

Abstract
Methionine aminopeptidase (MetAP) enzymes play a critical role in bacterial cell survival by cleaving formyl-methionine initiators at N-terminal of nascent protein, a process which is vital in proper protein folding. This makes MetAP an attractive and novel antibacterial target to unveil promising antibiotics. In this study, the crystal structure of R. prowazekii MetAP was used in structure-based virtual screening of drug libraries such as Asinex antibacterial library and Comprehensive Marine Natural Products Database (CMNPD) to identify promising lead molecules against the enzyme. This shortlisted three drug molecules; BDE-25098678, BDE-30686468 and BDD_25351157 as most potent leads that showed strong binding to the MetAP enzyme. The static docked conformation of the compounds to the MetAP was reevaluated in molecular dynamics simulation studies. The analysis observed the docked complexes as stable structure with no major local or global deviations noticed. These findings suggest the formation of strong intermolecular docked complexes, which showed stable dynamics and atomic level interactions network. The binding free energy analysis predicted net MMGBSA energy of complexes as: BDE-25098678 (-73.41 kcal/mol), BDE-30686468 (-59.93 kcal/mol), and BDD_25351157 (-75.39 kcal/mol). In case of MMPBSA, the complexes net binding energy was as; BDE-25098678 (-77.47 kcal/mol), BDE-30686468 (-69.47 kcal/mol), and BDD_25351157 (-75.6 kcal/mol). Further, the compounds were predicted to follow the famous Lipinski rule of five and have non-toxic, non-carcinogenic and non-mutagenic profile. The screened compounds might be used in experimental test to highlight the real anti- R. prowazekii MetAP activity.
Keywords: Rickettsia prowazekii, Methionine Aminopeptidase Enzyme, Asinex Library, Molecular Dynamics Simulation, WaterSwap
1. Introduction
Rickettsia prowazekii is an intracellular, obligate Gram-negative coccobacillus from the genus Rickettsia (Helgren et al., 2017). The bacterium is transmitted to humans by louse and is responsible for causing epidemic typhus (Sexton et al., n.d.). The disease is accompanied with high fever, dry cough, headache, myalgias, rash and delirium (Oberoi and Singh, 2010). So far, no other reservoir is reported for R. prowazekii except humans (Azad and Beard, 1998). The flying squirrel is considered as the main reservoir in United States (Fournier and Raoult, 2020). The reported mortality rate of epidemic typhus varies among people (Kim, 2022). The mortality rate is higher in malnourished patients and elderly people (Álvarez-López et al., 2021). The mortality rate is 60% among patients when the primary infection is untreated (Doppler and Newton, 2020). R. prowazekii is tagged as category B pathogen and a bioterrorism agent according to classification by Center for Disease Control and Prevention (CDC) (Ferreira et al., 2023). The human body louse, being the prime vector of R. prowazekii infection, harbors the bacterium inside the gut epithelium (Osterloh, 2021, Reece and Kaufmann, 2019). Here, the pathogen multiplies, followed by detachment, and finally is released into the feces (Ganta, 2022). Once R. prowazekii enters into the host cells, it escapes the phagosome and starts the multiplication in the cytoplasm, which continues till the cell bursts releasing all the cell contents into the extracellular space (Walker, 2019). The damage to the endothelial cells results into permeability and vasodilation of the vascular endothelium. The former often leads to hypotension, hypovolemia, interstitial edema, and hypoalbuminemia (Walker et al., 2007). The hypovolemia causes hyponatremia. Further, the vascular permeability is responsible for non-cardiogenic pulmonary edema. All together, these events cause a severe multi-organs failure (Dantas-Torres, 2007).
The epidemic typhus is primarily treated with 100 mg dose of doxycycline that typically last for 7–10 days (Botelho-Nevers et al., 2012). In alternative treatment, chloramphenicol of 500 mg dose is given orally/intravenously four times/day upto ten days (Wareham and Wilson, 2002). Azithromycin therapy has been reported with clinical failures (Blanton and Walker, 2016). As the repertoire of clinically effective antibiotics for treating intracellular R. prowazekii infections is small, together with the evolving resistance against chloramphenicol and tetracycline, this warrants the identification of novel drug targets for anti-rickettsia therapeutics development (Helgren et al., 2017). The bacterial protein synthesis begins at the N-terminal formyl-methionine residue of the nascent polypeptide which then undergoes a proper folding, a vital event for proteins functioning (Helgren, 2016, Helgren et al., 2017). The removal of the N-terminal formyl-methionine residue is accomplished through methionine aminopeptidases (MetAP) enzyme that is highly conserved among bacterial species due to their vital importance in functional protein making (R Helgren et al., 2016). The enzyme has been targeted in previous studies for discovery novel chemical scaffolds. For example, in one study, an inhibitor extracted from of Burkholderia pseudomallei showed growth inhibition of B. thailandensis by blocking the function of MetAP1 (Wangtrakuldee et al., 2013). In another work, furoic acids, 1,2,4-triazoles and quinolinols were disclosed to inhibit R. prowazekii MetAP1 (Helgren et al., 2017).
In this study, diverse chemical scaffolds of Asinex antibacterial and Comprehensive Marine Natural Products Database (CMNPD) libraries were used in structure-based virtual screening studies to identify promising binders of R. prowazekii MetAP (Helgren et al., 2017, Lionta et al., 2014, Lyu et al., 2021, Navid et al., 2021). Then, the best docked complexes were subjected to molecular dynamics simulation analysis to simulate real time cellular environment behavior of the complexes (Karplus, 2002). The atomic level binding free energies of complexes were then determined based on the simulation trajectories. For this purpose, molecular mechanics energies combined with the Poisson–Boltzmann or generalized Born and surface area continuum solvation (MMPBSA and MMGBSA) methods were employed (Massova and Kollman, 2000, Wang et al., 2019). Both methods are considered powerful in predicting intermolecular interaction energies at good accuracy with the modest requirement of computational speed. The selected compounds were also evaluated for absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties to shed light on their pharmacokinetic properties, which might be helpful to optimize compounds structure–activity relationship and get safe and effective chemical structures (Jia et al., 2019). The shortlisted compounds in the study might be used in a thorough experimental validation against R. prowazekii MetAP.
