

Summary
Cytotoxic CD8+ T cells recognize and eliminate infected or cancerous cells. A subset of CD8+ memory T cells called tissue-resident memory T cells (TRM) resides in peripheral tissues, monitors the periphery for pathogen invasion, and offers a rapid and potent first line of defense at potential sites of re-infection. TRM cells are found in almost all tissues and are transcriptionally and epigenetically distinct from circulating memory populations, which shows their ability to acclimate to the tissue environment to allow for long-term survival. Recent work and the broader availability of single-cell profiling has highlighted TRM heterogeneity among different tissues, as well as identified specialized subsets within individual tissues, that are time and infection dependent. TRM cell phenotypic and transcriptional heterogeneity has implications for understanding TRM function and longevity. This review aims to summarize and discuss the latest findings on CD8+ TRM heterogeneity using single-cell molecular profiling and explore the potential implications for immune protection and the design of immune therapies.
Keywords: Tissue-resident memory T cell, CD8 T cell, immune memory, TRM heterogeneity
Introduction
The immune system plays a crucial role in protecting the body from pathogens and cancerous cells. Upon antigen encounter, CD8+ T cells proliferate rapidly and differentiate into a heterogeneous population of short-lived terminal effector cells (TE) and memory-precursor cells (MP) that mediate pathogen clearance. Following the resolution of infection, the population of effector T cells contracts. However, a small proportion of CD8+ T cells persists and forms memory cells, which provide long-lived protection. CD8+ memory cells have classically been studied in the blood and lymphoid organs. These long-lived circulating CD8+ memory cells can be subdivided into central memory cells (TCM) and effector memory cells (TEM) based on their surface marker expression, trafficking patterns, and functional capabilities upon reinfection1. While TCM cells are predominately found in the blood and in secondary lymphoid tissues and are characterized by a high proliferative capacity, TEM cells patrol the blood and can transiently enter tissues during acute infections.
However, during acute infection, a subset of CD8+ T cells migrates into tissues and gives rise to tissue-resident memory CD8+ T cells (TRM)2–4 that provide local protection. TRM constitute a large portion of the CD8+ T cell memory population and can remain lodged in tissues for the lifetime of the organism, without recirculating5,6. TRM cells scan tissues for pathogens and offer a rapid and potent first line of defense due to their enhanced effector functions and proximity to sites of reinfection5.
Defining tissue-residency
The strongest evidence that TRM reside in tissues without re-entering circulation and are a distinct memory T cell population arose from studies using parabiotic surgery in which the circulatory systems of two mice are joined. This procedure results in an equilibrium of circulatory T cells from the two animals. However, tissue-resident T cells did not reach homeostasis and remained tissue-specific in each mouse2,7–10. Studies in humans have exploited human leukocyte antigen (HLA)-mismatched allografts that have different HLA alleles between the organ donor and recipient to longitudinally follow donor-derived TRM cells in transplants. Using this approach, it has also been proven in humans that TRM cells can reside in organs for extended periods of time without draining to the lymph nodes or entering circulation11–13. Considering the experimental burden of performing parabiosis experiments in mice, many studies instead use an intravascular staining approach to mark T cells in the vasculature (thus inferred to be recirculating) with a fluorochrome-labelled antibody. T cells residing in tissues are not marked by the intravenous injection of the antibody, which has shown to faithfully reflect the residency of most antigen-specific TRM observed via parabiosis14. This strategy efficiently labels cells in the vasculature, but cannot discriminate between cells that reside in the tissue permanently and cells that only transiently enter the tissue. Nonetheless, for the purposes of this review, tissue-residency will be inferred via the absence of intravenous labelling.
Common adaptations of memory T cells to tissue-residency
Although TRM cells share some transcriptional features with TCM and TEM cells, they are transcriptionally and epigenetically distinct from circulating memory populations15–18. A transcriptomic analysis of TRM across many tissues compared to their recirculating counterparts allowed the definition of “core” tissue-residency and circulatory gene expression signatures, which have helped define the tissue residency state15–17,19. Tissue residency is achieved by upregulation of adhesion and retention molecules and chemokine receptors (CD103, CD69, CCR9) and by reduced expression of lymphoid homing molecules (S1PR1, CCR7, CD62L) that help T cells to leave non-lymphoid tissues.
Multiple transcription factors that orchestrate the tissue-adaptation process have been identified. For example, CCR7, CD62L, and S1PR1 are controlled by the transcription factor KLF2, the expression of which is downregulated during TRM differentiation3. S1PR1 binds to sphingosine-1-phosphate (S1P), which is highly abundant in the blood and promotes tissue egress. Downregulation of S1PR1 is, hence, a necessity for tissue-residency. CD69 is a c-type lectin protein that can form a complex with S1PR1 and inhibit binding of S1P to S1PR1, thus hindering egress from tissues of TRM cells3. A recent study showed that downregulation of S1PR5, another receptor for S1P, is essential for TRM formation in the skin20,21, where S1PR5-deficiency leads to enhanced TRM formation. Interestingly, S1PR5 does not interact with CD6921. In contrast to S1PR1, which is directly controlled by KLF2, S1PR5 is regulated via T-bet and Zeb220, demonstrating how tissue-residency is the result of multiple coordinated adaptation processes by T cells. Downregulation of S1P-receptors is accompanied by the downregulation of the T-box transcription factors T-bet and Eomes, which subsequently renders TRM cells responsive to TGF-β22,23. TGF-β signaling induces the expression of CD103, thus promoting TRM cell retention, especially in epithelial tissues24,25. Furthermore, transcription factors that facilitate expression of the tissue-residency signature, while suppressing circulatory-associated genes, include Blimp115, Hobit15, and Runx316. In summary, while there is not a single TRM-specific “marker,” TRM cells are commonly characterized by expression of two surface molecules: CD103 and CD69. However, the expression of these two canonical TRM cell-surface proteins in tissues is neither uniform within a tissue, nor uniform across different tissue-resident T cells17.
However, TRM cells do not constitute a homogenous memory T cell population. In fact, recent work has highlighted TRM heterogeneity within tissues and among tissues with differences in surface marker expression, transcriptional changes, functionality, and longevity17,26. TRM cells have been identified in almost every human and murine tissue and infiltration and long-term maintenance of these cells in non-lymphoid tissues requires T cells to acclimate to the specific environments that may differ in a broad range of ways, including the availability of nutrients, metabolite composition, cytokine milieu, cell composition, and matrix proteins. It is therefore not surprising that, although TRM cells share a common residency gene-expression signature, they also require tissue-specific acclimatization to persist in and patrol these unique and specialized environments. In addition to heterogeneity of TRM cells observed across different tissues, heterogeneity is additionally apparent within a single tissue, where TRM subsets resembling those observed in circulatory memory populations exist and change in their relative abundance over the course of an infection27. Furthermore, distinct infections can result in formation of different TRM populations; for example, TRM cells in the intestine that persist after Yersinia pseudotuberculosis or intravenous Listeria monocytogenes infection show different expression of cell surface proteins when compared to TRM cells that arise after acute lymphocytic choriomeningitis virus (LCMV) infection28–30.
Single-cell RNA sequencing (scRNA-seq) is a powerful approach that can reveal heterogeneity within cell populations and has been used extensively in recent years to probe the dynamic gene expression patterns of a wide range of immune cell types in health and disease1,31–33. scRNA-seq combined with reporter- and fate-mapping approaches has led to important advances in the understanding of TRM intra- and inter-tissue heterogeneity with implications for TRM cytotoxicity, function, longevity, and plasticity. In this review, we will summarize and discuss the latest findings on CD8+ TRM heterogeneity using single-cell molecular profiling and highlight the different levels of TRM heterogeneity as well as the potential implications for immune protection and the design of immune therapies.
Intertissue heterogeneity
Although most reports studying TRM cells focus on epithelial tissues like the intestine or the skin, TRM cells can be found in almost every organ. Besides the small intestine and the skin, TRM cells have, for example, been described in the kidney17,34, liver12,15, salivary gland (SG)17,35,36, adipose tissue17, pancreas2, stomach2, female reproductive tract6, lung37,38, and colon2,26. Although, TRM cells in these organs vary in their durability6 – ranging from a half-life of 82 days in the uterus to no decay in the salivary gland – they lodge in these tissues for long periods of time, highlighting the necessity of recently migrating T cells to specifically acclimate to their new environment as they become long-lived TRM39. Much work has focused on understanding how TRM cells become resident. Common adaptations include the downregulation of tissue egress molecules, such as S1PR1, and the upregulation of retention signals. However, considering that all organs fulfill distinct physiological roles, provide specialized tissue architectures, and have a unique composition of cells and structural tissue, it seems very likely that, in addition to common adaptations to tissue retention in general, TRM cells also need to make tissue-specific adjustments in gene expression, metabolic state, and homeostatic dependencies. In addition to a lineage CD8+ T cell-defining signature and memory T cell signature, CD8+ TRM cells need to acquire a “common” residency signature as well as tissue-specific adaptations that mediate long term survival and function in that environment (Figure 1).
Figure 1.
