 10 cm2 dishes, split twice a week, and plated when they reached around 80% confluence.
P301L‐expressing SH‐SY5Y human neuroblastoma cells were generated using lentiviral gene transfer (Krupka et al, 2010). To the culture medium, 4.5 μg/ml blasticidin was added to select cell clones stably expressing the full‐length human hTau40 bearing the P301L mutation and a green fluorescent protein (GFP) tag, or cells expressing wild‐type tau (full‐length human hTau40, wtTau), or cells expressing the GFP‐vector only (vector cells). Of note, this specific cellular model stably expressing wild‐type tau (wtTau cells) or P301L‐tau (P301L cells) was used to avoid artifacts due to a transient protein expression which can be a cellular stress affecting mitochondrial network morphology.
Tau knockout (Tau KO) SH‐SY5Y cells were generated by disruption of the MAPT gene encoding for tau by the CRISPR‐Cas9 method, using the gRNAs 232 targeted exon 2 containing the initiating ATG (ENST00000344290.9) as previously described (Sola et al, 2020). Tau KO cells were characterized, and the absence of tau was confirmed by fluorescent confocal microscopy and immune protein blotting (Sola et al, 2020).
All cell lines were regularly tested for Mycoplasma contamination using the MycoAlert® PLUS Mycoplasma Detection Kit.
Human iPSCs culture
Human mutant (MAPT P301L/WT) and CRISPR/Cas9‐edited, isogenic control (MAPT WT/WT‐iso) iPSCs were kindly provided by Dr. Celeste Karch and the Tau Consortium Stem Cell Group (Karch et al, 2019). The iPSCs were maintained under feeder‐free conditions and cultured on Cellartis DEF‐CS COAT‐1‐coated plates in the Cellartis DEF‐CS basal medium, containing the Cellartis DEF‐CS additives GF‐1 (1:333) and GF‐2 (1:1,000). The iPSCs were kept in a humidified incubator at 37°C and 5% CO2 with medium replacement every day and passaged twice a week using TrypLE Select, as described in the manufacturer's protocol (Takara). To minimize cell death while passaging or plating, additionally, the Cellartis DEF‐CS additive GF‐3 was added (1:1,000) to the medium. For mitochondria isolation, as well as pregnenolone and cholesterol level determination, iPSCs were plated each into two wells of a Cellartis DEF‐CS COAT‐1‐coated 6‐well cell culture plate. After reaching 90% confluency, iPSCs were washed once with PBS and detached using TrypLE Select. Then, the samples were centrifuged for 5 min at 4°C at 600 rpm, followed by aspiration of the supernatant. Subsequently, the cell pellets were resuspended in PBS, centrifuged again for 5 min at 4°C at 600 rpm, followed by aspiration of the supernatant, and successive freezing of the cell pellet. Each cell pellet contained between 7 and 8 × 106 cells.
Mouse brain lysates
After removing the cerebellum, mouse brain hemispheres were crushed in 250 μl of ice‐cold buffer (138 mM NaCl, 5.4 mM KCl, 0.17 mM Na2HPO4, 0.22 mM KH2PO4, 5.5 mM glucose, 58.4 mM saccharose, 5× Roche complete protease inhibitor) with a glass homogenizer (5 strokes, 400 rpm). After this homogenization step, 75 μl of the preparation was transferred in a 500‐μl Eppendorf tube for analysis of the brain lysates (i.e., for cholesterol and pregnenolone quantification) and sonicated (2 s). The rest of the preparation was kept for mitochondrial isolation.
After the determination of protein concentration, brain lysates were normalized on 5 mg/ml of protein using PBS before measurement of the pregnenolone concentration and on 0.1 mg/ml for cholesterol content determination using the reaction buffer from the Amplex® Red Cholesterol Assay Kit.
Mitochondrial isolation
SH‐SH5Y cells were plated in T150 flasks at a density of 2 × 106 cells per flask and allowed to grow for 48 h. After washing with PBS, cells were mechanically detached using a cell scrapper and put in 1 ml of mitochondria isolation buffer (MIB: 210 mM mannitol, 70 mM sucrose, 10 mM Hepes, 1 mM EDTA, 0.45% BSA, 0.5 mM DTT, and 5× Roche complete protease inhibitor). Mouse brain lysates and iPSC pellets were also put in 750 μl of MIB. Cells and brain lysates were homogenized with a glass homogenizer (10–15 strokes, 400 rpm), transferred in 2 ml tubes, and centrifuged at 1,450 g for 7 min at 4°C to remove nuclei and tissue particles. Supernatants were transferred in new 2 ml tubes, while the homogenization/centrifugation steps were repeated on pellets (in 1 ml MIB). Combined supernatants were again centrifuged at 1,450 g for 3 min at 4°C. The resulting supernatant fractions were centrifuged at 10,000 g for 5 min at 4°C to pellet mitochondria. The resulting pellets were resuspended in 1 ml of MIB and centrifuged at 1,450 g for 3 min at 4°C to remove debris. The mitochondria‐enriched supernatants were centrifuged at 10,000 g for 5 min at 4°C to obtain the mitochondrial fraction. This fraction was resuspended in 80 μl of PBS, followed by determination of the protein content. Isolated mitochondria were normalized on 0.03 mg/ml of protein before cholesterol content determination using the reaction buffer from the Amplex® Red Cholesterol Assay Kit.
Cholesterol level
SH‐SY5Y cells were plated in DMEM +1% penicillin/streptomycin +1% GlutaMax +10% FBS +5% HS in 6 cm dishes at a density of 3 × 105 cells per dish. After 48 h growing, cells were washed with PBS, harvested in 250 μl PBS using a cell scrapper, and lysed by sonication (2 s). After the determination of protein concentration, cell lysates were normalized on 0.1 mg/ml. Determination of cholesterol concentration was performed in SH‐SY5Y cell lysates, iPSC lysates, and mouse brain lysates, as well as on isolated mitochondria. Determination of cholesterol concentration was performed using the Amplex® Red Cholesterol Assay Kit according to the instructions of the manufacturer. Briefly, 50 μl of cholesterol standard or the diluted samples (brain/cell lysates, isolated mitochondria) was pipetted into separate wells of a 96‐well microplate. The reaction was initiated by adding 50 μL of a mix containing the 300 μM Amplex® Red reagent, 2 U/ml HRP, and 2 U/ml cholesterol oxidase to each well. Plates were incubated at 37°C for 15 min, and the fluorescence intensity was measured (excitation: 530 nm—emission: 590 nm) using Cytation 3 Cell Imaging Multi‐Mode reader (BioTek, Luzern, Switzerland). Background fluorescence (from no‐cholesterol wells) was subtracted, and cholesterol concentration was calculated using the cholesterol standard curve.
