

Abstract
α-Synuclein is an abundant protein at the neuronal synapse that has been implicated in Parkinson’s disease for over 25 years and characterizes the hallmark pathology of a group of neurodegenerative diseases now known as the synucleinopathies. Physiologically, α-synuclein exists in an equilibrium between a synaptic vesicle membrane-bound α-helical multimer and a cytosolic largely unstructured monomer. Through its membrane-bound state, α-synuclein functions in neurotransmitter release by modulating several steps in the synaptic vesicle cycle, including synaptic vesicle clustering and docking, SNARE complex assembly, and homeostasis of synaptic vesicle pools. These functions have been ascribed to α-synuclein’s interactions with the synaptic vesicle SNARE protein VAMP2/synaptobrevin-2, the synaptic vesicle-attached synapsins, and the synaptic vesicle membrane itself. How α-synuclein affects these processes, and whether disease is due to loss-of-function or gain-of-toxic-function of α-synuclein remains unclear. In this review, we provide an in-depth summary of the existing literature, discuss possible reasons for the discrepancies in the field, and propose a working model that reconciles the findings in the literature.
Keywords: membrane binding, neuronal survival, Parkinson’s disease, synaptic vesicle, neurotransmitter release
Introduction
α-Synuclein is an abundant presynaptic protein that binds to synaptic vesicles and functions in neurotransmitter release by modulating synaptic vesicle pools and chaperoning SNARE complex assembly through its interaction with vesicle-associated membrane protein 2 (VAMP2)/synaptobrevin-2. Pathologically, increased levels as well as mutations in α-Synuclein are linked to Parkinson’s disease and related synucleinopathies, and SNARE dysfunction is a common occurrence in synucleinopathies. Here, we discuss the subcellular pools and functional conformations of α-Synuclein, its binding modes on synaptic vesicles, its role in chaperoning SNARE complex assembly and clustering synaptic vesicles, how this activity affects neurotransmission and the long-term functioning of neurons, and finally, we examine the role of SNARE dysfunction in synucleinopathies.
α-Synuclein exists in different subcellular pools
α-Synuclein protein is comprised of 140 amino acids and is encoded by the SCNA gene. It was initially identified in a screen for synaptic vesicle proteins, and named for its synaptic and nuclear localizations,1 though subsequent studies have demonstrated that in the brain, it is found mainly in the presynaptic terminal of neurons.2–3 There has been increased interest in α-Synuclein since the turn of the century because of two key findings implicating it in human disease: (1) α-Synuclein is abundant in Lewy bodies in sporadic cases of Parkinson’s Disease (PD),4 and (2) the missense mutation A53T in α-Synuclein causes familial PD.5 Subsequently, additional mutations in α-Synuclein, including A30G,6 A30P,7 E46K,8 H50Q,9 G51D,10–11 A53V,12 A53E,13 T72M,14 and E83Q15 (Figure 1), as well as gene duplication and triplication,16–18 have been implicated in familial PD. Furthermore, genome-wide association studies show that common variability in the SNCA locus is associated with PD risk.19
Figure 1.
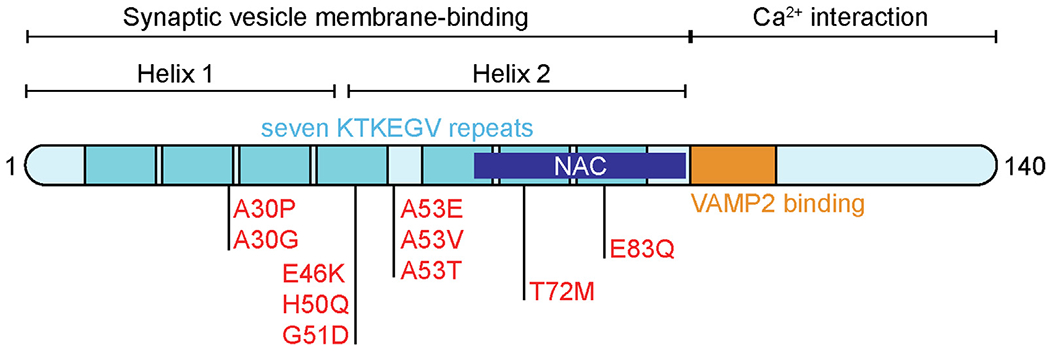
Structure of α-synuclein. The N-terminal domain is comprised of seven imperfect KTKEGV repeats that form two amphipathic α-helices upon binding to synaptic vesicle membranes. This region also contains the aggregation-prone NAC domain, as well as all disease-linked mutations. The C-terminal domain of α-synuclein is not involved in membrane binding and mediates interactions with calcium as well as the SNARE protein VAMP2/synaptobrevin-2.
α-Synuclein is intrinsically disordered in that it does not have a fixed three-dimensional structure in aqueous solution20–21 (Figure 2). The N-terminal domain is positively charged, lysine-rich, and includes a seven 11-residue repeat sequence that forms an amphipathic α-helix upon lipid-binding22 and membrane-binding23 (Figure 1 and 2). It also contains the non-amyloid-beta component (NAC) domain which was identified in Alzheimer’s disease (AD) neuropathology24 that is aggregation-prone.25–26 The C-terminal domain is negatively charged and glutamate-rich,27–28 and is involved in interactions with proteins, DNA, ions, polyamines and metals29–34 (Figure 1). The C-terminal domain also contains multiple phosphorylation sites, including at tyrosine residues 125, 133 and 136 and serine residue 129, of which S129 has been associated with pathology.35–37
Figure 2.
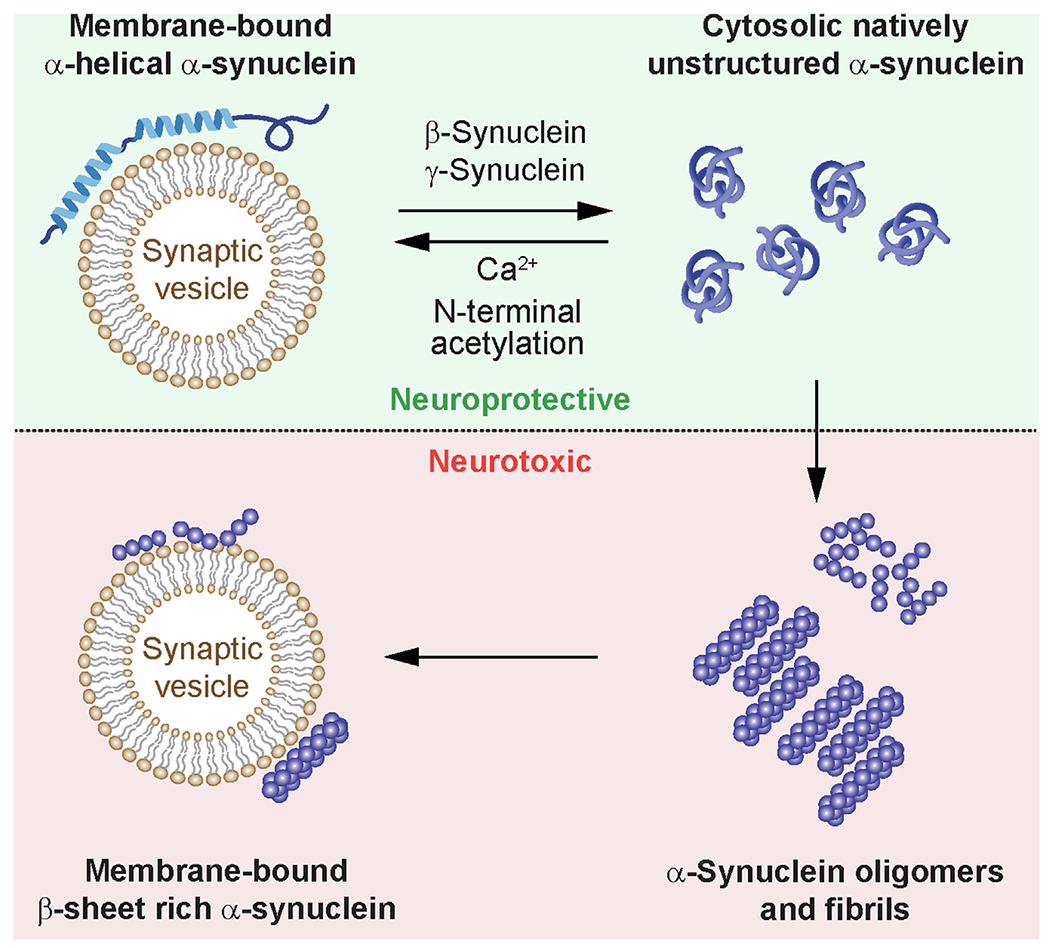
Implications of membrane binding of α-synuclein for function and disease. Under physiological conditions, α-synuclein exists in equilibrium between a membrane-bound α-helical conformation on synaptic vesicles and a natively unstructured state in the cytosol. Membrane binding is facilitated by calcium and N-terminal acetylation, whereas the cytosolic pool is increased in presence of β-synuclein or γ-synuclein. α-Synuclein oligomers and fibrils form from the unstructured cytosolic state, leading to neuropathology. Membrane binding of α-synuclein is neuroprotective in its α-helical multimeric state, while association of β-sheet rich oligomeric or fibrillar α-synuclein with synaptic vesicles is linked to neurotoxicity.
