

Abstract
Necroptosis is a form of programmed cell death executed by receptor-interacting serine/threonine protein kinase 1 (RIPK1), RIPK3, and mixed lineage kinase domain-like (MLKL). Platelets are circulating cells that play central roles in haemostasis and pathological thrombosis. In this study we demonstrate seminal contribution of MLKL in transformation of agonist-stimulated platelets to active haemostatic units progressing eventually to necrotic death on a temporal scale, thus attributing a yet unrecognized fundamental role to MLKL in platelet biology. Physiological agonists like thrombin instigated phosphorylation and subsequent oligomerization of MLKL in platelets in a RIPK3-independent but phosphoinositide 3-kinase (PI3K)/AKT-dependent manner. Inhibition of MLKL significantly curbed agonist-induced haemostatic responses in platelets that included platelet aggregation, integrin activation, granule secretion, procoagulant surface generation, rise in intracellular calcium, shedding of extracellular vesicles, platelet-leukocyte interactions and thrombus formation under arterial shear. MLKL inhibition, too, prompted impairment in mitochondrial oxidative phosphorylation and aerobic glycolysis in stimulated platelets, accompanied with disruption in mitochondrial transmembrane potential, augmented proton leak and drop in both mitochondrial calcium as well as ROS. These findings underscore the key role of MLKL in sustaining OXPHOS and aerobic glycolysis that underlie energy-intensive platelet activation responses. Prolonged exposure to thrombin provoked oligomerization and translocation of MLKL to plasma membranes forming focal clusters that led to progressive membrane permeabilization and decline in platelet viability, which was prevented by inhibitors of PI3K/MLKL. In summary, MLKL plays vital role in transitioning of stimulated platelets from relatively quiescent cells to functionally/metabolically active prothrombotic units and their ensuing progression to necroptotic death.
Subject terms: Cell biology, Kinases
Introduction
Platelets are enucleate circulating cells derived from megakaryocytes known for their central role in haemostasis and thrombosis. Whereas platelets are instrumental in preventing blood loss by sealing breach in the endothelium of injured vessels, they can also be the cause for life-threatening vaso-occlusive pathologies like ischemic stroke and myocardial infarction. Upon agonist exposure platelets undergo intense metabolic rewiring and execute energy-intensive activities to yield a prothrombotic phenotype that terminates with their necrosis. Contrasting this, life span of unstimulated platelets (10–12 days for human and 5 days for murine) is delimited by death through apoptosis [1–4].
Necroptosis is a form of programmed necrosis characterized by release of immunogenic cellular components that induce inflammation in neighbouring cells [5, 6]. Mostly, there are three key players that execute the process of necroptosis, namely RIPK (receptor-interacting serine/threonine protein kinase) 1, RIPK3 and MLKL (mixed lineage kinase domain-like). MLKL is the terminal effector molecule of necroptosis programme composed of an N-terminal four-helix bundle (4HB) or ‘killer’ domain and a C-terminal pseudokinase domain (psKD). The latter is phosphorylated at Ser-358/Thr-357 in its activation loop by RIPK3 [7] though its phosphorylation at Tyr-376 by TAM kinase has also been reported [8]. The precise mechanism of MLKL activation differs between species [9, 10]. The phosphorylation events at psKD are communicated to the 4HB domain through a connecting two-helix linker, thus providing an edge for ensuing conformational changes in MLKL. Phosphorylated MLKL can form oligomers where either 3, 4, 6 or 8 monomer units are coupled by disulfide bonds. The formed oligomers traffic from cytoplasm to insert themselves in surface as well as intracellular membranes through interaction with phosphatidylinositol or cardiolipin moieties that eventually compromises with membrane integrity leading to cell death [7].
In an elegant study using mice with megakaryocyte-specific knockout of MLKL Joseffson and co-workers [11] have demonstrated the existence of functional necroptosis signaling in megakaryocytes, the progenitors of platelets, though the pathway was dispensable for platelet production. They also demonstrate prolonged tail re-bleeding times in these mice and impaired agonist-induced activation of their platelets. Another significant report has indicated an MLKL-independent role for RIPK3 in thrombosis pathology [12]. Interestingly, agonist-stimulated platelet has also been demonstrated to have a critical role in RIPK1- and MLKL-dependent neutrophil necroptosis and venous thrombosis [13] However, the regulation and role of MLKL in human platelets remains largely unexplored. In the present study we demonstrate that, thrombin instigates phosphorylation and subsequent oligomerization of MLKL in platelets in a RIPK3-independent manner. MLKL plays vital role in temporal progression of thrombin-stimulated platelets from quiescence to functionally active haemostatic units that culminates in lytic death. In view of its fundamental contributions in platelet physiology, MLKL could be an attractive therapeutic target against thrombotic disorders.
Materials and methods
Materials
Antibodies against MLKL were purchased either from Sigma-Aldrich (# M6697) for fluorescence microscopy or from GeneTex (# GTX107538) for Western analysis studies. Antibody against phosphorylated (Ser-358) MLKL (p-MLKL) was procured from Abcam (# ab187091). MLKL inhibitors necrosulfonamide (NSA) (# 480073) was from Sigma-Aldrich and TC13172 (TC) (# HY-101524) was from MedChemExpress. Anti-CD62P (# 550561), anti-PAC-1 (# 340507), anti-human CD41a, anti-human CD14 antibodies and sheath fluid were from BD Biosciences. PE-annexin V (# 640908) was procured from BioLegend. Anti-actin antibody (# A2066), skimmed milk powder, thrombin, LY-294002 (LY), wortmannin (Wort), ethylene glycol tetraacetic acid (EGTA), ethylenediaminetetraacetic acid (EDTA), thrombin receptor-activating peptide (TRAP, # S1820) and DMSO were from Sigma. AKT inhibitor VIII (AKTi, # 14870), a potent allosteric inhibitor of AKT1 and AKT2, was bought from Cayman Chemical. RIPK3 inhibitors, GSK’843 (# AOB4898) and GSK’872 (# 5.30389), were products of AOBIOUS and Sigma, respectively. Fura-2/AM (# 344905) was purchased from Calbiochem. Collagen (# 385) and Chrono-lume luciferin luciferase reagent (# 395) were products of Chrono-log. Polyvinylidene fluoride (PVDF) membrane and enhanced chemiluminescence (ECL) kit were from Millipore. Bovine serum albumin was acquired from Amresco. Fibrinogen (Alexa fluor 488-conjugated) (# F13191), goat-anti-rabbit IgG (Alexa fluor 488-conjugated) (# A11008), MitoTracker Red (# M7512), MitoSOX Red (# M36008), Rhod-2/AM (# R1244), Fluo-4/AM (# F14201) and Restore Western Blot Stripping Buffer (# 21059) were from Invitrogen. HRP-conjugated goat anti-rabbit IgG was from Bangalore Genei. Triton X-100, Tween-20, CaCl2 and reagents for electrophoresis were purchased from Merck. CellTiter-Glo (# G7570), and CytoTox-Glo (# G9291) were purchased from Promega. Permanox Cell Culture Slide (# 160005) for microscopy and Calcein/AM (C3100MP) were acquired from Thermo Fisher. All other reagents were of analytical grade. Type I deionized water (18.2 MΩ.cm, Millipore) was used for preparation of solutions. Experiments were carried out strictly as per the guidelines of Institutional Ethical Committee.
Study design
To estimate the sample size, no calculations were made. At least three separate independent runs of each experiment were conducted, or the sample size was determined by the extent of the effect seen in the pilot experiments. No inclusion and exclusion criteria were set for experimental units or data points. The results are biological replicates (paired observations made on whole blood and/or platelet populations taken from several healthy people), and they apply to all in vitro and ex vivo investigations. The results were successfully reproduced with each biological replicate. All attempts at reproducing results were successful.
Ethics statement
Fresh venous blood from healthy volunteers was collected in acid citrate dextrose (ACD) under informed consent, strictly as per the recommendations of the Institutional Ethical Committee of Banaras Hindu University (Approval No. Dean/2015-16/EC/76). The study methodologies conformed to the standards set by the Declaration of Helsinki.
Platelet preparation
Platelets from human blood were isolated by differential centrifugation as described earlier [14]. Briefly, blood was centrifuged at 100 × g for 20 min to obtain platelet-rich plasma (PRP). PRP was then supplemented with 1 µm PGE1 and 2 mM EDTA and centrifuged at 800 × g for 7 min to sediment platelets. Pellet was washed in Tyrode buffer A (20 mM HEPES, 134 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 0.34 mM NaH2PO4, 12 mM NaHCO3, pH 6.2, supplemented with 5 mM glucose, 0.35% BSA and 1 µM PGE1) and centrifuged at 800 × g for 7 min. Finally, platelets were resuspended in Tyrode buffer B (pH 7.35), which was same as buffer A without PGE1 and BSA. The final cell count was adjusted to 2–4 × 108 cells/ml with automated cell counter (Multisizer 4, Beckman Coulter). Leukocyte contamination in platelet preparation was found to be less than 0.015%. All steps were executed under sterile conditions and precautions were taken to maintain the cells in resting state.
Platelet aggregation
Washed human platelets were stirred (12000 rpm) at 37 °C in a whole blood/optical lumi-aggregometer (Chrono-log model 700-2) for 1 min, followed by addition of agonists (thrombin, TRAP or collagen) either in presence or absence of MLKL inhibitors. Aggregation was recorded as percent light transmitted through the sample as a function of time while blank (buffer) represented 100% light transmission. Platelet aggregation in whole blood induced by collagen was recorded as change in electrical resistance (impedance) as a function of time.