Pregnenolone quantification
SH‐SY5Y cells were plated in DMEM +1% penicillin/streptomycin +1% GlutaMax +10% FBS +5% HS in 6 cm dishes at a density of 3 × 105 cells per dish. After 48 h growing, cells were washed with PBS, harvested in 250 μl PBS using a cell scrapper, and lysed by sonication (2 s). Pregnenolone concentration was quantified in from SH‐SY5Y cell lysates, iPSC lysates, and mouse brain lysates with a pregnenolone direct ELISA according to instructions of the manufacturer. Briefly, 50 μl of samples or pregnenolone standard was pipetted in the 96‐well ELISA plates. After adding 100 μl of the pregnenolone‐HRP conjugate solution, plates were incubated on a shaker (200 rpm) for 1 h at room temperature. Wells were washed three times with 300 μl of wash buffer, before adding 150 μl of TMB substrates. After 15‐min incubation, 50 μl of stop solution was added and absorbance was read at 450 nm using a Cytation 3 Cell Imaging Multi‐Mode reader (BioTek, Luzern, Switzerland). Pregnenolone concentration was calculated using the pregnenolone standard curve.
Targeted metabolomics on cholesterol‐related metabolites
Targeted metabolomics was performed by Biocrates Life Science (Innsbruck, Austria). Cells were plated in DMEM +1% penicillin/streptomycin +1% GlutaMax +10% FBS in 10 cm dishes until they reached 80% confluency. The cell medium was then removed, and cells were washed with cooled 0.9% NaCl solution. After removal, 1–1.5 ml of 0.9% NaCl solution was added to the plate and cells were quickly detached with a cell scrapper. The cell suspension was then transferred into a 2‐ml Eppendorf tube. A small aliquot was kept for cell counting and protein determination, and cells were centrifuged at 500 g for 5 min at 4°C. The supernatant was removed, and the cell pellet was rapidly frozen in liquid nitrogen and stored at −80°C until shipment to Biocrates. The number of cells per sample was between 1 and 3 × 106 cells. For measurement of metabolite concentrations, 50 μl extraction buffer was added to each sample, which was then vortexed and sonicated twice for 3 min on ice followed by freezing in liquid nitrogen for 30 s. After a third round of sonication, samples were centrifuged, and the supernatant was collected for analysis.
Biocrates' commercially available MxP® Quant 500 kit was used for the quantification of several endogenous metabolites of various biochemical classes, including cholesteryl esters and bile acids. With this kit, lipids and hexoses were measured by flow injection analysis‐tandem mass spectrometry (FIA‐MS/MS) using a triple quadrupole mass spectrometer with an electrospray ionization (ESI) source, and small molecules are measured by liquid chromatography–tandem mass spectrometry (LC–MS/MS) also using triple quadrupole mass spectrometry. The experimental metabolomics measurement technique is described in detail by patents EP1897014B1 and EP1875401B1. Briefly, a 96‐well‐based sample preparation device is used to quantitatively analyze the metabolite profile in the samples. This device includes prepipetted internal standards, and a predefined sample amount is added to the device. Next, a phenyl isothiocyanate (PITC) solution is added to derivatize some of the analytes (e.g., amino acids), and after the derivatization, the target analytes are extracted with an organic solvent, followed by a dilution step. The obtained extracts are then analyzed by FIA‐MS/MS and LC–MS/MS methods using multiple reaction monitoring (MRM) to detect the analytes. Concentrations are calculated using appropriate mass spectrometry software (Sciex Analyst®), and data are imported into Biocrates' MetIDQ™ software for further analysis.
Free oxysterols were extracted from samples with methanol using Biocrates kit filter plate loaded with an internal standard mixture beforehand: The metabolites were determined by UHPLD‐MS/MS with multiple reaction monitoring (MRM) in positive mode using SCIEX API 5500 QTRAP® (AB SCIEX, Darmstadt, Germany) instrument with electrospray ionization (ESI). Metabolite concentrations were then normalized on the protein content of each sample, and data analysis was performed using MetaboAnalyst 5.0. In total, 55 cholesterol‐related metabolites were assessed in SH‐SY5Y cell lysates, including 19 oxysterols, 22 cholesteryl esters, and 14 bile acids (Dataset EV1). Only 17 metabolites were found above the limit of detection (LOD) of the assay. Variables with missing values (< LOD) were excluded LOD.
Total RNA isolation, cDNA synthesis, and quantitative real‐time PCR
To characterize the ER‐mitochondria interaction, the expression levels of genes coding for proteins enriched at MAMs were investigated. The different cell lines were seeded in 6‐well plates at a density of 150,000 cells per well. After 48 h, the cells were washed once with PBS, lysed with RLT buffer, and subsequently homogenized for at least 30 s at 400 min−1 using the rotor‐stator homogenizer Potter S (B. Braun Biotech). Total RNA was isolated from homogenized cell lysates using the RNeasy Mini Kit according to the manufacturer's instructions. RNA concentrations were assessed with a Nanodrop 1000 spectrophotometer (Thermo Scientific). To synthesize cDNA, the volume containing 1 μg of RNA was reversely transcribed with the GoScript™ Reverse Transcription Mix, Oligo(dT) as described by the manufacturer. After reverse transcription, all cDNA samples were diluted 1:5 in nuclease‐free water. For the amplification of cDNA samples, the GoTaq® Probe qPCR Master Mix was used with the addition of 500 nM CXR reference dye. Along with the GoTaq® Probe qPCR Master Mix, each reaction contained 250 nM hydrolysis probe, 900 nM forward and reverse primer (see Table 1 below), which were combined in 17 μl, as well as 3 μl diluted cDNA template. Afterward, the plate was centrifuged briefly with an MPS 1000 Mini plate spinner (Labnet). Quantitative real‐time PCR reactions were performed in technical duplicate using the StepOnePlus™ Real‐Time PCR System (Applied Biosystems) under the following conditions: for 2 min at 95°C, for 3 s at 95°C followed by 30 s at 60°C, for a total of 40 cycles. The final quantification of relative fold gene expression was calculated based on the method with GAPDH as an endogenous reference.
Table 1.
