

SUMMARY
Environmental exposures are a major risk factor for developing colorectal cancer, and the gut microbiome may serve as an integrator of such environmental risk. To study the microbiome associated with of premalignant colon lesions, such as tubular adenomas (TA) and sessile serrated adenomas (SSA), we profiled stool samples from 971 participants undergoing colonoscopy and paired these data with dietary and medication history. The microbial signatures associated with either SSA or TA are distinct. SSA associates with multiple microbial antioxidant defense systems, while TA associates with a depletion of microbial methanogenesis and mevalonate metabolism. Environmental factors, such as diet and medications, link with the majority of identified microbial species. Mediation analyses found Flavonifractor plaudit and Bacteroides stercoris transmit the protective or carcinogenic effects of these factors to early carcinogenesis. Our findings suggest that the unique dependencies of each premalignant lesion may be exploited therapeutically or through dietary intervention.
eTOC BLURB
Lee et al. investigate the gut microbiome among patients with different types of pre-malignant colorectal adenomas. Each adenoma subtype is associated with a distinct microbial profile that is also correlated with environmental factors. Further, specific microbial species that likely mediate the protective or carcinogenic effects of these factors are identified.
Graphical Abstract
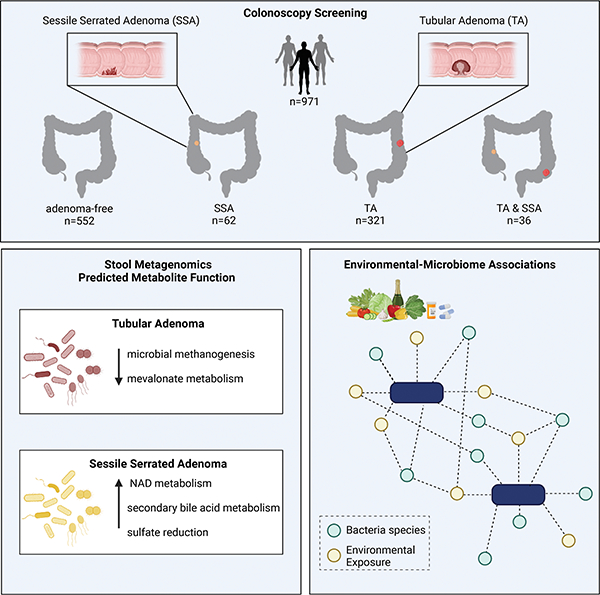
INTRODUCTION
Colorectal cancer (CRC) remains the second leading cause of cancer-related deaths in the United States1. Malignant changes in the intestinal tract typically develop from a dysplastic precursor lesion that can progress to colorectal cancer through the accumulation of multiple genetic mutations2. There are two primary premalignant lesions, the tubular adenomas (TA) (which includes tubular, tubulovillous and villous adenomas) and the sessile serrated adenomas (SSA) (which includes sessile serrated adenomas and traditional serrated adenomas). While TAs are more common, up to one in five CRCs arise through serrated polyps, further indicating that SSAs are important precursors of CRC3. CRCs arising from serrated lesions are thought to account for a large proportion of interval cancers and may represent the main cause of cancer screening failure.
Environmental exposures, such as diet and lifestyle, are important risk factors for CRC. Up to 50% of the CRC cases in the United States are attributable to modifiable risk factors, such as smoking, obesity, high consumption of red and processed meat and low consumption of dietary fiber4,5. Furthermore, systematic reviews have found that other modifiable environmental risk factors, such as smoking, alcohol, and body mass index are more strongly associated with SSAs than TAs6. A strong association between red meat consumption and risk of SSAs was also shown in a colonoscopy-based case–control study7. Chemoprevention has emerged as an approach to reduce colorectal cancer risk, and treatment with aspirin, non-steroidal anti-inflammatory drugs (NSAIDs), metformin, calcium and vitamins, folic acid as well as statins have been proposed8.
The composition of the gut microbiome, in turn, is largely shaped by such environmental risk factors, particularly diet, lifestyle and medications9,10. Although the precise mechanisms through which these factors may influence CRC risk are likely unique to each individual, there is emerging evidence to support the gut microbiome as a key integrator of environmental risk factors with host physiology to regulate CRC pathogenesis11. The gut microbiome plays an important role in nutrient processing and synthesis, and may thereby influence CRC development through metabolite-mediated changes in immune and metabolic signals12. Although there have been numerous previous reports of the microbiome in CRC, most only compared patients with established CRC against healthy individuals13,14. It has been proposed, however, that microbiome changes occur in the early stages of colorectal carcinogenesis15. For instance, the initiation of carcinogenesis may be driven by diffuse microenvironmental factors in response to external perturbations (such as diet); once past a given threshold, the local microenvironment becomes amenable to—and potentially driven by—cancer-associated microbes (i.e. F. nucleatum16, B. fragilis17, and E. coli18). However, there have only been few studies13 powered to study the uniquely gut microbiome changes associated with early adenomas and premalignant colorectal lesions. Few studies19–21 have previously profiled microbiome differences between traditional adenomas and serrated polyps, but were limited by sample size, targeted amplicon sequencing methods, and lack of discrimination between hyperplasia and dysplasia amongst serrated polyps.
In this study, we sought to determine whether the interplay of the gut microbiome with environmental risk factors differs between the two major precursors of CRC, TAs and SSAs. We sequenced stool samples from patients who participated in our Gastrointestinal Disease Endoscopy Registry (GIDER), a cross-sectional study that profiles the microbiota of stool samples from individuals undergoing routine endoscopic procedures, curated with paired dietary intake and medication history. Our findings indicate that distinct microbial populations are associated with specific CRC precursor lesions, and the majority of the significant microbial species associated with either adenoma subtypes can be linked to environmental risk factors, including diet and commonly prescribed medications. This study identified 19 and 8 microbial species, as well as 270 and 140 unique microbial enzymes associated with tubular and serrated adenoma respectively, which may provide further insights into the use of the microbiome as an early biomarker of CRC risk, as well as identify alterable points for cancer prevention.
RESULTS
Participant characteristics
Our study cohort included a total of 971 colonoscopy-screened individuals, composed of 552 adenoma-free controls, 321 cases with tubular or tubulovillous adenomas (TAs), 62 cases with sessile serrated adenomas (SSAs), and 36 cases with both TAs and SSAs (Table1, Fig. 1A). Among the participants with SSAs, more than half of the SSAs (57.1%, n=56) were considered advanced, and almost all of the SSAs were located in the proximal colon (n=90, 91.8%). In contrast, about one-third (n=133, 37.3%) of TAs were considered advanced, with either 3 or more concomitant adenomatous polyps (n=106; 29.7%) or adenomas greater than 10mm in size (n=57, 23.7%) (Fig. 1B,C). Participants with TAs were older than controls (64±9.6 vs 59.4±10.0years old, p<0.01), while participants with SSAs were similar in age (mean 60.2±10.8 years old). There was also an increased proportion of male patients with TAs compared to controls (60.8% vs 43.5%, p < 0.01). Of note, obesity, a common risk factor associated with adenomatous polyps, was not found to be significantly different between controls and participants with either TAs or SSAs. More participants with TAs were taking aspirin (36.7% vs 27.9%, p<0.01), accompanied by a corresponding higher proportions of concomitant cardiovascular diseases (8.1% vs 3.8%, p<0.01), compared to controls. In contrast, a significantly small proportion of participants with SSAs were taking aspirin (20.4%, p=0.03). Participants with either TAs or SSA, were both found to have lower dietary intake of vegetables (p<0.01) (Fig. 1D, Table S1).
Table 1:
Demographics of the GIDER cohort; demographics and endoscopic findings stratified by adenoma subtype
| Levels | No Adenoma (n=552) | TA only (n=321) | SSA only (n=62) | Both TA & SSA (n=36) | Total (N=971) | p value | |
|---|---|---|---|---|---|---|---|
| Demographics | |||||||
| Age | Mean (SD) | 59.6 (9.8) | 64.5 (9.4) | 57.5 (12.1) | 63.4 (8.5) | 61.2 (10.0) | <0.001 |
| Sex | Female | 241 (57.5) | 104 (46.0) | 23 (57.5) | 9 (39.1) | 377 (53.2) | 0.019 |
| Male | 178 (42.5) | 122 (54.0) | 17 (42.5) | 14 (60.9) | 331 (46.8) | ||
| BMI | Mean (SD) | 26.9 (5.5) | 27.4 (5.5) | 27.4 (6.1) | 27.3 (4.4) | 27.1 (5.5) | 0.712 |
| Smoking | No | 267 (67.9) | 120 (57.1) | 26 (65.0) | 16 (69.6) | 429 (64.4) | 0.275 |
| Stopped | 114 (29.0) | 82 (39.0) | 13 (32.5) | 6 (26.1) | 215 (32.3) | ||
| Yes | 12 (3.1) | 8 (3.8) | 1 (2.5) | 1 (4.3) | 22 (3.3) | ||
| Family history of CRC | No | 327 (79.4) | 188 (84.3) | 35 (87.5) | 21 (95.5) | 571 (81.9) | 0.371 |
| First Degree | 74 (18.0) | 31 (13.9) | 4 (10.0) | 1 (4.5) | 110 (15.8) | ||
| Second Degree | 11 (2.7) | 4 (1.8) | 1 (2.5) | 0 (0.0) | 16 (2.3) | ||
| Endoscopic findings | |||||||
| Adenoma Clinical Risk | None | 419 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 419 (59.2) | <0.001 |
| Low | 0 (0.0) | 156 (69.0) | 17 (42.5) | 5 (21.7) | 178 (25.1) | ||
| High | 0 (0.0) | 70 (31.0) | 23 (57.5) | 18 (78.3) | 111 (15.7) | ||
| Count | No Adenoma | 419 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 419 (59.2) | <0.001 |
| 1–2 Adenomas | 0 (0.0) | 181 (80.1) | 30 (75.0) | 6 (26.1) | 217 (30.6) | ||
| >=3 Adenomas | 0 (0.0) | 45 (19.9) | 10 (25.0) | 17 (73.9) | 72 (10.2) | ||
| Size | No Adenoma | 419 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 419 (59.2) | <0.001 |
| <1cm | 0 (0.0) | 178 (78.8) | 20 (50.0) | 13 (56.5) | 211 (29.8) | ||
| >=10cm | 0 (0.0) | 48 (21.2) | 20 (50.0) | 10 (43.5) | 78 (11.0) | ||
| Location | No Polyp | 419 (100.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 419 (59.2) | <0.001 |
| Right | 0 (0.0) | 126 (55.8) | 27 (67.5) | 15 (65.2) | 168 (23.7) | ||
| Left | 0 (0.0) | 30 (13.3) | 4 (10.0) | 0 (0.0) | 34 (4.8) | ||
| Pancolon | 0 (0.0) | 70 (31.0) | 9 (22.5) | 8 (34.8) | 87 (12.3) | ||
Figure 1: Descriptive demographics, environmental exposures and adenoma characteristics within the GIDER cohort.
