

Key Points
-
•
Nfix deficiency results in a loss of long-term hematopoietic stem cells and an accumulation of megakaryocyte and myelo-erythroid progenitors.
-
•
NFIX interacts with PU.1 at super-enhancers to regulate gene targets involved in cellular respiration and hematopoietic differentiation.
Visual Abstract
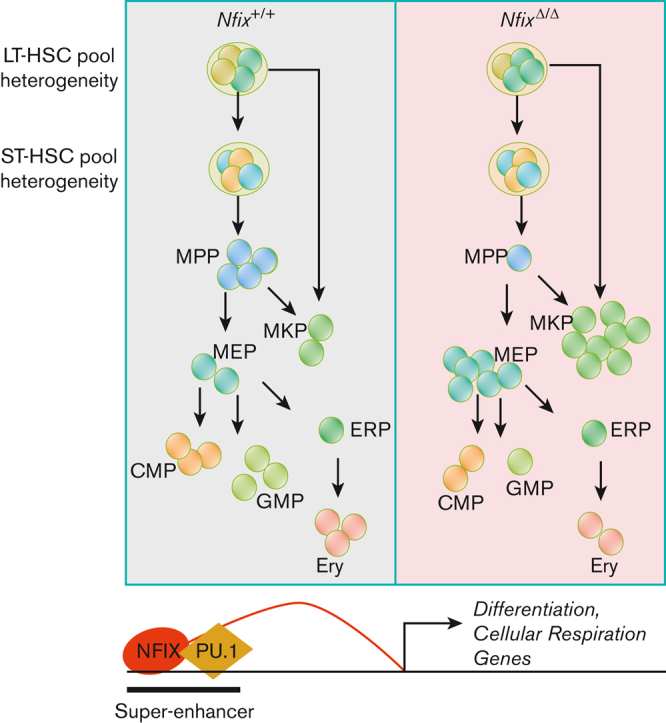
Abstract
The transcription factor (TF) nuclear factor I-X (NFIX) is a positive regulator of hematopoietic stem and progenitor cell (HSPC) transplantation. Nfix-deficient HSPCs exhibit a severe loss of repopulating activity, increased apoptosis, and a loss of colony-forming potential. However, the underlying mechanism remains elusive. Here, we performed cellular indexing of transcriptomes and epitopes by high-throughput sequencing (CITE-seq) on Nfix-deficient HSPCs and observed a loss of long-term hematopoietic stem cells and an accumulation of megakaryocyte and myelo-erythroid progenitors. The genome-wide binding profile of NFIX in primitive murine hematopoietic cells revealed its colocalization with other hematopoietic TFs, such as PU.1. We confirmed the physical interaction between NFIX and PU.1 and demonstrated that the 2 TFs co-occupy super-enhancers and regulate genes implicated in cellular respiration and hematopoietic differentiation. In addition, we provide evidence suggesting that the absence of NFIX negatively affects PU.1 binding at some genomic loci. Our data support a model in which NFIX collaborates with PU.1 at super-enhancers to promote the differentiation and homeostatic balance of hematopoietic progenitors.
Introduction
Hematopoietic stem cells (HSCs) are responsible for blood maintenance and can reconstitute the hematopoietic hierarchy when transplanted into a host whose hematopoietic system has been ablated. The previous 2 decades have witnessed an increased trend in the survival of people diagnosed with hematopoietic diseases and treated with HSC transplantation (HSCT).1 However, there remain many complications associated with HSCT, such as engraftment failure, infection, and mortality. To better understand the molecular regulation of HSCT, we have sought to identify novel molecular regulators of HSC repopulating activity. We recently identified nuclear factor I-X (NFIX) as required for optimal HSC in vivo repopulating activity after transplantation.2
NFIX belongs to the Nfi gene family, which also includes NFIA, NFIB, and NFIC.3,4 The NFI family function as transcriptional activators or repressors and bind to a palindromic DNA sequence (TGG(N6-7)GCCA) as homodimers or heterodimers. NFI family members are expressed in a variety of cell types, including multiple adult stem cell compartments.5, 6, 7, 8, 9 NFIC regulates the differentiation, proliferation, and apoptosis of dental follicle stem cells.6 NFIB is expressed by epithelial hair follicle stem cells, promoting proliferation and differentiation.7 NFIA functions as a transcriptional switch in multiple stem and progenitor cell compartments. It promotes gliogenesis in the developing chick neural tube while inhibiting further neurogenesis of ventricular zone progenitor cells and regulates the granulocytic/erythroid fate choice of human hematopoietic progenitors during in vitro differentiation.8,9 NFIX itself regulates the quiescence, adhesion, migration, and differentiation of neural stem cells.10, 11, 12 Recently, NFIX mutations have been associated with acute erythroid leukemia.13
We have shown that NFIX regulates HSC survival after transplantation: Nfix-deficient hematopoietic stem and progenitor cells (HSPCs) exhibit increased apoptosis acutely after transplantation and reduced colony-forming potential in vitro.2 In contrast, enforced NFIX expression protects hematopoietic cells from apoptosis and prolongs hematopoietic cell growth in vitro in a thrombopoietin-dependent manner.14 NFIX is well characterized as a vital regulator of transcription in immature cells; however, little is known about the role of NFIX in hematopoiesis. Furthermore, it is currently unknown whether NFIX collaborates with other characterized transcriptional regulators of hematopoiesis.
To explore the NFIX-regulated transcriptome in HSPCs, we generated a transcriptional, cellular atlas of wild-type and Nfix-deficient bone marrow (BM) HSPCs. Here, we report that the loss of Nfix results in perturbations in the relative frequencies of specific hematopoietic progenitor subsets. Via chromatin immunoprecipitation (ChIP)-seq and assay for transposase-accessible chromatin (ATAC)-seq, we show for the first time that NFIX partners with additional key hematopoietic transcription factors (TFs), including PU.1, especially at superenhancers. Moreover, we provide evidence suggesting the absence of NFIX negatively affects PU.1 binding at some genomic loci. We also computationally predict target genes of NFIX and PU.1 across the hematopoietic hierarchy and investigate their regulatory functions during hematopoietic differentiation.