2. Materials and methods
The complete methods flow of the study is given in Fig. 1.
Fig. 1.

Computational framework of methodology used in the study. The study begins from a target structure retrieval from protein database which then subjected to different chemoinformatics and biophysics studies including structure-based virtual screening studies, molecualr dynamics simulation and binding free energies analysis.
2.1. Preparation of R. prowazekii methionine aminopeptidase
The 3D structure of R. prowazekii MetAP was obtained from protein data bank by using PDB code:3MR1 (Helgren et al., 2017, Sussman et al., 1998). The enzyme structure was resolved using X-ray diffraction method with resolution value of 2.0 Å. The R-value free, R-value work and R-value observed for the enzyme was 0.212, 0.171 and 0.173, respectively. The total structure weight of the enzyme is 112.61 kDa having atom count of 8,868. After retrieval, the enzyme structure was subjected to a preparation phase in UCSF Chimera 1.16 (Kaliappan and Bombay, 2018). The co-crystalized ligands were discarded. The enzyme was then used in energy minimization process to optimize the structure. This was achieved by energy minimization tool of UCSF Chimera 1.16 where first missing hydrogen atoms were added, followed by charge assignment using Gasteiger charge calculation method. The energy minimization used were; steepest descent and conjugate gradients. Both the algorithms were applied for 1000 steps to ensure maximum possible structure optimization of the enzyme and remove steric clashes. Once energy minimization is completed, the enzyme is saved as.pdb for downward processing.
2.2. Preparation of inhibitor library
For virtual screening, two drug libraries were used: Asinex antibacterial library containing 5968 compounds and Comprehensive Marine Natural Products Database (CMNPD) library which offers more than 40 thousand compounds (Lyu et al., 2021, Navid et al., 2021). The Asinex antibacterial library provides a unique collection of compounds based on natural product like scaffolds; hence, providing maximum skeletal diversity. The structural diversity of compounds is further enriched by adding multiple stereogenic centers and polar functional groups. The CMNPD database is a dedicated database containing diverse chemical scaffolds from marine organisms. The marine environment harbors diverse biological life and thus could serve as a rich source of diverse chemical scaffolds. The libraries were fetched in.sdf format and imported to PyRx 0.8 (Dallakyan and Olson, 2015) to be energy minimized. The energy minimization was done using MM2 force field (Halgren, 1996), and afterward converted into.pdbqt.
2.3. Virtual screening
Virtual screening is an efficient method in modern drug discovery to examine drug libraries and highlight those which have a strong binding affinity to a biological target (Lionta et al., 2014). The virtual screening squeezes the library to minimum number of ideal compounds that can be tested against a biological target. The virtual screening was done in PyRx 0.8 (Dallakyan and Olson, 2015), where the grid box was set at His77 (x-axis: 0.64 Å, y-axis: 5.55 Å and z-axis:11.85 Å) (Helgren et al., 2017). The grid box dimensions used were 25 Å along XYZ dimensions. The number of exhaustiveness for each compound set is 100. The binding conformation of each compound was ranked based on the binding energy in kcal/mol. The binding energy score of the docking software predicts the binding affinity of docked ligand-receptor complex. However, the one with lowest energy score was screened as the best binding conformation with methionine aminopeptidase enzyme. The Lamarckian genetic algorithm was employed for ranking the ligands that switches between phenotypic and genotypic space (Yadava, 2018, Yadava et al., 2013). The visualization of docked conformation and interactions with the enzyme was done using UCSF Chimera 1.16 (Kaliappan and Bombay, 2018). Additionally, Discovery Studio 2021 was employed for atomic level interactions (Biovia, 2017). The validation of the docking procedure was conducted by docking a control compound (3-((4-fluorobenzyl)thio)-4H-1,2,4-triazole)) to the enzyme and then the docked pose was compared to the one already reported in the literature (R Helgren et al., 2016).
2.4. Molecular dynamics simulation (MDS)
The MDS was carried for the shortlisted complexes and control complex to decipher intermolecular binding stability and understand compounds binding mechanism and atomic level interactions during simulation time (Hansson et al., 2002). The MDS was done using AMBER simulation package v 22 (Case et al., 2022). The antechamber program was applied to process the complexes. The Ff14SB force field and GAFF force field were employed to generate parameters for the enzyme and compounds, respectively (He et al., 2020, Maier et al., 2015). The complexes were solvated in OPC solvation box (marginal distance of 10 Å between the protein and box edges). The protein residues were treated in a way to assign their standard ionization states at pH 7. To get neutral charge on the systems, counter ions were added. The preprocessing was conducted to energy minimize the complexes. The hydrogen atoms energy minimization, water box energy minimization, water molecules, and non-heavy atoms energy minimization were done in a step-wise fashion. The systems were heated gradually upto 310 K and maintained using via Langevin algorithm (Izaguirre et al., 2001). The gamma value set in the process was 1.0. The complexes were then subjected to equilibration for 100 ps with time step of 2 fs under constant number of particles, volume and temperature (NPT) ensemble guided by Berendsen temperature coupling algorithm (Berendsen et al., 1984). This was followed by another round of equilibration under constant number of particles, pressure and temperature (NPT) ensemble. The pressure equilibration was done under NPT ensemble for 300 ps and at 1 atm. The production run was accomplished for 100 ns under NPT ensemble. During the production run, the long term electrostatic interactions were calculated through particle mesh Ewald algorithm (Petersen, 1995). The non-bounded interactions were defined by cut-off value of 8 Å. The SHAKE algorithm was used to maintain hydrogen bonds (Kräutler et al., 2001). The simulation trajectories were analyzed using CPPTRAJ module (Roe and Cheatham III, 2013). For plotting purposes, XMGRACE v 5.1 was used (Turner, 2005).
