

Abstract
The growing concerns and cases of COVID-19 with the appearance of novel variants i.e., BA.2.75. BA.5 and XBB have prompted demand for more effective treatment options that could overcome the risk of immune evasion. For this purpose, discovering novel small molecules to inhibit druggable proteins such as PLpro required for viral pathogenesis, replication, survival, and spread is the best choice. Compounds from the Dark chemical matter (DCM) database is consistently active in various screening tests and offer intriguing possibilities for finding drugs that are extremely selective or active against uncommon targets. Considering the essential role of PLpro, the current study uses DCMdatabase for the identification of potential hits using in silico virtual molecular screening and simulation approaches to inhibit the current and emerging variants of SARS-CoV-2. Our results revealed the 10 best compounds with docking scores between −7.99 to −7.03 kcal/mol better than the control drug (GRL0617) among which DC 5977–0726, DC 6623–2024, DC C879-0379 and DC D135-0154 were observed as the best hits. Structural-dynamics properties such as dynamic stability, protein packing, and residue flexibility demonstrated the pharmacologically favorable properties of these top hits in contrast to GRL0617. The hydrogen bonding half-life revealed that Asp164, Arg166, Tyr264, and Tyr268 have major contributions to the hydrogen bonding during the simulation. However, some of the important hydrogen bonds were missing in the control drug (GRL0617). Finally, the total binding free energy was reported to be −34.41 kcal/mol for GRL0617 (control), −41.03 kcal/mol for the DC5977-0726-PLpro, for the DC6623-2024-Plpro complex the TBE was −48.87 kcal/mol, for the for DCC879-0379-Plpro complex the TBE was −45.66 kcal/mol while for the DCD135-0154-PLpro complex the TBE was calculated to be −40.09 kcal/mol respectively, which shows the stronger potency of these compounds against PLpro and further in in vivo and in vitro test are required for the possible usage as potential drug against SARS-CoV-2.
Keywords: SARS-CoV-2 variants, PLpro, Drugs, Free energy, Conformational dynamics
1. Introduction
The members of the subfamily Orthocoronavirinae (Coronaviridae family) are placed within four genera (alpha, beta, gamma, and delta coronaviruses). However, the pathogens of genus beta (β) coronavirus posed a serious threat to humanity in the 21st century by causing three major outbreaks (SARS-CoV1, MERS-CoV, and SARS-C0V2) in 2003, 2012, and 2019(Wang et al., 2020, Rotondo et al., 2021). SARS-CoV2 appeared in Wuhan, China, and penetrate across the globe with unprecedented speed. Therefore WHO (world health organization) declared a pandemic situation and advised to adopt stringent measures to cope with this alarming situation. The number of patients reached to 675 million whereas 6.7 million deaths are reported till 31 January 2023. To curtail the threat posed by SARS-CoV2 become very difficult due to fast mutations in all parts of the virus genome and especially in spike protein regions. These mutations lead to the appearance of numerous variants such as Alpha, Beta, gamma, Iota, Mu, Delta, Omicron, and finally the Omicron-BA.2. These mutations are instrumental and alter the drug and vaccine efficacy (Khan et al., 2021a, Khan et al., 2021b). The infections and deaths inflicted by this pathogen compelled the researchers to find new therapeutic targets for drugs and vaccines to combat this disease by using various structures (Spike protein, membrane protein, nucleocapsid protein, and envelop protein) and non-structure proteins (NSP 1–16). The proteins such as RNA-dependent RNA-polymerase (RdRp), 3-CLpro, and papain-like protease encoded by the ORF region (Fontanet et al., 2021, V’kovski et al., 2021) are important targets to control the COVID 19. The researchers and scientists followed systematic approaches for the identification of novel druggable targets for the development of novel anti-viral drugs. The experiments on respiratory diseases such as influenza suggest that arbidol or umifenovir are effective against the disease, however in severe cases arbidol is not very effective(Seneviratne et al., 2020). Whereas Remdesivir and Masitinib (a tyrosine-kinase inhibitor) are very effective against RdRP and 3CLpro respectively, however, they are in the clinical trials phase. Ritonavir and lopinavir are effective to reduce the severity of infectious diseases by co-targeting the 3CLpro (main protease) and RdRp (Beigel et al., 2020, Grein et al., 2020, Wu et al., 2020).
SARS-CoV-2′s effective control depends on the proper search for novel vaccines and drug targets to design safe, cheap, and effective vaccines and drugs. Spike protein is involved in the entry of the pathogen to the host cell and inhibition of this step is very important for the control of disease, therefore spike protein is a valuable drug target and numerous studies on HIV, HCV, and other infectious disease was conducted to target the spike protein (Khan et al., 2020a, Khan et al., 2020b). Similarly, the replication and maturation step depends on the cleavage of pp1a and pp1ab from the main protease. PLpro and 3CLpro have a vital role in converting pp1a and pp1ab into mature non-structure proteins, which facilitate replication. Therefore, targeting of PLpro and 3CLpro for anti-viral drugs will be an effective strategy. PLpro importance in replication and evasion of the host immune system makes it an effective drug target; therefore, we adopted a virtual screening approach for the identification of small molecules by searching ultra large drugs-library. Previously 3CLpro, RBD, RdRp, and other druggable proteins were targeted for the control of diseases caused by viruses through molecular screening approaches (Khan et al., 2020a, Khan et al., 2020b, Baildya et al., 2021, Khan et al., 2022). Therefore, PLpro will be a promising target for drug designing against COVID-19 by inhibiting the cleavage of pp1a and pp1ab. The inhibition of this step will help to stop the replication of pathogens. PLpro is highly conserved in all variants and this phylogenetic conserveness will be beneficial to design an effective drug against all variants of SARS-CoV-2. Papain-like protease (PLpro) active site is composed of a catalytic triad and it processes the viral polypeptides in a functional protein. These functional proteins are involved in hijacking an enzyme ubiquitin (Ubl) vital for host immune defense and thus contribute to evading the host immune system(Fu et al., 2021, Calleja et al., 2022). In all the variants of SARS-CoV-2, similar mutations in PLpro have been reported thus inhibition of any particular variant would demonstrated the pharmacological potential of any therapeutics against all.
