
Figure 2.
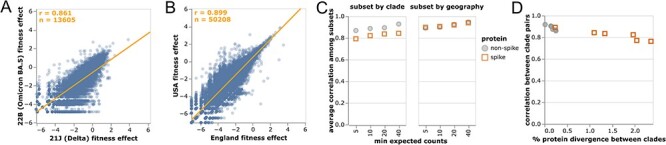
Correlations of mutation fitness effect estimates made using subsets of natural sequences. Correlations between estimates made (A) just using sequences from the Delta or Omicron BA.5 clades or (B) just from the USA or England. Each point is an amino acid mutation, the orange line is a least-squares regression, and orange text at upper left shows the number of mutations and Pearson’s correlation coefficient. Only mutations with at least 10 expected counts are shown, which is why panels have different numbers of mutations shown (sequence subsets vary in size). Different subset size are also the reason why the regression line in (A) deviates from the identity x = y. (C) Correlations between clade or geography subsets become higher with an increasingly large threshold for minimum expected counts. Spike mutations have a worse correlation when subsetting by viral clade (plot shows average correlation over all pairwise combinations of Delta, BA.1, BA.2, and BA.5), but not when subsetting by geography (USA or England). (D) Correlations in estimated mutation-effects decline for clades with higher protein divergence, with the effect most noticeable for spike since spike is more diverged among SARS-CoV-2 clades than other viral proteins. See https://jbloomlab.github.io/SARS2-mut-fitness/clade_corr_chart.html and https://jbloomlab.github.io/SARS2-mut-fitness/subset_corr_chart.html for versions of A and B that include all viral clades with at least 500,000 total expected counts (summed across all mutations) and have other interactive options.