Detection of MLKL and phosphorylated (Ser-358) MLKL by Western analysis
Platelets were pre-supplemented with 1 mM Ca2+ followed by stimulation with thrombin (0.5 U/ml) under non-stirring condition for either 5 min or 2 h at 37 °C. Proteins from platelet lysate were separated on 10% SDS-PAGE and electrophoretically transferred onto polyvinylidene difluoride (PVDF) membranes by employing Trans-Blot Turbo Transfer System (Bio-Rad) at 20 V/1.3 A for 20 min. Nonspecific protein binding sites were blocked with 5% skimmed milk in 1X-TBST (10 mM Tris-HCl, 150 mM NaCl, pH 8.0, 0.05% Tween 20) for 1 h at RT. Following 3 washings with TBST for 5 min each, membranes were incubated overnight with antibodies against either p(Ser-358)-MLKL or MLKL (1:1000 dilution each) at 4 °C with gentle agitation. After similar washing steps, blots were incubated with HRP-conjugated secondary antibodies (1:5000) for 1 h at RT. Antibody binding was detected using enhanced chemiluminescence detection kit (Millipore). Whenever required, blots were stripped using Restore Western Blot Stripping Buffer (Invitrogen) and reprobed for MLKL or β-actin. Images were acquired on multispectral imaging system (UVP BioSpectrum 800) and quantified using VisionWorks LS software (UVP).
Secretion from platelet α-granules and dense bodies
Secretion from platelet α-granules in response to a stimulus was quantified by surface expression of P-selectin (CD62P). Washed human platelets were pre-treated with MLKL inhibitors for 10 min at RT, followed by stimulation with thrombin (0.5 U/ml) for 5 min 37 °C. Cells were stained with PE- labelled anti-CD62P antibody (5% v/v) for 30 min at RT in dark. Samples were suspended in sheath fluid and analyzed on a flow cytometer (FACSCalibur, BD Biosciences). Forward and side scatter voltages were set at E00 and 350, respectively, with a threshold of 52 V. An amorphous gate was drawn to encompass platelets separate from noise and multi-platelet particles. All fluorescence data were collected using 4-quadrant logarithmic amplification for 10000 events in platelet gate from each sample and analyzed using CellQuest Pro Software. Secretion of adenine nucleotides from platelet dense granules was measured employing Chrono-lume reagent (stock concentration, 0.2 μM luciferase/luciferin). Luminescence generated was monitored in a lumi-aggregometer contemporaneous with platelet aggregation.
Determination of integrin αIIbβ3 activation and fibrinogen binding
Platelet stimulation induces conformational switch in integrins αIIbβ3 that allows high-affinity binding of fibrinogen leading to cell-cell aggregate formation. Washed human platelets (2.5 × 108/ml) were pre-treated either with the MLKL inhibitor TC (40 μM) or vehicle, followed by exposure to thrombin (0.5 U/ml) for 5 min at 37 °C. NSA could not be employed in flow cytometry-based experiments because of its intrinsic fluorescence in FITC channel (FL-1) [15]. Cells were incubated with either FITC-labelled PAC-1 antibody (5% v/v) that specifically recognizes the ‘active’ conformation of αIIbβ3, or with Alexa fluor 488-conjugated fibrinogen (10 μg/ml) for 30 min at RT in dark. Lastly, samples were suspended in sheath fluid, and analyzed by flow cytometry.
Isolation and analysis of platelet-derived extracellular vesicles (PEVs)
PEVs were isolated and characterized as described previously [16]. Platelets were pre-incubated either with NSA (40 μM) or TC (40 μM) for 10 min, followed by stimulation with thrombin (0.5 U/ml). Cells were sedimented at 800 × g for 10 min, and then at 1200 × g for 2 min at 22 °C to obtain PEVs cleared of platelets. Next, PEVs were analyzed with Nanoparticle Tracking Analyzer (NTA) where a beam from solid-state laser source (635 nm) was allowed to pass through the sample. Light scattered by rapidly moving particles in suspension in Brownian motion at room temperature was observed under 20X microscope. This revealed hydrodynamic diameters of particles, calculated using Stokes Einstein equation, within range of 10 nm to 1 µm and concentration between 107–109/ml. The average distance moved by each EV in x and y directions were captured with CCD camera (30 frames per sec) attached to the microscope. Both capture and analysis were performed using NanoSight LM10 (Malvern) and NTA 2.3 analytical software, which provide an estimate of particle size and counts in sample.
Intracellular calcium measurement using Fura-2/AM
Intracellular calcium was measured as described [17, 18]. Briefly, PRP isolated from fresh human blood was incubated with Fura-2/AM (2 μM) at 37 °C for 45 min in dark. Fura-2-loaded platelets were isolated, washed and finally resuspended in buffer B. Fluorescence of calcium-sensitive dye was recorded in 400 µl aliquots of platelet suspensions at 37 °C under non-stirring condition in Hitachi fluorescence spectrophotometer (model F-2500). Excitation wavelengths were set at 340 and 380 nm, with emission at 510 nm. Ratiometric measurement of intracellular free calcium concentration, [Ca2+]i, was monitored from fluorescence ratio (340 nm/380 nm) employing Intracellular Cation Measurement Program in FL Solutions software. Fmax was determined by lysing the cells with 40 μM digitonin in the presence of saturating CaCl2. Fmin was determined by the addition of 2 mM EGTA. Intracellular free calcium was calibrated according to the derivation of Grynkiewicz et al. [19].
Flow cytometric analysis of [Ca2+]i using Fluo-4/AM
Washed platelets (2 × 106/ml) were incubated with Fluo-4/AM for 30 min at RT in dark. After appropriate gating of platelets, events were analyzed in FL1 channel of flow cytometer (Becton Dickinson, model Accuri C6 + ), within the time lapse 1.5–5.0 min (in upper right quadrant). Baseline calcium levels were recorded for 60 sec, followed by addition of thrombin (0.1 U/ml).
Measurement of annexin V binding
Phosphatidylserine (PS) externalization, a characteristic of procoagulant platelets, was evaluated from annexin V binding. Washed human platelets pre-treated with different reagents were stimulated either with thrombin (0.5 U/ml), or cocktail of thrombin (0.5 U/ml) and collagen (5 μg/ml) at 37 °C for 15 min without stirring. PE-labeled annexin V (5% v/v) was added to each sample along with 5 mM CaCl2 and incubated for 30 min in dark at RT. Cells were analyzed by flow cytometry as described above.
Analysis of leucocyte-platelet interaction in whole blood
Leucocyte-platelet interaction was measured as described earlier [20, 21]. Fresh human blood (20 µl) was added to a cocktail containing 10 µl each from APC-anti-CD41a (platelet-specific) and FITC-anti-CD14 (leukocyte-specific) antibodies and mixed gently. Samples were treated with either NSA (80 μM), TC (80 μM) or vehicle for 10 min, followed by incubation with TRAP (2 μM) for 15 min at RT. RBCs were lysed with 800 μl FACS lysis solution (1X, BD Biosciences) for 10 min at RT. Leucocyte-platelet interaction was analyzed on a flow cytometer. Side scatter voltage was set at 350 with a threshold of 52 V. A dot plot of side scatter (SSC) versus log FITC-CD14 fluorescence was created using the CellQuest Pro software. Amorphous gates were drawn for monocyte (high fluorescence and low SSC) and neutrophil (low fluorescence and high SSC) populations. All fluorescence data were collected using 4-quadrant logarithmic amplification for 1000 events in either neutrophil or monocyte gate from each sample and analyzed with CellQuest Pro Software.
Lactate release assay
Lactate release rate in thrombin (0.5 U/ml)-stimulated platelets as a measure of glycolysis was determined with YSI 2900D Multiparameter Bioanalytical System (YSI Life Sciences) that employs immobilized enzyme electrodes and electrochemistry-based biosensing [22].
High-resolution respirometry
Mitochondrial respiration was measured using a high-resolution respirometer (Oxygraph-2k, Oroboros Instruments) at 37 °C under stirring conditions (750 rpm). Washed human platelets (1.5 × 108/ml, in buffer B supplemented with 5 mM glucose) were transferred into oxygraph chamber. Respiration was first allowed to stabilize at the routine state, i.e., in the physiological coupling state controlled by cellular energy demands for oxidative phosphorylation. Then, platelets were treated with either MLKL inhibitor (NSA) or DMSO (control) for 5 min followed by addition of thrombin (0.25 U/ml), and changes in oxygen flux were recorded in real time at high resolution (sampling at 2 sec intervals). Oxidative phosphorylation (OXPHOS) as well as leak respiration were measured following addition of oligomycin (1 μg/ml) to cell suspension. Calibration of air saturation was performed each day before starting experiments by letting Millipore water/buffer B stir with air in the oxygraph chamber until equilibration was reached and a stable signal obtained. All experiments were performed at an oxygen concentration in the range of 100–205 μM O2. Data were recorded and analyzed using DatLab 7.4 software (Oroboros Instruments).
Measurement of mitochondrial transmembrane potential (Δψm)
Washed human platelets (2.5 × 108/ml) were pre-treated with either NSA or TC followed by stimulation with thrombin (0.5 U/ml) for 5 min at RT. Cells were labelled with MitoTracker Red (500 nM) for 30 min at RT. Fluorescence from labelled platelets were analyzed by flow cytometry as described above.