List of PCR primers used in this study.
| Genes | Primers/ Probes | Sequences |
|---|---|---|
| ATAD3A |
Forward Reverse Probe |
5′‐GAGCCACCGAGAAGATAAG‐3′ 5′‐GACCAGCATGAACTTGTTG‐3′ 5′‐FAM‐CCACACTGAACGCCTTCCTG‐MGB‐Q530‐3′ |
| BAP31 |
Forward Reverse Probe |
5′‐CTCCTAAAAGATGGCAGAAGA‐3′ 5′‐GGCATCGATGACCAACAG‐3′ 5′‐FAM‐TTTCAAGTCCCGGCTGGT‐MGB‐Q530‐3′ |
| CANX |
Forward Reverse Probe |
5′‐AGCCTCTCATTGTTCAGTATG‐3′ 5′‐GATCCAGGTTGAGTTCTGG‐3′ 5′‐FAM‐TTCACATAGGCACCACCACA‐MGB‐Q530‐3′ |
| CYPD |
Forward Reverse Probe |
5′‐GACAGTTGTTTAGAGGCTCTTG‐3′ 5′‐CAGCCAATGCTTGATCATATTC‐3′ 5′‐FAM‐CCATCCTTGAGCTCTGCGGT‐MGB‐Q530‐3′ |
| DRP1 |
Forward Reverse Probe |
5′‐TCACGAGACAAGTCTTCTAAAG‐3′ 5′‐CCTCCAGATGCAACCTTG‐3′ 5′‐FAM‐TCAGCCTCAGCAGAAGCAGC‐MGB‐Q530‐3′ |
| FACL4 |
Forward Reverse Probe |
5′‐CTGCAAATGCCATGAAATTG‐3′ 5′‐GCTCCTTCCTTTTCAGTTTG‐3′ 5′‐FAM‐CAAACCAGTTTCAGGGGTCC‐MGB‐Q530‐3′ |
| FIS1 |
Forward Reverse Probe |
5′‐GCTCAAGGAATACGAGAAGG‐3′ 5′‐AGTCCATCTTTCTTCATGGC‐3′ 5′‐FAM‐AGCCGCTCCAGTTCCTTGG‐MGB‐Q530‐3′ |
| GAPDH |
Forward Reverse Probe |
5′‐CATGGTTTACATGTTCCAATATGA‐3′ 5′‐GGATCTCGCTCCTGGAAG‐3′ 5′‐FAM‐CATGGCACCGTCAAGGCTG‐MGB‐Q530‐3′ |
| GRP75 |
Forward Reverse Probe |
5′‐GGCGGGATTATGCATCAG‐3′ 5′‐CTCCAGCACCTTTGCTTG‐3′ 5′‐FAM‐ACCTTCCATAACTGCCACGC‐MGB‐Q530‐3′ |
| GRP78 |
Forward Reverse Probe |
5′‐CGAGAACACGGTCTTTGA‐3′ 5′‐GGGTAACCTTCTTTCCCAAA‐3′ 5′‐FAM‐CCAAGCGGCTCATCGGC‐MGB‐Q530‐3′ |
| IP3R |
Forward Reverse Probe |
5′‐CTTGGCAGCAGAGGTAGA‐3′ 5′‐CCATCTTTTCTTGGGCATTC‐3′ 5′‐FAM‐ACCTACTTCGAGAGGCGTCC‐MGB‐Q530‐3′ |
| MFN1 |
Forward Reverse Probe |
5′‐CAGTCACCAAGTAAAACAACAAA‐3′ 5′‐GGGTAATCTAGCAATTTCTTCTTC‐3′ 5′‐FAM‐TGCTCGCCTGTGCCAAC‐MGB‐Q530‐3′ |
| MFN2 |
Forward Reverse Probe |
5′‐CAGGATTCAGAAAGCCCAG‐3′ 5′‐GATGCACTCCTCAAATCTCC‐3′ 5′‐FAMCGCTCTCGCAGAAGGCTT‐‐MGB‐Q530‐3′ |
| OPA1 |
Forward Reverse Probe |
5′‐CCAGGTGTGATTAATACTGTGA‐3′ 5′‐CCATCTTGAATACACAGTATGATG‐3′ 5′‐FAM‐AGGCATGGCTCCTGACACAA‐MGB‐Q530‐3′ |
| PACS2 |
Forward Reverse Probe |
5′‐GCCAAGTCCACGGATAAC‐3′ 5′‐CTTCCCCACGTCAAAGTC‐3′ 5′‐FAM‐TCCTCGTCCAAGTCCTGCC‐MGB‐Q530‐3′ |
| PERK |
Forward Reverse Probe |
5′‐GTGCATATAGTGGAAAGGTGAG‐3′ 5′‐ACCGAAGTTCAAAGTGGC‐3′ 5′‐FAM‐TCATTGCCACTGCGAGGTCC‐MGB‐Q530‐3′ |
| PTPIP51 |
Forward Reverse Probe |
5′‐GGGTATATATTTCCAAGTGCTACA‐3′ 5′‐GTCCTTCTGGATAGCCAAATC‐3′ 5′‐FAM‐CCTTCGTGACATCTGGCAGC‐MGB‐Q530‐3′ |
| SIGMA1R |
Forward Reverse Probe |
5′‐TCTCGGATACCATCATCTCTG‐3′ 5′‐AAGGGTCCTGGGTAGAAG‐3′ 5′‐FAM‐TTGGTGGTGCCCTCTCTCCA‐MGB‐Q530‐3′ |
| SOAT |
Forward Reverse Probe |
5′‐ACAGGATGTTCTATAAGGATTGG‐3′ 5′‐ACCAGAGAAAGTCCTTGTAAG‐3′ 5′‐FAM‐AGCCAGTCATGGACCACCACATTCC‐MGB‐Q530‐3′ |
| StAR |
Forward Reverse Probe |
5′‐CATCGACCTCAAGGGGTG‐3′ 5′‐CAGGTGGTTGGCAAAATCC‐3′ 5′‐FAM‐CCAAGAGCATCATCAACCAGGTCCT‐MGB‐Q530‐3′ |
| TSPO |
Forward Reverse Probe |
5′‐CACTCAACTACTGCGTATG‐3′ 5′‐CGGGCACTCACTCTG‐3′ 5′‐FAM‐ACAACCATGGCTGGCGT‐MGB‐Q530‐3′ |
| VAPB |
Forward Reverse Probe |
5′‐GCCTCAATTAATGTATCTGGAAGA‐3′ 5′‐GGGCTAATGCTGAAATGGG‐3′ 5′‐FAM‐TCTTCCTCATCCGCAGTCCA‐MGB‐Q530‐3′ |
| VDAC |
Forward Reverse Probe |
5′‐CGGAATAGCAGCCAAGTATC‐3′ 5′‐GTCAGTTTAATACCTGGCTTTAGA‐3′ 5′‐FAM‐TGACCCTGACGCCTGCTTCTCG‐MGB‐Q530‐3′ |
ATAD3, ATPase family AAA domain‐containing protein 3; BAP31, b‐cell receptor‐associated protein 31; CALNX, calnexin; CypD, cyclophilin D; DRP1, dynamin‐related protein 1; ER, endoplasmic reticulum; FACL4, fatty‐acid‐coenzyme A ligase, Long‐Chain 4; FIS1, fission protein 1; GRP75, glucose‐regulated protein 75; IP3R, inositol trisphosphate receptor; MFN1, mitofusin 1; MFN2, mitofusin 2; OPA1, optic atrophy 1; P450scc, cholesterol side‐chain cleavage cytochrome P450; PACS2, phosphofurin acidic cluster sorting protein 2; PTPIP51, protein tyrosine phosphatase‐interacting protein 51; σ1R, sigma 1 receptor; SOAT1, sterol o‐acyltransferase 1; StAR, steroidogenic acute regulator protein; TSPO, translocator protein; VAPB, vesicle‐associated membrane protein‐associated protein B; VDAC, voltage‐dependent anion channel.
