

Abstract
Chronic lymphocytic leukemia (CLL) is a common type of adult leukemia that occurs in Western countries, and its incidence has gradually increased in China in recent years. The characteristics of CLL are highly heterogeneous. Despite promising response rates achieved with targeted therapy, new targets still need to be expanded due to the heterogeneous of disease. Bruton’s tyrosine kinase inhibitor (BTKi) has been used in the treatment of TP53 mutation. In this report, we present a case with myeloid differentiation primary response 88 (MYD88) mutation who developed a TP53 mutation after application of BTKi. Here, the patient was CLL unmutated (U-CLL) with MYD88 (L265P) mutation before initial treatment. After traditional treatment, the effect was not good, and BTKi was used for treatment, then TP53 mutation appeared. It is well known that immunoglobulin heavy chain unmutated (IGHV-U) and TP53 mutation in CLL indicate poor prognosis. The case suggests that whenever TP53 mutation occurs, BTKi is the best choice. This result is considered to be related to signal pathways. We aim to add to the collective knowledge by highlighting this rare cases of CLL with MYD88 (L265P) mutation in an Asian patient.
Keywords: Pathogenesis of chronic lymphocytic leukemia, signal pathways, MYD88 (L265P) mutation, TP53 mutation, diagnosis, prognosis
Introduction
Chronic lymphocytic leukemia, which is a clonal proliferative tumour of mature B lymphocytes that occurs mainly in middle-aged and elderly people, is characterised by the aggregation of lymphocytes in the peripheral blood, bone marrow, spleen, and lymph nodes [1]. Because high frequencies of the hotspot MYD88 (L265P) mutation is observed in B-cell tumors such as extranodal diffuse large B-cell lymphoma and Waldenström macroglobulinaemia (WM), this mutation can potentially serve as a marker of these diseases [2]. In the last decade, substantial advances have been made in the field of CLL research, including the identification of recurrent mutations [3]. Although the MYD88 mutation is helpful for identifying WM [4], it is not specific to this disease and in fact plays an important role in the pathogenesis of CLL. MYD88 is a typical junction protein that connects the downstream inflammatory signalling pathway to members of the toll-like receptor (TLR) and interleukin-1 (IL-1) receptor families [5,6]. It activates nuclear factor-kappa B (NF-κB) and its related signalling pathway, promoting the proliferation and survival of B cells [2]. The NF-κB pathway is one of the main pathways in the pathogenesis of CLL. Sequencing studies have identified mutations in the MYD88 as being important oncogenic drivers in B-cell lymphomas [2]. Mutations in different signalling pathways have been identified in CLL, some of which are associated with the disease prognosis [7]. For example, mutations in tumour protein 53 (TP53), ataxia telangiectasia mutated (ATM), neurogenic locus notch homolog protein 1 (NOTCH1), splicing factor 3B subunit 1 (SF3B1), and baculoviral IAP repeat containing 3 (BIRC3) are associated with a poor outcome [7,8]. MYD88 mutations in CLL have not been commonly described, and their prognostic impact is not clearly defined [7,8].
Genetic lesions in CLL can be categorised according to several biological pathways, such as those related to inflammation and B-cell receptor (BCR), NOTCH1 signalling, and NF-κB signalling [3]. The coordination of these deregulated biological pathways drive CLL leukaemogenesis in humans [9]. MYD88 not only acts as a signal transducer in the typical NF-κB pathway but also affects the signal transduction of the BCR pathway [2]. The TP53 pathway is a complex cellular stress response network. Some researchers have elucidated many other pathways that TP53 participates in, including DNA damage, cell metabolism, autophagy, ferroptosis, and pathways that involve the generation of reactive oxygen species, with the first two receiving more attention [10]. BTKi, which inhibits BCR-initiated signalling pathways, which promotes cell apoptosis, helps overcome patients with TP53 mutation [11].
In the patient reported herein, the monoclonal B lymphocytosis in the peripheral blood was less than 5 × 109/L, the lymph nodes were enlarged in many places, and there was no hepatosplenomegaly [1]. Gene hotspot detection revealed a MYD88 (L265P) mutation which was unique to this patient. This case report mainly focuses on the pathogenesis of CLL with MYD88 mutations and the prognosis of this patient who acquired a TP53 mutation after BTK inhibitor treatment. There are not many reports on the prognosis of patients who acquire a TP53 mutation after BTK inhibitor treatment.
