

Abstract
Alcohol-associated liver disease due to harmful alcohol use and NAFLD associated with metabolic syndrome are the 2 most common liver diseases worldwide. Control of respective risk factors is the cornerstone in the long-term management of these diseases. Furthermore, there are no effective therapies. Both diseases are characterized by metabolic derangements; thus, the focus of this review was to broaden our understanding of metabolic targets investigated in NAFLD, and how these can be applied to alcohol-associated liver disease. Conserved pathogenic pathways such as dysregulated lipid metabolism, cell death pathways including apoptosis and activation of innate immune cells, and stellate cells mediate both alcohol and NAFLDs, resulting in histological abnormalities of steatosis, inflammation, fibrosis, and cirrhosis. However, pathways such as gut microbiome changes, glucose metabolism and insulin resistance, inflammatory signaling, and microRNA abnormalities are distinct in these 2 diseases. In this review article, we describe conserved and distinct pathogenic pathways highlighting therapeutic targets that may be of potential in both diseases and those that are unique to each disease.
INTRODUCTION
Hepatic steatosis, or fatty liver, is the most common liver disease worldwide. The 2 most common etiologies of fatty liver are alcohol-associated liver disease (ALD) and NAFLD.[1,2] In the general US population, ~7%–10% engage in harmful alcohol use and 30%–40% are overweight or obese, which is the most common component of metabolic syndrome and the predominant risk factor for NAFLD.[3,4] With the current US population of about 330 million, it is estimated that there are about 10 million overweight or obese individuals with harmful alcohol use.
ALD and NAFLD are heterogeneous diseases, with the disease spectrum ranging from steatosis to progressive steatohepatitis, with or without fibrosis, to end-stage liver disease of cirrhosis and its complications. Alcoholic hepatitis (AH) is a unique clinical syndrome in patients with ALD and is characterized by acute onset of severe liver inflammation, potential for high short-term mortality of up to 80% in most severe forms, and acute-on-chronic liver failure with antecedent failure of multiple organs.
The earliest histological and imaging abnormality in either ALD or NAFLD is steatosis. Due to the conserved nature of lipid metabolic pathways, it is not surprising that the pathways that lead to steatosis demonstrate the most overlap between ALD and NAFLD. Nutrient overload mediates metabolic abnormalities and steatosis in NAFLD, whereas in ALD, these abnormalities are mediated by the direct effect of alcohol on the liver tissue or through real or functional deficiencies. In addition, harmful alcohol use mediates direct toxicity to the liver through its metabolism in hepatocytes releasing reactive oxygen species.[5] In NAFLD, direct lipotoxicity of certain lipid species plays a greater role.[6] Liver injury and inflammation are furthered by indirect effects of the gut-liver axis, gut microbiome, intestinal permeability, and innate immune cells.[7] Furthermore, AH is a unique phenotype in ALD and is not seen in patients with NAFLD, where the same pathways mediate the disease pathogenesis, and neutrophils are the major cell type mediating hepatic and systemic inflammation (Figure 1). In contrast, there are distinct changes in the gut microbiome, gut-liver axis, and macrophage-mediated inflammation, which characterize progressive NAFLD (Figure 2).[6,8,9] NAFLD is not a severe illness and is pathophysiologically distinct from AH; therefore, throughout this review, as we extrapolate NAFLD therapies to ALD, the term ALD will be used to refer to the less severe form of alcohol-induced liver disease and exclude AH.
FIGURE 1.
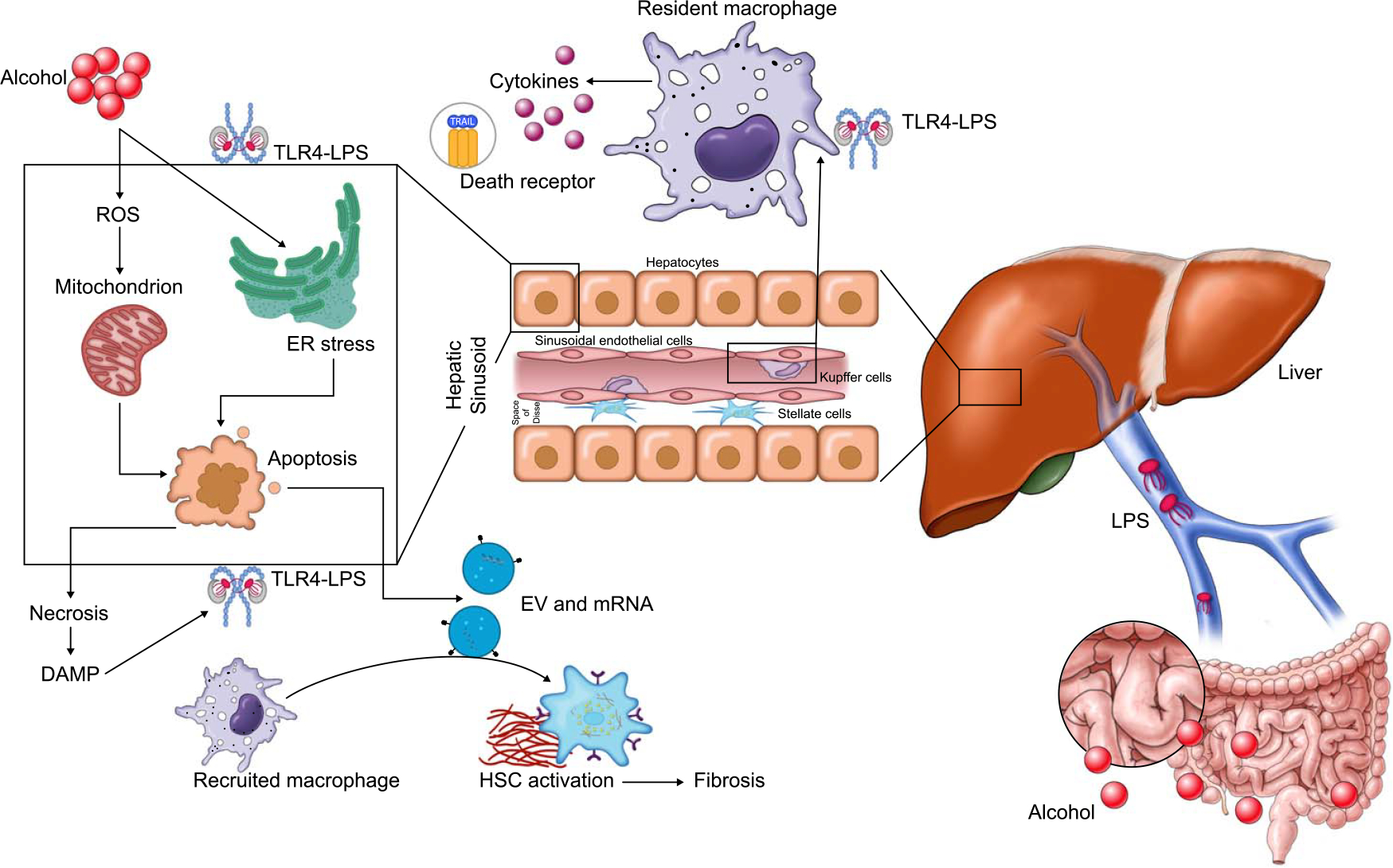
Pathogenesis of alcohol-associated liver disease and alcoholic hepatitis. Abbreviations: DAMP, damage-associated molecular patterns; ER, endoplasmic reticulum; EV, extracellular vesicles; LPS, lipopolysaccharide; ROS, reactive oxygen species; TLR-4, toll-like receptor-4.
FIGURE 2.