In situ proximity ligation assay
To visualize and quantify the impact of tau on the ER‐mitochondria interaction, the in situ proximity ligation assay (PLA) technology was used, allowing the detection of protein interactions with high specificity and sensitivity (Tubbs & Rieusset, 2016). The different SH‐SY5Y cell lines were seeded on precoated collagen coverslips in 12‐well plates at a density of 100,000 cells per well and the iPSCs on Cellartis DEF‐CS COAT‐1‐coated coverslips in 12‐well plates at a density of 150,000 cells per well. Fixation of cells was carried out 24‐h postseeding for standard PLAs, 24‐h post‐treatment for PLAs with GSK3β inhibition, or 48‐h post‐transfection for PLAs with siRNA transfections. For that, cells were first washed with PBS and then fixed in 10% formaldehyde for 10 min at room temperature. To stop the reaction, 1 M glycine in PBS was added as a quenching agent. Afterward, the cells were washed with PBS and incubated for 15 min at room temperature with 100 mM glycine in PBS. To permeabilize cells, they were first washed with PBS and then incubated with 0.1% Triton X‐100 in PBS for 15 min at room temperature under agitation. Blocking was conducted with the Duolink® Blocking Solution, provided by the manufacturer, for 1 h at 37°C in a humidity chamber. The cells were then incubated overnight in a humidity chamber at 4°C with two primary antibodies raised in different species. The following primary antibodies were used for SH‐SY5Y cell lines: anti‐IP3R1 (rabbit) 1/150, anti‐VDAC1 (mouse) 1/20, anti‐GRP75 (mouse) 1/20, anti‐VAPB (mouse) 1/200, anti‐PTPIP51 (rabbit) 1/50, and anti‐Tau (phospho T231; rabbit) 1/50 and the following for iPSCs: anti‐VAPB (mouse) 1/150, anti‐PTPIP51 (rabbit) 1/25. The next day, the PLA was conducted according to the Duolink® PLA fluorescence protocol using the Duolink® In Situ Red Starter Kit Mouse/Rabbit for standard PLAs and PLAs after GSK3β inhibition. For PLAs after siRNA transfection and PLAs using iPSCs, the Duolink® In Situ Detection Reagents Far Red were applied. Briefly, cells were incubated in a humidity chamber for 1 h at 37°C with PLA probes, then for 30 min at 37°C to perform the ligation, and lastly for 100 min at 37°C for the amplification step of the signals. To visualize the cell shape of the WT and Tau KO cells, additionally, a counterstaining step was included with the conjugated Alexa Fluor® 488 mouse anti‐β‐tubulin, class III antibody 1/100. Moreover, for PLAs after MFN2 siRNA transfection and using iPSCs, the conjugated Alexa Fluor® 555 rabbit anti‐TOMM20 antibody 1/250 (for SH‐SY5Y cell lines) and 1/150 (for iPSCs) was used as a positive control for the confirmation of transfection by assessing mitochondrial morphology (MFN2 siRNA transfection PLAs), as MFN2 KD induces mitochondrial fragmentation. For both counterstainings, the cells were incubated in a humidity chamber for 2 h at room temperature. Following the final washing steps, the coverslips were mounted on glass slides with the Vectashield H‐1,000 mounting medium and sealed with nail polish.
Cholesterol and organelles staining
Cells were plated in 12‐well plates containing coverslips coated with collagen (0.05 mg/ml) at a density of 100,000 cells per well. After 48‐h plating, cells were washed with PBS and fixed with Cell‐Based Assay Fixative Solution (Cholesterol Assay Kit [Cell‐Based] for 10 min). Cells were then washed 3 times for 5 min with Cholesterol Detection Wash Buffer and incubated with Filipin III (1/100) or antibodies targeting different organelles (mitochondria: TOMM20 Alexa fluor® 555 dilution 1/250, lysosomes: M6PR [cation independent] Alexa Fluor® 647 dilution 1/50, early endosomes: EEA1 Alexa Fluor® 647 dilution 1/100, Golgi apparatus: GM130 Alexa Fluor® 647 dilution 1/50) in Cholesterol Detection Assay Buffer in the dark for 4 h at room temperature.
For staining of lipid droplets, the LipidSpot™ 610 Lipid Droplet Stain (1/1,000) was used instead of an antibody. For ER staining, cells were transfected with an ER‐RFP DNA plasmid using the Xfect transfection reagent. Filipin staining was performed 48 h after transfection.
Staining was followed by two washes of 5 min each with Cholesterol Detection Wash Buffer, and coverslips were mounted on slides with the Vectashield H‐1000 mounting medium and sealed with nail polish.
Phospho‐Tau staining in cells
Cells were plated in 12‐well plates containing coverslips coated with collagen (0.05 mg/ml) at a density of 100,000 cells per well. After 48‐h plating, cells were washed with PBS and fixed with a solution of 4% paraformaldehyde for 10 min. Cells were then washed three times for 5 min with PBS and incubated with 0.15% TritonX100 for 10 min. After three washings, cells were incubated with the mouse anti‐phospho‐tau antibodies AT180 or AT8 overnight at 4°C. Cells were washed three times for 5 min with PBS and incubated with a goat anti‐mouse Alexa fluor® 568 secondary antibody for 1 h at room temperature. After three final washes with PBS, coverslips were mounted on slides with the Vectashield H‐1000 mounting medium and sealed with nail polish.
Staining of mouse brain sections
Mouse brains were cut in a Vibratome (30‐μm brain sections) in ice‐cold PBS and stored in cryoprotective solution (defrost conditions). Brain sections were washed three times for 10 min with PBS to remove the cryoprotectant. Sections were permeabilized with 0.2% Triton in PBS for 1 h, washed three times for 10 min, and blocked with 5% BSA for 1 h. Primary antibodies were diluted in PBS (anti‐GFP (chicken) 1/500, anti‐AT180 (mouse) 1/500, anti‐calnexin (rabbit) 1/300) and incubated overnight at 4°C. After 3 washes of 10 min with PBS, brain sections were incubated with secondary antibodies (anti‐chicken 488, anti‐mouse 568, anti‐rabbit 647) diluted 1/500 in PBS at room temperature for 1 h. Sections were then incubated for 5 min with DAPI (1/1,000 in PBS), washed once for 3 min, and mount onto superfrost plus slide. Sections were dried until almost translucent, mounted in Vectashield mounting medium (H‐1000), and sealed with nail polish.