Case presentation
A 65-year-old woman who was diagnosed with a bilateral shoulder mass more than seven months prior was admitted to our hospital on 16 July 2020. The following detailed medical history was obtained: body temperature, 36.5°C; pulse, 70 bpm; respiration, 18 bpm; blood pressure, 104/68 mmHg; conscious; and no yellow staining of the skin and mucosa. Multiple enlarged lymph nodes could be palpated in the bilateral neck, the right supraclavicular region, the submandibular area, both armpits, and the groin. The largest node, which measured approximately 5 cm in size, was located in the right armpit region. The liver and spleen were not palpable.
The laboratory findings at the time of admission are presented in Table 1. Her serum levels of vascular endothelial growth factor (VEGF) and IgM were elevated. The lymph node pathology, bone marrow cytology, and biopsy results are shown in Figure 1 and the positron emission tomography (PET)/computed tomography (CT) scans in Figure 2. Flow cytometric analysis showed positivity for the cell markers CD19, CD20, CD22, and FMC7. Peripheral blood tested negative for IGHV mutation. Fluorescence in situ hybridization (FISH) analysis performed on the left cervical lymph node revealed cell positivity for CEP12 and negativity for D13S319, AT-rich interaction domain 2 (ATM; gene symbol ARID2), and TP53 (Figure 3). Her karyotype was normal. We also found an MYD88 (L265P) mutation frequency of 35% and B-cell lymphoma 6 protein (BCL-6), forkhead box protein 1 (FOXO1), and F-box and WD repeat domain containing 7 (FBXW7) mutations. On the basis of these findings, the patient was diagnosed with CLL (Binet B, Rai I).
Table 1.
The laboratory findings
| CRP | CRP | 9.23 mg/L↑ |
| CBC | WBC | 3.36 × 109/L↓ |
| LY | 1.42 × 109/L | |
| RBC | 3.43 × 1012/L↓ | |
| HB | 109.00 g/L↓ | |
| PLT | 224.00 × 109/L | |
| ESR | ESR | 44 mm/h↑ |
| Coagulation | Fbg | 2.46 g/L |
| DD | 0.39 mg/L FEU | |
| Coombs test | Direct Coombs test | Negative |
| Indirect Coombs test | Negative | |
| β2 Microglobulin | β2-MG | 2.41 µg/mL |
| Biochemistry | TP | 72 g/L |
| Alb | 37.4 g/L↓ | |
| Glob | 29.9 g/L | |
| ALP | 57.3 U/L | |
| LDH | 134.7 U/L | |
| Mb | 24 ng/mL | |
| FBG | 5.6 mmol/L | |
| K | 4.4 mmol/L | |
| Mg | 0.9 mmol/L | |
| TG | 0.92 mmol/L | |
| HDL | 1.03 mmol/L↓ | |
| Apo A1 | 1.00 g/L↓ | |
| VEGF | VEGF | 247.8 pg/mL↑ |
| Lymphocyte immune analysis | CD3+ | 72.59% |
| CD3+CD4+ | 29.33%↓ | |
| CD3+CD8+ | 40.77%↑ | |
| CD4/CD8 | 0.72↓ | |
| NK cells | 12.49%↓ | |
| B cells | 14.01%↑ | |
| Immunoglobulin | IgM | 11.2 g/L↑ |
| Serum protein electrophoresis | GAMMA | 16.3% |
Notes: ↑Denotes above the upper limit of the reference range; ↓denotes below the lower limit of the reference range. Abbreviations: CBC, complete blood count; CRP, C-reactive protein; WBC, white blood cell; LY, lymphocyte; RBC, red blood cell; Hb, haemoglobin; PLT, platelet; ESR, erythrocyte sedimentation rate; Fbg, fibrinogen; DD, D-dimer; TP, total protein; Alb, albumin; Glob, globulin; ALP, alkaline phosphatase; LDH, lactate dehydrogenase; Mb, myoglobin; FBG, fasting blood glucose; TG, triglyceride; HDL, high-density lipoprotein; Apo A1, Apolipoprotein A1; VEGF, vascular endothelial growth factor; NK, natural killer cell.
Figure 1.