Measurement of mitochondrial reactive oxygen species (mROS)
Mitochondrial superoxide generation in platelets was analyzed as described earlier [21, 23]. Briefly, platelets pre-treated with inhibitors were challenged with thrombin (0.5 U/ml) for 10 min at RT. Platelets were labelled with MitoSOX Red (5 μM) for 15 min at RT in dark. Level of mROS was studied in labelled platelets by flow cytometry as described above.
Flow cytometric analysis of platelet mitochondrial calcium
Platelets (2.5 × 106/ml) were incubated with mitochondrial calcium indicator dye Rhod-2/AM (5 μM) for 30 min in the dark at RT. Mitochondrial calcium levels are reflective of Rhod-2 fluorescence changes, which were recorded using flow cytometer (Becton Dickinson, model Accuri C6 + ) with a 585/20 band pass filter. Fluorescence from unstimulated platelets was acquired for at least 1 min following which thrombin (0.5 U/ml) was added into the tube using a gel loading tip. Fluorescence was recorded for 5 min for each sample. Mean fluorescence over 30 sec in unstimulated as well as thrombin-stimulated platelets was determined using the BD Accuri C6+ software.
Platelet thrombus formation on immobilized collagen matrix under arterial shear
Platelet adhesion and thrombus growth on immobilized collagen matrix was quantified employing BioFlux (Fluxion Biosciences) microfluidics platform as described earlier [24]. Briefly, High-shear plates were coated with 50 µl collagen (100 μg/ml) at 10 dynes/cm2 for 30 sec and blocked with 1% bovine serum albumin at 10 dynes/cm2 for 15 min, following 1 h incubation at RT. Calcein green (2 μg/ml)-stained platelets (2 × 108/ml) were perfused over collagen at physiological arterial shear rate (1500 sec−1) for 5 min. Platelet adherence and thrombus formation in a fixed field over time was recorded. Representative images from 5 different fields were captured and analyzed using ImageJ (National Institutes of Health). Total area occupied by thrombi at 5 min was estimated by the average accumulation of platelets in 5 representative fields as mentioned earlier.
Cell viability assay
Cell viability was evaluated using a luciferase-coupled ATP quantitation assay kit (CellTiter-Glo viability assay, Promega), which determines the number of viable platelets based on quantitation of ATP, a measure of metabolically active cells [11]. The CellTiter-Glo reagent was added to equal volume of thrombin (0.5 U/ml for 2 h)-stimulated platelet suspension (2.5 × 108/ml) supplemented with 1 mM Ca2+ in 96-well white-walled microtiter plates. The reagent lyses the cells generating luminescent signal, which is proportional to the amount of ATP present in the lysate. After 15 min incubation at RT luminescence was measured in a multimodal microplate reader (BioTek model Synergy H1).
Cytotoxicity assay
Loss of membrane integrity was evaluated by using a luminescence-based kit (CytoTox-Glo cytotoxicity assay, Promega) that measures activity of a distinct intracellular protease (dead-cell protease) leaked extracellularly from membrane-compromised cells [25]. The CytoTox-Glo reagent was added to equal volume of thrombin-stimulated platelets supplemented with 1 mM Ca2+ in 96-well plates and Luminescence was measured as described above. The amount of luminescence directly correlates with the percentage of cells undergoing cytotoxic stress.
Detection of MLKL oligomerization
Platelets pre-supplemented with 1 mM Ca2+ were stimulated with thrombin (0.5 U/ml) for 2 h at 37 °C under non-stirring condition, followed by lysis with equal volume of 2X RIPA buffer (100 mM HEPES, 150 mM NaCl, 2 mM EGTA, 2 mM EDTA, 0.2% SDS and 2% Triton-X100, in presence of protease and phosphatase inhibitors) on the ice. The lysate was subjected twice to freeze and thaw cycle. Cell lysates were mixed with non-reducing sample buffer that lacked 2-mercaptoethanol and boiled at 95 °C for 5 min. Proteins were separated on 10% SDS-PAGE and transferred onto PVDF membrane. Nonspecific protein binding sites were blocked with 5% skimmed milk in 1X-TBST. Membranes were incubated with anti-MLKL antibody (1:1000) overnight. Finally, blots were incubated with HRP-conjugated secondary antibody (1:2500) for 1 h at RT and protein bands were visualized by applying ECL.
Analysis of MLKL accumulation at platelet plasma membrane
Washed human platelets were exposed to thrombin (0.5 U/ml) for 2 h at 37 °C. Cells were fixed on microscope slides with 2% paraformaldehyde for 12 min at RT. Next, cells were washed thrice with 1X PBS and permeabilized using 0.1% Triton X-100 in PBS supplemented with 0.02% BSA for 2 min. Following three subsequent washing steps with PBS, cells were blocked for 10 min with 2% BSA in PBS, and incubated overnight at 4 °C with primary antibody against MLKL (1:50) in 0.2% BSA. Slides were washed three times with PBS and incubated with anti-rabbit Alexa Fluor 488-conjugated secondary antibody (1:100) for 1 h at RT. Slides were examined under Zeiss LSM 700 laser scanning confocal microscope with 63X oil objective (numerical aperture, 1.40) and 1 AU pinhole size. Representative images from 5 to 10 different fields were captured and processed using ZEN imaging software. ImageJ (National Institutes of Health) was employed to analyze the number of cells in a field. Results were representative of at least three independent experiments.
Computational prediction of MLKL phosphorylation at Ser-358 by potential kinases
The online bioinformatics tool Group-based Prediction System (GPS), version 5.0 was employed to predict kinase-specific phosphorylation sites [26]. For this, the canonical sequence of MLKL was retrieved from UniProt (https://www.uniprot.org/) and input to GPS, with the threshold set at ‘All’. Scores for phosphorylation potential at different serine residues were recorded.
Statistical methods
The statistical analyses were performed using Prism 8.4.0 (GraphPad) software. Multiple t test and RM one-way analysis of variance (ANOVA) with Sidak’s multiple comparison test was used for evaluation. Tests were considered significant at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001). Data are presented as mean ± SEM of at least three individual experiments from different blood donors.
Result
Thrombin prompts phosphorylation of MLKL in platelets in phosphoinositide 3-kinase (PI3K)/AKT-dependent manner
MLKL is the terminal effector in pro-inflammatory necroptotic cell death program. Here we demonstrate that, MLKL is abundantly expressed in human platelets. As MLKL activation entails its phosphorylation within the pseudokinase domain, we asked whether the peptide is phosphorylated in platelets exposed to physiological agonists like thrombin. Thrombin (0.5 U/ml) evoked rise (by 34.03%) in MLKL phosphorylation in platelets when probed using an antibody directed at Ser-358 in the activation loop. Platelets were subsequently pre-treated with pharmacological inhibitors of either RIPK3 (GSK’843, 30 μM; GSK’872, 25 μM), MLKL (NSA, 40 μM; TC, 40 μM), PI3K (LY, 80 μM; wort, 500 nM), or AKT1/AKT2 isoforms (AKTi, 2.5 μM) for 10 min at RT, followed by thrombin challenge. Remarkably, thrombin-induced MLKL phosphorylation was significantly averted (by 21.8%, 21.75% and 45.9%, respectively, in cells preincubated with LY, wort and AKTi) (Fig. 1A, C) while RIPK3 inhibition hardly elicited any effect (Fig. 1B, D; please refer to Supplementary Fig. 4A, B for corresponding uncropped original blots), thus positioning PI3K-AKT axis, and not RIPK3, upstream of MLKL phosphorylation in platelets. In consistence, kinase-specific phosphorylation site prediction employing the online bioinformatics tool GPS 5.0 validated Ser-358 residue in MLKL as a potential substrate for AKT2 (Fig. 1E, F) in addition to the known specificity RIPK3 towards this residue. As expected, MLKL inhibitors that block MLKL polymer formation by conjugating with cysteine residues did not affect its phosphorylation, which was in line with the earlier reports [27, 28].
Fig. 1. Thrombin induced phosphorylation of MLKL in PI3K/AKT-dependent manner.

A, B Phosphorylation of MLKL in platelets pre-treated with either vehicle (DMSO), or specific inhibitors of PI3K, AKT1/AKT2, MLKL or RIPK3 as indicated, followed by thrombin (0.5 U/ml)-stimulation for 5 min at 37 ˚C. C (n = 7) and D (n = 5), corresponding densitometric analysis of p-MLKL normalised with MLKL. E Bioinformatics analysis with online tool GPS 5.0 demonstrating Ser-358 in MLKL primary structure as potential phosphorylation target for AKT2 and RIPK3. F The graphical sequence logo suggestive of putative phosphorylation targets of AKT2 in MLKL primary structure. The size of alphabets denotes the probability of amino acids as potential AKT2 substrates at the specific location. Position zero refers to the residue 358 in MLKL structure. Data are presented as mean ± SEM and are representative of at least three different experiments. *p < 0.05 as compared to resting platelets; #p < 0.05 as compared to thrombin-stimulated platelets.