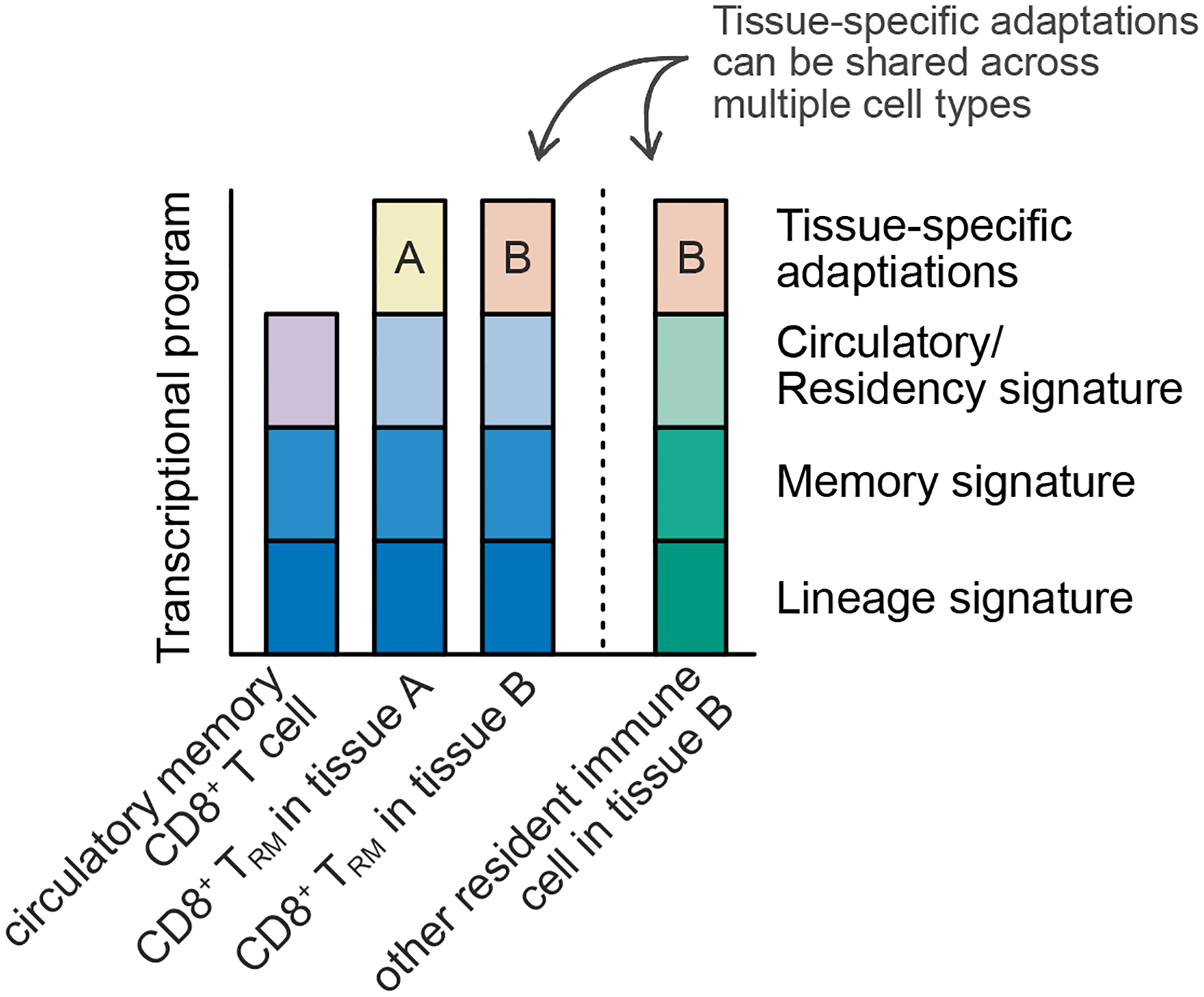
In addition to a shared lineage CD8+ T cell-defining signature and a memory T cell signature, TRM cells need to acquire a transcriptional residency signature. Long term survival and function within the tissue are mediated by tissue-specific adaptations of TRM cells that allow them to optimally acclimatize to their tissue of residence. These tissue-specific adaptations can be shared across multiple cell types.
Tissue-specific adaptations of other immune cell types that establish residency, such as tissue-resident macrophages, are well established and provide a basis for considering such changes for CD8+ TRM40–42. Specialized macrophages with distinct functions can be found in the spleen, skin (Langerhans cells), serosal tissues, lung, liver (Kupffer cells), gastro-intestinal tract, bones (osteoclasts), and the central nervous system (microglia)40. These tissue-specific adaptations are, by necessity, not reflected in the common TRM residency signatures observed when identifying the common adaptations of TRM originating from many tissues15,16. A careful comparison of gene expression across tissues in the same infection setting and the development of single-cell RNA sequencing (scRNA-seq) approaches has broadened our understanding of the differences in TRM populations, allowing differentiation between ubiquitous tissue-specific changes to the TRM cell population and changes in the abundance of shared, heterogeneous TRM cell populations17. How these specific adaptations are regulated is not completely understood, but may be initiated by distinct signals from cytokines, chemokines, cell-cell interactions, or metabolites, and could be reflected by unique patterns of transcription factor expression and lead to distinct expression patterns of surface proteins.
Cytokines required for Trm differentiation
In accordance with different transcriptional acclimatization of TRM cells to tissues, TRM cells require distinct cytokines to both start and sustain their differentiation process into tissue-resident memory cells. For example, TGF-β, a pleiotropic cytokine, has been well established in its importance for TRM formation in the skin, intraepithelial lymphocyte (IEL), and salivary gland, but it is not required for liver or kidney TRM formation17,22,23,25,36,43. Furthermore, it has been shown, that IL-15, a common gamma chain cytokine that promotes homeostatic proliferation and survival of memory cells44,45, is required for TRM survival in the skin22,46, liver47, salivary gland, and kidney48 but is dispensable for TRM homeostasis in the pancreas, female reproductive tract, and the small intestine in mice48,49. However, even if some TRM populations do not require IL-15 for maintenance, these cells still proliferate in response to IL-15, possibly to transiently amplify memory populations50. Not only do the different requirements for TRM development highlight the necessity for tissue-specific adaptation, but they might also indicate the existence of distinct TRM subsets that are driven and maintained by distinct cytokines and cell interactions.
TRM cells in different organs are dependent on distinct transcription factors
In line with the idea of tissue-specific adaptations of TRM cells, the transcription factors required for TRM differentiation differ among tissues. While some transcriptional adaptations (e.g., downregulation of KLF2) are shared among TRM populations, differential transcription factor dependence has been observed. For example, the transcription factor Hobit is required for skin, but not lung TRM generation15. Another example for tissue-specific transcription factors is the transcriptional repressor hypermethylated in cancer 1 (Hic1). Hic1 is induced during human iTreg differentiation51 and was shown to regulate homeostasis of intestinal lymphocyte populations in mice, thereby preventing the development of intestinal inflammation52. In LCMV infection, Hic1 expression by T cells is largely restricted to the small intestine (Figure 2), and knockdown of Hic1 specifically reduces TRM cells in the small intestine, while other organs remain unaffected17. In line with that, overexpression of Hic1 leads to a specific accumulation of TRM cells in the small intestine17. Mechanistically, this can partially be explained by the finding that Hic1 regulates expression of P2RX717, an extracellular ATP receptor that enhances metabolic fitness53 and contributes to small intestinal TRM survival53,54.
Figure 2.
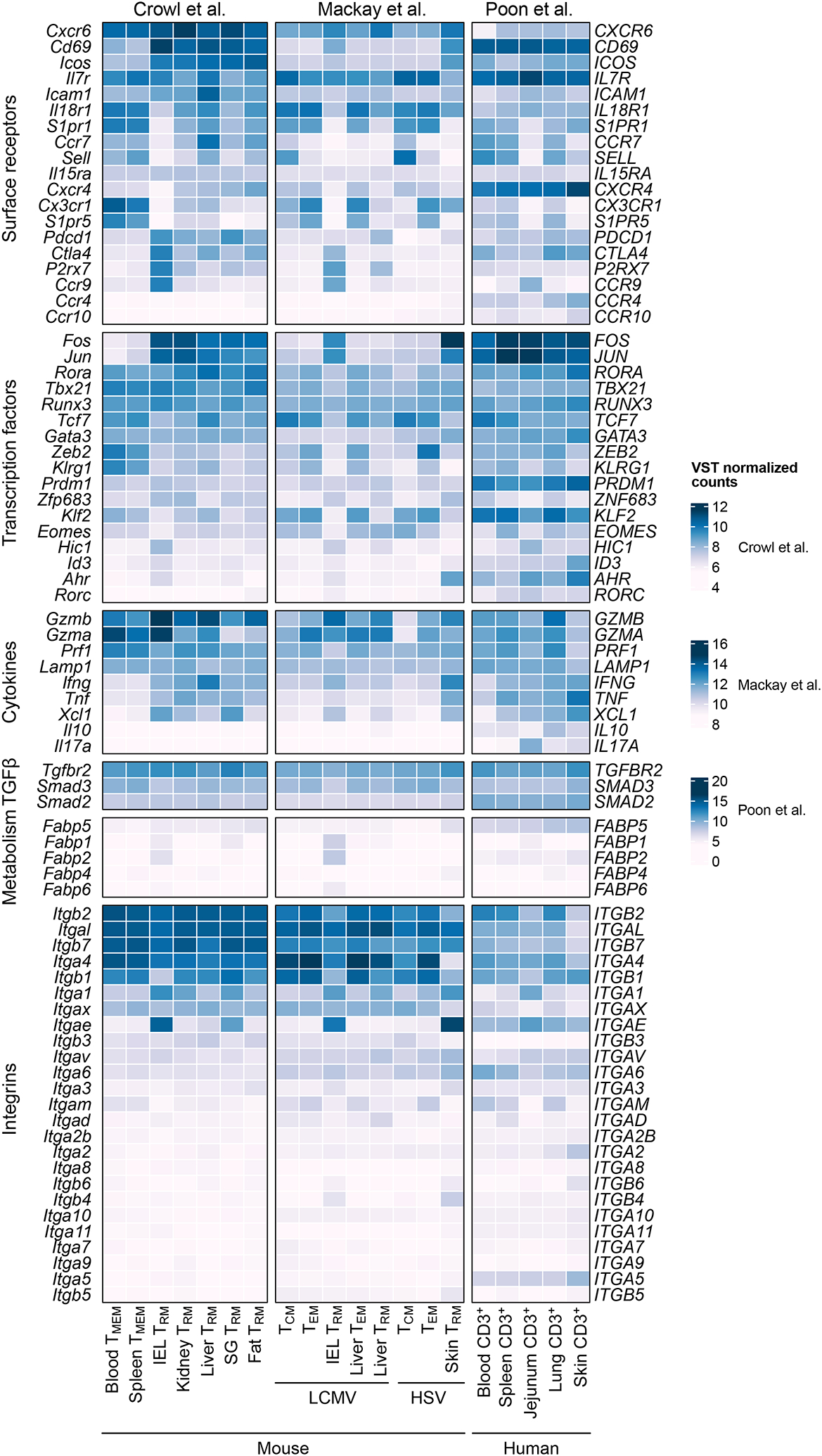
Expression of TRM molecules, lineage defining transcription factors, cytokines and integrins. Although TRM share common adaptation signatures, expression of TRM associated genes is heterogeneous across tissues and infections. Data from Crowl et al., 2022 (CD8+ P14 T cells isolated from spleen and blood (TMEM) and IV− CD8+ P14 T cells from IEL, kidney, liver, SG, and fat (TRM) at day 30 post infection with LCMV Armstrong, TMEM: all P14 cells); Mackay et al., 2016 (gp33 and np396 tetramer LCMV-specific CD8 T cell populations day 40 post LCMV Armstrong infection, and gBT-I cells 40 days post HSV infection); and Poon et al., 2023 (scRNA-seq of CD3+ T cells of two donors, aggregated to pseudo-bulk samples per donor). Mean expression values are plotted.