In its native state, α-Synuclein exists as a largely unstructured monomer in the cytosol.38–39 Upon membrane binding, it undergoes a conformational change to adopt an α-helical structure (Figure 2). α-Synuclein interacts with lipid membranes by association with negatively charged, acidic phospholipids, and its conformation upon binding depends on membrane curvature. A single α-helix forms on lower curvature membranes,40 while a broken helix forms on higher curvature membranes,41–42 mediating its physiologically relevant association with synaptic vesicle membranes. Membrane-bound α-Synuclein forms multimers, a conformation that clusters synaptic vesicles and promotes SNARE complex assembly43–44 (Figure 3).
Figure 3.
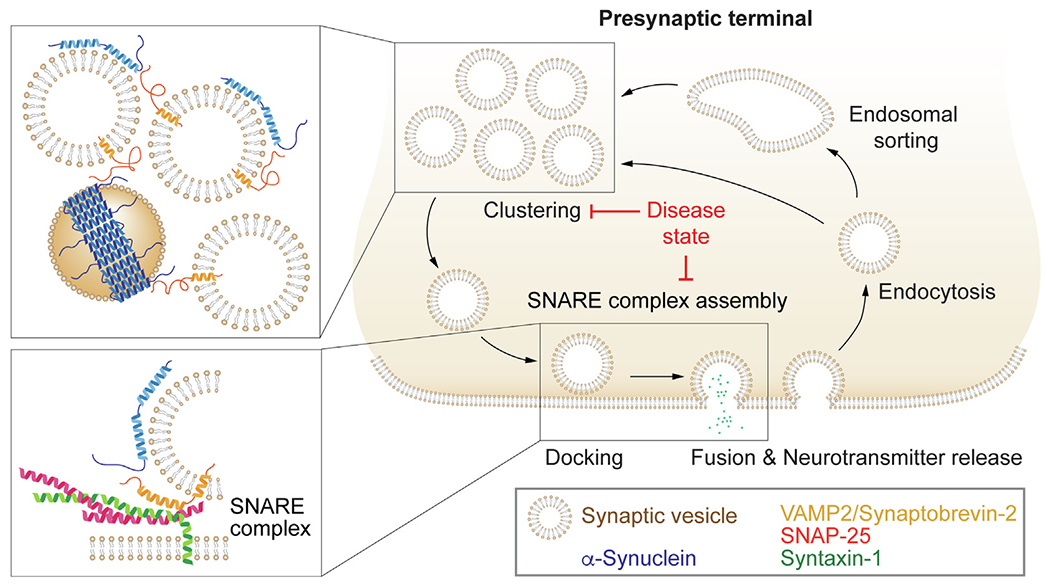
SNARE-dependent effects of α-synuclein on the synaptic vesicle cycle. Upon initiation of an action potential in the presynaptic terminal, synaptic vesicles docked to the plasma membrane fuse to release neurotransmitters to propagate the signal to the next neuron. Synaptic vesicle fusion is mediated by formation of the synaptic SNARE complex, composed of syntaxin-1 and SNAP-25 on the presynaptic plasma membrane, and VAMP2/synaptobrevin-2 on the synaptic vesicle. Following fusion, synaptic vesicle constituents are retrieved via endocytosis. Simultaneous binding of α-synuclein multimers to synaptic vesicle membranes and the synaptic vesicle protein VAMP2/synaptobrevin-2 clusters synaptic vesicles (top inset) and chaperones SNARE complex assembly (bottom inset). In diseased states, pathological α-synuclein or lack of physiologically functional α-synuclein due to recruitment into pathological α-synuclein aggregates results in impaired or dysfunctional synaptic vesicle clustering and reduced SNARE complex assembly. This imbalance in the synaptic vesicle cycle affects long-term neuron function and survival.
The equilibrium between the cytosolic and membrane-bound pool of α-Synuclein is highly relevant for pathological and physiological conditions, although the role of membrane binding in pathology is debated. Membrane binding of α-Synuclein has been shown to be protective against aggregation, whereas the soluble, cytosolic pool of α-Synuclein is prone to aggregation45 (Figure 2). Notably, all disease-associated α-Synuclein missense mutations are in the membrane-binding region, with some shown to decrease lipid binding46–49 (Figure 1). However, membrane binding has also been shown to induce aggregation and pathology.50–53 In pathological conditions, α-Synuclein is found in oligomeric conformations with β-sheet secondary structure and in fibrillar conformations,54–55 which are believed to drive the neurotoxicity leading to neuronal cell death and clinical manifestations of PD (Figure 2).
What are the reasons underlying the apparent contradictions about the role of α-Synuclein in membrane binding? Early reports demonstrated that in a cell-free system, α-Synuclein aggregation occurred in the membrane fraction but not in the cytosol fraction, and that addition of the cytosolic fraction accelerated aggregation.50 Further, α-Synuclein oligomers induced by in vitro chemical crosslinking associated preferentially with lipid droplets and cell membranes in cell culture.53 In contrast, in presence of monomeric α-Synuclein, membrane-binding inhibited α-Synuclein oligomerization.56 Understanding the conformational state of α-Synuclein is important to reconciling these results. Follow-up studies showed that induction of the α-helical conformation of α-Synuclein, which is associated with membranes in physiological states, was strongly correlated with inhibition of aggregation and fibril formation.57 To address the interplay of membrane binding and neuropathology directly, introduction of α-Synuclein mutations that reduce or block membrane binding led to increased aggregation in situ, and virus-mediated striatal expression of these mutants led to accelerated motor dysfunction and neurotoxicity of dopaminergic neurons in vivo compared to neurons expressing wildtype α-Synuclein.45,58 Taken together, α-Synuclein membrane binding is protective in physiological states, but correlates with aggregation after pathological changes in α-Synuclein such as oligomerization have already occurred (Figure 2).
α-Synuclein functions on the synaptic vesicle membrane
In development, α-Synuclein is initially somatic and concentrates in presynaptic terminals over time.59–62 Of all the synaptic proteins, it is one of the last to become enriched in synaptic terminals, suggesting a role in maintenance of synaptic connections rather than development of synapses. Localization of α-Synuclein in the mature brain is primarily presynaptic.3 While there is also evidence for localization on mitochondria and other organelles including the nucleus, this occurs primarily in overexpression conditions.63–65 α-Synuclein lacks a transmembrane domain, and in fractionation experiments, α-Synuclein dissociates easily from membranes and was therefore not present in early isolations of synaptic fractions.66 However, in a follow-up study with more sensitive detection techniques, α-Synuclein was detected both in isolated synaptosomes and on purified synaptic vesicles.67
How is synaptic vesicle binding mediated? Interactions at both the N-terminal and C-terminal domains of α-Synuclein are important. As discussed above, ionic interactions between negatively charged acidic lipid head groups and positively charged lysine residues in the N-terminal domain68–70 induce a conformational change in α-Synuclein that is favorable for association with synaptic vesicles.41 Certain post-translational modifications, including N-terminal acetylation of α-Synuclein, induce α-helicity and lead to an increase in its membrane binding.71 In contrast, heteromerization of the N-terminal lipidbinding domain of α-Synuclein with those of β-synuclein or γ-synuclein reduces synaptic vesicle binding of α-Synuclein in a concentration-dependent manner.72
The C-terminal domain of α-Synuclein interacts with proteins, DNA, ions, polyamines, and metals.29–34 C-terminal domain calcium-binding increases α-Synuclein-synaptic vesicle interactions and vesicle clustering at the synaptic terminal.73 The C-terminal domain also binds to VAMP2/synaptobrevin-2,74–75 and this interaction is important for synaptic vesicle clustering and SNARE complex assembly45,58,74–77 which is discussed further below. Functional consequences of the other interactions remain unclear and require further studies.
