

Abstract
Despite the capability of extracellular vesicles (EVs) derived from Gram‐negative and Gram‐positive bacteria to induce potent anti‐tumour responses, large‐scale production of bacterial EVs remains as a hurdle for their development as novel cancer immunotherapeutic agents. Here, we developed manufacturing processes for mass production of Escherichia coli EVs, namely, outer membrane vesicles (OMVs). By combining metal precipitation and size‐exclusion chromatography, we isolated 357 mg in total protein amount of E. coli OMVs, which was equivalent to 3.93 × 1015 particles (1.10 × 1010 particles/μg in total protein amounts of OMVs) from 160 L of the conditioned medium. We show that these mass‐produced E. coli OMVs led to complete remission of two mouse syngeneic tumour models. Further analysis of tumour microenvironment in neoantigen‐expressing tumour models revealed that E. coli OMV treatment causes increased infiltration and activation of CD8+ T cells, especially those of cancer antigen‐specific CD8+ T cells with high expression of TCF‐1 and PD‐1. Furthermore, E. coli OMVs showed synergistic anti‐tumour activity with anti‐PD‐1 antibody immunotherapy, inducing substantial tumour growth inhibition and infiltration of activated cancer antigen‐specific stem‐like CD8+ T cells into the tumour microenvironment. These data highlight the potent anti‐tumour activities of mass‐produced E. coli OMVs as a novel candidate for developing next‐generation cancer immunotherapeutic agents.
Keywords: bacterial extracellular vesicles, combination immunotherapy, immunotherapeutic agents, large‐scale mass production, outer membrane vesicles
1. INTRODUCTION
The 2018 Nobel Prize in Physiology or Medicine was awarded jointly to Drs. James P. Allison and Tasuku Honjo, who have contributed to discover cancer therapy by inhibiting negative immune regulation (Motofei, 2022). Their discovery leads to development of the mainstream cancer immunotherapy inhibiting immune checkpoints, which could cure advanced solid malignancies that had been intractable by conventional therapies (Nafaji et al., 2022). Among immune checkpoint targets, monoclonal antibodies directed against programmed cell death protein‐1 (PD‐1) or programmed cell death‐ligand 1 (PD‐L1) were globally employed for cancer immunotherapy (L. Chen & Han, 2015). The global sales of anti‐PD‐1/anti‐PD‐L1 antibodies rocketed to over 20 billion USD in 2021, and more than 1000 clinical trials are ongoing for application of these drugs in single or combination therapies (Upadhaya et al., 2022). However, in spite of these initial successes, only 10%–20% of patients suffered with each type of cancer were responsive to these drugs (S. Chen et al., 2021; Jiang et al., 2020). Thus, it is important to select the patients who will get benefit from anti‐PD‐1 or anti‐PD‐L1 antibody therapy before the treatment in clinical circumstances (Egen et al., 2020).
Anti‐cancer immune responses are hindered by insufficient activation of cancer‐specific T cells and immunosuppressive tumour microenvironments (Dersh et al., 2021; Rabinovich et al., 2007). Immune evasion by cancer resulted in persistent antigen presence, which leads to repeated stimulation of cancer‐specific T cells. PD‐1‐PD‐L1/2 interactions are normal defence mechanisms to prevent excessive immune activation through repeated stimulation, leading to exhaustion of the antigen‐specific T cells (Wherry et al., 2007). Reinvigoration or selective proliferation of exhausted T cells are the primary mechanism and underlying efficacy of PD‐1/PD‐L1 targeted immunotherapy (Im et al., 2016). Thus, cancer‐specific T cells should have been activated and recruited into tumour tissues (hot tumour) to make cancers respond well to anti‐PD‐1 or anti‐PD‐L1 antibody therapy (P. Chen et al., 2016; Ren et al., 2020; Tumeh et al., 2014). Cancers with insufficient T cell activation (cold tumour) or defective recruitment of activated T cells (altered tumour) comprise 80%–90% of cancers, which would not respond well to anti‐PD‐1 or anti‐PD‐L1 antibody therapy (Galon & Bruni, 2019). Thus, it is urgent to develop novel therapies, which can efficiently activate T cells in the context of immunosuppressive tumour microenvironment, to rescue cancer patients who do not respond well to anti‐PD‐1 or anti‐PD‐L1 antibody therapy (Lei et al., 2020).
The immune system has evolved to combat various microbes, such as bacteria and viruses, and thus microbes are the most efficient stimuli for activating the immune systems. On this basis, William Coley injected mixtures of extracts of heat‐inactivated bacteria, known as Coley's toxin, to cancer patients in the late 19th century, and found significant tumour regression from the patients (Carlson et al., 2020). However, Coley's toxin had been criticized due to safety issues and inconsistent efficacy (Carlson et al., 2020). Recently, we have revolutionized Coley's toxin immunotherapy, by using extracellular vesicles (EVs) derived from Gram‐negative and Gram‐positive bacteria, which are spherical, lipid‐bilayered, and nano‐sized entities (J. H. Kim et al., 2015). Gram‐negative and Gram‐positive bacterial EVs effectively induced robust and sustained immune responses, mediated by IFN‐γ and CXCL10, which eliminated established tumours without significant adverse effects (O. Y. Kim et al., 2017). Furthermore, engineered Gram‐negative bacterial EVs such as genetically engineering the surface of EVs to express PD‐1 ectodomain (Li et al., 2020) or the lumen of EVs to harbour PD‐1 plasmid (Pan et al., 2022), showed potent anti‐tumour responses by relieving the effector function of T cells from immune inhibitory axis of PD‐1/PD‐L1.
To further delineate mechanisms of actions of cancer immunotherapy using bacterial EVs and translate the immunotherapy to clinical trials, we developed strategies for large‐scale production of Escherichia coli EVs, namely, outer membrane vesicles (OMVs), by combining metal precipitation and size‐exclusion chromatography. Following mass production, we confirmed the purity and the yield of OMVs and performed proteomic analysis on OMVs. Then, we investigated the anti‐tumour activity of the mass‐produced E. coli OMVs. Intratumorally administered OMVs induced complete remission of two syngeneic cancer models. In addition, OMVs per se, which do not contain cancer antigens, significantly activated and recruited total and cancer antigen‐specific CD8+ T cells into the tumour tissue. Intratumoral CD8+ T cells induced by OMV treatment revealed increased stem‐like properties, which provided responsiveness to anti‐PD‐1 antibody immunotherapy and synergistic anti‐tumour activity with the therapy.
2. MATERIALS AND METHODS
2.1. Large‐scale production of E. coli OMVs
E. coli BL21 (DE3) ΔmsbB was constructed using CRISPR‐Cas9 gene deletion system (GenScript, Piscataway, NJ). Master cell bank of the bacterial clone was established with good manufacturing practice‐compliant operations (Binex, Busan, Republic of Korea) and characterized to ensure the purity, safety, functionality, and genetic stability (BioReliance, Rockville, MD). The seed culture was performed by inoculating E. coli BL21 (DE3) ΔmsbB in 2 L of animal‐free lysogeny broth based on vegetable peptone (VP‐LB; Oxoid, Basingstoke, Hampshire, UK). The main culture was conducted by inoculating the 2 L of seed culture in 200 L of VP‐LB using a 300 L‐scale fermentation system (9‐2428, Chemap AG, Männedorf, Switzerland). The main culture was maintained at 30°C, 350 rpm, and pH 7.2 (± 0.2), with continuous supplying 90 L/min of air and 10 L/min of O2, until the bacterial culture enters a stationary growth phase. To collect conditioned medium from the 200 L culture, the cells were removed by a continuous centrifugation system (GF‐150, Lilong, Liaoyang, China) at 10,000 × g for 20 min at 4°C. The cell‐free conditioned medium from the 200 L culture was filtrated with a 0.2 μm pore‐sized filter (Sartopore 2 Maxicaps 5447307H3, Sartorius, Göttingen, Germany), and then concentrated in 10‐folds by tangential flow filtration system with a 100 kDa molecular weight cut‐off (MWCO) membrane (3021466807E‐ Sartorius): feed flux and membrane pressure were maintained at 10.5 L/min and less than one bar, respectively. The concentrate was subsequently filtrated with a 0.2 μm pore‐sized filter (5445307H9, Sartorius) and then treated with 240,000 units of Benzonase® endonuclease (1.01695, Merck, Darmstadt, Germany) at room temperature for 6 h before OMV isolation.
OMVs were purified from the 16 L of filtrated concentrate of 160 L culture cell‐free conditioned medium by ExoLutE® isolation technologies (SL Bigen, Incheon, Republic of Korea), which comprises metal precipitation and size‐exclusion chromatography. Using a dynamic dialysis system (SpectraFlo™, Repligen, Waltham, MA), the pool of fractions containing OMVs from 4 runs of S300 size‐exclusion chromatography (743 mL of column volume) at a flow rate of 10.0 mL/min with a formulation buffer (2% sucrose, 104 mM sodium chloride, 10 mM L‐histidine, pH 7.4) were dialyzed against the formulation buffer using a 100 kDa MWCO membrane (Spectrum™ 131420T, Thermo Fisher Scientific, Waltham, MA) at 4°C for 18 h, with refreshing the buffer every 6 h. The dialyzed OMVs were sterilized by a sequential filtration with 0.2 μm pore‐sized filter (5445307H8, Sartorius) and 0.1 μm pore‐sized filter (5445358K8, Sartorius). Total protein concentration of E. coli OMVs purified from the 160 L culture conditioned medium was measured by Bradford assay (5000006, Bio‐Rad Laboratories, Hercules, CA). The aliquots of OMVs were stored at −80°C until necessary.