For generation of the heatmaps, data was downloaded from GEO (GSE182274, GSE70813, and GSE206507). For bulk RNA Seq data (GSE182274 and GSE70813), fastq files were processed using the nextflow pipeline (nf-core/rnaseq: 3.10.199). Variance stabilizing transformation (VST) was performed using DESeq2100. Mean VST values for selected genes were plotted using the R package ComplexHeatmap. For GSE206507 raw counts were downloaded from GEO and pseudobulk samples were created using the aggregateAcrossCells function from scuttle101 in R with the groups tissue and donor. Pseudobulk samples were then processed in DESeq2 and visualized with ComplexHeatmap as described above.
In addition to tissue-specific transcription factors, the dependency of TRM on TRM-defining transcription factors varies among tissues. Deletion of Runx3 reduces TRM cell numbers in multiple non-lymphoid organs, including in kidney and salivary gland, but its deletion leads to the strongest reduction of TRM cells in the small intestinal epithelium and affects CD103+ CD69+ TRM cells more compared to CD103− TRM cells16. Similarly, Blimp1 deletion impairs TRM formation in the IEL and SG more than the kidney17. Hence, tissue-specificity is not only achieved by the expression of tissue-defining transcription factors, but also by modulation of the expression of TRM-inducing transcription factors. It is likely that an interplay of both mechanisms leads to the optimal adaptation to tissues that TRM require for persistence. Therefore, further understanding and identification of tissue-specific transcriptional networks will be the basis for engineering tissue-programmed T cells.
TRM in different tissue sites are phenotypically distinct
Phenotypic comparisons of TRM in distinct tissues reveal tissue-specific patterns of expression of cell-surface receptors by TRM cells. While CD103 and CD69 expression, as well as loss of IL-18R expression, are commonly associated with and used for identification of TRM cells, substantial differences in expression of these molecules can be observed among different TRM populations. CD103 is an alpha integrin that, upon heterodimerization with a beta integrin, can bind to E-cadherin and facilitate retention in epithelial tissues. Thus, CD103 expression is mainly restricted to TRM cells associated with epithelial barrier tissues, including skin, small intestine, and salivary gland17,22,26,36. However, the integrin superfamily contains numerous subunits (with 18 alpha and 8 beta subunits)55, and a recent study shows that expression of the beta integrins differs substantially among tissues (Figure 2). For example, there are multiple differences in alpha- and beta-integrins between the small and large intestine: αE integrin expression is higher in the small intestine compared to the colon, whereas expression of α4β1 is higher in TRM cells from the colon26. These findings indicate that tissue-retention modules are specifically adapted to distinct regions of the intestine. CD69, which antagonizes S1PR1 and thereby hinders tissue egress, is widely used to identify TRM cells. However, the expression levels of CD69 in TRM cells and the functional relevance of CD69 for formation or maintenance of TRM cells varies vastly among tissues. In murine acute LCMV infection, CD69 expression is not needed for TRM formation in the small intestine, but necessary for kidney TRM cells, and forced expression of CD69 increases TRM formation in the kidney56. Downregulation of IL-18R is associated with the establishment of TRM cells in the kidney34. However, similar to CD103 and CD69, its expression varies among TRM cells from different tissues: IL-18R expression is lost in IV-negative TRM cells from the IEL after LCMV infection in mice but only partially downregulated in TRM cells obtained from kidney and SG, and it is still expressed in visceral-adipose tissue and liver TRM cells17. Ly6C expression in reduced in TRM cells compared to TCM cells49,57,58, but the expression of Ly6C also greatly depends on the studied tissue17,57. These findings demonstrate that the functional relevance of surface proteins commonly used for TRM identification largely depends on the tissue.
TRM cell in different tissues exhibit specific metabolic adaptations
Another level of specialization of TRM cells to their given tissue environment is reflected in their usage of fatty acid binding proteins (FABP)59 (Figure 2). TRM cells rely on fatty acid uptake for their survival60. However, the FABP family exists as a large family with many different isoforms that are expressed in a tissue-specific manner. Interestingly, TRM in different tissues express different isoforms of FABP, highlighting their specific adaptation to the tissue microenvironment59. Upon relocation, TRM cells adapt their FABP expression profile to the new location. Thus, isoform usage is determined by tissue-derived factors, and TRM cells actively and continuously sense their environment and adapt to it59.
TRM heterogeneity among organs in humans
Most studies of TRM heterogeneity among tissues have used murine models. Further, most studies rely on the adoptive transfer of T cell receptor (TCR)-transgenic antigen-specific T cells, and thus do not assess the contribution of a polyclonal endogenous T cell response and the relevance of the TCR clonotypes to TRM cells. A recent report, however, addresses these limitations and extends our knowledge of human tissue-resident T cells61,62. In this study, lymphocytes from the lung, jejunum, abdominal skin, their draining lymph nodes (LN) (pulmonary LN, mesenteric LN, and inguinal LN), and from blood and spleen were collected and used for a comprehensive profiling with CyToF, TCR-sequencing, and single-cell RNA sequencing. In accordance with findings from the murine TRM models, the authors found a unique composition of CD4+ and CD8+ TRM cells in the assessed barrier tissues: T cells lodged in the skin had high expression of CCR4, CCR10 and CXCR4, and the Th2 lineage-defining transcription factor GATA3; T cells that reside in the intestine exhibited a high expression of integrins (ITGAE, ITGA1, ICAM1), and T cells isolated from the lung displayed a site-specific expression pattern of CTLA4, IL10, and PDCD161 (Figure 2). In addition to location-specific expression patterns, the authors identified common adaptations among all barrier tissues and expression patterns that are shared among only some tissues. For example, T cells in the jejunum and lung were characterized by high expression of CXCR6 and Th17 signatures genes (CCL20, RORA, RORC, and IL17A)61, and TRM cells in the lung and skin showed increased expression of genes encoding matrix- and adhesion-associated molecules as well as PRDM1 encoding for BLIMP1, a transcription factor associated with tissue-residency and effector functions. These transcriptomic changes may indicate the specific needs of T cells to maintain immunity against various types of pathogens in threatened tissues throughout human life. Importantly, many of these site-specific adaptations are by human and mouse T cells; for example, expression of ITGAE, CCR9 or HIC1 and downregulation of KLF2 in the intestine or high expression of AHR in the skin. These findings indicate that some of these transcriptional profiles are conserved across species (Figure 2). However, expression of some genes is not conserved; for example, T cells in the jejunum exhibit elevated expression of IL17A in humans, but not in mice. These differences might arise from differential exposure of humans to pathogens (in contrast to mice in a specific-pathogen free environment), but also might reflect variance due to polyclonal CD3+ T cells for human versus TCR transgenic CD8+ T cells for mice or additional differences. Considering an individual’s age and history of infection also adds a new layer of complexity to understanding TRM phenotype, function, differentiation, and maintenance in humans. Insight into these topics is essential to improve targeted and tissue-adapted immune responses at barrier tissues in humans.
Clonal relationships between circulatory and tissue-resident memory T cells
How T cell clones that contribute to circulatory memory populations are proportional to tissue-resident T cell populations and whether or not the TCR clones found in TRM population are unique to the TRM populations or even the specific tissue are all topics of great interest. Using TCR sequencing, a recent report described that certain TCR clones expand more in barrier sites compared to circulatory memory populations, indicating that the TCR pool in circulation is not representative of that seen in tissues61 — an observation that has important implications in numerous areas, such as vaccine design and boosting strategies63. This finding is corroborated by a report using clonal tracing of T cell clones that showed that graft versus host disease (GvHD) in a murine model is maintained by expansion of a tissue-residing TCF1+ subpopulation and not by recruitment of T cells from secondary lymphoid organs or the blood64. Furthermore, in humans, TCR clones for CD4+ TRM cells segregated between the barrier tissues with only minimal overlap between the skin, lung, and jejunum, where CD4+ T cell clones are less disseminated and more site-specific compared to CD8+ T cell clones61. It is possible that this clonal segregation is a result of different infections or pathogens that T cells encounter at these three distinct barrier sites, but the possibility that certain TCR clones preferentially give rise to TRM cells in a specific organ cannot yet be ruled out. However, even within a naive T cell population that expresses the same TCR, a clonal population that possesses a heightened potential to form TRM cells was shown to exist65. In this study, barcode labelled naive OT-I T cells were transferred into recipient mice, and TRM cells were induced by vaccination or herpes simplex virus (HSV) infection. While T cell clones contributed proportionally to circulatory and skin effector T cells, a disparity in T cell clone distribution was seen at memory timepoints65, suggesting that distinct T cell clones do preferentially give rise to TRM cells. Interestingly, there is evidence that these T cell clones might become preconditioned towards their TRM fate even before entering the tissue65,66.
