

SUMMARY
Impaired phosphodiesterase (PDE) function and mitochondrial Ca2+ (i.e., [Ca2+]m) lead to multiple health syndromes by an unknown pathway. Here, we fluorescently monitor robust [Ca2+]m efflux mediated by the mitochondrial Na+/Ca2+ exchanger NCLX in hippocampal neurons sequentially evoked by caffeine and depolarization. Surprisingly, neuronal depolarization-induced Ca2+ transients alone fail to evoke strong [Ca2+]m efflux in wild-type (WT) neurons. However, pre-treatment with the selective PDE2 inhibitor Bay 60-7550 effectively rescues [Ca2+]m efflux similarly to caffeine. Moreover, PDE2 acts by diminishing mitochondrial cAMP, thus promoting NCLX phosphorylation at its PKA site. We find that the protection of neurons against excitotoxic insults, conferred by PDE2 inhibition in WT neurons, is NCLX dependent. Finally, the administration of Bay 60-7550 enhances new object recognition in WT, but not in NCLX knockout (KO), mice. Our results identify a link between PDE and [Ca2+]m signaling that may provide effective therapy for cognitive and ischemic syndromes.
Graphical Abstract
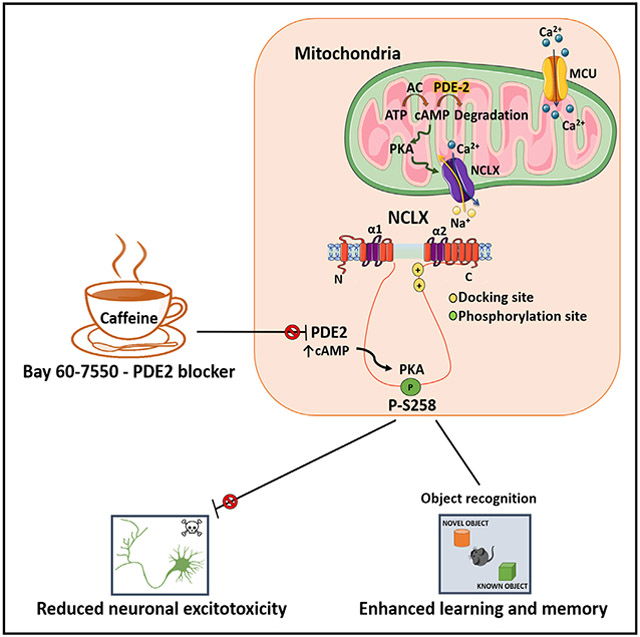
In brief
Rozenfeld et al. report that caffeine and the PDE2 blocker Bay-607550 stimulate mitochondrial Ca2+ removal in neurons. Inhibition of PDE2 stimulates PKA, which phosphorylates the regulatory domain of the mitochondrial exchanger NCLX, leading to its activation. Upregulation of NCLX prevents excitotoxic neuronal death and enhances new object learning in mice.
INTRODUCTION
Mitochondrial Ca2+ (i.e., [Ca2+]m) signaling plays a double-edged role, with its uptake primarily meditated by mitochondrial Ca2+ uniporter (MCU) (Kirichok et al., 2004; Baughman et al., 2011), a specific Ca2+ channel that conducts [Ca2+]m permeation powered by the steep (s−180 mV) mitochondrial membrane potential (Kirichok et al., 2004). Ca2+ is then pumped out of the mitochondria by an electrogenic mitochondrial Na+/Ca2+ exchanger NCLX (Palty et al., 2010; De Stefani et al., 2011). The latter is much slower than MCU and therefore constitutes a rate-limiting step in the [Ca2+]m cycle (Traaseth et al., 2004). The rise in [Ca2+]m activates several enzymes of the Krebs cycles (Rutter, 1990; Traaseth et al., 2004), and thereby the link between global Ca2+ rise and the energy supply is required for many subsequent signaling and molecular events. By modulating the local Ca2+ regulation at the cell membrane and at the endoplasmic reticulum (ER), mitochondria allosterically regulate the activity of receptors and ion channels (Poburko et al., 2009). Impaired balance between [Ca2+]m influx and efflux or change in [Ca2+]m buffering, however, can lead to uncontrolled [Ca2+]m rise, the so-called [Ca2+]m overload, which triggers the opening of the mitochondrial permeability pore (mPTP) and mitochondrial swelling (Rasola and Bernardi, 2011). The latter is an early hallmark event in ischemic and neurodegenerative syndromes. Remarkably the conditional knockout (KO) of NCLX leads to profound [Ca2+]m overload and heart failure or Alzheimer’s disease (AD) symptoms in a mouse AD model (Luongo et al., 2017; Jadiya et al., 2019). PKA-dependent phosphorylation of NCLX upregulates [Ca2+]m efflux and promotes the survival of Parkinson disease-associated PINK 1-deficient dopaminergic neurons (Kostic et al., 2015; Kostic et al., 2018). Still, the identity of key molecular players in the PKA pathway that control NCLX physiologically and thereby [Ca2+]m efflux (or failure thereof) in pathophysiological syndromes remain elusive.
Recent studies suggest that caffeine consumption lowers the risk of cardiovascular and neurodegenerative syndromes (Bhatti et al., 2013). Caffeine is a potent, albeit nonselective, inhibitor of phosphodiesterases (PDEs), but how the health-related effects of caffeine are linked to PDEs is unknown. PDEs, by degrading cAMP, control the PKA pathway and are of major therapeutic importance (Technikova-Dobrova et al., 2001; Acin-Perez et al., 2009). For example, inhibitors of PDE5 are effective drugs for treating erectile dysfunction or pulmonary hypertension (Corbin, 2004; Hemnes and Champion, 2006), and PDE4 blockers are used to treated psoriasis and alcoholic addiction (Acin-Perez et al., 2011; Gooderham and Papp, 2015; Logrip, 2015). PDE2, commonly termed PDE2A, degrades both cAMP and cGMP and is allosterically regulated by cGMP. Three variants of this enzyme termed PDE2A1, PDE2A2, and PDE2A3 were identified. While PDE2A1 is mainly found in the cytosol and PDE2A3 is found in the plasma membrane, PDE2A2 is mainly targeted to the mitochondria, directed into this organelle by its N-terminal domain (Acin-Perez et al., 2011; Monterisi et al., 2017). Numerous studies indicate that selective inhibitors of PDE2 protect against heart failure (Zaccolo and Movsesian, 2007), promote neuronal survival, and improve learning and memory in mice (Boess et al., 2004; Monterisi et al., 2017). Still, how PDE2 controls these processes and whether it is linked to the mitochondria is poorly understood.