Additionally, we isolated OMVs by combining ultracentrifugation and buoyant density gradient ultracentrifugation as reported (O. Y. Kim et al., 2017). Briefly, 300 mL of filtrated concentrate of 3 L culture cell‐free conditioned medium was subjected to ultracentrifugation at 150,000 × g for 3 h at 4°C. The pellet was suspended in 50% iodixanol (Sigma‐Aldrich, St. Louis, MO) followed by adding 40% and 10% iodixanol, and then centrifuged at 200,000 × g for 2 h at 4°C. The fraction of E. coli OMVs were collected between 10% and 40% iodixanol. Finally, OMVs were sterilized by passing through a 0.22 μm pore‐sized syringe filter (16532‐K, Sartorius).
2.2. Characterization of E. coli OMVs
Particle number of OMVs was determined by Nanosight LM10‐HS system with Nanoparticle Tracking Analysis software version 2.3 (Malvern Instruments Ltd., Malvern, UK), as reported (Dinh et al., 2020). Briefly, OMVs were diluted with HEPES‐buffered saline (HBS; 20 mM HEPES, 150 mM NaCl (pH 7.4)) and analyzed with the following parameters: injection volume = 0.5 mL; laser wavelength = 405 nm; camera level = 12; number of measurement = 3; measurement duration = 30 s; viscosity = 0.89 cP; chamber temperature = 25°C; detection threshold = 5; blur = auto; minimum track length = 10; minimum expected size = auto; and range of detection = (0.8‐2.5) × 109 particles/mL.
The purified OMVs were placed on 400‐mesh copper grid (FCF400‐Cu‐50, Electron Microscopy Sciences, Matfield, PA). The OMV‐loaded grid was rinsed with distilled water and then stained with 1% uranyl acetate (19481, Ted Pella, Redding, CA). Transmission electron microscopy images were obtained by JEM1011 electron microscope (JEOL, Tokyo, Japan) with an accelerating voltage of 80 kV.
The size distribution of OMVs was measured by Zetasizer Nano ZS with Zetasizer Software version 6.34 (Malvern Instruments Ltd.). OMVs were diluted with HBS (1 × 109 particles/mL, final volume 1 mL), and analyzed with the following parameters: laser wavelength = 633 nm; number of measurement = 5; measurement duration = 30 s; chamber temperature = 25°C.
Whole cell lysates were prepared from E. coli BL21 (DE3) ΔmsbB bacterial cells harvested by centrifuging the bacterial culture at 6000 × g for 30 min at 4°C. The harvested cells were resuspended in buffer A (50 mM Tris‐HCl, 2% sodium dodecyl sulfate (SDS), 0.1% bromophenol blue, 10% glycerol (pH 6.8)), and heated at 95°C for 5 min. The heated cells were cooled, then were subjected to sonication on ice (amplitude: 20–40%; 10 s × 5 times). After centrifugation at 10,000 × g for 10 min at 4°C, the supernatants were harvested to be used as whole cell lysates.
Protein samples (5 μg in total protein amounts) from E. coli BL21 (DE3) ΔmsbB whole cell lysates and OMVs were analyzed by 10% resolving gel SDS‐PAGE followed by staining with Coomassie Brilliant Blue G‐250 (LC6060, Invitrogen, Waltham, ME). To detect OmpA and FtsZ, protein samples from E. coli BL21 (DE3) ΔmsbB whole cell lysates and OMVs (both 1 μg in total protein amounts) were subjected to 10% resolving gel SDS‐PAGE, and then transferred to a polyvinylidene difluoride membrane. The membrane was blocked with 5% skim milk, incubated with rabbit anti‐OmpA (lab‐made; 1:1000 dilution) or anti‐FtsZ polyclonal antibody (AS10 715, Agrisera, Vännäs, Sweden; 1:1000 dilution), and then followed by horseradish peroxidase‐conjugated goat anti‐rabbit IgG (sc‐2004, Santa Cruz Biotechnology, Santa Cruz, CA; 1:2000 dilution). Immunoreactive bands were visualized with a chemiluminescent substrate (37074, Thermo Fisher Scientific).
2.3. Proteomic analysis of E. coli OMVs
Protein of OMVs (50 μg) were precipitated by treating methanol/chloroform (Choi et al., 2021). Precipitated proteins were conducted by in‐gel digestion as previously described (S. Lee et al., 2021). The tryptic peptides were analyzed in LC‐MS/MS with Orbitrap Eclipse™ Tribrid™ mass spectrometer (Thermo Fisher Scientific) coupled with Ultimate™ 3000 UHPLC system (Thermo Fisher Scientific). Peptides were dissolved in 20 μL of 0.1% formic acid in water and 5 μL was injected onto Acclaim PepMap C18 nano Viper 100 (75 μm × 2 cm, 3 μm) trap column (Thermo Fisher Scientific), at a flow rate of 5 μL/min with 0.095% formic acid in water for 4 min. Loaded peptides were separated on PepMap RSLC C18 ES803A (75 μm × 50 cm, 2 μm) analytical column (Thermo Fisher Scientific), by 180 min gradient from 5% to 90% of 0.1% formic acid in acetonitrile at a flow rate of 300 nL/min. The Orbitrap Eclipse™ Tribrid™ mass spectrometer was operated in a data‐dependent Top 20 scans mode switching between MS and MS2. Peptides were analyzed with the following parameters: 10 ppm of mass accuracy, 1850 V of ion spray voltage, 275°C of capillary temperature, m/z 375–1575 of resolution of full scans, and 120,000 higher‐energy collisional dissociation activation scans with 35% normalized collision energy. The quadrupole isolation window was 1.4 Da, and MS/MS spectra were detected on Orbitrap with resolution of 30,000.
The raw files were converted into mzML using MSConvert (http://proteowizard.sourceforge.net, version 3.0.10107). The converted mzML files were searched against UniProt (http://www.uniprot.org) E. coli K12 protein database (release 2022_04, 4,403 entries) using X! Tandem in Trans‐Proteomic Pipeline (TPP) (version 4.6). The tolerance was 5 ppm for precursor ions and 0.05 Da for fragment ions. Two potential missed cleavages were permitted for trypsin digestion. The following modifications were used: fixed modification for the carbamidomethylation of cysteine (15.995 Da) and variable modifications for the oxidation of methionine (15.995 Da), deamination of N‐terminal glutamine (−17.027 Da), and dehydration of N‐terminal glutamic acid (−18.011 Da). All identified proteins statistically processed via TPP. Further analyses were performed only with proteins with PeptideProphet ≥ 0.99 and ProteinProphet ≥ 0.99. Relative protein abundance of OMVs was calculated by weighted spectral counting using the absolute protein expression (APEX) tool (Lu et al., 2007). Subcellular localizations of OMV vesicular proteins were analyzed using UniProt (release 2022_04) (The UniProt Consortium, 2017; The UniProt Consortium, 2019) and DAVID (Huang et al., 2009; Sherman et al., 2022) databases.
2.4. Uptake and immuno‐stimulatory effects of E. coli OMVs
PBS or OMVs (100 μg) was mixed with 10 μM of Cy7 Mono NHS ester (Cytiva, Marlborough, MA), and incubated at room temperature for 30 min. After adding of 5 mM of ethanolamine (Sigma‐Aldrich), the reaction mixture went through a size‐exclusion spun column (column S of ExoLutE® Conditioned Medium Exosome Isolation Kit (SL Bigen)) to remove residual Cy7. RAW264.7 cells (2 × 105 cells/well) were seeded overnight, then incubated with the eluents from column S (Cy7‐labeled OMVs, 1, 10, 100, and 1000 ng/mL in total protein concentrations; free Cy7, the equivalent volumes of the eluents) at 37°C for 24 h. Subsequently, the cells were washed with PBS, and subjected to flow cytometry using CytoFLEX LX (Beckman Coulter, Brea, CA). Data were analyzed and acquired with BD FlowJo™ 10 software (BD Biosciences, San Jose, CA).
To study the immuno‐stimulatory effects of OMVs, RAW264.7 cells (5 × 104 cells/well) were seeded overnight, then incubated with the eluents from column S (Cy7‐labeled OMVs, 0.1, 0.3, 1, 3, 10, 30, 100, 300, and 1000 ng/mL in total protein concentrations; free Cy7, the equivalent volumes of the eluents) at 37°C for 24 h. Quantification of mouse IL‐6 and TNF‐α in the conditioned media was conducted using DuoSet ELISA kits (R&D Systems, Minneapolis, MN), according to the manufacturer's instructions.
2.5. Mice
C57BL/6 mice and BALB/c mice were purchased from Koatech (Pyeongtaek, Republic of Korea). Female, 8‐week‐old mice were used for experiments. All animal experiments were performed at the animal facility of Seoul National University College of Medicine with the approval of the Institutional Animal Care and Use Committee at Seoul National University (Approval No. SNU‐220307‐4‐5). Experiments complied with the ethical guidelines set by the Institutional Animal Care and Use Committee at Seoul National University and animals were killed at certain end points in a humane manner.
2.6. Cell culture
Murine cancer cell lines (MB49, EMT6, MC38, and B16F10) and macrophages (RAW264.7) were acquired from American Type Culture Collection (Manassas, VA). As cancer antigen‐specific immune response models, we established ovalbumin (OVA)‐expressed MC38 and B16F10 cancer cell lines by the infection of lentivirus harbouring the gene encoding whole OVA protein in the cytosol, generating MC38‐OVA and B16F10‐OVA stable cancer cell lines, respectively. MB49 and B16F10 cancer cell lines as well as RAW264.7 cells were cultured in Dulbecco's Modified Eagle Medium with 10% (v/v) foetal bovine serum and 1% (v/v) antibiotic‐antimycotic solution while 4 ng/mL puromycin was supplemented for B16F10‐OVA cell culture. EMT6 and MC38 cancer cell lines were cultured in Roswell Park Memorial Institute 1640 media with 10% (v/v) foetal bovine serum and 1% (v/v) antibiotic‐antimycotic solution while 4 ng/mL puromycin was supplemented for MC38‐OVA cell culture. Cells were cultured under sterile conditions at 37°C in a 5% CO2 atmosphere. All cancer cell lines were tested for Mycoplasma contamination in a regular basis and none of the cancer cell lines were determined to be positive throughout the entire study.