The medicinal phytocompounds from Eurycoma harmandiana, Sophora flavescens and Andrographis paniculate have been explored against the PLpro from SARS-CoV-2 using molecular screening and simulation approaches. Multi-factorial investigation revealed that canthin-6-one 9-O-beta-glucopyranoside is a potential natural hit to be used against for the inhibition of SARS-CoV-2 targeting the PLpro(Verma et al., 2021). Moreover, using the computational methods a drug repurposing approach was applied where all the FDA-approved drugs were tested against the PLpro active site revealing rac5c as a potential hit (Huynh et al., 2021). Interestingly, using the structure-based pharmacophore approach four novel hits were reported that inhibit PLpro, while considering the GRL017 as a positive control. The results demonstrated that four hits among the all demonstrated better binding affinity than the native drug used to generate the pharmacophore model(Tian et al., 2022). In addition, repurposing the anti-bacterial drugs and screening of natural compounds databases also shortlisted potential hits for the inhibition of SARS-CoV-2 targeting PLpro (Dhote et al., 2022, Patel et al., 2022, Sencanski et al., 2022, Sayaf et al., 2023). Considering the essential role of PLpro the current study uses molecular screening of Dark Chemical Matters (DCM) using in silico virtual molecular screening and simulation approaches to identify potential hits to inhibit the current and emerging variants of SARS-CoV-2. Although several databases are available for the screening against different diseases such as using high-throughput computational screening and molecular simulation approaches but DCM provides a unique chemical space for the discovery of potential drugs against human diseases. Moreover, the unexplored biological activities, structural diversity, optimization, and the potential to use it as repurposed and in polypharmacology concept makeit a unique platform for therapeutics identification and development (Wassermann et al., 2015). DCM also presents remarkable potential for discovering drugs with selectivity or action against rare targets. The innovative aspect of the work is the use of the DCM database, the first time ever screened against the SARS-CoV-2, and the combination of in silico virtual screening, and simulation methods to find possible hits against both the existing and novel SARS-CoV-2 variants. Furthermore, the study aimed to identify compounds with stronger potency against PLpro compared to the control drug GRL0617, thus providing a foundation for potential drug development against SARS-CoV-2.Using the binding free energy approach, the final hits were validated that need further in vitro and in vivo validations to determine the clinical potency of these molecules against SARS-CoV-2 for the treatment of COVID-19.
2. Materials and methods
2.1. Structures retrieval, preparation and molecular screening
A protein databank (Rose et al., 2016) was used for retrieval of the SARS-CoV-2 PLpro 3D structure with ID 7CJM. The dark Chemical Matters (DCM) database was screened for small molecules search (Wassermann et al., 2015). Open Babel and FAF-drugs4 web tools were used for small molecule preparation and to check the violation of Lipinski’s rule (O'Boyle et al., 2011). For the ligands in the database open Babel was used to prepare each ligand. The gen3D method was used to generate the three-dimensional (3D) coordinates for each molecule using a distance geometry to generate reasonable 3D structures. Next, each molecule was protonated by adding the explicit hydrogen while partial charges were set using the Gasteiger model which is important for understanding molecular interactions and properties. To minimize each ligand molecular MMFF94 force field was used to optimize the geometry of each geometry and eliminate steric clashes. Using the systematic search methods implemented in open Babel the lowest energy conformations were generated while counterions, solvents, or undesired substructures were also removed using the integrated function. The compounds violating Lipinski’s rule were removed from the analysis and the shortlisted compounds were subjected to molecular screening with the Smina docking tool with Autodock scoring function. Smina is considered as a fork of Autodock vina which also apply the receptor_prepare.py to convert the receptor file to the PDBQT format. Moreover, the ligand files prepared for the docking using open Babel were used as input for the screening. We determined the XYZ coordinates of an inner box of 10 Å × 10 Å × 10 Å and an automatic outer box of 29 Å × 29 Å × 29 Å for the screening of the DCM database against the receptor active site residues. This tool supports numerous ligand molecular formats, multi-ligand file, and automatic box creation options (Koes et al., 2013, Masters et al., 2020). To determine the Lipinski, Ghose, Veber, Egan, Muegge, PAINS, and bioavailability scores of the top four hits SWISSADME was used while for the Hepatotoxicity, carcinogenicity, immunogenicity, mutagenicity, and cytotoxicity ProTox-II (http://tox.charite.de/protox_II) webserver was used (Daina et al., 2017, Banerjee et al., 2018).
2.2. Molecular docking simulation
Re-docking of top hits was performed for avoiding false-positive prediction using AutoDockFR(Ravindranath et al., 2015). For the validation of the docking protocol, we considered re-docking of GRL0617 and compared it with the crystallographic coordinates. GRL0617 ligand was used for active site identification and residual cross-checking was performed for accuracy. Receptor flexibility is important feature in determining accurate ligand binding. ADFR improves the search space with increased false-positive results. It is based on AutoDock4 scoring with better accuracy and speed than the other tools. Asp164, Tyr264, Tyr268, and Gln269 were used to generate the active site pocket (grid). The location of these residues determined by XYZ coordinates of an inner box of 10 Å × 10 Å × 10 Å and an automatic outer box of 29 Å × 29 Å × 29 Å were built.
2.3. Molecular dynamics simulation of protein–ligand complexes
A molecular simulation was performed to assess the inhibition potential and validation of the dynamic behavior of top lead molecules. The stability, flexibility, and packing of complexes (compounds and PLpro) were checked on the AMBER21 package (Pearlman et al., 1995, Case et al., 2005, Salomon-Ferrer et al., 2013). For the receptor (protein) FF19SB force field was utilized while for the ligand GAFF (general amber force field) was used. The solvation, coordinate, and topology of the drug were estimated by the optimal point charge (OPC) water model and antechamber algorithm respectively. Neutralization was accomplished by the addition of Na + counter ions. The minimization of complexes was performed for cumulative 20,000 steps for optimized and improved structure. Heating and equilibration of the improved complex were performed for 50 ns. Both the NPT and NVT ensembles were considered to understand the interacting behaviour of each system with the environment and itself. Long-range contacts were obtained from Particle Mesh Ewald (PME) algorithm. Van der Waals (vdW) interactions of 1.4 nm and short-range Columbic were considered. A total run of 1000 ns and a complex production run of 200 ns was performed at a time step of 2 fs. Trajectories of simulations were obtained by CPPTRAJ and PTRAJ (Roe and Cheatham III 2013). The GRL0617-PLpro complex was also evaluated as a control to compare the results of the novel hits. Structural stability was checked by Root mean square deviation (RMSD) analysis by using the following equation.
| (1) |
The position difference between atoms is denoted as d, and the indices i are used to represent the original and superimposed structure. The calculation of the root mean square fluctuation (RMSF) of the B-factor is crucial in determining the flexibility of protein residues. The RMSF values, which can be obtained using the following equation, serve as a fundamental constraint for this analysis.
| (2) |
Moreover, hydrogen bonds over the simulation time and half-life of each hydrogen bond established by each ligand with the active site residues were estimated to reveal the real-time potency of the binding.