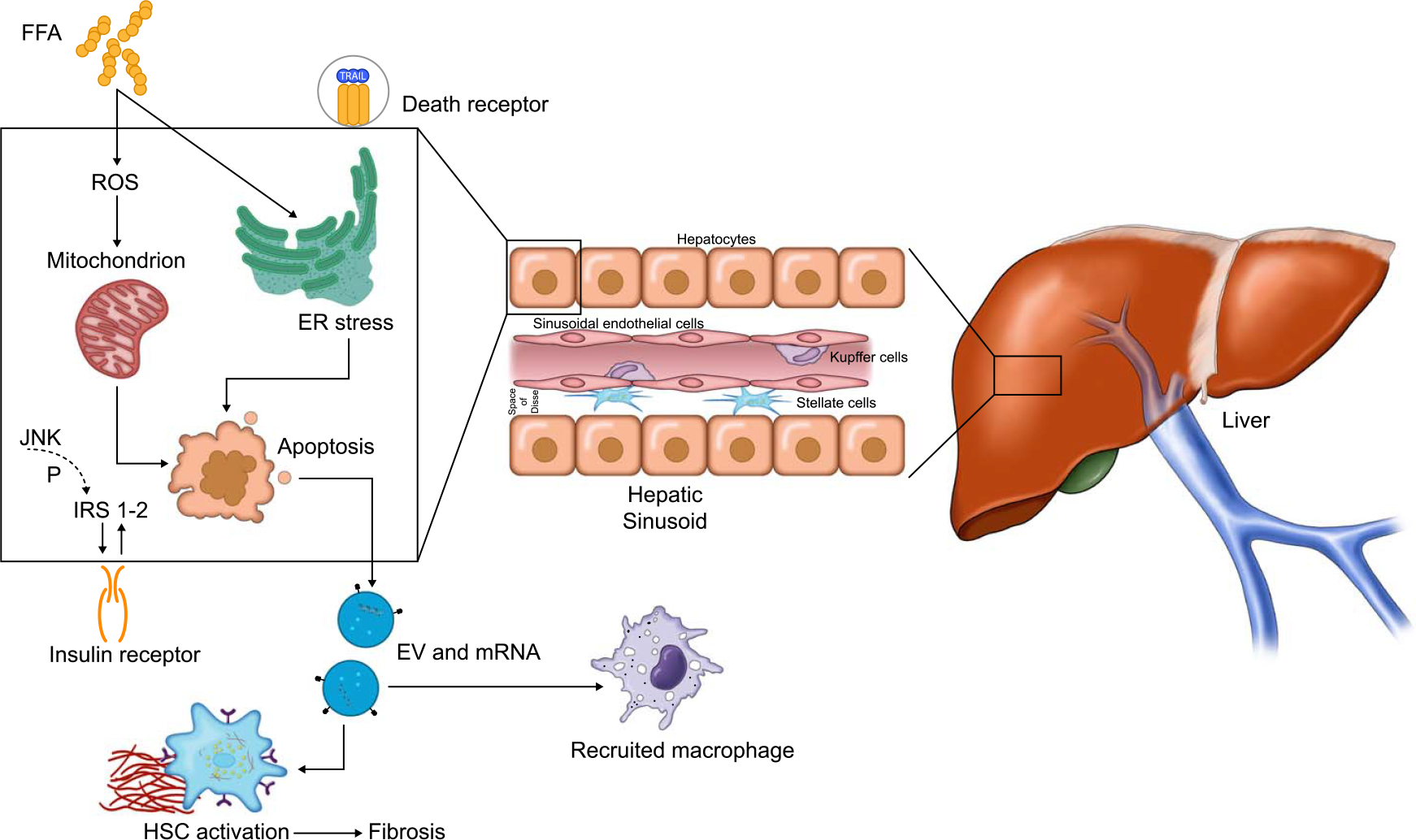
Pathogenesis of NAFLD. Abbreviations: ER, endoplasmic reticulum; EV, extracellular vesicles; FFA, free fatty acids; IRS, insulin receptor substrate; ROS, reactive oxygen species.
Over the last decade, several therapeutic targets have been identified and tested in phase 2 and 3 studies for both ALD and NAFLD. However, none of these have met endpoints for Food and Drug Administration (FDA) approval for use in routine clinical practice.[10,11] Of several ongoing or completed phase 2 and 3 clinical trials among patients with NASH, several but not all the drugs may have benefit in ALD with the potential of being examined among patients with ALD (Table 1). Throughout this article, we will briefly describe the available preclinical data in an animal model of ALD for the specific target/s, strongly justifying their assessment in clinical trials on patients with ALD. Although FDA-approved surrogate endpoints exist for NAFLD trials, in ALD and AH clinical trials, there is a significant unmet need to develop regulatory endpoints. Past trials in AH have relied on clinical prognostic scores and mortality in severe AH.[34] However, there exists an opportunity to define patient stratification and relevant endpoints in both patients with ALD and those with AH.
TABLE 1.
Treatment landscape for compounds targeted to treat NAFLD and NASH
| Drug | Target | Indication | Clinical trial name | Primary outcome(s) | Current status | Applicability in ALD |
|---|---|---|---|---|---|---|
| Lipid metabolism | ||||||
| Firsocostat (GS-0976)[12] | Acetyl-CoA carboxylase inhibition | NASH F1-F3 | 2-GS-0976 | Overall safety (adverse events) | Phase 2: Improved liver fat content, liver chemistry, and serum markers of fibrosis | Yes |
| Aramchol[13] | SCD1 inhibitor | NASH F0-F3 | 2-ARREST | Change in liver on MRS | Phase 2b: Reduced liver fat, improved histology, and liver chemistry | Yes |
| TVB-2640[14] | FASN inhibitor | NASH | FASCINATE-1 | Liver fat content | Phase 2a: Improvement in liver fat content | Yes |
| PXL065[15] | MPC inhibitor | NASH | NCT04321343 | Liver fat content | Phase 2: Active not recruiting | |
| Tesamorelin | GHRH analog | NAFLD | NCT03375788 | Liver fat content | Phase 2: Active and recruiting | |
| Elafibranor (GFT-505)[16] | PPAR-α/δ agonist | NASH F1-F3 | 2-Golden 3-Resolve-IT | NASH resolution without worsening of fibrosis | Phase 2b: Resolution of NASH without fibrosis worsening No effect on NASH resolution |
Yes |
| Saroglitazar[17] | NAFLD with ALT > 50 IU/L NASH with fibrosis |
NCT05011305 | ALT and liver fat content NASH resolution without worsening of fibrosis | Phase 2b: Improved ALT, liver fat, and metabolic profile Phase 2: Recruiting |
No | |
| Lanifibranor[18] | NASH with fibrosis | Decrease of ≥ 2 points in SAF score without fibrosis worsening | Phase 2b: ≥ 2 points SAF score improvement higher with 1200 mg dose vs. placebo (55% vs. 33%, p = 0.007) | Yes | ||
| Resmetirom (MGL-3196)[19] | Selective THR-β agonist | NASH F1-F3 | 2-MGL-3196 | Change from baseline in fat fraction on MRI-PDFF Histopathology was the secondary endpoint with paired biopsy at 36 wk |
Phase 2: Improved fat fraction and lipid profile, liver chemistry, and NASH histology | No |
| Resmetirom (MGL-3196) | Selective THR-β agonist | NASH F2-F3 | 3-MAESTRO (NCT03900429) | NASH resolution on histology All-cause mortality and liver-related events |
Phase 3: Active recruiting | No |
| VK2809 | THR-β agonist | NASH F1-F3 | NCT04173065 | Liver fat content and LDL | Phase 2: Active recruiting | |
| Glucose metabolism and insulin resistance | ||||||
| Pioglitazone | T2DM and NASH | NCT04501406 | NASH improvement without fibrosis worsening | Phase 2b: Recruiting | No | |
| Liraglutide[20] | GLP-1 analog | NASH F0-F4 | 2-LEAN | Improvement of NASH | Phase 2: NASH resolution on histology | No but may be of use in alcohol use disorder |
| Semaglutide[21] | GLP-1 analog | NASH F2-F3 | 2-Semaglutide | NASH resolution without worsening of fibrosis | Phase 2b: Semaglutide resulted in a higher percentage of patients with NASH resolution without fibrosis worsening | |
| Semaglutide | NASH F2-F3 | NCT04822181 | NASH resolution without fibrosis worsening Fibrosis improvement without NASH worsening Time to first liver-related clinical event |
Active and recruiting | ||
| Tirzepatide | Dual GIP and GLP-1 agonist | NASH F2-F3 | NCT04166773 | NASH resolution without fibrosis worsening | Active and recruiting | |
| Efinopegdutide | NAFLD >10% liver fat content |
NCT04944992 | Decrease in liver fat content by MR-PDFF Safety | Active and recruiting | ||
| ION224[12] | DGAT2 inhibitor | Type 2 diabetes and NAFLD | Safety Liver fat content by MR-PDFF |
Phase 2: Safe and reduced liver fat content | ||
| Dapagliflozin | SGLT2 inhibitor | Type 2 diabetes and NASH | NCT03723252 | NASH improvement on histology | Phase 3: Active recruiting | No |
| Inflammation, apoptosis, and regeneration | ||||||
| Cenicriviroc[22,23] | Chemokine receptor 2/5 antagonist | NASH F1-F3 | Improved NASH without fibrosis worsening Improvement in fibrosis without NASH worsening |
Phase 2b: No improvement in NASH, but improved fibrosis without NASH worsening | Yes | |
| Cenicriviroc | NASH F2-F3 | NCT03028740 | Improvement in fibrosis without NASH worsening Time to first liver-related event |
Phase 3: Completed and results awaited | ||
| Selonsertib (GS-4997)[24] | Apoptosis signal-regulating kinase 1 inhibitor | NASH F3 | 3-STELLAR 3 | Fibrosis improvement without worsening of NASH |
Phase 3: No benefit in fibrosis improvement | Yes, completed phase 2 clinical trial in severe AH patients |
| NASH F4 | 3-STELLAR 4 | Fibrosis improvement without worsening of NASH | Phase 3: No benefit in fibrosis improvement, infection, or survival | |||
| NASH F2-F3 | 3-MAESTRO | NASH resolution on histology | No data available | |||