2.7. Flow cytometry
To analyze tumour‐infiltrating lymphocytes, tumour tissue was isolated from the mice after they were euthanized in CO2 chamber. Tumour tissue was chopped using Dorco razor blade and chopped tumour tissue was incubated for 1 h at 37°C in Dulbecco's Modified Eagle Medium with 2% horse serum, 0.5 mg/mL collagenase D, and 40 μg/mL DNase I. After passing through 40‐μm pore‐sized filters to make single‐cell suspension, the sample was resuspended in 30% Percoll and then was layered on top of 70% Percoll. After centrifugation at 2200 rpm for 20 min at 25°C in lowest acceleration/deceleration speed, lymphocytes were isolated by collecting the interphase in between 30%/70% percoll density gradients. Cells were resuspended in fluorescence‐activated cell sorting buffer (2% horse serum, 0.05% NaN3 in Hanks’ Balanced Salt Solution) after RBC lysis buffer (00‐4300‐54, Invitrogen) treatment to remove red blood cells. FcγRII/III receptors were blocked using anti‐mouse CD16/CD32 antibodies (clone 2.4G2, lab‐made; 1:200 dilution) for 15 min at 4°C, and cells were stained for 30 min at 4°C with the following surface antibodies: CD3 (clone 17A2, 740739, 1:200 dilution), CD4 (clone GK1.5, 563933, 1:1,000 dilution), CD8α (clone 53–6.7, 553035, 1:1,000 dilution), CD25 (clone PC61, 563037, 1:500 dilution), PD‐1 (clone 29F.A12, 568577, 1:500 dilution), and Tim‐3 (clone RMT3‐23, 568429, 1:500 dilution) from BD Bioscience, and CD39 (clone Duha59, 143805, 1:500 dilution) from BioLegend (San Diego, CA).
To stain intracellular cytokines, cells were fixed with 4% paraformaldehyde for 10 min at room temperature and washed with 0.1% bovine serum albumin in PBS with 0.5% Triton X‐100. Cells were then stained for 1 h at room temperature with the following antibodies: IFN‐γ (clone XMG1.2, 505810, 1:500 dilution), granzyme B (clone QA16A02, 372206, 1:200 dilution), perforin (clone S16009A, 154306, 1:1,000 dilution), and TNF‐α (clone MP6‐XT22, 506323, 1:500 dilution) from BioLegend.
To stain intracellular transcription factors, cells were fixed and permeabilized using the transcription factor staining buffer set (00‐5523‐00, eBioscience, San Diego, CA). Cells were stained for 1 h at room temperature with the following antibodies: TCF‐1 (clone S33‐966, 564217, 1:500 dilution), and Foxp3 (clone 3G3, 566881, 1:200 dilution) from BD Bioscience, and Ki67 (clone SolA15, 12‐5698‐82, 1:100 dilution) from eBioscience.
Cells were analyzed using BD LSRFortessa X‐20, and data were acquired and analyzed with BD FACSDiva and FlowJo 10 software.
2.8. Tumour implantation and treatments
Murine cancer cells were resuspended in culture medium and 2 × 105 cells per mouse were implanted subcutaneously on the right flank. Tumour size was monitored by a blinded investigator in 2 days interval with digital calipers. Tumour volume was calculated with the formula of Volume = (Length × Width × Width)/2.
2.9. Toxicity of E. coli OMVs
C57BL/6 mice were subcutaneously administered with various amounts of E. coli OMVs (n = 10 per group). The survival rates, body weights, and possible adverse effects (piloerection, eye exudates, and diarrhoea) were monitored until 12 days after OMV administration.
2.10. Immunofluorescence
After tumour‐bearing mice were euthanized in CO2 chamber, tumour tissue was isolated from the mice, and was fixed in 4% paraformaldehyde at 4°C. Fixed tumour tissue was then infiltrated with sucrose solution in gradual manner from 15% to 30% and was frozen in an optimal cutting temperature compound. After cutting the tissue at a thickness of 50 μm, the sections were transferred to a 12‐well plate, rinsed with PBS, blocked with 10% normal goat serum and 0.3% Triton X‐100 in PBS, and then incubated overnight with the following primary antibodies: anti‐cluster of differentiation 31 (CD31)/platelet endothelial cell adhesion molecule‐1 (NB100‐2284, 1:200 dilution), anti‐CD8 (NBP1‐49045, 1:500 dilution), and anti‐CD11b (NB600‐1327, 1:500 dilution), from Novus Biologicals (Centennial, CO). Followed by several washing with PBS, the sections were incubated for 2 h at room temperature with Alexa Fluor 594 dye‐conjugated anti‐rabbit IgG (ab150080, Abcam, Cambridge, UK; 1:1,000 dilution) and Alexa Fluor 488 dye‐conjugated anti‐rat IgG (ab150157, Abcam, Cambridge, UK; 1:1,000 dilution): nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole. After the sections were mounted with fluorescent mounting medium (S3023, Agilent, Santa Clara, CA), images were acquired with an inverted microscope (DMi8; Leica, Wetzlar, Germany).
2.11. Statistical analysis
All statistical analyses were performed using GraphPad Prism 8.0 (GraphPad Software, San Diego, CA). To compare two groups and more than two groups, P values were acquired using two‐tailed unpaired Student's t‐tests and two‐way ANOVA with Bonferroni correction, respectively. P values less than 0.05 were considered as indication of statistical significance.
2.12. Data availability
We have submitted all relevant data of our experiments to EV‐TRACK knowledgebase (EV‐TRACK ID: EV230601). In addition, the mass spectrometry proteomics data have been deposited to ProteomeXchange Consortium via PRIDE (Perez‐Riverol et al., 2022) partner repository with identifier PXD043119.
3. RESULTS
3.1. Large‐scale production and characterization of E. coli OMVs
OMVs were isolated from 160 L of conditioned media of E. coli BL21 (DE3) ΔmsbB in a 300 L‐scale bioreactor (Figure 1a). It has been well‐known that E. coli ΔmsbB produce endotoxin (lipopolysaccharide) with attenuated toxicity in E. coli (S. Kim et al., 2009; Low et al., 1999; Somerville et al., 1999), and release OMVs that harbour endotoxin with reduced toxicity (O. Y. Kim et al., 2017). After pre‐clearing the conditioned medium by centrifugation and filtration, OMVs were concentrated by ultrafiltration. OMVs were isolated by metal precipitation and size‐exclusion chromatography, and then subsequently subjected to filtration and dialysis. From 160 L of the conditioned medium, we isolated 357 mg in total protein amounts of OMVs, which was equivalent to 3.93 × 1015 particles (1.10 × 1010 particles/μg in total protein amounts of OMVs).
FIGURE 1.
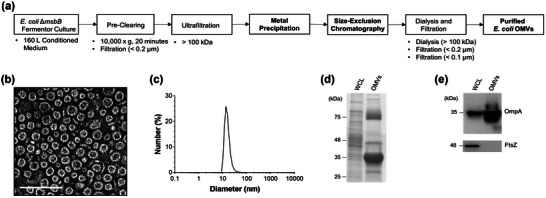
Large‐scale production and characterization of Escherichia coli OMVs. (a) Schematic workflow of large‐scale production of E. coli OMVs. (b) Transmission electron microscopic images of OMVs. Scale bars = 100 nm. (c) The size distribution of OMVs determined by dynamic light scattering (n = 5). (d) Coomassie Brilliant Blue staining of whole cell lysates (WCL) and OMVs, each 5 μg in total protein amounts. Molecular weight standards are indicated on the left (kDa). (e) Western blot using anti‐OmpA or anti‐FtsZ. A total of 1 μg of protein samples from whole cell lysates (WCL) and from OMVs were loaded to detect OmpA (upper panel) or FtsZ (lower panel).
OMVs have homogenously spherical and lipid‐bilayered structures, as visualized by transmission electron microscopy (Figure 1b). The average diameter of OMVs was 24.38 ± 5.81 nm, as measured by dynamic light scattering (Figure 1c). Although it has been reported that the size of Gram‐negative bacterial OMVs ranges from 20 to 400 nm in diameter (J. H. Kim et al., 2015; Toyofuku et al., 2019), it is worthwhile to note that some strains of E. coli release OMVs ranged in diameter from 20 to 40 nm with the highest peak at approximately 30 nm as observed in this study (Jang et al., 2015; O. Y. Kim et al., 2017; E. Lee et al., 2007; J. Lee et al., 2018; Park et al., 2010). SDS‐PAGE demonstrated that OMVs had distinct protein compositions when compared to whole cell lysates (Figure 1d). In addition, outer membrane proteins such as OmpA were enriched in OMVs while cytosolic proteins such as FtsZ were de‐enriched (Figure 1e and Figure S1), as previously reported (J. Lee et al., 2018).
3.2. Proteomic analysis of E. coli OMVs
After in‐gel digestion of methanol/chloroform‐precipitated OMV proteins with trypsin, the peptides were analyzed by LC‐MS/MS (technical replicate n = 3). The MS/MS files were searched with E. coli K12 proteome from UniProt database using X!Tandem and Trans‐Proteomic Pipeline (version 4.6; PeptideProphet > 0.99, ProteinProphet > 0.99). The relative abundance of identified vesicular proteins were calculated by (Lu et al., 2007). A total of 792, 802, and 782 vesicular proteins were identified from each replicate, resulting in identifying a total of 882 non‐redundant vesicular proteins (Figure 2a and Table S1). A total of 882 vesicular proteins were quantitated with high confidence (false discovery rate < 0.002 from all technical replicates), and their subcellular localizations were determined with UniProt and DAVID databases. Then, the abundance of whole and top 100 abundant vesicular proteins was depicted with their abundance ranks, marking different subcellular localizations with different colors (Figure 2b,c). Among whole vesicular proteins (total abundance = 100,000), outer membrane proteins were located at high abundance ranks (Figure 2b and Table S1). In fact, among top 100 abundant vesicular proteins (top 100 summed abundance = 54,730; 100.0%), there were 32 outer membrane proteins (summed abundance = 30,220; 55.2%), 12 periplasmic proteins (summed abundance = 3,946; 7.21%), 18 inner membrane proteins (summed abundance = 7,251; 13.2%), and 40 cytosolic proteins (summed abundance = 14,111; 25.8%) (Figure 2c; Table S2 and S3).
