
Figure 5: Activation of NF-κB signaling.
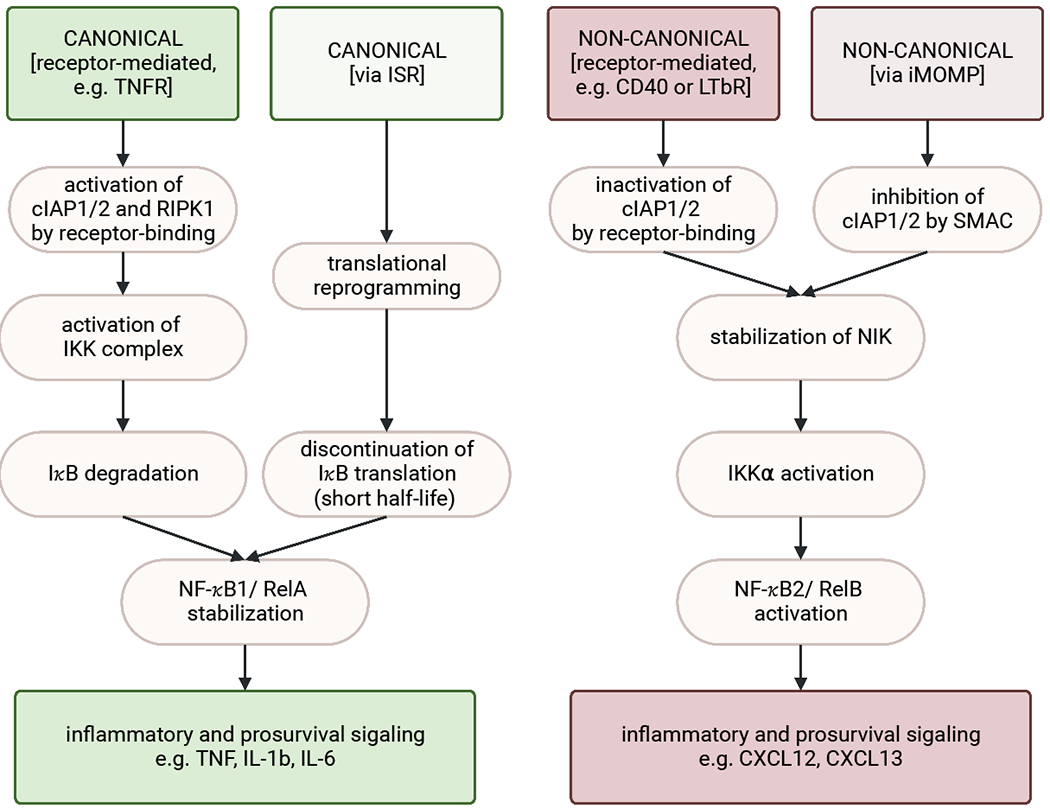
The canonical NF-κB signaling pathway can be promoted via receptor-mediated activation (e.g. via TNF-receptor) or engagement of the ISR. Receptor activation leads to the recruitment of adaptor proteins to form and activate a complex including cIAP and RIPK1. In turn, RIPK1 engages the IKK complex, which induces the proteasomal degradation of IkB. This then allows for the stabilization and nuclear localization of the NF-κB1/RelA heterodimer. Nuclear localization leads to the transcription of proinflammatory cytokines such as TNF, IL-1b, and IL-6. Activation of the ISR followed by translational reprogramming can lead to NF-κB stabilization due to discontinuation of IκB generation, which is a short-lived NFκB-inhibitor.
The non-canonical NF-κB signaling pathway can be engaged by the inhibition of cIAPs, which occurs either upon receptor engagement (e.g. via CD40 or LTbR) or by SMAC, which is released during MOMP. This results in the stabilization of NIK, which is then free to activate IKKα, resulting in NF-κB2/RelB activation and transcription of noncanonical NF-κB associated genes, such as CXCL12 and CXCL13.
Conclusively, upon specific receptor activation cIAPs engage the canonical NF-κB signaling pathway while inhibiting the non-canonical. Vice versa, inactivation of cIAPs promotes non-canonical NF-κB signaling through NIK stabilization while inhibiting the canonical pathway.