

Summary
A sustainable synthesis of interesting glycine betaine derivatives from cyclic 3°-amines viz. N-methyl morpholine (NMM), N-methyl piperidine (NMP), and 1,4-diazabicyclo[2.2.2]octane (DABCO) with numerous aryl diazoacetates 1 in water and under blue LED is reported. Generally, 3°-amines and metal carbenoids (from diazoacetates with transition metal catalysts) provide C-H insertion at the α-position of the amines. Computational comparison of the metal carbenoid with the singlet carbene (metal free and generated under blue LED) realized the difference in reactivity. Next, experimental results corroborated the preliminary findings. The products were isolated either by precipitation of the solid or gel-like final products from the aqueous reaction mixture without any chromatographic purification. The reaction mechanism was realized by control experiments. These compounds exhibit selective bactericidal properties against Gram-positive S. aureus, induce lipid droplets (LDs) formation in HePG2 cells and single crystal X-ray diffraction study of their halogenated analogs reveal interesting Hal … Hal contacts.
Subject areas: Chemistry, Chemical reaction, Organic chemistry
Graphical abstract

Highlights
-
•
Aqueous photochemical synthesis of glycine betaine derivatives
-
•
Quaternary ammonium compound synthesis
-
•
Antimicrobial properties of glycine betaine derivatives
-
•
Sustainable organic synthesis
Chemistry; Chemical reaction; Organic chemistry
Introduction
The discovery of strategies for introducing nitrogen functionality among organic molecules has been one of the key research targets in synthetic chemistry due to the ubiquitous presence of amino compounds in natural products, synthetic intermediates, and pharmaceutical agents.1 The intramolecular and intermolecular NH-insertion reactions of appropriate 1°/2°- and aromatic amines on metal carbenoids (generated from the reactions of transition metal [TM] catalysts and diazo compounds) has evolved as a powerful tool to create C-N bonds.2,3,4,5,6,7,8 Various metals, such as copper, iron, silver, iridium, and rhodium, have been studied as catalysts to facilitate the N-H insertion process. It is noteworthy that such reactions are less developed with nucleophilic amines since they could bind to the metal catalysts to poison them. More recently the TMs are replaced with blue LED or simple thermal decomposition to facilitate such reactions in more environment-friendly conditions.9,10 In this direction there have been numerous reports on TM-catalyzed thermal reactions between tertiary amines and diazoacetates, where instead of N-insertion onto the carbenoids the substituted alkoxy carbonyl methylene moiety of the diazoacetates get inserted into the C-H bond α- to the amines (Scheme 1A).11,12,13,14,15 However, our density functional theory (DFT) based computational analysis of metal carbenoids and electrophilic singlet carbenes (Scheme 2) stipulates that the latter could facilitate N-ylide formation with cyclic 3°-amines under blue LED. The DFT calculations further indicated that N-methyl morpholine (NMM) could be an ideal substrate for such reactions (Scheme 3). Accordingly, the reactions with 3°-amines such as NMM, N-methyl piperidine (NMP), and 1,4-diazabicyclo[2.2.2]octane (DABCO)and aryl diazo esters under the blue LED (5 W) in water, afforded N-ylide mediated novel zwitterionic glycine betaine derivatives 2–4 in good to excellent yields (Schemes 1B and 4). Herein we report an environment-friendly blue LED-driven synthesis of glycine betaine derivatives under metal-free conditions with water as a solvent. The products are easily isolable, the protocol generates low waste, and the reagents are readily available and economical. The betaine structure was confirmed by single-crystal X-ray diffraction (SCXRD) study from the crystals of the represented compounds generated from water. The control experiments realized the reaction mechanism. It is noteworthy that morpholine has been implicated in numerous drug molecules and natural products and DABCO is used as catalysts and base for many organic reactions.16,17 Their ubiquitous presence among bioactive compounds prompted us to investigate their biological properties. Accordingly, they were screened against E. coli and S. aureus to demonstrate selective antimicrobial activity against S. aureus with low micromolar IC50. They also induced lipid droplet (LD) formation in HePG2 liver cancer cells.
Scheme 1.

The difference between (a) reported works and (b) our aqueous reaction of tertiary amines with diazoacetates.
Scheme 2.

Direct comparison between Rh-carbenoid and blue LED induced singlet carbene and their difference in reactivity
(A) Davies's work on C-H activation by metal carbenoid.
(B) Our proof of concept C-N bond formation with singlet carbene.
(C) HOMO-LUMO interaction to facilitate N-ylide formation.
Scheme 3.

Reaction of (a) NMM, (b) NEM, and NBM with 1a. (c) Prediction of organic geometries and energetics.
Scheme 4.

Library of NMM, NMP, and DABCO based C2-aryl glycine betaines.
Results and discussion
Background and proof of concept
It is noteworthy that in general the metal-catalyzed reactions of aryl diazoacetates with dimethyl aniline derivatives, result in aryl insertion (via CH-activation) and with N-heteroarenes and the 1°/2°-amines afford NH-insertion at the carbenoid carbon.18,19,20,21 There are few reports (including ours) that such reactions could also be performed under metal-free conditions with singlet carbenes (generated from diazo compounds) in presence of blue LED.22,23,24,25,26 As mentioned in the previous section, 3°-amines with diazo compounds in presence of TM catalysts result in C-H activation of the carbon α-to the nitrogen (Scheme 1A). In one of the reports, Davies and co-workers demonstrated Rh-carbenoid mediated C-H activation for the cyclic and acyclic 3°-amines adjacent to the N atom, where, the bulky Rh-complex avoided reacting at the tri-valent N atom. The reaction proceeded through the possible formation of three-membered transition states, and the “build up” of positive charge developed over carbon atom was stabilized by adjacent heteroatom (Scheme 2A). This was further “counterbalanced” by the sterically demanding Rh-complex.13 We were intrigued by these outcomes which prompted us to explore the reactions between cyclic 3°-amines with aryl diazoacetates under blue LED (refer SI for the specifications). Subsequently, NMM was reacted to 4-bromophenyl diazoacetate 1a (at a ratio of 1: 1.5), in water under blue LED at room temperature (Scheme 2). To our pleasant surprise, instead of the expected C-H activation product, N-ylide formation happened at the 3°-amine (Scheme 2A) followed by ester hydrolysis of the carbene to afford the zwitterionic aryl glycine betaine derivative 2a in 78% yield in 8 h. The pure product was isolated by precipitation from the aqueous reaction mixture without any chromatography (Scheme 2A). The betaine structure was confirmed from the SCXRD study of the compound 2a (Scheme 2A). In a bid to investigate the N-ylide formation of the singlet carbenes as against the C-H activation of the metal carbenoids with the 3°-amines, we compared computationally the metal carbenoids and the singlet carbenes (Schemes 2A and 2C) (refer SI). Metal carbenoid and metal-free singlet carbene behave differently as the singlet carbene has an unshared lone pair of electrons located in the sp2 orbital, whereas in metal carbenoid the lone pair is co-ordinated with metal. Additionally, the singlet carbene possesses a non-bonded lone pair of electrons over carbene carbon (Scheme 2C) and during the reaction with 3°-amines the lone pair seek stabilization through available interactions. While studying the orbital delocalization of NMM through DFT calculations (refer supplementary information) using the B3LYP/6-311G(d,p) level of theory, it was found that the HOMO (highest occupied molecular orbital) lobe is primarily delocalized around N-atom (lone pair of N) and the LUMO (lowest unoccupied molecular orbital) is delocalized around the methyl group of NMM (Figure 2C). This could be the perfect pocket for carbene to react, as the lone pair of N- will interact with carbene’s empty p-orbital while the LUMO (lowest unoccupied molecular orbital) around methyl group of NMM would stabilize the lone pair of the carbene (Scheme 2C).
Figure 2.

