

Abstract
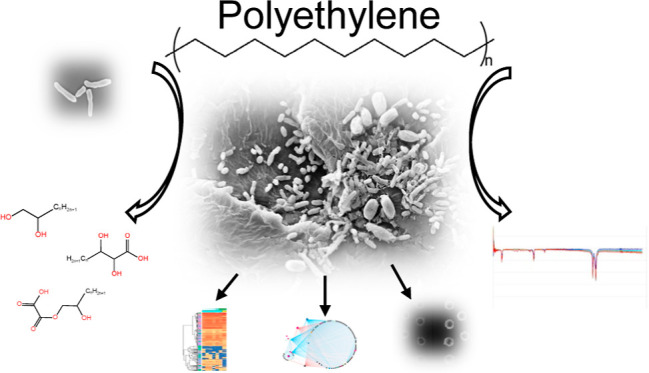
Polyethylene (PE) is the most widely produced synthetic polymer and the most abundant plastic waste worldwide due to its recalcitrance to biodegradation and low recycle rate. Microbial degradation of PE has been reported, but the underlying mechanisms are poorly understood. Here, we isolated a Rhodococcus strain A34 from 609 day enriched cultures derived from naturally weathered plastic waste and identified the potential key PE degradation enzymes. After 30 days incubation with A34, 1% weight loss was achieved. Decreased PE molecular weight, appearance of C–O and C=O on PE, palmitic acid in the culture supernatant, and pits on the PE surface were observed. Proteomics analysis identified multiple key PE oxidation and depolymerization enzymes including one multicopper oxidase, one lipase, six esterase, and a few lipid transporters. Network analysis of proteomics data demonstrated the close relationships between PE degradation and metabolisms of phenylacetate, amino acids, secondary metabolites, and tricarboxylic acid cycles. The metabolic roadmap generated here provides critical insights for optimization of plastic degradation condition and assembly of artificial microbial communities for efficient plastic degradation.
Keywords: polyethylene biodegradation, plastic-degrading enzyme, Rhodococcus sp., proteomics, proteomic network analysis
Short abstract
Minimal research exists on in-depth exploration of the mechanisms underlying the microbial polyethylene (PE) degradation process. This study offers crucial insights into key enzymes and metabolic pathways for optimized PE degradation and waste management strategies.
Introduction
Due to its durability, convenience in use especially for disposable and individual packaging, and stability under the natural setting, annual worldwide plastic production has been rapidly and continuously growing since its introduction in the 1950s. The annual production worldwide has increased over 200 folds since then and reached >390 million metric tons in 2021 (https://www.statista.com/statistics/282732/global-production-of-plastics-since-1950/), and the cumulative world production of plastic has reached 9.5 billion tons. The resulting waste accumulation has been an environmental issue of great concern. However, there are no ideal methods so far for managing this growing plastic waste. From 1950 to 2015, about 55% of the global plastic wastes was landfilled, 30% is still in use, 8% was incinerated, and 6% was recycled (https://ourworldindata.org/plastic-pollution#how-do-we-dispose-of-our-plastic). Among different types of plastics, high density polyethylene (HDPE) and polyethylene terephthalate (PET) are the plastic types with the highest recycling rates. Incineration of plastic wastes produces toxic byproducts and greatly contributes to the already problematic climate change. Simply discarding them in landfill is also not sustainable, and it takes up to a few hundred years for plastic to degrade in natural environment. Fortunately, recent research studies have shown that microorganisms, including bacteria and fungi as well as insects and the associated gut microorganisms, can degrade various types of plastics such as polyethylene (PE), polystyrene (PS), polypropylene (PP), polyvinyl chloride (PVC), polyurethane (PUR), and PET.1−3 Despite the increasing number of plastic-degrading environmental isolates uncovered so far, only a very small number of enzymes have been identified that can degrade plastic or are associated with plastic degradation. Specifically, degradation of PET by a bacteria strain Ideonella sakaiensis 201-F6 and the associated key enzyme PETase have been extensively studied.4,5 Recent multi-omics study of a bacterial strain Klebsiella sp. provided evidence of function of catalase-peroxidase in PVC degradation.6 However, in most cases plastic-degrading enzymes or those associated with degradation are yet to be identified.
PE is the most abundant plastic waste globally, and its recalcitrance to biodegradation is largely due to its high-molecular weight and hydrophobicity. Introduction of oxygen into the alkane structure and depolymerization are the two key limiting steps for its biodegradation. Over the past two decades, bacteria and fungi strains and a few enzymes associated with biodegradation of PE have been reported.1,7 However, the PE biodegradation rates, measured as weight loss (%), varied considerably (∼1.8 to 36%) due to the differences of PE materials, incubation length, and characterization methods.8 Key enzymes, especially those in the first few steps of PE oxidation and depolymerization, are still unknown. Transcriptomic analysis of Rhodococcus opacus R7 uncovered the potential key genes involved in depolymerization and oxidation of PE such as alkane monooxygenase and cytochrome P450 hydroxylase.9 Comparative genomics analysis of Rhodococcus genomes indicated the distribution of these potential key plastic degradation-associated genes in Rhodococcus genomes.10 Despite these findings, in-depth studies on PE-degrading microorganisms and the underlying plastic degradation mechanisms are still lacking.
In this study, we hypothesize that plastic-degrading bacteria attached to naturally weathered plastic waste can be enriched in the laboratory in carbon-free basal medium (CFBM) using plastic waste as the sole carbon source and subsequently isolated from the enrichment cultures. Toward the goal of uncovering PE-degrading microorganisms and the key enzymes in PE degradation, we isolated 87 bacterial strains from the 609 day enrichment cultures. Among these, strain A34 had high lipase activity and no growth in CFBM. It had high relative abundance (4.3%) in the microbial community and was identified as Rhodococcus strain based on 16S rRNA sequence. The genes potentially involved in PE degradation were annotated in its genome sequences. Time series proteomics analysis of A34 in CFBM with PE powder as the sole carbon source demonstrated induction of enzymes involved in PE oxidation and depolymerization such as catalase-peroxidase KatG, multicopper oxidase, hydrolase, monooxygenase, lipase, esterase, alcohol dehydrogenase, and aldehyde dehydrogenase. Network analysis of the proteomics data also demonstrated the close relationships between oxidation of PE, fatty acids β oxidation, as well as metabolisms of phenylacetate, amino acids, and secondary metabolites. Finally, a metabolic pathway for PE biodegradation was proposed.