RESULTS
We first aimed to monitor mild versus strong [Ca2+]m signals triggered in wild-type (WT) and NCLX KO mouse primary hippocampal neurons pre-loaded with the [Ca2+]m sensor Rhod2-AM (we achieve spatially specific mitochondrial loading, as reported previously [Kostic et al., 2015]). Consistent with previous studies (Walz et al., 1995), application of caffeine triggered multiple small [Ca2+]m transients (Figures 1A and 1B). Subsequent application of Ringer’s solution containing high K+ (50 mM KCl) induces neuronal depolarization and drives substantial Ca2+ entry that results in a much stronger [Ca2+]m response (Figure 1A). In NCLX KO neurons, the caffeine-induced [Ca2+]m signal was less spike-like, leading to a consequent gradual and continuous Ca2+ accumulation in mitochondria (Figures 1A-1C). Furthermore, while neuronal depolarization triggered an influx phase followed by fast [Ca2+]m efflux in the WT neurons, in NCLX KO neurons, depolarization was triggered by a similar mitochondrial Ca2+ influx but a profound inhibition of [Ca2+]m efflux (Figures 1A-1D). The depolarization-dependent [Ca2+]c response showed no significant differences between WT and NCLX KO neurons (Figures 1E and 1G), while cytosolic Ca2+ oscillations were still significantly different (Figures 1E and 1F). Rhod2 is a well-established selective mitochondrial Ca2+ dye (Figures 1 and 2), but it may also spill out to other cell compartments. To address this concern, and to ascertain that Ca2+ oscillations are mitochondrial related, we expressed, by viral delivery, a mitochondrial Ca2+ reporter 2mtGcAMP6, selectively targeted to the mitochondria (Figure S1A). We repeated the same experimental paradigm described in Figure 1A and found that pre-incubation with H89 triggered a modest increase in mitochondrial Ca2+ influx but a robust stimulation of mitochondrial Ca2+ efflux (Figures S2A-S2C). Note that we observed some differences in Ca2+ response rates between experiments, and although the reason for these differences is unclear, they might be related to minor differences in primary neuron confluency or the neuron:glia ratio. The results of this part indicate that caffeine-dependent spike-like signals and depolarization-dependent transient [Ca2+]m rises are controlled by NCLX in hippocampal neurons.
Figure 1. Mitochondrial Ca2+ extrusion is caffeine dependent in WT neurons but not in NCLX KO neurons.

(A) Representative fluorescence traces of mitochondrial Ca2+ [Ca2+]m changes monitored in WT and NCLX KO primary cultured hippocampal neurons. Neurons were pre-loaded with Rhod2-AM and initially superfused with Ringer’s solution. The [Ca2+]m signals were first triggered by caffeine containing (2 mM) Ringer’s solution and then neuronal depolarization trigged by Ringer’s solution containing high K+ (50 mM).
(B) Quantification of [Ca2+]m oscillations (per 300 s) shown in (A) for WT (n = 162) and NCLX KO neurons (n = 126).
(C) Quantification of [Ca2+]m influx rates shown in (A) for WT (n = 38) and NCLX KO neurons (n = 54).
(D) Quantification of [Ca2+]m efflux rates shown in (A) for WT (n = 164) and NCLX KO neurons (n = 106).
(E) Representative fluorescence traces of [Ca2+]c changes in WT and NCLX KO neurons pre-loaded with Fura 2-AM (1 μM) evoked by caffeine and high-K+ Ringer’s solution as in (A).
(F) Quantification of [Ca2+]c oscillations (per 300 s) shown in (E) for WT (n = 170) and NCLX KO (n = 166) neurons.
(G) Quantification of [Ca2+]c influx amplitude of the data shown in (E) for WT (n = 166) and NCLX KO neurons (n = 99).
(H) Representative fluorescence traces of [Ca2+]m changes monitored in WT and NLCX KO. [Ca2+]m signals were evoked only by high-K+ Ringer’s solution.
(I) Quantification of [Ca2+]m influx rates of data shown in (H) for WT (n = 160) and NCLX KO neurons (n = 114).
(J) Quantification of [Ca2+]m efflux rates shown in (H) for WT (n = 138) and NCLX KO neurons (n = 145).
(K) Representative fluorescence traces of mitochondrial Mg2+ [Mg2+]m tracked in WT primary cultured neurons (days in vitro [DIV] 10–15). To track the mitochondrial Mg2+ transport, neurons were pre-loaded with magnesium green (2 μM) and MitoTracker deep red (200 nM), and analysis focused on regions that showed MitoTracker deep red staining. Cells were initially superfused with Ringer’s solution at pH 7.4 and then caffeine containing (2 mM) Ringer’s solution.
(L) Quantification of basal and caffeine evoked [Mg2+]m rise shown in (K) for WT (n = 48).
All summary data represent mean ± SEM. ***p < 0.001, ****p < 0.0001, N.S., nonsignificant.
Figure 2. H89 inhibits Ca2+ efflux in WT neurons, whereas PKA inhibitor cancels caffeine effect.

(A) Representative fluorescence traces of [Ca2+]m changes monitored in control and H89 pre-treated WT primary cultured hippocampal neurons. Neurons were loaded with Rhod2-AM (1 μM) and initially superfused with caffeine, followed by Ringer’s solution containing high K+ as in Figure 1A.
(B) Quantification of [Ca2+]m oscillations shown in (A) for control (n = 105) and H89 pre-treated (n = 133) WT neurons.
(C) Quantification of [Ca2+]m influx rates shown in (A) for control (n = 164) and H89 pre-treated (n = 127) WT neurons.
(D) Quantification of [Ca2+]m efflux rates shown in (A) for control (n = 164) and H89 pre-treated WT (n = 30) neurons.
(E) Representative fluorescence traces of [Ca2+]c changes in control and H89 pre-treated WT neurons done as in Figure 1E.
(F) Quantification of [Ca2+]c influx amplitude changes during the maximum phases of the data shown in (E) for control (n = 138) and H89 pre-treated WT (n = 124) neurons.
(G) Representative fluorescence traces of [Ca2+]m changes in neurons pre-loaded with Rhod2-AM (1 μM) and initially superfused with or without caffeine (control or KT 5720 treated) as indicated, followed by Ringer’s solution containing high K+ as in Figure 1A.
(H) Quantification of [Ca2+]m efflux rates shown in (G) for control (n = 21), KT 5720- and caffeine-treated (n = 31) and caffeine-only-treated (n = 37) WT neurons.
All summary data represent mean ± SEM. **p < 0.01, ***p < 0.001 ****p < 0.0001, N.S., nonsignificant.
We next sought to compare the [Ca2+]m response triggered only by the high-K+-dependent depolarization (Figures 1H-1J) in WT and NCLX KO neurons, reasoning that it will show the same gap in [Ca2+]m efflux between WT and NCLX KO neurons. Surprisingly, without the initial caffeine exposure, we no longer saw any difference between [Ca2+]m efflux rates in WT versus KO-derived neurons following depolarization (Figures 1H and 1J) versus the major difference seen with caffeine pre-treatment. We also show a slightly smaller [Ca2+]c influx in the NCLX KO neurons (Figures S1E-S1G), consistent with greater mitochondrial uptake when the main efflux pathway is knocked out. Thus, our results suggest that caffeine, following its Ca2+ ER release, had an additional effect of augmenting [Ca2+]m efflux in neurons via NCLX activation. As Mg2+ is emerging as a key element in mitochondrial signaling and physiology (Daw et al., 2020), we therefore also measured Mg2+ transport in response to caffeine application and monitored mitochondrial and cytosolic Mg2+ responses. Neurons were pre-loaded with the Mg2+-sensitive dye magnesium green (see STAR Methods) and loaded with MitoTracker deep red to track mitochondrial Mg2+ levels (Figures 1K, 1L, S1H, and S1I). The application of caffeine was followed by a rise of cytosolic and mitochondrial Mg2+ influx (Figures 1K, 1L, S1H, and S1I), but considering the cationic selectivity of NCLX, Mg2+ does not transport this cation, and we therefore did not seek to monitor if Mg2+ efflux is mediated by this transporter (Palty et al., 2012).