2.4. The binding free energy calculations
The binding free energy of complexes was calculated with the MMPBSA.PY script by calculating all energy terms which contribute to total binding energy(Sun et al., 2014). The following equation was used for the free energy estimation of complexes.
| (3) |
Free binding energy was represented by ΔGbind in the equation. The specific energy terms which contribute to total free energy were obtained by equation.
| (4) |
3. Results
3.1. Structural retrieval and analysis
The growing concerns and cases of COVID-19 with the appearance of novel variants i.e., BA.2.75. BA.5 and XBB have prompted demand for more effective treatment options that could overcome the risk of immune evasion. For this purpose, discovering novel small molecules that could inhibit druggable proteins that are essential for viral pathogenesis, replication, survival, and spread is the best choice. Hence, PLpro is among the essential protein that leads to reduced viral burden upon inhibition. Due to its capacity to cleave ubiquitin and ubiquitin-like interferon-stimulated gene (ISG) 15 posttranslational modifications, PLpro functions as an effective regulator of host cell signaling cascades (Calleja et al., 2022). PLpro is one of the two necessary cysteine proteases that controls host immunological sensing, breaks down viral polyproteins, and targets host interferon and NF-β pathways. It also produces a viable replicase complex to perform its crucial function in viral replication and immune evasion. Consequently, it is the best choice to design novel potent inhibitors and act as potential viable drug targets for the effective treatment of COVID-19. The SARS-CoV-2 PLpro comprises of palm, fingers, thumb, and Ubl of which the interface of palm and thumb makes the active pocket including Asp164, Tyr264, Tyr268, and Gln269 residues. PLpro corresponds to region 2764–3263 in a long polyprotein of 7096 amino acids that perform several essential functions after being disintegrated into different functional proteins. The structure of PLpro in complex with GRL0617 inhibitor is shown in Fig. 1a while the three domains are mentioned in different colours. The residues that make up the catalytic triad including Cys111, His272, and Asp268 are also shown in Fig. 1b. For instance, the re-docking of GRL0617 was performed to validate the docking protocol and also see the binding pattern. As given in Fig. 1c, our protocol demonstrated an RMSD difference of 0.029 Å only between the crystallographic and docking conformation of GRL0617. The docking score for GRL0617 was reported to be −6.28 kcal/mol. This shows the accuracy of our method in predicting novel potential inhibitors for PLpro. The binding mode of GRL0617 reported hydrogen bonding interactions involving Asp164, Tyr264, Tyr268, and Gln269. The interaction pattern of GRL0617 is shown in Fig. 1d.
Fig. 1.

Structural organization of PLpro with different domains and catalytic triad. (a) Shows the domain organization of PLpro while (b) shows the catalytic triad comprised of Cys111, His272, and Asp286. (c) Show the docking validation result of the experimental and docking conformation of GRL0617, while (d) demonstrate the interaction pattern of GRL0617 with the active site residues of PLpro.
3.2. Molecular screening of the DCM database
Early-phase drug development relies heavily on high-throughput screening (HTS), which is the main source of novel drug products and the basis for medicinal chemistry. Virtual drug screening of large databases is the prime aim of pharmaceutical companies to design potential drug candidates against a wide range of targets. Dark chemical matter (DCM), or substances that are consistently active in various screening tests, has recently drawn more attention. One of the reasons is that a lot of DCM compounds might not be completely pharmacologically inert, but they offer intriguing possibilities for finding drugs that are extremely selective or active against uncommon targets. Therefore, in the current study, we screened the DCM library which contains 18,500 compounds from various sources against the active site of PLpro. Pre-filtering of the DCM database for R5 or Lipinski rule of five revealed that 1321 compounds did not obey the R5 rules and cannot be screened. The remaining 17,179 compounds demonstrated docking scores between −6.52 kcal/mol to −3.87 kcal/mol. We only selected those compounds that have a docking score > -6.00 kcal/mol. This criterion yielded only 166 compounds while the rest 17,013 compounds were discarded. The selected 166 compounds were subjected to induced-fit docking (IFD) for the second round of screening to avoid false-positive predictions. Our results revealed the 10 best compounds with docking scores between −7.99 to −7.03 kcal/mol respectively. Based on the interactions with the key residues and docking scores only four compounds were selected for molecular dynamics simulation-based validation. In contrast to the control drug GRL0617 the docking score for the top hits re-docking using the IFD protocol the potency of these molecules that can inhibit the function of PLpro. The selected top four compounds are given in Table 1 with their docking scores, 3D structures, database IDs, and names.
Table 1.
Top four hits from the DCM database with their docking scores, 3D structures, database IDs, and names.
| S. No | 2D structures | Names | Database IDs | Docking scores |
|---|---|---|---|---|
| 1. | 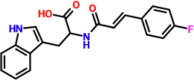 |
(2S)-2-[3-(4-fluorophenyl)prop-2-enoylamino]-3-(1H-indol-3-yl)propanoic acid | DC5977-0726 | −7.37 |
| 2. |  |
Cbz-Gly-Pro-D-Ser-OMe | DC6623-2024 | −7.99 |
| 3. | 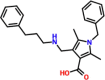 |
1-benzyl-2,5-dimethyl-4-{[(3-phenylpropyl)amino]methyl}sh-1H-pyrrole-3-carboxylic acid | DCC879-0379 | −7.07 |
| 4. | 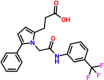 |
3-[1-(2-oxo-2-{[3-(trifluoromethyl)phenyl]amino}ethyl)-5-phenyl-1H-pyrrol-2-yl]propanoic acid | DCD135-0154 | −7.68 |
3.3. Binding mode of DC 5977–0726
DC 5977–0726 is a (2S)-2-[3-(4-fluorophenyl)prop-2-enoylamino]-3-(1H-indol-3-yl)propanoic acid which reported a docking score of −7.37 kcal/mol. This compound established four hydrogen bonds involving Asp164, Arg166, Glu167, and Tyr268. The oxygen atom acting as an acceptor interact with Tyr268 through a hydrogen bond, while NH connects with Asp164 while a single OH group establishes two hydrogen bonds with Asp164 and Arg166 respectively. For instance, these residues are essential in terms of catalytic function as they make up the catalytic triad and also the active site. Since this compound also blocks these essential residues, this shows the potential of DC 5977–0726 as an excellent inhibitor of PLpro from SARS-CoV-2. The binding mode of DC 5977–0726 is shown in Fig. 2. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
Fig. 2.