α-Synuclein chaperones SNARE complex assembly and clusters synaptic vesicles
Despite the body of research concerning α-Synuclein’s localization and binding activity at the presynaptic terminal, there is an ongoing debate over its physiological function. Neurotransmission requires a tightly coordinated sequence of events at the nerve terminal (reviewed in78), canonically involving (1) clustering of a reserve pool of synaptic vesicles near synapses which are crosslinked by synapsins or by liquid–liquid phase separation79 and released upon phosphorylation of synapsin80, (2) docking of synaptic vesicles at active zones81 that constitute the readily releasable pool82, (3) formation of the ternary synaptic SNARE complex, that is composed of syntaxin-1, SNAP-25, and VAMP2/synaptobrevin-2,83 (4) fusion of synaptic vesicles with the presynaptic plasmamembrane following calcium influx,84 and (5) disassembly of the SNARE complex mediated by SNAPs and NSF, and endosomal recycling of synaptic vesicles (Figure 3).85 The readily releasable pool undergoes exocytosis first upon stimulation, and is replenished by the reserve pool of synaptic vesicles upon depletion in the setting of sustained activity.86–89
The synuclein genes have only been identified in vertebrates, suggesting that they are not required for neurotransmission. However, α-Synuclein has been shown to modulate several steps in the synaptic vesicle cycle. Specifically, it has been implicated in synaptic vesicle clustering,44,77,90 synaptic vesicle docking and SNARE complex formation,43,74 and recycling pool homeostasis44,75–76,91 (Figure 3). These functions are mediated by α-Synuclein’s interactions with membranes,43,58,77 with VAMP2/synaptobrevin-274–75,77, and synapsins.92–93
How is α-Synuclein exerting these functions on a molecular level? α-Synuclein has been shown to chaperone SNARE complex assembly via binding to the SNARE protein VAMP2/synaptobrevin-2, where amino acids 96–140 of α-Synuclein interact with amino acids 1–28 of VAMP2/synaptobrevin-2.74 However, another study found that the C-terminus, specifically amino acids 110–140, were not required for the synaptic function of α-Synuclein.91 A subsequent study reconciled these differences, confirming the importance of the functional interaction between α-Synuclein and VAMP2/synaptobrevin-2, and demonstrated that amino acids 96–104 are required for binding to α-Synuclein.75 In cultured neurons, visualization of fluorescently tagged α-Synuclein in photobleaching experiments showed a similar abundance of α-Synuclein per vesicle as VAMP2/synaptobrevin-2, and binding of recombinantly expressed α-Synuclein to VAMP2/synaptobrevin-2.94 Taken together, these results suggest that α-Synuclein-V AMP2/synaptobrevin-2 binding is essential for the maintenance of synaptic vesicle function.74–75
While the interaction between α-Synuclein and VAMP2/synaptobrevin-2 is established, the direction of the effect of this interaction on SNARE complex assembly is debated. In an in vitro liposome fusion setting, α-Synuclein oligomers and monomers cooperatively inhibited SNARE-mediated vesicle membrane fusion by inducing liposome clustering.95 Taken in light of previous findings showing that physiological α-Synuclein promotes SNARE complex formation while pathological oligomers have a neurotoxic effect, these results support the hypothesis that the SNARE complex attenuation may represent a neurotoxic gain-of-function by α-Synuclein oligomers. This interaction may furthermore alternately enhance or inhibit SNARE complex formation depending on the presence of phosphatidylserine and either v-SNARE or t-SNARE proteins on the membrane of interest.96
In contrast, other data indicate that α-Synuclein’s synaptic function is mediated only by its membrane-binding domain in a concentration-dependent manner, rather than by its binding to VAMP2/synaptobrevin-2. Attenuation of SNARE-dependent lipid membrane mixing correlated positively with α-Synuclein concentrations in vitro, while in vivo experiments using C-terminally truncated and point mutated α-Synuclein demonstrated that this activity is reliant on α-Synuclein’s membrane binding ability, rather than its VAMP2/synaptobrevin-2 binding.97 However, another in vitro study found that large α-Synuclein oligomers preferentially bound to the N-terminal domain of VAMP2/synaptobrevin-2, just like the monomer, and prevented SNARE complex formation, while α-Synuclein monomers had a negligible effect.98 These results may help explain why oligomers seem to have a neurotoxic effect on dopaminergic neurons, while the monomeric form seems to have no such effect. Another study found that α-Synuclein inhibited SNARE-mediated vesicle fusion through modulating properties of the lipid bilayer, acting to increase the membrane fusion energy barrier by a variety of lipid-interaction mechanisms such as thinning membranes and increasing membrane rigidity without a direct interaction between α-Synuclein and any of the SNARE proteins.99 Supportively, α-Synuclein was found to block SNARE complex formation without directly binding to SNARE proteins by buffering arachidonic acid, which has been shown to promote SNARE complex formation and decrease its availability to SNARE proteins.100 While these disparate experimental findings seem contradictory, many of these studies were done in vitro, using purified proteins and liposomes at non-physiological concentrations. The applicability of these findings to neurons remains unclear.
Importantly, the effects of α-Synuclein on SNARE complex formation and synaptic vesicle clustering are mediated by synaptic vesicle membrane-bound α-Synuclein, and specifically by α-helical α-Synuclein multimers which assemble upon membrane binding.43–44 These data support a model where synaptic vesicle clustering could be induced by the interaction of α-Synuclein multimers with VAMP2/synaptobrevin-2 on neighboring vesicles.44,75 Simultaneous binding of α-Synuclein to VAMP2/synaptobrevin-2 and synaptic vesicle phospholipids has been shown to trigger clustering of synaptic vesicles.44,77 Supportively, electron microscopy and cryoelectron tomography demonstrated that deletion of synucleins decreased the inter-linking of synaptic vesicles, and increased vesicle tethering to the active zone,101 suggesting that α-Synuclein may modulate pools of synaptic vesicles at the synapse in part through synaptic vesicle clustering (Figure 3). Overexpression of α-Synuclein in a range predicted for gene multiplication in humans reduced the size of the reserve pool and the number of docked vesicles, suggesting that increased expression without overt toxicity leads to a physiologic defect in synaptic vesicle recycling.91 Antibody-mediated disruption of α-Synuclein lead to a dose-dependent loss of synaptic vesicles in the reserve pool, as well as depletion of the docked readily releasable pool.90 Taken together, these results suggest that α-Synuclein regulates vesicle recycling and docking in a dose-dependent manner, as both α-Synuclein loss-of-function and gain-of-function can impair vesicle recycling.
Synaptic vesicle clustering incorporates further interactions with additional proteins, including synapsins. Canonically, synapsin is essential for synaptic vesicle clustering as antibody-mediated blockade of synapsin function depletes the reserve pool while leaving the readily releasable pool intact.86 Using synapsin triple-knockout mice, it was shown that synapsins may enhance clustering by targeting α-Synuclein to synapses, and that this interaction is also required for α-Synuclein-mediated effects on endocytosis.92 Upon stimulation, synapsins dissociate from synaptic vesicles and disperse, followed by reclustering after the cessation of synaptic activity.102 In a similar manner, α-Synuclein is highly mobile at the synapse, and disperses from the nerve terminal in response to activity and following exocytosis.103 Synapsin regulates themobility of synaptic vesicle pools,104 and further, a synapsin liquid phase has been shown to mediate synaptic vesicle clustering.105–106 This liquid phase recruits α-Synuclein,107 with a concentration-dependent effect on synaptic vesicle clustering and requiring a specific ratio of α-Synuclein and synapsin-1.93
Overall, a model emerges in which α-Synuclein induces synaptic vesicle clustering and crosslinking through a combination of lipid membrane and VAMP2/synaptobrevin-2 binding, which then reduces SNARE complex formation acutely by decreasing the number of synaptic vesicles available for docking at the presynaptic membrane. In the long term or during sustained activity, however, this activity of α-Synuclein may indirectly promote SNARE complex formation by providing a large reserve pool of vesicles, allowing for sustained SNARE complex formation and transmitter release. This model is supported by the activity-dependence of SNARE complex assembly in synuclein knockout mice.74
More research is required to fully characterize the nature and function of α-Synuclein-SNARE interactions and their effect on synaptic vesicle clustering and neurotransmitter release. Currently, this functionality appears to depend on a wide variety of factors, including but not limited to α-Synuclein and SNARE protein concentrations, α-Synuclein conformations, α-Synuclein post-translational modifications, lipid membrane structure and composition, and the experimental system used to study this interaction. The disparate and in places conflicting findings from the literature reviewed here may be explained by several factors. Foremost, because α-Synuclein is an intrinsically disordered protein, its concentration and local environment can greatly affect its structure and function. This is compounded by its ability to form both oligomers and aggregates, which may further alter its function, potentially leading to both gain- and loss-of-function. Many of the assays performed in recent studies are in vitro and may not accurately reflect α-Synuclein’s physiological state. Similarly, overexpression studies may result in pathological oligomeric α-Synuclein species, which may obscure any functionally relevant readout. Therefore, future studies focusing on physiologically relevant settings with advances in microscopy are expected to improve our understanding of this interaction. As argued above, such future studies may be able to better accommodate the apparent conflicts in the literature.