(A and B) Mass experiment for the detection of intermediates A and B.
(C) Mass data of the deuterated product 2gʼ.
(D-1) The optimized reaction in presence of TEMPO. (D-2) The optimized reaction in D2O.
(E-1) The reaction in dark at r. t. (E-2) The reaction in dark at 50°C.
As a follow-up study when the NEM and NBM were reacted with 1a under the standard condition they failed to generate the desired glycine betaine derivatives as obtained from NMM (Scheme 3A and 3B). It could be that the related initial intermediates B1 and B2 (Scheme 3B) for NEM and NBM were energetically difficult to form. To investigate this, the gas phase geometry optimizations were performed (for the details refer to the supplementary information) using the B3LYP/6-311G(d,p) level of theory suggested for the reliable prediction of the molecular geometries and energetics (Scheme 3C. The Gaussian 09 software was used for all the theoretical calculations.27,28 From the computed reaction mechanism, it was evident that the formation of the 2b was more facile as its intermediate step 1 (Int-1) and the corresponding reaction energy was stable by −30.42 kcal/mol compared to the same for NEM and NBM reaction energies which were −26.93 and −23.60 kcal/mol, respectively. The energy diagram further depicted that the reaction energy of the TS2 for the NMM was lowest (14.24 kcal/mol) upon reacting with the water molecules, which facilitated the hydrogen abstraction from water. Furthermore, the higher TS2 of NEM and NBM, i.e., 15.91 and 17.95 kcal/mol, respectively, could restrict the advancement of the individual reactions to afford the products (Scheme 3C). This stipulated the role of water as a hydrolyzing agent in this transformation. The hydrolysis of the ester moiety (which is most facile for methyl ester) under water could afford the final betaine derivatives. When the reaction was performed at a ratio of 1:1 NMM:1a in ethyl acetate, similar result was observed. However, in the presence of dry solvents the reaction yields were substantially low (33 → 41% in CH2Cl2, CH3CN, and C2H4Cl2) which further emphasized the role of water in the reaction.
The reaction when performed in the absence of blue LED generated no product. The reaction under various light source viz. red, green, and white LED was not as efficient as the blue LED. In general, the reaction is operationally simple to perform, scalable, and works under open air at room temperature. The superior nucleophilicity of the N-methyl moiety could have prompted the formation of the quaternary ammonium ylide which in turn facilitated the hydrolysis of ester to attain stability for the resulting betaine derivatives (Scheme 2). It is noteworthy that the reaction occurred at room temperature and the desired product automatically precipitated from the reaction mixture during the reaction. However, under dark condition (without blue LED), the reaction between NMM and 1a did not yield any product. The starting material remained intact.
Synthesis of compounds
With the optimized procedure in hand, the robustness of the protocol was assessed by reacting numerous aryl diazoacetates 1a–o with NMM, DABCO, and NMP to provide the desired C2 aryl glycine betaines 2a–o, 3a–j, and 4a–j in excellent yield (Scheme 4). Aryl diazoacetates with electron-rich substituents 1c, 1d, 1f, and strong electron-withdrawing fluoro 1e, 1j, trifluoromethyl 1k, other halogen-substituted moieties 1a, 1g, 1i, 1l, and heteroaromatic 3-thienyl moieties 1o reacted fluently with NMM to generate the final compounds 2a–o in 65 → 89% yield (Scheme 4). Next, the bis tertiary cyclic amine, DABCO also worked as an efficient substrate for the formation of C2-aryl glycine betaine 3a–j with aryl diazo esters 1b–e, 1g, 1i–l, and 1n under the standard reaction conditions (Scheme 2). Like NMM, here too the aryl diazoacetates worked efficiently irrespective of the electronic nature of the aromatic ring and generated the desired compounds in 72 → 95% yield (Scheme 4). As observed during the preliminary studies all the NMM-based aryl glycine betaine derivatives 2, got precipitated as solid compounds from the aqueous reaction mixture and were isolated by simple filtration and subsequent washing by ethyl acetate. On the contrary, the DABCO-based derivatives 3 (all of them) formed gel-like semisolid globules that got stuck at the surface of the reaction vessel. Once the starting material got consumed (the reactions were monitored by HPLC) the water was decanted and the gel-like product was washed with ethyl acetate. Finally, the products were dissolved in CD3OD for the NMR analysis. Finally, we explored the utility of our protocol with NMP. Accordingly, the aryl diazo esters 1a–b, 1d–g, 1m, and 1p–r were reacted and the desired compounds 4a–j were obtained in good yield (Scheme 4). The pure solid products precipitated from the aqueous reaction mixture during the reaction and were isolated by filtration followed by extensive washing of the filtrate with ethyl acetate (Scheme 4).
It is noteworthy that the direct isolation of the pure products 2 and 3 (after ethyl acetate washing of the solid precipitate (2) and gel (3)) renders this strategy chromatography free. Hence no chromatographic wastes (in the form of silica/alumina) were generated. Additionally, the process is devoid of the usage of volatile organic compounds (VOC) (ethyl acetate used is an eco-compatible organic solvent). Since the yield of the reaction varies from good to excellent and the stoichiometric ratios of the starting materials, i.e., NMM, DABCO or NMP:aryl diazoester is 1:1, the aqueous wastes contain minimal organic compounds. The mildly intense blue LED of 5W conserves more energy compared to the reactions conducted under thermal energy. Finally, the reactions are completely devoid of the usage of acids, bases, and metal catalysts. Interestingly, the reaction with triethyl amine did not generate any product. The detailed experiments for the synthesis of the compounds 2–4 are described in the experimental file.
It is noteworthy that the reaction works with any kind of diazo esters (ethyl or benzyl derivatives of 1). However, as depicted in Scheme 3B, the N-ethyl or N-benzyl derivatives failed to generate the desired zwitterionic aryl glycine derivatives.
Hence, a water-based, blue LED-induced metal-free synthesis of glycine betaine derivatives was devised where the products were isolated without chromatographic separation. The waste generation is also low.
To demonstrate the scalability of the aqueous reaction, NMM was reacted in a gram scale with 0.5 g of 4-bromophenyl (1a) diazoacetate under the optimized condition to afford the desired final compounds 2a in 0.45 g (yield = 73%) yields which were comparable to the sub-millimolar scale reaction (refer SI).
It is worth mentioning that the diazo compounds 1a–r were perfectly safe and stable to handle. They could be purified over a chromatographic column or could be used directly. It is important that the columns are performed in the dark as prolonged exposure to light promotes nitrogen liberation for the diazo esters.
Comparison of the rate of reaction between electron rich and electron poor aryl diazoacetates
Next, the rate of reaction of electron-rich 1c (para-t-butyl) and electron-poor 1e (para-fluoro) with DABCO was measured to assess the influence of the respective functionalities on the aryl diazoacetates. Accordingly, 1c, 1e, and DABCO were stirred in a reaction vessel under optimized reaction conditions and the reaction mixture was irradiated with blue LED and was monitored by HPLC for 120 min to understand the comparative rate of formation of the desired products 3b and 3c. The reaction rate and kinetics were analyzed from the HPLC data (Figure 1A). From the plot in Figure 1B, it was observed that the rate of formation of 3b was faster than that of 3c indicating that the electron-rich 1c favored the reaction over the electron-poor counterpart. Figure 1C depicted the initial rate of formation of 3b and 3c, where the histogram depicted that the relative rate of formation of 3b was nearly thrice compared to that of 3c.
Figure 1.

Reaction kinetics for the reaction of 1e/c with NMM
(A) HPLC stack chromatogram showing the relative formation of 3b and 3c under different reaction conditions after 90 min.
(B) Percentage degradation of 1e and 1c and formation of 3c and 3b over the time under the blue LED in water.
(C) Bar diagram showing the calculated initial rate of formation of 3b and 3c under the optimized reaction condition.
Control experiments
Next to understand the reaction mechanism, a series of control experiments were performed (Figure 2). At the outset, two different ester-bearing diazo moieties, 1g and 1gʼ (methyl and benzyl derivatives, respectively) were reacted separately with NMM under the optimized reaction conditions. In both cases, the mass analysis of the crude reaction mixture through high-resolution mass spectroscopy (HRMS) indicated the formation of methyl and benzyl ester nitrogen ylides A and B as intermediates when crude reaction mixtures were analyzed during the reaction (Figures 2A and 2B). It is noteworthy that the reaction of NMM with 1g (methyl ester derivative) was faster than 1gʼ (benzyl ester). This stipulated that the nucleophilic attack of the NMM onto the carbene generated from the blue LED irradiation on 1g and 1gʼ could be the first step in the transformation and the steric bulk of the benzyl ester impedes the rate of the reaction with 1gʼ. Next, the reaction was performed under standard reaction conditions in the presence of the radical scavenger TEMPO (2, 2, 6, 6-tetramethyl-1-piperidinyloxy) (Figures 2 and 2D-1). There was no change in the extent formation or in the rate of formation of 2g. This indicated that the reaction does not operate via radical pathway (Figure 2D-1). When the reaction was performed in D2O instead of H2O, we observed formation of 2gʼ through HRMS analysis (Figure 2C and 3D-2). This could be possible due to the deuterium exchange at the carbene carbon from the solvent. Next, the reaction was performed in the dark at room temperature (Figure 2E-1) and heating at 50°C (Figure 2E-2). In both cases, no formation of 2g was observed. This further emphasized the importance of blue LED in the reaction. Finally, the UV-vis measurements revealed that 1g absorbed in the region of blue LED (450–470 nM) as expected. However, no new absorption peak was observed when it was mixed with NMM (Figure S2, refer supplementary information). This observation eliminates the possibility of formation of any adduct with 1g and NMM that could absorb in the blue LED region. This further supported the fact that the reaction proceeds through carbene generation under blue LED from 1g, which forms nitrogen ylide by reacting with NMM.
Mechanistic hypothesis of the reaction
The putative mechanism as depicted in Scheme 5 was devised through the control experiments (Figure 2), initial proof of concept reactions, and the reaction optimization studies discussed earlier. The water played an important role in the transformation, which could be primarily to generate the carboxylate anion for the final compounds and to provide the proton to quench the ylide B (as indicated by the D2O experiment in Figure 2C/D-2). Accordingly, the blue LED induced the formation of the singlet carbene A. Subsequently, the nucleophilic attack of the NMM on A afforded B which could be stabilized via resonance to Bʼ (Scheme 5). B could abstract a proton from the water to provide the quaternary ammonium intermediate C, which is then hydrolyzed by hydroxyl moiety to afford the desired product 2b through D along with the liberation of methanol (Scheme 5).
Scheme 5.

Plausible mechanism for the synthesis of 2–4.
Crystal structures
Single crystals of 2a and 2g the halogen derivatives were grown from water and subjected to the X-ray diffraction experiment for the determination of their crystal structures (Figure 3). Details of the crystal structure determination are given in the supplementary information. Crystallographic data and the refinement parameters are listed in Table S1. Interestingly, the bromo (2a) and chloro (2g) derivatives are isomorphous (similarity index: 0.007).29 The molecules in both the structures pack in a similar fashion (Figure 3). Further investigation on the formation of unusual triangular intermolecular halogen bonds (Br … Br = 3.64 Å and Cl … Cl = 3.57 Å) representing the 3-fold symmetry of the structures is in progress. A study highlighting the importance of halogen bonds in medicinal chemistry has also been undertaken from a structure-property relationship viewpoint.30
Figure 3.