Results
Isolation and Characterization of Bacteria Strain A34
Enrichment cultures were established by incubating naturally weathered plastic in CFBM (Figure 1a). Bacterial isolates were obtained from enrichment cultures after 609 days of incubation. Lipase activity assay was conducted as the initial screening of the potential PE-degrading bacterial clones since lipase was considered associated with plastic degradation.11 Over 70% of the tested bacterial isolates showed lipase activities. Among these, one strain A34 demonstrated strong lipase activity (Figure 1b). This A34 cell was rod-shaped with about 2 μm in length (Figure 1c). The size of the A34 colonies was small and was stained as Gram positive, and the optimal growth temperature was 30 °C on tryptic soy agar (see Figure S1, Supporting Information). When grown in liquid media containing varying levels of nutrients at 30 °C (Figure 1d), A34 grew well in rich medium such as tryptic soy broth (TSB), Luria–Bertani broth (LB), and half LB with or without supplementation of glucose. The final biomass yield decreased about 50% in R2A, a relatively nutrient-poor medium. In CFBM supplemented with yeast extract (YE, 0.01%) and glucose (0.05%), no growth was observed, suggesting that glucose is not a good carbon source for A34. As CFBM was the medium for enrichment cultures and the sole carbon source was plastic, no growth of A34 in CFBM + 0.01% YE + 0.05% glucose suggested that A34 had the potential of using plastic as a sole carbon source.
Figure 1.

Isolation and physiological and morphological characterization of Rhodococcus strain A34. (a) Original plastic waste, enrichment culture, and the associated microbial communities on the plastic surface. (b) Lipase activity of strain A34. E. coli was used as negative control. (c) Morphology of A34 cells under a scanning electron microscope. (d) Growth of A34 in liquid media with various levels of nutrients.
Full-length 16S rRNA amplicon sequencing and NCBI blast demonstrated that A34 was closely related (nucleotide sequence identity 100%, 1371 bp) to Rhodococcus qingshengjii strains CCM4446, dji-6, and dji-6-2, which were isolated from soils contaminated by fungicide carbendazim or organic pollutants,12 or carbendazim-contaminated wastewater treatment facility.13Rhodococcus strains have been reported to possess plastic degradation capability,9,14,15 but only limited number of whole genome sequences was available in NCBI, and genome sequences of other known plastic-degrading bacterial strains were collected and compared with A34 genome sequence to uncover the genome-level similarities and differences. The genome-level phylogenetic tree (see Figure S2a, Supporting Information) revealed that strain A34 is closely related to Rhodococcus ruber, a previously reported plastic degrading strain.14 The plastic waste type in the enrichment culture was LDPE based on Fourier-transform infrared spectroscopy (FTIR) spectrum (see Figure S3a, Supporting Information). The microbial community composition in the enrichment was relatively simple and dominated by Proteobacteria (41.46%), Firmicutes (29.71%), Actinobacteriota (15.86%), and Gemmatimonadota (8.25%) (see Figure S3b, Supporting Information). Genus level abundance of Rhodococcus was 4.3% (see Figure S3c, Supporting Information). Together with the growth phenotype of A34, the plastic type in the enrichment culture, and the close phylogenetic relationship with plastic degrading Rhodococcus strains suggested that A34 could be a promising PE degrader.
Genetic Potential of A34 in PE Degradation
The genome sequence of A34 was obtained via Illumina Hiseq. A total of 433 contigs (152–845,194 bp) were obtained from the qualified reads processed from 11,817,076 raw 150 bp reads. The estimated genome size was 7.44 Mb with GC content 62.2%. A total of 7525 open reading frames were annotated in A34 genome. Among these, 3080 genes, representing 41% of the genome, were hypothetical proteins or uncharacterized proteins, suggesting the complexity and unknown potential of the genome. Among genes with annotated function, 201 genes in oxidation and depolymerization of PE, 133 genes in fatty acid β-oxidation, 366 genes in transportation such as ATP-binding cassette (ABC) transporter, major facilitator superfamily-type transporter, and mammalian cell entry (MCE)-family protein, 462 transcriptional regulators and two-component system genes, 166 oxidoreductase genes, and 81 genes encoding mobile element protein or transposase were annotated. The gene annotation results demonstrated the genetic potential of A34 in PE degradation as well as the versatility of its metabolism (see Figure S2b, Supporting Information).
Degradation of PE Powder by A34
To assess the plastic degradation capability of A34, a biodegradation test was performed with commercially available PE powder (Figure 2a). After 30 days of incubation at 30 °C, the average weight loss of PE in the treatment group (T1–T4, 0.0421 ± 0.0028 g) was significantly higher than that of control group (C1–C4, 0.0375 ± 0.0019 g) (p < 0.0001, unpaired t-test) (Figure 2b). The weight loss in the control group represents the loss of PE powder during the PE collection, washing, and weighing procedures. The net weight loss was about 5 mg in the treatment group (T), about 1% weight loss considering the initial weight of PE (500 mg). This was comparable to previously reported PE degradation efficiencies by bacteria pure cultures with known absolute weights of PE at both the beginning and end of incubation with16 or without pre-treatment17 over a 30 day incubation period. Both the weight average molecular weight (Mw) and the number average molecular weight (Mn) decreased significantly, with about 45% reduction of Mn (p = 0.024) and 23% reduction of Mw (p = 0.008) (unpaired t-test) in the treatment group compared to the control group (Figure 2c). The top molecular mass and starting molecular mass of PE powders in treatment samples were significantly decreased compared to control samples (see Figure S4, Supporting Information). The HT-GPC results indicated the depolymerization of PE powder.
Figure 2.

Degradation of PE powder by Rhodococcus strain A34. (a) Experiment flowchart. PE weight loss and molecular-weight changes after 30 days of incubation are shown in (b,c), respectively. T, with A34 (T1–T4); C, without A34 (replicates C1–C4). (d) FTIR spectrum showed PE functional group changes in the T group compared to the C group and untreated PE. (e) Unique GC–MS peak was detected in the T group. (f) Unique GC–MS peak was identified as palmitic acid compared to the NIST library data and experimental data of standard palmitic acid. (g) PE surface changes after 30 days of incubation with A34 (T1–T4) or without A34 (C1–C4).
The functional group changes of the PE powder at the end of incubation period were examined by FTIR. Compared to the untreated PE and C groups, C–O stretch at wavenumber 1196 cm–1 and C=O stretch at wavenumber 1730 cm–1 were observed in the treatment samples (Figure 2d), indicating the incorporation of oxygen in the polymer. Furthermore, gas chromatography–mass spectrometry (GC–MS) of the supernatant samples collected from the treatment (T) and control (C) groups demonstrated a product at retention time of ∼27.49 min was more abundant in the T group compared to the C group. The product was identified as palmitic acid (CH3(CH2)14COOH) based on the match probability (68.5%) in the NIST library and the identical retention time to the palmitic acid standard (Figure 2e,f). Moreover, SEM images showed pronounced pits on the surfaces of the PE powder in the T group samples, in contrast to the smooth surfaces in the C group samples (Figure 2g). The PE weight loss, reduction in molecular weight, functional group changes, small-molecule products in supernatant, as well as surface alternations together provided solid evidence of PE degradation by A34.