Transfection of siRNA
To modulate the interaction of MAMs in our cell models, the effect of siRNA‐mediated knockdown of mitofusin 2 (MFN2) expression was investigated. The different cell lines were seeded 24 h prior to transfection on either precoated collagen coverslips in 12‐well plates at a density of 100,000 cells per well (for microscopy experiments) or on 10 cm2 dishes at a density of 750,000 cells per dish (for assessment of pregnenolone level). On the following day, shortly before transfection, the medium was exchanged with Opti‐MEM™ Reduced Serum Medium containing 5% FBS. Transfection of cells with siRNAs was conducted with the Xfect™ RNA Transfection Reagent according to the manufacturer's protocol. For MFN2 knockdown a 1:1 mix of Hs_MFN2_5 FlexiTube siRNA and Hs_MFN8 FlexiTube siRNA was used, while for control transfection the AllStars Negative Control FlexiTube siRNA. The target sequence of Hs_MFN2_5 FlexiTube siRNA consisted of CTGCACCGCCACATAGAGGAA and for Hs_MFN8 FlexiTube siRNA of AAGACTATAAGCTGCGAATTA. For transfections in 12‐well format, 50 pmol of total siRNA was diluted in Xfect Reaction Buffer and mixed by vortexing. For transfections in a 10‐cm dish format, 500 pmol of total siRNA was used. Next, the Xfect RNA Transfection Polymer was diluted in Xfect Reaction Buffer, vortexed well, and added to the diluted siRNA, followed by vortexing for at least 10 s. The samples were incubated for 10 min at room temperature to allow nanoparticle complexes to form. Afterward, the entire volume of nanoparticle complex solution was pipetted drop‐wise to the cells. All siRNAs were diluted to a final concentration of 45 nM. The cells were incubated with the transfection complexes for 4 h at 37°C in a humidified 5% CO2 incubator. Subsequently, the transfection complexes were removed and fresh medium was added. Experiments with transfected cells were performed 48‐h post‐transfection. Of note, MFN2 expression was assessed by RT–qPCR and a decreased expression of 90% was measured (Fig EV3O).
Treatment with GSK3β inhibitor
As a further approach to influence the ER‐mitochondria association, the impact of the GSK3β inhibitor CHIR 99021 was examined on our cellular models. Stock solutions of CHIR 99021 were prepared in DMSO with a concentration of 10 mM. The different cell lines were seeded on either precoated collagen coverslips in 12‐well plates at a density of 100,000 cells per well (for microscopy experiments) or in 10 cm2 dishes at a density of 750,000 cells per dish. On the next day, the cells were treated with either DMEM supplemented with 1% P/S (vehicle control condition) or with a final concentration of 100 nM CHIR99021. All experiments were performed after 24 h of treatment.
Cell viability assay
Cell viability was investigated using a 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl‐tetrazolium bromide (MTT) cell proliferation assay. After treatment, the cells were incubated with MTT in DMEM for 3 h. MTT is reduced to a violet formazan derivative by mitochondrial enzymatic activity. At the end of the reaction, the cells were dissolved in DMSO 100%. MTT absorbance was measured at 595 nm using the multimode plate reader Cytation 3.
Image acquisition
Image acquisition of brain sections was performed using a Zeiss LSM710 confocal microscope running the Zen Black software (2012). Single optical sections were obtained using a 63× 1.4 NA oil objective with the following parameters: 1,024 × 1,024 pixel resolution, zoom 2.5, scan speed 7, line scan average of 2, and scan mode bidirectional. All acquisition settings were kept constant during imaging. DAPI signal was detected using a blue laser diode (Ex: 405 nm), with 5% transmission, gain 550. Green fluorescent protein (GFP) signal was detected using an argon laser (Ex 488 nm) with 10% transmission, gain 600. Alexa Fluor® 568 signal was detected using a 561 nm laser with 15% transmission, gain 650. Alexa Fluor® 647 signal was detected using a 633 nm laser with 20% transmission, gain 650. Images were collected on two independent occasions for brain section (= 1 animal), with at least three fields of view on each brain section. Representative examples were used for figure preparation with the aid of Fiji.
For colocalization experiments between Filipin III (cholesterol) and intracellular organelles, images were acquired using an Applied Precision Widefield DeltaVision Core microscope fitted with an Olympus Plan Apo N 60× oil 1.25 NA lens (with extra 1.6 magnification lens) and a CoolSnap HQ2 camera (Photometrics) and fluorescence optics. The software used was Software: SoftWorx (version 4.1.2). Datasets of 1,024 × 1,024 pixels with up to 20 z‐sections spaced by 200 nm were collected. Images were acquired with an LED‐transmitted light source (solid‐state light). All acquisition settings were kept constant during imaging.
Filipin III was detected using a DAPI filter set (Ex 390/18 nm, Em 435/48 nm), exposure time 1 s with 100% transmission. TOMM20 Alexa fluor® 555 and ER‐RFP were detected using a TRITC filter set (Ex 542/27 nm, Em 597/45 nm), exposure time 100 ms with 100% transmission. M6PR Alexa Fluor® 647, EEA1 Alexa Fluor® 647, GM130 Alexa Fluor® 647, and LipidSpot were detected using a Cy5 filter set (Ex 632/22 nm, Em 679/34 nm), exposure time 2 s with 100% transmission. Following the acquisition, images were deconvolved using Huygens Deconvolution Software and processed. Images were collected on three independent occasions for each condition, with at least 10 (and typically 15) fields of view on each occasion. Representative examples were used for figure preparation with the aid of Fiji.
For the PLA experiments, the acquisition of confocal images was conducted using an inverted confocal microscope (Leica Microsystems TCS SPE DMI4000) attached to an external light source for enhanced fluorescence imaging (Leica EL6000) with the Leica Application Suite Advanced Fluorescence (Leica LAS AF) software. Images were taken using a HCX PL APO oil objective with a magnification of 63× 1.10–0.60 and by applying Type F immersion oil (Leica Microsystems). Pinhole settings were chosen in such a way that axially each cell was entirely present within the confocal volume. To obtain the images the following parameters were set: 1024 × 1024 pixel resolution, scan speed 400 Hz, scan mode bidirectional, zoom 2, and frame average 4. Z‐stacks were obtained with 10 steps and a z‐step size of 0.29 μm. During imaging, the acquisition settings were all kept constant. Green fluorescent protein (GFP) and β‐tubulin Alexa Fluor® 488 signals were detected using a 488 nm laser with 30% transmission, gain 700, and an emission bandwidth of 496–537 nm. TOMM20 Alexa Fluor® 555 signal was detected using a 532 nm laser with 30% transmission, gain 700, and an emission bandwidth of 555–600 nm. PLA signal in red was detected using a 532 nm laser with 30% transmission, gain 600, and an emission bandwidth of 594–753 nm. PLA signal in deep red was detected using a 635 nm laser with 30% transmission, gain 700, and an emission bandwidth of 640–800 nm. Images were collected on at least three independent occasions for each condition, with typically 15 fields of view each time. Representative examples were used for figure preparation with the aid of Fiji.