For 2a (left) and 2g (right), ORTEPs are plotted with 50% ellipsoids (top), and the molecular packing diagrams viewing down the c-axis are shown using capped sticks model and with major conformers only (bottom). H-atoms are omitted for clarity. The triangular halogen … halogen contacts are highlighted using dotted green lines.
Antimicrobial screening
Nearly a hundred years ago, it was discovered that quaternary ammonium salts (QACs) possessed antimicrobial properties.31 They are one of the most potent antibiotics and disinfectants used for numerous clinical activities including preoperative antiseptics, disinfectants of noncritical surfaces, etc.32,33,34,35,36,37,38,39,40 The general bactericidal mechanism of action of the QACs involve the electrostatic interactions between the positively charged head group of the QACs along with the negatively charged bacterial cellular membrane that facilitates the side chain of the QAC to permeate into the intramembrane region of the microbe leading to the cellular lysis via leakage of the cytoplasmic material.40 Similar to QACs, glycine betaines have also demonstrated antimicrobial properties as osmoporters.8 Based on these reports we envisaged our library of glycine betaine derivatives could also have bactericidal properties. Hence, a representative member of our library of compounds, 2a, 2c, 2g–k, 2o, and 3a–i were screened against Gram-negative (E. coli MG1655) and Gram-positive pathogens (S. aureus UAMS-1). For the preliminary screening against E. coli MG1655, compounds were used at a concentration of 150 and 300 μg/mL and the bacterial growth was measured by spectrophotometric method. As evident from Table 1, preliminary studies indicated that compounds were not able to inhibit the growth of E. coli even at 300 μg/mL concentration thereby rendering them ineffective against Gram-negative bacteria. Next, the antimicrobial screening of the same compounds against S. aureus UMS-1 at 100 and 200 μg/mL (Table 1) revealed strong growth inhibition among most of the compounds. Hence their inhibitory activity was further assessed in a dose-response manner against S. aureus and our results demonstrated that compounds 2a, 2o, 3d, 3f, and 3g were able to inhibit S. aureus growth below concentration 50 μg/mL. An interesting structure-activity relationship (SAR) was realized from the analysis of the results (Table 1). It was observed that the DABCO-based glycine betaine derivatives 3 were, in general, more potent than the NMM-based compounds 2. Among the DABCO-based molecules 3 the para-substituted analogs were more potent than the meta-substituents as demonstrated by the 4-fold improvement of the IC50 values of the analogs 2h → 2a containing the bromo substituent. Apart from the 3-thienyl and 3-bromo derivatives 2o and 2h, other meta-substituted compounds such as chloro, fluoro, and trifluoromethyl analogs 2i–k were inactive (Table 1). It is noteworthy that the 3-chloro and 3-trifluromethyl DABCO variants 3e and 3f of the glycine betain derivatives were potent (10 and 80 μg/mL, respectively) (Table 1). The highest activity was demonstrated by the 3-anisolyl derivative 3d with an IC50 of 5 μg/mL. In comparison, the 4-anisolyl derivative is completely inactive. It is noteworthy that none of the NMP-based products, i.e., 4 were active against the bacterial strains. Though the 3-fluoro analog 3i was inactive, from this preliminary SAR it became apparent that the meta-substituted DABCO variants 3d and 3e were in general potent antimicrobial agents against Gram-positive S. aureus bacteria (Table 1) and it would be pragmatic to develop these initial hit compounds to the desired lead candidates. To ensure that our compounds were not employing their antimicrobial potency via cytotoxicity, we screened them against baby hamster kidney 21 (BHK-21), a commonly used healthy cell line. For each of the potent compounds, the dose that was required to exhibit cytotoxicity was at least 100-fold higher than their respective IC50 concentrations.
Table 1.
Antimicrobial screening of compound 2 and 3 against E. coli and S. aureus
| Entry | Compound. # | Substituents |
Inhibitory screening |
Dose response screening |
||||
|---|---|---|---|---|---|---|---|---|
| NMM | DABCO | E. coli @ 150 μg/mL | E. coli @ 300 μg/mL | S. aureus @ 100 μg/mL | S. aureus @ 200 μg/mL | S. aureus IC50 (μg/mL) | ||
| 1 | 2a | 4-BrPh | – | – | – | +++ | +++ | 20 |
| 2. | 2c | 4-t-BuPh | – | – | – | +++ | +++ | <200 |
| 3. | 2g | 4-ClPh | – | – | – | +++ | +++ | 80 |
| 4. | 2h | 3-BrPh | – | – | – | +++ | +++ | 90 |
| 5. | 2i | 3-ClPh | – | – | – | +++ | +++ | <200 |
| 6. | 2j | 3-FPh | – | – | – | +++ | +++ | <200 |
| 7. | 2k | 3-CF3Ph | – | – | – | +++ | +++ | <200 |
| 8. | 2o | 3-thienyl | – | – | – | +++ | +++ | 40 |
| 9. | 3a | – | 4-anisolyl | – | – | +++ | +++ | <200 |
| 10. | 3b | – | 4-t-BuPh | – | – | +++ | +++ | <200 |
| 11. | 3c | – | 4-FPh | – | – | +++ | +++ | <200 |
| 12. | 3d | – | 3-anisolyl | – | – | +++ | +++ | 5 |
| 13. | 3e | – | 3-ClPh | – | – | +++ | +++ | 80 |
| 14. | 3f | – | 3-CF3Ph | – | – | +++ | +++ | 10 |
| 15. | 3g | – | 4-ClPh | – | – | +++ | +++ | 20 |
| 16. | 3i | – | 3-FPh | – | – | +++ | +++ | <200 |
| 17 | CPC | – | – | – | – | +++ | +++ | ∼20041 |
--- represents no inhibition effect of compounds on growth as O.D.600nm above 1 was observed.
+++ represents the inhibition effect of compounds on the growth of S. aureus cells.
Mention about the statistical analysis – IC50 for each compound against S. aureus was obtained by calculating the concentrations corresponding to percentage inhibition mean values.
Finally, to understand the effect of our compounds on the cellular morphology of S. aureus, it was treated with compounds 2a, 2g, 2o, 3d, 3f, and 3g and subsequent imaging by phase contrast microscopy revealed morphological aberrations. As evident from Figure 4, S. aureus cells when treated with the aforementioned compounds were enlarged in size indicating defects in cell division when compared to untreated control cells. Moreover, lysed cells were also observed in compound treatment conditions indicating reduced cell viability.
Figure 4.

Morphological aberrations in S. aureus treated with compounds 2g, 2a, 3d, 3f, 3g, and 2o
S. aureus cells at 0.2 O.D.600nm were treated with our compounds at their IC50 concentration and imaged after 8 h via phase contrast microscopy. WT are wildtype untreated cells of S. aureus. Arrowheads represent lysed cells.
To further support the morphological aberration as depicted in Figure 4, next S. aureus cells were treated with compounds 2a, 2g, 2o, 3d, 3f, and 3g and the reduction in bacterial cell viability was estimated further by staining the bacteria with Live/Dead Kit using SYTO 9 (green fluorescent nucleic acid stain) and propidium iodide (red fluorescent nucleic acid stain) (Figure 5). Stain SYTO 9 penetrates the membrane of all dead and alive cells, whereas propidium iodide penetrates only inside the membrane of dead cells, causing a reduction in SYTO 9 fluorescent signal. Therefore, the fluorescence signal intensity of SYTO 9 and propidium iodide could estimate bacterial cell viability. As depicted in Figure 5 upon treatment with compounds 2a, 2g, 2o, 3d, 3f, and 3g the viability of S. aureus UAMS-1 cells was fitness compromised, as the propidium iodide-stained dead cluster of cells were detected (Figure 5).
Figure 5.

S. aureus viability and morphology visualized by confocal microscopy after staining the treated cells with Live/Dead stain
UAMS-1 cells treated with compounds 2g, 2a, 3d, 3f, 3g, and 2o were stained using a 1:1 ratio of SYTO 9:propidium iodide solution. The first panel corresponds to the DIC (differential interference contrast) image. The second panel corresponds to the SYTO 9 stained live cells, the third panel corresponds to the propidium iodide-stained dead cells, and the fourth panel corresponds to the overlay of both green and red fluorescent signals. Scale bar - 1μm.
Compound 2a-induced intracellular lipid droplets in HepG2 cells
In recent years, LDs have evolved as storage organelles, which play crucial roles in various cellular events and energy homeostasis.42,43,44,45,46,47,48 Cells preserve lipids in the form of triacylglycerols (TAGs), sterol, and retinyl esters. These neutral lipids provide energy in cells, membrane synthesis, and molecular signaling at the time of requirement. LDs also play a key role in energy homeostasis, lipid metabolism, and act as fatty acid traffic hubs in cells. Consequently, LDs dysfunction could lead to various diseases, including type 2 diabetes, cardiovascular disease, Alzheimer’s disease, obesity, fatty liver disease, and cancer.49,50,51,52,53 Cruz et al. demonstrated that small molecules could facilitate the accumulation of LDs in cells and exploited the enzymes available in LDs as a potential therapy for cancer.54
To demonstrate the multifaceted applications of our glycine betaine derivatives, compound 2a was used to investigate if it could generate LDs in hepatic cancerous cells (HepG2) which could be a potential therapy for cancer treatment. Accordingly, compound 2a was treated in HepG2 for 37 h along with Nile red, a commercial LD marker (Figure 6). Interestingly, compound 2a treated cells showed a significant number of new LDs from confocal fluorescence image analysis, as shown in Figure 6. In contrast, the control (without treatment of molecule 2a) almost did not reveal new LD formation. This observation was interesting as it provided insightful insights for investigating the mechanism of how compound 2a induces LD formation in HePG2 cell lines (Figure 6). Consequently, it could offer therapeutic approaches against liver diseases, including obesity, fatty liver disease, and cancer. Detailed investigations are presently ongoing in our lab.
Figure 6.