Caffeine has a well-documented regulatory effect on the PKA pathway and is known to inhibit PDEs (Ku et al., 2011; Zeitlin et al., 2011). Since our previous studies indicated that PKA plays a key role in regulating NCLX activity (Kostic et al., 2015; Kostic et al., 2018), we reasoned that the stimulating effect of caffeine on [Ca2+]m efflux by NCLX may be due to increased cAMP and, therefore, is PKA dependent. Hence, we first compared the [Ca2+]m responses triggered by caffeine followed by Ringer’s solution containing high K+ in control versus PKA inhibitor H89 pre-treated WT neurons (Figures 2A-2D). Notably, PKA inhibition mimicked the NCLX KO (Figures 1A and 1B) in its strong reduction of caffeine-dependent [Ca2+]m spike-like activity and dramatic slowdown of [Ca2+]m efflux rate following neuronal depolarization (Figures 2A, 2B, and 2D) while not changing influx rates (Figure 2C). In addition, and consistent with previous studies (Sang et al., 2016), PKA inhibition also reduced peak [Ca2+]c response (Figures 2E and 2F). Interestingly, and consistent with data we show in Figure 1, application of only Ringer’s solution containing high K+ did not mimic the caffeine effect (Figures S2A-S2C). Although H-89 is a well-established PKA inhibitor, it may have several off-target effects (Davies et al., 2000). To ascertain that the caffeine effect is mediated by PKA, we performed the paradigm described in Figure 2E using the selective PKA inhibitor KT-5720 (100 nM) (Figures 2G, 2H, and S1G) and found that inhibition of PKA did not modulate mitochondrial Ca2+ influx but robustly inhibited the caffeine effect on mitochondrial Ca2+ efflux. Taken together, our results suggest that caffeine is upregulating NCLX activity through the PKA pathway and that this regulation is critical for activation of [Ca2+]m efflux by this exchanger in neurons.
Caffeine regulation of PKA is primarily mediated by inhibition of PDEs (Ribeiro and Sebastiao, 2010), with the highest affinity to PDE2 (Technikova-Dobrova et al., 2001; Acin-Perez et al., 2009). Remarkably, PDE2 is the only PDE form that is targeted to the mitochondria matrix (Acin-Perez et al., 2011). We therefore asked if PDE2 is linked to NCLX regulation. We applied Ringer’s solution containing high K+ to induce mitochondrial depolarization in the absence or the presence of the selective PDE2 blocker Bay 60-7550. Addition of Bay 60-7550 rescued the depolarization-dependent [Ca2+]m efflux compared with control and similarly to caffeine (Figures 3A-3C). No major effect on mitochondrial influx and a minor effect on [Ca2+]c responses were monitored (Figure 3B). In contrast to the strong effect of PDE2 inhibition on WT, in NCLX KO neurons, the application of Bay 60-7550 had no effect on [Ca2+]m and [Ca2+]c responses (Figures 3D-3F, S2D, and S2E). This set of results suggest that PDE2 is regulating [Ca2+]m efflux by NCLX in neurons.
Figure 3. PDE2-selective inhibitor mimics caffeine effect by accelerating Ca2+ efflux.

(A) Representative fluorescence traces of [Ca2+]m changes monitored in control and Bay 60-7550 pre-treated WT neurons loaded with Rhod2-AM. [Ca2+]m signals were evoked only by Ringer’s solution containing high K+.
(B) Quantification of [Ca2+]m influx rates changes shown in (G) for control (n = 150) and Bay 60-7550 pre-treated (n = 60) WT neurons.
(C) Quantification of [Ca2+]m efflux rates changes shown in (G) for control (n = 155) and Bay 60-7550 pre-treated (n = 61) WT neurons.
(D) Representative fluorescence traces of [Ca2+]m changes monitored in control NCLX KO and Bay 60-7550 pre-treated NCLX KO neurons loaded with Rhod2-AM. [Ca2+]m signals were evoked only by high-K+ Ringer’s solution.
(E) Quantification of [Ca2+]m influx rates changes shown in (D) for control NCLX KO (n = 107) and Bay 60-7550 pre-treated (n = 52) NCLX KO neurons.
(F) Quantification of [Ca2+]m efflux rates changes shown in (D) for control NCLX KO (n = 90) and Bay 60-7550 pre-treated (n = 50) NCLX KO neurons.
(G) Representative fluorescence traces of [Ca2+]m changes monitored in control and Bay 60-7550 pre-treated WT neurons. Neurons were preloaded with Rhod2-AM, initially superfused with Ringer’s solution, and then electrically stimulated by 5 consecutive pulses, each of 20 Hz frequency.
(H) Quantification of [Ca2+]m influx rate changes during the maximum phases shown in (G) for control (n = 18) and Bay 60-7550 pre-treated (n = 23) WT neurons.
(I) Quantification of [Ca2+]m efflux rates shown in (G) for control (n = 19) and Bay 60-7550 pre-treated (n = 24) WT neurons.
(J) Representative Rhod2-AM fluorescent traces of [Ca2+]m changes monitored in control and Bay 60-7550 pre-treated WT neurons. Neurons were washed with recording solution followed by digitonin containing (0.002%) Ringer’s solution. To trigger [Ca2+]m influx and efflux, permeabilized neurons were treated with intracellular solution containing Ca2+ (60 μM), followed by the addition of Na+ (20 mM) (see STAR Methods).
(K) Representative fluorescent traces of [Ca2+]m changes as in Figure 3J except that Bay 60-7550 was co-added with the permeabilization buffer.
(L) Quantification [Ca2+]m efflux rates shown in (J) and (K) in control (n = 206), Bay 60-7550 pre-treated (n = 205), and Bay 60-7550-treated (n = 266) WT neurons.
All summary data represent mean ± SEM. **p < 0.01, ****p < 0.0001, N.S., nonsignificant.
To further test the role of PDE2 using the neuronal-activity-dependent strategy, we monitored [Ca2+]m efflux in electrically stimulated (5 pulses, 20 Hz stimulation) neurons and found that Bay 60-7550 was similarly effective in rescuing the [Ca2+]m efflux in such stimulated neurons while not changing the [Ca2+]m influx (Figures 3G-3I). This set of experiments further indicate that inhibition of PDE2 is required for activation of [Ca2+]m efflux by NCLX in neurons. PDE2 is found not only in the mitochondria but other isoforms of this enzyme are also found in cell membrane and cytosol. To determine if the PDE2 effect is mediated by the mitochondrial PDE2 isoform, we performed a well-established paradigm of cellular digitonin permeabilization in which the cell membrane is permeabilized and the cytosol is washed out while keeping the mitochondria intact (Vercesi et al., 1991) (Figures 3J-3L). Firstly, we used the same experimental paradigm of Bay 60-7550 pre-incubation followed by the permeabilization (Figure 3J). We then applied Ca2+ (60 μM), which triggered, as expected, mitochondrial Ca2+ rise. Only after Na+ (20 mM) addition did we monitor mitochondrial Ca2+ efflux, indicating, consistent with previous studies (Pannaccione et al., 2020), that the major mitochondrial Ca2+ efflux pathway in excitable cells is the Na+/Ca2+ exchanger (Figure 3J). Hence, Bay 60-7550 was effective in faster activation of NCLX when the cell membrane and cytosolic content were removed, likely with the intact composition of nonmitochondrial PDE2 (Figure 3L). Pre-incubation with Bay 60-7550 may still have an indirect effect during the pre-incubation period. To ascertain that Bay 60-7550 acts directly on the mitochondrial NCLX, we added Bay 60-7550 only during permeabilization and subsequent imaging (Figure 3K). We monitored a similar activation effect of mitochondrial Na+-dependent Ca2+ efflux (Figures 3K and 3L). This set of experiments strongly indicate that the activation of NCLX is mediated by a mitochondrial PDE2.