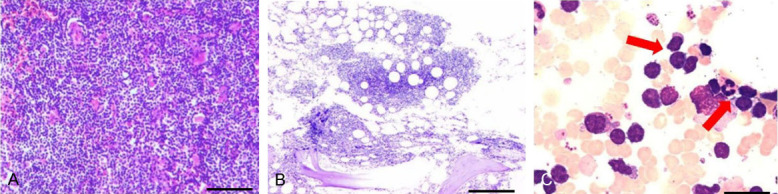
Immunostaining of specimen and bone marrow (BM) morphology. A: ×100. The immunostaining of left cervical lymph node. Heteromorphic lymphoid cells are diffused and infiltrated, with medium to small cells, sparse cytoplasm, slightly irregular round and oval nuclei, granular chromatin, inconspicuous nucleoli and hard to find mitosis. Bar = 100 μm. B: ×40. The immunostaining of BM biopsy specimen. Patchy atypical small lymphocyte infiltration is common in bone marrow stroma. Bar = 160 μm. C: ×1000. Morphology of BM. This kind of cells are small in size, mostly quasi-round or irregular, less cytoplasm, and pseudopod-like protuberance can be seen at the edge. Bar = 10 μm.
Figure 2.

Images of computed tomography and PET/CT (Blue arrow: After treatment of zanubrutinib. Red arrow: Before treatment of zanubrutinib). A-C: Images of computed tomography. It can be seen that the lymph nodes are obviously reduced, and some tend to be normal. D-F: Images of PET/CT. Extensive lymph node enlargement significantly reduced and metabolism of lymph node decreased after chemotherapy.
Figure 3.

FISH specimen. A: Normal. B: Initial diagnosis of lymph node FISH (before treatment): CEP12 (+). C: After eight cycles chemotherapy and zanubrutinib for over 6 months of treatment, bone marrow FISH: CEP12 (-). Bar = 10 μm.
The patient received four cycles of bendamustine (100 mg, days 1-2), followed by four cycles of a BR regimen (bendamustine 100 mg, days 1-2; rituximab 500 mg, day 0). After these eight cycles of treatment, the follow-up CT scan indicated that the lymph nodes had not decreased obviously (Figure 2). Therefore, the patient was treated with the BTKi zanubrutinib for over 6 months (Figures 2, 3 and 4). Subsequent analysis revealed obviously decreased bone marrow cells to the MYD88 (L265P) mutation, but mutations in the TP53 and ARID2 genes had been acquired. After 5 months of zanubrutinib treatment, TP53 gene and MYD88 (L265P) mutation showed a downward trend. Abnormally mature monoclonal B cells are still visible on the detection of tiny residual cells in the bone marrow. The patient was followed up recently, the results showed β2-MG and VEGF were negative. Complete blood count and biochemistry were normal. Echocardiography showed no obvious abnormality. Ultrasound indicated that there was little change in lymph node size from the last review. At the time of this report, the patient was stable.
Figure 4.

The changes of WBC, β2-MG, IgM, GAMMA, Hb, PLT, LDH, ESR, VEGF. The series of WBC, β2-MG, IgM, GAMMA are drawn on the primary axis (on left axis), and the series of Hb, PLT, LDH, ESR, VEGF are drawn on the secondary axis (on right axis).
Discussion
CLL represents a heterogeneous disease with a highly variable clinical course [7]. MYD88 mutations in CLL are not well characterized. Earlier reports yielded conflicting results in terms of clinicopathologic presentation and prognostic impact of MYD88 mutations in CLL patients [8]. MYD88 is a key molecule in innate immunity [5], which is located on chromosome 3p22.2 [7], where it not only acts as a signalling adaptor in the canonical NF-κB pathway but also plays an important role in BCR signalling [2]. In normal physiology, stimulation of the BCR activates NF-κB as well as the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) and nuclear factor of activated T cells (NFAT) pathways [2,9]. Subsequently, BTK is activated through its phosphorylation, whereupon it activates phospholipase Cγ2 (PLCγ2), thereby initiating activation of the NF-κB pathway [2]. As the MYD88 (L265P) mutant constitutively activates the NF-κB pathway, it is regarded as an important oncogenic driver in B-cell non-Hodgkin lymphoma [9]. Previous studies on the prognostic impact of MYD88 mutations in CLL also have been discordant [7,8].