Intratissue heterogeneity
The heterogeneity of cell surface receptor expression and overall gene expression indicated by scRNA-seq within a population of TRM cells from a given organ varies greatly. For example, TRM cells in the dermis are heterogeneous in their expression of CD69, with only approximately 70% of the dermal TRM cells expressing CD69, whereas almost all TRM cells in the epidermis express CD6922. In contrast, the majority of TRM cells in the dermis do not express CD103, but the expression pattern of CD103 on epidermal TRM is heterogeneous22. The heterogeneous expression of surface proteins of TRM within a tissue is not limited to the skin and has also been observed in other tissues. TRM in the salivary gland show a heterogeneous expression for both CD69 and CD10317,36, and kidney TRM are characterized by an intermediate expression of CD69 but almost no expression of CD10317,34 (Figure 3a). Similarly, liver-resident TRM exhibit low to intermediate expression of CD69 and no expression of CD10317,36. Similar findings are observed in human studies—whereas most of the intestinal T cells are positive for CD103 and CD69, expression of these two tissue-residency promoting molecules is heterogeneously expressed by T cells resident in the lung or skin and also varies between CD8+ and CD8− T cells (Figure 3b). While most small intestinal intraepithelial TRM cells in mice express CD103 and CD69 after acute LCMV infection, only about 60% of small intestinal TRM cells from the lamina propria, and approximately 30% of colonic TRM cells, express CD103 and CD6926. In addition to CD69 and CD103, other TRM markers and transcription factors including Il18r, Cd49a, Ly6c, Tcf1, T-bet, and Eomes have been shown to exhibit variable expression within the tissue as well as among tissues17,23,26,34,36. The expression levels of these canonical TRM molecules are further subject to changes over time: e.g., expression of CD103 and CD69 in small intestinal TRM cells increases from the effector phase to memory timepoints, whereas expression of IL-18R decreases. Hence, the time after infection adds a new dimension to TRM heterogeneity. This idea is further supported by the finding that the importance of TRM-driving factors can vary depending on the time after infection. For example, the inducible costimulator (ICOS) receptor is essential for TRM establishment, but not maintenance67, and the transcription factor EOMES is not required for formation of small intestinal TRM, but is needed for their long-term maintenance26. In summary, these findings support the hypothesis that different subtypes of TRM exist and that the composition of the TRM pool might vary over time.
Figure 3.
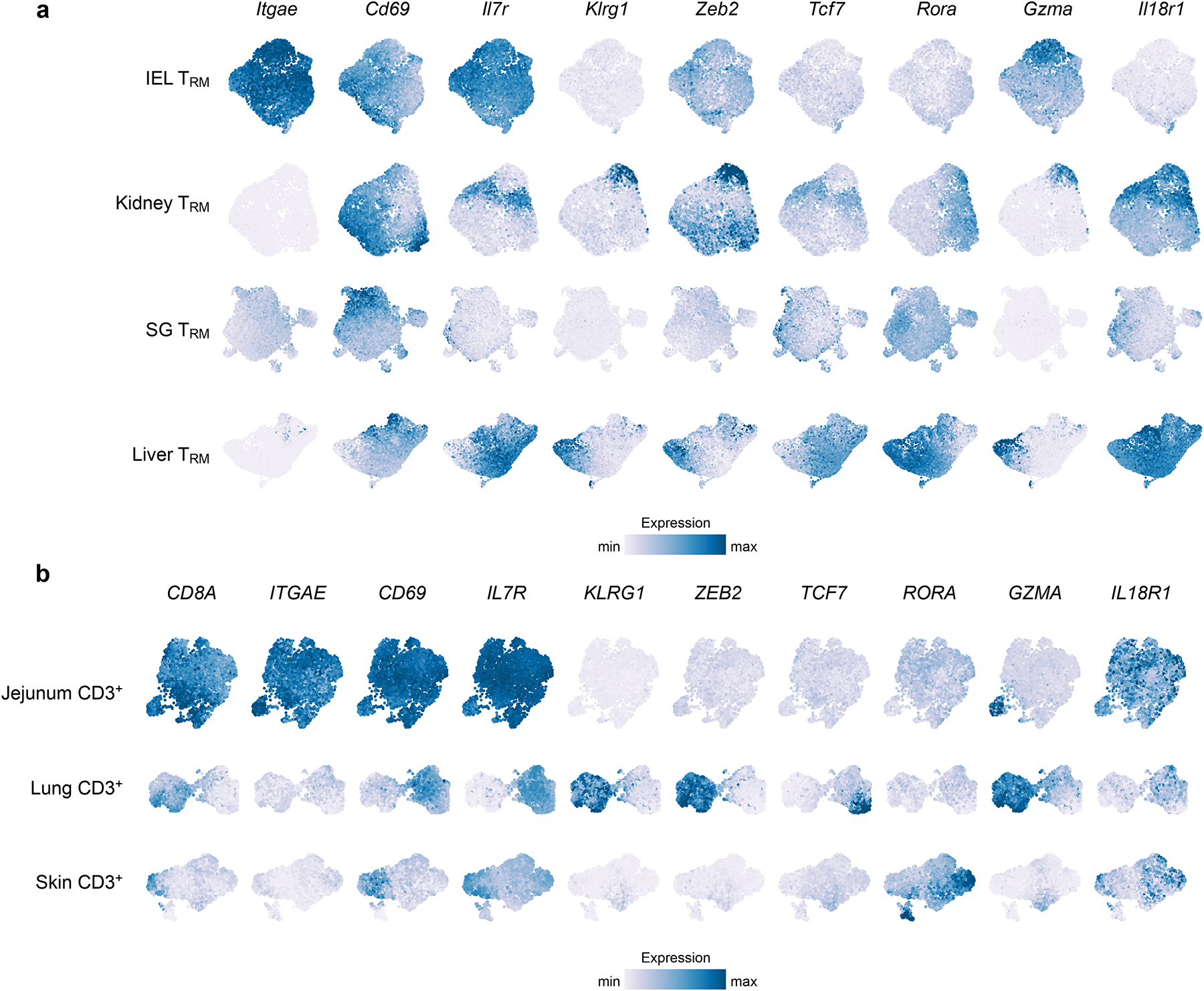
scRNA-seq reveals intertissue and intratissue heterogeneity. TRM cells within a single tissue display varying expression of genes associated with effector and memory potential, showing that distinct subsets of TRM cells exist. Scales for each gene are consistent across tissues to allow for comparison within and among tissues. Data from (a) Crowl et al. 2022 (scRNA-seq of murine IV− CD8+ P14 T cells from IEL, kidney, SG, and liver at day 32 post infection with LCMV Armstrong) and (b) Poon et al., 2023 (scRNA-seq of human CD3+ T cells of two donors). Donor D551: Jejunum and Lung; Donor D492: Skin.
For visualizing the intra-tissue heterogeneity, each tissue dataset (downloaded from GEO GSE182275 and GSE206507) was normalized separately using sctransform with method = “glmGamPoi” in Seurat102. Dimensional reduction was performed using the RunPCA function in Seurat following by RunUMAP using the first 20 PCAs. In addition, data imputation was performed using MAGIC103 using the default settings and the exact solver. Manually selected genes to highlight inter- and intratissue heterogeneity were plotted. Color scale was limited to the 98th expression percentile.
Parallel subsets between circulatory memory and tissue-resident memory T cells
The circulatory memory T cell compartment is phenotypically and functionally heterogeneous, with effector and memory T cell populations consisting of phenotypic and functional subsets that evolve over time. Terminally differentiated, short-lived effector cells (CD127−KLRG1+) and memory-precursor cells (CD127+KLRG1−) are predominantly found during the effector phase of infection68,69. These cells continue to differentiate over time and form circulatory memory T cells that can be broadly divided into TCM and TEM, which derive from memory precursor cells and t-TEM cells that come from KLRG1hi effector cells. Recent data show that intratissue heterogeneity of TRM populations reflects TRM subsets that functionally resemble those observed in circulation27,31,70.
Two distinct subsets of TRM cells that mirror those seen in circulatory memory populations have been identified27. While small intestinal T cells showed an enrichment for effector gene-signatures early after an infection, gene-expression at later timepoints was enriched for memory signatures, highlighting a continuous differentiation of TRM cells over time. Among the transcriptional regulators with differential expression over the course of infection were Blimp1 (Prdm1) and Id327. Id3 is an inhibitor of E protein transcription factors. It regulates long-lived circulatory memory formation in CD8+ T cells71, and is most highly expressed in small intestinal TRM cells at late time points after infection. In contrast, Blimp1, which marks terminally-differentiated T cells, displayed an inverse expression pattern in studies, peaking at day 4 of infection and subsequently declining in small intestinal TRM cells27 and skin TRM cells15. Using Blimp1 and Id3 reporter mice, two distinct subsets of small intestinal TRM cells were observed: Blimp1hiId3lo TRM cells expressed canonical effector genes (i.e., Cx3cr1, Zeb2, Klrg1, Gzma and Gzmb), and, in contrast, Id3hiBlimp1lo TRM cells expressed genes reminiscent of memory-precursor cells (i.e., Bach2, Tcf7, and Cd27)27. Furthermore, Id3hi TRM cells have shown elevated degranulation capacity and cytokine production of IFNγ, TNF, and IL-2 and yield a greater frequency of both circulating and resident secondary effector T cell populations after rechallenge compared to the Id3lo subsets27. Thus, Id3hi small intestinal TRM cells exhibit multifunctionality and memory potential, which is consistent with the greater proliferation capacity of Id3hi circulatory memory T cells.