MLKL drives transformation of thrombin-stimulated platelets to haemostatically ‘active’ phenotype independent of necroptosis induction
Thrombin is a potent physiological agonist that provokes platelet aggregation through cognate PAR receptors. Thrombin-induced aggregation of washed human platelets was significantly mitigated (by 23.54% and 41.75%, respectively) when cells were pre-treated with the MLKL inhibitors, NSA or TC (Fig. 2A, B), which also significantly restrained aggregation instigated by other strong agonists like thrombin receptor-activating peptide (TRAP) and collagen (Fig. 2C–F). TRAP-induced aggregation exhibited significant reversal after reaching a peak amplitude in the presence of NSA and TC consistent with destabilization of aggregates (Fig. 2C, D). This could be attributable to conformational recoil of platelet surface integrins αIIbβ3 resulting in decreased affinity towards fibrinogen and collapse of the aggregates, and lack of ability of disintegrated platelet aggregates to congregate again [29]. Collagen-mediated platelet aggregation in whole blood analyzed from electronic impedance, too, was retarded by NSA and TC by 42.1% and 56.14%, respectively (Fig. 2G, H). Platelet aggregation involves fibrinogen bridging between adjacent platelets through its binding to active conformers of αIIbβ3 integrins on platelet surface. Keeping with above observations, TC significantly constrained binding of PAC-1 (antibody targeted against conformationally active αIIbβ3), as well as fibrinogen to thrombin-stimulated platelets by 76.76% and 74.61%, respectively (Fig. 2I–L).
Fig. 2. MLKL has a potential role in regulating thrombin-induced platelet functions.

A, C, E Traces denote aggregation of washed human platelets (2.5 × 108/ml) induced by thrombin (Thr, 0.25 U/ml), TRAP (2 µM), and collagen (Coll, 2 µg/ml) in the absence (tracing 1) or presence of either NSA (40 µM) (tracing 2) or TC (40 µM) (tracing 3). Tracings 4, 5 and 6 in A represents secretion of adenine nucleotides from platelet dense granules induced by thrombin in the presence of vehicle, NSA or TC, respectively. B (n = 6), D (n = 11), and F (n = 9), corresponding bar diagrams denoting mean platelet aggregation. G Platelet aggregation in whole blood stimulated by collagen (0.5 µg/ml) in the presence of vehicle (tracing 1), NSA (80 µM) (tracing 2) or TC (80 µM) (tracing 3). H (n = 4), corresponding bar picture representing mean platelet aggregation. I, K, M Histograms representing binding of FITC-PAC-1, fluorescent fibrinogen and PE-anti-P-selectin antibody, respectively, to platelets pre-treated with thrombin (0.5 U/ml) for 5 min in the presence or absence of MLKL inhibitors, as indicated. J (n = 7), L (n = 7) and N (n = 11), corresponding bar diagrams representing mean fluorescence intensity of binding of FITC-PAC-1, fluorescent fibrinogen and PE-anti-P-selectin antibody, respectively. O (n = 5), bar representing percent secretion of adenine nucleotides from platelet dense granules derived from A (tracings 4–6). Data are presented as mean ± SEM and are representative of at least three different experiments. *p < 0.05 as compared to resting platelets; #p < 0.05 as compared to agonist-stimulated platelets.
Secretion of granule contents is a hallmark of strongly activated platelets. Surface externalization of P-selectin and release of adenine nucleotides (contents of alpha and dense granules, respectively) in thrombin-stimulated platelets, were significantly abrogated in the presence of NSA (by 44.02% and 58.33%) and TC (by 70.40% and 61.27%, respectively) (Fig. 2A, M, N, O). As P-selectin possesses high affinity towards P-selectin glycoprotein ligand-1 (PSGL-1) expressed on leucocyte membranes [30], we next queried possible inhibition of platelet-leucocyte interaction in presence of MLKL inhibitors. TRAP (2 µM) prompted a surge in platelet-monocyte as well as platelet-neutrophil aggregate formation (by 3.12- and 3.53- folds, respectively), which was significantly impaired in the presence of NSA (by 35.77% and 34.90%, respectively), as well as TC (by 33.08% and 37.63% respectively) (Fig. 3J–M).
Fig. 3. MLKL drives transformation of agonist-stimulated platelets to procoagulant phenotype.

A, B (n = 11), phosphatidylserine exteriorization in thrombin (0.5 U/ml)-stimulated platelets. C, D (n = 9), shedding of extracellular vesicles (PEVs) from thrombin (0.5 U/ml)-stimulated platelets. E, F (n = 9), intracellular calcium flux in Fura-2-loaded platelets stimulated with thrombin (0.1 U/ml) in the presence of 1 mM Ca2+. Tracings 1 and 2 represent [Ca2+]i in absence or presence of TC, respectively. G, H Intracellular calcium flux in a population of Fluo-4-loaded platelets against time following exposure to thrombin in presence or absence of TC. I (n = 6), bar diagram representing mean of Fluo-4-positive events in platelet populations over a period of 4 min following addition of vehicle or reagents as indicated. Flow cytometric analysis of monocyte-platelet aggregates (J, K, n = 7) and neutrophil-platelet aggregates (L, M, n = 7) in whole blood stained with anti-CD41a-APC (specific for platelets) and anti-CD14-FITC (specific for neutrophils/monocytes), followed by addition of TRAP (2 μM) in the presence or absence of NSA and TC, as indicated. Amorphous gates were drawn for monocyte (high fluorescence and low SSC) and neutrophil (low fluorescence and high SSC) populations. Data are presented as mean ± SEM and are representative of at least three different experiments. *p < 0.05 as compared to resting platelets; #p < 0.05 as compared to agonist-stimulated platelets.
A sub-population of stimulated platelets externalizes phosphatidylserine (PS) on surface membranes, thus rendering them ‘procoagulant’ at site of vascular injury [31]. Platelets challenged with thrombin were found to have 1.95-fold higher exposure of PS generating procoagulant surface, which was significantly averted upon preincubation of platelets with NSA or TC (by 14.16% and 38.25%, respectively) (Fig. 3A, B). PS exposure was further boosted (by 2.79-fold) when platelets were stimulated with combination of agonists (thrombin plus collagen). However, neither NSA nor TC had any significant inhibitory effect on annexin V binding on platelets exposed to dual agonists (Supplementary Fig. 1A, B), which underscores a relatively redundant albeit facilitatory role of MLKL in platelet PS exposure, and is in agreement with an earlier observation [11]. Phosphorylated MLKL associates with membranes bringing about substantial changes in membrane architecture [28]. We next interrogated whether it also serves to facilitate generation of EVs, which involves budding from plasma membrane consequent upon redistribution of aminophospholipids between membrane leaflets [32, 33]. Upon stimulation platelets shed extracellular vesicles (PEVs) [16, 17, 34, 35], which are pro-coagulant in nature and contribute to haemostatic responses [36]. Remarkably, thrombin-induced rise in PEVs was significantly thwarted upon preincubation with NSA or TC (by 52.77 and 50%, respectively) (Fig. 3C, D). Taken together, above observations strongly underscore the vital role played by phosphorylated MLKL in thrombin-induced haemostatic responses involving platelet membrane, which include platelet aggregation, fibrinogen binding to surface integrins, PS exposure and shedding of EVs. Haemostatic role served by MLKL in platelets was independent of necrosis, as no change in membrane permeability was noted early in the course of platelet stimulation (Fig. 6A).
Fig. 6. Role of MLKL in eliciting membrane permeabilization upon sustained stimulation of platelets.

A (n = 5), extent of leakage of cytosolic proteases from human platelets when cells were stimulated for different time points ranging from 5 min to 2 h at 37 °C with thrombin (0.5 U/ml). B (n = 14), release of cytosolic proteases from thrombin-stimulated platelets at 2 h in the presence of inhibitors of either MLKL or PI3K. Platelets treated with 0.1% Triton X-100 were used as positive control. C (n = 10), cell viability in thrombin (0.5 U/ml)-stimulated platelets in presence of inhibitors of either MLKL or PI3K. Data are presented as mean ± SEM and are representative of at least three different experiments. *P < 0.05 as compared to resting platelets; #P < 0.05 as compared to thrombin-stimulated platelets. Multiple t test, mixed-effect model and one-way ANOVA tests were used to analyse data.
Intracellular Ca2+ being a critical regulator of platelet activation [18], we next evaluated the role of MLKL in calcium dynamics in thrombin-stimulated platelets. As expected, thrombin provoked a rise in cytosolic free calcium in Fura-2-loaded washed platelets (in presence of 1 mM extracellular Ca2+) by 88.94%, which was retarded by 41.24% following exposure to TC (Fig. 3E, F). Notably, when cells were pre-incubated with 2 mM EGTA, rise in intracellular calcium upon thrombin stimulation was restricted to only 20 %, which remained further unaffected in presence of TC (Supplementary Fig. 2A–E). These findings strongly suggest a putative role of MLKL in facilitating calcium entry from outside but not on calcium mobilization from intracellular stores following platelet stimulation.
In order to analyze cell population with raised intracellular calcium, Fluo-4-loaded platelets (2 × 106/ml) were subjected to flow cytometry. After appropriate gating events were analyzed within the time lapse 1.5–5.0 min. Addition of thrombin to platelet suspension pre-supplemented with 1 mM calcium triggered an impressive rise in mean fluorescence of platelets (by 2.3-fold) compared to the cells before stimulation. Percentage of platelets with fluorescence above the threshold (indicated by the horizontal line) rose from 13.4% in unstimulated cells to 67.2% of Fluo-4 positive events upon thrombin stimulation (Fig. 3G, upper left and right quadrants, respectively). As expected, reduced fluorescence (by 26.4%) was recorded in thrombin-stimulated platelets preincubated with TC (Fig. 3H, I). Above series of findings are collectively suggestive of non-canonical role of MLKL in regulating platelet activation independent of necroptosis.