Michele et al. [12] found that Bcl-6 expression was limited mainly to follicle center lymphomas (FCL), whereas the great majority of CLL lacked immunoreactivity for Bcl-6. In CLL, a fragment of the first intron of BCL6 is a target of the somatic hypermutation process [13]. In early-stage patients with CLL, Jantus Lewintre et al. [13] found no correlation between expression and the mutations or polymorphism in BCL6, but high levels of BCL6 can discriminate patients with a worse prognosis. FOXO1 is highly expressed in normal B cells of the germinal centre as well as in non-Hodgkin lymphomas [14]. The main regulator of germinal center (GC) formation BCL6 is also a FOXO target [15]. The mutation of FOXO1 is caused by the FOXO1-Grb2-associated binder 1 (GAB1)-protein activator of interferon-induced protein kinase (PACT) axis induced by CLL cells [16]. Additionally, FOXO1 was reported to be a regulator of B-cell death, and its inactivation via B-cell receptor signalling to AKT was found to be critical to the survival of mature B cells [14]. There is an interdependent balance between AKT activity and the FOXO1-GAB1 axis [16]. BTK inhibitors affect this balance and inhibit catatonic AKT phosphorylation, thereby inhibiting cell proliferation [16].
In the case of this patient, genetic screening of the bone marrow lymphoma cells after zanubrutinib treatment revealed that the expression of MYD88 (L265P) mutation decreased but was positive for TP53 and ARID2 mutations. It is known that zanubrutinib can inhibit BTK and NF-κB signal transduction and is an effective molecular targeted therapy for chronic lymphoblastic leukaemia [1]. BTKi potently inhibits BCR signalling, downregulate NF-κB signalling, and rapidly reduce tumour burdens through a combination of tumour proliferation inhibition and increased apoptosis [17-19]. However, they also increase the expression of miRNA targets, including tumour suppressors, and cause decreased cell proliferation [19]. Among the miRNAs that have been tested, miR-22, miR-34a, miR-146b, and miR-181b were found to be significantly downregulated in patient CLL cells following ibrutinib treatment [19]. This leads to increased expression of multiple tumour suppressors, including ARID1B, ARID2, ATM, CYLD lysine 63 deubiquitinase (CYLD), FOXP1, histone deacetylase 1 (HDAC1), inhibitor of Bruton’s tyrosine kinase (IBTK), phosphatase and tensin homologue (PTEN), and suppressor of mothers against decapentaplegic 4 (SMAD4) [19]. TP53 mutated is the most important biomarker of CLL. miR-34a, of which TP53 is a direct target, has been reported to be altered in CLL and is associated with the disease progression and outcomes [20,21]. TP53 modulates gene products through the induction of miRNAs that are controlled by p53 at the transcriptional level. The activation process of TP53 is regulated by modifications such as phosphorylation and acetylation, which mediate a variety of cellular functions of p53, including apoptosis, ferroptosis, cell cycle arrest, metabolism, autophagy, stem cell differentiation, DNA repair, and senescence. All these cellular pathways are thought to contribute to the tumor suppressor function of p53 [22]. Recent studies have reduced the TP53 mutation cutoff to at least 5% VAF, but this must be evaluated in prospective clinical trial cohorts [23]. Clonal expansion of low-burden TP53 mutations in CLL is associated with intense chemoimmunotherapy but not with targeted therapy [24]. With regard to the TP53 gene mutation, regardless of whether it is a mutation or deletion, it is the most important marker of poor prognosis for CLL [24]. It is also a reliable marker for chemotherapy-resistant diseases [24]. At present, the patient was still positive for serum immunostasis electrophoresis. Bone marrow minimal residual disease (MRD) was positive, but the MYD88 (L265P) mutation rate was still decreasing. After relevant examination evaluation, BTKi non-target events, such as hypertension and atrial fibrillation, did not occur. Although the TP53 mutation appeared after the application of BTK inhibitors, its mutation rate also descended. The patient was diagnosed with U-CLL. U-CLL patients show a significantly poorer outcome [25]. Whether there is a link between the patient’s current status and the absence of IGHV genes mutation. There is still no relevant report. We found that the emergence of the new mutations post BTK inhibitor therapy had no adverse effect on our patient. Further confirmation of the specific targets of the CLL pathogenesis pathway corresponding to each mutated gene would help toward the development of future therapeutic strategies for this malignant disease.
Disclosure of conflict of interest
None.