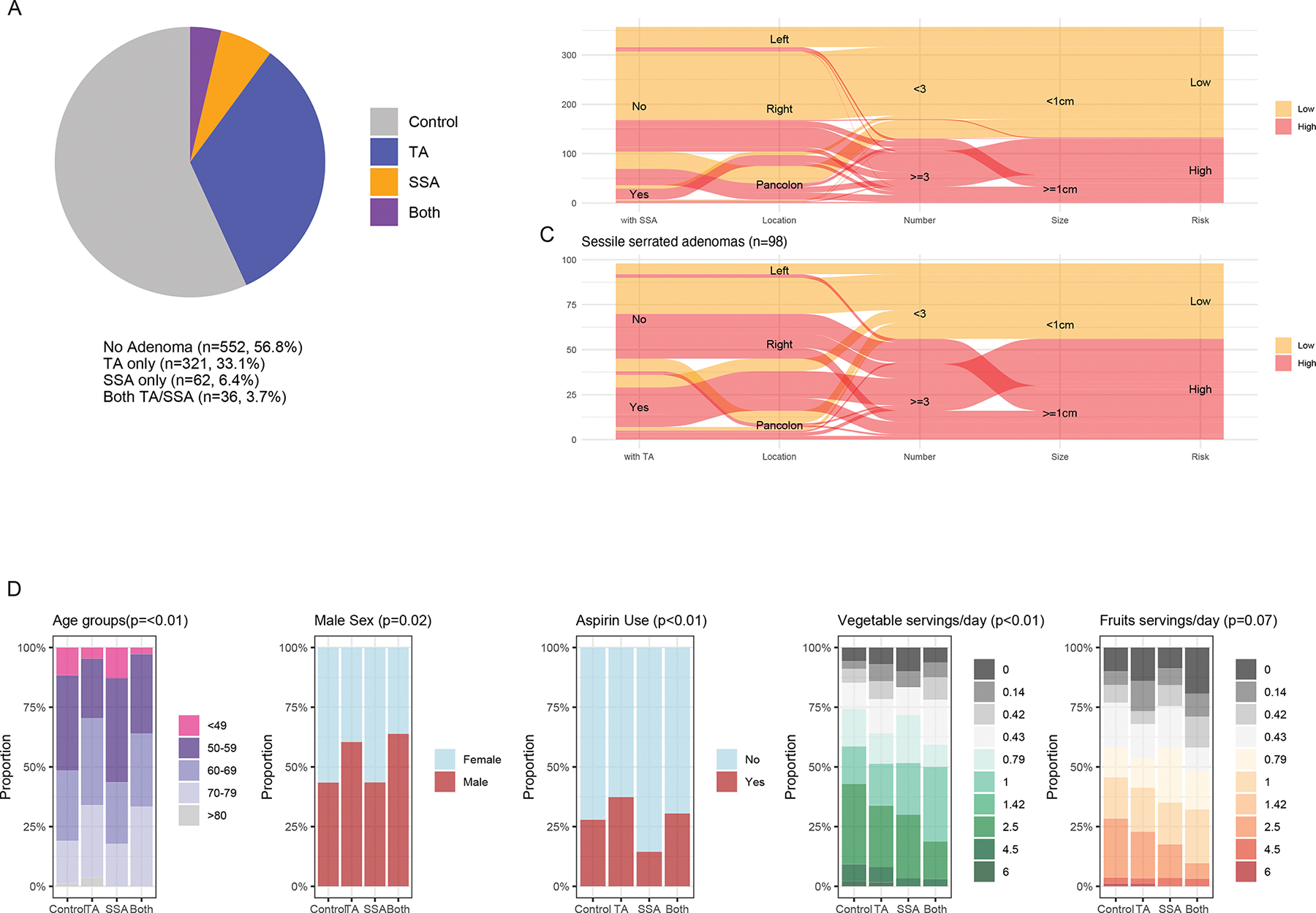
(A) Pie chart of the 917 participants within GIDER, grouped by their adenoma histological subtypes (i.e., no adenoma, serrated adenomas, tubular adenomas). (B, C) Number of patients with TA or SSA risk stratified as mid- or high-clinical risk depending on the total number of adenomas and size. (D) Significant demographic factors and environmental exposure differences between SSA and TA - participants with TA were of older age, more likely to be male, more likely to use aspirin, and self-reported higher daily servings of vegetables and lower daily servings of processed meat.
Global gut microbiota shifts in relation to colorectal polyps
Using Dirichlet multinomial mixture (DMM) modeling22, we identified 5 microbial meta-community types among all the GIDER participants (Fig. 2A). The 5 microbial meta-communities were mostly driven by the relative abundances of 137 bacterial species (Fig. 2B). Using the most abundant unique microbial species from each respective DMM meta-community, we demonstrated that these five clusters are congruent with the previously well described enterotypes23. Although there were no significant differences in the proportions of participants with either TAs or SSAs stratified by the 5 DMM meta-communities (Fig. S1), the proportions of SSAs were highest among participants DMM types 1 and 2, which were predominantly abundant in Firmicutes (Faecalibacterium prausnitzii and Eubacterium rectale, respectively). DMM types 3 and 4 are predominantly abundant in Bacteroidetes, such as Bacteroides uniforms and Bacteroides no vulgatus, respectively. Finally, DMM type 5 was driven by increased abundances of Prevotella copri (Fig. 2C).
Figure 2: Summary of GIDER community microbiome sub-communities.
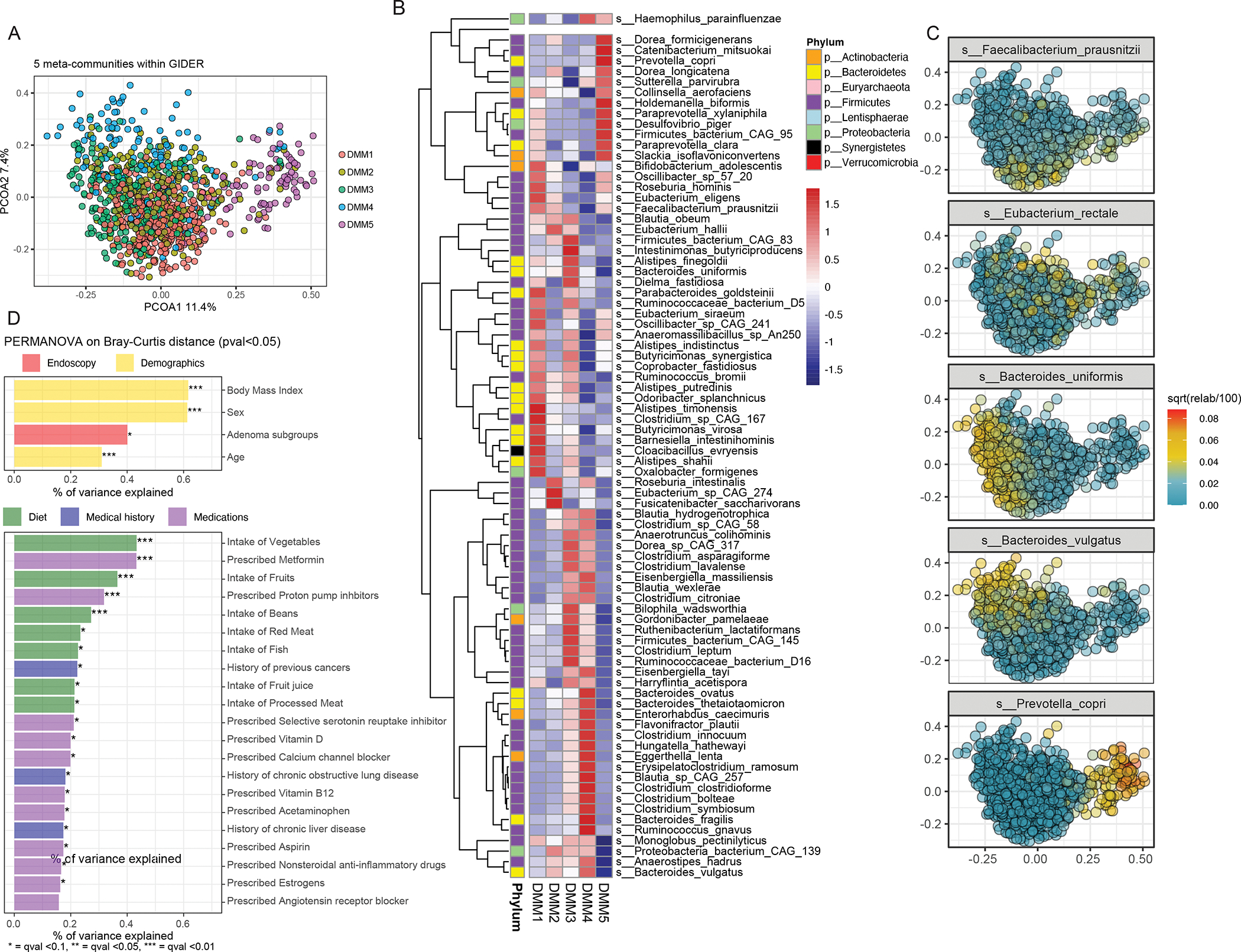
(A) Principal coordinate analysis (PCoA) plot coloured by five microbial clusters (meta-communities) identified by Dirichlet multinomial distribution. (B) The five clusters are determined by 137 significant (qval <0.05) differential abundant microbial species, as shown in this heatmap, whereby each cell represents the normalized average relative abundances. (C) The most abundant microbial species of each cluster is plotted on this density dot plot, to highlight that the five microbial clusters correspond predominant with Faecalibacterium prausnitzii (DMM1), Bacteroides uniformis (DMM3), Eubacterium rectale (DMM2), Bacteroides vulgatus (DMM4) and Prevotella copri (DMM5). (D) The significant (pval <0.05) host and environmental variables (yellow - demographics, green - diet, blue - medical history, purple - medications) explaining the microbiome variance determined by PERMANOVA on Bray-Curtis dissimilarity distances are shown in the barplot.