We next ask if PDE2 activity, invoked by the [Ca2+]m transients, regulates mitochondrial cAMP content. To monitor mitochondrial cAMP, we targeted the cAMP sensor Pink Flamindo (PF) (Harada et al., 2017) to the mitochondria by adding a mitochondrial targeting domain (see STAR Methods and Figure 4A) and found that the reconfiguration of this reporter led to its mitochondrial targeting (Figure 4B). To determine if Bay 60-7550 has an effect on mitochondrial cAMP levels we fluorescently monitored mitochondrial cAMP in neuronal SH-SY5Y cells and primary hippocampal neurons expressing this mitochondrial targeted PF (mtPF) by triggering ATP-dependent purinergic or depolarization-dependent (Ringer’s solution containing high K+) Ca2+ signals, respectively (Figures 4C-4F). In SH-SY5Y cells, ATP evoked a rise in mitochondrial cAMP (Figures 4C and 4D), and consistent with previous studies (Bubb et al., 2014), SH-SY5Y cells pre-treated with Bay-60-7550 showed a larger rise in mitochondrial cAMP. In primary neurons, depolarization triggered a fast drop in mitochondrial cAMP that was largely suppressed in Bay 60-7550 pre-treated neurons (Figures 4E and 4F). Moreover, we asked if caffeine would modulate mitochondrial cAMP levels by monitoring mitochondrial cAMP changes in SH-SY5Y cells expressing the mtPF (Figures 4G and 4H). We show that the application of caffeine also increased mitochondrial cAMP content, thus indicating its role in regulating mitochondrial cAMP. Next, we asked if the mitochondrial NCLX phosphorylation, at its S258PKA phosphorylation site, is the target of PDE2. Here, we focused on the SH-SY5Y cells because of the restricted selectivity of the pS258 antibody to human NCLX (Kostic et al., 2018) (see STAR Methods). We found that the inhibition of PDE2 by Bay 60-7550 enhanced NCLX phosphorylation at the S258 PKA-dependent position (Figures 4I and 4J). Our results indicate that the activation of NCLX by PDE2 inhibition is mediated by a rise in mitochondrial cAMP by triggering NCLX phosphorylation at its PKA S258 phosphorylation site. If PDE2 modulates the PKA-dependent NCLX phosphorylation, then the phosphomimetic NCLX mutants should be insensitive to Bay 60-7550. To address this hypothesis, we exogenously expressed S258A NCLX while at the same time silencing the expression of the endogenous exchanger and found that, as expected, Bay 60-7550 activated the WT NCLX, but it failed to activate mitochondrial Ca2+ efflux of the S258A NCLX mutant (Figures 4I and 4J). This result, consistent with NCLX phosphorylation western blot (WB) data (Figures 4K, 4L, S2I, and S2J), suggests that PDE2 acts by selectively targeting the same NCLX phosphorylation site as PKA. Additionally, caffeine enhanced NCLX phosphorylation at the same PKA phosphorylation site and had a similar effect to application of Forskolin. Moreover, we show that the co-application of Bay-60-7550 and caffeine had a similar stimulatory effect on mitochondrial Ca2+ efflux, indicating that PKA and PDE2 act on the same pathway (Figures S2F-S2H).
Figure 4. Bay 60-7550 increases NCLX activity by stimulating its phosphorylation.

(A) Scheme of the mitochondrial PF construct.
(B) Representative images of SH-SY5Y cells expressing the [Ca2+]m reporter CEPIA 2-AM in green and mtPF in red and primary cultured hippocampal neurons from WT mice infected with mtPF in red and expressing the [Ca2+]m reporter Mito-View 405 (100 nM) in blue.
(C) Representative fluorescence traces of mitochondrial cAMP changes in control and Bay 60-7550 pre-treated SH-SY5Y cells expressing mtPF. Cells were superfused with Ringer’s solution followed by addition of ATP containing Ringer’s solution (100 μM) to evoke a Ca2+ response.
(D) Quantification of cAMP changes shown in (C) of control (n = 71) and Bay 60-7550 pre-treated (n = 46) SH-SY5Y cells.
(E) Representative fluorescence traces of cAMP changes in WT and Bay 60-7550 pre-treated WT primary hippocampal neurons previously expressing the mtPF. Neurons were superfused with Ringer’s solution followed by Ringer’s solution containing high K+ to trigger a Ca2+ transient.
(F) Quantification of cAMP changes shown in (E) for control (n = 59) and Bay 60-7550 pre-treated (n = 40) WT neurons.
(G) Representative fluorescence traces of mitochondrial cAMP changes in control and caffeine pre-treated SH-SY5Y cells expressing mtPF. Cells were superfused with Ringer’s solution, followed by the addition of caffeine (2 mM) containing Ringer’s solution.
(H) Quantification of cAMP changes shown in (G) of control (n = 95) and caffeine pre-treated (n = 70) SH-SY5Y cells.
(I) SH-SY5Y cells ectopically expressing the WT or the NCLX s258A mutation while silencing the endogenous NCLX(Kostic et al., 2015) (see STAR Methods) were superfused with 100 μM ATP containing Ringer’s solution to evoke mitochondrial Ca2+ response in the presence or the absence of Bay 60-7550 as indicated.
(J) Quantification of mitochondrial Ca2+ efflux rates shown in (I) of WT (n = 46), WT + Bay 60-7550 (n = 178), s258A NCLX (n = 29) mutant, and s258A NCLX + Bay 60-7550 (n = 15) in SH-SY5Y cells.
(K) Immunoblot of SH-SY5Y cells, reacted with anti-pNCLX antibody (Ab), showing NCLX phosphorylation at its PKA-dependent S258 NCLX phosphorylation site. SH-SY5Y cells were pre-treated either with Bay 60-7550 (1 μM, 1 h), ATP (100 μM, 1 min), or both. Actin was used as loading control.
(L) Densitometry analys is of the immunoblot shown in (K), showing the rise (%) in NCLX phosphorylation in treated versus nontreated (control) SH-SY5Y cells. Immunoblot was done in 3 independent repetitions.
All summary data represent mean ± SEM. *p < 0.05, ****p < 0.0001, N.S., nonsignificant.
We next interrogated the role of PDE2 in controlling the [Ca2+]m transients evoked by the neuromodulators Gq-coupled vasopressin (VP) and Gs-coupled norepinephrine (NE). Their application evoked cytosolic Ca2+ transients (Figure S3), followed by mitochondrial influx, but not substantial efflux of Ca2+ (Figure 5). In contrast, pre-treatment of WT neurons with Bay-60-7550 revealed VP- and NE-dependent [Ca2+]m efflux in WT, but not in NCLX KO, neurons (Figures 5A-5D and 5E-5H), indicating that both NCLX and inhibition of PDE2 were required to activate neurotransmitter-dependent [Ca2+]m efflux. [Ca2+]c transients mimicked the results shown for [Ca2+]m (Figures S3A-S3H). Interestingly, co-application of VP and NE was sufficient to rescue [Ca2+]m efflux (Figures 5I and S3I-S3L), indicating that, consistent with our previous studies (Assali et al., 2020), PKA activation by the Gs-coupled NE receptor enhanced the mitochondrial Ca2+ efflux, which is primarily triggered by the Gq-coupled VP receptor. When Bay 60–70550 was co-applied with NE and VP, it further strongly augmented [Ca2+]m efflux, resulting in a large drop in [Ca2+]m that was well below the resting set point in WT neurons (Figures 5I and 5J). No efflux was monitored in NCLX KO neurons in the presence or the absence of Bay-60-7550 (Figures 5K and 5L). We suggest that inhibition of PDE2 is required to augment the neurotransmitter-dependent [Ca2+]m efflux. Furthermore, NE and VP play a synergistic role in amplifying [Ca2+]m efflux that is fully unmasked upon PDE2 inhibition.