FIGURE 2.
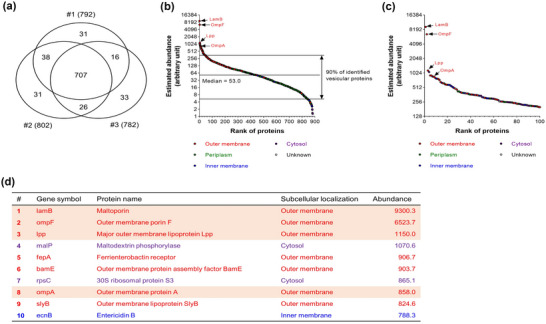
Proteomic analysis of Escherichia coli OMVs. (a) Venn diagram of the numbers of vesicular proteins identified (technical replicate n = 3). A total of 882 proteins was identified from OMVs. (b, c) The distribution of estimated abundance of whole (b) and top 100 abundant (c) vesicular proteins according to their abundance ranks. Proteins with different subcellular localizations were marked with different colours: outer membrane—red; periplasm—green; inner membrane—blue; cytosol—purple; and unknown localizations—white. Note that classical OMV marker proteins, such as LamB, OmpF, Lpp, and OmpA, were identified with high abundance. (d) Top 10 abundant vesicular proteins according to their abundance ranks were listed with their gene symbols, protein names, subcellular localizations, and abundance. Classical OMV marker proteins are highlighted.
We identified some classical OMV marker proteins, such as LamB, OmpF, Lpp, and OmpA. These proteins ranked top 10 out of a total of 882 vesicular proteins (Figure 2d). Furthermore, we also identified several inner membrane and cytosolic proteins among vesicular proteins as previously reported (Hong et al., 2019; O. Y. Kim et al., 2017; E. Lee et al., 2007; E. Lee et al., 2008; J. Lee et al., 2016). In addition, as LC‐MS/MS is very sensitive, proteins which were not detectable in Western blot, such as cytosolic protein FtsZ, can be identified from LC‐MS/MS.
3.3. Dose‐dependent tumour growth inhibition induced by E. coli OMVs
To evaluate therapeutic efficacy and anti‐tumour responses induced by mass‐produced OMVs, we intratumorally administered indicated dosages of OMVs for four times in 3‐day interval to mice bearing either MB49 or EMT6 tumour (Figure 3a). For both MB49 and EMT6 tumour, OMVs induced significant tumour regression in a dose‐dependent manner. Complete remission of MB49 and EMT6 tumour was observed in the majority of mice treated with 3.52 × 109 of OMVs. When treated with 2.20 × 108 and 8.80 × 108 of OMVs, 63% and 94% of tumour growth inhibition was achieved in MB49 tumour, respectively, whereas 56% and 85% of tumour growth inhibition was achieved in EMT6 tumour, respectively (Figure 3b,c and Figure S2, 3).
FIGURE 3.
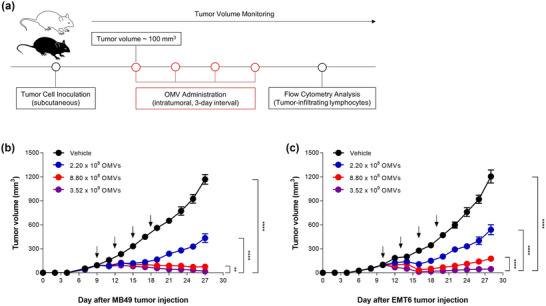
Dose‐dependent tumour growth inhibition induced by Escherichia coli OMVs. (a) Experimental procedure to evaluate OMV‐induced anti‐tumour responses. C57BL/6 and Balb/c mice were subcutaneously inoculated with MB49 and EMT6 cells, respectively. When the tumour volume reached about 100 mm3, indicated dosage of OMVs was intratumorally administered four times in 3 days interval. (b, c) Tumour volume was measured every 2 days. n = 11 for each dosage of OMVs. **p < 0.01, ****p < 0.0001 calculated by two‐way ANOVA with Bonferroni correction for multiple comparisons.
We next evaluated toxic effects of E. coli OMVs by subcutaneously administering various amounts of OMVs into C57BL/6 mice. No mice were dead upon administering 1.10 × 1011 and 3.30 × 1011 of OMVs (Figure S4A), which was about 30 times and 100 times higher than the effective dose of 3.52 × 109 OMVs that caused to complete remission of MB49 tumour as shown in Figure 3b, respectively. However, a total of four mice out of ten mice were dead upon administering 1.10 × 1012 of OMVs (> 300 times higher than the effective dose of 3.52 × 109 OMVs). The mice experienced temporal loss of body weights in a dose‐dependent manner, but they were recovered at most 10 days after OMV administration (Figure S4B). In addition, we observed that some mice, especially those administered with 1.10 × 1012 of OMVs, showed piloerection, eye exudates, and diarrhoea temporally, but the effects disappeared about a week of OMV administration.
3.4. E. coli OMVs induced activation and recruitment of cancer antigen‐specific T cells
To investigate whether OMVs per se, which do not contain cancer antigens, could directly activate cancer antigen‐specific T cell responses, mice were inoculated with MC38‐OVA or B16F10‐OVA cancer cells generated to express whole OVA protein as a cancer neoantigen in the cytosol. OMV treatment significantly increased infiltration of total CD3+ T cells and CD8+ T cells into the tumour tissue in MC38‐OVA tumour model while increasing infiltration of total CD3+ T cells, CD4+ T cells, and CD8+ T cells in B16F10‐OVA tumour model (Figure 4a). In both neoantigen‐expressing tumour models, OMV treatment decreased the proportion of regulatory T (Treg) cells in the tumour microenvironment, thus significantly increasing CD8/Treg ratio (Figure 4a).
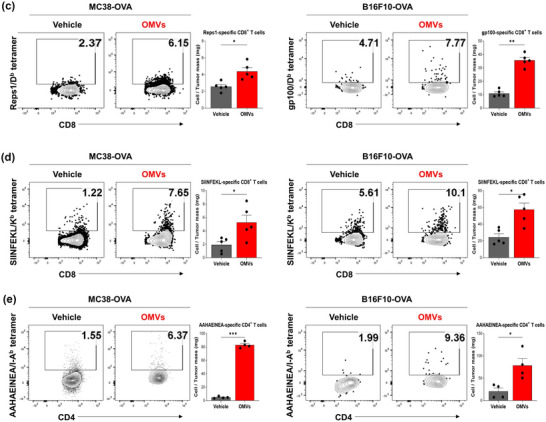
FIGURE 4.
Escherichia coli OMV‐induced recruitment and activation of cancer antigen‐specific T cell. C57BL/6 mice were subcutaneously inoculated with MC38‐OVA and B16F10‐OVA tumour cells. When the tumour volume reached about 100 mm3, 2.20 × 1010 of OMVs was intratumorally administered four times in 3 days interval. Mice were sacrificed 3 days post‐administration to analyze the immune profile of the tumour microenvironment via flow cytometry. (a) Flow cytometry analysis of tumour‐infiltrating T cell populations. Data are shown as frequency of CD3+, CD4+, and CD8+ T cells per milligram of tumour tissue, and as percentage of regulatory T cells from CD45+ T cells in tumour (n = 5). (b) Flow cytometry analysis of cytotoxic molecules expression on tumour‐infiltrating CD8+ T cell. Data are shown as frequency of Granzyme B+, TNFα+, Perforin+, and IFNγ+ CD8+ T cells per milligram of tumour tissue (n = 5). (c) Representative tetramer staining of Reps1‐specific CD8+ T cells and gp100‐specific CD8+ T cells. Reps1‐specific CD8+ T cells were analyzed using Reps1/Db tetramer and gp100‐specific CD8+ T cells were analyzed using gp100/Db tetramer. Data are shown as frequency of Reps1‐specific CD8+ T cells and gp100‐specific CD8+ T cells per milligram of tumour tissue (n = 5). (d, e) Representative tetramer staining of OVA‐specific CD8+ and CD4+ T cells. OVA‐specific CD8+ T cells were analyzed using SIIINFEKL/Kb tetramer and OVA‐specific CD4+ T cells were analyzed using AAHAEINEA/I‐Ab tetramer. Data are shown as frequency of OVA‐specific CD8+ (n = 5) and CD4+ T cells per milligram of tumour tissue (n = 4). *p < 0.05, **p < 0.01, ***p < 0.001 calculated by two‐tailed unpaired Student's t‐tests.