We identified 25 demographic and environmental risk factors (pval <0.05) with significant weight on the overall microbiome variation in the population (Fig. 2D). Overall, the 4 variables with the greatest effect on the community microbiome variance were age, sex, body mass index, and the presence of either TAs or SSAs. Of which, the presence of either TAs or SSAs could explain 0.4% of the microbial variance (p<0.05, FDR = 0.08). Despite the fact that the information about the participants’ diet, medical history and medications were obtained through basic food-frequency questionnaires, and self-reported surveys, this information still provided valuable insight into the connection between diet, medications and the microbiome in a large group of patients. Dietary components had significant effects on the community microbial variance, including fiber-rich foods (1.29 %; i.e. fruits, vegetables, beans), red and processed meats (0.4%), as did common medications, such as NSAIDs, aspirin (0.3 %), metformin (0.4%), and proton pump inhibitors (0.3%). To further reinforce the correlation between diet and microbial taxonomic variation, we also performed Mantel statistics, which reaffirmed that diet accounted for a small (0.6%) but significant (p<0.01) proportion of the microbial taxonomic variation between subjects.
Microbial features associated with colorectal polyps
Exploring specific taxonomic signatures, at the species level, associated with either TAs or SSAs, was done by applying multivariable linear models to each taxonomic feature present in at least 10 samples with 0.1% relative abundance, while correcting for host demographics, such as age, sex and BMI. Two unique consortiums of microbial species were associated with either TAs (n=19) or SSAs (n=8), while there were no shared microbial signatures among patients with either TA or SSA (Fig. 3A, Table S2). Additional analysis, which included both linear regression, while correcting for age, sex and body mass index, as well as pairwise Dunn’s test, comparing between samples with both TA and SSA against controls, did not identify any unique microbial signature for subjects with both TA and SSA. However, by using pairwise comparisons of a group mean dispersion, we were able to conclude that the average microbial communities of subjects with both TA and SSA were significantly different compared to subjects with TA (p=0.02), but not significantly different compared to subjects with SSA only (p=0.3). This suggests that the pattern in distribution with variation of microbiome amongst patients with both TA and SSA are more like patients with SSA only.
Figure 3: Association of gut microbiome taxonomic features associated with adenoma subtypes.
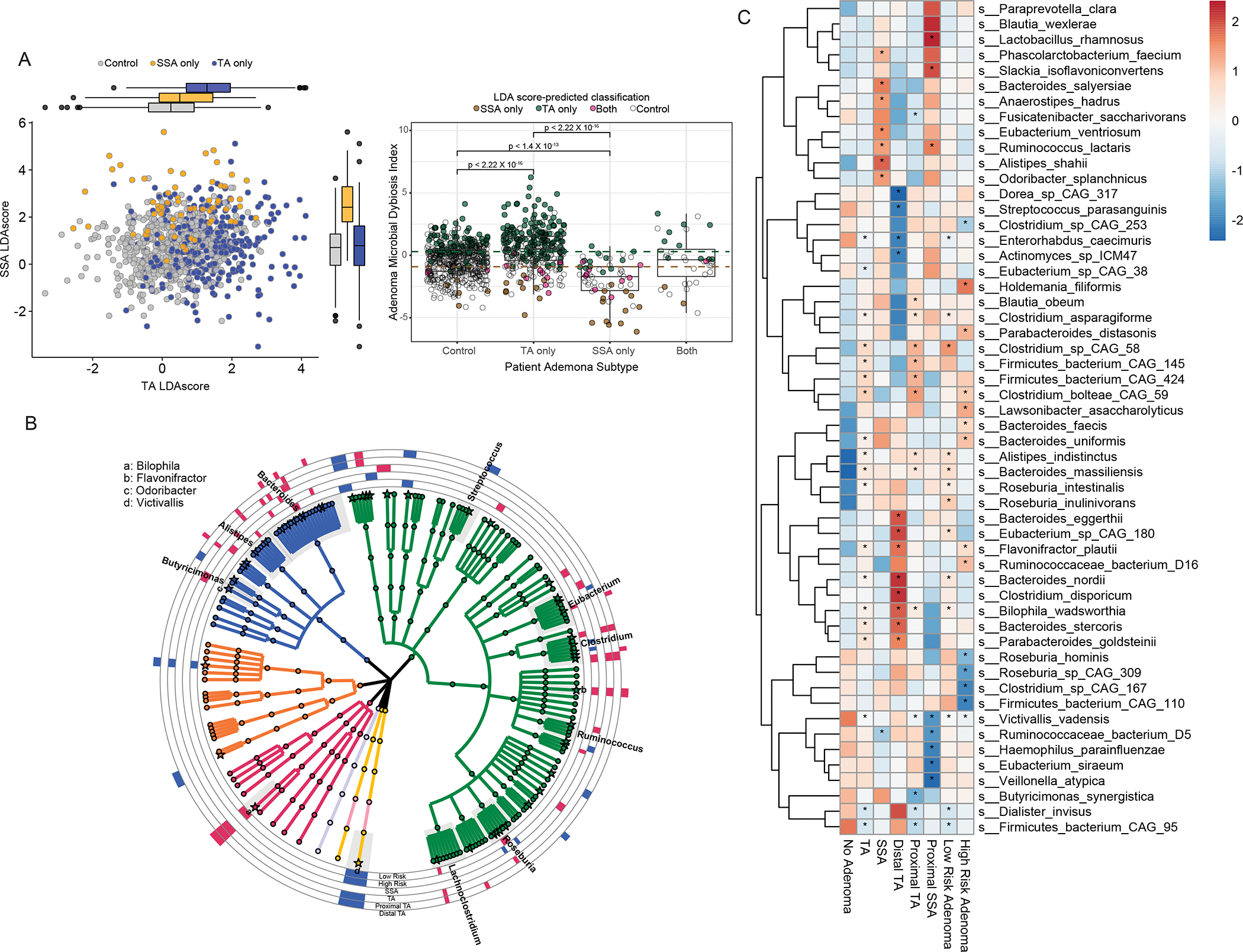
(A) Dotplot with both axes indicating the respective TA and SSA linear discriminant analysis (LDA) scores, demonstrating the segregation of the subjects by the presence of TA (blue), SSA(orange) or neither (gray). Boxplot of the Adenoma Microbial Dysbiosis Index (ADMI), a function of both LDA scores, which is significantly higher in cases with TA and lower in cases with SSA, whereby subjects are coloured as green, brown or pink, respectively, if they are predicted by LDA score to either be TA, SSA or both TA and SSA. The ADMI cut-offs to determine if subjects’ microbiome were TA-like or SSA-like are the green and brown dashed lines. (B) Phylogenetic tree (generated using the software GraPhlAn) constructed on 170 bacterial species detected in at least >10% of the GIDER cohort. Colored tree leaves indicate the species belong to the same phylum, and the outer rings indicate adenoma subgroup specific results with those microbial species enriched in the subgroup colored as red, and those diminished coloured as blue (C) Heatmap of microbial species significantly associated with TA, SSA, proximal TA, distsal TA, low and high risk adenomas, whereby each cell represents the average relative abundance of the microbial species, specific to the subgroup of interest, and * depicting those p<0.05, FDR <0.2.
Linear discriminant analysis (LDA) was used to make microbiome based classifiers for either TA or SSA compared to controls. Using the both LDA for TA and SSAs, we were able to discriminate the presence of any colonic adenoma with an accuracy of 70.2% (95%CI: 67.2–73.8). Using the LDA, the positive likelihood ratio of TA or SSA was 2.47 (95%CI 2.09–2.91) and 8.32 (95%CI 5.09–13.61), respectively. We further produced an Adenoma Microbial Dysbiosis Index (ADMI) using both LDA indices and found subjects with TA have significantly increased ADMI (0.5±1.6 vs −0.4±1.3, p<0.01), while subjects with SSA have significantly lower ADMI (−2.0±1.6 vs −0.4±1.3, p <0.01) when compared to controls. The ADMI also distinguishes between subjects with TA or SSA (p<0.01). We then applied the ADMI on subjects with both TA and SSA, and 12 were classified as more SSA-like and 10 were more TA-like, and the remaining 14 were classified as controls. Of note, among the subjects with both TA and SSA, those with higher ADMI (more TA-like) all had 3 or more TAs found on colonoscopy. Conversely, subjects with lower ADMI (more SSA-like) had adenomas exclusively in the proximal right colon only.
Microbial species more abundant in cases with SSA included prominent short-chain fatty acids (SCFA) producing members of the human gut microbiome, such as Ruminococcus lactaris, Eubacterium ventriosum, Odoribacter splanchnicus, Anaerostipes hadrus, and Alistipes shahii. Microbial species enriched in subjects with TA include Flavonifractor plautii, Bilophila wadsworthia, Bacteroides stercoris, Clostridium asparagiforme, Clostridium bolteae CAG 59, and Roseburia intestinalis. However, these SCFA producing microbes, associated with SSA in our study, have also pro-inflammatory microbial metabolic activities with subsequent immunomodulatory effects; Ruminococcus lactaris have been found to be enriched in rheumatoid arthritis24, Eubacterium ventriosum which harbors the highly immunomodulatory flagellin protein fliC (K02406) was previously described in patients with Lynch syndrome15, and Odoribacter splanchnicus, a prominent sulfate reducing bacteria which can generate genotoxic hydrogen sulfide, and has been shown to cause epithelial DNA damage25 and increased CRC risk26. Of note, 17 microbial species were identified to be increased in abundance in subjects with either low-risk (n=9) or high-risk adenomas (n=8), of which increased in Bacteroides species, such as Bacteroides faecis, Bacteroides massiliensis, Bacteroides nordii, and Bacteroides uniformis, with a corresponding decrease in Firmicutes species, such as Clostridium sp CAG167, Clostridium sp CAG 253, Firmicutes bacterium CAG 110 and Firmicutes bacterium CAG 95.