All data were acquired within the dynamic range of the camera, and all quantitation was performed on the raw images. Images presented in the figures of the present manuscript have been adjusted to aid clarity, with all comparable images treated identically.
Mitochondrial morphology analysis
The analysis of mitochondrial morphology was conducted blinded and performed with an automated image processing and morphometry macro in Fiji as previously described (Merrill et al, 2017). Briefly, images were background‐subtracted (rolling ball radius = 50 pixels) and uneven labeling of mitochondria was improved through local contrast enhancement using contrast‐limited adaptive histogram equalization (“CLAHE”). To segment mitochondria, the “Tubeness” filter was applied. After setting an automated threshold, the “Analyze Particles” plugin was used to determine the area and perimeter of individual mitochondria, and the “Skeletonize” function was used to measure mitochondrial length. The average metrics obtained reflect:
The mitochondrial elongation or form factor (FF): the form factor value describes the particle's shape complexity of the mitochondria, as the inverse of the circularity.
Area‐weighted form factor (AWFF): a variant of form factor with a bias toward larger mitochondria or mitochondrial networks. AWFF provides more realistic results in cases where highly elongated mitochondria are overlapping.
Mitochondrial length: the length reports the mitochondrial length or elongation in pixels, after the mitochondria are reduced to a single‐pixel‐wide shape (“Skeletonize” function on ImageJ).
Colocalization analysis
Colocalization analysis was conducted blinded and performed with the JACoP plugin in Fiji as previously described (Bolte & CordeliÈRes, 2006). A Costes randomization was performed on 16‐bit images with 200 randomized rounds and a bin width of 0.001. Colocalization data are represented using the Manders' overlap coefficient, with M1 being defined as the ratio of the “summed intensities of pixels from the image in the first channel for which the intensity in the second channel is above zero” to the “total intensity in the first channel.” M2 is defined conversely. M1 and M2 are good indicators of the proportion of the signal in the first channel coincident with a signal in the second channel over its total intensity.
Image analysis: analysis of PLA signals
The analysis of PLA signals was conducted blinded and performed using the Fiji software. For all channels, the z‐projection of z‐stacks was generated using the maximum intensity projection type. To quantify the PLA signals per cell, first, a region of interest (ROI) was drawn around the cell to obtain the cell shape by means of its GFP (Vector and P301L) or β‐tubulin (WT and Tau KO) signal. Then, the background of the PLA signal image was subtracted using a rolling ball radius of two pixels and additional noise was removed from the image by applying the “despeckle” command. Subsequently, the image was thresholded with the otsu algorithm. Next, the image with the selected ROI was analyzed using the “analyze particles” command, limited to the threshold. Lastly, based on the measured parameters, the contact points per cell were calculated according to the equation below.
Statistical analysis
GraphPad Prism (version 9.3.0) was used for statistical analysis and data presentation. Gaussian distribution of data was assessed using the D'Agostino‐Pearson normality test. Student's t‐test was used to compare two different conditions, and one‐way ANOVA with Tukey's multiple comparisons test was used to compare more than two different conditions. P‐values < 0.05 were considered as statistically significant. Statistical parameters can be found in the figure legends.
The analysis of targeted‐metabolomic data was performed using MetaboAnalyst 5.0. Data normalization was performed using the Autoscaling option (mean‐centered and divided by standard deviation of each variable) to make features more comparable. One‐way ANOVA was performed for univariate statistics, followed by post hoc Fisher's least significant difference method (Fisher's LSD) to provide a preliminary overview about features that are significant in discriminating the conditions WT vs P301L vs Tau KO. Principal component analysis (PCA) was performed to determine the directions that best explain the variance in our data set without referring to class labels. Partial least squares‐discriminant analysis (PLS‐DA) was used to measure the variable importance in projection (VIP) score, which identifies important features driving the difference between data sets. Hierarchical cluster analysis (shown as heat map) was performed using the Euclidean distance and Ward's linkage.
Author contributions
Leonora Szabo: Formal analysis; investigation; methodology; writing – original draft. Nadia Cummins: Investigation. Paolo Paganetti: Resources; methodology. Alex Odermatt: Supervision. Andreas Papassotiropoulos: Resources; supervision. Celeste Karch: Resources; methodology. Jürgen Götz: Resources; methodology. Anne Eckert: Resources; supervision; funding acquisition. Amandine Grimm: Conceptualization; formal analysis; supervision; funding acquisition; investigation; visualization; methodology; writing – original draft; project administration.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Dataset EV1
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank Fides Meier for general laboratory coordination and Alexia Loynton‐Ferrand and Kai Schleicher for training, microscopy assistance, and technical advice. This study was supported by grants from the Swiss National Science Foundation (# P2BSP3_172045) to AG, the Dementia Research, Synapsis Foundation Switzerland (#2017‐CDA02) to A.G., the Universität Basel Forschungsfonds (#3PE1050) to A.G., and the Swiss National Science Foundation (#31003A_179294, and #31003A_149728) to A.E., the Novartis Foundation for Medical‐Biological Research (#22A007) to A.G., and the Novartis Foundation for Medical‐Biological Research (# 18C143) to A.E. Confocal microscopy on brain samples was facilitated by the Queensland Brain Institute's Advanced Microscopy Facility, supported by the ARC LIEF Grant (LE130100078). Wide‐field microscopy was facilitated by the Imaging Core Facility (IMCF), Biozentrum, University of Basel. Open access funding provided by Universitat Basel.
EMBO reports (2023) 24: e57499
Data availability
The corresponding raw images presented in this study are available on BioImage Archive S‐BSST1053 (https://www.ebi.ac.uk/biostudies/studies/S-BSST1053).