Methods
Fluorescence-activated cell sorting (FACS) and cellular indexing of transcriptomes and epitopes by high-throughput sequencing (CITE-seq)
BM was extracted from the femurs, pelvic bones, tibias, and spines of tamoxifen (TAM)-treated Nfixflox/floxRosa26CreERT2+/+ and Nfixflox/floxRosa26CreERT2+/T mice via crushing. c-Kit+ cells were isolated via magnetic enrichment using anti-CD117 microbeads (Miltenyi Biotec, Carlsbad, CA) and an autoMACS magnetic cell separator (Miltenyi Biotec, Carlsbad, CA). For c-Kit+ cells, HSPCs (Lineage-Sca1+ c-Kit+), and long-term hematopoietic stem cells (LT-HSCs) (Lineage-Sca1+ c-Kit+CD48−CD150+) isolation, cells were stained with anti-Lineage BV605 cocktail, anti-Sca-1-PerCP-Cy5.5 (E13-161.7), anti-c-Kit-APC-e780 (2B8), anti-CD48-Alexa Fluor 700 (HM48-1), and anti-CD150-PE-Cy7 (TC15-12F12.2). For common myeloid progenitor (CMP) (Lineage−Sca-1−c-Kit+CD34+CD16/32med) isolation, cells were stained with anti-Lineage BV605 cocktail, anti-Sca-1-PerCP-Cy5.5 (E13-161.7), anti-c-Kit-APC-e780 (2B8), anti-CD34-FITC (RAM34), and anti-CD16/32-Alexa Fluor 700 (2.4G2). In addition to FACS antibodies, cells were labeled with cellular indexing of transcriptomes and epitopes by high-throughput sequencing (CITE-seq) antibodies (see supplemental Table 1). 4′,6-diamidino-2-phenylindole (Sigma-Aldrich, St Louis, MO) was used for dead cell exclusion. All cell isolations were performed using cell sorting on FACSAria III (BD Biosciences, San Jose, CA). Then, 7000 Lineage−cKit+ BM, 1000 HSPCs, 1000 CMPs, and 1000 LT-HSCs were pooled together for 10x genomics (see supplemental Methods).
Single-cell RNA-seq (scRNA-seq) analysis
Single-cell data were analyzed using cellranger count (version 6.0.0)15 with default parameters for CITE-seq (--feature-ref). The transcriptome database was cellranger-mm10-3.0.0. Cells were filtered based on: (1) number of expressed genes (ie, nFeature_RNA) between 200 and 6000 and (2) percentage of mitochondrial reads (ie, percent.mt) less than 10%. The weighted nearest neighbor Uniform Manifold Approximation and Projection (UMAP) and cell clusters were generated using Seurat (v4.0.4),16 following the reference guide of the weighted nearest neighbor analysis of CITE-seq, RNA + ADT. Specifically, principal component analysis (PCA) was used to generate embeddings for both the RNA and the protein space (ie, NormalizeData() %>% FindVariableFeatures() %>% ScaleData() %>% RunPCA()). Then the 2 spaces were integrated using the FindMultiModalNeighbors method with default parameters. Finally, cell clusters were identified using RunUMAP with default parameters and FindClusters with algorithm = 3 and resolution = 1. Cell cycle prediction was done using CellCycleScoring. The 2D kernel density estimation (KDE) was done using MASS (v7.3-54)17 and was plotted using ggplot2 (v3.3.5, the contour plot)18 and rayshader (v0.26.2, the 3D view).19 P value for the density difference comparing Nfix+/+ with NfixΔ/Δ for each cluster was calculated using the kde.test function (with parameter of binned = T) from the ks R package (version 1.13.2).20 The cluster sizes were fixed to 500 cells by sampling with replacement. The kernel density test was repeated 1000 times for each cluster, and the mean P value was used. To show that density differences were not a bias of parameter settings, we conducted: (1) KDE analysis with a range of bandwidth from 0.5 to 5 (supplemental Figure 4A-F) and (2) differential abundance analysis (DAseq)21 with a range of scales from 20 to 5000 (supplemental Figure 4G-H). The DA cells were generated using the getDAcells function based on the PCA embedding and UMAP embedding. The clustering of DA cells and differential test were done using the getDAregion function with default parameters and min.cell = 50. Differential gene expression analysis was done using FindMarkers with “slot = data, logfc.threshold = 0.2, test.used = wilcox, pseudocount.use = 1.” Genes were further filtered by false discovery rate ≤0.01, which was calculated using the Benjamini-Hochberg method in the R package.
Motif and peak analysis and motif distance analysis
NFIX and PU.1 overlapped peaks were determined using bedtools (eg, bedtools intersect -u) (v2.25.0).22 NFIX and PU.1 motifs were downloaded from the Homer motif database.23 Motif scanning was performed using FIMO (version 4.11.2)24 with default P value cutoff of 10−4. NFI motif was only searched on the positive strand. Motif distance was calculated using “bedtools closest -a NFIX -b PU.1 -k 1 -d.” Only unique NFIX and PU.1 occurrences were used in the calculation. Background sequences were randomly sampled from mm9 with “bedtools random -l 10000 -n 743.” The reason to set 10 000 for sequence length is to increase the chance of NFIX and PU.1 co-occurrence. Random sampling was performed 100 times. DeepTools (version 3.2.0)25 was used to draw footprint plots with a bin size of 2bp.
Prediction of NFIX-PU.1 cobinding peaks
Catchitt was used to identify lineage-specific NFIX-PU.1 cobinding peaks.26 The input contains: (1) training labels of NFIX-PU.1 cobinding peaks from HPC5 (generated by the “labels” subcommand); (2) NFIX and PU.1 motifs (generated by the “motif” subcommand); and (3) a cell-type–specific ATAC-seq signal (generated by the “access” subcommand with d = “Bigwig”). ATAC-seq data for LT-HSC, short-term HSC (ST-HSC), and multipotent progenitors (MPP) were downloaded from PRJNA335747. ATAC-seq data for the remaining lineages was downloaded from the mouse VISION project GSE143270.27,28 ATAC-seq data were normalized using S3norm.29 Chromosome 19 was a hold-out set for crossvalidation, with an area under the receiver operating characteristic of 0.95. Cobinding labels on all other chromosomes were put into the iterative learning model (command is “itrain i = 5 abb = 1 aba = 4 P = .01”). The top 1000 predicted cobinding peaks in each blood lineage (eg, LT-HSC, ST-HSC, MPP, megakaryocyte progenitor [MKP], CMP, granulocyte-macrophage progenitor [GMP], myelo-erythroid progenitor [MEP], erythroid progenitors, and erythroid cells [Ery]) were selected and used for downstream analyses.
Target gene assignment
Direct target genes were only assigned to the predicted NFIX-PU.1 peaks if they met both of the following requirements: (1) colocalized within the same topologically associating domain of the peaks or NFIX-PU.1 peaks interacted with the gene promoter based on promoter capture-HiC30 and (2) was a differentially expressed gene (DEG) between Nfix+/+ and NfixΔ/Δ in each cluster. To assess the quality of our approach, we randomly selected 1000 regions and applied the same target-finding pipeline. We hypothesized that cell-type–specific NFIX-PU.1 peaks would better explain DEG than random genomic loci, and thus the number of target genes for NFIX-PU.1 peaks would be higher than the number of target genes from a random approach. To test this hypothesis, we performed a paired t test. Specifically, for each of the 11 clusters, we had 2 values (ie, number of target genes): one obtained using NFIX-PU.1 peaks and the other obtained using random regions. We repeated the procedure 100 times. The average P value was 0.0049, confirming that our analysis was robust.