We were also interested in exploring microbiome profile differences between proximal and distal polyps. Of note, SSA, which is primarily present in the proximal colon, accounts for a disproportionate fraction of cancers identified after colonoscopy. Thus, to examine the regional microbiome signatures while adjusting for adenoma subtype, we decided to compare proximal TA (n=203), proximal SSA (n=45) and distal TA (n=50) against controls, excluding patients with pancolonic polyps and those with both TA and SSA. There were 34 microbial species differentially abundant with either left-sided (distal) or right-sided (proximal) adenomas (Fig3b). Bacteroides eggerthii, Bacteroides nordii, and Bacteroides stercoris, were more abundant in cases with left-sided TAs, which also reaffirms previous stool microbiome studies demonstrating such Bacteroides species to be associated with systemic inflammation and CRC (Feng et al., 2015). Proximal SSAs showed a consistent trend of increased abundance of Firmicutes species, such as Ruminococcus lactaris and Blautia wexlerae. Microbiome features associated with TA could be further differentiated to location-specific microbial TA signatures: Flavonifractor plautii was more abundant in left-sided TA, Clostridium sp CAG 58 was more abundant in right sided TA, and Bilophila wadworthia was more abundant in both proximal and distal TAs.
Of note, our study, similar to previous metagenomic cohort studies, which included a total of 176 subjects with adenomas and 480 controls across 8 geographically diverse cohorts15,27, did not identify Fusobacterium nucleatum (Fig. S2) as a major component of either adenoma subtype, as this microbial species was not substantively detected in stool amongst our cohort. Next, we performed a targeted search for Streptococcus gallolyticus, a previously established driver of asymptomatic colon cancers via bacteriocin production28, or through putative type VII secretion system29. However, S. gallolyticus was also not substantively detected in stool amongst our cohort. We also looked specifically for both enterotoxic Bacteroides fragilis as well as Clb+ Escherichia coli, through a targeted search for Bacteroides fragilis toxins (i.e., bft-1, bft-2, bft-3) and colibactin (i.e. ClbB-H) via ShortBRED30. Of interest, both markers were not prominent in cases with SSA; bft+ B. fragilis was not detected, and there was only 1 case of Clb+ E coli in subjects with SSA. However, in keeping with previous reports which found that right-sided colorectal tumors are more densely encased with biofilms with such mucus-invasive microbes 31, we were able to identify via stool samples, a modest increase in both bft+ B. fragilis and Clb+ E coli in subjects with proximal TA compared to controls. Both bft+ B. fragilis (4.9% vs 2.7%) and Clb+ E coli (4.9% vs 4.0%) were not present in cases with distal adenomas.
Metagenomic predicted functions and metabolites associated with colorectal polyps
To further understand the functional consequences of microbial composition changes associated with adenoma, we profiled the gene families in all metagenomes using HUMAnN3, and then summed their abundances according to the EC number annotations. We first undertook the same linear modeling, as previously described for taxonomic analysis, on enzyme abundance data to associate microbial species associated with adenoma, revealing 363 enzymes that were most differentially abundant (q<0.2) in stool samples with either TAs or SSAs (Table S3). These differential abundant enzymes could be further binned to their higher pathway function (Fig. 4A), and functional analysis demonstrated a decreased abundance of enzymes involved in methane metabolism (Fig. 4B) and increased metabolism of amino acids and lipids, compared to those without adenomas. The findings of decreased abundance of methane microbial metabolism is supported by a corresponding loss of keystone methanogenic Archaea species, such as Methanobrevibacter smithii (Fig. 4C).
Figure 4: Association of gut microbiome functional features associated with adenoma subtypes.
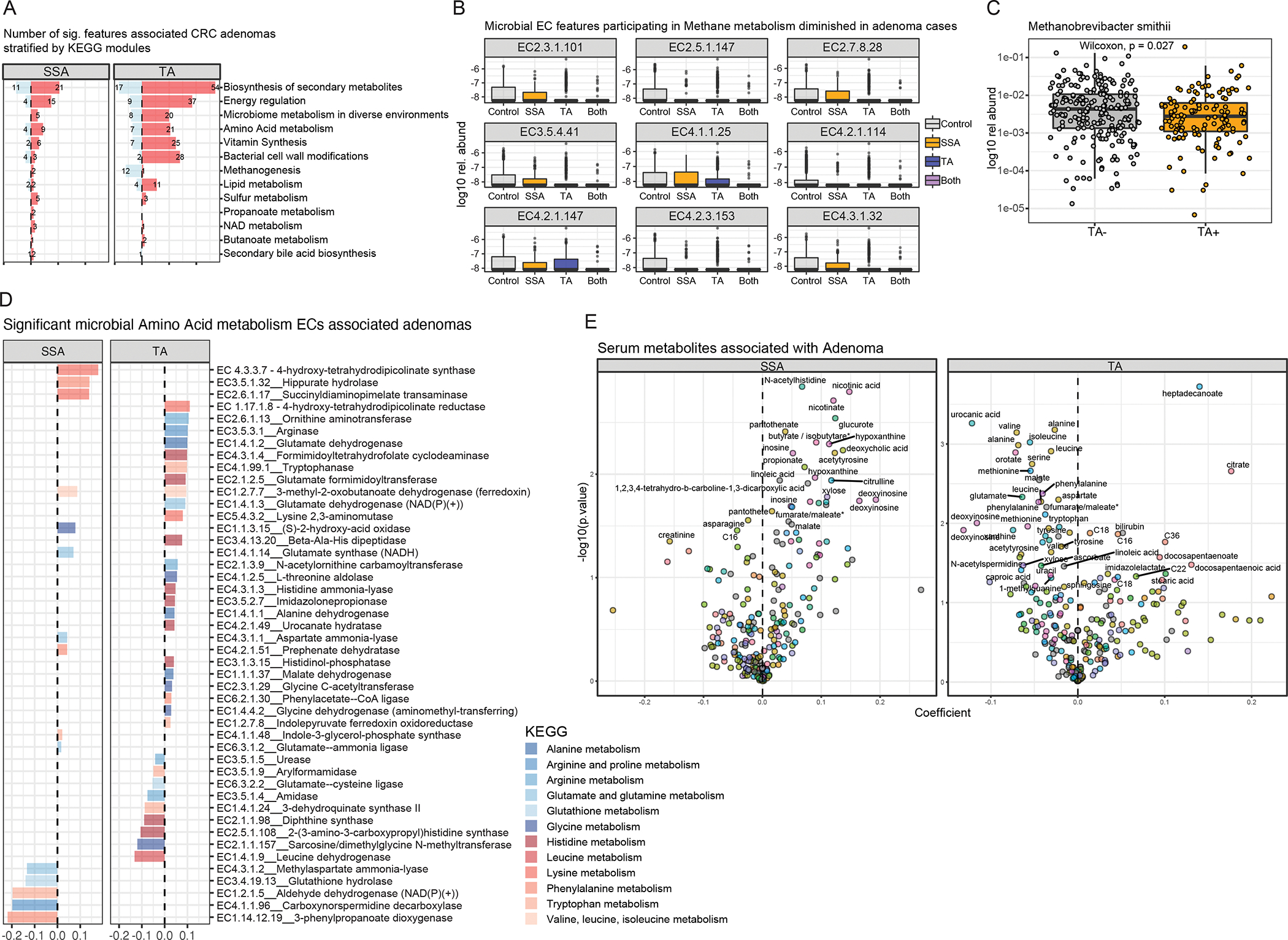
(A) Barplot of 363 significant microbial EC functional features (FDR <0.2), agglomerated to their parent metabolic pathway function, associated with either SSAs or TAs. (B) Patients with tubular adenomas also have significant negative associations with microbial features associated with methanogen metabolism, with (C) corresponding lower abundance of methanogenic microbial species - Methanobrevibacter smithii. (D) Participants with either SSAs or TAs have the most numbers of positive associations with amino acid metabolism, whereas amino-acid related pathways include both essential and non-essential amino-acids (E) Volcano plots highlighting predicted metabolites with increased or decreased abundance in either TA or SSA.
Another key pathway recently implicated in CRC pathogenesis is microbial sulfidogenesis. To better characterize the genes for microbial sulfur metabolism, we undertook a targeted search of sulfidogenic genes highlighted by Wolf, Cowley and colleagues32. We identified that LuxS and anaerobic sulfite reductase (Asr) A and C are more abundant in cases with SSA, compared to that of controls, demonstrating that Asr is likely a important contributor to sulfate reduction in SSA pathogenesis. We further stratified microbial metabolism of amino acids, a key pathogenic process in both SSA and TA CRC pathogenesis33 Both essential (e.g., EC4.1.99.1 Tryptophanase) and non-essential amino acid turnover (e.g., EC3.5.3.1 Arginase) were prevalent in both subtypes of adenomas, although there was a trend of increased abundance of essential amino acid metabolism pathways in SSA, and a mixed pattern of both non-essential and essential amino acid metabolism in TA (Fig. 4D).