| Vitamin E[25] | Antioxidant | Nondiabetic NASH | PIVENS | Improvement in NASH histology | Vitamin E better than placebo in improving steatosis and NASH but not fibrosis | Yes |
| Vitamin E[26] | Vitamin E with pioglitazone | Type 2 diabetes and NASH | Improvement in NASH without fibrosis worsening | Combination was better in meeting the primary endpoint | ||
| Tocoretinol | Antioxidant | NASH cirrhosis with MELD 8–17 | Change in MELD score | Phase 2: Active and recruiting | ||
| Bile acid metabolism | ||||||
| Obeticholic acid (INT-747)[27] | FXR agonist | NASH F2-F3 | 3-REGENERATE | NASH resolution without worsening of fibrosis Fibrosis improvement without worsening of NASH (on histology) |
Phase 3: histologic improvement in fibrosis without worsening of NASH (interim analysis) | Yes ongoing phase 2 trial |
| NASH F4 | 2-REVERSE | Fibrosis improvement without worsening of NASH (on histology) | No data available | |||
| EDP-305[28] | NASH F2-F3 | NCT04378010 | Change in ALT Change in liver fat content |
Phase 2 completed with reduction in ALT and liver fat | ||
| MET642 | NASH and liver fat ≥ 10% | NCT04773964 | Safety and pharmacokinetics | Active but not yet recruiting | ||
| Tropifexor (TXR, LJN452) | Nonsteroidal FXR agonist | NASH F1-F3 | 2-FLIGHT-FXR (NCT02855164) | Change in transaminases and fat fraction on MRI | Phase 2: Improved liver chemistry and fat content | Yes |
| Cilofexor (GS-9674)[29] | Nonsteroidal FXR agonist | NASH F1-F3 | 2-GS-9674 | Overall safety | Improved liver chemistry, serum bile acids, and liver fat content | Yes |
| Efruxifermin[30] | FGF-21 | NASH with fibrosis NASH CC |
Liver fat by MR-PDFF Safety and tolerability |
Phase 2b: Improvement in liver fat content Phase 2a: safely reduced noninvasive markers of injury and fibrosis |
Yes | |
| Pegbelfermin | FGF-21 | NASH F3 NASH CC |
FALCON 1 FALCON 2 |
Improvement of fibrosis without NASH worsening Improvement in NASH without fibrosis worsening |
Phase 2b: Recruiting Phase 2b: Recruiting |
Yes |
| BMS-986036[31] | Pegylated FGF-21 | NASH | F1-F3 and fat fraction > 10% | Safety and liver fat by MR- PDFF | Phase 2a: reduced fat fraction on MR-PDFF | Yes |
| Aldafermin[32] | FGF-19 | NASH F2-F3 | ALPINE 2/3 | Improvement in fibrosis without NASH worsening | Phase 2b: Safe but did not meet primary endpoint | Yes |
| Aldafermin[32] | FGF-19 | NASH CC | ALPINE 4 | Safety and improvement in ELF score | Phase 2b: Active not recruiting | Yes |
| Gut-liver axis | ||||||
| Fibrogenesis | ||||||
| Belapectin[33] | Galectin-3 inhibitor | NASH cirrhosis (HVPG > 6 mm Hg) | Reduction in HVPG or fibrosis | Phase 2b: No effect on HVPG or fibrosis. However, reduced variceal development in subgroup without esophageal varices at baseline | ||
Abbreviations: ALD, alcohol-associated liver disease; ALT, alanine aminotransferase; DGAT2, diacylglycerol acyltransferase-2; ELF, Enhanced Liver Fibrosis; FASN, fatty acid synthase; FXR, farnesoid X receptor; GIP, gastric inhibitory polypeptide; GLP-1, glucagon-like peptide 1; GNRH, growth hormone-releasing hormone; MELD, Model For-End-stage Liver Disease; MRE, magnetic resonance elastography; MR-PDFF, magnetic resonance proton density fat fraction; MRS, magnetic resonance spectroscopy; SAF, steatosis, activity, fibrosis; SCD1, stearoyl-CoA desaturase 1; T2DM, type 2 diabetes mellitus; THR-β, thyroid hormone receptor β.
PATHOPHYSIOLOGICAL MECHANISMS
Lipid metabolism
Hepatic steatosis
A common and initial pathology in ALD and NAFLD is defined as > 5% of hepatocytes containing fat droplets.[5,35] Hepatic steatosis in NAFLD is multifactorial, arising from increased free fatty acid flux from adipose tissue lipolysis due to systemic insulin resistance, diet-derived free fatty acids, and de novo lipogenesis (Figure 3). In contrast, alcohol mediates hepatic steatosis and abnormalities in lipid metabolism through several pathways: (a) increase in NADH-to-NAD ratio through its metabolism, impairing mitochondrial beta-oxidation of fatty acids; (b) inducing master regulators of de novo lipogenesis (SREBP-1c, ChREBP, and PPAR-γ); (c) increased adipose tissue lipolysis, leading to greater delivery and influx of free fatty acids into hepatocytes; (d) increased expression of the fatty acid transporter, CD36; (e) inhibition of AMPK, a regulator of metabolism in cells and inhibitor of lipogenesis; (f) suppression of peroxisome proliferator–activated receptor α (PPARα) activity; (g) impaired assembly and secretion of VLDL particles; and (h) intersection with lipid droplet proteins, variants of which influence susceptibility and progression of ALD.[36,37] Furthermore, alcohol leads to qualitative and quantitative changes in the content of complex lipids in hepatocytes. For example, the activity of sphingomyelinases has been shown to be increased up to 3-fold in humans in response to chronic alcohol use and declined within 1 week of abstinence from alcohol.[38,39] A similar finding of increased sphingomyelinase activity was shown in rodent models after exposure to alcohol,[40] and this effect was blunted in animals pretreated with N-acetylcysteine, suggesting a role of oxidative stress as a mechanism of activation of sphingomyelinase.[41] In another study, sphingomyelinase-knockout mice were resistant to alcohol-mediated fatty liver and apoptosis.[42] Furthermore, alcohol increases the accumulation of ceramides and sphingolipids by increasing the activity of serine palmitoyltransferase, the rate-limiting step in sphingolipid biosynthesis and of ceramide synthase.[43,44] Of the 3 types of ceramide synthases (1, 5, and 6), subtype 6 is the most relevant in the development of alcohol-associated fatty liver, with increased activity in zone 3 hepatocytes in both experimental as well as subjects with alcohol-associated steatosis.[44]
FIGURE 3.
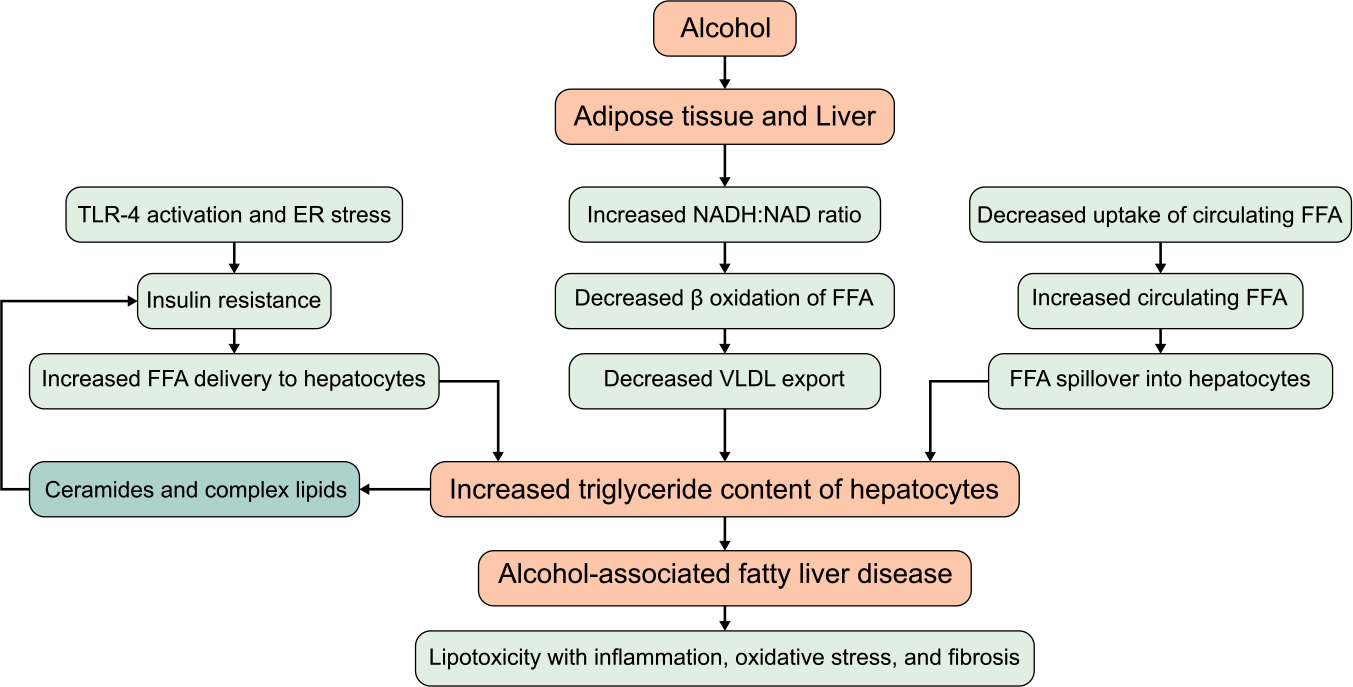
Mechanisms of alcohol induced increased hepatic triglyceride content and steatosis. Abbreviations: ER, endoplasmic reticulum; FFA, free fatty acids; TLR-4, toll-like receptor-4.