2.5. MM (GB/PBSA) analysis
MM (GB/PBSA) is a prime method to evaluate binding free energies of protein–ligand complexes. Binding free energies consider the sum of all intermolecular interactions presents between the ligands and receptor biomolecule. The assay was done using MMPBSA.py module of AMBER v22 (Miller et al., 2012, Wang et al., 2019, Zhang et al., 2017). The MM (GB/PBSA) equation was performed on 1000 frames of simulation trajectories collected from equal time interval of simulation time. The analysis was conducted using the following equation;
In the above equation, ΔG can be split into entropic term (TΔS) and enthalpic term (ΔEgas phase and ΔEsolv phase). The ΔEgas comprises van der Waals energy (Evdw), intramolecular energy (Eint) and electrostatic enenergy (Eele). The ΔEsolv phase contains non-electrostatic energy (Esurf) and electrostatic (Egb).
Further, the AMBER normal mode analysis was applied separately to predict entropy energy contribution to complexes, which was done on 10 frames of simulation trajectories (Genheden et al., 2012).
2.6. WaterSwap absolute binding free energy calculation
Another round of the absolute binding free energy calculation was done using WaterSwap method (Woods et al., 2014, Woods et al., 2011). The method swaps water cluster of equal size and volume of the bounded ligand to the protein. The WaterSwap absolute binding free energy was estimated using Bennett’s acceptance ratio (BAR), thermodynamic integration (TI) and free energy perturbation (FEP) (Ahmad et al., 2019). The WaterSwap analysis was conducted for default 1000 steps.
2.7. ADMETlab 2.0 analysis
Drug safety and efficacy are the two main issues that often cause drug failure. Therefore, absorption, distribution, metabolism, excretion and toxicity (ADMET) evaluation of drugs at an early stage of the drug development plays a critical role in leads identification (Jia et al., 2019, Pires et al., 2015). The AMDET profiling of drugs was done using ADMETlab 2.0 which is available online via: https://admetmesh.scbdd.com/(Xiong et al., 2021).
3. Results
3.1. Molecular docking studies
The drug libraries were screened against R. prowazekii MetAP enzyme using structure-based virtual screening process. For comparative analysis, a control molecule was used. As a result of this analysis, three compounds, namely, BDE-25098678, BDE-30686468 and BDD_25351157 were unveiled as promising molecules to bind the enzyme with high affinity (See Table 1). The IUPAC naming of the control, BDE-25098678, BDE-30686468 and BDD_25351157 is 3-((4-fluorobenzyl)thio)-4H-1,2,4-triazole, 4,7-dimethyl-2-(1-(3-phenoxybenzoyl)-1H-pyrrol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-1,3-diium-6-olate, 3-(1-((5-(2-carboxyphenyl)furan-2-yl)methyl)piperidin-2-yl)-5-(hydroxymethyl)-1H-pyrazol-2-ium and 3-carbamoyl-5-(1-(3-fluorophenyl)-5-oxopyrrolidin-3-yl)-7-hydroxy-1,3a-dihydropyrazolo[1,5-a]pyrimidin-4-ium, respectively. The binding energy score of the control and lead molecules to the receptor enzyme was as: control (-9.12 kcal/mol), BDE-25098678 (-11.54 kcal/mol), BDE-30686468 (-12.38 kcal/mol) and BDD_25351157 (-12.07 kcal/mol). The control molecule 3-(methylthio)-4H-1,2,4-triazole chemical moiety was seen primarily responsible for stable interactions with the enzyme. This chemical region formed hydrogen bonds with Cys68, Thr96 and Asp105 at bond distance length of 1.8 Å, 2.6 Å and 2.41 Å, respectively (Fig. 2A). Besides, the compound produced weak van Waals interactions with Tyr60, Phe174, His168, Glu233, Val95 and Asp94. The control compound was observed in different hydrophilic and hydrophobic interactions from all sides of the active pocket. BDE-25098678 compound formed a strong hydrogen bond with Thr96 through its 4,7-dimethyl-7H-pyrrolo[2,3-d]pyrimidin-1,3-diium-6-olate. The (3-phenoxyphenyl)(1H-pyrrol-1-yl)methanone makes two hydrogen bonds each with His77 and His175. The compound achieved a stable deep binding at the active cavity and accomplished several van der Waals, pi-pi stacked, pi-pi T shaped, alkyl and pi-alkyl interactions (Fig. 2B). The compound BDE-30686468 formed a cluster of hydrogen bonds with His168, His175, His77, and Glu201. Mostly, the compound is engaged at the active site through 2-(furan-2-yl)benzoic acid and 5-(hydroxymethyl)-1H-pyrazol-2-ium (Fig. 2C). The BDD_25351157 3-carbamoyl-7-hydroxy-1,3a-dihydropyrazolo[1,5-a]pyrimidin-4-ium segment was observed heavily in a strong hydrogen bonding network. This chemical moiety formed bonding with Asp105, Asp94, Gly233, Glu201, His168, and His175. The other chemical moiety of the compound (1-(3-fluorophenyl)pyrrolidin-2-one) generated a hydrogen bond with Lys61 (Fig. 2D). All the three shortlisted compounds and control compound made significant hydrophilic and hydrophobic interactions with methionine aminopeptidase enzyme which can be considered vital in overall intermolecular conformational stability.
Table 1.