Analysis of TRM differentiation in tissues has largely relied on bulk RNA sequencing and a small number of phenotypic markers, limiting the ability to discern functional differences and intermediate states of differentiation that might arise within the TRM population15,16. scRNA-seq overcomes this limitation and has identified clear functional and phenotypical subsets among TRM cells within the same tissues and highlighted similarities to the memory differentiation of memory CD8+ T cells in blood and secondary lymphoid organs. A longitudinal scRNA-Seq study31 of the T cell response to the Armstrong strain of LCMV (LCMV Armstrong) analyzed small intestinal TRM in comparison to circulatory memory cells and found a transcriptional memory signature that is used by both circulatory and tissue-resident subtypes of memory CD8+ T cells throughout their differentiation31. In addition to this shared signature, intestinal TRM cells exhibited a distinct signature, which may contribute to their adaptation to the intestinal environment (Figure 1, tissue-specific adaptations). Importantly, by focusing the analysis on TRM cells, the study also showed that the TRM population is heterogenous throughout the course of an infection and exists in multiple clusters, many of which were present at several time points. These findings suggest that multiple subtypes of TRM cells exist. For example, at day 60 of infection, two distinct types of clusters existed within the small intestinal TRM population: one with higher expression of memory-associated transcription factors, such as Id3, Jun, Fos, and Klf2, and higher expression of Il7r (encoding for CD127), and another with expressed transcription factors, such as Zeb2, that might indicate a more terminally differentiated cell state and have been shown to promote terminal differentiation of circulatory effector and memory T cells72 and lower expression of Il7r31. The difference between these two TRM subsets was also highlighted by higher cytokine production after restimulation with a cognate antigen in the CD127high TRM cells compared to CD127low TRM cells. Therefore, these subsets seem to phenotypically and functionally mirror the Id3hi and Blimp1hi subsets that were identified using reporter mice and confirm the presence of functionally relevant TRM subsets that change in their relative abundance over time.
A recent study using flow-cytometry and scRNA-seq to analyze donor-derived TRM cells from intestinal transplant recipients reveals substantial heterogeneity within the TRM compartment in a single tissue can be observed in humans as well13. Remarkably, by exploiting HLA-mismatches between the organ donor and recipient, the authors prove that donor-derived TRM can survive in the organ graft for at least 5 years following intestinal transplant13. Much like the mouse TRM compartment, two distinct populations were observed among the donor-derived intestinal TRM cells: a first population expressed higher levels of ITGAE, IL7R, KLRB1 and CCR6, indicating a more memory-like cell state, and a second population expressed higher levels of KLRG1, cytotoxic granules and the transcription factor ZEB2, thus resembling more terminally differentiated effector T cells13. Extending the characterization of intestinal TRM to the disease state, a different study analyzing intestinal T cells from patients with ulcerative colitis and healthy controls revealed enrichment of TRM-like cells with inflammatory properties (higher expression of ZEB2, TBX21, and PRF1) compared to controls70. These data suggest, that TRM cells normally exist in an equilibrium of multiple distinct differentiation states and that an imbalance of these TRM differentiation states might be associated with, or even causative for, autoimmune-mediated diseases such as ulcerative colitis.
A recent complementary study highlights heterogeneity within the human skin TRM population61. Using scRNA-seq of skin-resident T cells from two individuals, the authors were able to identify clusters of skin TRM cells that were transcriptionally poised towards Th1-like (TBX21), Th17-like (RORA), and cytotoxic (GZMA, GZMK, NKG7, PRF1, IFNG) responses61. The concept of skin TRM heterogeneity is further corroborated by previous studies that identified functionally and phenotypically discrete populations of resident and recirculating memory T cells in the skin73,74. CD103− TRM cells mainly localized in the dermis and a higher proliferative capacity compared to CD103+ TRM cells, which were enriched in the epidermis and had higher effector functions73.
In summary, these studies support the idea that multiple subsets within the TRM population with distinct functional capacities and transcriptomic programs exist in both mice and humans. This heterogeneity observed within the TRM population is also likely reflected in the TRM-mediated protection from secondary infection. More cytotoxic TRM cells might preferentially act on infected cells, and their reduced proliferation capacity might limit immunopathology caused by activated TRM cells. In contrast, TRM cells with higher stemness or “memory potential” might replenish and sustain the TRM pool over longer time periods. In line with this idea, recent studies have identified that CD103− TRM cells have the capacity to replenish and augment the CD103+ TRM pool upon a secondary infection75–77. Given the strong evidence for distinct subsets within intestinal TRM cells, it will be interesting to see which factors drive and maintain differentiation of these subsets. It is tempting to speculate that these cells might be localized in different areas of the tissue and that the tissue microenvironment, the level of inflammation, the abundance of antigens, the presence of cytokines, chemokines, and metabolites, and the interactions of T cell partners might govern the TRM cell adaptation to the tissue to allow for optimal tissue surveillance.
Infection-mediated variation in tissue-resident memory T cells
Infection type dictates the TRM phenotype
In addition to the environmental influences derived from different tissue that may regulate TRM formation and maintenance within a tissue5,49, the type of infection also shapes the outcome of TRM differentiation. For example, LCMV Armstrong causes an acute and systemic infection that leads to formation of TRM populations in many tissues. In this infection setting, the TRM population within the small intestine almost entirely expresses CD69 and CD103. In contrast, following Yersinia pseudotuberculosis infection, two distinct TRM populations form in the small intestine28: a CD103+ TRM population and a CD103− TRM population. The CD103+ subset constitutes the majority of TRM cells in the intraepithelial layer, whereas both populations are equally abundant in the lamina propria. Unlike CD103− TRM cells that develop in the absence of TGF-β, cluster around areas of bacterial infection, and are enriched in the lamina propria, CD103+ cells require TGF-β for their development and are scattered throughout the intestine28. Importantly, CD103− TRM cells in this setting are not precursor cells of CD103+ TRM but represent a distinct population that is stable over time. It has been shown that a single infection can give rise to two independent CD8+ TRM populations and that the phenotypic composition of the TRM population induced by different pathogens may reflect the unique signals associated with the infection type.
Route of infection alters the TRM phenotype
The phenotypic and transcriptional features of TRM cells are not only a result of the infection model used, but also depend on how the pathogen is administered. For example, oral infection with a mouse-adapted Listeria monocytogenes strain that contains a mutation in the internalin A protein to facilitate invasion of murine epithelial cells efficiently induced TRM cells in the small intestine30. However, mice infected with the same bacterial strain via an intranasal method, had fewer intestinal TRM cells, and these cells did not fully convert to a memory phenotype78. Similarly, infection of mice intranasally with an influenza virus expressing ovalbumin did not induce memory cells in the intestine30. Thus, the route of infection, and thereby the location of T cell priming, dictates where and to what extent TRM cells form in peripheral tissues.
Another study comparing how T cell priming in the spleen versus T cell priming in the mesenteric lymph node contributes to T cell differentiation into CD103+ intestinal TRM cells29,79 provides further evidence that the location of T cell priming regulates TRM formation. Comparing intravenous infection, where T cells were primed in the spleen, to foodborne infection with Listeria monocytogenes, where T cells were primed in the mesenteric lymph node, revealed that only mesenteric lymph-node-primed T cells give rise to CD103+ intestinal TRM cells29. This difference in TRM licensing is attributed to the presence of retinoic acid in the mesenteric lymph node, which regulates expression of intestinal homing receptors, such as CCR9 and α4β7-integrin, as well as genes that are part of the TRM gene-expression signature (Hic1, Xcl1, Itgae, and P2rx7)29.
These findings are of direct therapeutic relevance. While intramuscular, intravenous, or intranasal administration of an mRNA vaccine targeting the influenza A virus nucleoprotein were all capable of establishing memory cells in the lung and draining lymph nodes, the combination of intramuscular priming with an additional intranasal boost archived the highest frequencies of lung TRM cells38. This “prime-and-spike” approach has also been successfully used in mouse models of COVID-19 infection63, but, surprisingly, it was not able to induce an effective immune response in a human phase I trial80. It is not entirely clear why these differences occur, but it might be partially explained by different dosages of the vaccine or the way the vaccine is administered80. However, these results highlight the need for a better understanding of how TRM can be induced at specific tissue sites for therapeutic purposes. Thus, both tissue microenvironment and type of infection, and, therefore, inflammatory microenvironments, significantly impact TRM development in response to acute infection and have direct implications for vaccination strategies to archive infection experience and induce optimal tissue protection.
Common adaptations of immune cells to tissue residency in specific tissues
In addition to CD8+ TRM cells, many other immune cells, including CD4+ T cells, innate lymphoid cells (ILCs), macrophages, and natural killer (NK) cells, form tissue-resident populations similar to those described for CD8+ T cells15,16,40,41,81–84. Interestingly, some of the transcriptional programs inducing tissue residency and inhibiting tissue egress seem to be shared among the different tissue-resident immune cell types. For example, Blimp1 and Hobit have well-established roles in both CD8+ TRM and NK and NKT tissue-resident memory cells15, and suppression of Klf2 and S1PR1 and upregulation of Hobit is shared between CD4+ and CD8+ TRM cells84. These findings suggest that mechanisms for immune residency are shared between different immune cells and could reflect the adaptation to the unique environment of a specific tissue.
Runx3, which drives TRM formation in CD8+ TRM cells through a TGF-β-dependent transcriptional mechanism, is not required for tissue residency of CD4+ TRM cells in the dermis, which instead relies on Runx116,85. This discrepancy might, in part, be explained by different localization and expression profiles of CD8+ versus CD4+ TRM cells in the skin. CD4+ TRM express less CD103 compared to CD8+ TRM cells, preferentially reside in the dermal layer of the skin, and display a higher dynamic movement compared to CD8+ TRM cells86. Notably, overexpression of Runx3 in CD4+ TRM cells changes the localization of CD4+ TRM in the skin with a preference to the epithelium, similar to CD8+ TRM cells, and enhances effector functionallity85. These findings show that ectopic expression of Runx3 in CD4 T cells induces a residency program similar to that seen in CD8 TRM cells, which is usually not acquired by CD4 TRM cells due to the lack of Runx3 expression.
In addition to a core residency program shared among tissues, organ-specific adaptations seen in TRM cells are also mirrored by other immune cell types (Figure 1). For example, the transcription factor Hic1, which was first identified as a tumor suppressor in human cancers87,88, is relatively specifically expressed in intestinal TRM cells17. Interestingly, the intestine-specific expression pattern of this transcription factor is also seen in other immune populations, including innate lymphoid cells (ILCs), CD4+, Treg, and macrophage resident cells17,52,89,90: Loss of Hic1 reduced accumulation of T cells and ILC3 in the lamina propria and intraepithelial lymphocyte compartments52,89. These findings suggest, that Hic1 could serve a broad role in establishing or maintaining tissue-residency in the small intestine and that its function is shared among many cell types. More generally, these findings support the idea that tissue-specific adaptations can be shared across multiple types of resident immune cells (Figure 1).