MLKL augments formation of platelet thrombi on immobilized collagen under high shear
Our results so far demonstrate contribution of MLKL in transformation of platelets to ‘activated’ phenotype upon agonist challenge. We next probed whether MLKL has a role in promoting platelet dynamic adhesion as well as thrombus formation on immobilized collagen under arterial shear (1500 s−1) employing BioFlux microfluidics platform. Washed human platelets were preincubated either with NSA, TC or vehicle (control), and allowed to flow over collagen-coated surface for 10 min. Remarkably, total surface area covered by platelet thrombi was significantly diminished in the presence of either NSA or TC (by 39.75% and 55.92%, respectively) (Fig. 4B, D), which underscores a central role of MLKL in regulation of platelet-mediated thrombogenicity.
Fig. 4. MLKL prompts formation of platelet thrombi on immobilized collagen under high shear.

Calcein-stained human platelets were perfused over immobilised collagen matrix for 10 min in a microfluidic chamber at a shear rate of 1500 s-1. A, C Confocal images showing accumulation of platelets over collagen at 1, 5, and 10 min following start of perfusion in control (DMSO) as well as NSA (40 μM)- and TC (40 μM)-preincubated cells. B (n = 4) and D (n = 4), bar diagrams denoting per cent surface area covered with platelet thrombi after 5 min of cell perfusion. Total thrombi calculated as the average surface area covered by platelets in 4 representative fields. Figures are representative of ≥3 individual experiments (mean ± SEM). #p < 0.05 as compared to collagen-stimulated platelets.
MLKL regulates mitochondrial function and bioenergetics in thrombin-stimulated platelets
Although anucleate, platelets possess well-coupled functional mitochondria at an average 4 to 8 units per cell [37–40]. Studies including ours have implicated aerobic glycolysis as well as mitochondrial OXPHOS in sustaining diverse functional states of platelets that could be highly energy-demanding [21, 22, 41–46]. Metabolic plasticity is the key to enabling platelets to meet their extraordinary energy demand with so few mitochondria. As MLKL emerges to be a critical player in agonist-induced platelet activation, we next queried its contribution to mitochondrial oxygen consumption rate (OCR) in resting as well as thrombin-stimulated platelets.
Oxygen flux was measured in platelet suspension under stirring at high resolution (sampling at 2 s intervals) employing a Clark amperometric electrode. This system in cuvette format closely resembles the platelet aggregometry of Born [22]. Pre-incubation with NSA was found to have little impact on routine respiration in unstimulated platelets. However, the surge in oxygen consumption evoked by thrombin (0.25 U/ml) was partially mitigated (by 22.22%) in platelets treated with NSA (Fig. 5A–C), thus underlining a role for MLKL in facilitating mitochondrial electron flow. Platelets exposed to oligomycin (1 µg/ml), a specific inhibitor of complex V (ATP synthase) activity, exhibited significant drop in OCR with negligible leak respiration (proton leak) (Fig. 5A, B, E). OXPHOS represents coupling between oxygen consumption and ATP synthase activity and is determined by excluding leak from routine respiration. Strikingly, thrombin-induced OXPHOS was significantly impaired (by 27.5%) in the presence of NSA (Fig. 5A, B, D) associated with parallel rise in proton leak. As leak respiration represents oxygen consumption driven by proton mobilization across inner mitochondrial membrane independent of complex V activity, above findings are overwhelmingly suggestive of a stabilizing effect of MLKL on inner mitochondrial membrane that restrains proton leak and thus upholds sustenance of well-coupled mitochondria.
Fig. 5. MLKL contributes to sustenance of well-coupled functional mitochondria in thrombin-stimulated platelets.

Polarograms representing rate of oxygen consumption (red trace) and oxygen concentration (blue trace) in thrombin (0.25 U/ml)-stimulated platelets treated either with vehicle (A) or NSA (40 μM) (B). C (n = 4), bar diagram representing OCR in unstimulated and stimulated cells. D (n = 4), E (n = 4) and F (n = 5), bar diagrams representing changes in OXPHOS, proton leak and lactate release rate, respectively. G, I Histogram overlays representing fluorescence of MitoTracker Red and MitoSOX Red, respectively, in thrombin (0.5 U/ml)-stimulated platelets pre-incubated with either vehicle or MLKL inhibitors, as indicated. H (n = 7) and J (n = 6), bar diagrams representing mean fluorescence intensities of MitoTracker Red- and MitoSOX Red-positive platelets, respectively. K–M Quadrant dot plots representing fluorescence of Rhod-2-positive cells in platelet populations preincubated with either NSA or TC followed by addition of thrombin (0.5 U/ml) in presence of 1 mM calcium. N (n = 8), bar diagram representing mean fluorescence intensity of Rhod-2-positive cells. Data are presented as mean ± SEM and are representative of at least three different experiments. *P < 0.05 as compared to resting platelets; #P < 0.05 as compared to thrombin-stimulated platelets.
Leakage of proton from intermembrane space to mitochondrial matrix would ensue dissipation of transmembrane potential (ΔΨm) and consequential drop in mitochondrial ROS generation [21, 47]. As expected, ΔΨm and mROS were significantly mitigated in presence of NSA (by 33.91% and 19.85%, respectively), and TC (by 28.62% and 29.58%, respectively) in thrombin (0.5 U/ml)-stimulated platelets (Fig. 5G–J). In parallel, rise in mitochondrial calcium influx elicited by thrombin, which is dependent on ΔΨm [48], was significantly alleviated in the presence of NSA and TC (by 23.53% and 20.51%, respectively) (Fig. 5K–N). Calcium being an important driver for TCA cycle leading to mitochondrial oxygen consumption [49], this could explain drop in thrombin-induced spike in OCR in the presence of MLKL inhibitors.
Recent studies from our lab and others have demonstrated a preferential switch in energy metabolism to Warburg’s aerobic glycolysis over OXPHOS in activated platelets due to regulated pyruvate flux into mitochondria [22, 50]. As inhibition of MLKL led to impaired platelet activation, we next investigated its effect on rate of glycolysis. MLKL inhibitors, NSA and TC, significantly reduced lactate release in thrombin (0.5 U/ml)-stimulated platelets (by 66.2% and 81.4%, respectively) (Fig. 5F), reflective of compromised aerobic glycolysis. Thus, simultaneous inhibition of mitochondrial respiration and glycolysis by MLKL inhibitors would deplete ATP level in platelets [43, 44], which would explain significant decline in energy-intensive platelet responses observed in our study.
Taken together, above findings suggest that MLKL reduces proton leak across inner mitochondrial membrane and thus bolsters mitochondrial respiration through ΔΨm-dependent calcium entry into the matrix. We have previously reported that CCCP, a protonophore, disrupts ΔΨm leading to impaired mROS generation and mitochondrial calcium influx, which in turn inhibits platelet proaggregatory and procoagulant activities, respectively [21]. Thus, by maintaining mitochondria in a well-coupled state MLKL sustains mitochondrial ATP production, ΔΨm, mROS level and mitochondrial calcium transients in stimulated platelets, which are essential for platelet activation responses to agonist challenge [21].
MLKL mediates membrane permeabilization upon sustained stimulation of platelets with thrombin
Upon activation and accomplishment of haemostatic responses, platelets fulfil their raison d’eˆtre by getting necrosed and cleared from the lesion site [51, 52]. As MLKL contributed significantly in evolution of platelets to functional ‘prothrombotic’ units, we next queried its ensuing role in induction of necrosis. Exposure of cells to thrombin (0.5 U/ml) for a period spanning from 5 min to 2 h at 37 °C prompted progressive permeabilization of platelet membrane allowing growing leakage of cytosolic proteases (Fig. 6A). The leakage was found to be maximum at 2 h thrombin stimulation, which was 93.67% higher than the control counterparts not exposed to thrombin. Preincubation of platelets with either NSA, TC or LY brought about significant drop in the extent of leakage (by 37.55%, 45,38% and 36.71%, respectively) at 2 h, thus implicating MLKL and its upstream kinase PI3K in membrane permeabilization (Fig. 6B). In parallel with loss in membrane integrity, sustained stimulation of platelets, too, led to progressive decline in cell viability (by 45.65% at 2 h), which was significantly restored in the presence of inhibitors of MLKL as well as PI3K (by 50.17% and 39.66%, respectively) (Fig. 6C). These observations underscore a critical role of MLKL in temporal choreography of events underlying agonist-induced platelet activation progressing towards cell death.
MLKL forms oligomers and accumulates as surface ‘clusters’ in thrombin-stimulated platelets
Following phosphorylation, MLKL oligomerizes and assembles into higher order species that translocate to membrane compartments and disrupt membrane integrity [7, 53, 54]. As MLKL is phosphorylated in PI3K/AKT-dependent manner in thrombin-stimulated platelets, we asked whether it, too, undergoes oligomerization. MLKL was found to form dimers (~100 KDa) on non-reducing gels in platelets challenged with thrombin for 2 h at 37 °C, which was reversed in presence of inhibitors of MLKL, PI3K, as well as AKT (Fig. 7; please refer to Supplementary Fig. 4C, for corresponding uncropped original blots).