The HepG2 cells were administrated with Nile red for 10 min after being treated without/with compound 2a at 37°C for 12 h
Images were acquired in a confocal microscope in the emission range (Em. 580−620 nm; green: pseudo color image) for the Nile red (200 nM; Ex. 560 nm). The image scale bar is 10 μm. Images were captured using a 100X oil emersion lens. The marked “a,” “b,” and “c” signify the zoomed-in picture of the respective images, respectively.
Conclusion
In summary, we have analyzed computationally the difference between metal carbenoids and electrophilic singlet carbenes and realized their difference in reactivity against 3°-amines. DFT calculations depicted the suitability of NMM as a substrate for such reactions. Based on these studies we developed an optimized protocol for the reactions between cyclic 3°-amines such as NMM, DABCO, and NMP with aryl diazoacetate 1 in water to generate zwitterionic aryl glycine betaine derivatives 2–4 via N-ylides in good to excellent yield. The products 2–4 were isolated either as a solid or gel by precipitation from the aqueous reaction mixture. These aryl betaine derivatives (hitherto unreported) synthesized, purified, and crystallized under water exhibited good antimicrobial inhibition selectively on Gram-positive S. aureus. Cell morphological studies indicated cell enlargement and lysis which indicated reduced cell viability. Compound 2a induced LDs in HePG2 cells. The single crystal structures of compounds 2a and 2g demonstrated interesting Hal … Hal contacts. The exact bactericidal mechanism of these compounds is presently being explored in our lab which will enable us to develop more potent analogs against S. aureus.
Limitations of the study
Unless otherwise specified, there are no limitations in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strain | ||
| Staphylococcus aureus (UAMS-1) | Duke University | N/A |
| Escherichia coli (MG1655) | Laboratory strain collection | N/A |
| Chemicals | ||
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D8418 |
| TSB - Tryptone Soya Broth (Soyabean Casein Digest Medium) | HIMEDIA | LQ009A/M011 |
| Agarose special, low EEO | HIMEDIA | MB002, CAS No – 9012-36-6 |
| Phosphate Buffered Saline (PBS), pH 7.4 | HIMEDIA | TS1101-10X |
| Nile Red | Thermofisher | Cat#N1142 |
| Dulbecco’s modified Eagle’s medium (DMEM) | HIMEDIA | SKU#AL066A |
| Fetal bovine serum (FBS) | HIMEDIA | SKU#RM10432 |
| Streptomycin antibiotics | HIMEDIA | SKU#A018 |
| Critical commercial assays | ||
| LIVE/DEAD™ BacLight™ Bacterial Viability Kit | Invitrogen | L13152 |
| 96-well sterile tissue culture multi-well plates | HIMEDIA | TPP26/TPP96 |
| Experimental model: cell line | ||
| HepG2 | ATCC | N/A |
| Software and algorithms | ||
| Phase Contrast Microscope | Nikon | Eclipse Ts2R |
| Confocal laser scanning Microscope | Nikon | A1R MP + Ti-E |
| ImageJ/Fiji – image processing | ImageJ | https://imagej.net/software/fiji/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Subhabrata Sen at subhabrata.sen@snu.edu.in.
Materials availability
All reagents were purchased from commercial sources and used without purification unless otherwise mentioned. Compounds 1a-1r were synthesized according to the reported procedure.25 All blue light batch reactions were carried out under air as specified in Photochemical Reactor Aldrich Micro Photochemical Reactor, blue LED lights (ALDKIT001-1EA). LED light is an IP68 double density 12 V DC waterproof blue light with a spectral range of 435–445 nm with a wall plug power supply of 500 mA with 5–6 W. The irradiation vessel material is borosilicate glass. The distance of the irradiation vessel from the light source is 2 cm. All 1H and 13C NMR spectra were recorded taking tetramethylsilane as an internal standard at ambient temperature unless otherwise indicated with Bruker 400 MHz instruments at 400 MHz for1 H and 100 MHz for 13C NMR spectroscopy. Splitting patterns are designated as singlet (s), doublet (d), triplet (t), doublet of doublets (dd), and triplet of doublets (td). Splitting patterns that could not be interpreted or easily visualized are designated as multiplet (m). MS analyses were carried out using an Agilent 6540 accurate-mass Q-TOF LC/MS (Agilent Technologies, U.S.A.). Mass spectra were acquired in the positive ion mode by scanning from 100 to 1500 in the mass-to-charge ratio (m/z). The mobile phase composition used for UHPLC-QTOF MS comprises H2O (A) and ACN (B), with optimized linear gradient elution. The injection volume was 5 μL. The flow rate was set at 0.3 mL/min. Accurate mass analysis calibration was carried out by ESI-low concentration tuning mix solution provided by Agilent Technologies, U.S.A. The accuracy error threshold was set at 5 ppm. HPLC experiments were carried out on a Waters Alliance System (Milford, MA) consisting of an e2695 separation module and a 2998 photodiode-array detector. The HPLC system was controlled with EMPOWER software (Waters Corporation, Milford, MA). The UV experiments were carried out on SHIMADZU UV-2600i UV-VIS Spectrophotometer.
Method details
Synthesis of aryl diazo esters (1a–r)
All aryl diazo acetates were prepared by the reported procedure.25 Aryl acetates (5 mmol) were dissolved in acetonitrile (10 mL) in a clean oven dried round bottom flask, and DBU (1,8-diazabicyclo[5.4.0] undec-7-ene) (0.9 mL, 1.2 equivalents, 6 mmol) was added to it and stirred for 10 min. Then p-ABSA (4-acetamidobenzenesulfonyl azide) (1.44 g, 1.2 equivalents, 6 mmol) was added and stirred for 4 h in the dark at r.t.; after completion acetonitrile was removed under vacuum, diluted with ethyl acetate (25 mL), and washed with water and the organic layer was dried with brine and sodium sulfate, purified with flash column chromatography on silica gel (100–200 mesh size) with 5% ethyl acetate in hexane to yield ∼98%.
Synthetic procedure for compounds 2 - 4
Aryl diazoacetates (1 eq.) and N-methyl morpholine or 1,4-diazabicyclo[2.2.2]octane (1 eq.) were dissolved in water (5 mL) in a clean oven-dried 15 mL reaction vial and irradiated with blue LED for 10 h. The desired compounds are precipitated from the reaction mixture which were filtered, washed with ethyl acetate and dried to obtain the pure products.
Biological assays
MIC determination assay
To measure antibacterial activity of various compounds Minimum inhibitory concentration (MIC) assay was performed. E. coli strains were cultured in Luria Bertani (LB) broth whereas S. Aureus strains were cultured in Tryptic Soy broth (TSB) media and were diluted in 1:100 dilution ratio. Overnight cultures of both E. Coli and S. Aureus strains were diluted in 1:100 ratio and seeded into 24 well plates to which various concentration of compounds (dissolved in 50% DMSO) at 5, 10, 20, 40, 80 and 100 μg/mL were added. Cultures with DMSO were used as control. Plates were incubated at 37°C for 14–16 h. After incubation, each well was diluted in 1:1 ratio with TSB media, and readings were taken at OD600nm in TECAN Spark plate reader. The assay was conducted in biological triplicates. The inhibition percentage was calculated using the following formula,
IC50 values were then estimated from the obtained results.
Microscopy analysis of associated morphological aberrations
For Phase Contrast Imaging, overnight culture of S. Aureus was diluted in 1:100 ratio. At mid-exponential phase, the cells were treated with compounds 2g, 2a, 3d, 3f, 3g and 2o at their respective IC50 values. After 8 h of treatment, cells were immobilized on 1% PBS agarose-pad and imaged using Phase contrast microscope on a Nikon Eclipse Ts2R microscope attached with a Nikon DS-Fi3 camera along with Nikon Plan Fluor 100X oil Ph3 objective. Untreated cells were used as control. Adobe Photoshop CS6 was used for image processing.
Cell culture and confocal imaging
Human liver cancer cell lines (HepG2 cells) were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum and 100 U penicillin/0.1 mg/mL streptomycin antibiotics under an atmosphere of 5% CO2 at 37°C. Next, these cells were used for cellular experimental studies. Cell imaging was carried out by using the confocal microscope.
Cellular experimental procedure
At first, a 10 mM stock solution of compound 2a in DMSO was prepared. The cells were administrated with Nile red for 10 min after being treated without/with compound 2a at 37°C for 12 h. In control experiment (without treatment of compound 2a), 0.5% of DMSO was added, and the images were acquired in the emission range (Em. 580−620 nm; green: pseudo color image) for the Nile red (200 nM; Ex. 560 nm). Subsequently, in another experiment, 50 μM of compound 2a was treated, and images were acquired in the emission range (Em. 580−620 nm; green: pseudo color image) for the Nile red (200 nM; Ex. 560 nm). The image scale bar is 10 μm. Images were captured using a 100X oil emersion lens.
Live/Dead staining
Overnight cultures of S. aureus UMS-1 were diluted at a 1:100 ratio and at 0.1 O. D600nm and treated with compounds 2g, 2a, 3d, 3f, 3g, and 2o. After 5 h of treatment, S. aureus suspensions were adjusted to 0.3 O. D600nm and washed twice with 1X PBS. Cells were pre-stained with the Soty 9/Propidium iodide from Live/Dead BacLight Bacterial Viability Kit (BacLight; Molecular Probes, Carlsbad, CA, USA) in a ratio of 1:1. The mixture was incubated in the dark for 25 min at RT. Cells were washed again with 1X PBS to remove excess stain. Cells treated with 50% DMSO were used as the positive control. About 5μL of stained culture was immobilized on a PBS agarose pad, and confocal images were captured using laser scanning Nikon A1R MP + Ti-E confocal microscope system provided with solid-state lasers with excitation/emission maxima 480/500 for SYTO 9 and 490/635 for propidium iodide.
Characterization data

2-(4-bromophenyl)-2-(4-methylmorpholino-4-ium)acetate, (2a)
By following above mentioned method, 2a was synthesized by using 1a (99.4 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 95.2 mg, 78% yield as white solid, melting point 168-170°C; 1H NMR (400 MHz, CD3OD) δH 7.68 (d, J = 8 Hz, 2H), 7.60 (d, J = 8 Hz, 2H), 4.98 (s, 1H), 4.13–4.05 (m, 1H), 4.05–3.93 (m, 4H), 3.92–3.85 (m, 1H), 3.78–3.62 (m, 2H), 3.39 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.4, 135.4, 133.3, 129.6, 126.1, 81.7, 61.7, 61.4, 60.0, 59.5, 42.4 ppm. HRMS (ESI-TOF) m/z:[M + H]+ calcd for C13H17BrNO3 314.0386, found 314.0380.
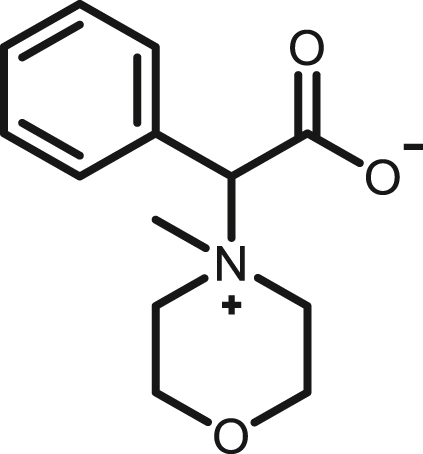
2-(4-Methylmorpholino-4-ium)-2-phenylacetate, (2b)
By following above mentioned method,2b was synthesized by using 1b (68.7 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 74.2 mg, 80% yield as white viscous oil; 1H NMR (400 MHz,CD3OD) δH 7.70–7.68 (m, 2H), 7.54–7.50 (m, 3H), 4.99 (s, 1H), 4.11–4.04 (m, 1H), 4.00–3.88 (m, 4H), 3.75–3.64 (m, 3H), 3.40 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.8, 133.6, 131.7, 130.3, 130.0, 82.6, 67.4, 61.7, 61.5, 59.9, 59.5, 56.2, 42.6 ppm. HRMS (ESI-TOF) m/z:[M + H]+ calcd for C13H18NO3 236.1281, found 236.1292.

7-(4-(tert-butyl)phenyl)-4-methyl-1,4-oxazepane-7-carboxylic acid, (2c)
By following above mentioned method, 2c was synthesized by using 1c (90 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 85 mg, 75% yield as white solid, melting point 205-207°C; 1H NMR (400 MHz, CD3OD) δH 7.61 (d, J = 8 Hz, 2H), 7.55 (d, J = 8 Hz, 2H), 4.95 (s, 1H), 4.10–4.06 (m, 1H), 4.04–3.92 (m, 4H), 3.89–3.86 (m, 1H), 3.74–3.62 (m, 2H), 3.40 (s, 3H), 1.34 (s, 9H) ppm 13C NMR (100 MHz, CD3OD) δC 170.0, 155.2, 133.4, 127.3, 127.0, 82.4, 61.8, 61.5, 59.8, 59.4, 35.7, 31.5 ppm. HRMS (ESI-TOF) m/z:[M + H]+ calcd for C17H26NO3 292.1907, found 292.1917.
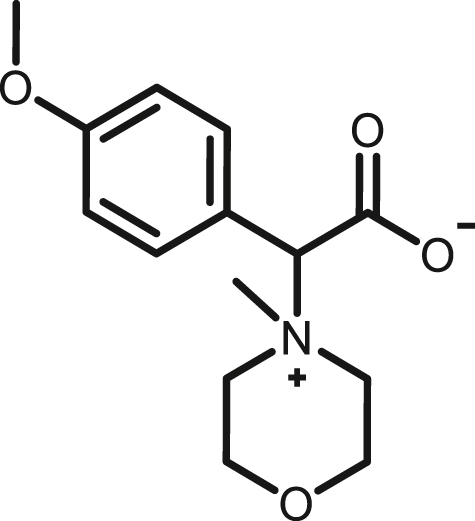
2-(4-methoxyphenyl)-2-(4-methylmorpholino-4-ium)acetate, (2d)
By following above mentioned method,2d was synthesized by using 1d (80.41 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 87.9mg, 85% yield as white solid, melting point 174-176°C; 1H NMR (400 MHz, CD3OD) δH 7.60 (d, J = 8 Hz, 2H), 7.03 (d, J = 8Hz, 2H), 4.93 (s, 1H), 4.10–4.03 (m, 1H), 3.97–3.86 (m, 5H), 3.84 (s, 3H), 3.69–3.58 (m, 2H), 3.36 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 170.1, 162.9, 135.0, 121.8, 115.3, 82.5, 61.8, 61.5, 59.6, 59.2, 55.9, 42.4 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20NO4 266.1387,found 266.1395.
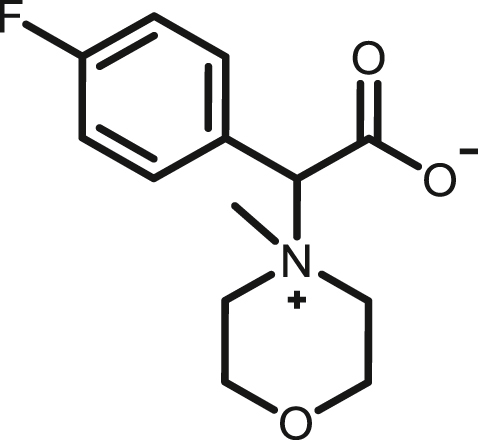
2-(4-fluorophenyl)-2-(4-methylmorpholino-4-ium)acetate, (2e)
By following above mentioned method,2e was synthesized by using 1e (75.7 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 70.1mg, 71% yield as white solid, melting point 184-186°C; 1H NMR (400 MHz, CD3OD) δH 7.73 (dd, J = 8,4 Hz, 2H), 7.25 (t, J = 10 Hz, 2H),5.00 (s,1H), 4.11–4.04 (m, 1H), 4.01–3.96 (m, 3H), 3.94–3.87 (m, 1H), 3.74–3.63 (m, 3H), 3.39 (s, 3H) ppm.13C NMR (100 MHz, CD3OD) δC 169.6, 165.4 (d, J = 249 Hz), 135.9(d, J = 9Hz), 126.5 (d, J = 3Hz), 116.9(d, J = 22Hz), 81.8, 67.4, 61.6 (d, J = 30Hz), 59.6 (d, J = 45Hz), 56.2, 42.4 ppm 19F NMR (376 MHz, CD3OD_SPE) δ −111.9 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17FNO3 254.1187,found 254.1179.