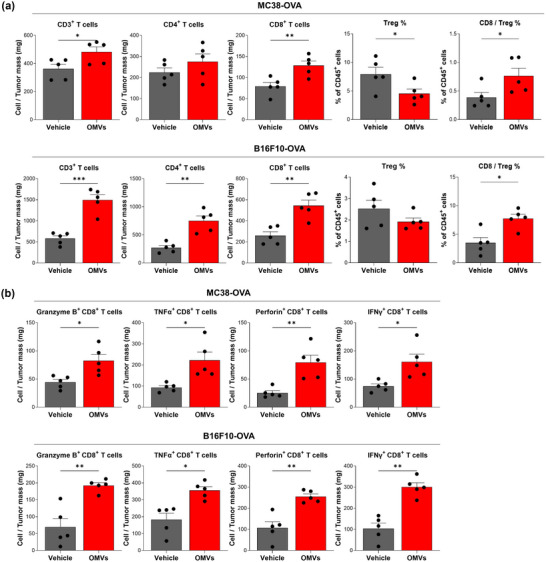
The infiltrated CD8+ T cells upon OMV treatment showed superior effector function compared to that of CD8+ T cells of vehicle treatment group. To assess cytolytic activity of CD8+ T cells, we analyzed frequency of CD8+ T cells expressing cytotoxic molecules (granzyme B, TNFα, perforin, and IFNγ) via intracellular flow cytometry. OMV treatment induced a substantial increase in the expression of cytotoxic molecules on CD8+ T cells in both MC38‐OVA and B16F10‐OVA tumour models compared to vehicle treatment (Figure 4b).
We next examined whether OMVs could induce activation and recruitment of cancer antigen‐specific T cells into the tumour microenvironment using tetramer staining of tumour associated antigens [MC38 (Reps1‐Db) and B16F10 (gp100‐Db)] and neoantigens [OVA (CD8+ T cells: SIINFEKL‐Kb) and (CD4+ T cells: AAHAEINEA‐Ab)] for both MC38‐OVA and B16F10‐OVA tumour models. Tumour associated antigen‐specific CD8+ T cells were increased in both MC38‐OVA and B16F10‐OVA tumour models: Reps1‐specific CD8+ T cells and gp100‐specific CD8+ T cells had highly increased after OMV treatment, respectively, compared to vehicle treatment (Figure 4c). Furthermore, OMVs also increased activation and recruitment of neoantigen OVA‐specific CD8+ T cells (SIINFEKL‐Kb+) and CD4+ T cells (AAHAEINEA‐Ab+) into the tumour microenvironment for both tumour models compared to vehicle treatment (Figure 4d,e).
Taken together, these results suggest that OMVs could activate cancer antigen‐specific T cells and induce recruitment of activated T cells into the tumour microenvironments without co‐administered cancer antigens and these tumour‐infiltrated T cells revealed enhanced cytolytic activity. Therefore, OMVs could act like antigen‐agnostic cancer vaccines (Saxena et al., 2021) and could change the tumour microenvironment thereby shifting from immunosuppressive into immune‐inflamed one.
3.5. E. coli OMVs enhanced TCF‐1+ stem‐like CD8+ T cells
To further characterize the tumour‐infiltrating T cells, we performed flow cytometry analysis to measure frequency of T cells that exhibit stem‐like properties (Eberhardt et al., 2021; Hudson et al., 2019). Evaluation of stem‐like TCF‐1+ CD8+ T cells demonstrated that OMVs significantly increased infiltration of stem‐like CD8+ T cells and neoantigen‐specific (SIINFEKL‐Kb+) stem‐like CD8+ T cells in MC38‐OVA tumour tissue compared to vehicle treatment (Figure 5a). Tumour‐infiltrating CD8+ T cells also revealed increased expression of surface PD‐1, TIM3, and CD39 molecules following OMV treatment compared to vehicle treatment (Figure 5b), indicating that cancer antigen‐specific CD8+ T cells were activated and recruited into the tumour microenvironment and became exhausted (Chow et al., 2023).
FIGURE 5.
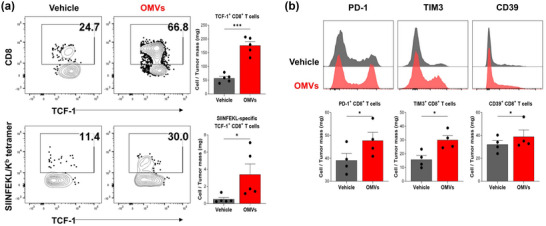
Escherichia coli OMV‐induced generation of TCF‐1+CD8+ T cells. (a) Representative staining of TCF‐1 on tumour‐infiltrating CD8+ T cells and on SIIINFEKL/Kb –specific CD8+ T cells. Data are shown as frequency of TCF‐1+CD8+ and TCF‐1+SIIINFEKL/Kb –specific CD8+ T cells per milligram of tumour tissue (n = 5). (b) Representative staining of T cell exhaustion markers (PD‐1, TIM3, CD39) expression on tumour‐infiltrating CD8+ T cells (n = 4). Data are shown as frequency of PD‐1+, TIM3+, and CD39+CD8+ T cells per milligram of tumour tissue. *p < 0.05, ***p < 0.001 calculated by two‐tailed unpaired Student's t‐tests.
3.6. Synergistic anti‐tumour activity of E. coli OMVs with anti‐PD‐1 immunotherapy
As stem‐like T cells are the responsible cells which proliferate following anti‐PD‐1 antibody therapy and exhibit anti‐tumour activity (Im et al., 2016), MC38‐OVA tumour model was used to evaluate the synergistic anti‐tumour effect of OMVs and anti‐PD‐1 combination therapy. Groups of MC38‐OVA tumour bearing mice were either untreated, treated with OMVs or anti‐PD‐1 antibody alone, or treated in combination of OMVs and anti‐PD‐1 antibody. Combination therapy resulted in significant tumour growth inhibition compared to those of groups treated with none or single therapy (Figure 6a and Figure S5).
FIGURE 6.
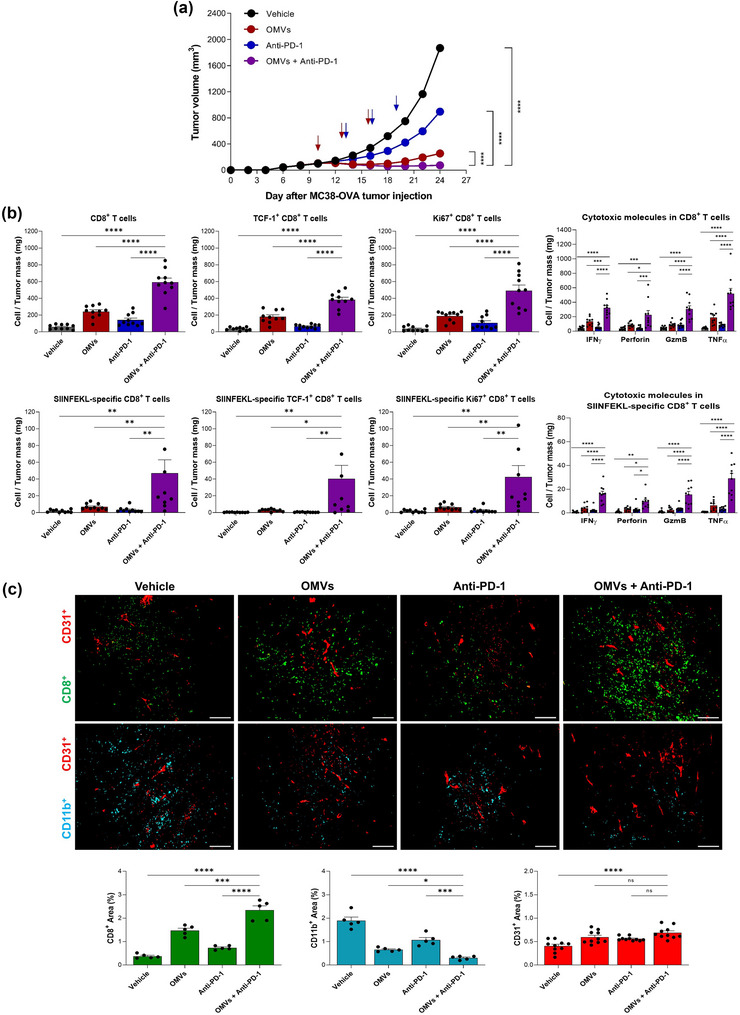
Synergistic anti‐tumour activity of Escherichia coli OMVs with anti‐PD‐1 immunotherapy. C57BL/6 mice were subcutaneously inoculated with MC38‐OVA cells. When the tumour volume reached about 100 mm3, mice were randomized into indicated groups. 2.20 × 1010 of OMVs was intratumorally administered three times in 3 days interval while 10 mg/kg anti‐PD‐1 antibody was intravenously administered three times in 3 days interval, 3 days after the first OMV administration. (a) Tumour volume was measured every 2 days. n = 22 for each group. (b) Flow cytometry analysis of total CD8+ T cell, TCF‐1+CD8+ T cell, Ki67+CD8+ T cell, and CD8+ T cells expressing cytotoxic molecules (IFNγ, Perforin, Granzyme B, TNFα); neoantigen‐specific CD8+ T cell, neoantigen‐specific TCF‐1+CD8+ T cell, neoantigen‐specific Ki67+CD8+ T cell, and neoantigen‐specific CD8+ T cells expressing cytotoxic molecules (IFNγ, Perforin, Granzyme B, TNFα). Data are shown as frequency of T cells per milligram of tumour tissue. GzmB, Granzyme B (n = 10). (c) Representative image of CD8+ T cells, CD11b+ myeloid cells, and CD31+ blood vessels in tumour tissue sections acquired via immunofluorescence (n = 5). Data are shown as percentage of leukocyte‐infiltrated area relative to the whole tumour, calculated using ImageJ software. n = 5 for each group. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 calculated by two‐way ANOVA with Bonferroni correction for multiple comparisons.
Flow cytometry analysis demonstrated that combination therapy led to further dramatic increase of tumour‐infiltrating CD8+ T cells and neoantigen‐specific CD8+ T cells that exhibit stem‐like memory phenotype compared to OMV only treatment (Figure 6b). It also enhanced proliferation and cytolytic ability of total CD8+ T cells and neoantigen‐specific CD8+ T cells compared to OMV only treatment (Figure 6b). Immunofluorescence microscopy confirmed that OMVs and anti‐PD‐1 combination therapy further increased CD8+ T cell infiltration while further decreased tumour‐infiltrating myeloid cell population (CD11b+) compared to OMV only treatment (Figure 6c). These results demonstrate that anti‐PD‐1 therapy synergistically enhances anti‐tumour effects induced by OMVs.