Enzymes Involved in PE Degradation and Other Cellular Pathways
To identify the key enzymes involved in PE degradation and the active metabolic pathways, a time series proteomics experiment using PE as the sole carbon source was conducted (Figure 3a). A total of 2989 proteins were detected across all samples at all timepoints. Out of these, 2624 proteins, detected in two or more replicate samples at each timepoint, were retained for further analysis. PCA analysis of the proteomes revealed the significant differences between intracellular proteomes [0 h (T0), 3 h (T3), 6 h (T6), 12 h (T12), 24 h (T24), and 48 h (T48)] and extracellular proteomes [proteins in the supernatant at 48 h (T48S)], and dramatic changes in T3 and T6 were compared to T0 (Figure 3b). The results indicate the acute responses of proteomes upon transition from nutrient-rich condition (TSB) to nutrient-poor condition (CFBM) where PE was the sole carbon source. Compared to T0, the numbers of intracellular protein were significantly higher at T3 (p = 0.007), T6 (p = 0.031), and T24 (p = 0.007), less at T48S (p = 0.018) (unpaired t-test) (Figure 3c). The number of proteins with significantly increased (up)/decreased (down) abundances compared to T0 were 154/43 in T3, 31/11 in T6, 6/5 in T12, 84/35 in T24, 0/0 in T48, and 58/176 in T48S, respectively, and 106/356 in comparison of T48S vs T48 (see Figure S6, Supporting Information).
Figure 3.

Proteins/enzymes responsive to the change of carbon source to PE powder in medium and the dynamic changes of proteomes over time. (a) General workflow of the time series proteomic incubation experiment. (b) PCA plot of the overall similarities of proteomes at different timepoints. (c) Numbers of proteins detected at each timepoints.
We hypothesize that enzymes involved in PE oxidation and depolymerization are secreted out of the cells to access the high-molecular-weight PE polymers in the supernatant. Among 1334 proteins in supernatant samples (T48S), 222 proteins were not detected at neither T0 nor T48 samples, indicating that these proteins could be induced and secreted out of the cells when PE was the sole carbon source, potentially playing important roles in PE oxidation and depolymerization. Examples of the extracellular proteins included multicopper oxidase, lipase, esterase, alcohol dehydrogenase, long-chain-fatty-acid-CoA ligase, lipid transporter MCE-family lipoprotein, etc. (Figure 4a; see Figure S6, Supporting Information). Among these proteins, only one putative esterase peg 6607 was present at time zero supernatant but not intracellular samples. At time zero, only 192 proteins were detected in supernatant samples (see Figure S5, Supporting Information). Protein domain analysis of the multicopper oxidase indicated the existence of three Cu-oxidase domains, suggesting its potential function in PE oxidation as laccase.18,19
Figure 4.

Temporal protein abundance (log 2 transformed) changes of A 34 are shown in the heatmaps [(a) *, PE oxidation; #, PE depolymerization; β, fatty acid β-oxidation]. (b) Networks generated from the temporal proteomics data were partitioned into 20 module. Positive or negative correlations between nodes were shown in blue or red, respectively. Color key for the nodes: A, oxidation of fatty acids or biosynthesis of fatty acids; B, metabolism of amino acid and secondary metabolites; C, metabolism of amino acids; D, metabolism of the secondary metabolites; E, TCA cycle; F, ABC transporter; G, protein degradation; H, purine metabolism; I, regulatory protein; J, sugar metabolism; K, DNA modification; and X, other pathways or hypothetical protein. (c) Protein abundance changes of the interested enzymes in each module (module # is shown in the left of the heatmap).
In addition to the extracellular enzymes, enzymes involved in the downstream steps of PE degradation and central metabolic pathways were also detected in high abundances in both intra- and extracellular protein extracts (Figure 4a). For instance, 19 long-chain-fatty-acid-CoA ligase, catalyzing the conversion of PE degradation product fatty acids to fatty acyl-CoA, was transported into the cells and entered fatty acid β-oxidation, 72 enzymes involved in all steps of β-oxidation. Both acyl-CoA oxidase and acyl-CoA dehydrogenase, converting fatty acyl-CoA to enoyl-CoA in the first step of β oxidation, were detected, indicating the presence of both pathways in A34. For acyl-CoA dehydrogenase, two very-long-chain acyl-CoA dehydrogenase (EC1.3.8.9), three medium-chain acyl-CoA dehydrogenase (EC1.3.8.8), and eight short-chain acyl-CoA dehydrogenase (EC1.3.8.1) were detected.
Enzymes catalyzing the reactions from the β oxidation intermediate, β-hydroxyacyl-CoA, to polyhydrohydroxyalkanates (PHAs) were highly abundant. Examples of other enzymes include those for fatty acid biosynthesis, production of unsaturated fatty acids, biosynthesis of pantothenate (vitamin B5, essential for synthesis of CoA) from α, β-dihydroxyisovalerate, and from pantothenate to CoA. All enzymes, except 3-oxoadipyl-CoA catalyzing the last step in phenylacetate catabolism, were present. Aromatic compounds are metabolized by bacteria through phenylacetate followed by oxidization, isomerization, hydrolytic ring cleavage, and β-oxidation.20 This indicates the metabolic potential of A34 in aromatic compound biodegradation.
All enzymes involved in the tricarboxylic acid (TCA) cycle and the associated bypass pathways such as pyruvate cycle, glyoxylate cycle, and γ-aminobutyric acid (GABA) shunt, were present. The pyruvate cycle operates routinely for energy production and regulation.21 The glyoxylate cycle allows cells to use two carbons compounds for biosynthesis of carbohydrates when simple sugars such as glucose or fructose are unavailable.22 The GABA shunt is important in carbon and nitrogen metabolism and stress responses in bacteria.23
For glycolysis and gluconeogenesis pathways, all enzymes except hexokinase and glucose 6-phosphatase, which catalyzes the interconversion between glucose and glucose 6-phosphate, were detected. Consistently, A34 could not grow with 3 mM glucose as the sole carbon source (Figure 1d). In contrast, all enzymes in the pentose phosphate pathway (PPP), a fundamental shunt important for carbon homoeostasis and biosynthesis of nucleotide and amino acid, were detected. Additionally, enzymes involved in biosynthesis of amino acids such as lysine via succinylase pathway, serine, glycine, tryptophan, cysteine, alanine, valine, leucine, isoleucine, and threonine were detected with high abundance.