4-Methyl-7-(p-tolyl)-1,4-oxazepane-7-carboxylic acid, (2f)
By following above mentioned method,2f was synthesized by using 1f (74.17 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 79.72 mg, 82% yield as white solid, melting point 188-190°C; 1H NMR (400 MHz, CD3OD) δH 7.56 (d, J = 8 Hz, 2H), 7.32 (d, J = 8 Hz, 2H), 4.94 (s, 1H), 4.11–4.04 (m, 1H), 4.03–3.85 (m, 5H), 3.72–3.60 (m, 2H), 3.38 (s, 3H), 2.39 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 170.0, 142.2, 133.5, 130.6, 127.2, 82.5, 61.8, 61.5, 42.5, 21.2 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20NO3 250.1438, found 250.1436.

2-(4-chlorophenyl)-2-(4-methylmorpholino-4-ium)acetate, (2g)
By following above mentioned method,2g was synthesized by using 1g (82.1 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 93.6 mg, 89% yield as white solid, melting point 181-183°C; 1H NMR (400 MHz,CD3OD) δH 7.67 (d, J = 8Hz, 2H), 7.52 (d, J = 8 Hz, 2H), 4.99 (s, 1H), 4.11–4.07 (m, 1H), 4.01–3.87 (m, 4H), 3.75–3.63 (m, 3H), 3.39 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.4, 137.9, 135.1, 130.2, 129.1, 81.7,67.4, 61.7, 61.4, 60.0, 59.5,56.2, 42.5 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17ClNO3 270.0891, found 270.0881.

2-(3-bromophenyl)-2-(4-methylmorpholino-4-ium)acetate, (2h)
By following above mentioned method,2h was synthesized by using 1h (99.4 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 94.3mg, 77% yield as white solid, melting point 163-165°C; 1H NMR (400 MHz, CD3OD) δH 7.89 (t, J = 4 Hz, 1H), 7.72–7.66 (m, 2H), 7.44 (t, J = 8 Hz, 1H), 4.99 (s, 1H), 4.11–4.07 (m, 1H), 4.06–3.92 (m, 4H), 3.91–3.83 (m, 1H), 3.79–3.65 (m, 2H), 3.41 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.2, 136.3, 134.8, 132.8, 132.4, 131.8, 123.8, 81.8, 61.7, 61.4, 60.1, 59.6, 42.3 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17BrNO3 314.0386, found 314.0399.

2-(3-chlorophenyl)-2-(4-methylmorpholino-4-ium)acetate, (2i)
By following above mentioned method,2i was synthesized by using 1i (82.1mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 82.6mg, 84% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.74 (t, J = 4Hz, 1H), 7.64–7.61 (m, 1H), 7.58–7.55 (m, 1H), 7.50 (t, J = 8 Hz, 1H), 4.99 (s, 1H), 4.12–4.05 (m, 1H), 4.02–3.93 (m, 4H), 3.91–3.87 (m, 1H), 3.78–3.66 (m, 2H), 3.42 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.2, 135.9, 133.4, 132.6, 132.0, 131.8, 131.6, 81.8, 61.7, 61.4, 60.1, 59.6, 42.4 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17ClNO3 [M + H]+ 270.0891, found 270.0903.
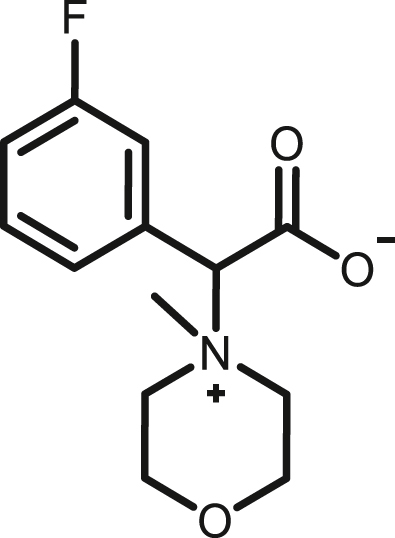
2-(3-fluorophenyl)-2-(4-methylmorpholino-4-ium)acetate,(2j)
By following above mentioned method,2j was synthesized by using 1j (75.7 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 68.1 mg, 69% yield as white solid, melting point 182-184°C; 1H NMR (400 MHz, CD3OD) δH 7.56–7.47 (m, 3H), 7.32–7.27 (m, 1H), 5.01 (s, 1H), 4.12–4.05 (m, 1H), 4.04–3.94 (m, 4H), 3.92–3.88 (m, 1H), 3.78–3.66 (m, 2H), 3.42 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.3,164.0 (d, J = 245 Hz), 132.8(d, J = 7 Hz), 131.9(d, J = 9 Hz), 129.7(d, J = 3 Hz), 120.3 (d, J = 23 Hz), 118.6 (d, J = 21 Hz), 81.8, 61.7, 61.4, 60.1, 59.6, 42.5 ppm 19F NMR (376 MHz, CD3OD_SPE) δ −113.41 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17FNO3 254.1187, found 254.1186.

4-Methyl-7-(3-(trifluoromethyl)phenyl)-1,4-oxazepane-7-carboxylic acid,(2k)
By following above mentioned method,2k was synthesized by using 1k (95.2 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 76.8 mg, 65% yield as white solid, melting point 166-168°C; 1H NMR (500 MHz, CD3OD) δH 8.03 (s, 1H), 7.96 (d, J = 5 Hz, 1H), 7.86 (d, J = 8 Hz, 1H), 7.73 (t, J = 8 Hz, 1H), 5.10 (s, 1H), 4.12–4.06 (m, 1H), 4.02–3.94 (m, 4H), 3.93–3.90 (m, 1H), 3.85–3.78 (m, 1H), 3.75–3.70 (m, 1H), 3.43 (s, 3H) ppm 13C NMR (100 MHz, CD3OD_SPE) δC 169.8, 137.9, 132.5, 132.0, 131.7, 129.1, 82.4, 62.4, 62.0, 60.8, 60.3, 56.7, 53.8, 42.9 ppm 19F NMR (376 MHz, CD3OD_SPE) δ −64.22 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H17F3NO3 304.1155, found 304.1168.

2-(2-bromophenyl)-2-(4-methylmorpholino-4-ium)acetate (2L))
By following above mentioned method,2L was synthesized by using 1L (99.4 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 95.5 mg, 78% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 8.04–8.02 (m, 1H), 7.98–7.95 (m, 1H), 7.87–7.85 (m, 1H), 7.73 (t, J = 8 Hz, 1H), 5.11 (s, 1H), 4.14–4.07 (m, 1H), 4.05–3.95 (m, 4H), 3.93–3.90 (m, 1H), 3.82–3.70 (m, 2H), 3.44 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.1, 137.3, 131.9, 131.1, 128.5, 81.8, 61.7, 61.4, 60.2, 59.6, 42.3 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H17BrNO3 314.0386, found 314.0401.

2-(2-methoxyphenyl)-2-(4-methylmorpholino-4-ium)acetate (2m)
By following above mentioned method,2m was synthesized by using 1m (80.41 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 89.9 mg, 87% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.37 (t, J = 8 Hz, 1H), 7.21–7.19 (m, 2H), 7.07–7.0 (m, 1H), 4.92 (s, 1H), 4.06–4.00 (m, 1H), 3.95–3.92 (m, 4H), 3.87–3.82 (m, 1H), 3.80 (s, 3H), 3.70–3.61 (m, 2H), 3.37 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.8, 161.3, 131.5, 131.1, 125.7, 119.2, 117.1, 82.6, 66.0, 62.4, 62.4, 62.3, 61.7, 61.5, 60.0, 59.5, 55.9, 42.6 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20NO4 266.1387,found 266.1394.

2-(3-methoxyphenyl)-2-(4-methylmorpholino-4-ium)acetate, (2n)
By following above mentioned method,2n was synthesized by using 1n (80.41 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 82.7 mg, 80% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.41 (t, J = 8 Hz, 1H), 7.25–7.23 (m, 2H), 7.11–7.08 (m, 1H), 4.96 (s, 1H) 4.12–4.02 (m, 1H), 3.98–3.95 (m, 4H), 3.91–3.87 (m, 1H), 3.83 (s, 3H), 3.74–3.65 (m, 2H), 3.41 (s, 3H) ppm 13C NMR (100 MHz, CD3OD) δC 169.8, 161.3, 131.5, 131.0, 125.7, 119.2, 117.1, 82.6, 61.7, 61.5, 61.5, 60.0, 59.5, 55.9, 42.6 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20NO4 266.1387,found 266.1392.