Results
An scatlas of Nfix-deficient hematopoietic progenitors
To illuminate the role of NFIX as a transcriptional regulator of HSPCs, we performed CITE-seq using Nfixflox/floxRosa26-CreERT2+/+ and Nfixflox/floxRosa26-CreERT2+/T mice treated with TAM (Figure 1A).31 Fourteen days of TAM treatment resulted in the efficient deletion of Nfix in BM HSPCs (supplemental Figure 1). TAM-treated Nfixflox/floxRosa26-CreERT2+/+ and Nfixflox/floxRosa26-CreERT2+/T HSPCs will be referred to as Nfix+/+ and NfixΔ/Δ, respectively, hereafter. After TAM treatment, BM was collected and simultaneously stained with CITE-seq and fluorophore-conjugated antibodies (Figure 1A; supplemental Table 1). To ensure adequate capture of each key hematopoietic progenitor population for sc profiling, rare populations (ie, LT-HSCs and CMPs) were collected separately via cell sorting and pooled with more abundant populations as follows: 7000 Lineage−c-Kit+ hematopoietic progenitors, 1000 HSPCs (Lineage−cKit+Sca1+), 1000 CMPs (Lineage−cKit+Sca1−CD34+CD16/32med), and 1000 LT-HSCs (Lineage− Sca1+ c-Kit+ CD150+CD48−). Pooled cells were then processed for scRNA-seq and cell-surface protein abundance using 10x genomics (Figure 1A). Although our sampling strategy may not reflect the true cellular abundance in the BM, our goal is to interrogate shifts in clusters found within these well-defined progenitor pools based on gene and surface marker expression (ie, CITE-seq).
Figure 1.

Single-cell atlas of NFIX-deficient BM hematopoietic progenitors. (A) Nfixflox/floxRosa26-CreERT2+/+ (Nfix+/+) and Nfixflox/floxRosa26-CreERT2+/T (NfixΔ/Δ) mice were treated with TAM for 14 days. Nfix+/+ and NfixΔ/Δ BM were collected and incubated with CITE-seq and FACS antibodies. Lineage−cKit+ BM cells, HSPCs, CMPs, and LT-HSCs were sorted and pooled 7:1:1:1 before processing for scRNA-seq and cell-surface protein abundance via 10x genomics. (B) Weighted nearest neighbor UMAP. Single cells from Nfix+/+ and NfixΔ/Δ BM clustered into 11 cell types based on cell-surface markers and RNA expression. (C) Protein abundance of 7 cell-surface markers representing the CITE-seq antibody signal (see also supplemental Figure 2). (D) Dot plot representing gene expression of specific cell marker genes across the 11 cell types. ERP, erythroid progenitors.
To identify distinct cell populations, we first performed dimensional reduction based on the weighted nearest neighbor workflow in Seurat version 416 and generated a UMAP visualization of Nfix+/+ (7341 cells) and NfixΔ/Δ (6608 cells) data based on a weighted combination of RNA and protein expression of surface markers (Figure 1B). We then performed clustering analysis using the smart local moving algorithm and identified 11 unique clusters (Figure 1B).32 Clusters were annotated based on cell surface and known gene markers (Figure 1C-D; supplemental Figure 2; supplemental Table 2).33 CD150, CD48, and SCA1 were used to identify phenotypic LT-HSCs, ST-HSCs, and MPPs (Figure 1C; supplemental Figure 2).34 Interestingly, LT-HSC.2 express high levels of CD41 and CD150 relative to LT-HSC.1 and fall in close proximity to the MKP cluster (Figure 1B; supplemental Figure 2). Thus, LT-HSC.2 likely represent megakaryocyte-primed HSCs.35 MKPs were annotated based on high CD41 expression (Figures 1C; supplemental Figure 2). CD71 and CD105, markers of committed progenitors and Ery, were used to identify CMPs, GMPs, erythroid progenitors, and Ery (Figure 1C; supplemental Figures 2 and 3).36 CMPs and GMPs were further annotated based on high CD16/32 expression (Figure 1C; supplemental Figure 2). The cell-type annotation based on surface markers can be further confirmed using gene expression markers. For example, high expression of erythroid gene markers, Gata2 and Apoe, indicated megakaryocytic and erythroid populations (Figure 1D; supplemental Figure 3).37,38 Annotated HSCs and MPPs expressed high levels of Mllt3, Hlf, Mecom, Meis1, and Mpl (Figure 1D; supplemental Figure 3).39, 40, 41, 42, 43 Cavin2 and Pbx1 were both highly expressed in annotated MKPs (Figure 1D; supplemental Figure 3).44,45 Hbb-bs and Hba-a1 were expressed in the Ery cluster (Figure 1D; supplemental Figure 3).46
We thus created an single-cell atlas of c-Kit+ mouse BM based on gene expression and cell-surface markers.
Loss of Nfix depletes a LT-HSC subpopulation and enriches for MKPs
We next examined our sincle-cell atlas to ask if the abundance of any BM HSPCs was perturbed by the loss of NFIX. Two-dimensional KDE confirmed a significant loss of LT-HSC.1 (P value = 1.2 × 10−3), MPPs (P value = 9.2 × 10−4), and GMPs (P value = 1.1 × 10−3) in NfixΔ/Δ HSPCs (Figure 2Ai-iii). NfixΔ/Δ HSPCs displayed a concomitant enrichment of LT-HSC.2 (P value = 3.9 × 10−3), ST-HSC.1 (P value = 9.9 × 10−3), MKP (P value = 3.5 × 10−3), and MEP (P value = 2.8 × 10−5) relative to Nfix+/+ HSPC (Figure 2Ai-iii). Consistently, we observed a significant decrease in the fraction of cells assigned to LT-HSC.1, MPP, and GMP clusters and an increase in those assigned to LT-HSC.2, ST-HSC.1, MKP, and MEP clusters in NfixΔ/Δ vs Nfix+/+ HSPCs (Figure 2B). To test the robustness of the cell population differences and to exclude possible estimation bias caused by parameter settings, we performed KDE analysis and DA analysis using a range of parameters. The KDE analyses with different bandwidths all show more crowded contour lines in the MKP and MEP clusters and less crowded lines in the LT-HSC.1 cluster for the NfixΔ/Δ sample (supplemental Figure 4A-F). The DA analysis result is another independent confirmation showing more enriched cells in ST-HSC.1, MEP, and MKP and more depleted cells in the LT-HSC1 cluster (supplemental Figure 4G-H). Consistent with our cluster annotations, the LT-HSC and ST-HSC clusters were largely quiescent. Thus, differences in cluster abundance did not correlate with changes in cell cycle status (supplemental Figure 4I).
Figure 2.