To further validate our metagenomic predicted microbial functions in TA and SSA — and provide further insights into potential bioactive microbial metabolites influencing TA and SSA formation — we undertook a computational approach to predict metabolites from metagenomes in the GIDER cohort, using Melonnpan34. In brief, we first trained a cross-validated elastic net regularized regression model on a set of paired metagenomes and metabolites abundances from an independent cohort35. We then subsequently applied this model on our GIDER metagenome to infer predictive metabolome, and subsequently identified putative metabolites associated with either TA or SSA using the same linear regression approach used for metagenomics, adjusting for age, gender and body-mass index. Here, we showed that TA was predicted to have increased polyunsaturated fatty acids (eg. heptadecanoate, docosapentaenoate, stearic acid) and decreased amino acids (eg. valine, alanine, isoleucine, phenylalanine), presumably reflecting a complex dietary pattern and altered fatty acid metabolism, with significant decrease in amino acids, in keeping with the putative microbial lipid and amino acid metabolisms enzymes identified with TA described above, as well as consistent with Liu and colleagues which demonstrated increased D-amino acid, sphingolipid and ether lipid metabolism in cases with CRC36. We have also validated the association of increased fatty acids associated with TAs in an independent cohort37 (Fig S3). Of note, SSA were predicted to have increased nicotinate and nicotinic acid (Fig4E); both are reaction products of NAD biosynthetic pathways (see Fig4A). Patients with SSA were also predicted to have increased proportions of secondary bile acids, such as deoxycholic acid, which are known in long-term high concentration states to lead to DNA damage, promoting carcinogenesis38.
Environmental-Microbiome associations with colorectal adenomas
Within GIDER, we uncovered 51 associations between 32 unique microbial taxa and 15 environmental exposures (dietary patterns and concomitant medications) (FDR <0.05), of which fruits and vegetable intake accounted for the most associations (10, and 8 associations, respectively) (Table S4). By focusing only on the significant dietary-microbe and medication-microbe correlations amongst microbial species significantly associated with either TAs or SSAs, we applied mediation analysis to better understand the dynamic interactions of environmental exposures through microbiome on early CRC carcinogenesis (Fig5A). We modeled specific dietary patterns or concomitant medications associated with either TA or SSA, where the microbiome was hypothesized to mediate the effect of environmental exposures and subtype (Fig5B). Our findings allow us to establish causal relationships, therefore differentiating between microbial features with potential mediation effect (i.e. Flavonifractor plautii and Bacteroides stercoris) or features which were ‘by-standers’ (such as Roseburia intestinalis). Both Flavonifractor plautii and Bacteroides stercoris were differentially more abundant in the presence of TA, but had significant reciprocal lower abundance in the setting of either increased vegetable intake or concomitant aspirin use. The plausible role of Flavonifractor plautii appears to be linked with the degradation of beneficial anticarcinogenic flavonoids, which was also found to be significantly correlated with the enzymes and modules involved in flavonoid degradation among individuals with colorectal cancer (CRC)39. It is also reported that Flavonifractor plautii is an important species in young CRC, where it is consistently the dominant population in young CRC cohorts40. In our GIDER cohort, we consistently found Flavonifractor plautii positively associated with TA, and we hypothesized that beneficial impact of increased vegetable consumption and aspirin use on lowering risk of TA and SSA, respectively, could be mediated by negating the effects of Flavonifractor plautii.
Figure 5: Interplay of environmental factors and gut microbiome features associated with adenoma subtypes.
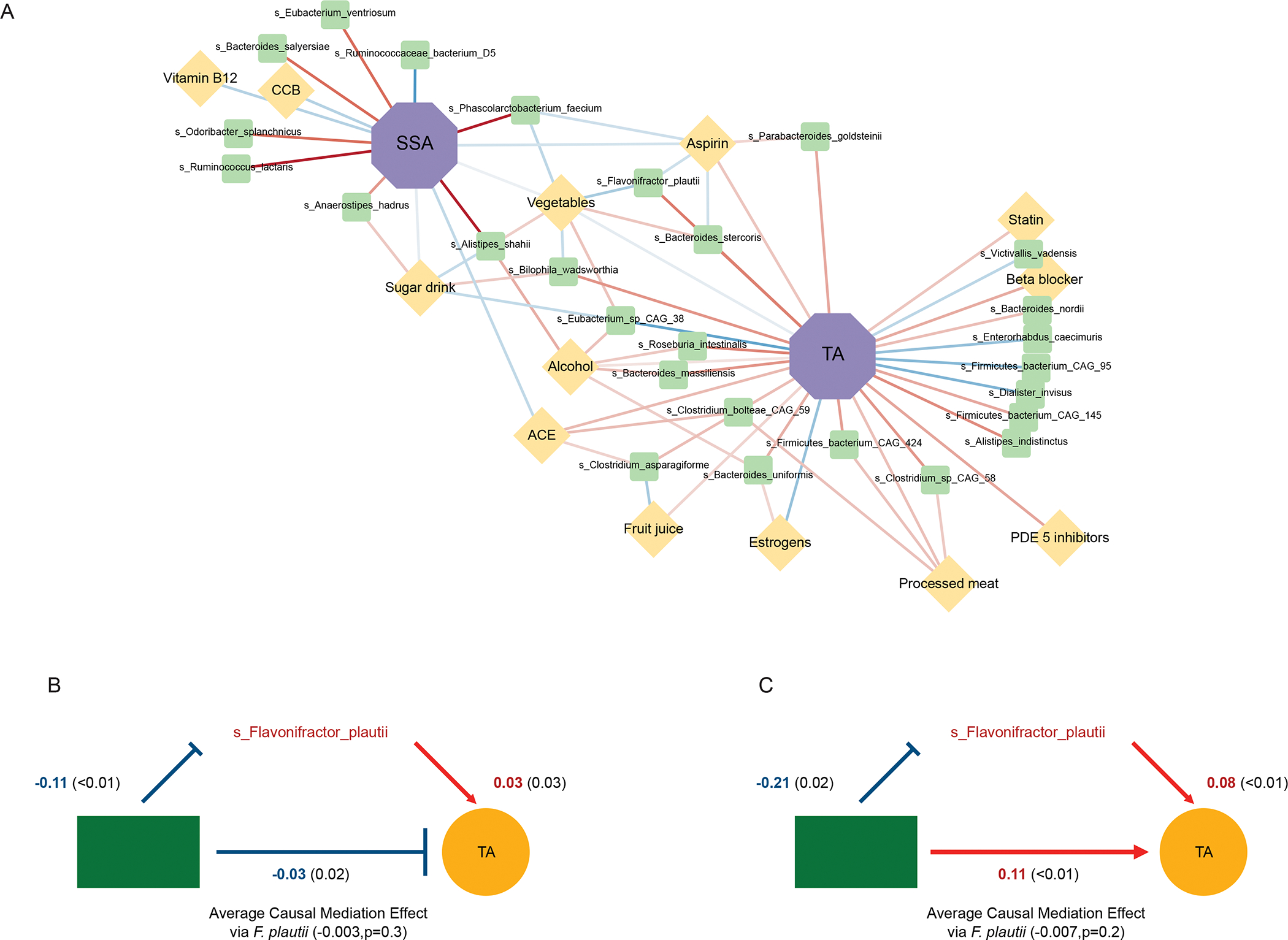
(A) Significant dietary-microbe and medication-microbe correlations associations limited to significant microbial species identified to be associated with either TAs (A) or SSAs (B) in GIDER, are plotted in this network correlation plot. Network shows all significant correlations (FDR < 0.05) between each pair of measurement types. Nodes coloured by class of environmental factor and sized by number of associations, lines by sign and strength of association (red for positive correlation, blue for negative correlation). (B) Mediation plots demonstrating statistically significant environmental exposure and microbe effects on tubular adenomas.
DISCUSSION
Our GIDER cohort is a cross-sectional study that profiles the microbiota of stool samples from individuals undergoing routine endoscopic procedures, curated with paired dietary intake and medication history. GIDER, to the best of our knowledge, is the largest colonoscopy cohort of TA and SSA that collected comprehensive dietary, demographic, medication history and endoscopy findings paired with fecal metagenomics. It is important to differentiate SSA from TA because they each have different biological and clinical characteristics, and this differentiation can have implications for colorectal cancer screening and surveillance. From GIDER, we were able to profile unique microbial taxonomic and functional signatures for either SSA or TA. SSA, associated with increased risk of colorectal cancer, particularly in the proximal colon, were associated with increased sulfate reduction and secondary bile acid metabolism. Through microbial-metabolite prediction, maintenance of antioxidant mechanisms through NAD-NADPH metabolism was a key microbial pathway associated with SSA carcinogenesis. On the other hand, TA, the most common type of polyp, was associated with diminished microbial methane metabolism and amino-acid driven lipogenesis.
Previous studies have shown the abundance of pathobionts such as F. nucleatum, enterotoxigenic B. fragilis, and pathogenic E coli in CRC. These pathobionts have been postulated to significantly impact neoplastic processes in colon cells, leading to CRC. In brief, virulence factors of F. nucleatum, such as FadA adhesin, induce proinflammatory cascades mediated by NF-KB, as well as activate Wnt/b-catenin signaling resulting in dysregulated cellular turnover and apoptosis. Enterotoxigenic B. fragilis, which encodes the metalloprotease bacteroides fragilis toxin (BFT), induces chronic inflammation and tissue damage by targeting intestinal cell tight junctions, cleaving E-cadherin. Pathogenic E coli with the pks pathogenicity island secretes colibactin (Clb), a genotoxin that induces interstrand crosslinks and double-strand DNA breaks in colon cells. However, in our cohort, we did not detect F. nucleatum associated with either TA or SSA, which is consistent with other cohorts13,15 with colorectal adenoma. This may suggest that such F. nucleatum may only be present in late stages of the CRC. We were able to detect the presence of other pathobionts, such as enterotoxigenic B. fragilis and Clb+ E coli, but only found them present in less than one-tenth amongst the stool of the GIDER subjects, whereas previous studies have shown these microbes o be prevalent (~50%) in the colon biofilms of subjects with CRCs18,41. Of note, there was still a modest increased representation of either bft+ B. fragilis and Clb+ E coli among subjects with proximal (right-sided) TA, compared to control, suggesting that stool samples may still offer insight on CRC-associated regional microbiome representation.