Fig. 7. Dimerization of MLKL in thrombin-stimulated platelets.
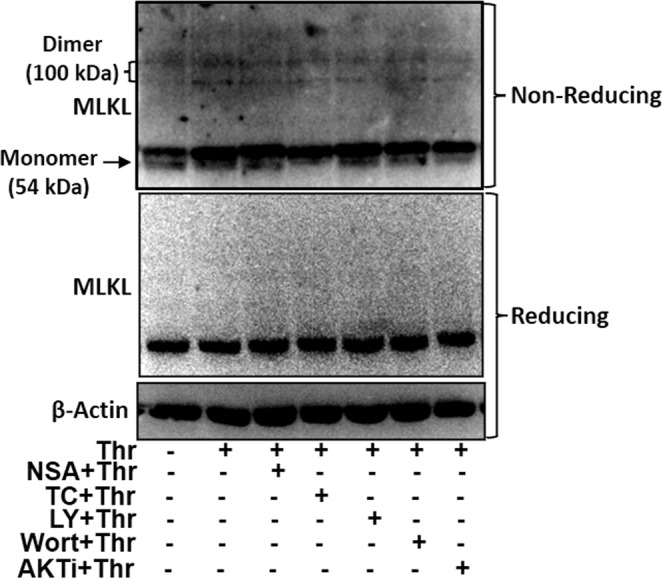
Immunoblots showing expression of MLKL as dimers and monomers in platelets exposed to thrombin for 2 h in presence or absence of inhibitors as indicated. SDS-PAGE was carried out either under non-reducing (to resolve MLKL monomers and dimers) as well as reducing conditions.
Next, we interrogated whether MLKL traffics to plasma membrane of stimulated platelets to form surface clusters that would trigger membrane rupture. In unstimulated cells MLKL was found to reside in small puncta that were distributed evenly throughout cytoplasm. Sustained platelet challenge with thrombin (0.5 U/ml) for 2 h in presence of 1 mM Ca2+ prompted remarkable focal clustering of MLKL at cell periphery that were readily detectable by immunofluorescence employing an anti-MLKL antibody (Fig. 8). An MLKL cluster was defined as a fluorescent particle (size >0.03 µ2) located at cell surface/ periphery, which was analysed using Fiji (Image J) software. MLKL clusters were found to be significantly higher in number in thrombin-stimulated platelets than within the control counterparts (Supplementary Fig. 3A). Surface translocation of MLKL was averted in cells pre-treated with inhibitors of either MLKL (NSA and TC), or PI3K (LY and Wort), or AKT (AKTi) (Fig. 8B), which also reduced the number of MLKL clusters on platelet surface (Supplementary Fig. 3B). Collectively, these findings were consistent with accumulation of phosphorylated MLKL at plasma membrane upon prolonged thrombin stimulation in PI3K/AKT-dependent manner, which eventually triggers membrane blowout and ensuing necroptotic cell death.
Fig. 8. Trafficking of MLKL to surface membrane of thrombin-stimulated platelets to accumulate as focal clusters.

A Confocal images representing staining of MLKL (shown by arrowheads) in resting, as well as thrombin-stimulated platelets stored for 2 h at 37 °C in presence of inhibitors as indicated. B (n = 6), bar diagram representing concentration of MLKL at cell periphery (calculated by dividing number of the cells with MLKL accumulation at periphery with the total number of cells from 5 different fields). Data are presented as mean ± SEM and are representative of at least three different experiments. *P < 0.05 as compared to resting platelets; #P < 0.05 as compared to thrombin-stimulated platelets. Mixed-effect model were used to analyse data.
Discussion
While platelets play a vital role in preventing unwanted blood loss at vessel injury, hyperactivity of these cells is associated with serious thrombotic consequences like acute myocardial infarction and ischemic stroke. Thus, it is imperative to thoroughly understand the mechanistic basis of platelet activation for designing effective therapeutic strategy for cardiovascular events. Platelets have a circulating life span of 10–12 days, following which they undergo apoptosis and are subsequently cleared by macrophages, if they do not encounter activating stimuli [1–4]. However, the major fate of procoagulant platelets generated upon vascular injury is necrotic cell death [51]. Although MLKL, the terminal executioner of necroptosis, is expressed in platelets [11], its relevance in these cells remains obscure. While investigating necroptosis signaling in human platelets, we unexpectedly came across the vital role played by MLKL in transformation of agonist-challenged platelets to prothrombotic phenotypes.
In order to unravel the role of MLKL in human platelets, we employed two pharmacologically unrelated inhibitors of MLKL, namely NSA and TC, which prevent MLKL oligomerization and disulfide bond formation by covalently binding to Cys-88 and Cys-86 residues, respectively [55]. Both NSA and TC prevent MLKL translocation to plasma membrane without affecting MLKL phosphorylation [55]. The two inhibitors exhibit specificity towards human MLKL and do not show activity against murine MLKL, which limits their use in mouse models in vivo [55]. NSA and TC were able to significantly retard thrombin-induced haemostatic responses in platelets, which include platelet aggregation, fibrinogen binding, secretion of granule contents, platelet-leucocyte interactions, shedding of EVs, rise in intracellular free calcium and formation platelet thrombi upon immobilized collagen. These results are consistent with the previous report showing impaired integrin activation and P-selectin externalization in MLKL-deficient murine platelets [11]. Notably, NSA and TC curbed phosphatidylserine exposure on human platelets challenged with thrombin but not with a stronger stimulus (combination of thrombin and collagen). This observation, too, is in agreement with the report by Moujalled et al. [11], which failed to demonstrate an effect of MLKL on procoagulant platelet function (using Mlkl − /− murine platelets). Considering the fact that murine platelets dominantly express high affinity PAR3 thrombin receptors as against to PAR1/PAR4 in human counterparts, the data overall support a relatively redundant albeit facilitatory role of MLKL in eliciting platelet PS exposure. The differences in the extent of inhibition exhibited by NSA and TC are likely attributable to their significant differences in pharmacodynamics as demonstrated earlier [55]. TC (EC50 2 ± 0.6 nM) has been found to be much more potent than NSA (EC50 447 ± 32 nM) in preventing MLKL-mediated necroptosis [55].
In parallel to promoting platelet prothrombotic activity, MLKL was found to stabilize inner mitochondrial membrane in platelets as MLKL antagonists disrupted mitochondrial transmembrane potential, accompanied with augmented proton leak, drop in OXPHOS and rise in both mitochondrial calcium as well as ROS. These observations underscored the seminal contribution of MLKL in sustaining mitochondrial energy generation that underlie platelet activation responses. The inhibitors, too, compromised aerobic glycolysis, the hallmark of agonist-stimulated platelets [22]. Thus, parallel impairment of mitochondrial respiration and glycolysis by MLKL inhibitors would exhaust ATP level in platelets [42–44], which would explain significant decline in energy-demanding platelet responses such as granule secretion observed in our study and attenuated JON/A binding and prolonged tail re-bleeding time in MLKL-deficient murine platelets reported earlier [11]. Raised intracellular calcium mediated by MLKL may also underscore degranulation [56] as well as cellular fragmentation observed in thrombin-stimulated platelets [57, 58]. Haemostatic role served by MLKL in platelets was independent of necrosis as no change in membrane permeability was noted early in the course of platelet stimulation. Platelet activation induced by collagen, another strong agonist, was also impeded in the presence of MLKL inhibitors.
Upon induction of necroptosis MLKL undergoes phosphorylation at Ser-358 by the upstream kinase RIPK3, which prompts MLKL oligomerization and localization to membranes [59, 60]. Intriguingly thrombin, in absence of known necroptosis inducers, was also able to incite MLKL phosphorylation at same serine residue in platelets in a RIPK3-independent manner. In silico analyses allowed us to identify PI3K and AKT, the downstream effectors of thrombin in platelets [26], as potential kinases for MLKL at Ser-358. Notably, PI3K has earlier been reported to function upstream of RIPK1-RIPK3-MLKL axis [61]. Keeping with above, inhibition of either PI3K or AKT precluded agonist-induced platelet responses, as reported earlier [62, 63] as well as MLKL phosphorylation, thus establishing the key role of thrombin-PI3K-AKT-MLKL axis in platelet activation. Zhang et al. [12]. have demonstrated an MLKL-independent role for RIPK3 in murine platelet activation and thrombus formation possibly mediated by RIPK3-AKT and/or RIPK3-Gα13 signaling axis as suggested in the article. Incidentally, the study does not report MLKL phosphorylation and activity in RIPK3-deficient platelets. It is reasonable here to speculate that parallel pathways operate in platelets leading to thrombogenesis, one driven by RIPK3 and its downstream effectors, and the other mediated through thrombin-PI3K-AKT-MLKL axis, though species-specific differences cannot be ruled out.