2-(4-Methylmorpholino-4-ium)-2-(thiophen-3-yl)acetate, (2o)
By following above mentioned method,2o was synthesized by using 1o (79.05 mg, 0.39 mmol) and NMM (39.45 mg, 0.39 mmol) in 62.1 mg, 66% yield as yellowish viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.85 (d, J = 4.0, 1H), 7.59–7.57 (m, 1H), 7.37–7.35 (m, 1H), 5.06 (s, 1H), 4.10–4.03 (m, 1H), 4.02–3.95 (m, 3H), 3.92–3.86 (m, 1H), 3.76–3.56 (m, 3H), 3.39 (s, 3H) ppm.13C NMR (100 MHz, CD3OD) δC 169.7, 132.4, 130.8, 130.6, 129.6,128.0, 77.6, 67.2,64.4,61.8, 61.5, 59.8, 59.3, 56.1,42.9 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C11H16NO3S 242.0845,found 242.0842.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(4-methoxyphenyl)acetate, (3a)
By following above mentioned method,3a was synthesized by using 1d (80.41 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 102.38 mg, 95% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.55 (d, J = 8 Hz, 2H), 7.03 (d, J = 8 Hz, 2H), 4.70 (s, 1H), 3.88–3.78 (m, 6H), 3.55–3.47 (m, 3H), 3.14 (t, J = 8 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 170.4, 162.7, 134.8, 122.0, 115.4, 80.9, 55.9, 51.5,47.1, 46.2 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H21N2 O3 277.1547,found 277.1546.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(4-(tert-butyl)phenyl)acetate, (3b)
By following above mentioned method 3b was synthesized by using 1c (80.41 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 102.38 mg, 95% yield as white viscous oil; 1H NMR (400 MHz,CD3OD) δH 7.57–7.52 (m, 4H), 4.72 (s, 1H), 3.87–3.80 (m, 3H), 3.58–3.51 (m, 3H), 3.15 (t, J = 8 Hz, 6H), 1.34 (s, 9H) ppm 13C NMR (100 MHz, CD3OD) δC 170.3, 155.0, 133.3, 127.5, 127.1, 80.9, 51.7, 46.2, 35.6, 31.5 ppm. HRMS (ESI-TOF) m/z: [M + H]+calcd for C18H27N2 O2 303.2067, found 303.2080.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(4-fluorophenyl)acetate (3c)
By following above mentioned method,3c was synthesized by using 1e (75.7 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 81.43 mg, 79% yield as yellowish viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.74–7.71 (m, 2H), 7.24 (t, J = 8 Hz, 2H), 4.79 (s, 1H), 3.86–3.79 (m, 3H), 3.58–3.50 (m, 3H), 3.15 (t, J = 8 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.9, 165.3 (d, J = 248 Hz),135.7 (d, J = 8 Hz), 126.7(d, J = 3 Hz), 117.0(d, J = 22 Hz), 80.2, 51.7, 46.1 ppm 19F NMR (376 MHz, CD3OD) δ −112.24 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18FN2 O2 265.1347, found 265.1348.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(3-methoxyphenyl)acetate (3d)
By following above mentioned method,3d was synthesized by using 1n (80.41 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 95.9 mg 89% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.40 (t, J = 8 Hz, 1H), 7.20–7.17 (m, 2H), 7.09–7.06 (m, 1H), 4.72 (s, 1H), 3.88–3.78 (m, 6H), 3.60–3.52 (m, 3H), 3.15 (t, J = 8 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 170.1, 161.4, 131.7, 131.1, 125.6, 118.9, 117.0, 81.1, 55.9, 51.9, 46.2 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H21N2 O3 277.1547,found 277.1546.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(3-chlorophenyl)acetate (3e)
By following above mentioned method,3e was synthesized by using 1e (82.1 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 94.1 mg, 86% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.70 (t, J = 4 Hz, 1H), 7.58–7.47 (m, 3H), 4.77 (s, 1H), 3.87–3.79 (m, 3H), 3.60–3.52 (m, 3H), 3.16 (t, J = 8 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.5, 135.9, 133.2, 132.8, 131.9, 131.6, 131.6, 80.2, 55.2, 55.2, 55.2, 51.9,46.1, 46.1 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18ClN2 O2 281.1051, found 281.1052.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(3-(trifluoromethyl)phenyl)acetate (3f)
By following above mentioned method 3f was synthesized by using 1k (95.2 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 91.93 mg, 75% yield as yellowish viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.98 (s, 1H), 7.91 (d, J = 4 Hz, 1H), 7.85 (d, J = 8 Hz, 1H), 7.72 (t, J = 8 Hz, 1H), 4.88 (s, 1H), 3.88–3.81 (m, 3H), 3.62–3.54 (m, 3H), 3.17 (t, J = 8 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.4, 137.2, 132.9, 132.5, 132.2, 132.0, 131.1, 130.1, 130.1, 128.3, 128.3, 126.5, 123.8, 80.1, 51.9, 46.2 ppm 19F NMR (376 MHz, CD3OD) δ −64.17 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H18F3N2 O2 315.1315, found 315.1316.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(4-chlorophenyl)acetate,(3g)
By following above mentioned method,3g was synthesized by using 1g (82.1 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 88.6 mg, 81% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.63 (d, J = 8 Hz, 2H), 7.51 (d, J = 8 Hz, 2H), 4.80 (s, 1H), 3.85–3.75 (m, 3H), 3.59–3.51 (m, 3H), 3.15 (t, J = 8Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.7, 137.6, 135.0, 130.2, 129.4, 80.1, 51.7, 46.1 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18ClN2 O2 281.1051, found 281.1048.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-phenylacetate,(3h)
By following above mentioned method,3h was synthesized by using 1b (68.7 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 77.8mg, 81% yield as white viscous oil;1H NMR (400 MHz, CD3OD) δH 7.65–7.63 (m, 2H), 7.53–7.50 (m, 3H), 4.79 (s, 1H), 3.87–3.80 (m, 3H), 3.60–3.52 (m, 3H), 3.18–3.10 (m, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 170.1, 133.5, 131.5, 130.6, 130.1, 81.1, 51.7, 46.1 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd. for C14H19N2 O2 247.1441, found 247.1451.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(3-fluorophenyl)acetate (3i)
By following above mentioned method,3i was synthesized by using 1j (75.7mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 74.21mg, 72% yield as yellowish viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.57–7.50 (m, 1H), 7.47–7.42 (m, 2H), 7.31–7.24 (m, 1H), 4.80 (s, 1H), 3.88–3.80 (m, 3H), 3.60–3.53 (m, 3H), 3.16 (t, J = 7.3 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.6, 164.0 (d, J = 245 Hz) 133.0 (d, J = 7 Hz), 132.0(d, J = 9 Hz), 129.6(d, J = 3 Hz), 120.1 (d, J = 22 Hz), 118.4 (d, J = 21 Hz), 80.2, 55.2, 55.2, 55.2, 52.0, 51.9, 46.2, 46.1 ppm 19F NMR (376 MHz, CD3OD) δ −113.36 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18FN2 O2 265.1347, found 265.1347.

2-(1,4-diazabicyclo[2.2.2]octan-1-ium-1-yl)-2-(2-bromophenyl)acetate,(3j)
By following above mentioned method,3j was synthesized by using 1L (99.4 mg, 0.39 mmol) and DABCO (43.74 mg, 0.39 mmol) in 111.6 mg, 88% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.88 (d, J = 8Hz,1H), 7.81 (d, J = 8.0Hz, 1H), 7.50 (t, J = 8, 8 Hz, 1H), 7.41 (t, J = 12, 4 Hz, 1H), 5.31 (s, 1H), 3.92–3.85 (m, 3H), 3.75–3.67 (m, 3H), 3.17 (t, J = 7.7 Hz, 6H) ppm 13C NMR (100 MHz, CD3OD) δC 169.2, 135.3, 134.0, 133.2, 130.8, 129.6, 129.3, 78.6, 55.2,52.0,46.1, 46.0 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H18BrN2 O2 325.0546, found 325.0558.

2-(1-Methylpiperidin-1-ium-1-yl)-2-phenylacetate, (4a)
By following above mentioned method,4a was synthesized by using 1b (99.4 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 72.7 mg, 80% yield as white solid, melting point 180-182°C; 1H NMR (400 MHz, CD3OD) δH 7.70–7.68 (m, 2H), 7.54–7.46 (m, 3H), 4.94 (s, 1H), 3.89–3.83 (m, 1H), 3.66–3.61 (m, 1H), 3.42–3.34 (m, 2H), 3.25 (s, 3H), 2.00–1.93 (m, 4H), 1.81–1.74 (m, 1H), 1.63–1.58 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 170.3, 133.5, 131.5, 130.8, 129.9, 80.5, 61.0, 60.8, 43.9, 22.3, 21.2, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H20N O2 234.1489, found 234.1488.

2-(4-methoxyphenyl)-2-(1-methylpiperidin-1-ium-1-yl) acetate, (4b)
By following above mentioned method,4b was synthesized by using 1d (80.41 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 86.26 mg, 84% yield as white solid, melting point 175-177°C; 1H NMR (400 MHz, CD3OD) δH 7.60 (d, J = 8 Hz, 2H), 7.02 (d, J = 8 Hz, 2H), 4.87 (s, 1H), 3.83 (s, 3H), 3.81–3.77 (m, 1H), 3.61–3.55 (m, 1H), 3.38–3.32 (m, 2H), 3.21 (s, 3H), 1.99–1.87 (m, 4H), 1.79–1.74 (m, 1H), 1.65–1.55 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 170.6, 162.8, 134.8, 122.2, 115.2, 80.2, 60.7, 60.6, 55.9, 43.7, 22.3, 21.2, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H22N O3 264.1594, found 264.1596.

2-(1-Methylpiperidin-1-ium-1-yl)-2-(p-tolyl)acetate, (4c)
By following above mentioned method,4c was synthesized by using 1f (74.17 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 80.06 mg, 83% yield as white solid, melting point 174-176°C; 1H NMR (400 MHz, CD3OD) δH 7.56 (d, J = 8 Hz, 2H), 7.30 (d, J = 8 Hz, 2H), 4.89 (s, 1H), 3.87–3.80 (m, 1H), 3.63–3.57 (m, 1H), 3.40–3.32 (m, 2H), 3.23 (s, 3H), 2.38 (s, 3H), 2.02–1.83 (m, 4H), 1.79–1.73 (m, 1H), 1.65–1.56 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 170.5, 142.0, 133.4, 130.5, 127.7, 80.3, 60.8, 60.7, 43.9, 22.3, 21.2, 21.2, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H22N O2 248.1645, found 248.1648.

2-(4-iodophenyl)-2-(1-methylpiperidin-1-ium-1-yl)acetate,(4d)
By following above mentioned method,4d was synthesized by using 1p (117.80 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 119 mg, 85% yield as white solid, melting point 194-196°C; 1H NMR (400 MHz, CDCl3) δH 7.66 (d, J = 8 Hz, 2H), 7.45 (d, J = 8.0 Hz, 2H), 5.51 (s, 1H), 3.91–3.84 (m, 1H), 3.71–3.64 (m, 1H), 3.47–3.44 (m, 2H), 3.22 (s, 3H), 1.84–1.71 (m, 5H), 1.64–1.46 (m, 1H). 13C NMR (100 MHz, CDCl3) δC 167.4, 137.9, 134.6, 130.0, 96.8, 80.1, 58.9, 58.6, 42.1, 21.3, 20.4, 20.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H19INO2 360.0455, found 360.0445.

2-(4-bromophenyl)-2-(1-methylpiperidin-1-ium-1-yl) acetate,(4e)
By following above mentioned method, 4e was synthesized by using 1a (99.4 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 96.1 mg, 79% yield as white solid, melting point 192-194°C; 1H NMR (400 MHz, CD3OD) δH δ 7.66 (d, J = 8 Hz, 2H), 7.60 (d, J = 8 Hz, 2H), 4.92 (s, 1H), 3.86–3.79 (m, 1H), 3.66–3.62 (m, 1H), 3.43–3.33 (m, 2H), 3.24 (s, 3H), 2.01–1.90 (m, 4H), 1.81–1.73 (m, 1H), 1.65–1.55 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 169.8, 135.2, 133.2, 130.1, 126.0, 79.8, 61.0, 60.9, 43.7, 22.3, 21.2, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H19BrN O2 312.0594, found 312.0589.