NFIX loss perturbs abundance of multiple hematopoietic progenitor cell clusters. (A)(i) UMAP plot comparing Nfix+/+ and NfixΔ/Δ cell clusters; (ii) Visualization of 2D KDE, with the resulting contour lines superimposed on UMAP plot shown in (i). ∗P value <.05; and (iii) Three-dimensional visualization of the KDE, with the resulting density map superimposed on top of the UMAP plot shown in panels i-ii. Arrowheads indicate significant differences between LT-HSC, MKP, GMP, and MEP clusters. Arrowhead colors correspond to clusters indicated in panels i-ii. (B) Bar plot of the percentage of cells assigned to each cell type. ∗P value <.05. (C) Representative GSEA results for LT-HSC.1 (top) and LT-HSC.2 (bottom). (D) Schematic of shifts in abundance of HSPC subsets following the loss of Nfix. GSEA, gene set enrichment analysis.
In sum, loss of Nfix results in a selective loss of LT- and ST-HSC subsets and MPP, as well as an accumulation of downstream MKP and MEP clusters, suggesting a possible block in differentiation. This is in line with a known role for a highly related gene, NFIA, in the granulocytic/erythroid fate choice of human hematopoietic progenitors.9
To better understand the perturbations in these populations, we next examined changes in gene expression using the gene set enrichment analysis across clusters (Figure 2C; supplemental Figure 5A-B). Differential gene expression analysis comparing NfixΔ/Δ and Nfix+/+ cells revealed enrichment of gene ontology (GO) terms associated with each cell cluster (supplemental Tables 3 and 4; supplemental Figure 5C). In LT-HSC.1, whose abundance is depleted in NfixΔ/Δ HSPCs, we observed decreased expression of genes associated with ribosome complex formation and translation (eg, macromolecule biosynthetic process) and cellular respiration (eg, cellular metabolic process; Figure 2C; supplemental Tables 3 and 4). Protein synthesis is tightly regulated in HSCs to preserve their function.47 These data suggest a novel role for NFIX in regulating metabolic processes. Multiple genes involved in modulating the electrochemical gradient and mitochondrion are downregulated in the absence of Nfix (Cox genes, Uqcr genes, and Atp synthase genes; supplemental Table 3; supplemental Figure 5B), consistent with previous studies showing mitochondrial membrane potential is important for HSPC self-renewal and differentiation.48 In addition, gene set enrichment analysis terms associated with differentiation were enriched (eg, regulation of cell differentiation; Figure 2C; supplemental Table 4), further supporting the altered hematopoiesis in NfixΔ/Δ HSPC.
We also observed an increase in LT-HSC.2 in NfixΔ/Δ HSPCs. Here, genes involved in the microenvironment (eg, response to stimuli and cell periphery) were perturbed (Figure 2C; supplemental Table 4). The BM niche is a rich microenvironment that supports the maintenance and survival of HSPCs.49 We again found GO terms related to differentiation (eg, cell differentiation and regulation of cell population proliferation) enriched in LT-HSC.2 DEG (Figure 2C; supplemental Table 4). These GO terms included the upregulated genes, Cd74 and Mcl1 (supplemental Table 3; supplemental Figure 6). Cd74 and Mcl1 play a role in HSC maintenance and survival.50,51 Activation of CD74 indirectly induces Bcl2 expression, a prosurvival factor.52 Consistently, we observe a significant increase of Bcl2 expression in NfixΔ/Δ MKPs (supplemental Table 3; supplemental Figure 5B), which is downstream of LT-HSC.2 and may explain its increased abundance in NfixΔ/Δ HSPCs (Figure 2B).
Taken together, we show that the loss of Nfix perturbs the abundance of BM HSPCs. Select LT-HSCs, MPPs, and GMPs are lost, which is accompanied by increases in a distinct LT-HSC subset, ST-HSCs, MKPs, and MEPs (summarized in Figure 2D). Our data are consistent with prior studies linking NFIX to HSPC survival and further implicate NFIX in HSPC differentiation and proliferation, ribosome biogenesis and translation, and cellular respiration.2,14,53
The scRNA-seq data we generated is illuminating and would pair well with functional studies that directly interrogate NFIX genetic targets. However, to our knowledge, there are no studies that describe how NFIX directly regulates hematopoiesis or identify potential transcriptional partners of NFIX.
Global profiling of NFIX shows co-occupancy with multiple hematopoietic factors
To investigate how NFIX regulates hematopoiesis, we sought to generate a global map of NFIX-binding sites. Currently, there are few commercial antibodies that can reliably discriminate between different NFI families because of high sequence homology. To address this, we generated a NFIX-specific monoclonal antibody (clone: 7B5.3). Our highly specific anti-NFIX monoclonal antibody exclusively immunoprecipitated NFIX-FLAG in 293T cells overexpressing NFIX-FLAG but not in cells overexpressing NFIA-FLAG (supplemental Figure 7A).
We then performed ChIP followed by high-throughput sequencing (ChIP-seq) using our novel anti-NFIX antibody and Nfix+/+ HPC5 cells as an in vitro surrogate for HSPCs.54 We identified 6783 high-quality NFIX peaks with an irreproducible discovery rate cutoff at 5% (supplemental Table 5; supplemental Figure 7C-E).55 To validate observed peaks as true NFIX-binding sites, we generated Nfix−/− HPC5 cells with sgRNAs combined with Cas9 targeting the third exon of Nfix (supplemental Figure 7B). NFIX ChIP-seq in Nfix−/− HPC5 cells detected only 155 peaks (Figure 3A; supplemental Figure 7C-E), which were excluded from further analysis. We performed motif discovery analysis and observed that the NFI consensus motif was the most overrepresented motif in NFIX peaks (supplemental Figure 7E), further confirming that these ChIP-seq peaks were directly occupied by NFIX. Genome-wide distribution reveals that most of the NFIX peaks (77%) were found in distal regulatory elements (Figure 3B). Previous studies found that NFIX primarily acts from enhancer regions.12,56 To study NFIX-occupied enhancers, we performed H3K27ac ChIP-seq in HPC5 cells and identified 2698 NFIX and H3K27ac overlapped peaks (Figure 3C). Motif enrichment analysis of these sites revealed enrichment for PU.1, NFE2, and RUNX2 motifs (Figure 3C), suggesting they are cofactors of NFIX. To validate co-occupancy of NFIX and the hematopoietic TFs suggested by motif analysis, we first compared global NFIX binding with ChIP-seq data from 27 published hematopoietic TFs in HPC7 cells.28,57,58 HPC7 cells have murine embryonic stem cell origins, whereas HPC5 cells are derived from murine adult BM, however, both cell lines overexpress the LIM-homeobox gene, Lhx2, and are cytokine-dependent.54,59 The genome-wide ChIP-seq signal correlation showed NFIX clustered with PU.1, as well as active histone marks such as H3K27ac, H3K9ac, and H3K4me3 (supplemental Figure 8). In contrast, there was no significant correlation between NFIX-binding signals and the repressive mark, H3K27me3 (supplemental Figure 8). For example, 1 distal regulatory element located ∼4kb upstream of the Il10ra gene is co-occupied by 10 different TFs in HPC7 (Figure 3D; supplemental Table 6), suggesting that NFIX regulates gene expression, such as Il10ra, via a protein complex. Consistently, NFIX occupancy signals are enriched in the binding sites of multiple TFs in HPC7 cells (Figure 3E).