Following their transformation to active haemostatic units as fulfilment of raison d’eˆtre, platelets are fated to get necrosed and cleared from the lesion site [51, 52] A subpopulation of agonist-stimulated platelets that exhibits procoagulant properties, too, become necrotic with loss of membrane integrity [51]. We interrogated if MLKL navigates platelets towards necrosis subsequent to its participation in prothrombotic events. MLKL trafficking and plasma membrane accumulation have been described as crucial necroptosis checkpoints. Phosphorylated MLKL forms oligomers (mostly dimers, trimers or tetramers through interaction within its N-terminal 4HB domains) that translocate to plasma membranes to orchestrate membrane permeabilization via an incompletely-understood mechanism [5]. We observed formation of MLKL dimers (~100 KDa) in platelets stimulated for 2 h with thrombin, which was reversed by inhibitors of MLKL as well as PI3K/AKT. In parallel, uniform distribution of MLKL in cytosol of quiescent platelets underwent major phenotypical alteration to form peripheral focal clusters upon sustained challenge with thrombin, which, too, was thwarted by inhibitors of either MLKL, PI3K or AKT. Although thrombin-induced platelet haemostatic responses were early membrane-based events, we did neither observe MLKL oligomerization nor peripheral focal clustering following 5 min stimulation with thrombin. Thus, it is fairly reasonable to believe that, phosphorylated MLKL translocates to membranes of agonist-challenged platelets to unleash a prothrombotic phenotype, supported by boosted energy generation, though its oligomerization in initial phases remains undetected.
Sustained stimulation with agonists, however, ensures enough peripheral accumulation of MLKL as focal clusters to trigger membrane blowout and ensuing necroptotic lysis of platelets. In support of this, we observed progressive permeabilization of platelet membrane for a period ranging from 5 min to 2 h following thrombin exposure. Inhibitors of MLKL as well as PI3K brought about significant drop in the extent of leakage. Concurrently, there was consistent decline in cell viability, which was significantly restored by inhibitors of either MLKL or PI3K. Accumulation of phosphorylated MLKL at intercellular junctions within platelet aggregate could also accelerate necroptosis in neighbouring platelets in a cell-extrinsic manner. These observations are suggestive of critical role played by PI3K-AKT-MLKL axis in successive stages in the choreography of events that underlie agonist-induced platelet activation and subsequent cell death (Fig. 9). The temporally-driven role of MLKL in platelets is relevant to mechanisms of thrombus formation and resolution in vivo, where thrombus contraction and lysis take place spanning over a timescale in hours, with two-fold increase in fibrin content due to persistent thrombin activity and 50% decline in platelet number in each hour due to disintegration/cell death [57]. Further organization and resolution of thrombus continue to progress over timescale spanning from 1 to 5 days within coronary arterial thrombi [64, 65].
Fig. 9. Scheme depicting the contribution of MLKL in agonist-induced platelet activation and necroptosis on a temporal scale.

Simplified schematic representation of thrombin-stimulated platelets shows that MLKL plays a key role in regulating activation and necroptotic signaling on a chronological scale.
Although there have been previous reports demonstrating the role of MLKL in murine platelet activation as well as haemostasis and venous thrombosis in mice, our study provides several novel insights into the regulation and role of MLKL in human platelets. We demonstrate RIPK3-independent regulation of MLKL in stimulated platelets that is mediated through PI3K-Akt signaling axis. We establish the role of MLKL in human platelet responses to agonist stimulation that include integrin activation, dense and alpha granule secretion, rise in cytosolic calcium, shedding of extracellular vesicles and platelet-leukocyte interactions using MLKL inhibitors. We show that MLKL is necessary for human platelet thrombus formation on immobilized collagen under arterial shear ex vivo using a microfluidics platform. We discover that MLKL is essential for maintaining well-coupled functional mitochondria as well as glycolytic flux in stimulated platelets that modulate platelet energy metabolism. We demonstrate progression of phosphorylated MLKL to its oligomerization, membrane localization and induction of necroptosis on a temporal scale upon prolonged stimulation of platelets with thrombin. Our findings could potentially unravel new avenues for developing anti-platelet therapies.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author contributions
DD designed and supervised the entire work; ME, PPK, VS, SNC, SKM and RNC provided samples and performed various experiments; DD, ME, and PPK analyzed results and wrote the manuscript.
Funding
This research was supported by a J.C. Bose National Fellowship and grants received by DD from Indian Council of Medical Research (ICMR) under Center for Advanced Research (CAR), and Science and Engineering Research Board (SERB), Government of India. ME and VS are recipients of CSIR-SRF and UGC-SRF, respectively.
Data availability
All relevant data that support the findings of this study are within the paper. Raw data incorporated in Microsoft Excel and/or GraphPad Prism files are available on request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
6/21/2023
In this article the Mohammad Ekhlak’s ORCID has been corrected.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-023-01181-6.
References
- 1.Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, et al. Programmed anuclear cell death delimits platelet life span. Cell. 2007;128:1173–86. doi: 10.1016/j.cell.2007.01.037. [DOI] [PubMed] [Google Scholar]
- 2.Nayak MK, Kulkarni PP, Dash D. Regulatory role of proteasome in determination of platelet life span. J Biol Chem. 2013;288:6826–34. doi: 10.1074/jbc.M112.403154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumari S, Chaurasia SN, Nayak MK, Mallick RL, Dash D. Sirtuin inhibition induces apoptosis-like changes in platelets and thrombocytopenia. J Biol Chem. 2015;290:12290–9. doi: 10.1074/jbc.M114.615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebois M, Dowling MR, Gangatirkar P, Hodgkin PD, Kile BT, Alexander WS, et al. Regulation of platelet lifespan in the presence and absence of thrombopoietin signaling. J Thromb Haemost. 2016;14:1882–7. doi: 10.1111/jth.13397. [DOI] [PubMed] [Google Scholar]
- 5.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim EH, Wong SW, Martinez J. Programmed Necrosis and Disease:We interrupt your regular programming to bring you necroinflammation. Cell Death Differ. 2019;26:25–40. doi: 10.1038/s41418-018-0179-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Tong A, Zhang Q, Wei Y, Wei X. The molecular mechanisms of MLKL-dependent and MLKL-independent necrosis. J Mol Cell Biol. 2021;13:3–14. doi: 10.1093/jmcb/mjaa055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM Kinases Promote Necroptosis by Regulating Oligomerization of MLKL. Mol Cell. 2019;75:457–68. doi: 10.1016/j.molcel.2019.05.022. [DOI] [PubMed] [Google Scholar]
- 9.Murphy JM. The Killer Pseudokinase Mixed Lineage Kinase Domain-Like Protein (MLKL). Cold Spring Harb Perspect Biol. 2020;12:a036376. [DOI] [PMC free article] [PubMed]
- 10.Petrie EJ, Sandow JJ, Jacobsen AV, Smith BJ, Griffin MDW, Lucet IS, et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat Commun. 2018;9:2422. doi: 10.1038/s41467-018-04714-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moujalled D, Gangatirkar P, Kauppi M, Corbin J, Lebois M, Murphy JM, et al. The necroptotic cell death pathway operates in megakaryocytes, but not in platelet synthesis. Cell Death Dis. 2021;12:133. doi: 10.1038/s41419-021-03418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Zhang J, Yan R, Tian J, Zhang Y, Zhang J, et al. Receptor-interacting protein kinase 3 promotes platelet activation and thrombosis. Proc Natl Acad Sci USA. 2017;114:2964–9. doi: 10.1073/pnas.1610963114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakazawa D, Desai J, Steiger S, Müller S, Devarapu SK, Mulay SR, et al. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 2018;4:6. doi: 10.1038/s41420-018-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulkarni PP, Ekhlak M, Singh V, Kailashiya V, Singh N, Dash D. Fatty acid oxidation fuels agonist-induced platelet activation and thrombus formation: Targeting β-oxidation of fatty acids as an effective anti-platelet strategy. Faseb j. 2023;37:e22768. doi: 10.1096/fj.202201321RR. [DOI] [PubMed] [Google Scholar]
- 15.Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P. Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis. Commun Biol. 2018;1:6. doi: 10.1038/s42003-017-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kushwaha G, Chaurasia SN, Pandey A, Dash D. Characterization of fibrinogen binding on platelet-derived extracellular vesicles. Thromb Res. 2018;172:135–8. doi: 10.1016/j.thromres.2018.10.021. [DOI] [PubMed] [Google Scholar]
- 17.Chaurasia SN, Kushwaha G, Kulkarni PP, Mallick RL, Latheef NA, Mishra JK, et al. Platelet HIF-2α promotes thrombogenicity through PAI-1 synthesis and extracellular vesicle release. Haematologica. 2019;104:2482–92. doi: 10.3324/haematol.2019.217463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallick RL, Kumari S, Singh N, Sonkar VK, Dash D. Prion protein fragment (106-126) induces prothrombotic state by raising platelet intracellular calcium and microparticle release. Cell Calcium. 2015;57:300–11. doi: 10.1016/j.ceca.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. doi: 10.1016/S0021-9258(19)83641-4. [DOI] [PubMed] [Google Scholar]
- 20.Barnard MR, Krueger LA, Frelinger AL, 3rd, Furman MI, Michelson AD. Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom. 2003;Chapter 6:Unit 6.15. doi: 10.1002/0471142956.cy0615s24. [DOI] [PubMed] [Google Scholar]