α-Synuclein’s SNARE complex chaperoning activity is important for the long-term functioning of the nerve terminal
One of the initial descriptions of α-Synuclein protein was in the zebra finch, termed synelfin, which showed changes in its gene expression during a critical song-learning period suggesting a role in synaptic plasticity.22 Later it was shown that αβγ-synuclein knockout mice have an age- and activity-dependent reduction in synaptic SNARE complex levels, suggesting a role for synucleins in the long-term function of the nerve terminal.74 The role of α-Synuclein in mediating these functions has been investigated using various models modulating α-Synuclein expression, and taken together, strongly suggest that α-Synuclein modulates synaptic function and neuronal survival.
Yet, the direction of α-Synuclein’s effects is controversial. α-Synuclein has been reported to have (1) no effect on neurotransmitter release,74,108 (2) to enhance synaptic transmission,109–113 or (3) to inhibit synaptic transmission.112,114–121 The cumulative evidence indicates that α-Synuclein plays a direct role in regulating synaptic vesicle pools in the presynapse, as detailed in the previous section, with downstream, indirect effects on neurotransmission itself depending on the model system and conditions interrogated. Indeed, given the absence of α-Synuclein in invertebrates and mild phenotypes in knockout models, α-Synuclein is not required for neurotransmission in general, nor dopaminergic neurotransmission specifically.122
Here, we will focus on knockout studies to exclude the confounding effect of potential toxicity from overexpression studies. The majority of studies suggest an inhibitory role on neurotransmitter release at dopaminergic synapses. Synapses of α-Synuclein knockout mice showed a faster recovery of dopamine release following stimulation-induced depression, indicating an inhibitory effect on the recovery of dopamine release.114 Of note, these mice had reduced levels of striatal dopaminewithout affecting the density of projections or dopamine reuptake, suggesting that the diminished recovery from depression observed in the knockout mice is due to a reduction of the readily releasable pool of dopamine-containing synaptic vesicles.114 In agreement, α-Synuclein knockout mice demonstrated strong facilitation of dopamine release with repeated stimulation that was not seen in wild-type mice, and deletion of all three members of the synuclein family showed greater dopamine release in dorsal striatal axons but not ventral striatal axons.121 However, studies of hippocampal synapses in α-synuclein knockout and αβγ-knockout mice showed either synaptic facilitation,109–113 or no effect at all.74,108
One explanation for these discrepancies is that α-synuclein may modulate synaptic function in an activity-dependent manner. Using αβγ-synuclein knockout mice, synuclein was shown to facilitate dopaminergic neurotransmission during bursts of activity separated by short intervals, and to depress neurotransmission during bursts of activity separated by long intervals.123 However, it remains puzzling that multiple lines of evidence suggest that there is no effect of synuclein knockout on glutamatergic neurotransmission.74,108,122 As it stands, there is no consensus for a mechanism on how α-synuclein directly acts on neurotransmitter release, in part due to the different regions interrogated, the interrogation of single synapses versus field recordings, the different stimulation protocols used, and the use of constitutive knockouts which may lead to compensation artifacts. Further experiments evaluating acute knockdown in different brain regions may be beneficial for clarification.
What is the role of α-synuclein over age and for neuronal survival? α-Synuclein knockouts have a reduction in dopaminergic neurons in the substantia nigra in some models,124 especially with aging,125 but these findings are not recapitulated in other α-synuclein knockouts.114–115 However, analysis of mice of ages ranging from only a few months to less than a year may have hampered detection of neurodegeneration, in addition to potential compensatory changes in constitutive knockout animals. Of interest, α-synuclein knockout mice are resistant to MPTP-induced dopaminergic neurodegeneration,126–128 suggesting that the normal function of α-synuclein is important to dopaminergic neuron viability and vulnerability in particular. Dopamine is thought to stabilize α-synuclein protofibrils and thus facilitate the selective degeneration of these neurons.129 Indeed, overexpression of α-synuclein in chromaffin cells and a pheochromocytoma cell line led to elevated catechol concentrations, and this effect was even more pronounced in presence of overexpressed pathogenic A30P and A53T α-synuclein.130 Thus, the toxic effects of α-synuclein likely rely on interactions with specific target neurons.
Importantly, constitutive removal of α-synuclein may lead to compensation over development, as knockdown of α-synuclein using virus-mediated expression of shRNA in the substantia nigra of adult animals led to rapid neuron loss beyond a certain threshold of reduced expression in both rodents and non-human primates.131–134 This effect was not seen in the hippocampus, although the degree of knockdown may not have been sufficient.135 Taken together, these results suggest that dopaminergic neurons are uniquely sensitive, and that while constitutive α-synuclein knockout leads to neuron loss in some paradigms, acute knockdown experiments demonstrate that α-synuclein is important for neuronal survival.
SNARE dysfunction in synucleinopathies
The hallmark of PD pathology is α-synuclein misfolding and aggregation and loss of dopaminergic neurons in the substantia nigra.136 Yet, how alterations in SNARE proteins affect the molecular mechanisms in PD remains unknown. In cerebral cortex brain homogenates from PD patients, a two-fold decrease in SNARE complex assembly was observed.137 In visual cortex tissue from patients with dementia with Lewy bodies (DLB), significant reductions in syntaxin-1 and SNAP-25 were found.138 This dysfunction may be clinically relevant, as in Parkinson’s disease dementia (PDD) and DLB, decreased levels of VAMP2/synaptobrevin-2 correlated with duration of dementia.139 In the striatum of a transgenic mouse model of PD, accumulation of α-synuclein in the synapses was accompanied by an age-dependent redistribution of the SNARE proteins SNAP-25, syntaxin-1 and VAMP2/synaptobrevin-2, as well as a reduction in dopamine release.140 These results suggest a correlation between synaptic SNARE proteins and the pathogenesis of synucleinopathies. The vulnerability of dopaminergic neurons in these diseases may be due to the unique properties of their synaptic vesicle cycling. There was a higher rate of stimulus-induced endocytosis of the vesicular monoamine transport VMAT2 compared to the glutamate transporter VGLUT1 suggesting a cell-specific synaptic vesicle recycling mechanism in dopaminergic neurons to sustain high rates of release.91
Altered SNARE complex levels have been directly linked to neurodegeneration. Lack of the SNAP-25 chaperone CSPα triggers SNARE complex formation defects which results in neurodegeneration.141–142 These deficits were rescued by expression of α-synuclein.74,143 Conversely, in the presence of α-synuclein aggregates, viral delivery of CSPα rescued the impaired synaptic vesicle recycling in a pheochromocytoma cell line, reduced synaptic α-synuclein aggregates, and restored normal dopamine release in mice overexpressing α-synuclein 1-120.144 SNARE complexes have also been proposed to mediate pathologically aggregated α-synuclein secretion via lysosomal exocytosis,145 which may contribute to the hypothesized cell-to-cell transmission of pathological aggregates. In addition, the SNARE complex-regulating protein Munc18-1/STXBP1 has been proposed to protect α-synuclein from aggregation.146 Interestingly, mutations in STXBP1 cause STXBP1 encephalopathies in humans,147–148 and dysfunction of some of the STXBP1 mutants have been linked to early onset parkinsonism,149–150 with co-aggregation of mutant Munc18-1 and α-Synuclein as a potential underlying mechanism.146
In Alzheimer’s disease (AD), postmortem brain tissue showed defects in synaptic SNARE complex assembly, although expression of individual proteins remained the same,137 while other studies have shown reduced expression of individual SNARE proteins.151–153 In a mouse model expressing human amyloid precursor protein and presenilin-1 double-transgene, SNARE complex levels were substantially reduced, mediated by Aβ oligomers binding to syntaxin-1 and blocking of SNARE complex assembly.154 Aβ peptides have also been shown to disrupt interactions between VAMP2/synaptobrevin-2 and SNAP-25 in neuronal cells, and Aβ42 was found to compete with VAMP2/synaptobrevin-2 for binding to synaptophysin at synapses.155
Is there a link to α-Synuclein in AD? Interestingly, α-Synuclein co-pathology has been reported in more than half of all autopsy-confirmed AD cases,156–158 and increased cerebrospinal fluid α-Synuclein levels were found in patients with or at risk of developing AD.159–160 Aβ and α-Synuclein have been shown to co-immunoprecipitate in the brains of patients with AD/PD and in transgenic mice.161 In mice, reducing endogenous α-Synuclein in an APP transgenic mouse model prevented the degeneration of cholinergic neurons and decreased corresponding behavioral deficits.162 However, in another study, overexpression of α-Synuclein in APP animals reduced Ab plaque deposition, but exacerbated deficits in spatial memory, increased extracellular Aβ oligomers, α-Synuclein oligomers, and exacerbated tau variants associated with AD.163 Ablating α-Synuclein improved memory retention in spite of increased plaque burden, prevented premature mortality, decreased extracellular Aβ oligomers, and rescued postsynaptic marker deficits.163 While these studies provide a possible link between SNARE complexes, α-Synuclein, and AD pathogenesis and progression, the molecular mechanisms on how α-Synuclein affects AD pathology remain unclear.