Figure 3.

NFIX colocalizes with multiple hematopoietic transcription factors. (A) Signal plot showing enrichment of NFIX peaks in Nfix+/+ HPC5 cells as compared with NFIX signals in Nfix−/− HPC5 cells. (B) Genomic annotation pie chart showing the distribution of NFIX peaks. (C) Top left, signal plot showing enrichment of H3K27ac peaks centered around NFIX peaks in Nfix+/+ HPC5 cells. Top right, schematic detailing focus on enhancer regions, where NFIX (red track) and H3K27ac (blue track) signals overlap. Below, HOMER motif discovery analysis identifies the primary motif as NFIX, as well as multiple secondary motifs. (D) Representative tracks showing the overlap of NFIX (red track) in Nfix+/+ HPC5 cells and additional hematopoietic factors (pink tracks) in HPC7 cells at the Il10ra gene locus. (E) Signal plots showing the enrichment of hematopoietic factors, PU.1, STAT3, RUNX1, and NFE2 (data from HPC7 cells), centered around NFIX peaks (data from HPC5 cells).
Collectively, these data support a model where NFIX collaborates with additional hematopoietic TFs to regulate gene expression at enhancers.
NFIX and PU.1 physically interact and cobind genomic regions
As PU.1 was implicated as an NFIX transcriptional partner by ChIP-seq, we then focused on validating if NFIX and PU.1 physically interact in a complex via coimmunoprecipitation. In cells overexpressing MYC-tagged NFIX and FLAG-tagged PU.1, both proteins were found in the same complex recovered during coimmunoprecipitation (Figure 4A). To further understand how NFIX and PU.1 interact at DNA in an endogenous chromatin environment, we performed PU.1 ChIP-seq in HPC5 cells and identified 743 peaks cobound by NFIX and PU.1 (supplemental Table 7). To better understand the mechanism of cobinding, we investigated the motif footprints of NFIX and PU.1 using high sequencing depth ATAC-seq data from HPC5 cells (Figure 4B). The median distance between NFI and PU.1 motifs is 147bp, which is significantly less than that expected by random chance (P value = 5 × 10−127), suggesting that NFIX and PU.1 co-occupied sites likely occur within 1 nucleosome or 2 adjacent nucleosomes (supplemental Figure 9). Overall, these data reveal that NFIX and PU.1 are capable of physically interacting and can bind DNA in close proximity, supporting a model of cooperativity between the 2 TFs.
Figure 4.

NFIX and PU.1 cobind regions of open chromatin, especially super-enhancers. (A) The ability of NFIX and PU.1 to physically interact was tested by co-IP following transient transfection in 293T cells. Proteins were either unexpressed (lane 1), expressed singly (lanes 2 and 3) or coexpressed (lane 4) as indicated at top, and expression was verified (inputs). IP of MYC-NFIX was selectively able to co-IP FLAG-PU.1 (middle panel, lower blot, lane 4) and reciprocally IP of FLAG-PU.1 was able to co-IP MYC-NFIX (bottom, upper blot, lane 4). (B) Transcription factor footprint plot for NFIX and PU.1 using HPC5 ATAC-seq data. Top: average Tn5 cutting frequency. Bottom: heatmap of Tn5 cutting frequency. (C) Genomic annotation pie chart for NFIX-PU.1 peaks. (D) Signal plots of ATAC-seq and H3K27ac for NFIX-PU.1 cobound peaks and NFIX background peaks in HPC5 cells. Background represents NFIX peaks not overlapping with PU.1. Top: average of normalized signal. Bottom: heatmap of epigenetic signals on NFIX-PU.1 peaks and background peaks. (E) Venn diagram showing overlaps between super-enhancers and NFIX-PU.1 peaks. (F) Representative tracks showing NFIX and PU.1 peak overlap near target genes, Pdcd1, Socs3, and Meis1. An asterisk (∗) is included to indicate differential PU.1 peak signals in Nfix−/− HPC5 ChIP-seq samples (∗P value <1 × 10−5). co-IP, coimmunoprecipitation.
Similar to the NFIX-binding patterns, 90% of NFIX and PU.1 cobound peaks are located 2 kilobases away from the transcription start site (TSS) (Figure 4C), suggesting that these NFIX-PU.1 cobound peaks are distal regulatory elements. Compared with other NFIX peaks, NFIX-PU.1 cobound peaks show higher chromatin accessibility and signals of H3K27ac (Figure 4D). In fact, 61% (n = 451) of NFIX-PU.1 cobound peaks overlap with predicted super-enhancers based on clustering of H3K27ac signals in ATAC-seq peaks (Figure 4E). To further glean the functional importance of NFIX with PU.1, we investigated changes in PU.1 binding in the absence of NFIX using the HPC5 Nfix−/− cell line. Here, we find significant differences in the PU.1 ChIP signal between Nfix+/+ and Nfix−/− HPC5 cells. Differential PU.1 peak analysis revealed 2916 differentially bound peaks between Nfix+/+ and Nfix−/− HPC5 cells (supplemental Table 8). For example, PU.1 binding was decreased at the Pdcd, Socs3, and Meis1 loci (Figure 4F).
Collectively, this functional genomic evidence indicates that NFIX and PU.1 interact and likely function at active distal super-enhancers.
NFIX and PU.1 regulate hematopoietic progenitor differentiation
To interrogate NFIX and PU.1 coregulatory function further, we sought to generate a genome-wide cobinding profile across blood lineages. However, because of limited cell numbers, it is not possible to perform conventional ChIP-seq assays to map NFIX-PU.1–binding sites in each primitive hematopoietic population. Thus, we used chromatin accessibility and TF motifs to predict NFIX and PU.1 co-occupancy using Catchitt (Figure 5A).26 Catchitt is an iterative machine-learning algorithm that combines open chromatin signals (ie, ATAC-seq) and DNA sequence features (ie, TF motifs) to predict TF-binding sites. We first used this algorithm to train a NFIX-PU.1 co-occupancy model using ATAC-seq signals and PU.1 and NFI motifs in HPC5 cells (Figure 5A). We then used the bona fide co-occupancy sites identified by ChIP-seq to assess the prediction accuracy on a hold-out chromosome (ie, excluded from training; Figure 5A; see “Methods”). The area under the receiver operating characteristic curve (see “Methods”) was close to 0.95, confirming the good performance of the algorithm. We then used publicly available ATAC-seq (from27,28) data from different primary primitive hematopoietic populations to predict NFIX-PU.1–binding sites (Figure 5A). We then selected the top 1000 predicted co-occupancy sites for each of the 11 scRNA-seq clusters for downstream analysis (supplemental Table 9). To correlate DEG patterns with NFIX-PU.1 sites, we performed target gene assignment for the DEG identified in each scRNA-seq cluster. The NFIX-PU.1 sites were assigned to the target genes if they fell within the same topologically associating domain or if their interactions were supported by promoter-capture HiC interactions.30 For all cell clusters, a combined total of 279 direct gene targets for NFIX-PU.1 putative target sites was found (Figure 5B; supplemental Table 10). We hypothesized that if our target gene assignments were accurate, the number of target genes assigned by NFIX-PU.1 sites should be higher than the number of target genes assigned by random genomic sites. Indeed, NFIX-PU.1 sites explained DEG significantly better than random sites (P value = .0049; see “Methods”; supplemental Figure 10).