4. DISCUSSION
In this study, we developed strategies for mass production of E. coli OMVs by combining metal precipitation and size‐exclusion chromatography. We isolated 357 mg (3.93 × 1015 particles) of OMVs from 160 L of E. coli BL21 (DE3) ΔmsbB culture. OMVs have similar morphology and diameter with other bacterial EVs. In addition, proteomic analysis on OMVs elucidated that vesicular proteome is enriched with outer membrane and periplasmic proteins, but de‐enriched with inner membrane proteins, when compared with the cellular proteome. Upon intratumoral administration, OMVs inhibited tumour growth in a dose‐dependent manner. OMVs induced activation and recruitment of antigen‐specific T cells into tumour microenvironment. Furthermore, OMVs induced stem‐like CD8+ T cell recruitment and showed synergistic effects with anti‐PD‐1 combination therapy.
In 2017, we reported for the first time that EVs derived from Gram‐negative and Gram‐positive bacteria could be the novel platform for cancer immunotherapy (O. Y. Kim et al., 2017). This study showed that bacterial EVs could effectively induce anti‐tumour immune responses, which could completely suppress established tumours without significant adverse effects. In addition, IFN‐γ, NK cells, and T cells played important roles in bacterial EV‐induced anti‐tumour immune responses. Unlike our pervious study, we here observed the anti‐tumour efficacy of E. coli OMVs when treated via in situ vaccination method. By administering OMVs directly into the tumour, we utilized antigen‐agnostic strategy where OMVs are used as immunostimulatory agents to modify the tumour microenvironment without the need for antigen prediction. As expected, OMVs alone substantially increased infiltration and activation of CD8+ T cells, especially cancer‐specific CD8+ T cells. More importantly, in situ vaccination of OMVs recruited cancer antigen‐specific CD8+ T cells with stem‐like features into the tumour microenvironment, suggesting capability of OMVs to mediate the crosstalk between innate and adaptive immunity: (1) potent initial activation of cancer‐specific CD8+ T cells, and (2) induction and maintenance of long‐standing adaptive immunity (Kallies et al., 2020).
T cell exhaustion resulting from persistent antigen stimulation is one of the key features of chronic infections and cancers (McLane et al., 2019). Exhausted T cells are characterized by the loss of effector functions and sustained upregulation of negative immune checkpoint molecules (McLane et al., 2019; Wherry & Kurachi, 2015). Since the interactions between PD‐1 and PD‐L1/2 play a major role in regulation of T cell exhaustion (Barber et al., 2006; Blackburn et al., 2008), PD‐1/PD‐L1 targeted immunotherapy have been widely implicated as potentially effective anti‐cancer therapy to reinvigorate and to induce selective proliferation of these exhausted T cells (J. Lee et al., 2015; Wherry & Kurachi, 2015). Thus, overcoming the immunosuppressive environment of cancer by recruiting activated cancer‐specific T cells is critical to improve therapeutic efficacy of PD‐1/PD‐L1 therapy (P. Chen et al., 2016; Tumeh et al., 2014). Moreover, recent studies indicated TCF‐1‐expressing progenitor of exhausted T cells (TPEX) as the primary responders to PD‐1/PD‐L1 therapy (Im et al., 2016; Miller et al., 2019; Sade‐Feldman et al., 2019; Siddiqui et al., 2019). These aspects further emphasize unmet needs to develop novel cancer immunotherapeutic agents that could shift immunosuppressive nature of cancer into immune‐inflamed one. In this study, we showed that in situ vaccination of OMVs caused not only effective infiltration of the activated cancer‐specific CD8+ T cells into the tumour microenvironment, but also upregulation of the expression level of exhaustion markers and TCF‐1 on these activated cancer‐specific CD8+ T cells. This explains synergism of combination therapy of OMVs and anti‐PD‐1, and enhanced anti‐tumour activity of engineered PD‐1‐OMVs over non‐engineered naïve OMVs (Li et al., 2020; Pan et al., 2022).
However, to develop as novel cancer immunotherapeutics, it is necessary to produce bacterial EVs in a large‐scale. The previous study employed ultracentrifugation, which is not suitable for mass production. In this study, we developed mass production strategies by the combination of metal precipitation and size‐exclusion chromatography. Using ultracentrifugation‐free strategies, we isolated E. coli OMVs in a large‐scale (160 L), yielding 2.23 mg in a total protein amount/2.45 × 1013 particles from one‐litre conditioned media with a purity of 1.10 × 1010 particles/μg in total protein amounts of OMVs. E. coli OMVs showed similar morphology and diameters with those of E. coli OMVs in other studies (E. Lee et al., 2007; J. Lee et al., 2018). Furthermore, E. coli OMVs were enriched with outer membrane and periplasmic proteins, and de‐enriched with inner membrane proteins, as previously reported (Hong et al., 2019; O. Y. Kim et al., 2017; E. Lee et al., 2007; E. Lee et al., 2008; J. Lee et al., 2016). To compare with well‐established conventional OMV isolation method, we isolated OMVs by combining ultracentrifugation and buoyant density gradient ultracentrifugation (O. Y. Kim et al., 2017): the yield was 0.497 mg of OMVs (9.26 × 109 particles/μg OMVs) from one‐litre conditioned media. Moreover, the yield of OMVs from one‐litre conditioned medium of small‐scale shake flask culture is 0.3 ∼ 5.0 mg in total protein amounts with a purity of 107–109 particles/μg in total protein amounts of OMVs (Bittel et al., 2021; Hong et al., 2019). Taken together, the yield and purity in this study is comparable or superior to conventional buoyant density gradient ultracentrifugation method and smaller‐scale preparations from shake flask culture. However, it could not be completely excluded that trace amount of contaminants is present in purified OMVs: these non‐vesicular proteins or contaminants may be contributed to the observed anti‐tumour activity or toxicity of E. coli OMVs.
To develop E. coli OMVs as next‐generation cancer immunotherapeutic agents, future research would be directed towards overcoming limitations of this study. First, critical process parameters and quality criteria of upstream and downstream processing should be established for reproducibility and quality control of large‐scale manufacturing of E. coli OMVs: (1) the effects of upstream oxygen concentration and cell density, (2) quantitation of critical quality attributor(s) such as highly abundant OMV marker protein (LamB, OmpF, Lpp, or OmpA) to determine OMV recovery from conditioned media, yield, and purity/impurity, (3) quality control criteria for purified OMVs by defining or measuring of yield and purity/impurity (Gimona et al., 2017; Gimona et al., 2021). Second, we have a plan for preclinical good laboratory practice (GLP) toxicology studies to evaluate in‐depth E. coli OMV‐mediated toxicity: we could not observe any serious adverse effects by administration of up to 100 times higher OMVs than their effective dose that caused to complete remission of MB49 and EMT6 tumour in mice. Third, in preclinical GLP toxicology studies, we will additionally carry out toxicokinetics and pharmacokinetics studies of E. coli OMVs. We observed that Cy7‐labeled OMVs were taken up by RAW264.7 macrophages (Figure S6A) and caused increased release of IL‐6 and TNF‐α in dose‐dependent manners (Figure S6B, C), suggesting that Cy7‐labeled OMVs would be applicable to their toxicokinetics and pharmacokinetics studies. Lastly, we are planning to further evaluate the effects of OMVs to orthotopically placed tumour models, which better mimic native tumour microenvironment compared to subcutaneous tumour models.
In conclusion, we developed mass production strategies to isolate OMVs derived from E. coli with high‐yield and low‐cost. Moreover, we demonstrated that these mass‐produced E. coli OMVs have potent anti‐tumour activities and activate cancer antigen‐specific stem‐like CD8+ T cells which enables an effective combination immunotherapy with anti‐PD‐1. Based on this study, we are now undergoing preclinical GLP toxicology studies and good manufacturing practice mass production for human clinical trial of E. coli OMVs as next‐generation cancer immunotherapeutic agents.
AUTHOR CONTRIBUTIONS
Solchan Won: Formal analysis; investigation; methodology; resources; validation; visualization; writing—original draft; writing—review and editing. Changjin Lee: Conceptualization; investigation; methodology; resources; validation. Seoyoon Bae: Formal analysis; investigation; validation; visualization; writing—review and editing. Jaemin Lee: Formal analysis; investigation; resources. Dongsic Choi: Data curation; investigation; supervision; validation. Min‐Gang Kim: Investigation; methodology; visualization. Sunghyun Song: Investigation; resources. Jaewook Lee: Data curation; formal analysis; investigation; software; validation; visualization; writing—original draft; writing—review and editing. Eunhye Kim: Investigation; visualization; writing—original draft. HaYong Shin: Investigation; visualization. Anita Basukala: Investigation; visualization. TaeRyong Lee: Conceptualization; project administration; supervision; validation. Dong‐Sup Lee: Conceptualization; funding acquisition; methodology; project administration; supervision; validation; writing—original draft; writing—review and editing. Yong Song Gho: Conceptualization; funding acquisition; methodology; project administration; supervision; validation; writing—original draft; writing—review and editing.
CONFLICT OF INTEREST STATEMENT
CL, JL, SS, JL and TRL are currently employed at SL Bigen in Incheon, Republic of Korea, developing E. coli OMVs as cancer immunotherapeutic agents. CL, JL, DC, SS, JL, SB, TRL and YSG are inventors of multiple EV‐associated patents for putative clinical utilization.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGEMENTS
This research was supported by National Research Foundation of Korea (NRF), grant funded by Korea government (MSIT) (2021R1A2C3005275 to Y.S.G and 2021R1A2C1011920 to D.‐S.L.) and Korea Health Technology R&D Project through Korea Health Industry Development Institute (KHIDI), grant funded by Ministry of Health & Welfare (MOHW) (HV20C0052 to Y.S.G and HV22C0228 to D.‐S.L). S.W., and E.K. were awarded fellowships by BK21 Four Biomedical Science Program through Seoul National University College of Medicine, Seoul 03080, Republic of Korea.