Altogether, there is accumulating evidence to suggest that there is a role for synaptic SNARE complex deficits in PD, DLB, and AD. With more evidence for a relationship between α-Synuclein and SNARE-dependent membrane fusion and/or SNARE complex formation, and with several studies showing SNARE dysfunction affecting disease pathology in AD, this leaves an exciting opportunity for further scientific advancements. It furthermore suggests that therapeutic strategies that aim at inhibiting aggregation of α-Synuclein or facilitating α-Synuclein clearance, such as small molecules and autophagy inhibitors,164–172 may benefit from targeting SNARE proteins as well.
Perspectives
There is a clear link between α-Synuclein and SNARE complex assembly. This connection is both functionally important and disease-relevant. However, it remains unclear if lack of SNARE complex assembly in synucleinopathies is due to α-Synuclein aggregation, if SNARE dysfunction in the context of α-Synuclein aggregation is involved in disease pathogenesis, or if α-Synuclein aggregation itself is the major driver of pathology. Whether α-Synuclein pathology is due to toxic gain-of-function or loss-of-function is an ongoing debate in the field. Several studies suggest that the physiological functions and pathological activities of α-Synuclein are mediated by different regions of α-Synuclein. Disruptions in the physiological functions of α-Synuclein do not always lead to pathological aggregation and cell death. However, loss-of-function and toxic gain-of-function cannot be studied in isolation. To resolve this long-standing question in the field, both have to be studied in a physiologically relevant context.
Acknowledgements
This work was funded by the Leon Levy Foundation (V.G.) and the NIH (3R01NS113960-02S1 to J.A.B., and R01NS121077, 1RF1NS126342, and 1R01NS113960 to J.B.).
Abbreviations:
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CSPα
cysteine string protein α
- DLB
dementia with Lewy Bodies
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NAC
non-amyloid-beta component
- NSF
N-ethylmaleimide sensitive fusion protein
- PD
Parkinson’s disease
- PDD
Parkinson’s disease dementia
- SNAP
soluble NSF-attachment protein
- SNAP-25
synaptosomal-associated protein of 25 kDa
- SNARE
soluble NSF-attachment protein receptor
- VAMP2
vesicle-associated membrane protein 2
- VGLUT1
vesicular glutamate transporter 1
- VMAT2
vesicular monoamine transporter 2
Footnotes
CRediT authorship contribution statement
Virginia Gao: Conceptualization, Writing – original draft, Writing – review & editing, Visualization, Funding acquisition. Juan A. Briano: Writing – original draft, Funding acquisition. Lauren E. Komer: Writing – original draft. Jacqueline Burré: Conceptualization, Writing – original draft, Writing – review & editing, Visualization, Supervision, Funding acquisition.
DECLARATION OF COMPETING INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Maroteaux L, Campanelli JT, Scheller RH, (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci 8, 2804–2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakajo S, Tsukada K, Omata K, Nakamura Y, Nakaya K, (1993). A new brain-specific 14-kDa protein is a phosphoprotein. Its complete amino acid sequence and evidence for phosphorylation. Eur. J. Biochem 217, 1057–1063. [DOI] [PubMed] [Google Scholar]
- 3.Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T, (1995). The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, (1997). Alpha-Synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- 5.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, et al. , (1997). Mutation in the alpha-Synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 6.Liu H, Koros C, Strohaker T, Schulte C, Bozi M, Varvaresos S, Ibanez de Opakua A, Simitsi AM, et al. , (2021). A Novel SNCA A30G Mutation Causes Familial Parkinson’s Disease. Mov. Disord 36, 1624–1633. [DOI] [PubMed] [Google Scholar]
- 7.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, et al. , (1998). Ala30Pro mutation in the gene encoding alpha-Synuclein in Parkinson’s disease. Nat. Genet 18, 106–108. [DOI] [PubMed] [Google Scholar]
- 8.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, et al. , (2004). The new mutation, E46K, of alpha-Synuclein causes Parkinson and Lewy body dementia. Ann. Neurol 55, 164–173. [DOI] [PubMed] [Google Scholar]
- 9.Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH, (2013). A novel alpha-Synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lesage S, Anheim M, Letournel F, Bousset L, Honore A, Rozas N, Pieri L, Madiona K, et al. , (2013). G51D alpha-Synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol 73, 459–471. [DOI] [PubMed] [Google Scholar]
- 11.Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, et al. , (2013). alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 125, 753–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshino H, Hirano M, Stoessl AJ, Imamichi Y, Ikeda A, Li Y, Funayama M, Yamada I, et al. , (2017). Homozygous alpha-Synuclein p. A53V in familial Parkinson’s disease. Neurobiol. Aging 57, 248 e7–248 e12. [DOI] [PubMed] [Google Scholar]
- 13.Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Poyhonen M, et al. , (2014). Novel alpha-Synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35 (2180), e1–e5. [DOI] [PubMed] [Google Scholar]
- 14.Fevga C, Park Y, Lohmann E, Kievit AJ, Breedveld GJ, Ferraro F, de Boer L, van Minkelen R, et al. , (2021). A new alpha-Synuclein missense variant (Thr72Met) in two Turkish families with Parkinson’s disease. Parkinsonism Relat Disord 89, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar ST, Mahul-Mellier AL, Hegde RN, Riviere G, Moons R, Ibanez de Opakua A, Magalhaes P, Rostami I, et al. , (2022). A NAC domain mutation (E83Q) unlocks the pathogenicity of human alpha-Synuclein and recapitulates its pathological diversity. Sci. Adv 8, eabn0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, et al. , (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841. [DOI] [PubMed] [Google Scholar]
- 17.Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, et al. , (2004). Causal relation between alpha-Synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171. [DOI] [PubMed] [Google Scholar]
- 18.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, et al. , (2004). Alpha-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169. [DOI] [PubMed] [Google Scholar]
- 19.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, et al. , (2009). Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr., (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, (1997). Evidence that the precursor protein of non-A beta component of Alzheimer’s disease amyloid (NACP) has an extended structure primarily composed of random-coil. Mol. Cells 7, 78–83. [PubMed] [Google Scholar]
- 22.George JM, Jin H, Woods WS, Clayton DF, (1995). Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 15, 361–372. [DOI] [PubMed] [Google Scholar]
- 23.Eliezer D, Kutluay E, Bussell R Jr., Browne G, (2001). Conformational properties of alpha-Synuclein in its free and lipid-associated states. J. Mol. Biol 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 24.Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, et al. , (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U S A 90, 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ulmer TS, Bax A, Cole NB, Nussbaum RL, (2005). Structure and dynamics of micelle-bound human alpha-Synuclein. J. Biol. Chem 280, 9595–9603. [DOI] [PubMed] [Google Scholar]
- 26.Hashimoto M, Takenouchi T, Mallory M, Masliah E, Takeda A, (2000). The role of NAC in amyloidogenesis in Alzheimer’s disease. Am. J. Pathol 156, 734–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bertini I, Gupta YK, Luchinat C, Parigi G, Peana M, Sgheri L, Yuan J, (2007). Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J. Am. Chem. Soc 129, 12786–12794. [DOI] [PubMed] [Google Scholar]
- 28.Wu KP, Kim S, Fela DA, Baum J, (2008). Characterization of conformational and dynamic properties of natively unfolded human and mouse alpha-Synuclein ensembles by NMR: implication for aggregation. J. Mol. Biol 378, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R, (1999). alpha-Synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem 274, 25481–25489. [DOI] [PubMed] [Google Scholar]
- 30.Paik SR, Shin HJ, Lee JH, Chang CS, Kim J, (1999). Copper(II)-induced self-oligomerization of alpha-Synuclein. Biochem. J 340 (Pt 3), 821–828. [PMC free article] [PubMed] [Google Scholar]
- 31.Nielsen MS, Vorum H, Lindersson E, Jensen PH, (2001). Ca2+ binding to alpha-Synuclein regulates ligand binding and oligomerization. J. Biol. Chem 276, 22680–22684. [DOI] [PubMed] [Google Scholar]
- 32.Cherny D, Hoyer W, Subramaniam V, Jovin TM, (2004). Double-stranded DNA stimulates the fibrillation of alpha-Synuclein in vitro and is associated with the mature fibrils: an electron microscopy study. J. Mol. Biol 344, 929–938. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez CO, Hoyer W, Zweckstetter M, Jares-Erijman EA, Subramaniam V, Griesinger C, Jovin TM, (2004). NMR of alpha-Synuclein-polyamine complexes elucidates the mechanism and kinetics of induced aggregation. EMBO J. 23, 2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brown DR, (2007). Interactions between metals and alpha-Synuclein-function or artefact? FEBS J. 274, 3766–3774. [DOI] [PubMed] [Google Scholar]