Fatty acids
Hepatocyte fat content is a balance between influx of free fatty acids (import from peripheral tissues due to lipolysis and de novo lipogenesis from ingested sugars and proteins) and their use through fatty acid oxidation. De novo lipogenesis contributes significantly to hepatic steatosis in both conditions. De novo lipogenesis involves 3 key enzymes, acetyl-CoA carboxylase (ACC), which converts acetyl-CoA to malonyl-CoA; fatty acid synthase, which converts malonyl-CoA to long-chain fatty acids; and stearoyl-CoA desaturase 1, which catalyzes the synthesis of monounsaturated fatty acids.[6] Lipogenesis is regulated by SREBP-1c activity with ACC being the rate-limiting enzyme, and fatty acid oxidation is regulated by the nuclear receptors PPAR-α and PPAR-δ, with the mitochondrial carnitine palmitoyltransferase being the rate-limiting enzyme.[36]
Neutral and complex lipids
Fatty acids are stored as simple lipids (ester linkage bond with alcohols like glycerol to form triglyceride) or complex lipids (ester linkage bond with phospholipids or sphingolipids). Present in plasma and cell membranes, sphingolipids comprise 10%–20% of membrane lipids and support specific membrane functions.[45] These complex lipids can be synthesized de novo in all cells, starting with the conjugation of amino acid serine and fatty acid palmitoyl CoA. Ceramides, a special class of sphingolipids are generated in the cells from dihydroceramide through the activity of dihydroceramide desaturase 1, from sphingomyelin through hydrolytic activity of sphingomyelinase, or as a result of acylation of sphingosine through ceramide synthase. The ceramide content is regulated within the cell by its conversion to sphingosine through ceramidases.
Ceramides can directly result in endoplasmic reticulum stress and mitochondrial dysfunction.[46] Phospholipids are also altered in ALD due to choline deficiency and decreased activity of phosphatidylethanolamine methyltransferase, which results in reduced conversion of phosphatidylethanolamine to phosphatidylcholine.[47] Decreased phosphatidylcholine to phosphatidylethanolamine ratio results in decreased export of fatty acids, leading to aggravation of steatosis and steatohepatitis. Phosphatidylcholine administration reduced fibrosis in a baboon model of ALD,[48] and betaine supplementation improved the activity of phosphatidylethanolamine methyltransferase in a mouse model with attenuation of steatosis.[49]
Glucose metabolism and insulin resistance
Insulin resistance is a central pathway in NAFLD, but the data on its role in ALD are scanty and emerging. Apart from inflammation, cell death, and oxidative stress, abnormalities in lipid metabolism and hepatic lipids also mediate insulin resistance (Figure 3). For example, ceramides can result in impaired insulin signaling and beta-oxidation of fatty acids through inhibition of serine-threonine kinase, a critical enzyme for intracellular effects of insulin.[50,51] This is achieved by ceramide-induced activation of protein kinase C, which phosphorylates and inhibits translocation of Akt/PKB, and by activation of protein phosphatase 2A, which is needed for dephosphorylation of Akt/PKB.[52]
Gut-liver axis and bile acid metabolism
Gut microbes include 1014 cells, including bacteria, fungi, viruses, archaea, and protozoa. The bacterial microbiome in healthy humans is dominated by beneficial bacterial phyla such as Bacteroides and Firmicutes, and a smaller proportion consists of Proteobacteria, Actinobacteria, and Verrucomicrobia.[53] The gut bacterial microbiome in patients with liver disease is characterized by dysbiosis, with an increase in harmful and a decrease in beneficial bacteria, and this abnormality worsens with an increase in disease severity and is also associated with liver and patient-related outcomes.[54] Mechanistically, recent studies have identified excess alcohol production by Klebsiella pneumoniae as a driver of hepatic steatosis in NAFLD.[55] Generation of shortchain fatty acids, which can serve as a substrate for de novo lipogenesis; decrease in expression of tight junction proteins; and regulation of food intake and energy expenditure by endocannabinoid signaling and gut hormones are additional effects of microbial metabolites in NAFLD. Altered bile acid metabolism is also associated with both ALD and NAFLD and has provided therapeutic opportunities.
Farnesoid X receptor (FXR) is a nuclear receptor that regulates cholesterol and bile acid metabolism. FXR can also be activated by FGF family members, especially by FGF-19 (FGF-15 in mice) subfamily.[56] FGF-19, expressed in ileal enterocytes reaches the liver through portal vein to activate the FGF4 membrane receptor, resulting in the inhibition of the rate-limiting enzyme CYP7A1 in the conversion of cholesterol to primary bile acids (cholic acid and chenodeoxycholic acid), with an upregulation of bile acid exporters at the biliary canaliculi and at the basolateral surface of enterocytes (Ost alpha-beta heterodimer).[56] FGF-21, another member of the FGF-19 subfamily, is synthesized in the liver and other extrahepatic tissues and activates FGF3 membrane receptor, leading to physiological effects of FXR activation.[56]
Hepatic inflammation and cell injury
Lipotoxicity
Mitochondrial dysfunction, endoplasmic reticulum stress, lysosomal membrane permeabilization, hepatocyte injury with apoptosis and other forms of cell death, inflammation, and the recruitment and proinflammatory activation of macrophages, are some of the recognized mechanisms of free fatty acid–induced lipotoxicity.[35] Although, lipotoxicity is well described and studied in NAFLD,[35] it also plays a role in ALD, as free fatty acids are elevated in liver biopsy samples from patients with ALD.[57] For example, preclinical data have shown a shift of intrahepatic fatty acids from saturated to unsaturated fatty acids,[58] with a benefit of saturated fatty acid diet in ameliorating ALD pathology in a mice model of ALD.[59]
Immune cells
Macrophages, neutrophils, and T cells are most studied in the context of NAFLD and ALD as key mediators of the inflammation associated with each disease. Death of resident macrophages (KCs) along with chemokines and cytokines creates an empty niche and a mechanism for the recruitment of monocyte-derived macrophages into the liver. These monocyte-derived macrophages contribute to the infiltration proinflammatory macrophages and restore the KC pool.[60] In AH, there is an increase in circulating neutrophils, which infiltrate the hepatic parenchyma and contribute to inflammation. The role of neutrophils in the pathogenesis of NAFLD remains less well studied, though it has been suggested that neutrophil elastase may be a therapeutic target as it mediates insulin resistance in NAFLD.[61]
Extracellular vesicles and microRNAs (miRs)
Surrounded by a lipid bilayer and containing bioactive cargoes, extracellular vesicles are secreted by hepatocytes into the extracellular space and taken up by surrounding cells, thus mediating cross-talk with adjacent hepatocytes and other liver cells including HSCs.[62–64] Extracellular vesicles may also contain short noncoding RNAs or miRs, which can regulate the expression of genes related to immunity, inflammation, and regeneration. Measurable in serum, distinct EV cargo and miRs have been recognized in ALD and NAFLD.[51,65,66] For example, miRs 34a and 122 are elevated in NAFLD with respective target effects of reducing fatty acid oxidation and fibrogenesis.[36,67] Another miR, let7d, which physiologically regulates fatty acid oxidation is deceased in NAFLD. In contrast, miR-155 is increased in ALD and mainly targets TNF-α, resulting in the induction of LPS sensitization, toll-like receptor-4 activation, and inflammation.[68,69] ALD is also characterized by elevated levels of miR-217, resulting in reduced fatty acid oxidation.[70]
Adipokines
Circulating soluble levels of hormones, especially adiponectin, are increased in ALD, which is the opposite of the observed reduction noted in patients with NAFLD.[35,71,72] Increase in adiponectin levels in ALD is associated with changes in other hormones such as leptin, visfatin, resistin, and omentin, all affecting insulin signaling and steatosis.[72] For example, alcohol induces an increase in leptin levels similar to NAFLD and obesity, which is also associated with an increase in leptin levels.[72,73] Leptin promotes HSC activation with inflammation of hepatocytes through TNF-α release from KCs and by the release of the chemokine CCL2 from HSCs.[73]
Apoptosis, pyroptosis, and ferroptosis
Cell death in ALD and NAFLD is linked to immune cell activation, and apoptosis is best studied in this regard. For example, a result of inflammatory signaling in ALD and NAFLD is activation of death receptors, leading to apoptosis,[74,75] mediated by death receptors especially TRAIL receptor 2.[76,77] Cross-talk between apoptotic bodies and immune cells leads to the formation of a proinflammatory loop. Other pathways mediating apoptosis are the activation of caspases and apoptosis signal-regulating kinase 1 enzyme, which results in the activation of JNK pathway.[78,79] Caspase 1 is also activated by inflammasomes in hepatocytes and inflammatory cells, mediating sterile inflammation, especially the release of IL-1β.[80,81] Pyroptotic cell death occurs following caspase 1–mediated cleavage of gasdermin, leading to the formation of plasma membrane pores and cell death. Due to its dependence on the inflammasome, which is known to be activated in NASH and ALD, pyroptotic cell death has been suggested to mediate NASH and may also play a role in ALD.[82] Cell-specific activation of the inflammasome, predominantly in KCs in ALD, and within the hepatocytes in NAFLD, may influence efficacy of caspase inhibitors or other drugs targeting this pathway.[36,80] Ferroptosis, iron-dependent cell death, has been implicated in NASH,[83] and may also play a role in alcohol-induced cell death.[84] Thus, many forms of cell death can trigger inflammation in NASH and correspondingly may play a role in ALD.