References
- Area‐Gomez E, Schon EA (2016) Mitochondria‐associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 38: 90–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Area‐Gomez E, de Groof A, Bonilla E, Montesinos J, Tanji K, Boldogh I, Pon L, Schon EA (2018) A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis 9: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Lee VMY, Trojanowski JQ (2007) Tau‐mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 8: 663–672 [DOI] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow EM, Mandelkow E (2001) Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta‐structure. J Biol Chem 276: 48165–48174 [DOI] [PubMed] [Google Scholar]
- Bolte S, CordeliÈRes FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, Tu WY, Kao TC, Hsu CM, Cheng JT et al (2012) GSK3beta‐mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS One 7: e49112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieri D, Vicario M, Vallese F, D'Orsi B, Berto P, Grinzato A, Catoni C, De Stefani D, Rizzuto R, Brini M et al (2018) Tau localises within mitochondrial sub‐compartments and its caspase cleavage affects ER‐mitochondria interactions and cellular Ca2+ handling. Biochim Biophys Acta Mol Basis Dis 1864: 3247–3256 [DOI] [PubMed] [Google Scholar]
- Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnóczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, Hajnóczky G (2010) Imaging interorganelle contacts and local calcium dynamics at the ER‐mitochondrial interface. Mol Cell 39: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins N, Tweedie A, Zuryn S, Bertran‐Gonzalez J, Götz J (2018) Disease‐associated tau impairs mitophagy by inhibiting parkin translocation to mitochondria. EMBO J 38: e99360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, Ravid R, Drose S, Brandt U, Muller WE et al (2005) Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem 280: 23802–23814 [DOI] [PubMed] [Google Scholar]
- Distl R, Meske V, Ohm TG (2001) Tangle‐bearing neurons contain more free cholesterol than adjacent tangle‐free neurons. Acta Neuropathol 101: 547–554 [DOI] [PubMed] [Google Scholar]
- Distl R, Treiber‐Held S, Albert F, Meske V, Harzer K, Ohm TG (2003) Cholesterol storage and tau pathology in Niemann‐pick type C disease in the brain. J Pathol 200: 104–111 [DOI] [PubMed] [Google Scholar]
- Duarte A, Poderoso C, Cooke M, Soria G, Cornejo Maciel F, Gottifredi V, Podesta EJ (2012) Mitochondrial fusion is essential for steroid biosynthesis. PLoS One 7: e45829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B, Gotz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo . Neuron 75: 618–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans HT, Taylor D, Kneynsberg A, Bodea L‐G, Götz J (2021) Altered ribosomal function and protein synthesis caused by tau. Acta Neuropathol Commun 9: 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes T, Resende R, Silva DF, Marques AP, Santos AE, Cardoso SM, Domingues MR, Moreira PI, Pereira CF (2021) Structural and functional alterations in mitochondria‐associated membranes (MAMs) and in mitochondria activate stress response mechanisms in an In vitro model of Alzheimer's disease. Biomedicine 9: 881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–E2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckner F, Ohm TG (2014) Tau pathology induces intraneuronal cholesterol accumulation. J Neuropathol Exp Neurol 73: 846–854 [DOI] [PubMed] [Google Scholar]
- Gomez L, Thiebaut PA, Paillard M, Ducreux S, Abrial M, Crola Da Silva C, Durand A, Alam MR, Van Coppenolle F, Sheu SS et al (2015) The SR/ER‐mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death Differ 23: 313–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Ittner LM (2008) Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci 9: 532–544 [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, Barmettler R, Nitsch RM (2001) Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 276: 529–534 [DOI] [PubMed] [Google Scholar]
- Grimm A (2021) Impairments in brain bioenergetics in aging and tau pathology: a chicken and egg situation? Cell 10: 2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm A, Biliouris EE, Lang UE, Götz J, Mensah‐Nyagan AG, Eckert A (2015) Sex hormone‐related neurosteroids differentially rescue bioenergetic deficits induced by amyloid‐β or hyperphosphorylated tau protein. Cell Mol Life Sci 73: 201–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm A, Lejri I, Hallé F, Schmitt M, Götz J, Bihel F, Eckert A (2019) Mitochondria modulatory effects of new TSPO ligands in a cellular model of tauopathies. J Neuroendocrinol 32: e12796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su T‐P (2009) MAM: more than just a housekeeper. Trends Cell Biol 19: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Wang Y, Gan X, Fang D, Zhong C, Wu L, Hu G, Sosunov AA, McKhann GM, Yu H et al (2015) Drp1‐mediated mitochondrial abnormalities link to synaptic injury in diabetes model. Diabetes 64: 1728–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering‐Brown S, Chakraverty S, Isaacs A, Grover A (1998) Association of missense and 5′‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 393: 702–705 [DOI] [PubMed] [Google Scholar]
- Issop L, Fan J, Lee S, Rone MB, Basu K, Mui J, Papadopoulos V (2015) Mitochondria‐associated membrane formation in hormone‐stimulated Leydig cell steroidogenesis: role of ATAD3. Endocrinology 156: 334–345 [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW (2004) The glamour and gloom of glycogen synthase kinase‐3. Trends Biochem Sci 29: 95–102 [DOI] [PubMed] [Google Scholar]
- Kaidanovich‐Beilin O, Woodgett JR (2011) GSK‐3: functional insights from cell biology and animal models. Front Mol Neurosci 4: 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, Steenvoorden E, Rynearson KD, Brouwers JF, Helms JB et al (2019) Cholesterol metabolism is a druggable Axis that independently regulates tau and amyloid‐β in iPSC‐derived Alzheimer's disease neurons. Cell Stem Cell 24: 363–375.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Kao AW, Karydas A, Onanuga K, Martinez R, Argouarch A, Wang C, Huang C, Sohn PD, Bowles KR et al (2019) A comprehensive resource for induced pluripotent stem cells from patients with primary tauopathies. Stem Cell Rep 13: 939–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaytor MD, Orr HT (2002) The GSK3β signaling cascade and neurodegenerative disease. Curr Opin Neurobiol 12: 275–278 [DOI] [PubMed] [Google Scholar]
- Krupka N, Strappe P, Götz J, Ittner LM (2010) Gateway‐compatible lentiviral transfer vectors for ubiquitin promoter driven expression of fluorescent fusion proteins. Plasmid 63: 155–160 [DOI] [PubMed] [Google Scholar]
- Law SM, Zheng JJ (2022) Premise and peril of Wnt signaling activation through GSK‐3β inhibition. iScience 25: 104159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, Feng Q, Wang Q, Ye K, Liu GP et al (2016) Human wild‐type full‐length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6: 24756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Cheung G, Espinoza N, Papadopoulos V (2022) Function, regulation, and pharmacological effects of pregnenolone in the central nervous system. Curr Opin Endocr Metab Res 22: 100310 [Google Scholar]
- Manczak M, Reddy PH (2012) Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer's disease. Hum Mol Genet 21: 5131–5146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manji HK, Zarate CA, Chen G, Du J (2009) Mood stabilizers. In Encyclopedia of neuroscience, Squire LR (ed), pp 967–974. Oxford: Academic Press; [Google Scholar]