- 35.Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, et al. , (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol 4, 160–164. [DOI] [PubMed] [Google Scholar]
- 36.Awa S, Suzuki G, Masuda-Suzukake M, Nonaka T, Saito M, Hasegawa M, (2022). Phosphorylation of endogenous alpha-Synuclein induced by extracellular seeds initiates at the pre-synaptic region and spreads to the cell body. Sci. Rep 12, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawahata I, Finkelstein DI, Fukunaga K, (2022). Pathogenic Impact of alpha-Synuclein Phosphorylation and Its Kinases in alpha-Synucleinopathies. Int. J. Mol. Sci 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burre J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC, (2013). Properties of native brain alpha-Synuclein. Nature 498, E4–6 discussion E6–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fauvet B, Fares MB, Samuel F, Dikiy I, Tandon A, Eliezer D, Lashuel HA, (2012). Characterization of semisynthetic and naturally Nalpha-acetylated alpha-Synuclein in vitro and in intact cells: implications for aggregation and cellular properties of alpha-Synuclein. J. Biol. Chem 287, 28243–28262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bussell R Jr., Eliezer D, (2003). A structural and functional role for 11-mer repeats in alpha-Synuclein and other exchangeable lipid binding proteins. J. Mol. Biol 329, 763–778. [DOI] [PubMed] [Google Scholar]
- 41.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC, (2003). A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem 278, 15313–15318. [DOI] [PubMed] [Google Scholar]
- 42.Ferreon AC, Gambin Y, Lemke EA, Deniz AA, (2009). Interplay of alpha-Synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. U S A 106, 5645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burre J, Sharma M, Sudhof TC, (2014). alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. U S A 111, E4274–E4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S, (2014). alpha-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol 24, 2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burre J, Sharma M, Sudhof TC, (2015). Definition of a molecular pathway mediating alpha-Synuclein neurotoxicity. J. Neurosci 35, 5221–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jo E, Fuller N, Rand RP, St George-Hyslop P, Fraser PE, (2002). Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-Synuclein. J. Mol. Biol 315, 799–807. [DOI] [PubMed] [Google Scholar]
- 47.Fares MB, Ait-Bouziad N, Dikiy I, Mbefo MK, Jovicic A, Kiely A, Holton JL, Lee SJ, et al. , (2014). The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-Synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet 23, 4491–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghosh D, Sahay S, Ranjan P, Salot S, Mohite GM, Singh PK, Dwivedi S, Carvalho E, et al. , (2014). The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-Synuclein aggregation and membrane binding. Biochemistry 53, 6419–6421. [DOI] [PubMed] [Google Scholar]
- 49.Mohite GM, Kumar R, Panigrahi R, Navalkar A, Singh N, Datta D, Mehra S, Ray S, et al. , (2018). Comparison of Kinetics, Toxicity, Oligomer Formation, and Membrane Binding Capacity of alpha-Synuclein Familial Mutations at the A53 Site, Including the Newly Discovered A53V Mutation. Biochemistry 57, 5183–5187. [DOI] [PubMed] [Google Scholar]
- 50.Lee HJ, Choi C, Lee SJ, (2002). Membrane-bound alpha-Synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem 277, 671–678. [DOI] [PubMed] [Google Scholar]
- 51.Perni M, Galvagnion C, Maltsev A, Meisl G, Muller MB, Challa PK, Kirkegaard JB, Flagmeier P, et al. , (2017). A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. U S A 114, E1009–E1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Limbocker R, Staats R, Chia S, Ruggeri FS, Mannini B, Xu CK, Perni M, Cascella R, et al. , (2021). Squalamine and Its Derivatives Modulate the Aggregation of Amyloid-beta and alpha-Synuclein and Suppress the Toxicity of Their Oligomers. Front. Neurosci 15, 680026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL, (2002). Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem 277, 6344–6352. [DOI] [PubMed] [Google Scholar]
- 54.Uversky VN, (2007). Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem 103, 17–37. [DOI] [PubMed] [Google Scholar]
- 55.Ono K, (2017). The Oligomer Hypothesis in alpha-Synucleinopathy. Neurochem. Res 42, 3362–3371. [DOI] [PubMed] [Google Scholar]
- 56.Narayanan V, Scarlata S, (2001). Membrane binding and self-association of alpha-synucleins. Biochemistry 40, 9927–9934. [DOI] [PubMed] [Google Scholar]
- 57.Zhu M, Fink AL, (2003). Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem 278, 16873–16877. [DOI] [PubMed] [Google Scholar]
- 58.Burre J, Sharma M, Sudhof TC, (2012). Systematic mutagenesis of alpha-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci 32, 15227–15242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Withers GS, George JM, Banker GA, Clayton DF, (1997). Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res 99, 87–94. [DOI] [PubMed] [Google Scholar]
- 60.Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M , Yoshimoto M, Thal LJ, Saitoh T, et al. , (1998). Expression pattern of synucleins (non-Abeta component of Alzheimer’s disease amyloid precursor protein/alpha-synuclein) during murine brain development. J. Neurochem 71 , 338–344. [DOI] [PubMed] [Google Scholar]
- 61.Bayer TA, Jakala P, Hartmann T, Egensperger R, Buslei R, Falkai P, Beyreuther K, (1999). Neural expression profile of alpha-synuclein in developing human cortex. NeuroReport 10, 2799–2803. [DOI] [PubMed] [Google Scholar]
- 62.Galvin JE, Schuck TM, Lee VM, Trojanowski JQ, (2001). Differential expression and distribution of alpha-, beta-, and gamma-synuclein in the developing human substantia nigra. Exp. Neurol 168, 347–355. [DOI] [PubMed] [Google Scholar]
- 63.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK, (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci 26, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, et al. , (2011). Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem 286, 20710–20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK, (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem 283, 9089–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, et al. , (2006). Molecular anatomy of a trafficking organelle. Cell 127, 831–846. [DOI] [PubMed] [Google Scholar]
- 67.Taoufiq Z, Ninov M, Villar-Briones A, Wang HY, Sasaki T, Roy MC, Beauchain F, Mori Y, et al. , (2020). Hidden proteome of synaptic vesicles in the mammalian brain. Proc. Natl. Acad. Sci. U S A 117, 33586–33596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE, (2000). alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- 69.Perrin RJ, Woods WS, Clayton DF, George JM, (2000). Interaction of human alpha-Synuclein and Parkinson’s disease variants with phospholipids. Structural analysis using site-directed mutagenesis. J. Biol. Chem 275, 34393–34398. [DOI] [PubMed] [Google Scholar]
- 70.Middleton ER, Rhoades E, (2010). Effects of curvature and composition on alpha-synuclein binding to lipid vesicles. Biophys. J 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maltsev AS, Ying J, Bax A, (2012). Impact of N-terminal acetylation of alpha-synuclein on its random coil and lipid binding properties. Biochemistry 51, 5004–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Carnazza KE, Komer LE, Xie YX, Pineda A, Briano JA, Gao V, Na Y, Ramlall T, et al. , (2022). Synaptic vesicle binding of alpha-synuclein is modulated by beta- and gamma-synucleins. Cell Rep 39, 110675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lautenschlager J, Stephens AD, Fusco G, Strohl F, Curry N, Zacharopoulou M, Michel CH, Laine R, et al. , (2018). C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC, (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun J, Wang L, Bao H, Premi S, Das U, Chapman ER, Roy S, (2019). Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. U S A 116, 11113–11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Scott D, Roy S, (2012). alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recyclingpool homeostasis. J. Neurosci 32, 10129–10135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Diao J, Burre J, Vivona S, Cipriano DJ, Sharma M, Kyoung M, Sudhof TC, Brunger AT, (2013). Native alpha-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. Elife 2, e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sudhof TC, (2004). The synaptic vesicle cycle. Annu. Rev. Neurosci 27, 509–547. [DOI] [PubMed] [Google Scholar]
- 79.Zhang M, Augustine GJ, (2021). Synapsins and the Synaptic Vesicle Reserve Pool: Floats or Anchors? Cells 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hosaka M, Hammer RE, Sudhof TC, (1999). A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 24, 377–387. [DOI] [PubMed] [Google Scholar]
- 81.Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC, (1994). The role of Rab3A in neurotransmitter release. Nature 369, 493–497. [DOI] [PubMed] [Google Scholar]
- 82.Rizzoli SO, Betz WJ, (2004). The structural organization of the readily releasable pool of synaptic vesicles. Science 303, 2037–2039. [DOI] [PubMed] [Google Scholar]
- 83.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE, (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. [DOI] [PubMed] [Google Scholar]