Hepatic fibrosis
The activation of HSCs is a key step in the development of fibrosis in any liver disease, including ALD and NAFLD.[85] TGF-β is a master regulator of fibrogenesis and is an important therapeutic target.[85] Cross-talk between the extracellular matrix and hepatic cells is a critical step in fibrogenesis. Integrins, which mediate this cross-talk, have attracted the attention as important targets of drug discovery targeting fibrosis. Lysyl oxidase is a family of 4 extracellular enzymes[1–4] that cross-link the collagen fibers and promote liver fibrosis. Other factors, such as sonic hedgehog released from inflamed and ballooned hepatocytes, upregulate the expression of TGF-β in HSCs.[85]
PHARMACOLOGICAL THERAPIES TAGETING METABOLISM
Therapies targeting lipid metabolism
Inhibitors of fatty acid synthesis
The various types of fat within hepatocytes are labile and change in response to intervention. AMP kinase agonists reduce de novo lipogenesis through inhibition of SREBP-1c, ChREBP, and ACC phosphorylation. Several AMP kinase agonists are in clinical trials. Resveratol, an agonist of AMP kinase, has shown protection from hepatic steatosis in an alcohol-fed mouse model.[86] Inhibiting ACC using firsocostat (ND-630 or GS-0976) in an animal model showed benefit in reducing hepatic steatosis.[87] In a phase 2b clinical trial, this drug reduced liver fat and levels of tissue inhibitor of metalloprotease-1, without improvement in aminotransferases or liver stiffness measurement.[88] Inhibition of ACC is also associated with enhanced beta-oxidation of fatty acids, which results in the elevation of serum triglycerides. Similarly, fatty acid synthase can be targeted using its inhibitor TVB-2640.[14] SCD1 can be inhibited by fatty acid/bile acid conjugate 3beta-arachidyl-amido, 7alpha-12alpha-dihydroxy, 5beta-cholan-24-oic acid (aramchol), leading to improved hepatic steatosis and insulin sensitivity.[89] Based on the encouraging data with the use of aramchol in phase 2 studies,[13,90] a phase 3/4 clinical trial is ongoing in patients with NAFLD (NCT 04104321). Fatty acids are stored in the liver and adipose tissue and esterified into triglycerides. The last and committed steps in triglyceride synthesis are mediated by the enzymes acyl-CoA diacylglycerol acyltransferase-1 and 2. Its inhibition in a high-fat diet animal model has been shown to reduce hepatic steatosis.[91] To ameliorate the elevation of serum triglycerides, the inhibition of diacylglycerol acyltransferase-2 is being studied in combination with an ACC inhibitor in an ongoing phase 2 clinical trial (NCT04399538) (Table 2).
TABLE 2.
Clinical trials with use of combination therapies in patients with NAFLD and NASH
| Drugs | Target | Indication | Clinical trial name | Primary outcome(s) | Current status |
|---|---|---|---|---|---|
| Saroglitazar+vitamin E | PPAR-γ Antioxidant | NAFLD | NCT04193982 | Change in NAFL fibrosis score | Phase 3: Recruiting |
| PF-06865571+PF-05221304 | DGAT2 ACC | NAFLD | NCT04399538 | Change in liver fat content | Phase 2: Recruiting |
| MET-409 +empaglifozin | FXR SGLT2 | NASH | NCT04702490 | Safety and tolerability of MET-409 | Phase 2: Active not yet recruiting |
| Tropifexor+licoglifozin | FXR SGLT2 | ELIVATE NASH F2-F3 | NCT04065841 | Improvement in fibrosis without worsening of NASH NASH resolution without fibrosis worsening |
Phase 2: Recruiting |
| Semaglutide+cilofexor +firscostat | GLP-1 FXR+ACC | NASH cirrhosis | NCT04971785 | Improvement in fibrosis without worsening of NASH NASH resolution without fibrosis worsening |
Phase 2: Recruiting |
| Tropifexor +cenicriviroc | FXR Chemokine receptor 2/5 | NASH F2-F3 | TANDEM NCT03517540 | Safety of combination | Phase 2: Completed and results awaited |
| Cilofexor+firscostat +selonsertib | FXR ACC+ASK-1 | NASH F3-F4 | ATLAS NCT03449446 | Adverse effects and safety of combination | Phase 2: Completed and results awaited |
Abbreviations: ACC, acetyl-CoA carboxylase; ASK-1, apoptosis signal-regulating kinase 1; DGAT2, diacylglycerol acyltransferase-2; FXR, farnesoid X receptor; GLP-1, glucagon-like peptide 1; PPAR, peroxisome proliferator-activated receptor; SGLT2, sodium-glucose co-transporter type 2.
PPAR α/δ agonists
In a mouse model, PPAR agonists have shown benefit in reducing the development of steatosis in response to ethanol feeding.[92] PPAR agonists are also in phase 2 and 3 trials in NASH, suggesting that they may be worth investigating in human clinical trials of patients with ALD. Examples include saroglitazar (dual PPAR α/γ), pemafibrate (K-877), seladelpar (MBX-8025), elafibranor (dual PPAR α/δ), and lanifibranor (IVA337, pan-PPAR). In this regard, it is interesting to note that increasing fatty acid oxidation improves NASH, rather than pose oxidative stress, likely by shifting the excess or balance of residual fatty acids. PPARs also regulate immune cell types, which may play a role in the efficacy of these drugs in NASH and ALD.[93]
Thyroid hormone receptor β agonists
Thyroid hormone receptor β is expressed in hepatocytes and regulates lipid metabolism, among its pleiotropic effects. Examples of thyroid hormone receptor β agonists are resmetirom (MGL-3196) and VK2809 (MB07811). Resmetirom administration led to a significant reduction in liver fat in patients with NASH in a phase 2a clinical trial, and there is an ongoing phase 3 clinical trial (Table 1). A phase 2 trial with VK2809 in patients with NASH is ongoing.
Inhibitors of ceramide synthesis
Pharmacological inhibition of synthesis of ceramides improves steatosis and glucose tolerance.[42,94] Of the 3 pathways involved in ceramide biosynthesis, effect on ceramide synthase inhibition and not hydrolysis of sphingomyelin was shown to be most relevant in improving insulin resistance, steatosis, glucose tolerance, and dyslipidemia in an animal model of alcohol-associated steatosis.[44] Inhibition or deletion of dihydroceramide desaturase has also been shown to improve hepatic steatosis in an animal model of NAFLD.[95] Although not yet studied in ALD, this may be an important therapeutic target if the experimental data are encouraging as in NAFLD.[96]
Therapies targeting glucose metabolism
Insulin sensitizers
PPAR-γ agonism with pioglitazone reduced steatosis, inflammation, and fibrosis in patients with NASH.[25,97] Incretin hormones from small bowel mucosa like glucagon-like peptide 1 and gastric inhibitory polypeptide mediate insulin secretion from islet cells of pancreas through binding to their G-protein–coupled receptors. Their agonists like liraglutide, semaglutide, HM15211, and coradudite (MED10382) have been successfully tried in patients with NASH. Sodium-glucose co-transporter type 2 inhibitors dapagliflozin, epagliflozin, canagliflozin, and licogliflozin reduce reabsorption of glucose across the renal tubules (Table 1).
Although insulin resistance can occur in ALD mediated by the effect of alcohol on liver and on adipose tissue,[94] insulin sensitizers currently do not seem to have a potential in patients with ALD.[36] However, data are emerging on the reprogramming of hepatocyte metabolism of glucose, especially in more severe forms of ALD with severe AH.[98] Reprogramming of hepatocytes leads to impaired use of glucose in generating energy, as glucose is trapped in the cells as glucose-6 phosphate. Hexokinase domain containing 1 is the most activated enzyme in patients with AH and also correlated with disease severity and patient survival. Targeting this enzyme and pathways involved in the transcriptomic and epigenetic reprogramming such as liver-enriched transcription factors especially hepatocyte nuclear factor 4-alpha may be of potential in the treatment of patients with AH.[99] Data are also emerging on the benefit of glucagon-like peptide 1 and gastric inhibitory peptide agonists in the management of alcohol use disorder with a reduction in craving and alcohol consumption.[100] The exact mechanism is unclear, but is thought to be centrally mediated through dopamine signaling.[101] It should be noted that insulin sensitizers will need to be used with caution, and after careful safety testing, in patients with ALD given the added risk of lactic acidosis with metformin in actively drinking patients,[102] and the risk of HCC with PPAR-γ agonists.[103]
Therapies targeting bile acid metabolism
FXR agonists
Obeticholic acid (OCA) is currently ongoing a large phase 3 clinical trial in patients with NASH and significant fibrosis. However, this drug is limited due to its adverse effect profile of an increase in cholesterol levels and potential for severe pruritus.[27] Many second-generation FXR agonists are currently in development in an attempt to overcome the adverse effects of OCA while retaining the histological benefit. These compounds differ in their chemical structure, their propensity for liver accumulation, and their preferential intestinal versus hepatic FXR agonism.[104] For example, EDP-305 in phase 2 clinical trial resulted in improved alanine aminotransferase and reduced intrahepatic fat, with a lower increase in LDL cholesterol.[28] The minimal increase in LDL cholesterol with EDP-305 may obviate the need for the coadministration of statins as needed with use of OCA or an FGF agonist aldafermin (also known as NGM-282).[105] Currently, a clinical trial is also underway examining the benefit of FXR agonist OCA in patients with moderate and severe alcohol-associated hepatitis (NCT02039219). Many other compounds such as tropifexor (LJN452), cilofexor nidufexor (LMB763), non–bile acid FXR agonists like EDP-297 and EDP-305, MET409, and EYP001a are under development for NASH (Table 1) and may have potential for exploring their use in patients with ALD.