2-(4-chlorophenyl)-2-(1-methylpiperidin-1-ium-1-yl)acetate, (4f)
By following above mentioned method,4f was synthesized by using 1g (82.1 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 91.8 mg, 88% yield as white solid, melting point 189-191°C; 1H NMR (400 MHz, CD3OD) δH 7.68 (d, J = 8 Hz, 2H), 7.50 (d, J = 8 Hz, 2H), 4.92 (s, 1H), 3.87–3.81 (m, 1H), 3.66–3.60 (m, 1H), 3.43–3.34 (m, 2H), 3.25 (s, 3H), 1.98–1.90 (m, 4H), 1.80–1.75 (m, 1H), 1.63–1.59 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 169.9, 137.8, 135.0, 130.1, 129.6, 79.7, 61.1, 60.9, 43.8, 22.3, 21.2, 20.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H19ClNO2 268.1099, found 268.1096.

2-(4-fluorophenyl)-2-(1-methylpiperidin-1-ium-1-yl)acetate,(4g)
By following above mentioned method,4g was synthesized by using 1e (75.7 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 73.50 mg, 75% yield as white solid, melting point 186-188°C; 1H NMR (400 MHz, CD3OD) δH 7.73 (dd, J = 10, 6 Hz, 2H), 7.23 (t, J = 8 Hz, 2H), 4.94 (s, 1H), 3.86–3.80 (m, 1H), 3.65–3.60 (m, 1H), 3.42–3.34 (m, 2H), 3.24 (s, 3H), 2.04–1.91 (m, 4H), 1.80–1.73 (m, 1H), 1.64–1.57 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 170.16, 166.5, 164.1, 135.7(d, J = 9 Hz), 126.9 (d, J = 4 Hz), 116.8(d, J = 22 Hz), 79.7, 60.9, 60.7, 43.7, 22.3, 21.2, 20.9. 19F NMR (376 MHz, CD3OD) δ −112.22 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C14H19FN O2 252.1394, found 252.1394.

2-(1-Methylpiperidin-1-ium-1-yl)-2-(o-tolyl)acetate, (4h)
By following above mentioned method,4h was synthesized by using 1q (74.17 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 82.95 mg, 86% yield as white viscous oil; 1H NMR (400 MHz, CD3OD) δH 7.81 (d, J = 8 Hz, 1H), 7.41–7.36 (m, 2H), 7.31–7.27 (m, 1H), 5.10 (s, 1H), 3.80–3.69 (m, 2H), 3.54–3.46 (m, 2H), 3.28 (s, 3H), 2.56 (s, 3H), 2.04–1.79 (m, 5H), 1.54–1.44 (m, 1H). 13C NMR (100 MHz, CD3OD) δC 170.8, 141.6, 132.8, 132.2, 131.3, 129.6, 127.2, 79.0, 60.2, 59.7,42.4, 22.3, 21.1, 20.9, 20.7. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H22N O2 248.1645, found 248.1643.

2-(2-methoxyphenyl)-2-(1-methylpiperidin-1-ium-1-yl)acetate,(4i)
By following above mentioned method,4i was synthesized by using 1m (80.41 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 84.2 mg, 82% yield as viscous oil; 1H NMR (400 MHz, CDCl3) δH 7.90 (d, J = 8 Hz, 1H), 7.35 (t, J = 8 Hz, 1H), 6.97–6.89 (m, 2H), 5.36 (s, 1H), 3.93–3.87 (m, 1H), 3.81 (s, 3H), 3.64–3.62 (m, 1H), 3.35–3.33 (m, 2H), 3.22 (s, 3H), 1.95–1.70 (m, 5H), 1.57–1.51 (m, 1H). 13C NMR (100 MHz, CDCl3) δC 167.2, 158.9, 133.1, 131.7, 121.0, 111.3, 77.1, 73.0, 59.6, 59.4, 55.8, 43.2, 21.4, 20.5, 20.2. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C15H22N O3 264.1594, found 264.1592.