Figure 5.

Predicted target genes coregulated by NFIX and PU.1 for each hematopoietic compartment. (A) NFIX-PU.1 cobinding site prediction pipeline. The input features are normalized to ATAC-seq signals and NFIX + PU.1 motifs. The input labels are NFIX-PU.1 peaks identified from ChIP-seq in HPC5 cells. The output is cobinding probability in each 50 bp genomic bin. The tracks show high correlation between the predicted cobinding signals and observed NFIX and PU.1 signals in HPC5 on the held-out chromosome, which was chromosome 19 (see “Methods”). (B) Graph representing the total number of predicted direct target genes for NFIX-PU.1. Red bars represent the number of upregulated genes (NfixΔ/Δ vs Nfix+/+) from our scRNA-seq data. Blue bars represent the number of downregulated genes from our scRNA-seq data. (C) Significant GO terms for DEG that are putatively regulated by NFIX and PU.1.
We then investigated the relationship between NFIX-PU.1 cobinding and gene expression changes in our cell clusters (Figure 2Ai). Interestingly, in HSC and MPP clusters, most of the NFIX-PU.1–predicted target genes were upregulated in NfixΔ/Δ HSPCs (Figure 5B). In contrast, NFIX-PU.1–predicted target genes were downregulated in committed progenitors (Figure 5B). These data suggest a developmentally regulated functional switch from a repressive to an activating NFIX-PU.1–containing transcriptional complex as HSC differentiate. NFIX has been shown to function as a switch during the genetic transition from embryonic muscle to fetal muscle.60
To glean how NFIX and PU.1 collaborate to regulate hematopoiesis, we performed GO analysis on all 279 genes predicted as targets of NFIX-PU.1 for each cell cluster. Consistent with our previous findings (Figure 2C), NFIX and PU.1 were predicted to positively regulate genes associated with cytoplasmic translation and oxidative phosphorylation in LT-HSC.1, suggesting a role in translation and cellular respiration (Figure 5C; supplemental Table 11). Also consistent with our previous findings (Figure 2C), NFIX and PU.1 were predicted to regulate genes related to HSC differentiation specifically in the NfixΔ/Δ MPP cluster. All together, we observed a similar enrichment of GO terms here as in our scRNA-seq data, confirming that NFIX and PU.1 likely work together to regulate cellular respiration and hematopoietic differentiation.
Discussion
Here, we investigated how NFIX regulates hematopoietic progenitors by interrogating its role in the hematopoietic hierarchy, genome-wide occupancy profiles, putative target genes, and transcriptional partners in hematopoietic cells. We find that NFIX binds active distal regulatory elements to directly regulate genes involved in ribosome biogenesis and translation, cellular respiration, and differentiation. In addition, NFIX cooperates with multiple hematopoietic transcriptional regulators, such as PU.1, at super-enhancers. Using integrative analysis (ie, scRNA-seq, ChIP-seq, and ATAC-seq), this study represents the first comprehensive description of the transcriptional targets and coregulatory partners of NFIX in hematopoietic progenitors as well as the effects of Nfix loss on the hierarchy of BM HSPCs.
NFIX is the first member of the NFI family linked to HSPC function in vivo, necessitating a more thorough understanding of its activity.2 We show that when Nfix is deleted, there is a significant loss of LT-HSC.1, MPPs, and GMPs that is concomitant with an increase in LT-HSC.2, ST-HSC.1, MKPs, and MEPs. Based on gene expression and cell-surface CD41 protein expression, LT-HSC.2 appear to be megakaryocyte-biased. Our data suggest a shift toward megakaryocyte commitment when NFIX is lost (Figure 2C). These changes do not result from bias in the sorting and pooling of cell populations (supplemental Figure 4A-B). Rather, changes in cell population frequency may represent a novel role for NFIX in regulating hematopoietic heterogeneity, especially within the LT-HSC compartment. Further investigation of the regulatory mechanisms affected by NFIX (eg, paired scRNA-seq and scATAC-seq) will clarify its precise role in hematopoietic differentiation.
The association of NFIX with genes involved in ribosome biogenesis, translation, cellular respiration, and hematopoietic differentiation is consistent with a role for NFIX in other cell contexts and adult stem cell populations.5, 6, 7, 8, 9 For example, Nfix−/− neural progenitors exhibit perturbed differentiation.10,11 The downregulation of multiple ribosomal RNA genes suggests that biosynthetic processes may be stunted, which can result in impaired lineage commitment.61 Indeed, characterization of Nfix−/− HPC5 cells revealed significantly increased apoptosis compared with Nfix+/+ HPC5 cells (supplemental Figure 11A). This is in contrast to our previous observations, where ectopic expression of Nfix in HSPCs results in protection from apoptosis.14 In addition, we observed a significant reduction in the number of colonies formed from Nfix−/− HPC5 cells compared with Nfix+/+ HPC5 cells (supplemental Figure 11B). These data when combined, indicate the importance of NFIX for the survival and regulation of hematopoietic cells.
NFIX concentrated at superenhancers with a master regulator of hematopoiesis, PU.1. Adam et al62 also observed NFI family member occupancy at superenhancers in adult stem cells. Likewise, PU.1 has an established role in binding to active superenhancers in multiple cellular contexts.63, 64, 65 These findings suggest that NFIX and PU.1 cooperate at super-enhancer elements to putatively coregulate genes associated with hematopoietic differentiation. We also show decreased binding of PU.1 in the absence of NFIX, suggesting PU.1 may require NFIX for efficient binding. In addition, our data suggest that NFIX likely function in multiprotein complexes that include PU.1, STAT3, FLI1, LYL1, RUNX1, and GATA2, all well-known hematopoietic TFs, to coregulate gene transcription. These data and observations consistently support a model in which NFIX collaborates with multiple key hematopoietic TFs to promote the survival of primitive hematopoietic cells. One caveat for this co-occupancy analysis is that NFIX ChIP-seq was performed using HPC5 cells; in contrast, previously published ChIP-seq for other TFs was performed using HPC7 cells. We did confirm the cobinding between NFIX and PU.1 by performing PU.1 ChIP-seq using HPC5 cells, and future work should include confirmation of other cobinding factors suggested by our analysis. Future work toward understanding the precise role of NFIX will be informative, especially its function in the molecular processes of HSPC transplantation and cooperation with other key hematopoietic factors.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank the flow cytometry core, especially Richard Ashmun, Stacie Woolard, and Luisheng He. The authors acknowledge the library preparation and sequencing performed by the Hartwell Genome Sequencing Facility and VUMC Vanderbilt Core Facilities.