FGF analogs
Although FGF activity has been shown to be increased in NAFLD, FGF receptors are resistant to its target effect.[56] In phase 2 randomized placebo-controlled clinical trial, 24-week use of an engineered FGF-19 analog aldafermin resulted in reduced intrahepatic fat and a trend on improvement in fibrosis. The drug was safe and none of the patients had to discontinue the medication due to adverse effects.[106] However, in another phase 2b study in patients with NASH with stage 2 or 3 fibrosis, use of aldafermin for 24 weeks did not result in meeting the primary endpoint of improvement of fibrosis by one stage without worsening of NASH.[32] Although the drug was well tolerated in both studies, increase in LDL cholesterol between 0.2 and 0.4 mmol/L occurred early at 4 weeks, as among patients treated with OCA. Increase in LDL cholesterol of the same milder magnitude has been observed with cilofexor with a reduced efficacy in lowering hepatic fat and alanine aminotransferase levels.[29,107] Although the role of nuclear receptors such as FGF-19 is not yet determined in patients with ALD, preclinical data in mice have shown that alcohol induces expression of FGF-15/19 or FGF-21 with favorable lipid metabolism and bile acid profiles, resulting in amelioration of alcohol-associated steatohepatitis changes and protection from the development of ALD.[108,109] Other drugs targeting the FGF pathway are being assessed in patients with NASH, such as FGF-21 analogs pegbelfermin (BMS-986036) and efruxifermin (AKR001) and FGF receptor–activating humanized monoclonal antibodies MK-3655 (NGM313) and BFKB8488A (Table 1) and may have the potential for use in patients with ALD. In a mouse model, FGF-21 activity was shown to be activated by alcohol, but this protected from the development of ALD.[110]
Therapies targeting inflammatory and cell death pathways
Of therapies that were assessed targeting this paradigm, several molecules (cenecriviroc, selonsertib, and belapectin) have made it to phase 3 studies in patients with NAFLD and are worthy of investigation in patients with ALD (Table 1). Of these, selonsertib (inhibitor of apoptosis-stimulating kinase 1) examined in a phase 2b study in patients with ALD did not show an efficacy signal and will probably not move further in a phase 3 study. Cenicriviroc, an inhibitor of chemokine ligands type 2 and 5, prevented and treated inflammation and fibrosis in a mouse model of ALD,[111] justifying assessment of this molecule in human studies of patients with ALD. Vitamin E as an antioxidant showed a reduction of hepatic steatosis and inflammation in patients with NASH (PIVENS trial), and is a potential therapeutic target in patients with ALD.[25] Similarly, caspase inhibitor emricasan has been used successfully in patients with NASH.[112] However, a clinical trial in patients with severe AH had to be halted due to issues with pharmacokinetics and drug availability in sick patients with liver failure (NCT01912404). The protein ROCK-1 and sphingolipid, sphingosine-1-phosphate, mediate the release of extracellular vesicles mediating inflammation and can potentially be targeted,[36] to treat patients with ALD.
As the focus of this review is on potential targets gleaned from the NASH world, specific drugs targeting inflammation and hepatic regeneration in ALD such as corticosteroids, granulocyte colony-stimulating factor, IL-22, and DUR-928 will not be discussed here.[5]
Therapies targeting fibrosis
Antagonism of TGF-β can be achieved using monoclonal antibodies (lerdilimumab) or inhibiting its target receptor TGF-β1 receptor. Simtuzumab, a monoclonal antibody that blocks a critical step in laying down of collagen mediated by lysyl oxidase, was ineffective in patients with NASH with bridging fibrosis or compensated cirrhosis.[113] Antibodies to integrins have shown antifibrotic effect in animal models of NASH, and a current clinical trial is ongoing in patients with NASH with advanced fibrosis.[114] Sonic hedgehog signaling inhibitors such as cyclopamine and vismodegib have been examined in preclinical studies in NASH animal models,[115] and could be of potential in ALD.
Drugs targeting alcohol use and food intake
As the main risk factors for ALD and NAFLD are excess and harmful alcohol use and food intake, respectively, there may be a rationale for therapies targeting pathways controlling alcohol use and food intake. Animal models and functional imaging studies have shown that central pathways in the frontal cortex and midbrain involving neurotransmitters (dopamine, serotonin, GABA, and opioids) mediate addictive behavior to drugs including alcohol, food, or any other activity.[116] Although peripheral pathways and gut-derived hormones (ghrelin, leptin, insulin, and neuropeptide YY) through their interaction with the central pathways are mainly involved to control food intake, data are emerging on the role of glucagon-like peptide 1 in mediating alcohol use behavior in humans.[117,118] Although several pharmacotherapies exist and are in development for alcohol use disorder,[119] the development of therapies targeting food addiction is limited due to their potential risk of adverse effects, especially mood disorders.[116]
FUNDING INFORMATION
Harmeet Malhi is supported by the National Institutes of Health (DK111378, AA21788). Vijay H. Shah is supported by the National Institutes of Health (AA21788).
Abbreviations:
- ACC
acetyl-CoA carboxylase
- AH
alcoholic hepatitis
- ALD
alcohol-associated liver disease
- ALT
alanine aminotransferase
- ASK-1
apoptosis signal-regulating kinase 1
- DAMP
damage-associated molecular patterns
- DGAT2
diacylglycerol acyltransferase-2
- ELF
Enhanced Liver Fibrosis
- ER
endoplasmic reticulum
- EV
extracellular vesicles
- FASN
fatty acid synthase
- FDA
Food and Drug Administration
- FFA
free fatty acids
- FXR
farnesoid X receptor
- GIP
gastric inhibitory polypeptide
- GLP-1
glucagon-like peptide 1
- GNRH
growth hormone–releasing hormone
- IRS
insulin receptor substrate
- LPS
lipopolysaccharide
- MELD
Model For End-stage Liver Disease
- miR
microRNA
- MRE
magnetic resonance elastography
- MR-PDFF
magnetic resonance proton density fat fraction
- MRS
magnetic resonance spectroscopy
- OCA
obeticholic acid
- PPAR
peroxisome proliferator–activated receptor
- ROS
reactive oxygen species
- SCD1
stearoyl-CoA desaturase 1
- SGLT2
sodium-glucose co-transporter type 2
- T2DM
type 2 diabetes mellitus
- THR-β
thyroid hormone receptor β
- TLR-4
oll-like receptor-4
Footnotes
CONFLICTS OF INTEREST
Ashwani K. Singal consults for Pleiogenix Pharma. He advises Durect. Vijay H. Shah consults for Durect Corporation, GENFIT, Korro Bio Inc., and Seal Rock Therapeutics Inc. He advises Akaza Bioscience Ltd, AgomAb Therapeutics, Intercept Pharmaceuticals Inc., Mallinckrodt Pharmaceuticals, Resolution Therapeutics Ltd, and Surrozen. Harmeet Malhi has no conflicts to report.