Networks Built from Proteomics Data
To uncover the relationships between enzymes involved in PE degradation pathways and other cellular pathways, networks were generated based on the changes of protein abundance over time and the Pearson correlation coefficient between each pair of nodes (protein). A total of 20 modules including 212 nodes were obtained with Pearson correlation coefficient cutoff at 0.97. We focused on the analyses of modules with more than three nodes (modules 1 to 12, Figure 4b,c).
The close relationships between PE oxidation and degradation and other metabolism pathways were demonstrated in seven modules. In module 1, PE oxidation enzymes were positively related to enzymes involved in phenylacetate metabolism, fructose, and mannose metabolism and TCA. Examples of enzymes included fatty acid oxidation enzymes peg 6219 (long-chain-fatty-acid-CoA ligase) and peg 2407 (Acyl-CoA thioesterase II), phenylacetate metabolism enzyme peg 3820 (1,2-phenylacetyl-CoA epoxidase), TCA cycle enzyme peg 1042 (succinate dehydrogenase), and fructose and mannose metabolism enzyme peg 7101 (GDP-mannose 4,6-dehydratase). Phenylacetate metabolism enzyme peg 3820 was linked to fatty acid oxidation enzymes peg 6420 (enoyl-CoA hydratase/isomerase) and peg 6419 (acyl-CoA dehydrogenase) in module 12. In module 2, three phenylacetate metabolism enzymes peg 3822, peg 3823, and peg 3824 were closely related to peg 7200, a methylmalonyl CoA epimerase involved in the catabolism of odd number carbon chain fatty acids. Other enzymes positively related to the hub peg 7200 included peg 1394 (dihydroxy-acid dehydratase), a hydrolase that cleaves carbon-oxygen bonds in PE oxidation and also participates in biosynthesis of valine, leucine, isoleucine, pantothenate, and coenzyme A (CoA), and an oxidoreductase peg 1160 (linoleoyl-CoA desaturase) involved in the conversion between saturated and unsaturated fatty acids. Module 6 indicated the negative relationships between PE oxidation enzyme peg 2908 (an aldehyde dehydrogenase) and an oleate hydratase peg 2097 that cleaves carbon-oxygen bonds in fatty acids, and other enzymes involved in metabolisms of secondary metabolites. Links in module 8 indicated the positive relationship between PE oxidation enzymes alcohol dehydrogenases peg 5134 and peg 5557 and enzymes involved in DNA modification. Links in module 9 indicate the positive relationships between PE oxidation and degradation enzymes peg 5123 (aldehyde dehydrogenase), peg 5089 (alcohol dehydrogenase), peg 3772 (long-chain-fatty-acid-CoA ligase), and peg 5113 (FMN-dependent dehydrogenase) and enzymes involved in metabolisms of amino acids or secondary metabolites. Links in module 10 indicated the close relationship between PE oxidation and degradation enzymes including peg 1577 (alcohol dehydrogenase), peg 3210 (secretory lipase), and peg 196 (fatty acid desaturase) as well as enzymes involved in other cellular pathways such as metabolism of amino acids.
Links in other modules indicated the close relationship between different cellular pathways. For instance, module 3 demonstrated the close relationship between enzymes involved in purine biosynthesis, metabolism of amino acids, and/or secondary metabolites. Module 4 indicated the close relationships between protein synthesis and multiple cellular pathways such as fatty acids β oxidation, TCA, and protein degradation. Module 5 contained negative links between peg 5920 (acyl-CoA carboxylase-like protein), that is potentially involved in biosynthesis of fatty acids, and enzymes involved in amino acid metabolism including a regulator GarA (peg 2735) regulating glutamate metabolism. Module 11 indicated relationships between an enzyme involved in fructose and mannose metabolism (peg 6928, GDP-mannose 4,6-dehydratase) and a regulatory protein peg 603, and an enzyme involved in RNA metabolism (peg 6828, DEAD-like helicase).
In conclusion, the network data provided evidence for the close relationships between different cellular pathways, especially between “PE oxidation and degradation” and “metabolism of phenylacetate, amino acids, purine, and secondary metabolites”.
Metabolic Pathways of A34 in Carbon-Free Basal Medium with PE as a Carbon Source
Based on the time series proteomics, FTIR, and GC–MS data, a conceptual model of PE degradation and the central metabolic pathways in A34 with PE as the sole carbon sources is proposed (Figure 5). In this model, a series of extracellular enzymes, including catalase-peroxidase KatG, multicopper oxidase (laccase), monooxygenase, lipase, and esterase, introduce oxygen into the hydrocarbon chain at either the polymer chain terminus (exo-attack) or somewhere along the polymer chain (endo-attack) and de-polymerize PE to lower-molecular-weight polymers. We induced protein expression of two abundant extracellular proteins (a multicopper oxidase peg 1726 and an esterase peg 6607) (Figure 4a) in E. coli host cells and incubated PE powder with crude enzymes for 1 week. PE functional group changes (appearance of C–O stretching ester and C–O stretch alcohol) (Figure S7) were observed in PE powders incubated with both peg 1726 and peg 6607 but not with either peg 1726 or peg 6607. The depolymerized products are further oxidized by alcohol dehydrogenase and aldehyde dehydrogenase. Subsequently, the long-chain fatty acids are converted to fatty acyl-CoA and transported into the microbial cells via lipid transporter MCE-family lipoprotein. Fatty acyl-CoA is then completely degraded via β oxidation and TCA cycle, providing materials for biosynthesis of amino acids, secondary metabolites, fatty acids, etc. FTIR results of the PE powder demonstrated the formation of C–O and C=O, and carboxylic acid [palmitic acid (C16H32O2)] was detected in the supernatant samples. Consistent to the lack of growth with glucose as a carbon source, genes for the phosphotransferase system for glucose, hexokinase, and glucose 6-phosphatase catalyzing the interconversion between glucose and glucose 6-phosphate are absent in the A34 genome. All other enzymes involved in glycolysis and glycogenolysis, as well as all enzymes for PPP, were found in high abundance in proteomics data, showing the feasibility of our hypothesized biodegradation pathway.
Figure 5.