Binding mode of DC 5977–0726 in complex with PLpro. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
3.4. Binding mode of DC 6623–2024
The scientific name of DC 6623–2024 is Cbz-Gly-Pro-D-Ser-OMe which has been predicted previously as a cysteine protease inhibitor. This compound reported a docking score of −7.99 kcal/mol while establishing eight essential hydrogen bonding contacts. Among the key essential interactions, Gly163 established a single hydrogen bond, Asp164 established a double hydrogen bond, Tyr 264 also reported a single hydrogen bond, Gln269 established double hydrogen bonds while Thr301 was also involved in a single hydrogen bond. The oxygen atom acting as an acceptor connects with Gly163 and Gln269 while the OH group acting as a donor interacts with Thr301 and in the same manner the NH group being a donor connect with Asp164 from the catalytic triad. From the interactions, it can be seen that this compound also blocks the same essential residues previously reported to be required for ligand recognition and other functionality of the PLpro enzyme (Fu et al., 2021). The extra contacts and more hydrogen bonds make this hit the best choice for further investigation. The binding mode of DC 6623–2024 is shown in Fig. 3. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
Fig. 3.

Binding mode of DC 5977–0726 in complex with PLpro. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
3.5. Binding mode of DC C879-0379
The scientific name of DC C879-0379 is 1-benzyl-2,5-dimethyl-4-{[(3-phenylpropyl)amino]methyl}sh-1H-pyrrole-3-carboxylic acid which has been shown to exhibit activities against Leukemia, Marburg virus, steroid receptors, anti-cancer and Small Molecule Inhibitors of Eukaryotic Translation Initiation. The activity of this compound against the Marburg virus makes it viable for usage against SARS-CoV-2. This compound reported a docking score of −7.07 kcal/mol with five hydrogen bonds. Among the key essential interactions Asp164, Arg166, Glu167, and Tyr268 are involved in single hydrogen bonds. Unlike the first two compounds, here the centroid of the benzene ring at the tail connects with Tyr268 and NH group of the backbone of the ligand molecule. Similarly, the interaction paradigm for the residues Asp164 and Arg168 connects with the ligand. The binding mode of DC C879-0379 is shown in Fig. 4. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
Fig. 4.

Binding mode of DC C879-0379 in complex with PLpro. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
3.6. Binding mode of DC D135-0154
The scientific name of DC D135-0154 is 3-[1-(2-oxo-2-{[3-(trifluoromethyl)phenyl]amino}ethyl)-5-phenyl-1H-pyrrol-2-yl]propanoic acid has been previously reported to act as Lactamase, Influenza NS1 Protein, anti-malarial and anti-cancerous agent. The activity of this compound against the influenza virus makes it viable for use against SARS-CoV-2. This compound reported a docking score of −7.68 kcal/mol with seven hydrogen bonds. Arg166 established double hydrogen bonds while Asp164, Glu167, Tyr264, and Gln269 established single hydrogen bonds each. Interestingly, the NH and OH groups act on the catalytic triad residues i.e. Asp164 and Arg168 while the centroid and oxygen atoms interact with Tyr268 and Gln269. These interactions with the essential residues determine the pharmacological potential of these compounds against the PLpro of SARS-CoV-2 which may block these residues from functionality and rescue the host immune system. The binding mode of DC D135-0154 is shown in Fig. 5. The left panel shows the 3D interactions while the right panel shows interactions in 2D. The ADMET and Toxicity results for the top four hits are given in Table 2.
Fig. 5.

Binding mode of DC D135-0154 in complex with PLpro. The left panel shows the 3D interactions while the right panel shows interactions in 2D.
Table 2.
ADMET and Toxicity results for the top four hits predicted by SWISSADME and ProTox-II.
| Compounds ID /Parameters | DC5977-0726 | DC6623-2024 | DCC879-0379 | DCD135-0154 |
|---|---|---|---|---|
| Lipinski | 0 violations | 0 violations | 0 violations | 0 violations |
| Ghose | 0 violations | 1 violation | 1 violation | 0 violations |
| Veber | 0 violations | 0 violations | 0 violations | 0 violations |
| Egan | 0 violations | 0 violations | 0 violations | 0 violations |
| Muegge | 0 violations | 1 violations | 0 violations | 1 violation |
| PAINS | 0 alerts | 0 alerts | 0 alerts | 0 alerts |
| Hepatotoxicity | Inactive | Inactive | Inactive | Inactive |
| Carcinogenicity | Inactive | Inactive | Inactive | Inactive |
| Immunogenicity | Inactive | Inactive | Inactive | Inactive |
| Mutagenicity | Inactive | Inactive | Inactive | Inactive |
| Cytotoxicity | Inactive | Inactive | Inactive | Inactive |
| Bioavailability Score | 0.56 | 0.55 | 0.85 | 0.55 |
3.7. Structural stability assessment of the top hits
The determination of the binding stability of the protein–ligand complexes elucidate the inhibition mechanism of a pharmacologically active compound. This approach has been widely applied to determine the structural stability of a ligand-bound complex in a dynamic environment. It also validates the pharmacological effects of novel hits identified through molecular search. In the current study, we also used the root mean square deviation (RMSD) as validating approach to determine the binding stability of our top hits. We calculated the RMSD for GRL0617 in complex with PLpro to further validate and confirm our results. The binding stability determines the pharmacological potential of any small molecule bound to a receptor. Hence, to see the binding stability RMSD over the simulation time was computed for the GRL0617-PLpro complex. The GRL0617-PLpro complex reported minor structural perturbation at different time intervals. The average RMSD for GRL0617-PLpro complex remained lower than 2.0 Å. However, it converges with the other complexes thus showing that all the complexes have attained similar atomic configurations and thus validates the accuracy of the method. The RMSD for GRL0617-PLpro complex is shown in Fig. 6a. Furthermore, as given in Fig. 6a, the DC 5977–0726-PLpro complex reported no significant dynamic structural perturbation except a minor increment in the RMSD between 83 and 95 ns. From the very beginning, the RMSD was below 2.0 Å which gradually increased with time but remained stable. The maximum RMSD was recorded between 83 and 95 ns after that the RMSD decreased back and followed a similar trend as the start of the simulation. An average RMSD was reported to be 1.98 Å. The current RMSD pattern shows a stable binding of DC 5977–0726 to PLpro which consequently induces pharmacological effects to inhibit PLpro. The DC 6623–2024 or Cbz-Gly-Pro-D-Ser-OMe in complex with PLpro demonstrated a continuous increase in the RMSD value at the beginning of the simulation. The RMSD gradually increased and reached up to 3.0 Å at 20 ns but then abruptly declined. Although the RMSD remained stable overall average RMSD remained higher than the previous complex. An average RMSD for this complex was calculated to be 2.50 Å. This implies that the stronger binding of this molecule causes the inhibition of PLpro. The RMSD pattern of DC 6623–2024 or Cbz-Gly-Pro-D-Ser-OMe in complex with PLpro is shown in Fig. 6b. Although the RMSD of DC 6623–2024 complex remained higher than the control but still it determined stable dynamics and therefore shows the potential of this molecule blocking the key active site residues. Moreover, the DC C879-0379 molecule in complex with PLpro demonstrated an alike pattern of RMSD as DC 5977–0726-PLpro complex. The complex reported a higher RMSD between 38 and 50 ns but then stabilized after 50 ns and continues to follow a stable uniform RMSD trend. An average RMSD for this complex was calculated to be 2.01 Å and presented in Fig. 6c. In contrast, the DC D135-0154 in complex with PLpro determined destabilizing effects with an average RMSD lower than 3.0 Å. The complex demonstrated structural perturbation in a dynamic environment until the end of the simulation. The RMSD pattern for DC D135-0154 in complex with PLpro is shown in Fig. 6d.