In a similar example of common adaptations of immune cells to a tissue, the tissue-specific isoforms of fatty acid binding proteins (FABP) of CD8+ TRM cells mimic the expression pattern seen in other resident immune cells for skin, small intestine, and liver59. Thus, even though resident immune cells exhibit distinct differentiation trajectories and are derived from distinct precursor cells, they can share common adaptation methods to the tissue they reside in. Intriguingly, understanding tissue-specific adaptations in one resident population might be extrapolated to other immune cells. Considering that an effective immune response requires coordination of many different immune cells, each with a specific function, the ability to boost tissue-acclimatization of multiple immune cells simultaneously within a given tissue could be a promising therapeutic concept. In contrast, inhibiting tissue influx of immune cells by inhibiting common adaptation processes or, conversely, directing regulatory T cells to a tissue, could be an effective therapy for autoimmune and immune-mediated diseases, highlighting the need to further understand how immune cells acclimatize to tissues.
TRM heterogeneity is reflected in T cell function
The pathognomonic function of memory cells is to protect from reinfection, where the memory cells re-encounter their cognate antigen. Among the memory cell population, TRM cells provide the first line of defense at barrier tissues. Upon activation, TRM cells not only produce cytokines, such as IFNγ or TNF, but also, and maybe even more importantly, they can alert the neighborhood and recruit circulating lymphoid and non-lymphoid cells to the tissue to aid in the response49,78,91. Importantly, TRM cells can rapidly proliferate, traffic to regional lymph nodes, and even contribute to secondary circulatory memory populations26,36,92–94. Thus, TRM cells exhibit features of effector cells as well as features that are reminiscent of TCM cells.
By comparing TRM cells from the small intestine to the colon after acute LCMV infection in mice, a recent study identified higher expression of granzyme A and B in small intestinal TRM cells compared to TRM cells residing in the colon26. However, the highest expression of IFNγ was observed in colon TRM cells and for both the small intestine and the colon, IFNγ was higher in intraepithelial TRM cells compared to TRM cells located in the lamina propria26. Importantly, this functional heterogeneity among intestinal TRM cells did not appear to be primarily driven by differences in CD69 and CD103 expression26 but reflected the specific functional adaptation of cells to their residing tissue.
The functional capacity of TRM cells is diverse among tissues and even among subsets within a distinct tissue. This diversity adds another layer of complexity to understanding how TRM adapt to barrier tissues and optimally respond to different types of antigens to sustain a long-lived memory population.
TRM in secondary infections
Memory T cells are functionally specialized to provide enhanced protection against subsequent infection. TRM cells locally surveil non-lymphoid tissues and provide the first line of defense upon reinfection at barrier sites. TRM can undergo in situ proliferation48,92–95 independent of help from CD4+ T cells93 and can autonomously amplify local immune surveillance and memory in various barrier sites in response to a range of stimuli93.
Secondary systemic adaptive immune responses are commonly attributed to circulatory memory cells. Recent reports suggest, however, that TRM cells themselves can undergo retrograde migration and substantially contribute to secondary circulatory memory cells, thus forming an “outside-in” immune response36,92,94. For example, in one experiment, congenically distinct OT-I TRM engrafted skin was transplanted to infection-matched mice, and TRM cells were then reactivated locally with their cognate peptide. After 2–3 weeks, ex-TRM cells were detectable in the draining lymph node and gave rise to circulating TCM and TEM cells. Interestingly, this egress from the non-lymphoid tissue could be reduced by treatment with FTY720, a S1P-receptor modulator, suggesting that the tissue egress is at least partially mediated by S1P92. In contrast, another report studying the recall response of skin TRM cells did not find a contribution of TRM cells to the circulatory memory response95. In this study, HSV-specific T cells were injected into mice, and skin-resident TRM cells were induced with DNFB, which acts as a non-specific stimulus to recruit T cells into the skin. Thereafter, these mice were rechallenged with local HSV infection95. In this experimental setting, local proliferation of TRM cells that provided localized protection from HSV-infection could be observed. However, skin TRM cells were constrained to their location and did not migrate towards the lymph node after HSV infection95. It is unclear, if these differences were a result of the different infections used or how TRM cells were induced, but these findings highlight the diversity of site-specific immunity.
Most studies focusing on TRM “stemness,” or ability to contribute to secondary responses and memory formation, rely on adoptive transfer experiments of TRM cells into new recipient mice. Using the transfer TRM has revealed that small intestinal intraepithelial TRM cells can lose the expression of their hallmark molecules CD69 and CD103 and transdifferentiate to secondary circulatory memory populations26,36,49,92,94. Phenotypically, these cells mostly give rise to TEM-like memory cells (as identified by CD127+CD62L−), but few ex-TRM cells also upregulate L-selectin (CD62L), a marker that is classically expressed by TCM cells49,92. After transfer of equal numbers of TRM, TEM, and TCM cells into freshly infected recipient mice, cell numbers of secondary circulatory memory cells derived from ex-TRM cells were higher than those derived from ex-TEM, but lower than those derived from ex-TCM cells. Thus, TRM cells can display “developmental plasticity” and are not terminally-differentiated92. This finding is also supported by a machine-learning approach trained on a whole-genome bisulfite sequencing dataset using naive T cells as a reference for the highest stemness and exhausted T cells as a reference for the lowest plasticity92. The authors found that TRM cells had an intermediate plasticity score between TEM and TCM cells. However, despite their ability to contribute to secondary memory populations, these ex-small-intestinal TRM cells continued to exhibit phenotypic traces of their tissue origin and had a higher expression of Ly6C and CCR9 compared to ex-TCM and ex-TEM cells. In line with this, and despite their phenotypic plasticity, small intestinal ex-TRM cells retain a bias to their tissue of origin92.
Additional studies comparing the capacity of TRM cells from different tissues to contribute to secondary memory responses found substantial differences between skin ex-TRM cells and liver ex-TRM cells: skin ex-TRM cells were unable to expand and repopulate the splenic or hepatic T cell pool to the same extent as ex-liver TRM cells36. This difference is partially explained by the finding that TRM cells from the skin depend on TGF-β, whereas liver TRM can form independently of TGF-β. Furthermore, using a tamoxifen-inducible knockout of TGFBRII in TRM cells after acute LCMV infection, has shown that TRM cells in the small intestine and salivary gland require ongoing TGF-β signaling, whereas TRM cells in the kidney, liver, and visceral adipose tissue can persist without TGF-β17. This finding suggests that TGF-β imprinting may be one underlying mechanism repressing TRM memory potential. This is further supported by the finding that TGF-β-dependent CD103+ salivary gland ex-TRM cells are disadvantaged in repopulating the splenic memory population compared to their CD103− counterparts, which do not require TGF-β36, and also by a report showing that TGF-β-dependent TRM formation limits the response to tumor vaccines96. Reflecting the intra-tissue heterogeneity of TRM cells, it has been shown that the Id3high TRM populations, which exhibit increased multifunctionality, yield a greater frequency of circulatory resident populations after rechallenge compared to their Blimp1high counterpart27. This study showed that the observed heterogeneity within the TRM populations is functionally relevant in primary and secondary immune responses and that different TRM subsets may be differently poised to contribute to secondary infection.
In conclusion, TRM cells need distinct signals, cytokines, and transcriptional programs to adapt to their specific niche in their tissue of residence. This leads to specialized TRM cells that exhibit different phenotypic traits, distinct functions, and also different potential to contribute to secondary infection response. Hence, by both studying the differences and commonalities between TRM from a broad range of tissues, we can infer what makes a TRM cell responsive to secondary infections. It will be interesting to see how tissue-specific adaptations by primary TRM cells influence their ability to respond to secondary infection and contribute to secondary memory and whether tissue-specific adaptations require a trade-off with future memory potential.
However, one main limitation of analyzing the contribution of TRM cells to secondary infections by transferring sorted populations into new hosts is that TRM cells are subjected to harsh methods of isolation that might alter their biology due to the necessity of isolating them from the tissue. Further insights were gained by studies focusing on secondary TRM responses in situ. Using intravital mucosal imaging, TRM cells were found to proliferate within their residing tissue upon antigen restimulation, and it was found that the pool of secondary TRM cells was mostly derived from primary TRM cells independently of TCM cells and proliferation in lymphoid tissues93. Furthermore, using reporter and fate-mapping mouse models has brought many new insights into how TRM cells within barrier sites respond to reinfection. These reporter mice models rely on genes that are uniquely expressed in TRM cells and are hence used to mark and track TRM cells during secondary infection without the need to isolate the memory cells, disrupt them from their residing environment, and transfer them into a new host.
For example, the transcription factor Hobit can be expressed in TRM cells in the liver, the small intestinal IEL, and LPL TRM cells, but not in circulatory or mesenteric lymph node memory T cells15. Using a Hobit-reporter mouse (tdTomato integrated into the Hobit locus) and a Hobit fate-mapping mouse (Hobit-Cre R26-LSL-eYFP), it has been confirmed that reinfection causes local expansion of TRM cells. These cells can also drain into lymph nodes and contribute to circulatory memory where they resemble TEM cells expressing KLRG1 and CX3CR1, and only a small proportion expresses the TCM marker CD62L94. These secondary ex-TRM cells downregulate the expression of Hobit, which is in line with the finding that IEL ex-TRM cells can lose expression of CD103 and CD69 after transfer and reinfection. These results underscore that at least certain parts of the TRM adaptation program are not permanently imprinted and that TRM cells can adapt to new environments and transdifferentiate into other T cell memory subsets.