Patients with TA have enrichment of microbial species, such as Clostridium bolteae, Clostridium asparagiforme, which are typically overabundant in patients with inflammatory bowel disease, suggesting a universal microbial architecture shared between mucosal inflammation disorders and TA carcinogenesis. Conversely, identification of beneficial bacteria depleted in adenomas may lead to implementation of dietary interventions or microbial-modulatory therapies, such as supplementation of Lactobacillus gallinarum42 and L. rueterin43, which have been shown to be CRC chemoprotective, and attenuate the development of CRC in preclinical models. Our findings also demonstrated that participants with TAs have lower abundances of Archaea species and correspondingly lower abundances of microbial pathways contributing to methanogenesis, and this is consistent with previous literature indicating that depletion of methanogens is associated with colorectal adenomas44. Archaea are known essential microbial anaerobic members of the human gut, playing an important role for effective digestion and removal of harmful metabolites45 such as trimethylamine during methanogenesis, and this process is mainly dependent on hydrogen. Reduction in hydrogen concentration by gut methanogens results in lower production of hydrogen sulfite by sulfite-reducing bacteria, thus reducing potential damage to colonic epithelial cells46. Further studies to determine mechanisms by which colonic Archaea species contribute to colorectal carcinogenesis are needed. Of interest, lipid metabolism was distinctly more present in TA. One other prominent pathway for TA identified is the mevalonate pathway, which studies have demonstrated is involved in the regulation of the innate immune response and immune defense, in particular activating inflammasomes in response to other bacterial toxins47. It also regulates tumorigenesis, whereby suppression of the mevalonate pathway was found to inhibit growth and proliferation of colon cancer cell lines48.
In contrast, we found patients with SSA to have predicted microbial functions prioritizing maintenance of multiple antioxidant defense systems. Amongst SSA subjects, there was enrichment of microbial enzymes synthesizing glutathione from glutamate, glycine, and cysteine. The increased abundance in microbial enzymes for glutathione synthesis is consistent with prior findings whereby cancer cells have increased glutathione levels to alleviate the effects of oxidative stress, a hallmark of CRC carcinogenesis49. The computationally predicted metabolite composition further reiterated importance of microbial antioxidant defense systems amongst SSA subjects. Here, we predict subjects with SSA to have increased abundances of microbial NAD-NADPH metabolites, whereby NADPH also acts as a potent antioxidant50. Our unique microbial signatures for SSA, whereby SSAs have high risks of developing into CRC and tend to be missed more often during colonoscopy, may lead to more effective identification and management strategies for patients with SSA.
Environmental factors, particularly diet and commonly prescribed medications, can have effects on the microbial composition11. Furthermore, there have been strong associations between dietary habits and colorectal cancer risk. We therefore investigated dietary patterns and microbiome profiles through network correlation analysis and mediation analysis. We modeled microbiome features as either mediators or bystanders of environmental influences, and we found the chemoprotective effective of well established protective factors, such as increased vegetable intake and concomitant aspirin use, to be mediated by specific microbial features (i.e. Flavonifractor plautii). Our results serve as the foundation for further prospective and interventional studies, thereby leading to the development and clinical translation of potential microbiota-based strategies for cancer prevention.
Although our study benefited from a large sample size, extensive metadata, and comprehensive bacterial profiling, we did not examine colorectal mucosal samples. Stool samples are easily obtainable and important for developing tools for risk stratification and screening for CRC, but mucosal samples would add to the discovery of bacteria associated with adenomas. Other limitations include a mostly white study population, limiting generalizability to other racial groups, and the cross-sectional design, which does not allow us to establish the temporality of the bacteria-adenoma relationship.
Colorectal cancer represents a heterogeneous groups of cancers arising through different combinations of genetic and epigenetic events; the predominant “conventional” pathway characterized by APC mutation, chromosomal instability, and paucity of CpG island hypermethylation present in TAs, and the “serrated” pathways characterized by BRAF mutation, chromosomal stability and high CpG island hypermethylation, evident in SSAs. Because colorectal cancer arises along different molecular pathways from distinct precursor lesions (TA, SSA), we have also demonstrated different microbiome features are involved in adenoma subtype. Our findings suggest CRC develops from different adenoma subtypes through a multi-hit model of carcinogenesis, characterized by heterogeneity in both the host genetic risk and the varying background exposure to environmental risk. The distinct host and environmental pressure that drive regional and multi-hit microbial heterogeneity results in distinct microbial profiles for varying adenoma subtypes. This wealth of data and accurate adenoma phenotyping within GIDER allows for significant contributions toward understanding the role of gut microbes in the etiology of colorectal cancer, particularly the how the gut microbiota may influence cancer susceptibility through harvesting nutrients and sources of energy from the diet and metabolism of xenobiotics — which may be both potentially beneficial or detrimental to colon carcinogenesis.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Ramnik J. Xavier (xavier@molbio.mgh.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
GIDERmetagenomics data is available in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) (SRA): PRJNA784939.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study cohort
The GI Disease and Endoscopy Registry (GIDER) enrolled 1197 consecutive participants from Massachusetts General Hospital in Boston between 2015 and 2022. Eligible participants were 18 years or older who were referred for an outpatient colonoscopy and able to provide informed written consent. For the current analysis, we excluded participants who were not able to provide stool samples (n=111), were on concomitant antibiotics or probiotics (n=44), found to have colitis (n=4), had a personal history of a hereditary cancer syndrome (n=29), and had suboptimal metagenomic read counts (n=30); exclusion due to these non-mutually exclusive criteria resulted in 971 individuals. Among these participants, the vast majority were receiving colonoscopy for routine screening and surveillance, while only 1.1% (n=8) had symptoms including abdominal pain, rectal bleeding, or change in bowel habit.
Upon provision of informed consent, detailed information was obtained about mucosal and histological subtyping of polyps (size, number, location, endoscopic appearance, histological subtype). Epidemiological risk factors pertinent to colorectal adenoma risk, such as age, gender, family history, body mass index, and smoking status were also included. Concomitant medications at baseline were noted, including aspirin, NSAIDs and antibiotic use. Dietary intake was assessed using a semi-quantitative food frequency questionnaire that was collected concordantly with the fecal sample. Stool samples were collected in 100% ethanol, aliquoted, and stored at −80C prior to DNA extraction and metagenomic sequencing. Stool samples were not collected at the time of colonoscopy to avoid potential effects of the bowel preparation.
Colorectal polyps were identified at colonoscopy, resected, and analyzed by pathology as per routine clinical care. Polyp-free controls were defined as those with no adenomas identified during the colonoscopy. Tubular adenoma (TA) cases were defined as those with at least one tubular or tubulovillous adenoma. Sessile serrated adenoma (SSA) cases were identified as having at least one SSA. We further classified cases as high clinical risk if polyps were ≥1cm and/or there were ≥3 adenomas present. Proximal adenomas were defined as adenomas located in the cecum, ascending colon, hepatic flexure, transverse colon or splenic flexure. Distal adenomas were defined as adenomas located in the descending colon, sigmoid or rectum.
Ethics Statement
All study procedures were approved by the Institutional Review Board at Massachusetts General Hospital under protocol number 2015P000275.
METHOD DETAILS
Metagenomic Sequencing
DNA was extracted from ~100 mg of stool using enzymatic treatment with 15 mg/ml lysozyme and proteinase K (Qiagen) for 10 min incubation followed by mechanical lysis using 0.1mm glass beads and 3 min bead beating on the “homogenize” setting of a Mini Bead beater-8 (Biospec Products). After lysis, samples were centrifuged for 5 minutes at 30,000 rpm to pellet debris and supernatant passed through a QIAshredder spin column (Qiagen) prior to proceeding with nucleic acid extraction using a Allprep DNA/RNA Mini Kit (Qiagen) following manufacturer protocol. Metagenomic libraries were constructed from 100–250 pg of DNA using the Nextera XT DNA Library Preparation Kit (Illumina) as per the manufacturer’s recommended protocol. Sequencing was performed on the HiSeq 2500 2× 101 PE platform (Illumina), targeting ~2.5 Gb of sequence per aliquot. Demultiplexing, BAM and FASTQ file generation were performed using the Picard suite (https://broadinstitute.github.io/picard).
QUANTIFICATION AND STATISTICAL ANALYSIS
Metagenomic profiling
Metagenomic taxonomic and functional profiles were generated using the bioBakery meta’omics workflow 51 in the Terra workspace. In brief, reads mapping to the human genome were first filtered out using KneadData (https://github.com/biobakery/kneaddata). Taxonomic profiles of shotgun metagenomes were generated using MetaPhlan3 53, which uses a library of clade-specific markers to provide pan-microbial profiling. Functional profiling was performed by HUMAnN3 54, whereby HUMAnN3 constructs a sample-specific reference database from the pangenomes of the subset of species detected in the samples by MetaPhlAn3. Sample reads are mapped against this database to quantify gene presence and abundance on a per-species basis. A translated search is then performed against a UniRef-based protein sequence catalog (UniRef release 202) for all reads that fail to map at the nucleotide level. The results are abundance profiles of gene families (UniRef90s), for both metagenomics, stratified by each species contributing to those genres, and further summarized to higher level gene groups such as Enzyme Commission numbers (ECs), KEGG orthology (KOs), or metaCYC pathways. This resulted in the generation of 586 microbial species (from 186 genera) and 2609 annotated EC features for downstream analyses. For subsequent analysis, read counts were transformed into relative abundances by normalization to the total number of reads per sample. Low-abundance filters were applied to discard taxonomic and functional features whose relative abundance did not reach 0.1% and 0.001% respectively, in at least 10% of the individuals.
Prediction of microbial metabolites
We predicted metabolite compositions from metagenomic sequencing data using MelonnPan v0.99. In brief, we reannotated the publicly available PRISM metagenomic data, using the updated HUMAnN3 database. Then, we retrained the MelonnPann elastic net regularization model, using 10 fold cross validation, to subsequently infer which HUMAnN3 Uniref90 features are predictive, and have these features estimate the composite metabolome based on the reference >8000 metabolite features described previously in the PRISM dataset.
Profiling of genes encoding Colibactin and Bacteroidetes Fragilis toxin
To obtain a more comprehensive database of bacteroides fragilis toxin (bft) and colibactin genes, for which the bft-1, bft-2, bft-3, ClbB homologues were selected from the UniProt database, to form the reference library. The shortBRED tool 30 shortbred_identify.py (v0.95) was then used to identify unique markers for bft, with the UniRef90 database (v202) used as negative control. The shortbred_quantify.py tool (v0.95) was then used to perform a quantification of these markers in the metagenomes.