2-(3,4-dimethylphenyl)-2-(1-methylpiperidin-1-ium-1-yl)acetate, (4j)
By following above mentioned method,4j was synthesized by using 1r (79.64 mg, 0.39 mmol) and NMP (38.67 mg, 0.39 mmol) in 88.68 mg, 87% yield as white solid, melting point 197-199°C; 1H NMR (400 MHz, CD3OD) δH 7.44 (s, 1H), 7.39 (dd, J = 7.6, 2.0 Hz, 1H), 7.24 (d, J = 8 Hz, 1H), 4.86 (s, 1H), 3.85–3.79 (m, 1H), 3.63–3.58 (m, 1H), 3.36–3.33 (m, 2H), 3.23 (s, 3H), 2.30 (d, J = 4 Hz, 6H), 2.01–1.89 (m, 4H), 1.80–1.72 (m, 1H), 1.63–1.56 (m,1H). 13C NMR (100 MHz, CD3OD) δC 170.6, 140.5, 138.4, 134.4, 131.0, 130.9, 128.0, 80.5, 60.8, 60.7, 43.8, 22.3, 21.2, 20.9, 19.8, 19.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H24N O2 262.1802, found 262.1801.
Scale-up reaction of 2a with NMM and 1a under the optimized condition
By following the optimized method, 2a was synthesized by using 1a (500 mg, 1.96 mmol) and NMM (198.1 mg, 1.96 mmol) in 442.6 mg (1.41 mmol) i.e., in 73% yield as white solid which was precipitated from the aqueous reaction mixture and was filtered and washed with ethyl acetate.
Acknowledgments
The author’s acknowledge Shiv Nadar Institution of Eminence Deemed to be University for the funding.
Author contribution
S.R., A.B., and D.M. synthesized the compounds; B.M., R.B., and Prof. Munshi performed SCXRD experiments, developed the DFT studies, and energy calculations; P.P., M.M., and Prof. Priyadarshini screened the compounds against bacterial strains and performed the morphological studies; S.M. and Prof. Samanta performed the lipid droplet experiments; Prof. Sen designed the project and wrote the manuscript with the help of Prof. Munshi.
Declaration of interests
All funding sources for the study are listed in the “acknowledgments” section of the manuscript. We the authors and our immediate family members have no financial interests to declare. We the authors and our immediate family members have no related patents to declare or positions to declare, and are not members of the journal’s advisory board. The authors declare no financial interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 13, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107285.
Contributor Information
Parthapratim Munshi, Email: parthapratim.munshi@snu.edu.in.
Subhabrata Sen, Email: subhabrata.sen@snu.edu.in.
Supplemental information
Data and code availability
-
•
This study does not generate new unique reagent.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.
References
- 1.Hili R., Yudin A.K. Making Carbon-Nitrogen Bonds in Biological and Chemical Synthesis. Nat. Chem. Biol. 2006;2:284–287. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 2.Aller E., Buck R.T., Drysdale M.J., Ferris L., Haigh D., Moody C.J., Pearson N.D., Sanghera J.B. N–H insertion reactions of rhodium carbenoids. Part 1. Preparation of α-amino acid and α-aminophosphonic acid derivatives. J. Chem. Soc., Perkin Trans. 1. 1996;1:2879–2884. doi: 10.1039/P19960002879. [DOI] [Google Scholar]
- 3.Qin G., Li L., Li J., Huang H. Palladium-Catalysed Formal Insertion of Carbenoids into Aminals via C–N Bond Activation. J. Am. Chem. Soc. 2015;137:12490–12493. doi: 10.1021/jacs.5b08476. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z., Yang Y., Song Q., Li L., Zanoni G., Liu S., Xiang M., Anderson E.A., Bi X. Chemoselective carbene insertion into the N−H bonds of NH3·H2O. Nat. Commun. 2022;13:7649. doi: 10.1038/s41467-022-35394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramakrishna K., Sivasankar C. Iridium catalysed acceptor/acceptor carbene insertion into N–H bonds in water. Org. Biomol. Chem. 2017;15:2392–2396. doi: 10.1039/C7OB00177K. [DOI] [PubMed] [Google Scholar]
- 6.Wang Z.J., Peck N.E., Renata H., Arnold F.H. Cytochrome P450-Catalyzed Insertion of Carbenoids into N-H Bonds. Chem. Sci. 2014;5:598–601. doi: 10.1039/C3SC52535J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y., Lu H., Zhang X., Zhu M., He K., Yuan H., Xing S. Silver carbenoids derived from diazo compounds: A historical perspective on challenges and opportunities. Chem Catal. 2021;175:599–610. doi: 10.1016/j.checat.2021.05.001. [DOI] [Google Scholar]
- 8.Doyle M.P., Taunton J., Pho H.Q. Conformational and electronic preferences in rhodium(ii) carboxylate and rhodium(ii) carboxamide catalysed carbon-hydrogen insertion reactions of n, n-disubstituted diazoacetoacetamides. Tetrahedron Lett. 1989;30:5397–5400. doi: 10.1016/S0040-4039(01)80577-3. [DOI] [Google Scholar]
- 9.Jurberg I.D., Davies H.M.L. Blue light-promoted photolysis of aryldiazoacetates. Chem. Sci. 2018;9:5112–5118. doi: 10.1039/C8SC01165F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen S.R., Spangler J.E., Hansen J.H., Davies H.M.L. Metal-Free N–H Insertions of Donor/Acceptor Carbenes. Org. Lett. 2012;14:4626–4629. doi: 10.1021/ol3020754. [DOI] [PubMed] [Google Scholar]
- 11.Kuninobu Y., Nishi M., Takai K. Iron-catalysed synthesis of glycine derivatives via carbon–nitrogen bond cleavage using diazoacetate. Chem. Commun. 2010;46:8860–8862. doi: 10.1039/C0CC03781H. [DOI] [PubMed] [Google Scholar]
- 12.Davies H.M.L., Hansen T., Hopper D.W., Panaro S.A. Highly Regio-Diastereo-and Enantioselective C−H Insertions of Methyl Aryldiazoacetates into Cyclic N-Boc-Protected Amines. Asymmetric Synthesis of Novel C2-Symmetric Amines and threo-Methylphenidate. J. Am. Chem. Soc. 1999;121:6509–6510. doi: 10.1021/ja9910715. [DOI] [Google Scholar]
- 13.Davies H.M.L., Hansen T., Churchill M.R. Catalytic Asymmetric C-H Activation of Alkanes and Tetrahydrofuran. J. Am. Chem. Soc. 2000;122:3063–3070. doi: 10.1021/ja994136c. [DOI] [Google Scholar]
- 14.Davies H.M.L., Venkataramani C. Catalytic Enantioselective Synthesis of β2-Amino Acids. Angew. Chem. Int. Ed. 2002;41:2197–2199. doi: 10.1002/1521-3757(20020617)114:12%3C2301::AID-ANGE2301%3E3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 15.Brol A., Olszewski T.K. Synthesis and stability of 1-aminoalkylphosphonic acid quaternary ammonium salts. Org. Biomol. Chem. 2021;19:6422–6430. doi: 10.1039/D1OB00703C. [DOI] [PubMed] [Google Scholar]
- 16.Pal’chikov V.A. Morpholines. Synthesis and biological activity. Russ. J. Org. Chem. 2013;49:787–814. doi: 10.1134/S1070428013060018. [DOI] [Google Scholar]
- 17.Mallavadhani U.V., Fleury-Bregeot N. Encyclopedia of Reagents for Organic Synthesis. John Wiley & Sons, Ltd; 2010. 1,4-Diazabicyclo [2.2.2]octane. [DOI] [Google Scholar]
- 18.Gao L., Liu S., Wang Z.-C., Mao Y., Shi S.L. Ligand- and Additive-Free CuCl2-Catalyzed para-C−H Alkylation of Aniline Derivatives via Carbene Insertion. Asian J. Org. Chem. 2022;11 doi: 10.1002/ajoc.202100723. e2012100723. [DOI] [Google Scholar]
- 19.Xu B., Li M.L., Zuo X.D., Zhu S.F., Zhou Q.L. Catalytic Asymmetric Arylation of α-Aryl-α-diazoacetates with Aniline Derivatives. J. Am. Chem. Soc. 2015;137:8700–8703. doi: 10.1021/jacs.5b05086. [DOI] [PubMed] [Google Scholar]
- 20.Yu Z., Ma B., Chen M., Wu H.H., Liu L., Zhang J. Highly Site-Selective Direct C–H Bond Functionalization of Phenols with α-Aryl-α-diazoacetates and Diazooxindoles via Gold Catalysis. J. Am. Chem. Soc. 2014;136:6904–6907. doi: 10.1021/ja503163k. [DOI] [PubMed] [Google Scholar]
- 21.Yang J.M., Cai Y., Zhu S.F., Zhou Q.L. Iron-catalyzed arylation of α-aryl-α-diazoesters. Org. Biomol. Chem. 2016;14:5516–5519. doi: 10.1039/C5OB02418H. [DOI] [PubMed] [Google Scholar]
- 22.Stivanin M.L., Fernandes A.A.G., Silva A.F., Okada C.Y., Jurberg I.D. Blue Light-Promoted N−H Insertion of Carbazoles, Pyrazoles and 1,2,3-Triazoles into Aryldiazoacetates. Adv. Synth. Catal. 2020;362:1106–1111. doi: 10.1002/adsc.201901343. [DOI] [Google Scholar]
- 23.Empel C., Patureau F.W., Koenigs R.M. Visible Light Induced Metal-Free Carbene N-Carbazolation. J. Org. Chem. 2019;84:11316–11322. doi: 10.1021/acs.joc.9b01753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hansen S.R., Spangler J.E., Hansen J.H., Davies H.M.L. Metal-Free N–H Insertions of Donor/Acceptor Carbenes. Org. Lett. 2012;14:4626–4629. doi: 10.1021/ol3020754. [DOI] [PubMed] [Google Scholar]
- 25.Maiti D., Das R., Sen S. Blue LED-Mediated N–H Insertion of Indoles into Aryldiazoesters at Room Temperature in Batch and Flow: Reaction Kinetics, Density Functional Theory, and Mechanistic Study. J. Org. Chem. 2021;86:2522–2533. doi: 10.1021/acs.joc.0c02649. [DOI] [PubMed] [Google Scholar]
- 26.Maiti D., Das R., Sen S. Photolytic amino etherification reactions of aryl diazoacetates with N-heterocycles and a stoichiometric amount of dioxane/tetrahydropyran in aqueous medium: synthesis of 1,4-dioxepane/1,4,7-dioxazonan-6-one systems. Green Chem. 2021;23:8533–8544. doi: 10.1039/D1GC02797B. [DOI] [Google Scholar]
- 27.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. Gaussian, Inc; 2009. Gaussian 09, Revision D.01. [Google Scholar]
- 28.Lu T., Chen F. Multiwfn: A Multifunctional Wavefunction Analyser. J. Comput. Chem. 2012;33:580–592. doi: 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- 29.Wilcken R., Zimmermann M.O., Lange A., Joerger A.C., Boeckler F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 30.Kálmán A., Argay G., Scharfenberg-Pfeiffer D., Höhne E., Ribár B. `Main-part' isostructuralism of several cardenolides and bufadienolides. Structures of three cardenolides: (21S)-methyldigitoxigenin, uzarigenin and sarmentogenin methanol solvate. Acta Crystallogr. B. 1991;47:68–77. doi: 10.1107/S0108768190008734. [DOI] [Google Scholar]
- 31.Domagk G. Eine neue Klasse von Desinfektionsmitteln. Dtsch. med. Wochenschr. 1935;61:829–832. [Google Scholar]
- 32.Tezel U., Pavlostathis S.G. Quaternary ammonium disinfectants: microbial adaptation, degradation and ecology. Curr. Opin. Biotechnol. 2015;33:296–304. doi: 10.1016/j.copbio.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 33.Gerba C.P. Quaternary Ammonium Biocides: Efficacy in Application. Appl. Environ. Microbiol. 2015;81:464–469. doi: 10.1128/AEM.02633-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morandini A., Leonetti B., Riello P., Sole R., Gatto V., Caligiuri I., Rizzolio F., Beghetto V. Synthesis and Antimicrobial Evaluation of Bis-morpholine Triazine Quaternary Ammonium Salts. ChemMedChem. 2021;16:3172–3176. doi: 10.1002/cmdc.202100409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonnell G., Russell A.D. Antiseptics and Disinfectants: Activity, Action, and Resistance. Clin. Microbiol. Rev. 1999;12:147–179. doi: 10.1128/cmr.12.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tischer M., Pradel G., Ohlsen K., Holzgrabe U. Quaternary Ammonium Salts and Their Antimicrobial Potential: Targets or Nonspecific Interactions? ChemMedChem. 2012;7:22–31. doi: 10.1002/cmdc.201100404. [DOI] [PubMed] [Google Scholar]
- 37.Babbs M., Collier H.O.J., Austin W.C., Potter M.D., Taylor E.P. Salts of Decamethylene-Bis-4-Aminoquinaldinium ("De-quadin"), a New Antimicrobial Agent. J. Pharm. Pharmacol. 1956;8:110–119. doi: 10.1111/j.2042-7158.1956.tb12138.x. [DOI] [PubMed] [Google Scholar]
- 38.Tyagi S., Tyagi V.K. Novel Cationic Gemini Surfactants and Methods for Determination of Their Antimicrobial Activity – Review. Tenside Surfactants Deterg. 2014;51:379–386. doi: 10.3139/113.110319. [DOI] [Google Scholar]
- 39.Buffet-Bataillon S., Tattevin P., Bonnaure-Mallet M., Jolivet-Gougeon A. Emergence of resistance to antibacterial agents: the role of quaternary ammonium compounds—a critical review. Int. J. Antimicrob. Agents. 2012;39:381–389. doi: 10.1016/j.ijantimicag.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Denyer S.P. Mechanisms of action of antibacterial biocides. Int. Biodeterior. Biodegrad. 1995;36:227–245. doi: 10.1016/0964-8305(96)00015-7. [DOI] [Google Scholar]
- 41.Dubovoy V., Nawrocki S., Verma G., Wojtas L., Desai P., Al-Tameemi H., Brinzari T.V., Stranick M., Chen D., Xu S., et al. Synthesis, Characterization, and Investigation of the Antimicrobial Activity of Cetylpyridinium Tetrachlorozincate. ACS Omega. 2020;5:10359–10365. doi: 10.1021/acsomega.0c00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaffer J.E. Lipotoxicity: when tissues overeat. Curr. Opin. Lipidol. 2003;14:281–287. doi: 10.1097/00041433-200306000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Fawcett D.W. Saunders); 1966. An Atlas of Fine Structure: The Cell, its Organelles, and Inclusions. [DOI] [Google Scholar]
- 44.Farese R.V., Walther T.C. Lipid Droplets Finally Get a Little R-E-S-P-E-C-T. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorger D., Athenstaedt K., Hrastnik C., Daum G. A yeast strain lacking lipid particles bears a defect in ergosterol formation. J. Biol. Chem. 2004;279:31190–31196. doi: 10.1074/jbc.M403251200. [DOI] [PubMed] [Google Scholar]
- 46.Sandager L., Gustavsson M.H., Ståhl U., Dahlqvist A., Wiberg E., Banas A., Lenman M., Ronne H., Stymne S. Storage lipid synthesis is non-essential in yeast. J. Biol. Chem. 2002;277:6478–6482. doi: 10.1074/jbc.M109109200. [DOI] [PubMed] [Google Scholar]
- 47.Petschnigg J., Wolinski H., Kolb D., Zellnig G., Kurat C.F., Natter K., Kohlwein S.D., Kohlwein S.D. Good Fat, Essential Cellular Requirements for Triacylglycerol Synthesis to Maintain Membrane Homeostasis in Yeast. J. Biol. Chem. 2009;284:30981–30993. doi: 10.1074/jbc.M109.024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velázquez A.P., Tatsuta T., Ghillebert R., Drescher I., Graef M. Lipid droplet–mediated ER homeostasis regulates autophagy and cell survival during starvation. J. Cell Biol. 2016;212:621–631. doi: 10.1083/jcb.201508102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh S., Zhao B., Bie J., Song J. Macrophage cholesteryl ester mobilization and atherosclerosis. Vasc. Pharmacol. 2010;52:1–10. doi: 10.1016/j.vph.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris C.A., Haas J.T., Streeper R.S., Stone S.J., Kumari M., Yang K., Han X., Brownell N., Gross R.W., Zechner R., Farese R.V. DGAT enzymes are required for triacylglycerol synthesis and lipid droplets in adipocytes[S] J. Lipid Res. 2011;52:657–667. doi: 10.1194/jlr.M013003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choudhary V., Golani G., Joshi A.S., Cottier S., Schneiter R., Prinz W.A., Kozlov M.M. Architecture of lipid droplets in endoplasmic reticulum is determined by phospholipid intrinsic curvature. Curr. Biol. 2018;28:915–926.e9. doi: 10.1016/j.cub.2018.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jornayvaz F.R., Shulman G.I. Diacylglycerol Activation of Protein Kinase Cε and Hepatic Insulin Resistance. Cell Metabol. 2012;15:574–584. doi: 10.1016/j.cmet.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Preuss C., Jelenik T., Bódis K., Müssig K., Burkart V., Szendroedi J., Roden M., Markgraf D.F. A New Targeted Lipidomics Approach Reveals Lipid Droplets in Liver, Muscle and Heart as a Repository for Diacylglycerol and Ceramide Species in Non-Alcoholic Fatty Liver. Cells. 2019;8:277. doi: 10.3390/cells8030277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cruz A.L.S., Barreto E.D.A., Fazolini N.P.B., Viola J.P.B., Bozza P.T. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020;11:105. doi: 10.1038/s41419-020-2297-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This study does not generate new unique reagent.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper can be obtained from the lead contact upon request.