Figure 5. Vasopressin (VP) and norepinephrine (NE) trigger mitochondrial Ca2+ influx followed by Bay 60-7550-enhanced, NCLX-mediated Ca2+ efflux.

(A) Representative fluorescence traces of [Ca2+]m changes monitored in VP-treated control and Bay 60-7550 pre-treated WT neurons. Neurons were pre-loaded with Rhod2-AM and initially superfused with Ringer’s solution. VP (1 μM) containing Ringer’s solution was added as indicated by the bar.
(B) Quantification of [Ca2+]m efflux rates shown in (A) for VP-treated control (n = 53) and Bay 60-7550 pre-treated WT (n = 41) neurons.
(C) Representative fluorescence traces of [Ca2+]m changes monitored in NE-treated (5 μM) control and Bay 60-7550 pre-treated WT neurons as in (A).
(D) Quantification of [Ca2+]m efflux rates shown in (C) for control (n = 62) and Bay 60-7550 pre-treated WT (n = 33) neurons.
(E) Representative fluorescence traces of [Ca2+]m changes in VP-treated control NCLX KO and Bay 60-7550 pre-treated NCLX KO neurons using the same experiment paradigm shown in (A).
(F) Quantification of [Ca2+]m efflux rates shown in (E) for VP-treated control KO (n = 70) and Bay 60-7550 pre-treated NCLX KO (n = 30) neurons.
(G) Representative fluorescence traces of [Ca2+]m changes NE-treated control NCLX KO and Bay 60-7550 pre-treated NCLX KO neurons as in (E).
(H) Quantification of [Ca2+]m efflux rates shown in (G) for NE-treated control NCLX KO (n = 51) and Bay 60-7550 pre-treated NCLX KO (n = 76) neurons.
(I) Representative fluorescence traces of [Ca2+]m changes monitored of WT neurons treated with VP and NE co-applied at time intervals indicated by the time bar (100 s) in the absence or the presence of Bay 60-7550 pre-treated WT neurons as described in (A).
(J) Quantification of [Ca2+]m efflux rates shown in (I) for control (n = 67) and Bay 60-7550 pre-treated WT (n = 90) neurons.
(K) Representative fluorescence traces of [Ca2+]m changes monitored in NCLX-KO neurons treated with VP and NE of control NCLX KO and Bay 60-7550 pre-treated NCLX KO neurons of the experiment performed as in (I).
(L) Quantification of [Ca2+]m efflux rates shown in (K) for NCLX KO (n = 50) and Bay 60-7550 pre-treated NCLX KO (n = 52) neurons.
All summary data represent mean ± SEM. **p < 0.01, ****p < 0.0001, N.S., nonsignificant.
Inhibition of PDE has emerged as promising strategy in preventing neuronal death (Ding et al., 2014). Considering the strong link of [Ca2+]m overload and neuronal survival, we asked if the reactivation effect of PDE2 blocker on [Ca2+]m efflux by NCLX will rescue neurons subject to an excitotoxic insult. We subjected cultured neurons to such an insult by pre-treating them overnight with the glutamate uptake transport inhibitor TBOA (Sharifullina and Nistri, 2006, Corsini et al., 2016; Yeh et al., 2017). As shown previously (Soares et al., 2017), Bay 60-7550 was effective in preventing TBOA-dependent neuronal death (Figures 6A-6C). Remarkably, the rescuing effect of Bay 60-7550 on neuronal survival was largely eliminated in NCLX KO neurons, indicating that the PDE2 blocker enhances survival by acting through the activation of NCLX.
Figure 6. Bay 60-7550 promotes neuronal survival and mouse learning via NCLX regulation.

(A) Representative images of WT and NCLX KO primary hippocampal neurons stained with Sytox green, cellular death indicator (1 μM), NeuN, specific neuronal marker (1:100), and DAPI for nuclei labeling (1: 10,000). Images present control groups, Bay 60-7550 (1 μM, 1-h pre-treatment), TBOA (80 μM, overnight pre-treatment), and TBOA + Bay 60-7550 (overnight and 1-h pre-treatment, respectively).
(B) Quantification of live WT neurons in the field shown in (A) presented as percentage (n = 43).
(C) Quantification of live NCLX KO neurons in the field shown in (A) presented as percentage (n = 20).
(D) Schematic illustration of the Bay 60-7550 treatment/object recognition experimental design.
(E) Discrimination index of vehicle- (control) versus Bay 60-7550-injected WT (n = 15/17) mice.
(F) Discrimination index of vehicle (control) versus Bay 60-7550 NCLX-KO (n = 11) mice.
All summary data represent mean ± SEM of one-way ANOVA analysis, Tukey’s test. **p < 0.01, ****p < 0.0001, N.S., nonsignificant.
Finally, numerous studies show that inhibition of PDE2 is effective in enhancing learning and memory (Boess et al., 2004; Lueptow et al., 2016), but the molecular link that couples PDE2 to these processes is unknown. Considering the role of mitochondria in controlling neuronal plasticity (Mattson, 2007), we reasoned that the well-established effect of PDE2 inhibition on enhancing learning and memory may be linked to [Ca2+]m extrusion rescue via NCLX. To address this hypothesis, we subjected control- and Bay 60–7050-injected WT and NCLX KO mice to a new object recognition test (Figures 6D-6F). Although the basal object recognition score of NCLX KO mice tended to be slightly higher (s10%) than in WT mice, Bay 60-7550 treatment enhanced discrimination between two objects in WT mice, as previously reported, but failed to evoke this effect in NCLX KO mice (Figure 6F). Altogether, these results indicate that NCLX is required for PDE2-dependent enhancement of new object recognition.
DISCUSSION
We studied the effect of caffeine on ER Ca2+ release in hippocampal neurons. To our surprise, we found that caffeine also had a profound and lasting effect in activating [Ca2+]m efflux in these neurons. The PDE-inhibitory activity of caffeine (Ribeiro and Sebastiao, 2010) and its regulation of NCLX led us to hypothesize that the regulation of [Ca2+]m efflux was mediated by PDE-dependent NCLX. Notably, among PDEs, PDE2 has the highest affinity to caffeine. Because it is only PDEA2 that is targeted to the mitochondria (Acin-Perez et al., 2009), we therefore reasoned that PDEA2 regulates NCLX in neurons, and our hypothesis is supported by the following findings: (1) caffeine upregulation of [Ca2+]m efflux is reproduced by the selective PDE2 inhibitor Bay 60-7550 (Figure 3); (2) the upregulation of [Ca2+]m efflux by Bay 60-7550 is blunted in neurons derived from NCLX-KO neurons, indicating that PDE2 controls NCLX-mediated [Ca2+]m efflux (Figures 3 and 5); (3) application of Bay 60-7550 enhances NCLX phosphorylation at its NCLX S258 site, indicating that PDE2 acts through the PKA phosphorylation site of NCLX (Figure 4); and (4) remarkably, the KO of NCLX eliminates hallmark physiological implications of PDE2 inhibition by enhancing neuronal survival and cognitive function (Figure 6). PDE2A1 and PDE2A3 are located in the compartments of cellular membrane and cytosol and are inhibited by Bay 60-7550. Therefore, it can be argued that they mediate NCLX regulation. Our following findings indicate that the mitochondrial PDE2A2 form is the major NCLX regulator: (1) NCLX is upregulated by Bay 60-7550 when neurons are permeabilized by digitonin, thereby largely eliminating the cell membrane and cytosolic PDE2 form. (2) The upregulation of NCLX by Bay 60-7550 is retained even when this PDE2 inhibitor is added during the permeabilization process, arguing against any indirect effect of the cytosolic or cell membrane PDE2 forms. (3) However, the PDE2-inhibitory effect is eliminated when the mitochondrial S258A NCLX mutant that mimics a non-PKA phosphorylated state of NCLX is expressed. Altogether, our findings indicate that NCLX is regulated by the mitochondrial PDE2A2 form.