Analysis of metagenomic diversity and metacommunities
Alpha diversity (richness and Shannon diversity) was calculated for taxonomic profiles at the species level, using the Vegan package in R. Subsequent analysis to determine the cluster of metagenomes (at the species level) associated with remission, utilized Dirichlet multinomial (DMM) algorithms, whereby DMM bins samples on the basis of microbial community structure, and the appropriate number of clusters was determined based on the lowest Laplace approximation score. Differential abundance analyses were performed using the post-hoc Dunn tests to identify significant species taxonomic features between DMM metacommunities with FDR correction threshold of 0.05. For association of metagenomic diversity and metacommunities with categorical and continuous clinical metadata, we applied Fisher’s exact tests, Mann-Whiteley U-tests, and linear regression models when appropriate. Quantification of variance within the metagenomic dataset was calculated using PERMANOVA as implemented by the ‘adonis’ function in the R package Vegan. Dietary distance matrices were calculated by ordering the dietary intake frequencies from less to more frequent, assigning integers to these levels and calculating the Manhattan distance. Quantification of covariation between dietary intake frequencies and microbial taxonomic abundances was done using the Mantel test.
Statistical analysis
We used linear models to identify putative differential abundance analysis of all omic measurement types. Features were log transformed to variance-stabilize the data. Zero values were additively smoothed by half of the minimal abundance for each feature. Abundance were then log-transformed and fitted with the following per-feature linear mixed-effects model:
That is, in each per-feature multivariable model, the transformed abundance of each feature was modeled as a function of the adenoma status (with no polyp controls used as the reference), while adjusting for age (continuous variable), body-mass index (continuous variables), as well as sex (with female as reference). Fitting was performed with the MaAsLin2 package in R, with nominal p-value adjusted for multiple hypothesis testing with a target FDR of 0.2.
Spearman’s correlation was used to assess relationships between microbial features that were associated with case/control status, dietary frequency and medications. Correlations were calculated using the ppcor R package, correcting for host demographic factors age, BMI and sex. Significant correlation coefficients (qval <0.2) were selected for network visualization using the Cytoscape 3.6.0.
Linear discriminant analysis (LDA) was first performed on the square-root transformed taxonomic relative abundances microbiome data, excluding subjects with both TA and SSA, using the MASS package in R. Two linear combinations of features that best separated subjects for the presence of either TA or SSA were identified. Both LDA models for TA and SSA were then used to predict in all subjects if they had the presence of TA or SSA, respectively. Subjects that were predicted to have both, would need to be classified as both TA and SSA present in both LDA models. Likelihood ratios were calculated using the confusion matrix against the ground truth, whilst factoring for a prevalence of 36.8% of TA and 10.1% of SSA. We further developed a computed Adenoma Microbial Dysbiosis Index (AMDI), but taking the different of the LDA scores for TA and SSA (i.e., LDA score for TA - LDA score for SSA). We performed receiver operating characteristics (ROC) analysis on the ADMI, calculated the optimal cut-off for the ADMI for either TA or SSA using the Youden Index (J), whereby this points on the ROC curve where the sum of sensitivity and specificity is maximized. The cut offs for TA and SSA were found to be 0.303 and −0.899, respectively, as they provided the highest J value. Using the ADMI, we then determined if subjects with TA or SSA were more TA-like or more SSA-like. Mediation analysis via the Mediation package in R, was used to investigate whether the identified environment-adenoma relations are mediated by microbial species. In brief, we employed the classical three-step method. We first satisfied the significant relationship between the independent exposure (i.e., specific dietary habits, or concomitant medication use) and the dependent outcome (i.e., TA or SSA) determined by binary logistic regression. To establish mediation, we then tested if there was significant relationship between the independent exposure and the mediating microbiome feature using linear regression, and whether the mediating microbiome feature is significantly related to the dependent outcome (i.e., TA or SSA), when adjusted by the independent exposure (i.e., specific dietary habit or concomitant medication use), using the mediation analysis function with 10,000 simulations. This mediation analysis approach was performed separately for each dependent outcome (i.e., TA or SSA), selected exposure (concomitant medications such as Aspirin, ACEi, and dietary intake of vegetables, sugar beverages, fruit juices, alcohol, processed meat), and all 27 significantly associated microbial features previously identified through the earlier described multi-regression analysis.
Supplementary Material
Table S1: Demographics of the GIDER cohort, Related to Figure1. Selected (p<0.05) demographic, dietary, medication and endoscopic characteristics of the 708 GIDER participants, stratified by their adenoma subtypes (i.e., no adenoma, tubular adenomas only, serrated adenoma only, both tubular and serrated adenomas).
Table S2: List of significant microbial taxonomic features, Related to Figure 3. Differential abundant microbial taxa between participants with either TA or SSA compared to controls (p<0.05, q<0.2).
Table S3: List of significant microbial functional features, Related to Figure 4. Differential abundant microbial EC between participants with either TA or SSA compared to controls (p<0.05, q<0.2).
Table S4: List of microbe-dietary interactions, Related to Figure 5. Significant microbial species associated with various dietary components (p<0.05, q<0.2).
Table S5: List of microbe-medication interactions, Related to Figure 5. Significant microbial species associated with various prescribed medications (p<0.05, q<0.2).
KEY RESOURCE TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Patients fecal samples | Massachusetts General Hospital and Harvard Medical School | GIDER |
| Critical Commercial Assays | ||
| Mini Bead beater-8 | Biospec Products | 693 |
| QIAshredder | Qiagen | 79654 |
| Proteinase K | Qiagen | 19131 |
| AllPrep DNA/RNA Mini Kit | Qiagen | 80204 |
| Nextera XT DNA Library Preparation kit | Illumina | FC-131–1096 |
| Deposited Data | ||
| Shotgun metagenomic sequences | Sequence Read Archive | PRJNA784939 |
| Software and Algorithms | ||
| bioBakery meta’omics workflow | (McIver et al., 2018) | https://app.terra.bio/#workspaces/rjxmicrobiome/mtx_workflow |
| Trimmomatic (v0.36) | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| KneadData (v0.7.2) | Huttenhower lab | http://huttenhower.sph.harvard.edu/kneaddata |
| Metaphlan 3 | (Beghini et al., 2020) | https://huttenhower.sph.harvard.edu/metaphlan |
| HUMAnN3 | (Franzosa et al., 2018) | https://huttenhower.sph.harvard.edu/humann |
| MaAsLin2 | Huttenhower lab | https://huttenhower.sph.harvard.edu/maaslin/ |
| MelonnPan | (Mallick et al., 2019) | https://huttenhower.sph.harvard.edu/melonnpan/ |
| ShortBRED | (Kaminski et al., 2015) | https://huttenhower.sph.harvard.edu/shortbred/ |
| R (v3.6.1) | packages for analytical part: vegan, finalfit, tidyverse, broom, MASS, qvalue, HMP, mediation | ttps://www.r-project.org/ |
HIGHLIGHTS.
The gut microbiome varies by type and location of colonic adenomas
Tubular adenomas associate with a decrease in enzymes that metabolize methane
Serrated adenomas exhibit increased NAD, bile acid, and sulfate metabolic potential
Most of the significant microbial species also correlate with diet or medications
ACKNOWLEDGEMENTS
The authors thank participating patients and GIDER research team (Ashwin Ananthakrishnan, Emily Bethea, Irun Bhan, V Alin Botoman, Kristin Burke, Peter Carolan, Andrew Chan, Raymond Chung, Francis Colizzo, Kathleen Corey, Peter Kelsey, Braden Kuo, Paul Lochhead, Jay Luther, Barbara Nath, Long Nguyen, Norman Nishioka, Daniel Pratt, Esperance Schaefer, Amandeep Singh, Molly Thomas, Chris Velez, Joseph Yarze, Danielle Bellavance, Benjamin Maxner) at the MGH. The authors also thank Luke Besse for project management and making data available through the Sequence Read Archive (SRA) and the Broad Institute Microbial ‘Omics Core for help with sequencing data generation. The authors are also grateful to Elizabeth Heppenheimer for editorial assistance with the manuscript and figures. The graphical abstract was created with biorender.com. The study was funded in part by the Center for Microbiome Informatics and Therapeutics at MIT (RJX), the Center for the Study of Inflammatory Bowel Disease (CSIBD; NIH grant DK43351) (RJX), NIH grant AI172147 (RJX), and NIH grant AG068393 (HK).
Footnotes
DECLARATION OF INTERESTS
RJX is a co-founder of Celsius Therapeutics and Jnana Therapeutics, a member of the scientific advisory board of Nestle, and a member of the board of directors at Moonlake Immunotherapeutics.
JLWJ is a co-founder of AMILI and serves as a member of the scientific advisory board.
DCC is a consultant to Guardant.
HK has received grant funding from Pfizer and is a consultant to Takeda.