Best ranked compounds that achieved stable binding conformation with the R. prowazekii MetAP.
| Rank | Compound ID | 2D Structure | IUPAC Name | Binding Energy Score (kcal/mol) |
|---|---|---|---|---|
| 1 | BDE-25098678 | 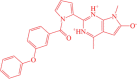 |
4,7-dimethyl-2-(1-(3-phenoxybenzoyl)-1H-pyrrol-2-yl)-7H-pyrrolo[2,3-d]pyrimidin-1,3-diium-6-olate | −11.54 |
| 2 | BDE-30686468 | 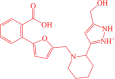 |
3-(1-((5-(2-carboxyphenyl)furan-2-yl)methyl)piperidin-2-yl)-5-(hydroxymethyl)-1H-pyrazol-2-ium | −12.38 |
| 3 | BDD_25351157 |  |
3-carbamoyl-5-(1-(3-fluorophenyl)-5-oxopyrrolidin-3-yl)-7-hydroxy-1,3a-dihydropyrazolo[1,5-a]pyrimidin-4-ium | −12.07 |
| 4 | CMNPD1125 | 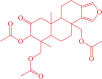 |
(7-acetoxy-6,9a-dimethyl-8-oxo-3b,4,5,5a,6,7,8,9,9a,9b,10,11-dodecahydrophenanthro[1,2-c]furan-3b,6-diyl)bis(methylene) diacetate | −10.10 |
| 5 | CMNPD150 | (2E,6E)-furan-3-ylmethyl 3,7-dimethyl-8-(4-methylfuran-2-yl)octa-2,6-dienoate | −9.63 | |
| 6 | CMNPD798 |  |
(Z)-7-(3-hydroxy-2-((E)-3-hydroxyoct-1-en-1-yl)-5-oxocyclopentyl)hept-5-enoic acid | −8.21 |
| 7 |  |
(Z)-6-bromo-1-(5-hydroxy-3-methylpent-3-en-1-yl)-2,5,5,8a-tetramethyldecahydronaphthalen-2-ol | −7.66 | |
| 8 | CMNPD148 | 3-((2E,4E,6E)-4,8-dimethylnona-2,4,6-trien-1-yl)furan | −7.11 | |
| 9 | CMNPD359 |  |
€-3-formyl-5-(2,6,6-trimethylcyclohex-2-en-1-yl)pent-2-en-1-yl acetate | −6.54 |
| 10 | CMNPD638 |  |
(E)-2-(1-bromohex-3-en-1-yl)-5-(3-bromopropa-1,2-dien-1-yl)hexahydrofuro[3,2-b]furan | −6.01 |
| 11 | Control | 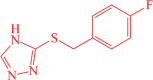 |
3-((4-fluorobenzyl)thio)-4H-1,2,4-triazole | −9.12 |
Fig. 2.

Docked binding mode of control/lead molecules against the targeted R. prowazekii MetAP enzyme. A. Control, B. BDE-25098678, C. BDE-30686468 and D. BDD_25351157.
3.2. MDS investigation
Molecular dynamics simulation is a theoretical approach to study the biological molecule’s structure–function relationship. It covers physical movements analysis of nucleic acids and proteins. During simulation, the interactions of atoms/molecules over specific time provides an insight into the dynamic evolution of the system. The simulation analysis mimics the structural conformational changes in biomolecules adopted during a given period of time. The MD analysis done herein includes root mean square deviation (RMSD), root mean square fluctuation (RMSF) and radius of gyration (Rg). These analyses are done based on C-α atoms of R. prowazekii methionine aminopeptidase enzyme. The first assay done was RMSD to investigate thermodynamic conformational stability of docked complexes during the simulation time (Fig. 3A). The frames collected from simulation trajectories were superimposed over each other to look for structure changes in docked complexes during the simulation time. The docked intermolecular conformation was used as a reference structure in this regard. The mean RMSD noticed for BDE-25098678, BDE-30686468, BDD_25351157 and control are 1.53 Å (standard deviation: 0.15), 1.66 Å (standard deviation: 0.233), 1.52 Å (standard deviation: 0.20) and 1.66 Å (standard deviation:0.23), respectively. Throughout the simulation time, the complexes were seen in good equilibrium with some minor conformational changes due to naturally flexible loop regions. The residue level structural stability information was obtained using RMSF, which sheds light on the individual residue in particular those involved in ligand binding and hence being vital for the protein functionality (Fig. 3B). The mean RMSF value of BDE-25098678, BDE-30686468, BDD_25351157 and control system was 0.92 Å (standard deviation: 0.45), 1.01 Å (standard deviation:0.39), 0.93 Å (standard deviation:0.41), and 1.02 Å (standard deviation:0.55), respectively. The average RMSF value of the systems depicts that in the presence of compounds the R. prowazekii MetAP enzyme remained structurally stable and no major global structure variations were noticed. However, the C-terminal and the central domain of the enzyme showed some local fluctuations, which were due to flexible loop regions providing space for structural accommodations gained by the compounds inside the active pocket. Further understanding of the intermolecular conformation was disclosed using Rg analysis (Fig. 3C). The Rg analysis demonstrates complex degree of compactness during simulation time. The high Rg value indicates high relax nature of the complex while lower value implies highly compact complex. The complex nature of receptor molecule is an indication of a strong intermolecular binding. The average Rg value of complexes were as; BDE-25098678 (46.87 Å), BDE-30686468 (44.78 Å), BDD_25351157 (46.91 Å) and control system (45.78 Å). In order to determine lead molecules and control stable dynamics, ligand RMSD was calculated throughout the length of simulation time as given in Fig. 4A. The mean RMSD of ligands were as; BDE-25098678 (0.98 Å), BDE-30686468 (1.01 Å), BDD_25351157 (0.561 Å) and control system (1.42 Å). These values clearly indicate that the ligand docked conformation with the enzyme was noticed in very stable state. Further, it shows that the ligands stability allows the receptor enzyme to be in a more conformational equilibrium. Next, hydrogen bonds analysis was performed in order to determine the number of hydrogen bonds formed between the ligands and MetAP enzyme. As can be seen in Fig. 4B, at least 2–4 hydrogen bonds were observed in most frames of the simulation time. This indicates that hydrogen bonds played a vital role in the ligand’s stability at the docked site of enzyme and overall complexes conformational stability.
Fig. 3.

MDS analysis based on C-α atoms. All the analyses were carried out in Angstrom (Å). A. RMSD, B. RMSF and C. Rg.