References
- 1.Hallek M, Al-Sawaf O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am J Hematol. 2021;96:1679–1705. doi: 10.1002/ajh.26367. [DOI] [PubMed] [Google Scholar]
- 2.de Groen RAL, Schrader AMR, Kersten MJ, Pals ST, Vermaat JSP. MYD88 in the driver’s seat of B-cell lymphomagenesis: from molecular mechanisms to clinical implications. Haematologica. 2019;104:2337–2348. doi: 10.3324/haematol.2019.227272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kikushige Y. Pathogenesis of chronic lymphocytic leukemia and the development of novel therapeutic strategies. J Clin Exp Hematop. 2020;60:146–158. doi: 10.3960/jslrt.20036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shekhar R, Naseem S, Binota J, Varma N, Malhotra P. Frequency of MYD88 L256P mutation and its correlation with clinico-hematological profile in mature B-cell neoplasm. Hematol Oncol Stem Cell Ther. 2021;14:231–239. doi: 10.1016/j.hemonc.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Deguine J, Barton GM. MyD88: a central player in innate immune signaling. F1000Prime Rep. 2014;6:97. doi: 10.12703/P6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Improgo MR, Tesar B, Klitgaard JL, Magori-Cohen R, Yu L, Kasar S, Chaudhary D, Miao W, Fernandes SM, Hoang K, Westlin WF, Kim HT, Brown JR. MYD88 L265P mutations identify a prognostic gene expression signature and a pathway for targeted inhibition in CLL. Br J Haematol. 2019;184:925–936. doi: 10.1111/bjh.15714. [DOI] [PubMed] [Google Scholar]
- 7.Shuai W, Lin P, Strati P, Patel KP, Routbort MJ, Hu S, Wei P, Khoury JD, You MJ, Loghavi S, Tang Z, Fang H, Thakral B, Medeiros LJ, Wang W. Clinicopathological characterization of chronic lymphocytic leukemia with MYD88 mutations: L265P and non-L265P mutations are associated with different features. Blood Cancer J. 2020;10:86. doi: 10.1038/s41408-020-00351-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putowski M, Giannopoulos K. Perspectives on precision medicine in chronic lymphocytic leukemia: targeting recurrent mutations-NOTCH1, SF3B1, MYD88, BIRC3. J Clin Med. 2021;10:3735. doi: 10.3390/jcm10163735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, Shaffer AL, Romesser P, Wright G, Powell J, Rosenwald A, Muller-Hermelink HK, Ott G, Gascoyne RD, Connors JM, Rimsza LM, Campo E, Jaffe ES, Delabie J, Smeland EB, Fisher RI, Braziel RM, Tubbs RR, Cook JR, Weisenburger DD, Chan WC, Staudt LM. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hernández Borrero LJ, El-Deiry WS. Tumor suppressor p53: biology, signaling pathways, and therapeutic targeting. Biochim Biophys Acta Rev Cancer. 2021;1876:188556. doi: 10.1016/j.bbcan.2021.188556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morabito F, Gentile M, Monti P, Recchia AG, Menichini P, Skafi M, Atrash M, De Luca G, Bossio S, Al-Janazreh H, Galimberti S, Salah Z, Morabito L, Mujahed A, Hindiyeh M, Dono M, Fais F, Cutrona G, Neri A, Tripepi G, Fronza G, Ferrarini M. TP53 dysfunction in chronic lymphocytic leukemia: clinical relevance in the era of B-cell receptors and BCL-2 inhibitors. Expert Opin Investig Drugs. 2020;29:869–880. doi: 10.1080/13543784.2020.1783239. [DOI] [PubMed] [Google Scholar]
- 12.Raible MD, Hsi ED, Alkan S. Bcl-6 protein expression by follicle center lymphomas. A marker for differentiating follicle center lymphomas from other low-grade lymphoproliferative disorders. Am J Clin Pathol. 1999;112:101–107. doi: 10.1093/ajcp/112.1.101. [DOI] [PubMed] [Google Scholar]
- 13.Jantus Lewintre E, Reinoso Martin C, Garcia Ballesteros C, Pendas J, Benet Campos C, Mayans Ferrer JR, Garcia-Conde J. BCL6: somatic mutations and expression in early-stage chronic lymphocytic leukemia. Leuk Lymphoma. 2009;50:773–780. doi: 10.1080/10428190902842626. [DOI] [PubMed] [Google Scholar]
- 14.Xie L, Ushmorov A, Leithauser F, Guan H, Steidl C, Farbinger J, Pelzer C, Vogel MJ, Maier HJ, Gascoyne RD, Moller P, Wirth T. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119:3503–3511. doi: 10.1182/blood-2011-09-381905. [DOI] [PubMed] [Google Scholar]