Conceptual model of metabolic pathways of A34 when grown in defined medium with PE as the carbon source. The pathways are illustrated based on the proteomics data, FTIR, and GC–MS data. Solid arrow represents one-step reaction; broken-line arrow represents the absence of the corresponding enzyme in proteomics data or unknown enzymes. The initial PE depolymerization step indicated by a broken-line arrow might involve multiple enzymes, such as multicopper oxidase, catalase peroxidase, esterase, lipase, and others. PE functional group changes were observed after incubation with crude enzymes of combination of a multicopper oxidase peg 1726 and an esterase peg 6607 (Figure S7). The orange frame highlighted the two types of extracellular key PE degradation enzymes. Color key for intracellular pathways: light brown, fatty acid β-oxidation; fatty acid biosynthesis, phenylacetate metabolism, biosynthesis of pantothenate, and CoA; light green, TCA cycle and the associated pyruvate cycle, glyoxylate cycle, and GABA skanehunt; light red, glycolysis, glycogenolysis, and pentose phosphate pathway; and light blue, biosynthesis of amino acids and secondary metabolites. PE oxidation and depolymerization enzymes: alkane hydroxylase* (genes exist but protein not detected); AD, alcohol dehydrogenase; ALD, aldehyde dehydrogenase; enzymes in fatty acid β-oxidation: 1, acyl-CoA oxidase or acyl-CoA dehydrogenase; 2, enoyl-CoA dehydratase; 3, 3-hydroxyacyl-CoAcdehydrogenase; and 4, β-ketoacyl-CoA thiolase. Enzymes in biosynthesis of pantothenate and CoA: 5, dihydroxy-acid dehydratase; 6, valinepyruvate aminotransferase; 7, ketopantoate hydroxymethyltransferase; 8, ketopantoate reductase PanG or ketol-acid reductoisomerase [NADP(+)]; 9, pantothenate synthetase; 10, pantothenate kinase; 11, phosphopantothenoylcysteine synthetase; 12, phosphopantothenoylcysteine decarboxylase; 13, phosphopantetheine adenylyltransferase; and 14, dephospho-CoA kinase. Enzymes in phenylacetate metabolism: 15, phenylacetate-coenzyme A ligase; 16, 1,2-phenylacetyl-CoA epoxidase; 17, 1,2-epoxyphenylacetyl-CoA isomerase; 18, 2-oxepin-2(3H)-ylideneacetyl-CoA hydrolase; 19, 3-oxo-5,6-dehydrosuberyl-CoA semialdehyde dehydrogenase; 20, 3-hydroxyacyl-CoA dehydrogenase; 21, 3-oxoadipyl-CoA those. Enzymes in fatty acid biosynthesis: 22, acetyl-coenzyme A carboxylase; 23, malonyl CoA-ACP acyltransferase; 24, 3-oxoacyl-ACP synthase of FASI, KASI, or KASII; 25, 3-oxoacyl-ACP reductase; 26, β-hydroxyacyl-ACP dehydratase; and 27, enoyl-ACP reductase. Enzymes in PHA biosynthesis: PhaC, PHA synthase. Enzymes in the TCA cycle and associated pyruvate cycle, glyoxylate cycle, and GABA shunt: PDH, pyruvate dehydrogenase; CS, citrate synthase; ACO, aconitate hydratase; ICDH, isocitrate dehydrogenase; OGDH, 2-oxoglutarate dehydrogenase; SCS, succinyl coenzyme A synthetase; SDH, succinate dehydrogenase; FUM, fumarate hydratase; MDH, malate dehydrogenase; PEPCK, phosphoenolpyruvate carboxykinase; PEPC, phosphoenolpyruvate carboxylase; PK, pyruvate kinase; MAE, NAD-dependent malic enzyme; ICL, isocitrate lyase; MS, malate synthase G; GOGAT, glutamate synthase; GDC, glutamate decarboxylase; GAT, GABA aminotransferase; and SSADH, succinic semialdehyde dehydrogenase. Enzyme in glycolysis, gluconeogenesis, and the pentose phosphate pathway: 28, glucose-6-phosphate isomerase; 29, 6-phosphofructokinase; 30, fructose-bisphosphate aldolase; 31, triosephosphate isomerase; 32, glyceraldehyde-3-phosphate ketol-isomerase; 33, phosphoglycerate kinase; 34, phosphoglycerate mutase; 35, enolase; 36, fructose-1,6-bisphosphatase; 37, glucose-6-phosphate dehydrogenase; 38, 6-phosphogluconolactonase; 39, 6-phosphogluconate dehydrogenase; 40, ribose-5-phosphate isomerase; and 41, transketolase. Enzymes in biosynthesis of amino acids: l-threonine, l-isoleucine, l-valine, and leucine: AST, aspartate aminotransferase; AK, aspartate kinase; ASD, aspartate-semialdehyde dehydrogenase; HD, homoserine dehydrogenase; HK, homoserine kinase; TS, threonine synthase; TD, threonine dehydratase; AHAS, acetolactate synthase; AHAIR, acetohydroxy acid isomeroreductase; DHAD, dihydroxy-acid dehydratase; TA, branched-chain amino acid aminotransferase; IPMS, isopropylmalate synthase; IPMI, α-isopropylmalate isomerase; IPMD, α-isopropylmalate dehydrogenase; tryptophan: 42, anthranilate synthase; 43, anthranilate phosphoribosyltransferase; 44, phosphoribosylanthranilate isomerase 45, tryptophan synthase; l-serine, glycine, and l-cysteine: 46, d-3-phosphoglycerate dehydrogenase; 47, phosphoserine aminotransferase, 48, phosphoserine phosphatase; 49, serine hydroxymethyltransferase; 50, serine acetyltransferase; 51, cysteine synthase; l-lysine: 52, aspartokinase; 53, aspartate-semialdehyde dehydrogenase; 54, 4-hydroxy-tetrahydrodipicolinate synthase; 55, 4-hydroxy-tetrahydrodipicolinate reductase; 56, 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase; 57, N-succinyl-l,l-diaminopimelate aminotransferase; 58, N-succinyl-l,l-diaminopimelate desuccinylase; 59, diaminopimelate epimerase; 60, diaminopimelate decarboxylase; l-valine, 61, alanine dehydrogenase; 62, alanine transaminase; and 63, alanine racemase.
Discussion
In pursuit of isolating PE-degrading bacteria and identifying key PE degradation enzymes, a long-term enrichment experiment using CFBM was established from naturally weathered plastic waste. Among the bacterial strains isolated from the 609 day enrichment cultures, a lipase-positive Rhodococcus strain (named A34 strain here) was obtained. Consistent to the existence of potential PE-degrading enzyme genes in Rhodococcus stains based on genome-based exploration,10 the A34 strain genome had abundant potential PE oxidation and degradation genes. Further time series proteomics data demonstrated the induction and high abundances of extracellular key PE oxidation and depolymerization enzymes such as catalase-peroxidase KatG, multicopper oxidase, lipase, and esterase. Along with the plastic functional group changes such as the appearance of C–O and C=O bonds and presence of palmitic acid in the supernatant, the results provided evidence for the roles of these enzymes in PE degradation and oxidation via both exo-attack at the polymer chain terminus and endo-attack along the polymer chain.24
A few studies have demonstrated the role of PE oxidation/degradation enzymes in PE degradation,2,18,19,25 and it has been proposed that lipase and esterase are involved in PE degradation, especially in degradation of intracellular short-chain-fatty-acids.2 However, evidence at the proteomics level was lacking. In this study, with the time series proteomics experiment design including both intracellular and extracellular protein samples, 12 esterase (four in intra- and extracellular, seven in extracellular only, and one at 0 h only) and three lipases (two in both intra- and extracellular and one in extracellular only) were detected, indicating some lipase and esterase function in degradation of high-molecular-weight PE outside the microbial cells. The multicopper oxidase (laccase) was identified exclusively in the extracellular portion, indicating its important role in PE oxidation and depolymerization as reported in PE degradation by R. ruber C20819 or fungi.26 In addition, nine MCE-family lipoproteins, which have been suggested as the potential lipid ABC transporters,27 were detected in high abundance with seven in both intra and extra-cellular and two in extracellular sample only.