Hobit expression is neither restricted to TRM cells from a specific organ nor specifically expressed in a functionally distinct TRM subset. Two new complementary studies addressed this by developing CD103+ fate-mapping mice that specifically and permanently mark CD103+ TRM cells, thereby allowing dissection of the differences between CD103+ and CD103− TRM populations75,76. Using a CD103-CreERT2 mouse crossed to a flox-STOP-flox fluorescent reporter mouse, both groups could selectively mark cells that express CD103 at a specific time by treatment with tamoxifen at memory timepoints, thus excluding cells that might only temporally express CD103 during early activation or the effector phase. Despite using different infection models (VSV, Listeria monocytogenes, LCMV, and Yersinia pseudotuberculosis) and different TCR transgenic mice, both studies found, that CD103+ TRM cells residing in the intestinal epithelium were poorly reactivated upon reinfection. Surprisingly, reporter positive CD103+ cells did not retreat to the draining mesenteric lymph nodes. Instead, the CD103− TRM population responded to secondary infection, showing an increase in TCR signaling (as measured by Nur77) and in proliferation compared to their CD103+ TRM counterparts75–77. Since CD8+ CD103+ TRM cells do not show an increase in TCR signaling activity and seem to respond in a TCR-independent manner after antigen reencounter, it will be interesting to determine what signals activate these cells. A recent report found that CD4+ TRM cells in the lung that formed after Bordetella pertussis infection in mice, were able to respond to non-cognate immune challenges97,98. These findings show that the TCR-independent activation of TRM can occur in both CD4+ and CD8+ TRM cells and suggest that TRM might acquire innate-like features to respond to secondary infection at barrier sites. The lack of proliferative capacity of CD103+ TRM cells might be a physiological adaptation to limit immunopathology.
Supporting the idea that CD103− TRM cells possess a higher proliferative capacity, these studies found that the percentage of reporter-positive cells within the CD103+ TRM population decreased after reinfection, suggesting that the pool of CD103+ cells is re-seeded by CD103+ TRM cells. To exclude a competition for antigen and space limitations, CD103-Cre-ERT2 mice were crossed to DTRfloxed mice, thus, after treatment with tamoxifen, all CD103− cells could be depleted with diphtheria toxin to efficiently eliminate competition between CD103+ and CD103− TRM cells, as well as circulatory memory cells during reinfection. Interestingly, even in these artificial conditions, CD103+ TRM cells only marginally contributed to circulatory memory populations, showing that the limited expansion capacity is an intrinsic feature of CD103+ TRM cells76. In summary, these studies suggest, that CD103− TRM cells in the small intestine might represent a precursor TRM cell population with the ability to replenish and sustain the CD103+ TRM pool in the tissue.
In previous reports, TRM cells were shown to locally proliferate upon antigen re-encounter92,93,95, and it is still unclear what causes these divergent results. This might be explained in part by different infection models, different induction of TRM, or different organs studied. However, these differences make clear that the heterogeneity within a TRM population in a single tissue results in different functional capacities and that CD103+ and CD103− TRM subsets need to be assessed separately. In terms of cytokine production, CD103+ cells predominately produced Granzyme A, whereas CD103− TRM cells secreted higher levels of IFNγ, TNF, and IL-2, which is in line with the higher production of these cytokines previously seen in Id3hi cells, which also exhibited a greater plasticity upon transfer and reinfection27,75. However, in contrast, these Id3hi cells exhibited more expression of CD103+, indicating that one marker alone might not be sufficient to delineate among the functionally distinct subsets of TRM cells in the small intestine.
Outlook
Localized tissue-immunity is an integral component of our body’s immune defenses. Understanding, how immune cells enter and persist within a tissue has direct therapeutic potential. Recent studies highlight that TRM cells need to acquire tissue-specific changes to allow for optimal acclimatization to these tissues. Currently, most vaccination strategies focus on generating pathogen-specific memory cells or antibodies. However, recent studies suggest that by directing these memory cells to the exposed tissue, protection against infection can be enhanced. A better understanding of how memory cells adapt to the tissue microenvironment will hopefully allow for tissue-tailored vaccination strategies. Additionally, releasing TRM cells from tissues could boost secondary systemic immune responses. Besides prevention of diseases, T cell therapies provide a novel treatment strategy for cancer and autoimmune diseases. Directing T cells to solid tumors remains a challenge, and mechanisms learned from tissue-resident cells could be extrapolated to improve tumor-infiltration and survival of T cells. Alternatively, allowing tissue egress of exhausted tumor-infiltrating T cells could render them more susceptible to checkpoint therapy, since they would be removed from the immunosuppressive tumor environment. Similarly, selectively targeting resident immune cells in autoimmune-diseases or after organ transplantation provides an exciting new treatment approach where alloreactive T cells could be depleted or functionally impaired16,17.
Single-cell RNA sequencing has helped to dissect TRM heterogeneity, but we are only beginning to understand the complexity of TRM cell populations and the underlying mechanisms resulting in these specialized T cell subsets. By studying common mechanisms across multiple TRM populations and dissecting individual adaptations of TRM from a broad range of tissues, we can infer what signal, cytokines, cell-cell interaction, or environmental sensing mechanisms are required to initiate TRM differentiation. Recent technological advances that allow the study of cellular transcriptomes in intact tissues, combined with computational methods creating cell-cell communication networks, will deepen our understanding of the signals driving T cell residency. This knowledge on how TRM differentiation is embedded in a cellular and environmental network will bring us one step closer to tissue-directed immune-cell therapies.
Acknowledgement
This work is supported in part by grants to A.W. Goldrath from the National Institutes of Health (AI132122 and AI145815) and the Tata Chancellor’s Endowed Chair. The authors thank A. Ferry and K. Takehara for insightful discussions and feedback on the manuscript.
Footnotes
Disclosures: A.W. Goldrath is a member of the SAB of ArsenalBio and Foundery Innovations.
References
- 1.Milner JJ, Nguyen H, Omilusik K, et al. Delineation of a molecularly distinct terminally differentiated memory CD8 T cell population. Proc Natl Acad Sci U S A. 2020;117(41):25667–25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinert EM, Schenkel JM, Fraser KA, et al. Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell. 2015;161(4):737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14(12):1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10(5):524–530. [DOI] [PubMed] [Google Scholar]
- 5.Masopust D, Soerens AG. Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol. 2019;37:521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijeyesinghe S, Beura LK, Pierson MJ, et al. Expansible residence decentralizes immune homeostasis. Nature. 2021;592(7854):457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Gavil NV, Koewler N, Masopust D, Jameson SC. Parabiosis in Mice to Study Tissue Residency of Immune Cells. Curr Protoc. 2022;2(5):e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity. 2004;20(5):551–562. [DOI] [PubMed] [Google Scholar]
- 9.Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, Kupper TS. Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature. 2012;483(7388):227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187(11):5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallett LJ, Burton AR, Amin OE, et al. Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J Exp Med. 2020;217(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pallett LJ, Maini MK. Liver-resident memory T cells: life in lockdown. Semin Immunopathol. 2022;44(6):813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.FitzPatrick MEB, Provine NM, Garner LC, et al. Human intestinal tissue-resident memory T cells comprise transcriptionally and functionally distinct subsets. Cell Rep. 2021;34(3):108661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson KG, Mayer-Barber K, Sung H, et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat Protoc. 2014;9(1):209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackay LK, Minnich M, Kragten NA, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352(6284):459–463. [DOI] [PubMed] [Google Scholar]
- 16.Milner JJ, Toma C, Yu B, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552(7684):253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crowl JT, Heeg M, Ferry A, et al. Tissue-resident memory CD8(+) T cells possess unique transcriptional, epigenetic and functional adaptations to different tissue environments. Nat Immunol. 2022;23(7):1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wakim LM, Woodward-Davis A, Liu R, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol. 2012;189(7):3462–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar BV, Ma W, Miron M, et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017;20(12):2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evrard M, Wynne-Jones E, Peng C, et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J Exp Med. 2022;219(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hallisey VM, Schwab SR. Blood-thirsty: S1PR5 and TRM. J Exp Med. 2022;219(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–1301. [DOI] [PubMed] [Google Scholar]
- 23.Mackay LK, Wynne-Jones E, Freestone D, et al. T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity. 2015;43(6):1101–1111. [DOI] [PubMed] [Google Scholar]
- 24.Bergsbaken T, Bevan MJ, Fink PJ. Local Inflammatory Cues Regulate Differentiation and Persistence of CD8(+) Tissue-Resident Memory T Cells. Cell Rep. 2017;19(1):114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, Bevan MJ. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39(4):687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin YH, Duong HG, Limary AE, et al. Small intestine and colon tissue-resident memory CD8(+) T cells exhibit molecular heterogeneity and differential dependence on Eomes. Immunity. 2023;56(1):207–223 e208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milner JJ, Toma C, He Z, et al. Heterogenous Populations of Tissue-Resident CD8(+) T Cells Are Generated in Response to Infection and Malignancy. Immunity. 2020;52(5):808–824 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. 2015;16(4):406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiu Z, Khairallah C, Chu TH, et al. Retinoic acid signaling during priming licenses intestinal CD103+ CD8 TRM cell differentiation. J Exp Med. 2023;220(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity. 2014;40(5):747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurd NS, He Z, Louis TL, et al. Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci Immunol. 2020;5(47). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kakaradov B, Arsenio J, Widjaja CE, et al. Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol. 2017;18(4):422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Savas P, Virassamy B, Ye C, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24(7):986–993. [DOI] [PubMed] [Google Scholar]