- 84.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC, (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. [DOI] [PubMed] [Google Scholar]
- 85.Koenig JH, Ikeda K, (1996). Synaptic vesicles have two distinct recycling pathways. J. Cell Biol. 135, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P, (1995). Distinct pools of synaptic vesicles in neurotransmitter release. Nature 375, 493–497. [DOI] [PubMed] [Google Scholar]
- 87.Rosenmund C, Stevens CF, (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207. [DOI] [PubMed] [Google Scholar]
- 88.Rizzoli SO, Betz WJ, (2005). Synaptic vesicle pools. Nat. Rev. Neurosci 6, 57–69. [DOI] [PubMed] [Google Scholar]
- 89.Kaeser PS, Regehr WG, (2017). The readily releasable pool of synaptic vesicles. Curr. Opin. Neurobiol 43, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fouke KE, Wegman ME, Weber SA, Brady EB, Roman-Vendrell C, Morgan JR, (2021). Synuclein Regulates Synaptic Vesicle Clustering and Docking at a Vertebrate Synapse. Front. Cell Dev. Biol 9, 774650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, et al. , (2010). Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Atias M, Tevet Y, Sun J, Stavsky A, Tal S, Kahn J, Roy S, Gitler D, (2019). Synapsins regulate alpha-synuclein functions. Proc. Natl. Acad. Sci. U S A 116, 11116–11118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hoffmann C, Sansevrino R, Morabito G, Logan C, Vabulas RM, Ulusoy A, Ganzella M, Milovanovic D, (2021). Synapsin Condensates Recruit alpha-Synuclein. J. Mol. Biol 433, 166961. [DOI] [PubMed] [Google Scholar]
- 94.Fakhree MA, Zijlstra N, Raiss CC, Siero CJ, Grabmayr H, Bausch AR, Blum C, Claessens MM, (2016). The number of alpha-synuclein proteins per vesicle gives insights into its physiological function. Sci. Rep 6, 30658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoo G, Yeou S, Son JB, Shin YK, Lee NK, (2021). Cooperative inhibition of SNARE-mediated vesicle fusion by alpha-synuclein monomers and oligomers. Sci. Rep 11, 10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lou X, Kim J, Hawk BJ, Shin YK, (2017). alpha-Synuclein may cross-bridge v-SNARE and acidic phospholipids to facilitate SNARE-dependent vesicle docking. Biochem. J 474, 2039–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lai Y, Kim S, Varkey J, Lou X, Song JK, Diao J, Langen R, Shin YK, (2014). Nonaggregated alpha-synuclein influences SNARE-dependent vesicle docking via membrane binding. Biochemistry 53, 3889–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi BK, Choi MG, Kim JY, Yang Y, Lai Y, Kweon DH, Lee NK, Shin YK, (2013). Large alpha-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. U S A 110, 4087–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.DeWitt DC, Rhoades E, (2013). alpha-Synuclein can inhibit SNARE-mediated vesicle fusion through direct interactions with lipid bilayers. Biochemistry 52, 2385–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Darios F, Ruiperez V, Lopez I, Villanueva J, Gutierrez LM, Davletov B, (2010). Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 11, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vargas KJ, Schrod N, Davis T, Fernandez-Busnadiego R, Taguchi YV, Laugks U, Lucic V, Chandra, (2017). Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep 18, 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chi P, Greengard P, Ryan TA, (2001). Synapsin dispersion and reclustering during synaptic activity. Nat. Neurosci 4, 1187–1193. [DOI] [PubMed] [Google Scholar]
- 103.Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH, (2005). Neural activity controls the synaptic accumulation of alpha-synuclein. J. Neurosci 25, 10913–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Orenbuch A, Shalev L, Marra V, Sinai I, Lavy Y, Kahn J, Burden JJ, Staras K, et al. , (2012). Synapsin selectively controls the mobility of resting pool vesicles at hippocampal terminals. J. Neurosci 32, 3969–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Milovanovic D, Wu Y, Bian X, De Camilli P, (2018). A liquid phase of synapsin and lipid vesicles. Science 361, 604–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pechstein A, Tomilin N, Fredrich K, Vorontsova O, Sopova E, Evergren E, Haucke V, Brodin L, et al. , (2020). Vesicle Clustering in a Living Synapse Depends on a Synapsin Region that Mediates Phase Separation. Cell Rep 30 2594–2602 e3. [DOI] [PubMed] [Google Scholar]
- 107.Brodin L, Milovanovic D, Rizzoli SO, Shupliakov O, (2022). alpha-Synuclein in the Synaptic Vesicle Liquid Phase: Active Player or Passive Bystander? Front Mol Biosci 9, 891508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, et al. , (2004). Double-knockout mice for alpha-and beta-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. U S A 101, 14966–14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu S, Ninan I, Antonova I, Battaglia F, Trinchese F, Narasanna A, Kolodilov N, Dauer W, et al. , (2004). alpha-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 23, 4506–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gureviciene I, Gurevicius K, Tanila H, (2007). Role of alpha-synuclein in synaptic glutamate release. Neurobiol. Dis 28, 83–89. [DOI] [PubMed] [Google Scholar]
- 111.Gureviciene I, Gurevicius K, Tanila H, (2009). Aging and alpha-synuclein affect synaptic plasticity in the dentate gyrus. J. Neural. Transm. (Vienna) 116, 13–22. [DOI] [PubMed] [Google Scholar]
- 112.Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, et al. , (2010). alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U S A 107, 19573–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Steidl JV, Gomez-Isla T, Mariash A, Ashe KH, Boland LM, (2003). Altered short-term hippocampal synaptic plasticity in mutant alpha-synuclein transgenic mice. NeuroReport 14, 219–223. [DOI] [PubMed] [Google Scholar]
- 114.Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, et al. , (2000). Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252. [DOI] [PubMed] [Google Scholar]
- 115.Cabin DE, Shimazu K, Murphy D, Cole NB, Gottschalk W, McIlwain KL, Orrison B, Chen A, et al. , (2002). Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci 22, 8797–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yavich L, Tanila H, Vepsalainen S, Jakala P, (2004). Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci 24, 11165–11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yavich L, Jakala P, Tanila H, (2006). Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. J. Neurochem 99, 724–732. [DOI] [PubMed] [Google Scholar]
- 118.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, et al. , (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci 26, 11915–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Senior SL, Ninkina N, Deacon R, Bannerman D, Buchman VL, Cragg SJ, Wade-Martins R, (2008). Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci 27, 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu N, Joshi PR, Cepeda C, Masliah E, Levine MS, (2010). Alpha-synuclein overexpression in mice alters synaptic communication in the corticostriatal pathway. J. Neurosci. Res 88, 1764–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, et al. , (2011). Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J. Neurosci 31, 7264–7274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sulzer D, Edwards RH, (2019). The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem 150, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Somayaji M, Cataldi S, Choi SJ, Edwards RH, Mosharov EV, Sulzer D, (2020). A dual role for alpha-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. U S A 117, 32701–32710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Robertson DC, Schmidt O, Ninkina N, Jones PA, Sharkey J, Buchman VL, (2004). Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem 89, 1126–1136. [DOI] [PubMed] [Google Scholar]
- 125.Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL, (2010). Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 31, 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dauer W, Kholodilov N, Vila M, Trillat AC, Goodchild R, Larsen KE, Staal R, Tieu K, et al. , (2002). Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U S A 99, 14524–14529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, et al. , (2005). Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. U S A 102, 3413–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schluter OM, Fornai F, Alessandri MG, Takamori S, Geppert M, Jahn R, Sudhof TC, (2003). Role of alpha-synuclein in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice . Neuroscience 118, 985–1002. [DOI] [PubMed] [Google Scholar]
- 129.Outeiro TF, Klucken J, Bercury K, Tetzlaff J, Putcha P, Oliveira LM, Quintas A, McLean PJ, et al. , (2009) . Dopamine-induced conformational changes in alpha-synuclein. PLoS ONE 4, e6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mosharov EV, Staal RG, Bove J, Prou D, Hananiya A, Markov D, Poulsen N, Larsen KE, et al. , (2006). Alpha-synuclein overexpression increases cytosolic catecholamine concentration. J. Neurosci 26, 9304–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gorbatyuk OS, Li S, Nash K, Gorbatyuk M, Lewin AS, Sullivan LF, Mandel RJ, Chen W, et al. , (2010). In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther 18, 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kanaan NM, Manfredsson FP, (2012). Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? J. Parkinsons. Dis 2, 249–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP, (2018). Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front. Mol. Neurosci 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Collier TJ, Redmond DE Jr., Steece-Collier K, Lipton JW, Manfredsson FP, (2016). Is Alpha-Synuclein Loss-of-Function a Contributor to Parkinsonian Pathology? Evidence from Non-human Primates. Front. Neurosci 10, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lewis J, Melrose H, Bumcrot D, Hope A, Zehr C, Lincoln S, Braithwaite A, He Z, et al. , (2008). In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kalia LV, Lang AE, (2015). Parkinson’s disease. Lancet 386, 896–912. [DOI] [PubMed] [Google Scholar]