Fig. 6.

Dynamic stability assessment of the GRL0617 (control) and top hits in complex with PLpro. (a) RMSD of the GRL0617 (control) and DC 5977–0726 in complex with PLpro. (b) RMSD pattern of the GRL0617 (control) and DC 6623–2024 in complex with PLpro. (c) RMSD of the GRL0617 (control) and DC C879-0379 in complex with PLpro. (d) RMSD pattern of the GRL0617 (control) and DC D135-0154 in complex with PLpro.
3.8. Protein size estimation through structural packing
Estimation of protein size or structural packing is an important notion to understand the pharmacological features of the binding molecule. It shows the binding and unbinding events that took place during the simulation. Therefore, to see how the packing of the protein is altered during the simulation we calculated the radius of gyration (Rg) as a function of time. For the GRL0617 (control) and PLpro complex, the Rg was calculated and presented in Fig. 7a. It can be seen that the average Rg for the GRL0617 (control) and PLpro complex remained higher and demonstrated an increase and decrease pattern that potentially show the maximum number of unbinding events experienced by this ligand during the simulation. As given in Fig. 7a, the DC 5977–0726 in complex with PLpro demonstrated a wave-like pattern. The Rg of DC 5977–0726 in the complex with PLpro started from 19.3 and increased slowly with time. The highest Rg was observed at 20 ns. Afterward, the Rg continues to decrease and at 80 ns the lowest Rg was recorded. Then the Rg was observed to increase again from 81 ns to 160 ns. An average Rg was estimated to be 19.60 Å. The increase and decrease in the protein size/Rg value are associated with the movement of different domains particularly the Ubl which owns higher flexibility. On the other hand, the Rg of the DC 6623–2024 in complex with PLpro started from 19.60 Å and reached the maximum at 10 ns. Afterward, an abrupt decline in the Rg was seen and a gradual decrease until 80 ns. After 80 ns the Rg continues to increase again and at 120 ns the Rg stabilized at 19.30 Å and continues to follow the same pattern until the end of the simulation. Moreover, the DC C879-0379 in complex with PLpro demonstrated a similar pattern as RMSD. A peak at 40 ns and then a gradual decrease in the Rg graph till the end of the simulation. An average Rg for the DC C879-0379 in complex with PLpro was calculated to be 19.54 Å. The Rg graph for DC C879-0379 in complex with PLpro is given in Fig. 7c. Furthermore, the DC D135-0154 in complex with PLpro also reported a similar pattern as RMSD. With the highest Rg value among all complexes, this complex demonstrated continuous increase and decrease in the Rg pattern during the 200 ns. An average Rg for this complex was estimated to be 20.06 Å. The Rg graph for DC D135-0154 in complex with PLpro is given in Fig. 7d. In contrast to the GRL0617 (control) and PLpro complex, the Rg for these top four hits remained very lower with no significant perturbation that causes minimal unbinding events and shows the potency of these molecules. Overall, the current findings convey essential information regarding the binding and unbinding events that were observed during the simulation. Interestingly the current findings strongly align with the previous hits against PLpro thus further validating the pharmacological potential of these DCM molecules and stressing the clinical evaluation of these hits.
Fig. 7.

Structural packing assessment of the top hits in complex with PLpro. (a) Rg of the GRL0617 (control) and DC 5977–0726 in complex with PLpro. (b) Rg pattern of the GRL0617 (control) and DC 6623–2024 in complex with PLpro. (c) Rg of the GRL0617 (control) and DC C879-0379 in complex with PLpro. (d) The rg pattern of the GRL0617 (control) and DC D135-0154 in complex with PLpro.
3.9. Estimation of residues flexibility through RMSF
Residue’s flexibility is important to decipher the role of essential residues in molecular interaction, switching on/off, protein residues networking, and communication. It has been a widely applicable approach to see how flexibility indexing determines the functionality of the protein. Considering the importance of residues flexibility, we also calculated the residues flexibility of these complexes. From Fig. 8, it can be seen that all the complexes demonstrated a similar pattern of RMSF which remains minimal mostly however the region 120–180 (correspond to 180–240 originally) which is the fingers region reported higher fluctuation. This shows the opening and closing of the binding cavity covered by the fingers’ domain movement. The RMSF of the control drug GRL0617 (control) and PLpro complex reported alike RMSF patterns as the others. The current findings show that the binding of these top-hit molecules stabilizes the internal flexibility and thus causes stabilized binding.
Fig. 8.

Residue’s flexibility analysis (RMSF) of GRL0617 (control) and top hits in complex with PLpro. Each complex is represented with a different colour aligned with the RMSD and Rg.