Fig. 4.

Ligands RMSD (A) and hydrogen bonds (B) analyses of docked complexes.
3.3. MM (GB/PBSA) binding free energies
The MM (GB/PBSA) method is a powerful post simulation approach done on simulation trajectories to estimate the free energy of binding of small molecules to a biomolecule receptor. This method splits the free energy into solvation energy term and molecular mechanics term. The difference between bound and unbound solvated molecules are compared and the energy differences are estimated. The estimation of free energy is done on equilibrium trajectories frames extracted from the simulation trajectories. The MM (GB/PBSA) estimated binding free energies for the studied systems are tabulated in Table 2. As can be observed that the systems exhibit robust intermolecular binding energies. The compounds docking with the R. prowazekii methionine aminopeptidase enzyme formed a strong network of van der Waals as well as of electrostatic interactions. The van der Waals energy was estimated the most favorable and noticed as −68.71 kcal/mol, −61.09 kcal/mol, −70.84 kcal/mol and 62.84 kcal/mol for BDE-25098678, BDE-30686468, BDD_25351157 and control molecule, respectively. The electrostatic interactions were also reported to play a high role in intermolecular stability. The electrostatic energy is −26.78 kcal/mol, –23.47 kcal/mol, −26.22 kcal/mol and −20.17 kcal/mol for BDE-25098678, BDE-30686468, BDD_25351157 and control molecule, respectively. Both the van der Waals and electrostatic energies term concluded their promising role in overall system conformation stability during simulation time. The net MMGBSA energy was found in the following order: BDE-25098678 (-73.41 kcal/mol), BDE-30686468 (-59.93 kcal/mol), BDD_25351157 (-75.39 kcal/mol) and control molecule (-64.64 kcal/mol). The BDE-25098678 and BDD_25351157 complexes were found the most promising docked compounds compared to BDE-30686468 and control. In case of MMPBSA, the complexes net binding energy was as; BDE-25098678 (-77.47 kcal/mol), BDE-30686468 (-69.47 kcal/mol), BDD_25351157 (-75.6 kcal/mol) and control molecule (-60.18 kcal/mol).
Table 2.
Free energies estimated for the systems based on MM (GB/PBSA) method. The values tabulated in the table are in kcal/mol.
| Method | Energy Parameter | BDE-25098678 | BDE-30686468 | BDD_25351157 | Control Complex |
|---|---|---|---|---|---|
| MM-GBSA | Van der Waals Energy | −68.71 | −61.09 | −70.84 | −62.84 |
| Electrostatic Energy | −26.78 | –23.47 | −26.22 | −20.17 | |
| Delta Gas Phase Energy | −95.49 | −84.56 | −97.06 | −83.01 | |
| Delta Solvation Energy | 22.07 | 24.63 | 21.67 | 18.37 | |
| Net Energy | −73.41 | −59.93 | −75.39 | −64.64 | |
| MM-PBSA | Van der Waals Energy | −68.71 | −61.09 | −70.84 | −62.84 |
| Electrostatic Energy | −26.78 | –23.47 | −26.22 | −20.17 | |
| Delta Gas Phase Energy | −95.49 | −84.56 | −97.06 | −83.01 | |
| Delta Solvation Energy | 18.02 | 15.09 | 21.46 | 22.83 | |
| Net Energy | −77.47 | −69.47 | −75.6 | −60.18 |
3.4. Entropy analysis
The presence of the entropy energy contributes to system’s disorder and randomness and, as a result, impacts the intermolecular interactions between R. prowazekii MetAP enzyme and the screened compounds. The entropy energy results for the studied systems are given in Table 3. The findings suggest less presence of entropy energy in the systems, demonstrating less energy is available to make the system in disorder state. The control system was found to have more entropy energy compared to the leads system. The control complex net entropy energy was 4.50 kcal/mol that implies a non-favorable contribution to overall complex conformational stability. Among the lead systems, the BDD_25351157 system revealed the most stable entropy energy (-3.48 kcal/mol), followed by BDE-30686468 (-2.35 kcal/mol) and BDE-25098678 (-1.14 kcal/mol).
Table 3.
Entropy energies in kcal/mol. The entropy results were generated using AMBER normal mode analysis.
| Complex | Translational | Rotational | Vibrational | DELTA S Total |
|---|---|---|---|---|
| BDE-25098678 | 11.15 (0.24) | 10.68 (0.50) | 2315.14 (2.85) | −1.14 |
| BDE-30686468 | 12.68 (0.32) | 12.09 (0.61) | 2761.23 (2.23) | −2.35 |
| BDD_25351157 | 9.66 (0.54) | 8.13 (0.66) | 2355.16 (3.64) | −3.48 |
| Control | 15.110 (1.05) | 16.04 (0.71) | 2391.55 (2.36) | 4.50 |
3.5. WaterSwap analysis
WaterSwap is an efficient theoretical method to estimate the absolute binding free energy of protein–ligand complexes. The WaterSwap functions by λ-coordinate construction, followed by connecting protein box (containing periodic water box of protein–ligand complex) to a Water box (containing water cluster). The λ-coordinate swap protein bounded ligand with water cluster of equal shape and volume. The free energy change are calculated using Bennett acceptance ratio method (BAR), free energy perturbation (FEP), and thermodynamic integration (TI) methods. The WaterSwap findings are documented in Fig. 5. The systems are well converged as the different of energy unit among the methods is less then 1 kcal/mol. Among the systems, the BDE-25098678 and BDE-30686468 complexes were found slightly more stable in terms of securing a stable absolute binding free energy compared to BDD_25351157 and control. The FEP, TI and Bennett’s energy value of BDE-25098678 is −43.6 kcal/mol, −42.94 kcal/mol and −43.61 kcal/mol, respectively.
Fig. 5.

Revalidation of MM (GB/PBSA) method by WaterSwap method. Three algorithms are provided, namely, Bennett’s, TI and FEP.