The unique distribution of PDE2 in brain regions associated to cognitive function indicates that the enzyme is linked to these processes. Indeed, numerous studies employing PDE2 inhibitors show that inhibition of PDE2 enhances (Gomez and Breitenbucher, 2013) cognitive function in an AD model during regular learning paradigm, and in particular, new object recognition was also clinically tested (Boess et al., 2004; Lueptow et al., 2016). Despite the intense interest in the neuronal PDE2 role, a major unsolved question that impedes the progress in the field is the identity of its cellular targets, which further mediate its neuronal effects. As described previously (Savai et al., 2010), we find that inhibition of PDE2 by Bay 60-7550 rescues cells against excitotoxic insults. The pro-survival effect of the PDE2 blocker is fully diminished in NCLX-KO neurons, indicating that this pathophysiological aspect of PDE2 is mediated by controlling NCLX. Remarkably, the well-documented enhancement of new object recognition triggered by PDE2 blocker is also NCLX dependent. These results indicate that NCLX is the long-sought mitochondrial target for PDE2.
Although we do not fully know how NCLX is linked to cognitive function, recent studies indicate that mitochondria are critical for providing the metabolic energy and anabolic production of molecules required for de novo protein synthesis (Weinberg et al., 2015). Such proteins are essential for synaptic plasticity linked to the mentioned processes. Our results suggest that the temporal control of [Ca2+]m transients play a role in facilitating this process, which may be governed by NCLX. Of particular interest is our observation that the [Ca2+]m response is temporally modulated by neuromodulators such as the Gs-coupled NE and Gq-coupled VP (Figure 5). Consistent with our previous study (Assali et al., 2020), the NE receptor, by activating PKA, upregulates NCLX activity. Our results suggest that inhibition of PDE2 further activates PKA, leading to additional amplification of mitochondrial Ca2+ efflux by NCLX. Considering the major metabolic and anabolic cost in providing the energy for neuronal transmission and for plasticity-linked new protein translation, the key role of Ca2+ in tuning mitochondrial activity constitutes the link between PDE2 and PKA regulation of NCLX and may play role in controlling such processes. Interestingly, we recently found that a human genetic mutation of NCLX, impairing its Ca2+ transport activity, is associated with severe mental retardation (Stavsky et al., 2021).
The brain may not be the only organ where the interaction between PDE2 and NCLX may have pathophysiological implication. The conditional knock down of cardiac NCLX leads to fatal heart failure (Jadiya et al., 2019), while the inhibition of PDE2 effectively prevents heart failure in a murine model (Vettel et al., 2017). Considering the link between PDE2 and NCLX, it will be of interest to determine in future studies if the cardio-protective effect of PDE2 may be modulated in the context of NCLX regulation.
To conclude, we show that in hippocampal neurons, [Ca2+]m efflux is tonically suppressed but can be activated by caffeine. The strong effect of PDE2 inhibition on [Ca2+]m efflux by attenuation of mitochondrial cAMP degradation is linked to NCLX phosphorylation at its PKA phosphorylation site. Additionally, we show that the PDE2 block is critical for enhancing neuronal survival following excitotoxic insult only when NCLX is active. Finally, we suggest that the enhancement of learning and memory functions mediated by PDE2 inhibition are NCLX dependent.
Limitations of the study
We show that caffeine, by inhibiting PDE2, enhances PKA phosphorylation leading to mitochondrial NCLX activation, thereby reducing neuronal excitotoxicity and enhancing learning in mice. Caffeine affects multiple cellular targets and may therefore have multiple additional effects that were not addressed in this study. It is unclear if caffeine has a similar effect on human neurons. Further studies are therefore required to determine if caffeine has a similar effect on the human brain.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Israel Sekler (sekler@bgu.ac).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL6 Wild-Type (WT) (Envigo, C57BL Cat #6JRCCH5B043) 0–3 days old mouse pups were maintained as previously described (Gitler et al., 2004a). NCLX KO mice were obtained from Jackson Laboratories (Cat #026242). This strain was generated at the Jackson Laboratory by Cas9 RNA injection and an 18-mer guide sequence ATACTGGAGACGGCGTCT, which resulted in a 13 bp deletion (AGACGGCGTCTGG) in exon 2 beginning at chromosome 5 positive strand position 120, 513, 241 bp (GRCm38), which is predicted to cause amino acid sequence changes after residue 21 and an early truncation 33 amino acids late (Stavsky et al., 2021).
Animals were treated with the approvals and in accordance with the guidelines of the Ben-Gurion University Institutional Committee for Ethical Care and Use of Animals (Reference Number: IL 07-02-2019C).
Briefly, mice were housed in specific pathogen free facilities. Rodent care practices were under sterile conditions, with sterile supplies. Rodents Hosing conditions are: 12:12 light:dark cycles at 20–24°C and 30–70% relative humidity. Animals are free fed auto-claved rodent chow and have free access to reverse osmosis filtered water. Rodents are housed in individually ventilated GM500 (Tecniplast, Italy) cages in groups of maximum 5 mice per cage.
Mouse pups used for generating primary neuronal cultures were chosen randomly out of the same litter. Mice used for behavioral testing were chosen at the certain age and gender (See section of Object recognition chapter in Method details).
Primary neuronal culture
Primary culture of hippocampal neurons was done for each independent experiment from mouse pups as previously described (Gitler et al., 2004b). Each mouse hippocampus was used to generate culture on 6 coverslips with 50,000–70,000 cells. Cultures were typically maintained at 37°C in a 5% CO2 humidified incubator for 10–15 days before experimentation. In fluorescent imaging related experiments neurons were easily distinguishable from glia: they appeared phase bright, had smooth rounded somata and distinct processes and laid just above the focal plane of the glial layer.
Cell cultures
SH-SY5Y (neuroblastoma subline (SK-N-SH) derived from a metastatic bone tumor) and HEK293 (human embryonic kidney cell line) cell lines were cultured (37°C, 5% CO2) in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 1% penicillin/streptomycin, as previously described (Palty et al., 2004).
METHOD DETAILS
Genotyping
NCLX+/+ (WT)or NCLX −/− (KO) mice were used after genotyping using PCR of DNA isolated from mouse tail biopsy samples. Primers 5′- TGGCTCTGATACTGGAGACG-3′ and 5′- CATGGCAGTCTGGTTGACAC-3′ developed by our group, amplified a 128 bp band from the wild-type allele whereas primers 5’ -TGCTCTGGGCTCCTGTCTTC-3′ and 5′- CATGGCAGTCTGGTTGACAC-3′ amplified a 115-bp band from the knockout allele. PCR products were run on 4% Nusieve agarose gel (Cat #50090, LONZA) on ice for higher resolution of bands separation results (Witt et al., 1993).