REFERENCESr
- 1.Wong RJ, Singal AK. Trends in liver disease etiology among adults awaiting liver transplantation in the United States, 2014–2019. JAMA Netw Open. 2020;3:e1920294. [DOI] [PubMed] [Google Scholar]
- 2.Cholankeril G, Ahmed A. Alcoholic liver disease replaces hepatitis C virus infection as the leading indication for liver transplantation in the United States. Clin Gastroenterol Hepatol. 2018;16:1356–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kranzler HR, Soyka M. Diagnosis and pharmacotherapy of alcohol use disorder: a review. JAMA. 2018;320:815–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018;67:328–57. [DOI] [PubMed] [Google Scholar]
- 5.Singal AK, Mathurin P. Diagnosis and treatment of alcohol-associated liver disease: a review. JAMA. 2021;326:165–76. [DOI] [PubMed] [Google Scholar]
- 6.Parthasarathy G, Revelo X, Malhi H. Pathogenesis of nonalcoholic steatohepatitis: an overview. Hepatol Commun. 2020;4:478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–42. [DOI] [PubMed] [Google Scholar]
- 8.Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–64. [DOI] [PubMed] [Google Scholar]
- 9.Wang XJ, Malhi H. Nonalcoholic fatty liver disease. Ann Intern Med. 2018;169:ITC65–80. [DOI] [PubMed] [Google Scholar]
- 10.Arora SS, Axley P, Ahmed Z, Satapathy SK, Wong R, Kuo YF, et al. Decreasing frequency and improved outcomes of hepatitis C-related liver transplantation in the era of direct-acting antivirals—a retrospective cohort study. Transpl Int. 2019;32:854–64. [DOI] [PubMed] [Google Scholar]
- 11.Negi CK, Babica P, Bajard L, Bienertova-Vasku J, Tarantino G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism. 2021;126:154925. [DOI] [PubMed] [Google Scholar]
- 12.Loomba R, Morgan E, Watts L, Xia S, Hannan LA, Geary RS, et al. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2020;5:829–38. [DOI] [PubMed] [Google Scholar]
- 13.Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. 2021;27:1825–35. [DOI] [PubMed] [Google Scholar]
- 14.Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN Inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology. 2021;161:1475–86. [DOI] [PubMed] [Google Scholar]
- 15.Harrison SA, Thang C, Bolze S, Dewitt S, Hallakou-Bozec S, Dubourg J, et al. Evaluation of PXL065—deuterium-stabilized (R)-pioglitazone in NASH patients: a phase 2 randomized placebo-controlled trial (DESTINY-1). J Hepatol. 2023. [DOI] [PubMed] [Google Scholar]
- 16.Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–159.e5. [DOI] [PubMed] [Google Scholar]
- 17.Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-alpha/gamma agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology. 2021;74:1809–24. [DOI] [PubMed] [Google Scholar]
- 18.Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- 19.Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of nonalcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with nonalcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387:679–90. [DOI] [PubMed] [Google Scholar]
- 21.Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113–24. [DOI] [PubMed] [Google Scholar]
- 22.Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018;67:1754–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ratziu V, Sanyal A, Harrison SA, Wong VW, Francque S, Goodman Z, et al. Cenicriviroc treatment for adults with nonalcoholic steatohepatitis and fibrosis: final analysis of the phase 2b CENTAUR study. Hepatology. 2020;72:892–905. [DOI] [PubMed] [Google Scholar]
- 24.Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: results from randomized phase III STELLAR trials. J Hepatol. 2020;73:26–39. [DOI] [PubMed] [Google Scholar]
- 25.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bril F, Biernacki DM, Kalavalapalli S, Lomonaco R, Subbarayan SK, Lai J, et al. Role of vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: a randomized controlled trial. Diabetes Care. 2019;42:1481–8. [DOI] [PubMed] [Google Scholar]
- 27.Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of nonalcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96. [DOI] [PubMed] [Google Scholar]
- 28.Ratziu V, Rinella ME, Neuschwander-Tetri BA, Lawitz E, Denham D, Kayali Z, et al. EDP-305 in patients with NASH: a phase II double-blind placebo-controlled dose-ranging study. J Hepatol. 2022;76:506–17. [DOI] [PubMed] [Google Scholar]
- 29.Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR Agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020;72:58–71. [DOI] [PubMed] [Google Scholar]
- 30.Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. 2021;27:1262–71. [DOI] [PubMed] [Google Scholar]
- 31.Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019;392:2705–17. [DOI] [PubMed] [Google Scholar]
- 32.Harrison SA, Abdelmalek MF, Neff G, Gunn N, Guy CD, Alkhouri N, et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. 2022;7:603–16. [DOI] [PubMed] [Google Scholar]
- 33.Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. 2020;158:1334–345.e5. [DOI] [PubMed] [Google Scholar]
- 34.Mathurin P, Thursz M. Endpoints and patient stratification in clinical trials for alcoholic hepatitis. J Hepatol. 2019;70:314–8. [DOI] [PubMed] [Google Scholar]
- 35.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–73. [DOI] [PubMed] [Google Scholar]
- 36.Greuter T, Malhi H, Gores GJ, Shah VH. Therapeutic opportunities for alcoholic steatohepatitis and nonalcoholic steatohepatitis: exploiting similarities and differences in pathogenesis. JCI Insight. 2017;2::e95354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.You M, Matsumoto M, Pacold CM, Cho WK, Crabb DW. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology. 2004;127:1798–808. [DOI] [PubMed] [Google Scholar]
- 38.Peterson K Biomarkers for alcohol use and abuse–a summary. Alcohol Res Health. 2004;28:30–7. [PMC free article] [PubMed] [Google Scholar]
- 39.Reichel M, Greiner E, Richter-Schmidinger T, Yedibela O, Tripal P, Jacobi A, et al. Increased acid sphingomyelinase activity in peripheral blood cells of acutely intoxicated patients with alcohol dependence. Alcohol Clin Exp Res. 2010;34:46–50. [DOI] [PubMed] [Google Scholar]
- 40.Deaciuc IV, Nikolova-Karakashian M, Fortunato F, Lee EY, Hill DB, McClain CJ. Apoptosis and dysregulated ceramide metabolism in a murine model of alcohol-enhanced lipopolysaccharide hepatotoxicity. Alcohol Clin Exp Res. 2000;24:1557–65. [PubMed] [Google Scholar]
- 41.Setshedi M, Longato L, Petersen DR, Ronis M, Chen WC, Wands JR, et al. Limited therapeutic effect of N-acetylcysteine on hepatic insulin resistance in an experimental model of alcohol-induced steatohepatitis. Alcohol Clin Exp Res. 2011;35:2139–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liangpunsakul S, Rahmini Y, Ross RA, Zhao Z, Xu Y, Crabb DW. Imipramine blocks ethanol-induced ASMase activation, ceramide generation, and PP2A activation, and ameliorates hepatic steatosis in ethanol-fed mice. Am J Physiol Gastrointest Liver Physiol. 2012;302:G515–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Longato L, Ripp K, Setshedi M, Dostalek M, Akhlaghi F, Branda M, et al. Insulin resistance, ceramide accumulation, and endoplasmic reticulum stress in human chronic alcohol-related liver disease. Oxid Med Cell Longev. 2012;2012:479348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams B, Correnti J, Oranu A, Lin A, Scott V, Annoh M, et al. A novel role for ceramide synthase 6 in mouse and human alcoholic steatosis. FASEB J. 2018;32:130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barron KA, Jeffries KA, Krupenko NI. Sphingolipids and the link between alcohol and cancer. Chem Biol Interact. 2020;322:109058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lei X, Zhang S, Emani B, Barbour SE, Ramanadham S. A link between endoplasmic reticulum stress-induced beta-cell apoptosis and the group via Ca2+-independent phospholipase A2 (iPLA2beta). Diabetes Obes Metab. 2010;12(suppl 2):93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duce AM, Ortiz P, Cabrero C, Mato JM. S-adenosyl-Lmethionine synthetase and phospholipid methyltransferase are inhibited in human cirrhosis. Hepatology. 1988;8:65–8. [DOI] [PubMed] [Google Scholar]
- 48.Lieber CS, Robins SJ, Li J, DeCarli LM, Mak KM, Fasulo JM, et al. Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology. 1994;106:152–9. [DOI] [PubMed] [Google Scholar]
- 49.Kharbanda KK, Mailliard ME, Baldwin CR, Beckenhauer HC, Sorrell MF, Tuma DJ. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J Hepatol. 2007;46:314–21. [DOI] [PubMed] [Google Scholar]
- 50.Ramirez T, Longato L, Dostalek M, Tong M, Wands JR, de la Monte SM. Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. Alcohol Alcohol. 2013;48:39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fukushima M, Dasgupta D, Mauer AS, Kakazu E, Nakao K, Malhi H. StAR-related lipid transfer domain 11 (STARD11)-mediated ceramide transport mediates extracellular vesicle biogenesis. J Biol Chem. 2018;293:15277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 2012;15:585–94. [DOI] [PubMed] [Google Scholar]
- 53.Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H. Gut microbiome, liver immunology, and liver diseases. Cell Mol Immunol. 2021;18:4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, et al. Fatty liver disease caused by high-alcohol-producing Klebsiella pneumoniae. Cell Metab. 2019;30:675–688.e7. [DOI] [PubMed] [Google Scholar]
- 56.Szczepanska E, Gietka-Czernel M. FGF21: a novel regulator of glucose and lipid metabolism and whole-body energy balance. Horm Metab Res. 2022;54:203–11. [DOI] [PubMed] [Google Scholar]
- 57.Mavrelis PG, Ammon HV, Gleysteen JJ, Komorowski RA, Charaf UK. Hepatic free fatty acids in alcoholic liver disease and morbid obesity. Hepatology. 1983;3:226–31. [DOI] [PubMed] [Google Scholar]