3.6. ADMET analysis
AMDET testing is considered a essential part in the present drug development and discovery process to delineate the compound’s pharmacodynamics and pharmacokinetics (Van De Waterbeemd and Gifford, 2003). The early ADMET profiling of drugs helps in reducing the drug’s failure in the later phases of drug discovery. Furthermore, the early ADMET profiling of the compounds minimizes the drug discovery time, cost and associated complications (Jia et al., 2019). The control and lead molecules were found to follow most the ADMETlab 2.0 druglike properties. The physicochemical properties of the control/lead molecules are presented in Fig. 6. Details about control, BDE-25098678, BDE-30686468 and BDD_25351157 physicochemical properties, medicinal chemistry, absorption, distribution, metabolism, excretion and toxicology are given in Appendix 1, Appendix 2, Appendix 3 and Appendix 4, respectively. Briefly, the BDE-25098678, BDE-30686468 and BDD_25351157 fulfill Lipinski rule of five (Lipinski, 2004), Pfizer rule (Ursu et al., 2011), and golden triangle (Zerroug et al., 2019). This suggests that these compounds can be promising lead molecules and have high chances to be successful in terms of pharmacokinetics and could be investigated as potential drug candidates. Similarly, the compounds were decoded to have no alert for pan-assay interference compounds (PAINS); thus, they might not give any false positive results (Whitty, 2011). This further implies that the compounds bind to a specific biomolecule rather than interacting with multiple biological targets. In addition, the compounds showed no alert for acute toxicity rule, genotoxic carcinogenicity rule, non-genotoxic carcinogenicity rule, skin sensitization rule, aquatic toxicity rule, non-biodegradable rule and sureChEMBL rule.
Fig. 6.

ADMET profiling of control/lead molecules against the targeted R. prowazekii MetAP enzyme. A. Control, B. BDE-25098678, C. BDE-30686468 and D. BDD_25351157. The upper and lower limits can be depicted by yellow and pink shaded regions, respectively. The blue line demonstrates compounds properties. The different compounds properties evaluated in the process are; MW (molecular weight), nRig (rings number), formal charge (fchar), rigid bonds number (nRig), rotatable bonds number (nRot), topological polar surface area (TPSA), heteroatoms number (nhet), atoms number in biggest ring (MaxRing), hydrogen bond acceptor number (nHA) and hydrogen bond donors’ number (nHD).
4. Discussion
Computational drug design is considered a powerful tool to identify new drug molecules against any given target. In parallel to the traditional drug design, the in silico approaches could save time, resources and speed up drug discovery pipeline (Macalino et al., 2015, Talele et al., 2010). This study filtered three promising molecules; BDE-25098678 (binding energy score of −11.54 kcal/mol), BDE-30686468 (binding energy score of −12.38 kcal/mol) and BDD_25351157 (binding energy score of −12.07 kcal/mol) as potential lead molecules against R. prowazekii MetAP enzyme, which is vital for protein folding process. These molecules showed robust intermolecular interactions with the enzyme and formed a stable binding conformation. The stable binding conformation is validated by several molecular dynamics simulation analyses. A priori, the computer-aided drug design methods have been employed to successfully identify several lead compounds. For example, captopril for hypertension, saquinavir for AIDS, zanamivir for influenza etc (Talele et al., 2010). Recently, Chang et al identified several oxadiazoles to inhibit penicillin binding protein 2a of methicillin resistant Staphylococcus aureus. In one study, Deepasree et al reported terpenoid compounds against phosphatidylinositol-specific phospholipase C from Listeria monocytogenes (Deepasree and Venugopal, 2023). The study found forty terpenoid compounds against the targeted enzyme using combine approach of molecular docking and molecular dynamics simulation. In another work, 55 dihydrophenanthrene derivatives were employed to build two 2D-QSAR models. The study recognized several lead molecules against SARS-CoV-2 main protease enzyme (Oubahmane et al., 2023). In short, the lead molecules identified herein are promising candidates to be evaluated in experimental studies.
5. Concluding remarks
The study concluded three potential molecules; BDE-25098678, BDE-30686468, and BDD_25351157 as the best binders of R. prowazekii MetAP, a vital enzyme for post-translation modifications of the newly synthesized proteins. The compounds were seen docked deep inside the MetAP active pocket and formed robust van der Waals and electrostatic interactions. Dynamically, the docked complexes are found to be stable with no major RMSF fluctuations, suggesting the formation of a strong intermolecular bonding and a stable binding mode. Additionally, the binding free energies indicate the formation of stable complexes with the domination of van der Waals energy. The compounds follow Lipinski rule of five and have no toxic chemical moieties. Despite the promising results herin, the study has few limitations that can be addressed in the future work. For example, advanced quantum mechanics/molecular mechanics (QM/MM) approach can be used to investigate the chemical reactivity and estimate the electrostatic interaction energies of the studied systems (Bharadwaj et al., 2021b, Bharadwaj et al., 2021a, Bharadwaj et al., 2021c, Bharadwaj et al., 2019). Experimental enzyme inhibition assays can be performed to disclose the compounds real potency against R. prowazekii MetAP. The outcomes of the study might be helpful in the designing of new chemical derivatives based on the parent scaffolds of the filtered compounds, which could bring about more biologically active agents.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through the project number (IF2/PSAU/2022/03/22532)