- 15.Shore AM, White PC, Hui RC, Essafi A, Lam EW, Rowe M, Brennan P. Epstein-Barr virus represses the FoxO1 transcription factor through latent membrane protein 1 and latent membrane protein 2A. J Virol. 2006;80:11191–11199. doi: 10.1128/JVI.00983-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seda V, Vojackova E, Ondrisova L, Kostalova L, Sharma S, Loja T, Mladonicka Pavlasova G, Zicha D, Kudlickova Peskova M, Krivanek J, Liskova K, Kren L, Benes V, Musilova Litzmanova K, Borsky M, Oppelt J, Verner J, Pospisilova S, Brychtova Y, Panovska A, Tan Z, Zhang S, Doubek M, Amruz Cerna K, Mayer J, Mraz M. FoxO1-GAB1 axis regulates homing capacity and tonic AKT activity in chronic lymphocytic leukemia. Blood. 2021;138:758–772. doi: 10.1182/blood.2020008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, Farooqui M, Wiestner A. Ibrutinib inhibits BCR and NF-κB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood. 2014;123:3286–3295. doi: 10.1182/blood-2014-02-548610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A, Saba N, Martyr S, Soto S, Valdez J, Gyamfi JA, Maric I, Calvo KR, Pedersen LB, Geisler CH, Liu D, Marti GE, Aue G, Wiestner A. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia. 2014;28:2188–2196. doi: 10.1038/leu.2014.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saleh LM, Wang W, Herman SE, Saba NS, Anastas V, Barber E, Corrigan-Cummins M, Farooqui M, Sun C, Sarasua SM, Zhao Z, Abousamra NK, Elbaz O, Abdelghaffar HA, Wiestner A, Calvo KR. Ibrutinib downregulates a subset of miRNA leading to upregulation of tumor suppressors and inhibition of cell proliferation in chronic lymphocytic leukemia. Leukemia. 2017;31:340–349. doi: 10.1038/leu.2016.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dufour A, Palermo G, Zellmeier E, Mellert G, Duchateau-Nguyen G, Schneider S, Benthaus T, Kakadia PM, Spiekermann K, Hiddemann W, Braess J, Truong S, Patten N, Wu L, Lohmann S, Dornan D, GuhaThakurta D, Yeh RF, Salogub G, Solal-Celigny P, Dmoszynska A, Robak T, Montillo M, Catalano J, Geisler CH, Weisser M, Bohlander SK. Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood. 2013;121:3650–3657. doi: 10.1182/blood-2012-10-458695. [DOI] [PubMed] [Google Scholar]
- 21.Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE, Valeri N, Calore F, Sampath D, Fanini F, Vannini I, Musuraca G, Dell’Aquila M, Alder H, Davuluri RV, Rassenti LZ, Negrini M, Nakamura T, Amadori D, Kay NE, Rai KR, Keating MJ, Kipps TJ, Calin GA, Croce CM. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol Ther. 2021;220:107720. doi: 10.1016/j.pharmthera.2020.107720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bomben R, Rossi FM, Vit F, Bittolo T, Zucchetto A, Papotti R, Tissino E, Pozzo F, Degan M, Polesel J, Bulian P, Marasca R, Reda G, Laurenti L, Olivieri J, Chiarenza A, Laureana R, Postorino M, Del Principe MI, Cuneo A, Gentile M, Morabito F, Fronza G, Tafuri A, Zaja F, Foà R, Di Raimondo F, Del Poeta G, Gattei V. Clinical impact of TP53 disruption in chronic lymphocytic leukemia patients treated with ibrutinib: a campus CLL study. Leukemia. 2023;37:914–918. doi: 10.1038/s41375-023-01845-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malcikova J, Pavlova S, Kunt Vonkova B, Radova L, Plevova K, Kotaskova J, Pal K, Dvorackova B, Zenatova M, Hynst J, Ondrouskova E, Panovska A, Brychtova Y, Zavacka K, Tichy B, Tom N, Mayer J, Doubek M, Pospisilova S. Low-burden TP53 mutations in CLL: clinical impact and clonal evolution within the context of different treatment options. Blood. 2021;138:2670–2685. doi: 10.1182/blood.2020009530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodríguez-Caballero A, Fuentes Herrero B, Oliva Ariza G, Criado I, Alcoceba M, Prieto C, Pérez Caro M, García-Montero AC, González Díaz M, Forconi F, Sarmento-Ribeiro AB, Almeida J, Orfao A. The hydropathy index of the HCDR3 region of the B-cell receptor identifies two subgroups of IGHV-mutated chronic lymphocytic leukemia patients with distinct outcome. Front Oncol. 2021;11:723722. doi: 10.3389/fonc.2021.723722. [DOI] [PMC free article] [PubMed] [Google Scholar]