Generation of the mitochondrial cAMP biosensor Pink Flamindo - mtPF
PF vector, a biosensor for cAMP in mammalian cells (Harada et al., 2017) was purchased from Addgene (Cat #102356). In order to direct this sensor into the mitochondria, the 2MT signaling peptide was genetically introduced into PF vector using the 2mtGCaMP6 vector gifted from Diego De Stefani (Patron et al., 2014). A vector containing the cAMP biosensor together with the 2MT segment was constructed as follows: The 2MT segment was subcloned by PCR amplification of the 2mtGCaMP6 vector utilizing the following primers: forward, ATGCGAATTCCACCATGTCCGTCCTGAG and reverse GCTAGGATCCAGAACCAAGCTTCCCCTCCG (Patron et al., 2014). The PCR product (237 bp) was then ligated into the PF vector between the ECOR1 and BamH1 sites to generate the mtPF (Ohta et al., 2018).
Transfection
Transfection of SH-SY5Y and HEK293 cells was performed using the calcium phosphate precipitation protocol as previously described (Palty et al., 2004). mtPF mitochondrial localization in SH-SY5Y cells was determined using the mitochondrial marker CEPIA2-mt (Addgene, Cat #58218) as previously described (Kostic et al., 2015).
Viral constructs
Viral particles were prepared as described previously (Tevet and Gitler, 2016). Briefly, cDNAs of interest (mtPF and 2mtGCaMP6) were subcloned by restriction/ligation into a plasmid containing adeno-associated virus 2 (AAV2) inverted terminal repeats flanking a cassette consisting of the 0.47 kb human synapsin 1 promoter (hSyn) (Kugler et al., 2003), the woodchuck post-transcriptional regulatory element and the bovine growth hormone polyA signal. Viral particles were produced in HEK-293T cells (ATCC, CRL-3216) using both the pD1 and pD2 helper plasmids (Groh et al., 2008), which encode the rep/cap proteins of AAV1 and AAV2, respectively. Primary cultures of hippocampal neurons were infected at 5–7 day in vitro (DIV) and incubated for at least 7 days before imaging. Virion titer was individually adjusted to produce 75–90% infection efficiency.
In neurons, mitochondrial localization of mtPF was verified by co-localized with Mito-View 405 labeling (Biotium, Cat #70070-T), and 2mtGCaMP6 localization was verified by co-localization with hSyn:mito-DsRed2 fluorescence.
Fluorescence microscopy
Ion imaging and cAMP experiments were performed on imaging system consisted of Olympus IX73 inverted microscope and Cell-sense division 1 Olympus software (Wendenstrasse, Hamburg, Germany) CooLed 4000 LED monochromator, and Q imaging cooled Retiga 6000 camera (Surrey, British Columbia, Canada). We used magnification 10x for cytosolic and 20x for mitochondrial measurements. For live cytosolic Ca2+ (Fura2-AM) imaging we used (Zeiss), Polychrome V monochromator (TILL Photonics, Planegg, Germany) and a SensiCam cooled charge-coupled device (PCO, Kelheim, Germany) as previously described (Kostic et al., 2015).
Fluorescence measurements of the cell survival assay and the mitochondrial calcium monitoring during electrical stimulation were performed using the imaging system consisted of Nikon TiE inverted microscope driven by the NIS-elements software package (Nikon). The microscope was equipped with an Andor sCMOS camera (Oxford Instruments), a 40x 0.75 NA Super Fluor and a 60X 1.4 NA oil-immersion apochromatic objective objectives (Nikon), EGFP and Cy3 TE-series optical filter sets (Chroma) as well as a BFP and Cy5 filter set (Semrock) as previously described (Stavsky et al., 2021). For field stimulation, cultured neurons on coverslips were placed in a stimulation chamber (RC-49MFSH, Warner Instruments) and stimulated at an intensity of 10 V/cm using a stimulus isolation unit (SIU-102B, Warner Instruments). Stimulation duration and frequency was controlled by an isolated pulse stimulator (2100, A-M Systems).
Live mitochondria co-localizations and whole cell images were taken on a NIKON C2Plus laser unit docked to a Nikon Eclipse Ti unit of the confocal microscope by using a 20X and 60X oil immersion objective respectively, as described (Leyton-Jaimes et al., 2019).
Live fluorescence imaging
In all live fluorescent experiments, cells were pre-loaded with the indicated ion specific fluorescent dye at the indicated concentrations for 30 min at room temperature or 37°C using Ringer’s solution containing (in mM): 126 NaCl, 5.4 KCl (Sigma-Aldrich, Cat #P9333), 0.8 MgCl2, 20 HEPES, 1.8 CaCl2, 15 glucose (Gerbu, Cat #2028), with pH adjusted to 7.4 with NaOH (Sigma-Aldrich, Cat #221465) and supplemented with 0.1% bovine serum albumin (BSA, VWR, Cat #0332). After loading, cells were quickly and thoroughly washed in fresh dye-free Ringer’s solution for 30 minutes.
At the beginning of each experiment, cells were perfused with Ca2+-containing Ringer’s solution supplemented with 0.1% BSA. To trigger ionic responses, cells were perfused with the caffeine (Sigma, Cat #C0750) (2 mM); High concentrated potassium Ringer’s solution (High K+) containing: (in mM): 70 NaCl, 50 KCl, 0.8 MgCl2, 20 HEPES, 1.8 CaCl2, 15 glucose, with pH adjusted to 7.4 with NaOH; Norepinephrine (Levophed; norepinephrine bitartrate 4 mg/4 mL, Cat #NDC 0409-3375-04, Hospira, Inc.) (5μM), Vasopressin (Santa Cruz, Cat #sc-356188) (1μM). [Ca2+]c was monitored in cells loaded with Ca2+ specific ratiometric dye Fura2-AM (Sigma Aldrich, Cat #F0888) (1 μM), which were illuminated alternately with 340 nm and 380 nm excitations and imaged with a 510 nm long pass filter (Palty et al., 2004). [Ca2+]m was measured in cells loaded with Ca2+ specific dye Rhod2-AM (Biotium, Cat #50024) (1 μM) that preferentially localizes in mitochondria. Rhod2-AM was excited at 552 nm wavelength light and imaged with a 570 nm-long pass filter as previously described (Kostic et al., 2015). [Mg2+]m was measured in cells double loaded with Mg2+ specific dye Magnesium Green (Invitrogen, Cat #M3735) (2 μM) in a 1 h pre-incubation in 37°C and Mito Tracker Deep red as described previously, for live mitochondrial colocalization, measuring only mitochondrial Mg2+ currents were done by choosing mitoTracker Deep Red positive ROIs. [Mg2+]total was measured in neurons loaded only with Magnesium Green as described (Daw et al., 2020). Magnesium Green was excited by 440 nm and imaged with 480 nm emission filter as done previously (Daw et al., 2020). cAMP levels were monitored in cells pre- transfected/infected by PF using excitation of 545 nm and imaged with a 570 nm emission filter as described before (Harada et al., 2017). Calcium oscillations were analyzed as previously described (Samanta et al., 2018).
Bay 60-7550 (Santa Cruz, Cat #sc-396772A), H89 (Sigma, Cat #B1427) and KT 5720 (Cayman chemical, Cat #10011011) pre-treatments were performed as 1 h incubations prior to fluorescent imaging experiments in concentration of 1 μM, 10 μM, and 100 nM (Davies et al., 2000) respectively (Kostic et al., 2015; Monterisi et al., 2017). 100 μM of ATP (Amresco, Cat #0220) used for mitochondrial Ca2+ imaging and western blot in SH-SY5Y.
Live fluorescent imaging of permeabilized neurons was done by using 0.002% of Digitonin (Sigma, Cat #D141) containing solution by the protocol described previously (Roy et al., 2017).