This research was supported by National Institutes of Health grants R01DK104028 (S.M.-F.) and R35GM133614 (Y.C.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: M.W., Y.L., A.M.-H., Q.Q., and C.P. conceptualized the study and were responsible for the methodology and validation; Y.L. worked on the software and curated the data; M.W. and Y.L. conducted the formal analysis; M.W. investigated; M.W., Y.L., S.B., and C.C. provided the resources; M.W., Y.L., W.K.C., S.M.-F., and Y.C. wrote the original draft, and reviewed and edited the manuscript; W.K.C., M.W., and Y.L. visualized; and W.K.C., S.M.-F., and Y.C. supervised.
Footnotes
∗M.W. and Y.L. contributed equally to this study.
The data sets generated and/or analyzed reported in this article have been deposited in the NCBI Gene Expression Omnibus database (accession number GSE166922).
All codes used in this study are available at https://github.com/YichaoOU/NFIX_integrative_analysis.
Data are available on request from the corresponding author, Shannon McKinney-Freeman (shannon.mckinney-freeman@stjude.org).
The full-text version of this article contains a data supplement.
Contributor Information
Yong Cheng, Email: yong.cheng@stjude.org.
Shannon McKinney-Freeman, Email: shannon.mckinney-freeman@stjude.org.
Supplementary Material
References
- 1.D'Souza A, Lee S, Zhu X, Pasquini M. Current use and trends in hematopoietic cell transplantation in the United States. Biol Blood Marrow Transplant. 2017;23(9):1417–1421. doi: 10.1016/j.bbmt.2017.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holmfeldt P, Pardieck J, Saulsberry AC, et al. Nfix is a novel regulator of murine hematopoietic stem and progenitor cell survival. Blood. 2013;122(17):2987–2996. doi: 10.1182/blood-2013-04-493973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249(1):31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 4.Piper M, Gronostajski R, Messina G. Nuclear factor one X in development and disease. Trends Cell Biol. 2019;29(1):20–30. doi: 10.1016/j.tcb.2018.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Harris L, Genovesi LA, Gronostajski RM, Wainwright BJ, Piper M. Nuclear factor one transcription factors: divergent functions in developmental versus adult stem cell populations. Dev Dyn. 2015;244(3):227–238. doi: 10.1002/dvdy.24182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang F, Liang M, Zhao C, Fu Y, Yu S. NFIC promotes the vitality and osteogenic differentiation of rat dental follicle cells. J Mol Histol. 2019;50(5):471–482. doi: 10.1007/s10735-019-09841-z. [DOI] [PubMed] [Google Scholar]
- 7.Chang C-Y, Pasolli HA, Giannopoulou EG, et al. NFIB is a governor of epithelial–melanocyte stem cell behaviour in a shared niche. Nature. 2013;495(7439):98–102. doi: 10.1038/nature11847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52(6):953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 9.Starnes LM, Sorrentino A, Pelosi E, et al. NFI-A directs the fate of hematopoietic progenitors to the erythroid or granulocytic lineage and controls beta-globin and G-CSF receptor expression. Blood. 2009;114(9):1753–1763. doi: 10.1182/blood-2008-12-196196. [DOI] [PubMed] [Google Scholar]
- 10.Heng YH, McLeay RC, Harvey TJ, et al. NFIX regulates neural progenitor cell differentiation during hippocampal morphogenesis. Cereb Cortex. 2014;24(1):261–279. doi: 10.1093/cercor/bhs307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heng YH, Zhou B, Harris L, et al. NFIX regulates proliferation and migration within the murine SVZ neurogenic niche. Cereb Cortex. 2015;25(10):3758–3778. doi: 10.1093/cercor/bhu253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martynoga B, Mateo JL, Zhou B, et al. Epigenomic enhancer annotation reveals a key role for NFIX in neural stem cell quiescence. Genes Dev. 2013;27(16):1769–1786. doi: 10.1101/gad.216804.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iacobucci I, Qu C, Varotto E, et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood. 2021;137(12):1628–1640. doi: 10.1182/blood.2020009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hall T, Walker M, Ganuza M, et al. Nfix promotes survival of immature hematopoietic cells via regulation of c-Mpl. Stem Cell. 2018;36(6):943–950. doi: 10.1002/stem.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8(1):14049–14060. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hao Y, Hao S, Andersen-Nissen E, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587.e3529. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venables WN, Ripley BD. Springer; 2002. Modern Applied Statistics With S. [Google Scholar]
- 18.Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 19.Morgan-Wall T. Create Maps and Visualize Data in 2D and 3D. 2023. https://www.rayshader.com/ Accessed 3 January 2023.
- 20.Duong T, Goud B, Schauer K. Closed-form density-based framework for automatic detection of cellular morphology changes. Proc Natl Acad Sci USA. 2012;109(22):8382–8387. doi: 10.1073/pnas.1117796109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao J, Jaffe A, Li H, et al. Detection of differentially abundant cell subpopulations in scRNA-seq data. Proc Natl Acad Sci USA. 2021;118(22) doi: 10.1073/pnas.2100293118. e2100293118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homer motif database. http://homer.ucsd.edu/homer/custom.motifs Updated October 2019.