Analysis of the time series proteomics data uncovered the comprehensive metabolic pathways of A34 when using PE as the sole carbon source. These pathways included the complete fatty acid β oxidation and fatty acid biosynthesis, TCA cycle, and the associated pyruvate cycle, glyoxylate cycle, and GABA shunt, amino acid biosynthesis pathway, PPP, nearly complete glycolysis and gluconeogenesis pathways, and phenylacetate catabolism pathway. In addition, many transcriptional regulators (160) and two-component system transcriptional response regulators (12) were detected in high abundances. Among these, 61 transcriptional regulators and four two-component system response regulators were induced by shifting from rich medium to CFBM with PE as the sole carbon source. AcrR/TetR family transcriptional regulators (31) accounted for half of the induced transcriptional regulators. AcrR/TetR family transcriptional regulators play important roles in regulating genes involved in metabolism, efflux pump, osmotic stress, antibiotic production, and pathogenesis.28
In summary, the metabolic roadmap constructed in the newly isolated Rhodococcus A34 holds crucial implications for the prospective assembly of artificial microbial communities and the refinement of plastic degradation conditions to optimize the plastic biodegradation efficiency. Notably, the identification of multiple extracellular proteins exclusively responsive to PE (Figure 4a) underscores the potential formation of an enzyme mixture (PEases) for effective PE depolymerization, as indicated by our crude enzyme incubation assay (Figure S7). To advance these perspectives, future endeavors include assessment of capability of A34 in degrading higher-molecular-weight PE and HDPE, other plastics such as PP and PS, and organic pollutants, alongside refining degradation conditions and enhancing the efficiency of the PEases through AI-driven or conventional protein engineering methodologies. Collectively, our study opens new avenues for comprehending bacterial PE degradation by elucidating critical enzymes and metabolic pathways integral to this process.
Materials and Methods
Establishment of Enrichment Cultures, Bacterial Strain Isolation, and Characterization
Naturally weathered plastic waste was harvested from a lakeside environment (Norman, OK, USA) and incubated in CFBM to enrich plastic degrading microorganisms. Bacterial strains were isolated by plating the enrichment cultures onto 1/2 LB plates. Detailed methods are described in the Supporting Information.
Lipase Activity Assay
The qualitative lipase activity assay was conducted by using agar plates supplemented with olive oil (Sigma-Aldrich, cat O1514) and rhodamine B (Sigma-Aldrich, cat R6626).29 Detailed methods are described in the Supporting Information.
Optimum Growth Medium
Six media containing different levels of nutrients were used, including LB broth, 1/2 LB broth, 1/2 LB + 0.1% glucose, TSB, Reasoner’s 2A (R2A), and CFBM + 0.01% YE + 0.05% glucose. Growth curves were generated using 100-well plates and BioScreen C Automated Growth Curve System with the culture volume of 300 μL per well and continuous shaking. The OD600 values were measured every 30 min for 48 h with four replicates per medium. Medium only wells were used as control (blank).
16S rRNA Gene Amplicon Sequencing and Whole Genome Sequencing
Detailed methods of 16S rRNA gene amplification and Sanger sequencing, whole genome sequencing library preparation, Illumina HiSeq, and sequence data analysis are described in the Supporting Information.
Genome-Level Phylogeny Analysis
The phylogenetic genome tree was constructed using the concatenated alignment of 120 ubiquitous single-copy proteins obtained from GTDB-Tk.30 The multiple sequence alignments containing spurious sequences and poorly aligned regions were filtered using trimAl.31 The phylogenetic tree was then reconstructed using FastTree31 with GTR + CAT parameters. Finally, the phylogenetic tree was visualized using the Interactive Tree of Life (iTOL, https://itol.embl.de/).32
PE Degradation Test and Characterization of PE Physical and Chemical Changes and Potential Degradation Products
PE powder (Sigma-Aldrich, cat: 427772-250G, 0.5 g per test) was used for the degradation test (Figure 2a). The physical and chemical changes in PE were determined using SEM and FTIR, and the potential PE degradation products were determined using GC–MS. Detailed methods are described in the Supporting Information.
Time Series Proteomics Analysis of A34 Using PE Powder as the Sole Carbon Source
The A34 strain was revived in rich medium TSB and washed with CFBM twice before resuspending in CFBM containing PE powder (Figure 3a). Detailed methods are described in the Supporting Information.
Network Analysis
The protein co-expression network was generated using the methodology outlined in our previous study.33 Briefly, normalized and log 2 transformed proteomic data from all time points were used to construct the network using the random matrix theory approach.34 A cut-off of the Pearson correlation coefficient of 0.97 was used to identify highly correlated gene pairs, which was determined by the network identification method.34 Submodules were then identified using fast greedy modularity optimization.35
Acknowledgments
This work was supported by the National Science Foundation (grant number 2132156 to A.Z., X.Z., and A.H.). We thank Justin Smolen and Yidan Shen and the Texas A&M University (TAMU) Laboratory for Synthetic-Biologic Interactions (RRID: SCR_022287) for the help of FTIR, Dr. Yohannes H. Rezenom at TAMU Laboratory for Biological Mass Spectrometry service, and collaboration facility for the help of GC–MS, and Dr. Peiran Wei at Soft Matter Facility for the help of HT-GPC at Texas A&M University. We thank Aaron Wang for help sampling the naturally weather plastic waste and Mu Peng for help setting up the enrichment culture.
Data Availability Statement
The whole genome sequence has been deposited in the NCBI database under accession number SAMN34156462. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Proteomics Identification Database) partner repository with the data set identifier PXD041981 and PXD044483.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.3c03778.