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsps.2023.101745.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Ahmad S., Raza S., Abro A., Liedl K.R., Azam S.S. Toward novel inhibitors against KdsB: a highly specific and selective broad-spectrum bacterial enzyme. J. Biomol. Struct. Dyn. 2019;37:1326–1345. doi: 10.1080/07391102.2018.1459318. [DOI] [PubMed] [Google Scholar]
- Álvarez-López D.I., Ochoa-Mora E., Heitman K.N., Binder A.M., Álvarez-Hernández G., Armstrong P.A. Epidemiology and clinical features of Rocky Mountain spotted fever from enhanced surveillance, Sonora, Mexico: 2015–2018. Am. J. Trop. Med. Hyg. 2021;104:190. doi: 10.4269/ajtmh.20-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad A.F., Beard C.B. Rickettsial pathogens and their arthropod vectors. Emerg. Infect. Dis. 1998;4:179. doi: 10.3201/eid0402.980205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Bharadwaj S., Lee K.E., Dwivedi V.D., Yadava U., Panwar A., Lucas S.J., Pandey A., Kang S.G. Discovery of Ganoderma lucidum triterpenoids as potential inhibitors against Dengue virus NS2B-NS3 protease. Sci. Rep. 2019;9:19059. doi: 10.1038/s41598-019-55723-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S., Dubey A., Kamboj N.K., Sahoo A.K., Kang S.G., Yadava U. Drug repurposing for ligand-induced rearrangement of Sirt2 active site-based inhibitors via molecular modeling and quantum mechanics calculations. Sci. Rep. 2021;11:10169. doi: 10.1038/s41598-021-89627-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S., Dubey A., Yadava U., Mishra S.K., Kang S.G., Dwivedi V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021;22:1361–1377. doi: 10.1093/bib/bbaa382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj S., Rao A.K., Dwivedi V.D., Mishra S.K., Yadava U. Structure-based screening and validation of bioactive compounds as Zika virus methyltransferase (MTase) inhibitors through first-principle density functional theory, classical molecular simulation and QM/MM affinity estimation. J. Biomol. Struct. Dyn. 2021;39:2338–2351. doi: 10.1080/07391102.2020.1747545. [DOI] [PubMed] [Google Scholar]
- Biovia D.S. San Diego; CA, USA: 2017. Discovery studio visualizer. [Google Scholar]
- Blanton L.S., Walker D.H. Treatment of tropical and travel related rickettsioses. Curr. Treat. Options Infect. Dis. 2016;8:42–56. [Google Scholar]
- Botelho-Nevers E., Socolovschi C., Raoult D., Parola P. Treatment of Rickettsia spp. infections: a review. Expert Rev. Anti Infect. Ther. 2012;10:1425–1437. doi: 10.1586/eri.12.139. [DOI] [PubMed] [Google Scholar]
- Case, D.A., Duke, R.E., Walker, R.C., Skrynnikov, N.R., Cheatham III, T.E., Mikhailovskii, O., Simmerling, C., Xue, Y., Roitberg, A., Izmailov, S.A., others, 2022. AMBER 22 Reference Manual.
- Dallakyan S., Olson A.J. Small-molecule library screening by docking with PyRx. Chem. Biol. Springer. 2015:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Dantas-Torres F. Rocky Mountain spotted fever. Lancet Infect. Dis. 2007;7:724–732. doi: 10.1016/S1473-3099(07)70261-X. [DOI] [PubMed] [Google Scholar]
- Deepasree, K., Venugopal, S., 2023. Molecular docking and dynamic simulation studies of terpenoid compounds against phosphatidylinositol-specific phospholipase C from Listeria monocytogenes. Informatics Med. Unlocked 101252.
- Doppler J.F., Newton P.N. A systematic review of the untreated mortality of murine typhus. PLoS Neglected Trop. Diseases. 2020;14:e0008641. doi: 10.1371/journal.pntd.0008641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira, C., Doursout, M.-F.J., Balingit, J.S., 2023. Bioterrorism, in: 2000 Years of Pandemics: Past, Present, and Future. Springer, pp. 325–340.
- Fournier, P.-E., Raoult, D., 2020. Epidemic Louse-Borne Typhus, in: Hunter’s Tropical Medicine and Emerging Infectious Diseases. Elsevier, pp. 577–579.
- Ganta R.R. Rickettsiaceae and coxiellaceae: Rickettsia and coxiella. Vet. Microbiol. 2022:377–380. [Google Scholar]
- Genheden S., Kuhn O., Mikulskis P., Hoffmann D., Ryde U. The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J. Chem. Inform. Model. 2012;52:2079–2088. doi: 10.1021/ci3001919. [DOI] [PubMed] [Google Scholar]
- Halgren, T. a, 1996. Merck Molecular Force Field. J. Comput. Chem. 17, 490–519. https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<520::AID-JCC2>3.0.CO;2-W
- Hansson T., Oostenbrink C., van Gunsteren W. Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2002;12:190–196. doi: 10.1016/s0959-440x(02)00308-1. [DOI] [PubMed] [Google Scholar]
- He X., Liu S., Lee T.-S., Ji B., Man V.H., York D.M., Wang J. Fast, accurate, and reliable protocols for routine calculations of protein–ligand binding affinities in drug design projects using AMBER GPU-TI with ff14SB/GAFF. ACS Omega. 2020;5:4611–4619. doi: 10.1021/acsomega.9b04233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgren T.R., Chen C., Wangtrakuldee P., Edwards T.E., Staker B.L., Abendroth J., Sankaran B., Housley N.A., Myler P.J., Audia J.P., et al. Rickettsia prowazekii methionine aminopeptidase as a promising target for the development of antibacterial agents. Bioorg. Med. Chem. 2017;25:813–824. doi: 10.1016/j.bmc.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Helgren, T., Wangtrakuldee, P., L Staker, B., J Hagen, T., 2016. Advances in bacterial methionine aminopeptidase inhibition. Curr. Top. Med. Chem. 16, 397–414. [DOI] [PMC free article] [PubMed]
- Helgren, T.R., 2016. The utilization of flavonoids as inhibitors of urease and as antimalarial agents and the discovery of bacterial methionine aminopeptidase inhibitors.
- Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001;114:2090–2098. [Google Scholar]
- Jia, C.-Y., Li, J.-Y., Hao, G.-F., Yang, G.-F., 2019. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov. Today. [DOI] [PubMed]
- Kaliappan, S., Bombay, I.I.T., 2018. UCSF Chimera-Overview.