3.10. Hydrogen bonding and fraction analysis
Hydrogen bonding estimation may accurately demonstrate the pharmacological potential of a lead small molecule. It is considered as a vital parameter to see how strongly the lead molecule binds to a target receptor. Therefore, in the current study, we also calculated hydrogen bonds over time using the simulation trajectories. As given in Fig. 9a-d, the average number of hydrogen bonds in each complex lies between 120 and 140. In the DC 5977-the 0726-PLpro complex average number of hydrogen bonds was 128, in the DC 6623–2024-PLpro complex average number of hydrogen bonds was 131, in the DC C879-0379-PLpro complex average number of hydrogen bonds was 134 while in the DC D135-0154-PLpro average number of hydrogen bonds were estimated to be 138 respectively. This shows that all the complexes have almost similar numbers of average hydrogen bonds which consequently causes the inhibition of PLpro. In contrast, the average number of hydrogen bonds in the GRL0617-PLpro complex remained 118 which is far less than the top-hit molecules bound in the cavity. Next, we evaluated the percentage consistency of each hydrogen bond in each complex using the simulation trajectories. The results are given in Table 3 which shows that the key residues such as Asp164, Arg166, Tyr264, and Tyr268 have major contributions to the hydrogen bonding during the simulation. For instance, these residues are previously reported to have an essential role in discovering anti-SARS-CoV-2 drugs targeting PLpro(Mitra et al., 2022). When compared with the GRL0617-PLpro complex the bonds such as Gly163, Gln269, and Thr301 were not observed. However, the other bonds such as Arg164, Arg166, Glu167, Tyr264, and Tyr268 determines a lower half-life than each of the top hit complex. This shows that our compounds occupy the binding cavity in larger volume by blocking more active residues than the control drug and the consistency is also much higher than the control. This further validates the pharmacological potential of our top hits and could be tested for clinical usage.
Fig. 9.

Hydrogen bonding half-life analysis of the GRL0617 (control) and top hits in complex with PLpro. (a) Hydrogen bonding half-life analysis of the GRL0617 (control) and DC 5977–0726 in complex with PLpro. (b) Hydrogen bonding half-life analysis of the GRL0617 (control) and DC 6623–2024 in complex with PLpro. (c) Hydrogen bonding half-life analysis of the GRL0617 (control) and DC C879-0379 in complex with PLpro. (d) Hydrogen bonding half-life analysis of the GRL0617 (control) and DC D135-0154 in complex with PLpro.
Table 3.
Hydrogen bonding residues half-life analysis during the simulation for each complex.
| Residues | GRL0617 (control) | DC5977-0726 | DC6623-2024 | DCC879-0379 | DCD135-0154 |
|---|---|---|---|---|---|
| Gly163 | – | 32% | 4% | 8% | 3% |
| Asp164 | 53% | 67% | 74% | 76% | 59% |
| Arg166 | 14% | 44% | 62% | 47% | 31% |
| Glu167 | 04% | 22% | 17% | 14% | 13% |
| Tyr264 | 11% | 72% | 68% | 71% | 76% |
| Tyr268 | 70% | 67% | 74% | 79% | 76% |
| Gln269 | – | 12% | 24% | 14% | 13% |
| Thr301 | – | 2% | 20% | 5% | 6% |
3.11. Binding free energy calculation
The Gibbs binding free energy approach may be used to re-evaluate the correct binding conformation and energy. This method is the most commonly used and accurate for re-ranking the most efficacious inhibitor against the corresponding receptor. It has the benefit over other rationale approaches in terms of computing cost. As a result, we approximated the binding free energy using the MM/GBSA technique, taking accuracy and adaptability into account. The vdW energy for the control (GRL0617)-PLpro complex was calculated to be −34.41 kcal/mol, for DC5977-0726-PLpro the vdW was calculated to be −39.36 kcal/mol, −45.13 kcal/mol for DC6623-2024-Plpro, for DCC879-0379-Plpro complex the vdW was −44.98 kcal/mol while the DCD135-0154-PLpro complex demonstrated a vdW of −39.34 kcal/mol kcal/mol respectively. Finally, the total binding free energy was reported to be −34.41 kcal/mol for GRL0617 (control), −41.03 kcal/mol for the DC5977-0726-PLpro, for the DC6623-2024-Plpro complex the TBE was −48.87 kcal/mol, for the for DCC879-0379-Plpro complex the TBE was −45.66 kcal/mol while for the DCD135-0154-PLpro complex, the TBE was calculated to be −40.09 kcal/mol respectively. It can be seen that the TBE of all the complexes is higher and could inhibit the PLpro more strongly in in vivo and in vitro conditions than the GRL0617 (control) and demands further experimental tests for the possible usage as a potential drug against SARS-CoV-2. The binding free energy results are shown in Table 4.
Table 4.
The binding free energy calculation results from molecular simulation using MM/GBSA analysis. All the results are calculated in kcal/mol.
| Parameters | GRL0617 (control) | DC5977-0726 | DC6623-2024 | DCC879-0379 | DCD135-0154 |
|---|---|---|---|---|---|
| vdW | −34.41 | −39.36 | −45.13 | −44.98 | −39.34 |
| EEL | −4.22 | −3.13 | −4.01 | −6.38 | −6.32 |
| EGB | 6.47 | 8.24 | 7.36 | 12.18 | 8.45 |
| ESURF | −4.25 | −6.78 | −7.09 | −6.48 | −5.88 |
| Delta Total | −36.41 | −41.03 | −48.87 | −45.66 | −43.09 |
4. Discussion
The growing concerns and cases of COVID-19 with the appearance of novel variants i.e. BA.2.75. BA.5 and XBB have prompted demand for more effective treatment options that could overcome the risk of immune evasion. For this purpose, discovering novel small molecules that could inhibit druggable proteins that are essential for viral pathogenesis, replication, survival, and spread is the best choice. Hence, PLpro is among the essential protein that leads to reduced viral burden upon inhibition. Due to its capacity to cleave ubiquitin and ubiquitin-like interferon-stimulated gene (ISG) 15 posttranslational modifications, PLpro functions as an effective regulator of host cell signaling cascades (Calleja et al., 2022). PLpro, or papain-like protease, is an important drug target in the context of viral infections, particularly coronaviruses. This protease is responsible for cleaving viral polyproteins into functional units, enabling the virus to replicate and spread. As such, inhibiting PLpro can prevent viral replication and halt the spread of infection. Virtual screening is an essential tool in the search for PLpro inhibitors. With this method, researchers use computer models to screen large databases of compounds for potential inhibitors of PLpro. This allows them to rapidly identify compounds that have the potential to bind to the target and block its activity, without having to synthesize and test each compound individually. One promising approach to virtual screening is the use of Dark Chemical Matter (DCM) compounds. DCM compounds are a class of molecules that are underrepresented in traditional drug discovery databases but are believed to have great potential as drug candidates. By using virtual screening to identify DCM compounds that are predicted to bind to PLpro, researchers can quickly identify new drug leads that might otherwise have gone undiscovered. Once potential PLpro inhibitors have been identified through virtual screening, they must be tested experimentally to confirm their activity and selectivity. This is typically done using assays that measure the ability of a compound to inhibit the enzymatic activity of PLpro. Promising compounds can then be further optimized through medicinal chemistry to improve their potency, pharmacokinetic properties, and safety.