- 34.Liao W, Liu Y, Ma C, et al. The downregulation of IL-18R defines bona fide kidney-resident CD8(+) T cells. iScience. 2021;24(1):101975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woyciechowski S, Hofmann M, Pircher H. alpha(4) beta(1) integrin promotes accumulation of tissue-resident memory CD8(+) T cells in salivary glands. Eur J Immunol. 2017;47(2):244–250. [DOI] [PubMed] [Google Scholar]
- 36.Christo SN, Evrard M, Park SL, et al. Discrete tissue microenvironments instruct diversity in resident memory T cell function and plasticity. Nat Immunol. 2021;22(9):1140–1151. [DOI] [PubMed] [Google Scholar]
- 37.Stolley JM, Johnston TS, Soerens AG, et al. Retrograde migration supplies resident memory T cells to lung-draining LN after influenza infection. J Exp Med. 2020;217(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kunzli M, O’Flanagan SD, LaRue M, et al. Route of self-amplifying mRNA vaccination modulates the establishment of pulmonary resident memory CD8 and CD4 T cells. Sci Immunol. 2022;7(78):eadd3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y, Ma C, Zhang N. Tissue-Specific Control of Tissue-Resident Memory T Cells. Crit Rev Immunol. 2018;38(2):79–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavin Y, Winter D, Blecher-Gonen R, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159(6):1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mass E, Nimmerjahn F, Kierdorf K, Schlitzer A. Tissue-specific macrophages: how they develop and choreograph tissue biology. Nat Rev Immunol. 2023:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mani V, Bromley SK, Aijo T, et al. Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate. Science. 2019;366(6462):eaav5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L. Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol. 2002;168(10):4827–4831. [DOI] [PubMed] [Google Scholar]
- 45.Goldrath AW, Sivakumar PV, Glaccum M, et al. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. 2002;195(12):1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adachi T, Kobayashi T, Sugihara E, et al. Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med. 2015;21(11):1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holz LE, Prier JE, Freestone D, et al. CD8(+) T Cell Activation Leads to Constitutive Formation of Liver Tissue-Resident Memory T Cells that Seed a Large and Flexible Niche in the Liver. Cell Rep. 2018;25(1):68–79 e64. [DOI] [PubMed] [Google Scholar]
- 48.Schenkel JM, Fraser KA, Casey KA, et al. IL-15-Independent Maintenance of Tissue-Resident and Boosted Effector Memory CD8 T Cells. J Immunol. 2016;196(9):3920–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masopust D, Vezys V, Wherry EJ, Barber DL, Ahmed R. Cutting edge: gut microenvironment promotes differentiation of a unique memory CD8 T cell population. J Immunol. 2006;176(4):2079–2083. [DOI] [PubMed] [Google Scholar]
- 50.Jarjour NN, Wanhainen KM, Peng C, et al. Responsiveness to interleukin-15 therapy is shared between tissue-resident and circulating memory CD8(+) T cell subsets. Proc Natl Acad Sci U S A. 2022;119(43):e2209021119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ubaid U, Andrabi SBA, Tripathi SK, et al. Transcriptional Repressor HIC1 Contributes to Suppressive Function of Human Induced Regulatory T Cells. Cell Rep. 2018;22(8):2094–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burrows K, Antignano F, Bramhall M, et al. The transcriptional repressor HIC1 regulates intestinal immune homeostasis. Mucosal Immunol. 2017;10(6):1518–1528. [DOI] [PubMed] [Google Scholar]
- 53.Borges da Silva H, Beura LK, Wang H, et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature. 2018;559(7713):264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borges da Silva H, Peng C, Wang H, et al. Sensing of ATP via the Purinergic Receptor P2RX7 Promotes CD8(+) Trm Cell Generation by Enhancing Their Sensitivity to the Cytokine TGF-beta. Immunity. 2020;53(1):158–171 e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8(5):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Walsh DA, Borges da Silva H, Beura LK, et al. The Functional Requirement for CD69 in Establishment of Resident Memory CD8(+) T Cells Varies with Tissue Location. J Immunol. 2019;203(4):946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beura LK, Wijeyesinghe S, Thompson EA, et al. T Cells in Nonlymphoid Tissues Give Rise to Lymph-Node-Resident Memory T Cells. Immunity. 2018;48(2):327–338 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41(6):886–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frizzell H, Fonseca R, Christo SN, et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci Immunol. 2020;5(46). [DOI] [PubMed] [Google Scholar]
- 60.Pan Y, Tian T, Park CO, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poon MML, Caron DP, Wang Z, et al. Tissue adaptation and clonal segregation of human memory T cells in barrier sites. Nat Immunol. 2023;24(2):309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maurice NJ, Jameson SC. Resident memory T cells develop regional dialects. Nat Immunol. 2023;24(2):209–210. [DOI] [PubMed] [Google Scholar]
- 63.Mao T, Israelow B, Pena-Hernandez MA, et al. Unadjuvanted intranasal spike vaccine elicits protective mucosal immunity against sarbecoviruses. Science. 2022;378(6622):eabo2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sacirbegovic F, Gunther M, Greco A, et al. Graft-versus-host disease is locally maintained in target tissues by resident progenitor-like T cells. Immunity. 2023;56(2):369–385 e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kok L, Dijkgraaf FE, Urbanus J, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med. 2020;217(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kok L, Masopust D, Schumacher TN. The precursors of CD8(+) tissue resident memory T cells: from lymphoid organs to infected tissues. Nat Rev Immunol. 2022;22(5):283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peng C, Huggins MA, Wanhainen KM, et al. Engagement of the costimulatory molecule ICOS in tissues promotes establishment of CD8(+) tissue-resident memory T cells. Immunity. 2022;55(1):98–114 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joshi NS, Cui W, Chandele A, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27(2):281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4(12):1191–1198. [DOI] [PubMed] [Google Scholar]
- 70.Boland BS, He Z, Tsai MS, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol. 2020;5(50). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang CY, Best JA, Knell J, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12(12):1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Omilusik KD, Best JA, Yu B, et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J Exp Med. 2015;212(12):2027–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watanabe R, Gehad A, Yang C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. 2015;7(279):279ra239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clark RA, Watanabe R, Teague JE, et al. Skin effector memory T cells do not recirculate and provide immune protection in alemtuzumab-treated CTCL patients. Sci Transl Med. 2012;4(117):117ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fung HY, Teryek M, Lemenze AD, Bergsbaken T. CD103 fate mapping reveals that intestinal CD103(−) tissue-resident memory T cells are the primary responders to secondary infection. Sci Immunol. 2022;7(77):eabl9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.von Hoesslin M, Kuhlmann M, de Almeida GP, et al. Secondary infections rejuvenate the intestinal CD103(+) tissue-resident memory T cell pool. Sci Immunol. 2022;7(77):eabp9553. [DOI] [PubMed] [Google Scholar]
- 77.Jensen IJ, Farber DL. Gutsy memory T cells stand their ground against pathogens. Sci Immunol. 2022;7(77):eade7168. [DOI] [PubMed] [Google Scholar]
- 78.Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346(6205):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Heeg M, Goldrath AW. License to kill: Retinoic acid programs T cells for tissue residency. J Exp Med. 2023;220(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Madhavan M, Ritchie AJ, Aboagye J, et al. Tolerability and immunogenicity of an intranasally-administered adenovirus-vectored COVID-19 vaccine: An open-label partially-randomised ascending dose phase I trial. EBioMedicine. 2022;85:104298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fuchs A, Vermi W, Lee JS, et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38(4):769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turner DL, Farber DL. Mucosal resident memory CD4 T cells in protection and immunopathology. Front Immunol. 2014;5:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McFarland AP, Yalin A, Wang SY, et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity. 2021;54(6):1320–1337 e1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beura LK, Fares-Frederickson NJ, Steinert EM, et al. CD4(+) resident memory T cells dominate immunosurveillance and orchestrate local recall responses. J Exp Med. 2019;216(5):1214–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fonseca R, Burn TN, Gandolfo LC, et al. Runx3 drives a CD8(+) T cell tissue residency program that is absent in CD4(+) T cells. Nat Immunol. 2022;23(8):1236–1245. [DOI] [PubMed] [Google Scholar]
- 86.Gebhardt T, Whitney PG, Zaid A, et al. Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature. 2011;477(7363):216–219. [DOI] [PubMed] [Google Scholar]
- 87.Wales MM, Biel MA, el Deiry W, et al. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat Med. 1995;1(6):570–577. [DOI] [PubMed] [Google Scholar]
- 88.Issa JP, Zehnbauer BA, Kaufmann SH, Biel MA, Baylin SB. HIC1 hypermethylation is a late event in hematopoietic neoplasms. Cancer Res. 1997;57(9):1678–1681. [PubMed] [Google Scholar]
- 89.Burrows K, Antignano F, Chenery A, et al. HIC1 links retinoic acid signalling to group 3 innate lymphoid cell-dependent regulation of intestinal immunity and homeostasis. PLoS Pathog. 2018;14(2):e1006869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DiSpirito JR, Zemmour D, Ramanan D, et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci Immunol. 2018;3(27):eaat5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schenkel JM, Fraser KA, Vezys V, Masopust D. Sensing and alarm function of resident memory CD8(+) T cells. Nat Immunol. 2013;14(5):509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fonseca R, Beura LK, Quarnstrom CF, et al. Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat Immunol. 2020;21(4):412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Beura LK, Mitchell JS, Thompson EA, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. 2018;19(2):173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Behr FM, Parga-Vidal L, Kragten NAM, et al. Tissue-resident memory CD8(+) T cells shape local and systemic secondary T cell responses. Nat Immunol. 2020;21(9):1070–1081. [DOI] [PubMed] [Google Scholar]
- 95.Park SL, Zaid A, Hor JL, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. 2018;19(2):183–191. [DOI] [PubMed] [Google Scholar]
- 96.Li G, Srinivasan S, Wang L, et al. TGF-beta-dependent lymphoid tissue residency of stem-like T cells limits response to tumor vaccine. Nat Commun. 2022;13(1):6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cornelis R, Shulman Z. Bystander activation of tissue-resident memory CD4 T cells: Getting by with a little help from unfamiliar T-cell friends. Eur J Immunol. 2023:e2350413. [DOI] [PubMed] [Google Scholar]
- 98.Curham LM, Mannion JM, Daly CM, et al. Bystander activation of Bordetella pertussis-induced nasal tissue-resident memory CD4 T cells confers heterologous immunity to Klebsiella pneumoniae. Eur J Immunol. 2023:e2250247. [DOI] [PubMed] [Google Scholar]
- 99.Ewels PA, Peltzer A, Fillinger S, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol. 2020;38(3):276–278. [DOI] [PubMed] [Google Scholar]
- 100.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McCarthy DJ, Campbell KR, Lun AT, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33(8):1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.van Dijk D, Sharma R, Nainys J, et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 2018;174(3):716–729 e727. [DOI] [PMC free article] [PubMed] [Google Scholar]