- Karplus, M., 2002. Molecular dynamics simulations of biomolecules. [DOI] [PubMed]
- Kim H.K. Rickettsia-host-tick interactions: Knowledge advances and gaps. Infect. Immun. 2022;90:e00621–e721. doi: 10.1128/iai.00621-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kräutler V., Van Gunsteren W.F., Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- Lionta, E., Spyrou, G., K Vassilatis, D., Cournia, Z., 2014. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr. Top. Med. Chem. 14, 1923–1938. [DOI] [PMC free article] [PubMed]
- Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lyu C., Chen T., Qiang B., Liu N., Wang H., Zhang L., Liu Z. CMNPD: a comprehensive marine natural products database towards facilitating drug discovery from the ocean. Nucleic Acids Res. 2021;49:D509–D515. doi: 10.1093/nar/gkaa763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macalino S.J.Y., Gosu V., Hong S., Choi S. Role of computer-aided drug design in modern drug discovery. Arch. Pharma. Res. 2015;38:1686–1701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massova I., Kollman P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000;18:113–135. [Google Scholar]
- Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Navid A., Ahmad S., Sajjad R., Raza S., Azam S.S. Structure based in silico screening revealed a potent acinetobacter baumannii Ftsz inhibitor from asinex antibacterial library. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2021;19:3008–3018. doi: 10.1109/TCBB.2021.3103899. [DOI] [PubMed] [Google Scholar]
- Oberoi A., Singh N. Rickettsiae infections-classification. JK Sci. 2010;12:57. [Google Scholar]
- Osterloh A. Vaccine design and vaccination strategies against rickettsiae. Vaccines. 2021;9:896. doi: 10.3390/vaccines9080896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oubahmane M., Hdoufane I., Delaite C., Sayede A., Cherqaoui D., El Allali A. Design of potent inhibitors targeting the main protease of SARS-CoV-2 using QSAR MODELING, MOLECULAR DOCKING, AND MOLECULAR DYNAMICS SIMULATIONS. Pharmaceuticals. 2023;16:608. doi: 10.3390/ph16040608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995;103:3668–3679. [Google Scholar]
- Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece S.T., Kaufmann S.H.E. Host defenses to intracellular bacteria. Clin. Immunol. Elsevier. 2019:375–389. [Google Scholar]
- Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Sexton, D.J., McClain, M.T., Edwards, M.S., n.d. Biology of Rickettsia rickettsii infection.
- Sussman J.L., Lin D., Jiang J., Manning N.O., Prilusky J., Ritter O., Abola E.E. Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D: Biol. Crystallogr. 1998;54:1078–1084. doi: 10.1107/s0907444998009378. [DOI] [PubMed] [Google Scholar]
- Talele T.T., Khedkar S.A., Rigby A.C. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010;10:127–141. doi: 10.2174/156802610790232251. [DOI] [PubMed] [Google Scholar]
- Turner, P.J., 2005. XMGRACE, Version 5.1. 19. Cent. Coast. Land-Margin Res. Oregon Grad. Inst. Sci. Technol. Beaverton, OR.
- Ursu O., Rayan A., Goldblum A., Oprea T.I. Understanding drug-likeness. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011;1:760–781. [Google Scholar]
- Van De Waterbeemd H., Gifford E. ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- Walker D.H. CRC Handbook of Viral and Rickettsial Hemorrhagic Fevers. CRC Press; 2019. The pathogenesis and pathology of the hemorrhagic state in viral and rickettsial infections; pp. 9–46. [Google Scholar]
- Walker D.H., Ismail N., Olano J.P., Valbuena G., McBride J. Pathogenesis, immunity, pathology, and pathophysiology in rickettsial diseases. Rickettsial Diseases. CRC Press. 2007:27–38. [Google Scholar]
- Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z.H., Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Wangtrakuldee P., Byrd M.S., Campos C.G., Henderson M.W., Zhang Z., Clare M., Masoudi A., Myler P.J., Horn J.R., Cotter P.A., et al. Discovery of inhibitors of Burkholderia pseudomallei methionine aminopeptidase with antibacterial activity. ACS Med. Chem. Lett. 2013;4:699–703. doi: 10.1021/ml400034m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareham D.W., Wilson P. Chloramphenicol in the 21st century. Hosp. Med. 2002;63:157–161. doi: 10.12968/hosp.2002.63.3.2061. [DOI] [PubMed] [Google Scholar]
- Whitty A. Growing PAINS in academic drug discovery. Future Med. Chem. 2011;3:797–801. doi: 10.4155/fmc.11.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C.J., Malaisree M., Hannongbua S., Mulholland A.J. A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies. J. Chem. Phys. 2011;134 doi: 10.1063/1.3519057. [DOI] [PubMed] [Google Scholar]
- Woods C.J., Malaisree M., Michel J., Long B., McIntosh-Smith S., Mulholland A.J. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014;169:477–499. doi: 10.1039/c3fd00125c. [DOI] [PubMed] [Google Scholar]
- Xiong G., Wu Z., Yi J., Fu L., Yang Z., Hsieh C., Yin M., Zeng X., Wu C., Lu A., et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49:W5–W14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava U. Search algorithms and scoring methods in protein-ligand docking. Endocrinol. Int. J. 2018;6:359–367. [Google Scholar]
- Yadava U., Singh M., Roychoudhury M. Pyrazolo [3, 4-d] pyrimidines as inhibitor of anti-coagulation and inflammation activities of phospholipase A 2: insight from molecular docking studies. J. Biol. Phys. 2013;39:419–438. doi: 10.1007/s10867-013-9299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerroug A., Belaidi S., BenBrahim I., Sinha L., Chtita S. Virtual screening in drug-likeness and structure/activity relationship of pyridazine derivatives as Anti-Alzheimer drugs. J. King Saud Univ. 2019;31:595–601. [Google Scholar]
- Zhang, X., Perez-Sanchez, H., C Lightstone, F., 2017. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 17, 1631–1639. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.