Detailed descriptions of materials and methods: bacterial strain isolation, morphological and physiological characterization; lipase activity assay; 16S rRNA gene amplicon sequencing and whole genome sequencing; PE powder degradation test; characterization of PE physical and chemical changes and potential degradation products; time series proteomics analysis of A34 using PE powder as the sole carbon source; heterologous protein expression and crude enzyme incubation experiment; morphology, gram staining, and optimal growth temperature of Rhodococcus strain A34; genome-level phylogeny of strain A34 and genetic potential of A34 genome; type of plastic and the microbial community compositions in the enrichment cultures; size distribution of PE powder significantly decreased in samples incubated with A34 (T) compared to samples without A34 (C) for 30 days; proteins detected in time zero samples of Rhodococcus strain A34 grown in rich medium TSB; volcano maps showing proteins with significant abundance changes at each timepoint compared to time zero or extracellular vs intracellular (T48S vs T48); and PE functional group changes after incubation with heterologous expressed crude enzymes (PDF)
Author Contributions
X.T., H.O., and A.Z. contributed equally to this work. A.Z. and X.T. designed the experiment. A.Z. isolated and characterized the bacteria strain. X.T. prepared the whole genome sequencing and proteomics experiment samples. H.O. performed FTIR and GC–MS experiments. A.Z., X.T., D.W., and C.P. analyzed the proteomics data. H.M. performed gram staining. J.S.M. participated in the plastic degradation experiment and crude enzyme incubation experiment. A.T.R. participated in proteomics sample preparation and crude enzyme incubation experiment. D.M. and X.T. performed the genome level phylogeny analysis. A.Z., X.T., and H.O. analyzed the data and wrote the manuscript. X.Z., A.H. and J.Z. provided technical guidance.
The authors declare no competing financial interest.
Supplementary Material
References
- Ru J.; Huo Y.; Yang Y. Microbial Degradation and Valorization of Plastic Wastes. Front. Microbiol. 2020, 11, 442. 10.3389/fmicb.2020.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatge S.; Yang Y.; Ahn J.-H.; Hur H.-G. Biodegradation of polyethylene: a brief review. Appl. Biol. Chem. 2020, 63, 27. 10.1186/s13765-020-00511-3. [DOI] [Google Scholar]
- Liu J.; Xu G.; Zhao S.; Chen C.; Rogers M. J.; He J. Mechanistic and microbial ecological insights into the impacts of micro-and nano-plastics on microbial reductive dehalogenation of organohalide pollutants. J. Hazard. Mater. 2023, 448, 130895. 10.1016/j.jhazmat.2023.130895. [DOI] [PubMed] [Google Scholar]
- Yoshida S.; Hiraga K.; Takehana T.; Taniguchi I.; Yamaji H.; Maeda Y.; Toyohara K.; Miyamoto K.; Kimura Y.; Oda K. A bacterium that degrades and assimilates poly(ethylene terephthalate). Science 2016, 351, 1196–1199. 10.1126/science.aad6359. [DOI] [PubMed] [Google Scholar]
- a Austin H. P.; Allen M. D.; Donohoe B. S.; Rorrer N. A.; Kearns F. L.; Silveira R. L.; Pollard B. C.; Dominick G.; Duman R.; El Omari K.; et al. Characterization and engineering of a plastic-degrading aromatic polyesterase. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E4350–E4357. 10.1073/pnas.1718804115. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lu H.; Diaz D. J.; Czarnecki N. J.; Zhu C.; Kim W.; Shroff R.; Acosta D. J.; Alexander B. R.; Cole H. O.; Zhang Y.; et al. Machine learning-aided engineering of hydrolases for PET depolymerization. Nature 2022, 604, 662–667. 10.1038/s41586-022-04599-z. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Peng H.; Yang D.; Zhang G.; Zhang J.; Ju F. Polyvinyl chloride degradation by a bacterium isolated from the gut of insect larvae. Nat. Commun. 2022, 13, 5360. 10.1038/s41467-022-32903-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanan N.; Montazer Z.; Sharma P. K.; Levin D. B. Microbial and Enzymatic Degradation of Synthetic Plastics. Front. Microbiol. 2020, 11, 580709. 10.3389/fmicb.2020.580709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ru J.; Huo Y.; Yang Y. Microbial Degradation and Valorization of Plastic Wastes. Front. Microbiol. 2020, 11, 442. 10.3389/fmicb.2020.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampolli J.; Orro A.; Manconi A.; Ami D.; Natalello A.; Di Gennaro P. Transcriptomic analysis of Rhodococcus opacus R7 grown on polyethylene by RNA-seq. Sci. Rep. 2021, 11, 21311. 10.1038/s41598-021-00525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampolli J.; Orro A.; Vezzini D.; Di Gennaro P. Genome-Based Exploration of Rhodococcus Species for Plastic-Degrading Genetic Determinants Using Bioinformatic Analysis. Microorganisms 2022, 10, 1846. 10.3390/microorganisms10091846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León-Zayas R.; Roberts C.; Vague M.; Mellies J. L. Draft Genome Sequences of Five Environmental Bacterial Isolates That Degrade Polyethylene Terephthalate Plastic. Microbiol. Resour. Announce. 2019, 8, e00237 10.1128/mra.00237-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Švec P.; Černohlávková J.; Busse H.-J.; Vojtková H.; Pantůček R.; Cnockaert M.; Mašlaňová I.; Králová S.; Vandamme P.; Sedláček I. Classification of strain CCM 4446T as Rhodococcus degradans sp. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 4381–4387. 10.1099/ijsem.0.000584. [DOI] [PubMed] [Google Scholar]; b Xu J.-L.; He J.; Wang Z.-C.; Wang K.; Li W.-J.; Tang S.-K.; Li S.-P. Rhodococcus qingshengii sp. nov., a carbendazim-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2007, 57, 2754–2757. 10.1099/ijs.0.65095-0. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Xu J.; Li Y.; Wang K.; Wang Y.; Hong Q.; Li W.-J.; Li S.-P. Rhodococcus jialingiae sp. nov., an actinobacterium isolated from sludge of a carbendazim wastewater treatment facility. Int. J. Syst. Evol. Microbiol. 2010, 60, 378–381. 10.1099/ijs.0.013219-0. [DOI] [PubMed] [Google Scholar]
- Orr I. G.; Hadar Y.; Sivan A. Colonization, biofilm formation and biodegradation of polyethylene by a strain of Rhodococcus ruber. Appl. Microbiol. Biotechnol. 2004, 65, 97–104. 10.1007/s00253-004-1584-8. [DOI] [PubMed] [Google Scholar]