- 58.Fernando H, Bhopale KK, Boor PJ, Ansari GA, Kaphalia BS. Hepatic lipid profiling of deer mice fed ethanol using (1)H and (3)(1)P NMR spectroscopy: a dose-dependent subchronic study. Toxicol Appl Pharmacol. 2012;264:361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen P, Torralba M, Tan J, Embree M, Zengler K, Starkel P, et al. Supplementation of saturated long-chain fatty acids maintains intestinal eubiosis and reduces ethanol-induced liver injury in mice. Gastroenterology. 2015;148:203–214.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Parthasarathy G, Malhi H. Macrophage heterogeneity in NASH: more than just nomenclature. Hepatology. 2021;74:515–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kostallari E, Valainathan S, Biquard L, Shah VH, Rautou PE. Role of extracellular vesicles in liver diseases and their therapeutic potential. Adv Drug Deliv Rev. 2021;175:113816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sehrawat TS, Arab JP, Liu M, Amrollahi P, Wan M, Fan J, et al. Circulating extracellular vesicles carrying sphingolipid cargo for the diagnosis and dynamic risk profiling of alcoholic hepatitis. Hepatology. 2021;73:571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakao Y, Fukushima M, Mauer AS, Liao CY, Ferris A, Dasgupta D, et al. A comparative proteomic analysis of extracellular vesicles associated with lipotoxicity. Front Cell Dev Biol. 2021;9:735001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase-dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol. 2016;64:651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kakazu E, Mauer AS, Yin M, Malhi H. Hepatocytes release ceramide-enriched pro-inflammatory extracellular vesicles in an IRE1alpha-dependent manner. J Lipid Res. 2016;57:233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jin X, Chen YP, Kong M, Zheng L, Yang YD, Li YM. Transition from hepatic steatosis to steatohepatitis: unique microRNA patterns and potential downstream functions and pathways. J Gastroenterol Hepatol. 2012;27:331–40. [DOI] [PubMed] [Google Scholar]
- 68.Babuta M, Szabo G. Extracellular vesicles in inflammation: focus on the microRNA cargo of EVs in modulation of liver diseases. J Leukoc Biol. 2021;111:75–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Babuta M, Furi I, Bala S, Bukong TN, Lowe P, Catalano D, et al. Dysregulated autophagy and lysosome function are linked to exosome production by micro-RNA 155 in alcoholic liver disease. Hepatology. 2019;70:2123–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. 2012;287:9817–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Kim C, Jogasuria A, Han Y, Hu X, Wu J, et al. Myeloid cell-specific lipin-1 deficiency stimulates endocrine adiponectin-FGF15 axis and ameliorates ethanol-induced liver injury in mice. Sci Rep. 2016;6:34117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parker R, Kim SJ, Gao B. Alcohol, adipose tissue and liver disease: mechanistic links and clinical considerations. Nat Rev Gastroenterol Hepatol. 2018;15:50–9. [DOI] [PubMed] [Google Scholar]
- 73.Ikejima K, Okumura K, Lang T, Honda H, Abe W, Yamashina S, et al. The role of leptin in progression of non-alcoholic fatty liver disease. Hepatol Res. 2005;33:151–4. [DOI] [PubMed] [Google Scholar]
- 74.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feldstein AE, Gores GJ. Apoptosis in alcoholic and nonalcoholic steatohepatitis. Front Biosci. 2005;10:3093–9. [DOI] [PubMed] [Google Scholar]
- 76.Idrissova L, Malhi H, Werneburg NW, LeBrasseur NK, Bronk SF, Fingas C, et al. TRAIL receptor deletion in mice suppresses the inflammation of nutrient excess. J Hepatol. 2015;62:1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malhi H, Barreyro FJ, Isomoto H, Bronk SF, Gores GJ. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–101. [DOI] [PubMed] [Google Scholar]
- 79.Zhang W, Kudo H, Kawai K, Fujisaka S, Usui I, Sugiyama T, et al. Tumor necrosis factor-alpha accelerates apoptosis of steatotic hepatocytes from a murine model of non-alcoholic fatty liver disease. Biochem Biophys Res Commun. 2010;391:1731–6. [DOI] [PubMed] [Google Scholar]
- 80.de Carvalho Ribeiro M, Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol. 2022;17:345–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beier JI, Banales JM. Pyroptosis: an inflammatory link between NAFLD and NASH with potential therapeutic implications. J Hepatol. 2018;68:643–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu CY, Wang M, Yu HM, Han FX, Wu QS, Cai XJ, et al. Ferroptosis is involved in alcohol-induced cell death in vivo and in vitro. Biosci Biotechnol Biochem. 2020;84:1621–8. [DOI] [PubMed] [Google Scholar]
- 85.Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75:473–88. [DOI] [PubMed] [Google Scholar]
- 86.Ajmo JM, Liang X, Rogers CQ, Pennock B, You M. Resveratrol alleviates alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci USA. 2016;113:E1796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155:1463–473.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scorletti E, Bhatia L, McCormick KG, Clough GF, Nash K, Hodson L, et al. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: results from the Welcome* study. Hepatology. 2014;60:1211–21. [DOI] [PubMed] [Google Scholar]
- 90.Safadi R, Konikoff FM, Mahamid M, Zelber-Sagi S, Halpern M, Gilat T, et al. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2014;12:2085–91.e1. [DOI] [PubMed] [Google Scholar]
- 91.Choi CS, Savage DB, Kulkarni A, Yu XX, Liu ZX, Morino K, et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses dietinduced hepatic steatosis and insulin resistance. J Biol Chem. 2007;282:22678–88. [DOI] [PubMed] [Google Scholar]
- 92.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003;278:27997–8004. [DOI] [PubMed] [Google Scholar]
- 93.Le Menn G, Neels JG. Regulation of immune cell function by PPARs and the connection with metabolic and neurodegenerative diseases. Int J Mol Sci. 2018;19:1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Correnti J, Lin C, Brettschneider J, Kuriakose A, Jeon S, Scorletti E, et al. Liver-specific ceramide reduction alleviates steatosis and insulin resistance in alcohol-fed mice. J Lipid Res. 2020;61:983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhu M, Jia Z, Yan X, Liu L, Fang C, Feng M, et al. Danhe granule ameliorates nonalcoholic steatohepatitis and fibrosis in rats by inhibiting ceramide de novo synthesis related to CerS6 and CerK. J Ethnopharmacol. 2022;295:115427. [DOI] [PubMed] [Google Scholar]
- 96.Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. 2019;365:386–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- 98.Massey V, Parrish A, Argemi J, Moreno M, Mello A, Garcia-Rocha M, et al. Integrated multiomics reveals glucose use reprogramming and identifies a novel hexokinase in alcoholic hepatitis. Gastroenterology. 2021;160:1725–740.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Argemi J, Latasa MU, Atkinson SR, Blokhin IO, Massey V, Gue JP, et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat Commun. 2019;10:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antonsen KK, Klausen MK, Brunchmann AS, le Dous N, Jensen ME, Miskowiak KW, et al. Does glucagon-like peptide-1 (GLP-1) receptor agonist stimulation reduce alcohol intake in patients with alcohol dependence: study protocol of a randomised, double-blinded, placebo-controlled clinical trial. BMJ Open. 2018;8:e019562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kruse Klausen M, Thomsen M, Wortwein G, Fink-Jensen A. The role of glucagon-like peptide 1 (GLP-1) in addictive disorders. Br J Pharmacol. 2022;179:625–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mueller L, Moser M, Prazak J, Fuster DG, Schefold JC, Zuercher P. Metformin’s role in hyperlactatemia and lactic acidosis in ICU patients: a systematic review. Pharmacology. 2023:1–11. doi: 10.1159/000528252. [Online ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ishtiaq SM, Arshad MI, Khan JA. PPARgamma signaling in hepatocarcinogenesis: mechanistic insights for cellular reprogramming and therapeutic implications. Pharmacol Ther. 2022;240:108298. [DOI] [PubMed] [Google Scholar]
- 104.Kremoser C FXR agonists for NASH: How are they different and what difference do they make? J Hepatol. 2021;75:12–5. [DOI] [PubMed] [Google Scholar]
- 105.Rinella ME, Trotter JF, Abdelmalek MF, Paredes AH, Connelly MA, Jaros MJ, et al. Rosuvastatin improves the FGF19 analogue NGM282-associated lipid changes in patients with non-alcoholic steatohepatitis. J Hepatol. 2019;70:735–44. [DOI] [PubMed] [Google Scholar]
- 106.Harrison SA, Neff G, Guy CD, Bashir MR, Paredes AH, Frias JP, et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2021;160:219–231.e1. [DOI] [PubMed] [Google Scholar]
- 107.Loomba R, Noureddin M, Kowdley KV, Kohli A, Sheikh A, Neff G, et al. Combination therapies including cilofexor and firsocostat for bridging fibrosis and cirrhosis attributable to NASH. Hepatology. 2021;73:625–43. [DOI] [PubMed] [Google Scholar]
- 108.Hartmann P, Hochrath K, Horvath A, Chen P, Seebauer CT, Llorente C, et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology. 2018;67:2150–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Desai BN, Singhal G, Watanabe M, Stevanovic D, Lundasen T, Fisher FM, et al. Fibroblast growth factor 21 (FGF21) is robustly induced by ethanol and has a protective role in ethanol associated liver injury. Mol Metab. 2017;6:1395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wagner-Skacel J, Horvath A, Grande P, Wenninger J, Matzer F, Fazekas C, et al. Association of fibroblast growth factor 21 with alcohol consumption and alcohol liver cirrhosis. Neuropsychiatr. 2021;35:140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR2/5 signaling prevents and reverses alcohol-induced liver damage, steatosis, and inflammation in mice. Hepatology. 2019;69:1105–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol. 2020;72:816–27. [DOI] [PubMed] [Google Scholar]
- 113.Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic steatohepatitis. Gastroenterology. 2018;155:1140–53. [DOI] [PubMed] [Google Scholar]
- 114.Slack RJ, Macdonald SJF, Roper JA, Jenkins RG, Hatley RJD. Emerging therapeutic opportunities for integrin inhibitors. Nat Rev Drug Discov. 2022;21:60–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Verdelho Machado M, Diehl AM. Role of hedgehog signaling pathway in NASH. Int J Mol Sci. 2016;17:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blumenthal DM, Gold MS. Neurobiology of food addiction. Curr Opin Clin Nutr Metab Care. 2010;13:359–65. [DOI] [PubMed] [Google Scholar]
- 117.Tufvesson-Alm M, Shevchouk OT, Jerlhag E. Insight into the role of the gut-brain axis in alcohol-related responses: emphasis on GLP-1, amylin, and ghrelin. Front Psychiatry. 2022;13:1092828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Klausen MK, Jensen ME, Moller M, Le Dous N, Jensen AO, Zeeman VA, et al. Exenatide once weekly for alcohol use disorder investigated in a randomized, placebo-controlled clinical trial. JCI Insight. 2022;7:e159863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Arab JP, Izzy M, Leggio L, Bataller R, Shah VH. Management of alcohol use disorder in patients with cirrhosis in the setting of liver transplantation. Nat Rev Gastroenterol Hepatol. 2022;19:45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]