KS has served as a consultant to Arena, Boston Pharmaceutics, Gelesis, GI Supply, and Takeda/Shire; he has received research support from Ironwood and Urovant.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA, and Jemal A (2020). Colorectal cancer statistics, 2020. CA Cancer J. Clin. 70, 145–164. [DOI] [PubMed] [Google Scholar]
- 2.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al. (2015). Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 6, 1–13. [DOI] [PubMed] [Google Scholar]
- 3.Fujita K, Yamamoto H, Matsumoto T, Hirahashi M, Gushima M, Kishimoto J, Nishiyama K-I, Taguchi T, Yao T, and Oda Y (2011). Sessile serrated adenoma with early neoplastic progression: a clinicopathologic and molecular study. Am. J. Surg. Pathol. 35, 295–304. [DOI] [PubMed] [Google Scholar]
- 4.Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I, et al. (2018). Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 68, 31–54. [DOI] [PubMed] [Google Scholar]
- 5.Song M, and Giovannucci E (2016). Preventable Incidence and Mortality of Carcinoma Associated With Lifestyle Factors Among White Adults in the United States. JAMA Oncol 2, 1154–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song Y (2020). Clinicopathological and Endoscopic Features of Sessile Serrated lesions with dysplasia: Systematic review and meta-analysis. 10.37766/inplasy2020.11.0061. [DOI] [Google Scholar]
- 7.Mosley D, Su T, Murff HJ, Smalley WE, Ness RM, Zheng W, and Shrubsole MJ (2020). Meat intake, meat cooking methods, and meat-derived mutagen exposure and risk of sessile serrated lesions. Am. J. Clin. Nutr. 111, 1244–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liang PS, Shaukat A, and Crockett SD (2021). AGA Clinical Practice Update on Chemoprevention for Colorectal Neoplasia: Expert Review. Clin. Gastroenterol. Hepatol. 10.1016/j.cgh.2021.02.014. [DOI] [PubMed] [Google Scholar]
- 9.Spector T, Asnicar F, Berry S, Valdes A, Franks P, Wolf J, Hadjigeorgiou G, Roy CL, Leeming E, Drew D, et al. (2020). Microbiome Signatures of Nutrients, Foods and Dietary Patterns: Potential for Personalized Nutrition from The PREDICT 1 Study. Current Developments in Nutrition 4, 1587–1587. [Google Scholar]
- 10.Jackson MA, Verdi S, Maxan M-E, Shin CM, Zierer J, Bowyer RCE, Martin T, Williams FMK, Menni C, Bell JT, et al. (2018). Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 9, 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song M, Chan AT, and Sun J (2020). Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 158, 322–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kau AL, Ahern PP, Griffin NW, Goodman AL, and Gordon JI (2011). Human nutrition, the gut microbiome and the immune system. Nature 474, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. (2019). Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cullin N, Azevedo Antunes C, Straussman R, Stein-Thoeringer CK, and Elinav E (2021). Microbiome and cancer. Cancer Cell. 10.1016/j.ccell.2021.08.006. [DOI] [PubMed] [Google Scholar]
- 15.Yan, Yan Y, Drew DA., Markowitz A., Lloyd-Price J., Abu-Ali G., Nguyen LH., Tran C., Chung DC., Gilpin KK., et al. (2020). Structure of the Mucosal and Stool Microbiome in Lynch Syndrome. Cell Host & Microbe 27, 585–600.e4. 10.1016/j.chom.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sears CL, and Garrett WS (2014). Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kordahi MC, Stanaway IB, Avril M, Chac D, Blanc M-P, Ross B, Diener C, Jain S, McCleary P, Parker A, et al. (2021). Genomic and functional characterization of a mucosal symbiont involved in early-stage colorectal cancer. Cell Host Microbe. 10.1016/j.chom.2021.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, Wu X, DeStefano Shields CE, Hechenbleikner EM, Huso DL, et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avelar-Barragan J, DeDecker L, Lu ZN, Coppedge B, Karnes WE, and Whiteson KL (2022). Distinct colon mucosa microbiomes associated with tubular adenomas and serrated polyps. NPJ Biofilms Microbiomes 8, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon H, Kim N, Park JH, Kim YS, Lee J, Kim HW, Choi YJ, Shin CM, Park YS, Lee DH, et al. (2017). Comparisons of Gut Microbiota Among Healthy Control, Patients With Conventional Adenoma, Sessile Serrated Adenoma, and Colorectal Cancer. J Cancer Prev 22, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G, Yuen E, Freiman H, Lustbader I, Salik J, et al. (2016). The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome 4, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes I, Harris K, and Quince C (2012). Dirichlet multinomial mixtures: generative models for microbial metagenomics. PLoS One 7, e30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. (2015). The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 21, 895–905. [DOI] [PubMed] [Google Scholar]
- 25.Attene-Ramos MS, Wagner ED, Gaskins HR, and Plewa MJ (2007). Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 5, 455–459. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Nguyen LH, Mehta RS, Song M, Huttenhower C, and Chan AT (2021). Association Between the Sulfur Microbial Diet and Risk of Colorectal Cancer. JAMA Netw Open 4, e2134308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, et al. (2019). Author Correction: Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 25, 1948. [DOI] [PubMed] [Google Scholar]
- 28.Proutière A, du Merle L, Périchon B, Varet H, Gominet M, Trieu-Cuot P, and Dramsi S (2021). Characterization of a Four-Component Regulatory System Controlling Bacteriocin Production in Streptococcus gallolyticus. MBio 12. 10.1128/mBio.03187-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taylor JC, Gao X, Xu J, Holder M, Petrosino J, Kumar R, Liu W, Höök M, Mackenzie C, Hillhouse A, et al. (2021). A type VII secretion system of Streptococcus gallolyticus subsp. gallolyticus contributes to gut colonization and the development of colon tumors. PLoS Pathog. 17, e1009182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput. Biol. 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, et al. (2014). Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. U. S. A. 111, 18321–18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolf PG, Cowley ES, Breister A, Matatov S, Lucio L, Polak P, Ridlon JM, Gaskins HR, and Anantharaman K (2022). Diversity and distribution of sulfur metabolic genes in the human gut microbiome and their association with colorectal cancer. Microbiome 10, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lieu EL, Nguyen T, Rhyne S, and Kim J (2020). Amino acids in cancer. Exp. Mol. Med. 52, 15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mallick H, Franzosa EA, Mclver LJ, Banerjee S, Sirota-Madi A, Kostic AD, Clish CB, Vlamakis H, Xavier RJ, and Huttenhower C (2019). Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun. 10, 3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, Reinker S, Vatanen T, Hall AB, Mallick H, McIver LJ, et al. (2019). Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu N-N, Jiao N, Tan J-C, Wang Z, Wu D, Wang A-J, Chen J, Tao L, Zhou C, Fang W, et al. (2022). Multi-kingdom microbiota analyses identify bacterial-fungal interactions and biomarkers of colorectal cancer across cohorts. Nat Microbiol 7, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim M, Vogtmann E, Ahlquist DA, Devens ME, Kisiel JB, Taylor WR, White BA, Hale VL, Sung J, Chia N, et al. (2020). Fecal Metabolomic Signatures in Colorectal Adenoma Patients Are Associated with Gut Microbiota and Early Events of Colorectal Cancer Pathogenesis. MBio 11. 10.1128/mBio.03186-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pai R, Tarnawski AS, and Tran T (2004). Deoxycholic acid activates beta-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol. Biol. Cell 15, 2156–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gupta A, Dhakan DB, Maji A, Saxena R, P K VP, Mahajan S, Pulikkan J, Kurian J, Gomez AM, Scaria J, et al. (2019). Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. mSystems 4. 10.1128/mSystems.00438-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang Y, Du L, Shi D, Kong C, Liu J, Liu G, Li X, and Ma Y (2021). Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat. Commun. 12, 6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kinzler KW, Pardoll DM, and Sears CL (2014). Microbiota organization is a distinct feature of proximal colorectal cancers. Proceedings of the. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugimura N, Li Q, Chu ESH, Lau HCH, Fong W, Liu W, Liang C, Nakatsu G, Su ACY, Coker OO, et al. (2021). Lactobacillus gallinarum modulates the gut microbiota and produces anti-cancer metabolites to protect against colorectal tumourigenesis. Gut 71, 2011–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bell HN, Rebernick RJ, Goyert J, Singhal R, Kuljanin M, Kerk SA, Huang W, Das NK, Andren A, Solanki S, et al. (2022). Reuterin in the healthy gut microbiome suppresses colorectal cancer growth through altering redox balance. Cancer Cell 40, 185–200.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coker OO, Wu WKK, Wong SH, Sung JJY, and Yu J (2020). Altered Gut Archaea Composition and Interaction With Bacteria Are Associated With Colorectal Cancer. Gastroenterology 159, 1459–1470.e5. [DOI] [PubMed] [Google Scholar]
- 45.Koskinen K, Pausan MR, Perras AK, Beck M, Bang C, Mora M, Schilhabel A, Schmitz R, and Moissl-Eichinger C (2017). First Insights into the Diverse Human Archaeome: Specific Detection of Archaea in the Gastrointestinal Tract, Lung, and Nose and on Skin. MBio 8. 10.1128/mBio.00824-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teigen LM, Geng Z, Sadowsky MJ, Vaughn BP, Hamilton MJ, and Khoruts A (2019). Dietary Factors in Sulfur Metabolism and Pathogenesis of Ulcerative Colitis. Nutrients 11. 10.3390/nu11040931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, Zhang X, Gavin RM, Forde SD, Germain G, et al. (2016). Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 17, 922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gong L, Xiao Y, Xia F, Wu P, Zhao T, Xie S, Wang R, Wen Q, Zhou W, Xu H, et al. (2019). The mevalonate coordinates energy input and cell proliferation. Cell Death Dis. 10, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaughn AE, and Deshmukh M (2008). Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 10, 1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, and Huttenhower C (2018). bioBakery: a meta’omic analysis environment. Bioinformatics 34, 1235–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Thomas AM, Manghi P, Valles-Colomer M, et al. (2020). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Cold Spring Harbor Laboratory, 2020.11.19.388223. 10.1101/2020.11.19.388223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Specieslevel functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Demographics of the GIDER cohort, Related to Figure1. Selected (p<0.05) demographic, dietary, medication and endoscopic characteristics of the 708 GIDER participants, stratified by their adenoma subtypes (i.e., no adenoma, tubular adenomas only, serrated adenoma only, both tubular and serrated adenomas).
Table S2: List of significant microbial taxonomic features, Related to Figure 3. Differential abundant microbial taxa between participants with either TA or SSA compared to controls (p<0.05, q<0.2).
Table S3: List of significant microbial functional features, Related to Figure 4. Differential abundant microbial EC between participants with either TA or SSA compared to controls (p<0.05, q<0.2).
Table S4: List of microbe-dietary interactions, Related to Figure 5. Significant microbial species associated with various dietary components (p<0.05, q<0.2).
Table S5: List of microbe-medication interactions, Related to Figure 5. Significant microbial species associated with various prescribed medications (p<0.05, q<0.2).
Data Availability Statement
GIDERmetagenomics data is available in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) (SRA): PRJNA784939.