Our results revealed that the top hits target the key essential residues and these residues have been previously blocked by the experimental ligand to dysfunction the role of PLpro in viral replication and immune evasion (Fu et al., 2021). We reported that among the DCM database, four hits exhibit significant binding and block the key active site and catalytic triad residues. For instance, using structure-based methods, a study reported the impact of blocking the key residues i.e. Asp164, Gln269, and Tyr273, and its association with the loss of the function of PLpro (Pang et al., 2021). Interestingly our shortlisted compounds also blocked these residues and thus inform the pharmacological potential of these hits from the DCM database. Nonetheless, our compounds also block the interactions with the other key residues along with the catalytic triad. From the simulation results, it was observed that identified shortlisted molecules potentially bound stably in the active pocket of PLpro and therefore need further validations for clinical approval. Our results are more significant than the other studies as they performed only 20 ns simulation while we validated our results by doing 200 ns simulation each but also demonstrated stable dynamics with no significant strutcural perturbations. Overall the current findings strongly align with the previous studies and also the stable binding of these molecules shows potential pharmacological inhibition of PLpro thus making it the first choice for further validation and clinical usage (Mitra et al., 2022). For instance, similar findings of stable RMSD for novel hits that potentially inhibit PLpro has been previously reported by various studies (Rao et al., 2021, Mitra et al., 2022). Not only the RMSD but the hydrogen bonding results strongly align with the previously reported results. Interestingly, Zhang et., al, reported potential inhibitors i.e. A3175, A3659, and A3777 which also exhibit similar scaffolds as our reported compounds, and the hydrogen paradigm was also reported to be similar to our results. Moreover, the findings also corroborate with their top hits, and mostly the benzene ring, the NH group, OH group, and Oxygen atoms are mostly involved in blocking the catalytic triad (Zhang et al., 2022). The results given in Table 2 show that the key residues such as Asp164, Arg166, Tyr264, and Tyr268 have a major contribution to the hydrogen bonding during the simulation. For instance, these residues are previously reported to have an essential role in discovering anti-SARS-CoV-2 drugs targeting PLpro (Mitra et al., 2022). Finally, the re-evaluation of the docking predictions was confirmed through the binding free energy calculation method. The Gibbs binding free energy approach may be used to re-evaluate the correct binding conformation and energy. These results further confirmed the validity of our results and the shortlisted potential hits as promising inhibitors for the inhibition of SARS-CoV-2 targeting the PLpro.
5. Conclusions
In conclusion, the emergence of novel variants of COVID-19, including BA.2.75, BA.5, and XBB, has heightened the need for effective treatments capable of overcoming immune evasion. In this study, the focus was on discovering small molecules that can inhibit druggable proteins, particularly PLpro, which plays a crucial role in viral pathogenesis, replication, survival, and spread. The utilization of Dark Chemical Matter (DCM) in virtual molecular screening and simulation approaches provided promising results in identifying potential hits against both current and emerging variants of SARS-CoV-2. Our results revealed DC 5977–0726, DC 6623–2024, DC C879-0379 and DC D135-0154 as the best hits. Structural-dynamics properties and the hydrogen bonding half-life revealed that Asp164, Arg166, Tyr264, and Tyr268 have major contributions to the hydrogen bonding during the simulation. Finally, the total binding free energy revealed the stronger potency of these compounds against PLpro. Overall, the discovery of novel small molecules with selective activity against crucial viral proteins like PLpro presents an encouraging avenue for the development of effective treatments against COVID-19, particularly against emerging variants. The compounds' potency, selectivity, and pharmacological characteristics can be improved further using medicinal chemistry approaches. Given that SARS-CoV-2 is still evolving, the study may be expanded to see whether the compounds work against other SARS-CoV-2 strains or variants. This would provide insights into the broad-spectrum activity and potential for long-term effectiveness of the identified compounds. Further in in vivo and in vitro tests are required for the possible usage as a potential drug against SARS-CoV-2.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding
This work was supported by the National Key R&D Program of China (Grant No. 2021YFA0911500) and Shanghai Pujiang Program (Grant No. 21PJ1405100). Dong-Qing Wei was supported by the National Science Foundation of China (Grant No. 32070662, 61832019, 32030063), the Science and Technology Commission of Shanghai Municipality (No. 19430750600), as well as SJTU JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2021ZD02).
Acknowledgment
The authors are thankful to the researchers supporting project number [RSP2023R335], King Saud University, Riyadh, Saudi Arabia.
Footnotes
Peer review under responsibility of King Saud University.
Contributor Information
Abbas Khan, Email: abbaskhan@sjtu.edu.cn.
Liaqat Ali, Email: Liaqat.ali@numspak.edu.pk.
Abdulrahman Alshammari, Email: Abdalshammari@ksu.edu.sa.
Abdullah F. Alasmari, Email: afalasmari@ksu.edu.sa.
Anwar Mohammad, Email: Anwar.mohammad@dasmaninstitute.org.
Dong-Qing Wei, Email: dqwei@sjtu.edu.cn.