- 137.Sharma M, Burre J, Sudhof TC, (2012). Proteasome inhibition alleviates SNARE-dependent neurodegeneration. Sci. Transl. Med 4, 147ra113. [DOI] [PubMed] [Google Scholar]
- 138.Mukaetova-Ladinska EB, Andras A, Milne J, Abdel-All Z, Borr I, Jaros E, Perry RH, Honer WG, et al. , (2013). Synaptic proteins and choline acetyltransferase loss in visual cortex in dementia with Lewy bodies. J. Neuropathol. Exp. Neurol 72, 53–60. [DOI] [PubMed] [Google Scholar]
- 139.Vallortigara J, Whitfield D, Quelch W, Alghamdi A, Howlett D, Hortobagyi T, Johnson M, Attems J, et al. , (2016). Decreased Levels of VAMP2 and Monomeric Alpha-Synuclein Correlate with Duration of Dementia. J. Alzheimers Dis 50, 101–110. [DOI] [PubMed] [Google Scholar]
- 140.Garcia-Reitbock P, Anichtchik O, Bellucci A, Iovino M, Ballini C, Fineberg E, Ghetti B, Della Corte L, et al. , (2010). SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 133, 2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sharma M, Burre J, Bronk P, Zhang Y, Xu W, Sudhof TC, (2012). CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 31, 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sharma M, Burre J, Sudhof TC, (2011). CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol 13, 30–39. [DOI] [PubMed] [Google Scholar]
- 143.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC, (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. [DOI] [PubMed] [Google Scholar]
- 144.Calo L, Hidari E, Wegrzynowicz M, Dalley JW, Schneider BL, Podgajna M, Anichtchik O, Carlson E, et al. , (2021). CSPalpha reduces aggregates and rescues striatal dopamine release in alpha-synuclein transgenic mice. Brain 144, 1661–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xie YX, Naseri NN, Fels J, Kharel P, Na Y, Burré J, Sharma M, (2021). Lysosomal Exocytosis Releases Pathogenic α-Synuclein Species from Neurons. BioRxiv.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chai YJ, Sierecki E, Tomatis VM, Gormal RS, Giles N, Morrow IC, Xia D, Gotz J, et al. , (2016). Munc18-1 is a molecular chaperone for alpha-synuclein, controlling its self-replicating aggregation. J. Cell Biol 214, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Abramov D, Guiberson NGL, Burre J, (2021). STXBP1 encephalopathies: Clinical spectrum, disease mechanisms, and therapeutic strategies. J. Neurochem 157, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stamberger H, Nikanorova M, Willemsen MH, Accorsi P, Angriman M, Baier H, Benkel-Herrenbrueck I, Benoit V, et al. , (2016). STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology 86, 954–962. [DOI] [PubMed] [Google Scholar]
- 149.Keogh MJ, Daud D, Pyle A, Duff J, Griffin H, He L, Alston CL, Steele H, et al. , (2015). A novel de novo STXBP1 mutation is associated with mitochondrial complex I deficiency and late-onset juvenile-onset parkinsonism. Neurogenetics 16, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lanoue V, Chai YJ, Brouillet JZ, Weckhuysen S, Palmer EE, Collins BM, Meunier FA, (2019). STXBP1 encephalopathy: Connecting neurodevelopmental disorders with alpha-synucleinopathies? Neurology 93, 114–123. [DOI] [PubMed] [Google Scholar]
- 151.Musunuri S, Khoonsari PE, Mikus M, Wetterhall M, Haggmark-Manberg A, Lannfelt L, Erlandsson A, Bergquist J, et al. , (2016). Increased Levels of Extracellular Microvesicle Markers and Decreased Levels of Endocytic/Exocytic Proteins in the Alzheimer’s Disease Brain. J. Alzheimers Dis 54, 1671–1686. [DOI] [PubMed] [Google Scholar]
- 152.Shimohama S, Kamiya S, Taniguchi T, Akagawa K, Kimura J, (1997). Differential involvement of synaptic vesicle and presynaptic plasma membrane proteins in Alzheimer’s disease. Biochem. Biophys. Res. Commun 236, 239–242. [DOI] [PubMed] [Google Scholar]
- 153.Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ, (2000). Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J. Neurol. Sci 175, 81–90. [DOI] [PubMed] [Google Scholar]
- 154.Yang Y, Kim J, Kim HY, Ryoo N, Lee S, Kim Y, Rhim H, Shin YK, (2015). Amyloid-beta Oligomers May Impair SNARE-Mediated Exocytosis by Direct Binding to Syntaxin 1a. Cell Rep 12, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sharda N, Pengo T, Wang Z, Kandimalla KK, (2020). Amyloid-beta Peptides Disrupt Interactions Between VAMP-2 and SNAP-25 in Neuronal Cells as Determined by FRET/FLIM. J. Alzheimers Dis 77, 423–435. [DOI] [PubMed] [Google Scholar]
- 156.Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O’Connell B, et al. , (1998). Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am. J. Pathol 153, 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lopez OL, Becker JT, Sweet RA, Martin-Sanchez FJ, Hamilton RL, (2006). Lewy bodies in the amygdala increase risk for major depression in subjects with Alzheimer disease. Neurology 67, 660–665. [DOI] [PubMed] [Google Scholar]
- 158.Arai Y, Yamazaki M, Mori O, Muramatsu H, Asano E , Katayama Y, (2001). Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: morphology and its relationship to tau aggregation. Brain Res. 888, 287–296. [DOI] [PubMed] [Google Scholar]
- 159.Vergallo A, Bun RS, Toschi N, Baldacci F, Zetterberg E , Blennow K, Cavedo E, Lamari F, et al. , (2018). Association of cerebrospinal fluid alpha-synuclein with total and phospho-tau181 protein concentrations and brain amyloid load in cognitively normal subjective memory complainers stratified by Alzheimer’s disease biomarkers. Alzheimers Dement 14, 1623–1631. [DOI] [PubMed] [Google Scholar]
- 160.Twohig D, Rodriguez-Vieitez E, Sando SB, Berge G, Lauridsen C, Moller I, Grontvedt GR, Brathen G, et al. , (2018). The relevance of cerebrospinal fluid alpha-synuclein levels to sporadic and familial Alzheimer’s disease. Acta Neuropathol. Commun 6, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tsigelny IF, Crews L, Desplats P, Shaked GM, Sharikov Y, Mizuno H, Spencer B, Rockenstein E, et al. , (2008). Mechanisms of hybrid oligomer formation in the pathogenesis of combined Alzheimer’s and Parkinson’s diseases. PLoS ONE 3, e3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Spencer B, Desplats PA, Overk CR, Valera-Martin E, Rissman RA, Wu C, Mante M, Adame A, et al. , (2016). Reducing Endogenous alpha-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci 36, 7971–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Khan SS, LaCroix M, Boyle G, Sherman MA, Brown JL, Amar F, Aldaco J, Lee MK, et al. , (2018). Bidirectional modulation of Alzheimer phenotype by alpha-synuclein in mice and primary neurons. Acta Neuropathol. 136, 589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vijiaratnam N, Simuni T, Bandmann O, Morris HR, Foltynie T, (2021). Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 20, 559–572. [DOI] [PubMed] [Google Scholar]
- 165.Hebron ML, Lonskaya I, Moussa CE, (2013). Tyrosine kinase inhibition facilitates autophagic SNCA/alpha-synuclein clearance. Autophagy 9, 1249–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Werner MH, Olanow CW, (2022). Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov. Disord 37, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pujols J, Pena-Diaz S, Lazaro DF, Peccati F, Pinheiro F, Gonzalez D, Carija A, Navarro S, et al. , (2018). Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. U S A 115, 10481–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chen KS, Menezes K, Rodgers JB, O’Hara DM, Tran N, Fujisawa K, Ishikura S, Khodaei S, et al. , (2021). Small molecule inhibitors of alpha-synuclein oligomers identified by targeting early dopamine-mediated motor impairment in C. elegans. Mol. Neurodegener 16, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pena-Diaz S, Pujols J, Conde-Gimenez M, Carija A, Dalfo E, Garcia J, Navarro S, Pinheiro F, et al. , (2019). ZPD-2, a Small Compound That Inhibits alpha-Synuclein Amyloid Aggregation and Its Seeded Polymerization. Front. Mol. Neurosci 12, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Paul A, Zhang BD, Mohapatra S, Li G, Li YM, Gazit E, Segal D, (2019). Novel Mannitol-Based Small Molecules for Inhibiting Aggregation of alpha-Synuclein Amyloids in Parkinson’s Disease. Front Mol Biosci 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Vittorio S, Adornato I, Gitto R, Pena-Diaz S, Ventura S, De Luca L, (2020). Rational design of small molecules able to inhibit alpha-synuclein amyloid aggregation for the treatment of Parkinson’s disease. J. Enzyme Inhib. Med. Chem 35, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Robustelli P, Ibanez-de-Opakua A, Campbell-Bezat C, Giordanetto F, Becker S, Zweckstetter M, Pan AC, Shaw DE, (2022). Molecular Basis of Small-Molecule Binding to alpha-Synuclein. J. Am. Chem. Soc 144, 2501–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]