- a Goudriaan M.; Morales V. H.; van der Meer M. T. J.; Mets A.; Ndhlovu R. T.; van Heerwaarden J.; Simon S.; Heuer V. B.; Hinrichs K.-U.; Niemann H. A stable isotope assay with 13C-labeled polyethylene to investigate plastic mineralization mediated by Rhodococcus ruber. Mar. Pollut. Bull. 2023, 186, 114369. 10.1016/j.marpolbul.2022.114369. [DOI] [PubMed] [Google Scholar]; b Mor R.; Sivan A. Biofilm formation and partial biodegradation of polystyrene by the actinomycete Rhodococcus ruber. Biodegradation 2008, 19, 851–858. 10.1007/s10532-008-9188-0. [DOI] [PubMed] [Google Scholar]; c Mor R.; Sivan A. Biofilm formation and partial biodegradation of polystyrene by the actinomycete Rhodococcus ruber: Biodegradation of polystyrene. Biodegradation 2008, 19, 851–858. 10.1007/s10532-008-9188-0. [DOI] [PubMed] [Google Scholar]; d Sandhu M.; Jha P.; Paul A. T.; Singh R. P.; Jha P. N. Evaluation of biphenyl-and polychlorinated-biphenyl (PCB) degrading Rhodococcus sp. MAPN-1 on growth of Morus alba by pot study. Int. J. Phytorem. 2020, 22, 1487–1496. 10.1080/15226514.2020.1784088. [DOI] [PubMed] [Google Scholar]
- Sudhakar M.; Doble M.; Murthy P. S.; Venkatesan R. Marine microbe-mediated biodegradation of low- and high-density polyethylenes. Int. Biodeterior. Biodegrad. 2008, 61, 203–213. 10.1016/j.ibiod.2007.07.011. [DOI] [Google Scholar]
- Harshvardhan K.; Jha B. Biodegradation of low-density polyethylene by marine bacteria from pelagic waters, Arabian Sea, India. Mar. Pollut. Bull. 2013, 77, 100–106. 10.1016/j.marpolbul.2013.10.025. [DOI] [PubMed] [Google Scholar]
- Yao C.; Xia W.; Dou M.; Du Y.; Wu J. Oxidative degradation of UV-irradiated polyethylene by laccase-mediator system. J. Hazard. Mater. 2022, 440, 129709. 10.1016/j.jhazmat.2022.129709. [DOI] [PubMed] [Google Scholar]
- Santo M.; Weitsman R.; Sivan A. The role of the copper-binding enzyme – laccase – in the biodegradation of polyethylene by the actinomycete Rhodococcus ruber. Int. Biodeterior. Biodegrad. 2013, 84, 204–210. 10.1016/j.ibiod.2012.03.001. [DOI] [Google Scholar]
- Teufel R.; Mascaraque V.; Ismail W.; Voss M.; Perera J.; Eisenreich W.; Haehnel W.; Fuchs G. Bacterial phenylalanine and phenylacetate catabolic pathway revealed. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14390–14395. 10.1073/pnas.1005399107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y. B.; Peng B.; Li H.; Cheng Z. X.; Zhang T. T.; Zhu J. X.; Li D.; Li M. Y.; Ye J. Z.; Du C. C.; et al. Pyruvate cycle increases aminoglycoside efficacy and provides respiratory energy in bacteria. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E1578–E1587. 10.1073/pnas.1714645115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov F. A.; Koonin E. V.; Morgunov I. G.; Finogenova T. V.; Kondrashova M. N. Evolution of glyoxylate cycle enzymes in Metazoa: evidence of multiple horizontal transfer events and pseudogene formation. Biol. Direct 2006, 1, 31. 10.1186/1745-6150-1-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feehily C.; Karatzas K. A. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J. Appl. Microbiol. 2013, 114, 11–24. 10.1111/j.1365-2672.2012.05434.x. [DOI] [PubMed] [Google Scholar]
- Wilkes R. A.; Aristilde L. Degradation and metabolism of synthetic plastics and associated products by Pseudomonas sp.: capabilities and challenges. J. Appl. Microbiol. 2017, 123, 582–593. 10.1111/jam.13472. [DOI] [PubMed] [Google Scholar]
- Jeon H. J.; Kim M. N. Comparison of the functional characterization between alkane monooxygenases for low-molecular-weight polyethylene biodegradation. Int. Biodeterior. Biodegrad. 2016, 114, 202–208. 10.1016/j.ibiod.2016.06.012. [DOI] [Google Scholar]
- Fujisawa M.; Hirai H.; Nishida T. Degradation of Polyethylene and Nylon-66 by the Laccase-Mediator System. J. Polym. Environ. 2001, 9, 103–108. 10.1023/A:1020472426516. [DOI] [Google Scholar]
- Isom G. L.; Davies N. J.; Chong Z.-S.; Bryant J. A.; Jamshad M.; Sharif M.; Cunningham A. F.; Knowles T. J.; Chng S.-S.; Cole J. A.; et al. MCE domain proteins: conserved inner membrane lipid-binding proteins required for outer membrane homeostasis. Sci. Rep. 2017, 7, 8608. 10.1038/s41598-017-09111-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Deng W.; Li C.; Xie J. The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell. Signalling 2013, 25, 1608–1613. 10.1016/j.cellsig.2013.04.003. [DOI] [PubMed] [Google Scholar]; b Cuthbertson L.; Nodwell J. R. The TetR Family of Regulators. Microbiol. Mol. Biol. Rev. 2013, 77, 440–475. 10.1128/MMBR.00018-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnath L.; Sithole B.; Govinden R. Identification of lipolytic enzymes isolated from bacteria indigenous to Eucalyptus wood species for application in the pulping industry. Biotechnol. Rep. 2017, 15, 114–124. 10.1016/j.btre.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chaumeil P.-A.; Mussig A. J.; Hugenholtz P.; Parks D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2019, 36, 1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Capella-Gutiérrez S.; Silla-Martínez J. M.; Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N.; Dehal P. S.; Arkin A. P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 2010, 5, e9490 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I.; Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A.; He Z.; Redding-Johanson A. M.; Mukhopadhyay A.; Hemme C. L.; Joachimiak M. P.; Luo F.; Deng Y.; Bender K. S.; He Q.; et al. Hydrogen peroxide-induced oxidative stress responses in Desulfovibrio vulgaris Hildenborough. Environ. Microbiol. 2010, 12, 2645–2657. 10.1111/j.1462-2920.2010.02234.x. [DOI] [PubMed] [Google Scholar]
- Luo F.; Yang Y.; Zhong J.; Gao H.; Khan L.; Thompson D. K.; Zhou J. Constructing gene co-expression networks and predicting functions of unknown genes by random matrix theory. BMC Bioinf. 2007, 8, 299. 10.1186/1471-2105-8-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Newman M. E. J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8577–8582. 10.1073/pnas.0601602103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Clauset A.; Newman M. E. J.; Moore C. Finding community structure in very large networks. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2004, 70, 066111. 10.1103/PhysRevE.70.066111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The whole genome sequence has been deposited in the NCBI database under accession number SAMN34156462. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Proteomics Identification Database) partner repository with the data set identifier PXD041981 and PXD044483.