- Martin LA, Kennedy BE, Karten B (2014) Mitochondrial cholesterol: mechanisms of import and effects on mitochondrial function. J Bioenerg Biomembr 48: 137–151 [DOI] [PubMed] [Google Scholar]
- Merrill RA, Flippo KH, Strack S (2017) Measuring mitochondrial shape with ImageJ. In Techniques to investigate mitochondrial function in neurons, Strack S, Usachev Y (eds), pp 31–48. New York, NY: Humana Press; [Google Scholar]
- Misgeld T, Kerschensteiner M, Bareyre FM, Burgess RW, Lichtman JW (2007) Imaging axonal transport of mitochondria in vivo . Nat Methods 4: 559–561 [DOI] [PubMed] [Google Scholar]
- Monte MJ, Marin JJG, Antelo A, Vazquez‐Tato J (2009) Bile acids: chemistry, physiology, and pathophysiology. World J Gastroenterol 15: 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillusson S, Stoica R, Gomez‐Suaga P, Lau DHW, Mueller S, Miller T, Miller CCJ (2016) There's something wrong with my MAM; the ER–mitochondria Axis and neurodegenerative diseases. Trends Neurosci 39: 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel P, Woodgett JR (2017) Chapter eight – glycogen synthase kinase 3: a kinase for all pathways? In Current topics in developmental biology, Jenny A (ed), pp 277–302. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Perreault S, Bousquet O, Lauzon M, Paiement J, Leclerc N (2009) Increased association between rough endoplasmic reticulum membranes and mitochondria in transgenic mice that express P301L tau. J Neuropathol Exp Neurol 68: 503–514 [DOI] [PubMed] [Google Scholar]
- Pinton P (2018) Mitochondria‐associated membranes (MAMs) and pathologies. Cell Death Dis 9: 413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43: 815–825 [DOI] [PubMed] [Google Scholar]
- Porcu P, Barron AM, Frye CA, Walf AA, Yang SY, He XY, Morrow AL, Panzica GC, Melcangi RC (2016) Neurosteroidogenesis today: novel targets for neuroactive steroid synthesis and action and their relevance for translational research. J Neuroendocrinol 28: 12351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS (2015) Mitochondria‐associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)‐voltage‐dependent Anion Channel 2 (VDAC2) interaction. J Biol Chem 290: 2604–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez S, Haddad‐Tóvolli R, Radosevic M, Toledo M, Pané A, Alcolea D, Ribas V, Milà‐Guasch M, Pozo M, Obri A et al (2022) Hypothalamic pregnenolone mediates recognition memory in the context of metabolic disorders. Cell Metab 34: 269–284.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Drose S, Brandt U et al (2009) Amyloid‐beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer's disease mice. Proc Natl Acad Sci USA 106: 20057–20062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetin K, Steullet P, Pachoud M, Perbet R, Parietti E, Maheswaran M, Eddarkaoui S, Bégard S, Pythoud C, Rey M et al (2020) Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer's disease. Nat Neurosci 23: 1567–1579 [DOI] [PubMed] [Google Scholar]
- Rusinol AE, Cui Z, Chen MH, Vance JE (1994) A unique mitochondria‐associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre‐Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269: 27494–27502 [PubMed] [Google Scholar]
- Schmitt K, Grimm A, Kazmierczak A, Strosznajder JB, Götz J, Eckert A (2012) Insights into mitochondrial dysfunction: aging, amyloid‐beta, and tau‐a deleterious trio. Antioxid Redox Signal 16: 1456–1466 [DOI] [PubMed] [Google Scholar]
- Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Muller WE, Leuner K (2012) A new link to mitochondrial impairment in tauopathies. Mol Neurobiol 46: 205–216 [DOI] [PubMed] [Google Scholar]
- Schumacher M, Weill‐Engerer S, Liere P, Robert F, Franklin RJM, Garcia‐Segura LM, Lambert JJ, Mayo W, Melcangi RC, Parducz A et al (2003) Steroid hormones and neurosteroids in normal and pathological aging of the nervous system. Prog Neurobiol 71: 3–29 [DOI] [PubMed] [Google Scholar]
- Sola M, Magrin C, Pedrioli G, Pinton S, Salvadè A, Papin S, Paganetti P (2020) Tau affects P53 function and cell fate during the DNA damage response. Commun Biol 3: 245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M (1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21: 428–433 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K‐F, Vizcay‐Barrena G, Lin W‐L, Xu Y‐F, Lewis J et al (2014) ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: 3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo L, Eckert A, Grimm A (2020) Insights into disease‐associated tau impact on mitochondria. Int J Mol Sci 21: 6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs E, Rieusset J (2016) Study of endoplasmic reticulum and mitochondria interactions by in situ proximity ligation assay in fixed cells. J Vis Exp 2016: 54899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck BJ, Miller LVC, Katsinelos T, Smith AE, Wilson EL, Keeling S, Cheng S, Vaysburd MJ, Knox C, Tredgett L et al (2022) Cholesterol determines the cytosolic entry and seeded aggregation of tau. Cell Rep 39: 110776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE (2003) Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res Mol Biol 75: 69–111 [DOI] [PubMed] [Google Scholar]
- Volgyi K, Juhász G, Kovacs Z, Penke B (2015) Dysfunction of endoplasmic reticulum (ER) and mitochondria (MT) in Alzheimer's disease: the role of the ER‐MT cross‐talk. Curr Alzheimer Res 12: 655–672 [DOI] [PubMed] [Google Scholar]
- Weill‐Engerer S, David J‐P, Sazdovitch V, Liere P, Eychenne B, Pianos A, Schumacher M, Delacourte A, Baulieu E‐E, Akwa Y (2002) Neurosteroid quantification in human brain regions: comparison between Alzheimer's and nondemented patients. J Clin Endocrinol Metabol 87: 5138–5143 [DOI] [PubMed] [Google Scholar]
- Wilson EL, Metzakopian E (2021) ER‐mitochondria contact sites in neurodegeneration: genetic screening approaches to investigate novel disease mechanisms. Cell Death Differ 28: 1804–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzig M, Grimm A, Schmitt K, Lejri I, Frank S, Brown SA, Eckert A (2020) Clock‐controlled mitochondrial dynamics correlates with cyclic pregnenolone synthesis. Cells 9: 2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodgett JR (2013) Glycogen synthase kinase‐3. In Encyclopedia of biological chemistry, Lennarz WJ, Lane MD (eds), 2nd edn, pp 434–438. Waltham, MA: Academic Press; [Google Scholar]
- Wu J‐H, Zhang S‐H, Gao F‐J, Lei Y, Chen X‐Y, Gao F, Zhang S‐J, Sun X‐h (2013) RNAi screening identifies GSK3β as a regulator of DRP1 and the neuroprotection of lithium chloride against elevated pressure involved in downregulation of DRP1. Neurosci Lett 554: 99–104 [DOI] [PubMed] [Google Scholar]
- Wu Y, Chen M, Jiang J (2019) Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 49: 35–45 [DOI] [PubMed] [Google Scholar]
- Yang K, Chen Z, Gao J, Shi W, Li L, Jiang S, Hu H, Liu Z, Xu D, Wu L (2017) The key roles of GSK‐3β in regulating mitochondrial activity. Cell Physiol Biochem 44: 1445–1459 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Li S, Li M, Ke D, Wang Q, Yang Y, Liu G‐P, Wang X‐C, Liu E, Wang J‐Z (2022) Human tau accumulation promotes glycogen synthase kinase‐3β acetylation and thus upregulates the kinase: a vicious cycle in Alzheimer neurodegeneration. EBioMedicine 78: 103970 [DOI] [PMC free article] [PubMed] [Google Scholar]