- 21.Kulkarni PP, Ekhlak M, Sonkar VK, Dash D. Mitochondrial ATP generation in stimulated platelets is essential for granule secretion but dispensable for aggregation and procoagulant activity. Haematologica. 2022;107:1209–13. doi: 10.3324/haematol.2021.279847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni PP, Tiwari A, Singh N, Gautam D, Sonkar VK, Agarwal V, et al. Aerobic glycolysis fuels platelet activation: small-molecule modulators of platelet metabolism as anti-thrombotic agents. Haematologica. 2019;104:806–18. doi: 10.3324/haematol.2018.205724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masselli E, Pozzi G, Vaccarezza M, Mirandola P, Galli D, Vitale M, et al. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int J Mol Sci. 2020;21:4866. [DOI] [PMC free article] [PubMed]
- 24.Tiwari A, Gautam D, Kulkarni PP, Ekhlak M, Sonkar VK, Agrawal V, et al. Noncanonical Sonic Hedgehog signaling amplifies platelet reactivity and thrombogenicity. Blood Adv. 2022;6:5024–40. doi: 10.1182/bloodadvances.2021006560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morgan JE, Prola A, Mariot V, Pini V, Meng J, Hourde C, et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat Commun. 2018;9:3655. doi: 10.1038/s41467-018-06057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Xu H, Lin S, Deng W, Zhou J, Zhang Y, et al. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genomics Proteom Bioinforma. 2020;18:72–80. doi: 10.1016/j.gpb.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci USA. 2017;114:E7450–e9. doi: 10.1073/pnas.1707531114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samson AL, Zhang Y, Geoghegan ND, Gavin XJ, Davies KA, Mlodzianoski MJ, et al. MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat Commun. 2020;11:3151. doi: 10.1038/s41467-020-16887-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filkova AA, Martyanov AA, Garzon Dasgupta AK, Panteleev MA, Sveshnikova AN. Quantitative dynamics of reversible platelet aggregation: mathematical modelling and experiments. Sci Rep. 2019;9:6217. doi: 10.1038/s41598-019-42701-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gerrits AJ, Frelinger AL, 3rd, Michelson AD. Whole blood analysis of leukocyte-platelet aggregates. Curr Protoc Cytom. 2016;78:6.15.1–6.0. doi: 10.1002/cpcy.8. [DOI] [PubMed] [Google Scholar]
- 31.Reddy EC, Rand ML. Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front Cardiovasc Med. 2020;7:15. doi: 10.3389/fcvm.2020.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Antwi-Baffour S, Adjei J, Aryeh C, Kyeremeh R, Kyei F, Seidu MA. Understanding the biosynthesis of platelets-derived extracellular vesicles. Immun Inflamm Dis. 2015;3:133–40. doi: 10.1002/iid3.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Russell AE, Sneider A, Witwer KW, Bergese P, Bhattacharyya SN, Cocks A, et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: an ISEV position paper arising from the ISEV membranes and EVs workshop. J Extracell Vesicles. 2019;8:1684862. doi: 10.1080/20013078.2019.1684862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94:3791–9. doi: 10.1182/blood.V94.11.3791. [DOI] [PubMed] [Google Scholar]
- 35.Lopez E, Srivastava AK, Burchfield J, Wang YW, Cardenas JC, Togarrati PP, et al. Platelet-derived- extracellular vesicles promote hemostasis and prevent the development of hemorrhagic shock. Sci Rep. 2019;9:17676. doi: 10.1038/s41598-019-53724-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dyer MR, Alexander W, Hassoune A, Chen Q, Brzoska T, Alvikas J, et al. Platelet-derived extracellular vesicles released after trauma promote hemostasis and contribute to DVT in mice. J Thromb Haemost. 2019;17:1733–45. doi: 10.1111/jth.14563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–83. doi: 10.1182/blood-2014-05-573543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shuster RC, Rubenstein AJ, Wallace DC. Mitochondrial DNA in anucleate human blood cells. Biochem Biophys Res Commun. 1988;155:1360–5. doi: 10.1016/S0006-291X(88)81291-9. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi T, Tanaka S, Hori Y, Hirayama F, Sato EF, Inoue M. Role of mitochondria in the maintenance of platelet function during in vitro storage. Transfus Med. 2011;21:166–74. doi: 10.1111/j.1365-3148.2010.01065.x. [DOI] [PubMed] [Google Scholar]
- 40.Melchinger H, Jain K, Tyagi T, Hwa J. Role of platelet mitochondria: life in a nucleus-free zone. Front Cardiovasc Med. 2019;6:153. doi: 10.3389/fcvm.2019.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holmsen H. Energy Metabolism and platelet responses. Vox Sang. 1981;40:1–7. doi: 10.1111/j.1423-0410.1981.tb00732.x. [DOI] [PubMed] [Google Scholar]
- 42.Holmsen H, Robkin L, Day HJ. Effects of antimycin A and 2-deoxyglucose on secretion in human platelets. Differential inhibition of the secretion of acid hydrolases and adenine nucleotides. Biochem J. 1979;182:413–9. doi: 10.1042/bj1820413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmsen H, Setkowsky CA, Day HJ. Effects of antimycin and 2-deoxyglucose on adenine nucleotides in human platelets. Role of metabolic adenosine triphosphate in primary aggregation, secondary aggregation and shape change of platetets. Biochem J. 1974;144:385–96. doi: 10.1042/bj1440385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaczara P, Sitek B, Przyborowski K, Kurpinska A, Kus K, Stojak M, et al. Antiplatelet effect of carbon monoxide is mediated by NAD(+) and ATP depletion. Arterioscler Thromb Vasc Biol. 2020;40:2376–90. doi: 10.1161/ATVBAHA.120.314284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ravi S, Chacko B, Sawada H, Kramer PA, Johnson MS, Benavides GA, et al. Metabolic plasticity in resting and thrombin activated platelets. PLoS One. 2015;10:e0123597. doi: 10.1371/journal.pone.0123597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verhoeven AJ, Mommersteeg ME, Akkerman JW. Metabolic energy is required in human platelets at any stage during optical aggregation and secretion. Biochim Biophys Acta. 1984;800:242–50. doi: 10.1016/0304-4165(84)90402-1. [DOI] [PubMed] [Google Scholar]
- 47.Cardenes N, Corey C, Geary L, Jain S, Zharikov S, Barge S, et al. Platelet bioenergetic screen in sickle cell patients reveals mitochondrial complex V inhibition, which contributes to platelet activation. Blood. 2014;123:2864–72. doi: 10.1182/blood-2013-09-529420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choo HJ, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol. 2012;32:2946–55. doi: 10.1161/ATVBAHA.112.300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- 50.Nayak MK, Ghatge M, Flora GD, Dhanesha N, Jain M, Markan KR, et al. The metabolic enzyme pyruvate kinase M2 regulates platelet function and arterial thrombosis. Blood. 2021;137:1658–68. doi: 10.1182/blood.2020007140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hua VM, Abeynaike L, Glaros E, Campbell H, Pasalic L, Hogg PJ, et al. Necrotic platelets provide a procoagulant surface during thrombosis. Blood. 2015;126:2852–62. doi: 10.1182/blood-2015-08-663005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood. 2010;116:2011–8. doi: 10.1182/blood-2010-01-261669. [DOI] [PubMed] [Google Scholar]
- 53.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci USA. 2014;111:15072–7. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yan B, Liu L, Huang S, Ren Y, Wang H, Yao Z, et al. Discovery of a new class of highly potent necroptosis inhibitors targeting the mixed lineage kinase domain-like protein. Chem Commun. 2017;53:3637–40. doi: 10.1039/C7CC00667E. [DOI] [PubMed] [Google Scholar]
- 56.Lopez E, Bermejo N, Berna-Erro A, Alonso N, Salido GM, Redondo PC, et al. Relationship between calcium mobilization and platelet α- and δ-granule secretion. A role for TRPC6 in thrombin-evoked δ-granule exocytosis. Arch Biochem Biophys. 2015;585:75–81. doi: 10.1016/j.abb.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 57.Kim OV, Nevzorova TA, Mordakhanova ER, Ponomareva AA, Andrianova IA, Le Minh G, et al. Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica. 2019;104:1866–78. doi: 10.3324/haematol.2018.202309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arce NA, Li R. The secret afterlife of platelets. Haematologica. 2019;104:1699–701. doi: 10.3324/haematol.2019.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garnish SE, Meng Y, Koide A, Sandow JJ, Denbaum E, Jacobsen AV, et al. Conformational interconversion of MLKL and disengagement from RIPK3 precede cell death by necroptosis. Nat Commun. 2021;12:2211. doi: 10.1038/s41467-021-22400-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johnston A, Wang Z. Necroptosis: MLKL Polymerization. J Nat Sci. 2018;4:e513. [PMC free article] [PubMed]
- 61.Hu S, Chang X, Zhu H, Wang D, Chen G. PI3K mediates tumor necrosis factor induced-necroptosis through initiating RIP1-RIP3-MLKL signaling pathway activation. Cytokine. 2020;129:155046. doi: 10.1016/j.cyto.2020.155046. [DOI] [PubMed] [Google Scholar]
- 62.Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–10. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yi W, Li Q, Shen J, Ren L, Liu X, Wang Q, et al. Modulation of platelet activation and thrombus formation using a pan-PI3K inhibitor S14161. PLoS One. 2014;9:e102394. doi: 10.1371/journal.pone.0102394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rittersma SZ, van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, et al. Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis: a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation. 2005;111:1160–5. doi: 10.1161/01.CIR.0000157141.00778.AC. [DOI] [PubMed] [Google Scholar]
- 65.Weisel JW, Litvinov RI. Visualizing thrombosis to improve thrombus resolution. Res Pr Thromb Haemost. 2021;5:38–50. doi: 10.1002/rth2.12469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data that support the findings of this study are within the paper. Raw data incorporated in Microsoft Excel and/or GraphPad Prism files are available on request.