References
- Baildya N., Khan A.A., Ghosh N.N., et al. Screening of potential drug from Azadirachta Indica (Neem) extracts for SARS-CoV-2: an insight from molecular docking and MD-simulation studies. J. Mol. Struct. 2021;1227 doi: 10.1016/j.molstruc.2020.129390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P., Eckert A.O., Schrey A.K., et al. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46:W257–W263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J.H., Tomashek K.M., Dodd L.E., et al. Remdesivir for the Treatment of Covid-19 - Final Report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja D.J., Lessene G., Komander D. Inhibitors of SARS-CoV-2 PLpro. Front. Chem. 2022;10 doi: 10.3389/fchem.2022.876212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D.A., Cheatham T.E., III, Darden T., et al. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhote A.M., Patil V.R., Lokwani D.K., et al. Strategic analyses to identify key structural features of antiviral/antimalarial compounds for their binding interactions with 3CLpro, PLpro and RdRp of SARS-CoV-2: in silico molecular docking and dynamic simulation studies. J. Biomol. Struct. Dyn. 2022;40:11914–11931. doi: 10.1080/07391102.2021.1965912. [DOI] [PubMed] [Google Scholar]
- Fontanet A., Autran B., Lina B., et al. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet. 2021;397:952–954. doi: 10.1016/S0140-6736(21)00370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Huang B., Tang J., et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grein J., Ohmagari N., Shin D., et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020;382:2327–2336. doi: 10.1056/NEJMoa2007016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh T., Cornell W., Luan B. In silico exploration of inhibitors for SARS-CoV-2's papain-like protease. Front. Chem. 2021;8 doi: 10.3389/fchem.2020.624163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Ali S.S., Khan M.T., et al. Combined drug repurposing and virtual screening strategies with molecular dynamics simulation identified potent inhibitors for SARS-CoV-2 main protease (3CLpro) J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1779128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Khan M., Saleem S., et al. Phylogenetic analysis and structural perspectives of RNA-dependent RNA-polymerase inhibition from SARs-CoV-2 with natural products. Interdisc. Sci., Comput. Life Sci. 2020;12:335–348. doi: 10.1007/s12539-020-00381-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Khan T., Ali S., et al. SARS-CoV-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021;112176 doi: 10.1016/j.biopha.2021.112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Heng W., Wang Y., et al. In silico and in vitro evaluation of kaempferol as a potential inhibitor of the SARS-CoV-2 main protease (3CLpro). Phytother. PTR Res. 2021 doi: 10.1002/ptr.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Randhawa A.W., Balouch A.R., et al. Blocking key mutated hotspot residues in the RBD of the omicron variant (B. 1.1. 529) with medicinal compounds to disrupt the RBD-hACE2 complex using molecular screening and simulation approaches. RSC Adv. 2022;12:7318–7327. doi: 10.1039/d2ra00277a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D.R., Baumgartner M.P., Camacho C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters L., Eagon S., Heying M. Evaluation of consensus scoring methods for AutoDock Vina, smina and idock. J. Mol. Graph. Model. 2020;96 doi: 10.1016/j.jmgm.2020.107532. [DOI] [PubMed] [Google Scholar]
- Mitra D., Verma D., Mahakur B., et al. Molecular docking and simulation studies of natural compounds of Vitex negundo L. against papain-like protease (PLpro) of SARS CoV-2 (coronavirus) to conquer the pandemic situation in the world. J. Biomol. Struct. Dyn. 2022;40:5665–5686. doi: 10.1080/07391102.2021.1873185. [DOI] [PubMed] [Google Scholar]
- O'Boyle N.M., Banck M., James C.A., et al. Open Babel: An open chemical toolbox. J. Cheminf. 2011;3:1–14. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J., Gao S., Sun Z., et al. Discovery of small molecule PLpro inhibitor against COVID-19 using structure-based virtual screening, molecular dynamics simulation, and molecular mechanics/Generalized Born surface area (MM/GBSA) calculation. Struct. Chem. 2021;32:879–886. doi: 10.1007/s11224-020-01665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R., Prajapati J., Rao P., et al. Repurposing the antibacterial drugs for inhibition of SARS-CoV2-PLpro using molecular docking, MD simulation and binding energy calculation. Mol. Divers. 2022;26:2189–2209. doi: 10.1007/s11030-021-10325-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlman D.A., Case D.A., Caldwell J.W., et al. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995;91:1–41. [Google Scholar]
- Rao P., Patel R., Shukla A., et al. Identifying structural–functional analogue of GRL0617, the only well-established inhibitor for papain-like protease (PLpro) of SARS-CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol. Divers. 2021:1–21. doi: 10.1007/s11030-021-10220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindranath P.A., Forli S., Goodsell D.S., et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput. Biol. 2015;11:e1004586. doi: 10.1371/journal.pcbi.1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Rose P.W., Prlić A., Altunkaya A., et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2016;gkw1000 doi: 10.1093/nar/gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotondo J.C., Martini F., Maritati M., et al. SARS-CoV-2 infection: New molecular, phylogenetic, and pathogenetic insights. efficacy of current vaccines and the potential risk of variants. Viruses. 2021;13:1687. doi: 10.3390/v13091687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon-Ferrer R., Case D.A., Walker R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2013;3:198–210. [Google Scholar]
- Sayaf A.M., Ahmad H., Aslam M.A., et al. Pharmacotherapeutic Potential of Natural Products to Target the SARS-CoV-2 PLpro Using Molecular Screening and Simulation Approaches. Appl. Biochem. Biotechnol. 2023 doi: 10.1007/s12010-023-04466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sencanski, M., V. Perovic, J. Milicevic, et al., 2022. Identification of SARS-CoV-2 Papain-like Protease (PLpro) Inhibitors Using Combined Computational Approach**. ChemistryOpen. 11, e202100248. 10.1002/open.202100248. [DOI] [PMC free article] [PubMed]
- Seneviratne S.L., Abeysuriya V., De Mel S., et al. Favipiravir in COVID-19. Int. J. Progressive Sci. Technol. 2020;19:143–145. [Google Scholar]
- Sun H., Li Y., Tian S., et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014;16:16719–16729. doi: 10.1039/c4cp01388c. [DOI] [PubMed] [Google Scholar]
- Tian X., Zhao Q., Chen X., et al. Discovery of Novel and Highly Potent Inhibitors of SARS CoV-2 Papain-Like Protease Through Structure-Based Pharmacophore Modeling, Virtual Screening, Molecular Docking, Molecular Dynamics Simulations, and Biological Evaluation. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.817715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma D., Mitra D., Paul M., et al. Potential inhibitors of SARS-CoV-2 (COVID 19) proteases PL(pro) and M(pro)/ 3CL(pro): molecular docking and simulation studies of three pertinent medicinal plant natural components. Curr. Res. Pharmacol. Drug Discov. 2021;2 doi: 10.1016/j.crphar.2021.100038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vkovski P., Kratzel A., Steiner S., et al. Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021;19:155–170. doi: 10.1038/s41579-020-00468-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Horby P.W., Hayden F.G., et al. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassermann A.M., Lounkine E., Hoepfner D., et al. Dark chemical matter as a promising starting point for drug lead discovery. Nat. Chem. Biol. 2015;11:958–966. doi: 10.1038/nchembio.1936. [DOI] [PubMed] [Google Scholar]
- Wu X., Yu K., Wang Y., et al. Efficacy and Safety of Triazavirin Therapy for Coronavirus Disease 2019: A Pilot Randomized Controlled Trial. Engineering (Beijing). 2020;6:1185–1191. doi: 10.1016/j.eng.2020.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.-C., Zhao H.-L., Liu J., et al. Design of SARS-CoV-2 Mpro, PLpro dual-target inhibitors based on deep reinforcement learning and virtual screening. Future Med. Chem. 2022;14:393–405. doi: 10.4155/fmc-2021-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]