Won, S. , Lee, C. , Bae, S. , Lee, J. , Choi, D. , Kim, M.‐G. , Song, S. , Lee, J. , Kim, E. , Shin, H. Y. , Basukala, A. , Lee, T. R. , Lee, D.‐S. , & Gho, Y. S. (2023). Mass‐produced gram‐negative bacterial outer membrane vesicles activate cancer antigen‐specific stem‐like CD8+ T cells which enables an effective combination immunotherapy with anti‐PD‐1. Journal of Extracellular Vesicles, 12, e12357. 10.1002/jev2.12357
Solchan Won, Changjin Lee, Seoyoon Bae, and Jaemin Lee authors equally contributed to the work.
Contributor Information
Tae Ryong Lee, Email: trlee@slbigen.com.
Dong‐Sup Lee, Email: dlee5522@snu.ac.kr.
Yong Song Gho, Email: ysgho@postech.ac.kr.
REFERENCES
- Barber, D. L. , Wherry, E. J. , Masopust, D. , Zhu, B. , Allison, J. P. , Sharpe, A. H. , Freeman, G. J. , & Ahmed, R. (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature, 439, 682–687. [DOI] [PubMed] [Google Scholar]
- Bittel, M. , Reichert, P. , Sarfati, I. , Dressel, A. , Leikam, S. , Uderhardt, S. , Stolzer, I. , Phu, T. A. , Ng, M. , Vu, N. K. , Tenzer, S. , Distler, U. , Wirtz, S. , Rothhammer, V. , Neurath, M. F. , Raffai, R. L. , Günther, C. , & Momma, S. (2021). Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe‐host‐communication in vivo . Journal of Extracellular Vesicles, 10, e12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn, S. D. , Shin, H. , Freeman, G. J. , & Wherry, E. J. (2008). Selective expansion of a subset of exhausted CD8 T cells by αPD‐L1 blockade. Proceedings National Academy of Science USA, 105, 15016–15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, R. D. , Flickinger, J. C., Jr. , & Snook, A. E. (2020). Talkin' toxins: From Coley's to modern cancer immunotherapy. Toxins (Basel), 12, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , & Han, X. (2015). Anti‐PD‐1/PD‐L1 therapy of human cancer: Past, present, and future. Journal of Clinical Investigation, 125, 3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P. , Roh, W. , Reuben, A. , Cooper, Z. A. , Spencer, C. N. , Prieto, P. A. , Miller, J. P. , Bassett, R. L. , Gopalakrishnan, V. , Wani, K. , De Macedo, M. P. , Austin‐Breneman, J. L. , Jiang, H. , Chang, Q. , Reddy, S. M. , Chen, W.‐S. , Tetzlaff, M. T. , Broaddus, R. J. , Davies, M. A. , … Wargo, J. A. (2016). Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer discovery, 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhang, Z. , Zheng, X. , Tao, H. , Zhang, S. , Ma, J. , Liu, Z. , Wang, J. , Qian, Y. , Cui, P. , Huang, D. , Huang, Z. , Wu, Z. , & Hu, Y. (2021). Response efficacy of PD‐1 and PD‐L1 inhibitors in clinical trials: A systematic review and meta‐analysis. Frontiers in oncology, 11, 562315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, D. , Rak, J. , & Gho, Y. S. (2021). Isolation of extracellular vesicles for proteomic profiling. Methods in Molecular Biology, 2261, 193–206. [DOI] [PubMed] [Google Scholar]
- Chow, A. , Uddin, F. Z. , Liu, M. , Dobrin, A. , Nabet, B. Y. , Mangarin, L. , Lavin, Y. , Rizvi, H. , Tischfield, S. E. , Quintanal‐Villalonga, A. , Chan, J. M. , Shah, N. , Allaj, V. , Manoj, P. , Mattar, M. , Meneses, M. , Landau, R. , Ward, M. , Kulick, A. , … Rudin, C. M. (2023). The ectonucleotidase CD39 identifies tumor‐reactive CD8+ T cells predictive of immune checkpoint blockade efficacy in human lung cancer. Immunity, 56, 93–106.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dersh, D. , Holly, J. , & Yewdell, J. W. (2021). A few good peptides: MHC class I‐based cancer immunosurveillance and immunoevasion. Nature Reviews Immunology, 21, 116–128. [DOI] [PubMed] [Google Scholar]
- Dinh, N. T. H. , Lee, J. , Lee, J. , Kim, S. S. , Go, G. , Bae, S. , Jun, Y. I. , Yoon, Y. J. , Roh, T.‐Y. , & Gho, Y. S. (2020). Indoor dust extracellular vesicles promote cancer lung metastasis by inducing tumour necrosis factor‐α. Journal of Extracellular Vesicles, 9, 1766821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt, C. S. , Kissick, H. T. , Patel, M. R. , Cardenas, M. A. , Prokhnevska, N. , Obeng, R. C. , Nasti, T. H. , Griffith, C. C. , Im, S. J. , Wang, X. , Shin, D. M. , Carrington, M. , Chen, Z. G. , Sidney, J. , Sette, A. , Saba, N. F. , Wieland, A. , & Ahmed, R. (2021). Functional HPV‐specific PD‐1+ stem‐like CD8 T cells in head and neck cancer. Nature, 597, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen, J. G. , Ouyang, W. , & Wu, L. C. (2020). Human anti‐tumor immunity: Insights from immunotherapy clinical trials. Immunity, 52, 36–54. [DOI] [PubMed] [Google Scholar]
- Galon, J. , & Bruni, D. (2019). Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nature Reviews Drug Discovery, 18, 197–218. [DOI] [PubMed] [Google Scholar]
- Gimona, M. , Brizzi, M. F. , Choo, A. B. H. , Dominici, M. , Davidson, S. M. , Grillari, J. , Hermann, D. M. , Hill, A. F. , De Kleijn, D. , Lai, R. C. , Lai, C. P. , Lim, R. , Monguió‐Tortajada, M. , Muraca, M. , Ochiya, T. , Ortiz, L. A. , Toh, W. S. , Yi, Y. W. , Witwer, K. W. , … Lim, S. K. (2021). Critical considerations for the development of potency tests for therapeutic applications of mesenchymal stromal cell‐derived small extracellular vesicles. Cytotherapy, 23, 373–380. [DOI] [PubMed] [Google Scholar]
- Gimona, M. , Pachler, K. , Laner‐Plamberger, S. , Schallmoser, K. , & Rohde, E. (2017). Manufacturing of human extracellular vesicle‐based therapeutics for clinical use. International Journal of Molecular Sciences, 18, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, J. , Dauros‐Singorenko, P. , Whitcombe, A. , Payne, L. , Blenkiron, C. , Phillips, A. , & Swift, S. (2019). Analysis of the Escherichia coli extracellular vesicle proteome identifies markers of purity and culture conditions. Journal of Extracellular Vesicles, 8, 1632099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, D. W. , Sherman, B. T. , & Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hudson, W. H. , Gensheimer, J. , Hashimoto, M. , Wieland, A. , Valanparambil, R. M. , Li, P. , Lin, J.‐X. , Konieczny, B. T. , Im, S. J. , Freeman, G. J. , Leonard, W. J. , Kissick, H. T. , & Ahmed, R. (2019). Proliferating transitory T cells with an effector‐like transcriptional signature emerge from PD‐1+ stem‐like CD8+ T cells during chronic infection. Immunity, 51, 1043–1058.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im, S. J. , Hashimoto, M. , Gerner, M. Y. , Lee, J. , Kissick, H. T. , Burger, M. C. , Shan, Q. , Hale, J. S. , Lee, J. , Nasti, T. H. , Sharpe, A. H. , Freeman, G. J. , Germain, R. N. , Nakaya, H. I. , Xue, H.‐H. , & Ahmed, R. (2016). Defining CD8+ T cells that provide the proliferative burst after PD‐1 therapy. Nature, 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, S. C. , Kim, S. R. , Yoon, Y. J. , Park, K. , Kim, J. H. , Lee, J. W. , Kim, O. Y. , Choi, E. J. , Kim, D. K. , Choi, D. S. , Kim, Y. K. , Park, J. , Di Vizio, D. , & Gho, Y. S. (2015). In vivo kinetic biodistribution of nano‐sized outer membrane vesicles derived from bacteria. Small, 11, 456–461. [DOI] [PubMed] [Google Scholar]
- Jiang, Y. , Zhao, X. , Fu, J. , & Wang, H. (2020). Progress and challenges in precise treatment of tumors with PD‐1/PD‐L1 blockade. Frontiers in immunology, 11, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallies, A. , Zehn, D. , & Utzschneider, D. T. (2020). Precursor exhausted T cells: Key to successful immunotherapy? Nature Reviews Immunology, 20, 128–136. [DOI] [PubMed] [Google Scholar]
- Kim, J. H. , Lee, J. , Park, J. , & Gho, Y. S. (2015). Gram‐negative and Gram‐positive bacterial extracellular vesicles. Seminars in cell & developmental biology, 40, 97–104. [DOI] [PubMed] [Google Scholar]
- Kim, O. Y. , Park, H. T. , Dinh, N. T. H. , Choi, S. J. , Lee, J. , Kim, J. H. , Lee, S.‐W. , & Gho, Y. S. (2017). Bacterial outer membrane vesicles suppress tumor by interferon‐γ‐mediated antitumor response. Nature Communications, 8, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Kim, K. , Lee, S. , Kim, E. , Kim, M. , Lee, E. , Gho, Y. S. , Kim, J. W. , Bishop, R. E. , & Chang, K. T. (2009). Structural modifications of outer membrane vesicles to refine them as vaccine delivery vehicles. Biochimica Et Biophysica Acta, 1788, 2150–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E. , Bang, J. Y. , Park, G. W. , Choi, D. , Kang, J. S. , Kim, H. J. , Park, K.‐S. , Lee, J.‐O. , Kim, Y. K. , Kwon, K. H. , Kim, K. P. , & Gho, Y. S. (2007). Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli . Proteomics, 7, 3143–3153. [DOI] [PubMed] [Google Scholar]