- 24.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27(7):1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramírez F, Dündar F, Diehl S, Grüning BA, Manke T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014;42(W1):W187–W191. doi: 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keilwagen J, Posch S, Grau J. Accurate prediction of cell type-specific transcription factor binding. Genome Biol. 2019;20(1):9. doi: 10.1186/s13059-018-1614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu X, Wu C, Bhavanasi D, Wang H, Gregory BD, Huang J. Chromatin dynamics during the differentiation of long-term hematopoietic stem cells to multipotent progenitors. Blood Adv. 2017;1(14):887–898. doi: 10.1182/bloodadvances.2016003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang G, Keller CA, Heuston EF, et al. An integrative view of the regulatory and transcriptional landscapes in mouse hematopoiesis. Genome Res. 2020;30(3):472–484. doi: 10.1101/gr.255760.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiang G, Keller CA, Giardine B, et al. S3norm: simultaneous normalization of sequencing depth and signal-to-noise ratio in epigenomic data. Nucleic Acids Res. 2020;48(8) doi: 10.1093/nar/gkaa105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C, Yu W, Tober J, et al. Spatial genome re-organization between fetal and adult hematopoietic stem cells. Cell Rep. 2019;29(12):4200–4211. e4207. doi: 10.1016/j.celrep.2019.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waltman L, van Eck NJ. A smart local moving algorithm for large-scale modularity-based community detection. Eur Phys J B. 2013;86(11):1–14. [Google Scholar]
- 33.Zhang X, Lan Y, Xu J, et al. CellMarker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res. 2018;47(D1):D721–D728. doi: 10.1093/nar/gky900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells. 2007;25(6):1339–1347. doi: 10.1634/stemcells.2006-0644. [DOI] [PubMed] [Google Scholar]
- 35.Gekas C, Graf T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood. 2013;121(22):4463–4472. doi: 10.1182/blood-2012-09-457929. [DOI] [PubMed] [Google Scholar]
- 36.Mori Y, Chen JY, Pluvinage JV, Seita J, Weissman IL. Prospective isolation of human erythroid lineage-committed progenitors. Proc Natl Acad Sci U S A. 2015;112(31):9638–9643. doi: 10.1073/pnas.1512076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doré LC, Crispino JD. Transcription factor networks in erythroid cell and megakaryocyte development. Blood. 2011;118(2):231–239. doi: 10.1182/blood-2011-04-285981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paul F, Arkin Ya, Giladi A, et al. Transcriptional heterogeneity and lineage commitment in myeloid progenitors. Cell. 2015;163(7):1663–1677. doi: 10.1016/j.cell.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 39.McKinney-Freeman S, Cahan P, Li H, et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell. 2012;11(5):701–714. doi: 10.1016/j.stem.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Komorowska K, Doyle A, Wahlestedt M, et al. Hepatic leukemia factor maintains quiescence of hematopoietic stem cells and protects the stem cell pool during regeneration. Cell Rep. 2017;21(12):3514–3523. doi: 10.1016/j.celrep.2017.11.084. [DOI] [PubMed] [Google Scholar]
- 41.Kataoka K, Sato T, Yoshimi A, et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med. 2011;208(12):2403–2416. doi: 10.1084/jem.20110447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pineault N, Helgason CD, Lawrence HJ, Humphries RK. Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp Hematol. 2002;30(1):49–57. doi: 10.1016/s0301-472x(01)00757-3. [DOI] [PubMed] [Google Scholar]
- 43.Ninos JM, Jefferies LC, Cogle CR, Kerr WG. The thrombopoietin receptor, c-Mpl, is a selective surface marker for human hematopoietic stem cells. J Transl Med. 2006;4(1):9. doi: 10.1186/1479-5876-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Wang J, Xie M, et al. Early megakaryocyte lineage-committed progenitors in adult mouse bone marrow. bioRxiv. Preprint posted online 6 August 2021 doi: 10.1097/BS9.0000000000000187. doi:2021.2008.2006.455158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiMartino JF, Selleri L, Traver D, et al. The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood. 2001;98(3):618–626. doi: 10.1182/blood.v98.3.618. [DOI] [PubMed] [Google Scholar]
- 46.Liu S, McConnell SC, Ryan TM. Erythropoiesis in the absence of adult hemoglobin. Mol Cell Biol. 2013;33(11):2241–2251. doi: 10.1128/MCB.01734-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Signer RA, Magee JA, Salic A, Morrison SJ. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature. 2014;509(7498):49–54. doi: 10.1038/nature13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morganti C, Bonora M, Ito K, Ito K. Electron transport chain complex II sustains high mitochondrial membrane potential in hematopoietic stem and progenitor cells. Stem Cell Res. 2019;40:101573. doi: 10.1016/j.scr.2019.101573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20(8):833–846. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Becker-Herman S, Rozenberg M, Hillel-Karniel C, et al. CD74 is a regulator of hematopoietic stem cell maintenance. PLoS Biol. 2021;19(3):e3001121. doi: 10.1371/journal.pbio.3001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Opferman JT, Iwasaki H, Ong CC, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 2005;307(5712):1101–1104. doi: 10.1126/science.1106114. [DOI] [PubMed] [Google Scholar]
- 52.Lantner F, Starlets D, Gore Y, et al. CD74 induces TAp63 expression leading to B-cell survival. Blood. 2007;110(13):4303–4311. doi: 10.1182/blood-2007-04-087486. [DOI] [PubMed] [Google Scholar]
- 53.Holmfeldt P, Ganuza M, Marathe H, et al. Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J Exp Med. 2016;213(3):433–449. doi: 10.1084/jem.20150806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinto do OP, Richter K, Carlsson L. Hematopoietic progenitor/stem cells immortalized by Lhx2 generate functional hematopoietic cells in vivo. Blood. 2002;99(11):3939–3946. doi: 10.1182/blood.v99.11.3939. [DOI] [PubMed] [Google Scholar]
- 55.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann Appl Stat. 2011;5(3):1752–1779. [Google Scholar]
- 56.Fraser J, Essebier A, Brown AS, et al. Common regulatory targets of NFIA, NFIX and NFIB during postnatal cerebellar development. Cerebellum. 2020;19(1):89–101. doi: 10.1007/s12311-019-01089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilson NK, Foster SD, Wang X, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7(4):532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- 58.Wilson NK, Schoenfelder S, Hannah R, et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 2016;127(13):e12–e23. doi: 10.1182/blood-2015-10-677393. [DOI] [PubMed] [Google Scholar]
- 59.Pinto do OP, Kolterud A, Carlsson L. Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. EMBO J. 1998;17(19):5744–5756. doi: 10.1093/emboj/17.19.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Messina G, Biressi S, Monteverde S, et al. Nfix regulates fetal-specific transcription in developing skeletal muscle. Cell. 2010;140(4):554–566. doi: 10.1016/j.cell.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 61.Khajuria RK, Munschauer M, Ulirsch JC, et al. Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell. 2018;173(1):90–103.e119. doi: 10.1016/j.cell.2018.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adam RC, Yang H, Ge Y, et al. NFI transcription factors provide chromatin access to maintain stem cell identity while preventing unintended lineage fate choices. Nat Cell Biol. 2020;22(6):640–650. doi: 10.1038/s41556-020-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carey HA, Hildreth BE, Geisler JA, et al. Enhancer variants reveal a conserved transcription factor network governed by PU.1 during osteoclast differentiation. Bone Research. 2018;6(1):8. doi: 10.1038/s41413-018-0011-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Staber PB, Zhang P, Ye M, et al. Sustained PU.1 levels balance cell-cycle regulators to prevent exhaustion of adult hematopoietic stem cells. Mol Cell. 2013;49(5):934–946. doi: 10.1016/j.molcel.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosenbauer F, Owens BM, Yu L, et al. Lymphoid cell growth and transformation are suppressed by a key regulatory element of the gene encoding PU.1. Nat Genet. 2006;38(1):27–37. doi: 10.1038/ng1679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.