- Lee, E. , Choi, D. , Kim, K. , & Gho, Y. S. (2008). Proteomics in Gram‐negative bacterial outer membrane vesicles. Mass Spectrometry Reviews, 27, 535–555. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Ahn, E. , Kissick, H. T. , & Ahmed, R. (2015). Reinvigorating exhausted T cells by blockade of the PD‐1 pathway. Forum on Immunopathological Diseases and Therapeutics, 6, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. , Kim, O. Y. , & Gho, Y. S. (2016). Proteomic profiling of Gram‐negative bacterial outer membrane vesicles: Current perspectives. Proteomics: Clinical Applications, 10, 897–909. [DOI] [PubMed] [Google Scholar]
- Lee, J. , Yoon, Y. J. , Kim, J. H. , Dinh, N. T. H. , Go, G. , Tae, S. , Park, K.‐S. , Park, H. T. , Lee, C. , Roh, T.‐Y. , Di Vizio, D. , & Gho, Y. S. (2018). Outer membrane vesicles derived from Escherichia coli regulate neutrophil migration by induction of endothelial IL‐8. Frontiers in Microbiology, 9, 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. , Choi, K. , Bang, G. , Park, S. , Kim, E. , Choi, J. , Chung, Y.‐H. , Kim, J. , Lee, S. G. , Kim, E. , & Kim, J. Y. (2021). Identification of nucleolin as a novel AEG‐1‐interacting protein in breast cancer via interactome profiling. Cancers (Basel), 13, 2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Q. , Wang, D. , Sun, K. , Wang, L. , & Zhang, Y. (2020). Resistance mechanisms of anti‐PD1/PDL1 therapy in solid tumors. Frontiers in Cell and Developmental Biology, 8, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhao, R. , Cheng, K. , Zhang, K. , Wang, Y. , Zhang, Y. , Li, Y. , Liu, G. , Xu, J. , Xu, J. , Anderson, G. J. , Shi, J. , Ren, L. , Zhao, X. , & Nie, G. (2020). Bacterial outer membrane vesicles presenting programmed death 1 for improved cancer immunotherapy via immune activation and checkpoint inhibition. ACS Nano, 14, 16698–16711. [DOI] [PubMed] [Google Scholar]
- Low, K. B. , Ittensohn, M. , Le, T. , Platt, J. , Sodi, S. , Amoss, M. , Ash, O. , Carmichael, E. , Chakraborty, A. , Fischer, J. , Lin, S. L. , Luo, X. , Miller, S. I. , Zheng, L.‐M. , King, I. , Pawelek, J. M. , & Bermudes*, D. (1999). Lipid A mutant Salmonella with suppressed virulence and TNFα induction retain tumor‐targeting in vivo . Nature Biotechnology, 17, 37–41. [DOI] [PubMed] [Google Scholar]
- Lu, P. , Vogel, C. , Wang, R. , Yao, X. , & Marcotte, E. M. (2007). Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nature Biotechnology, 25, 117–124. [DOI] [PubMed] [Google Scholar]
- McLane, L. M. , Abdel‐Hakeem, M. S. , & Wherry, E. J. (2019). CD8 T cell exhaustion during chronic viral infection and cancer. Annual Review of Immunology, 37, 457–495. [DOI] [PubMed] [Google Scholar]
- Miller, B. C. , Sen, D. R. , Al Abosy, R. , Bi, K. , Virkud, Y. V. , LaFleur, M. W. , Yates, K. B. , Lako, A. , Felt, K. , Naik, G. S. , Manos, M. , Gjini, E. , Kuchroo, J. R. , Ishizuka, J. J. , Collier, J. L. , Griffin, G. K. , Maleri, S. , Comstock, D. E. , Weiss, S. A. , … Haining, W. N. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nature Immunology, 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motofei, I. G. (2022). Nobel Prize for immune checkpoint inhibitors, understanding the immunological switching between immunosuppression and autoimmunity. Expert Opinion on Drug Safety, 21, 599–612. [DOI] [PubMed] [Google Scholar]
- Nafaji, S. , Majidpoor, J. , & Mortezaee, K. (2022). The impact of microbiota on PD‐1/PD‐L1 inhibitor therapy outcomes: A focus on solid tumors. Life Sciences, 310, 121138. [DOI] [PubMed] [Google Scholar]
- Pan, J. , Li, X. , Shao, B. , Xu, F. , Huang, X. , Guo, X. , & Zhou, S. (2022). Self‐blockade of PD‐L1 with bacteria‐derived outer‐membrane vesicle for enhanced cancer immunotherapy. Advanced Materials, 34, e2106307. [DOI] [PubMed] [Google Scholar]
- Park, K. , Choi, K. , Kim, Y. , Hong, B. S. , Kim, O. Y. , Kim, J. H. , Yoon, C. M. , Koh, G. Y. , Kim, Y. K. , & Gho, Y. S. (2010). Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE, 5, e11334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol, Y. , Bai, J. , Bandla, C. , Garcia‐Seisdedos, D. , Hewapathirana, S. , Kamatchinathan, S. , Kundu, D. J. , Prakash, A. , Frericks‐Zipper, A. , Eisenacher, M. , Walzer, M. , Wang, S. , Brazma, A. , & Vizcaíno, J. A. (2022). The PRIDE database resources in 2022: A hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Research, 50, D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich, G. A. , Gabrilovich, D. , & Sotomayor, E. M. (2007). Immunosuppressive strategies that are mediated by tumor cells. Annual Review of Immunology, 25, 267–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, D. , Hua, Y. , Yu, B. , Ye, X. , He, Z. , Li, C. , Wang, J. , Mo, Y. , Wei, X. , Chen, Y. , Zhou, Y. , Liao, Q. , Wang, H. , Xiang, B. , Zhou, M. , Li, X. , Li, G. , Li, Y. , Zeng, Z. , & Xiong, W. (2020). Predictive biomarkers and mechanisms underlying resistance to PD1/PD‐L1 blockade cancer immunotherapy. Molecular cancer, 19, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade‐Feldman, M. , Yizhak, K. , Bjorgaard, S. L. , Ray, J. P. , de Boer, C. G. , Jenkins, R. W. , Lieb, D. J. , Chen, J. H. , Frederick, D. T. , Barzily‐Rokni, M. , Freeman, S. S. , Reuben, A. , Hoover, P. J. , Villani, A.‐C. , Ivanova, E. , Portell, A. , Lizotte, P. H. , Aref, A. R. , Eliane, J.‐P. , … Hacohen, N. (2019). Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell, 176, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena, M. , van der Burg, S. H. , Melief, C. J. M. , & Bhardwaj, N. (2021). Therapeutic cancer vaccines. Nature Reviews Cancer, 21, 360–378. [DOI] [PubMed] [Google Scholar]
- Sherman, B. T. , Hao, M. , Qiu, J. , Jiao, X. , Baseler, M. W. , Lane, H. C. , Imamichi, T. , & Chang, W. (2022). DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Research, 50, W216–W221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui, I. , Schaeuble, K. , Chennupati, V. , Fuertes Marraco, S. A. , Calderon‐Copete, S. , Pais Ferreira, D. , Carmona, S. J. , Scarpellino, L. , Gfeller, D. , Pradervand, S. , Luther, S. A. , Speiser, D. E. , & Held, W. (2019). Intratumoral Tcf1+PD‐1+CD8+ T cells with stem‐like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity, 50, 195–211.e10. [DOI] [PubMed] [Google Scholar]
- Somerville, J. E., Jr. , Cassiano, L. , & Darveau, R. P. (1999). Escherichia coli msbB gene as a virulence factor and a therapeutic target. Infection and Immunity, 67, 6583–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium . (2017). UniProt: The universal protein knowledgebase. Nucleic Acids Research, 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium . (2019). UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research, 47, D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyofuku, M. , Nomura, N. , & Eberl, L. (2019). Types and origins of bacterial membrane vesicles. Nature Reviews Microbiology, 17, 13–24. [DOI] [PubMed] [Google Scholar]
- Tumeh, P. C. , Harview, C. L. , Yearley, J. H. , Shintaku, I. P. , Taylor, E. J. M. , Robert, L. , Chmielowski, B. , Spasic, M. , Henry, G. , Ciobanu, V. , West, A. N. , Carmona, M. , Kivork, C. , Seja, E. , Cherry, G. , Gutierrez, A. J. , Grogan, T. R. , Mateus, C. , Tomasic, G. , … Ribas, A. (2014). PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhaya, S. , Neftelinov, S. T. , Hodge, J. , & Campbell, J. (2022). Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nature Reviews Drug Discovery, 21, 482–483. [DOI] [PubMed] [Google Scholar]
- Wherry, E. J. , Ha, S. , Kaech, S. M. , Haining, W. N. , Sarkar, S. , Kalia, V. , Subramaniam, S. , Blattman, J. N. , Barber, D. L. , & Ahmed, R. (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity, 27, 670–684. [DOI] [PubMed] [Google Scholar]
- Wherry, E. J. , & Kurachi, M. (2015). Molecular and cellular insights into T cell exhaustion. Nature Reviews Immunology, 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
We have submitted all relevant data of our experiments to EV‐TRACK knowledgebase (EV‐TRACK ID: EV230601). In addition, the mass spectrometry proteomics data have been deposited to ProteomeXchange Consortium via PRIDE (Perez‐Riverol et al., 2022) partner repository with identifier PXD043119.