Cell survival assay (TBOA test) and immunostaining
Triplicates of hippocampal neurons grown on coverslips were incubated with 80μM DL-threo-beta-benzyloxyaspartate (TBOA) (Tocris, Cat #1223) overnight as previously described (Yeh et al., 2017). Next day, randomly chosen coverslips were incubated for 1 hour in Neurobasal-medium with or without Bay 60-7550 (1μM). Following the Bay 60-7550 treatment, live cells were stained with 5 mM Sytox green (1:5,000) (Invitrogen, Cat #S7020) at 37°C and washed in Dulbesco Phospate Buffered Saline - DPBSx1 (Biological Industries, Cat# 02-020-1A). The following steps were performed at RT. Neurons were then fixed for 10 minutes with 4% paraformaldehyde (PFA, Electron microscopy sciences, Cat #BN15710), permeabilized with 0.1% TritonX-100 (Sigma Aldrich, Cat# T8787) for 2 minutes and blocked with 5% powdered milk (Sigma-Aldrich, Cat #70166) in DPBSx1 for 1 hour. Coverslips were incubated for 1 hour with a primary antibody (mouse monoclonal anti NeuN 1:100 EMD Millipore, Cat #MAB377), rinsed, incubated for 1 hour with a secondary antibody (anti-mouse IgG NorthenLights NL557, 1:1000, R&D systems, Cat #NL007) and rinsed. Coverslips were incubated with 1:10,000 DAPI (Sigma, Cat #D9542) for 15 min, rinsed and finally mounted with Epredia™ Immu-Mount (Thermo Fisher Scientific, Cat #9990402).
Random 5–10 images were taken from each coverslip. Neurons positively stained by NeuN were marked as areas of interest and counted manually. Counting was performed on 4–10 images from each repetition in both genotypes including presence or absence of treatment. Neurons were considered live if Sytox showed a weak fluorescence signal or no signal at all, or dead if Sytox green showed a strong fluorescence signal. The cell number of the imaged fields was counted blindly 3 times and averaged. The quantification represents percentage of live out of total number of cells in the field in all images taken.
For 2mtGCaMP6 mitochondrial localization, neurons were treated similarly and immunolabeled with a rabbit polyclonal anti β3-tubulin primary antibody (1:1000, Synaptic Systems, Cat #302-302) and an anti-rabbit IgG AlexaFluor 405 (1:1000, Abcam, Cat #ab175652) secondary antibody.
Immunoblotting
pS258-NCLX determination from whole-cell lysates, as previously described (Kostic et al., 2018). Briefly, SH-SY5Y cells were first washed with DPBSx1 and lysed for 20 min in lysis buffer [Hepes 50 mM, Ph = 7.4, NaCl 150 mM, EGTA 1 mM, Glycerol 10%, TritonX100 1%, MgCl2 10 mM, protease inhibitor (1:50) (Sigma cat # P2714) and PhosSTOP (1:10) (Roches, Cat #04 906 845 001)], all ice-cold. Lysates were then centrifuged at 4°C for 15 min at 1,000*g and supernatant was collected. Equal amounts of protein (20 μg) from lysates were resolved by SDS-PAGE (Molecular Biology, Cat #001981232300), transferred to the nitrocellulose membrane and Immunoblot analysis was performed. Forskolin (50 μM, 30 min) (Cayman, Cat #11018) was used as a positive control for Ps258-NCLX phosphorylation.
Object recognition test
Male C57Bl/6 (WT) mice and NCLX KO mice aged 8–14 weeks were tested for the effect of Bay 60-7550 on learning and memory using the Object Recognition (OR) test as previously described (Lueptow et al., 2016). Each genotype group was randomly divided into 2 groups treated with either the PDE2 inhibitor Bay 60-7550 or the vehicle control. The number of mice tested is WT control (17), WT Bay 60-7550 (16), NCLX KO, vehicle and Bay 60-7550 (11). Mice were tested individually in a transparent Plexiglas box (20 × 40 cm) and filmed from above for offline analysis using Ethovision© (Noldus, The Netherlands). Behavior in the object recognition test (ORT) was assessed as previously described (Lueptow et al., 2016). On the first day of the experiment, each mouse was placed individually in the apparatus without objects for 10 minutes to habituate to the novel environment (T0: habituation). On the second day, mice were given intraperitoneally (IP) injections of Bay 60-7550 (1 mg/kg) or vehicle [The PDE2 inhibitor Bay 60-7550 solution was dissolved in 85% saline, 10% kolliphor (Sigma, Cat #C5135-500G), 5% ethanol. The vehicle solution was dissolved in 90% saline, 10% kolliphor] 30 minutes prior to training and held in a separate cage in a different room. After 30 min, the mouse was placed in the arena in which two identical objects assembled from Lego-like blocks were placed in the test box at a distance of 16 cm from one another allowing the mouse to explore the object from all sides for 10 minutes (T1: training). Twenty-four hours later, the mice were tested in the same arena for 10 minutes with two objects: one familiar object (i.e., object used in T1) and a novel Lego object that had the same texture but differed in shape and size (T2: testing). Placement of the novel object was alternated between the two locations and counterbalanced within and between the groups. Between trials, the apparatus and each object were carefully cleaned with 80% ethanol to remove odor cues. The experimenter was blind to the treatment when testing the mice. All testing was done under low light conditions. It was expected that the mouse would explore the novel object more than the familiar object and that this difference would be greater in mice treated with Bay 60-7550, indicating that they better remembered the familiar object. Learning was assessed as the preference for the novel object over the familiar object on the third day (T3), as follows and as previously described (Lueptow et al., 2016), briefly: Discrimination Index (DI) was calculated for each mouse during T2: [DI= (novel object exploration time - familiar object exploration time)/(novel object exploration time+familiar object exploration time)]. The data were derived from analyzing the mouse’s exploration using the Ethovision® program (Noldus, The Netherlands). A zone surrounding each object was defined by a square within a perimeter of 1 cm above the object. The time spent in the Familiar object and Novel object zone was measured for each mouse. Sitting on the object was included; however, all the films were screened to determine that no mouse spent more than 10 sec sitting on an object or engaged in other behavior that was not explorative (e.g., self-grooming, sleeping). No mice were omitted on this basis. Two mice in the WT-vehicle group, which spent less than 30 sec in the New Object zone, were eliminated from the analysis. The data were analyzed by ANOVA for the effects of genotype and drug (vehicle vs Bay 60-7550) using the Statistica ® program.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all data shown in results section, at least 3 repetitions from independent cell cultures were conducted. In each of them traces were obtained from at least 3 samples of 3 regions of interest of at least 5 cells, in a non-blind manner and plotted using KaleidaGraph. All independent data points except for data shown in Figure 6, were included in the statistical analysis using 2 tailed unpaired Student’s t test. Only for data shown in Figure 6, individual data points were analyzed for normality using Shapiro-Wilk test followed by statistical analysis using 1-way ANOVA analysis (Tukey test). All experimental data was considered for statistical analysis. The rate of Ca2+ transport was derived by a linear fit of the absolute change in the fluorescence (ΔF) over a period of 10–20 s following initiation of apparent efflux/influx (ΔF/dt) from each graph. Rates from n- independent experiments (as described in legends to the figures) were averaged and displayed in bar graph format (mean ± SEM). GraphPad Prism 6 software was used for statistical analysis and Adobe Illustrator for the graphic design of the figures. All bar graphs were presented as averaged single sample values of (n) independent measurements ± SEM. A value of p < 0.05 accepted as statistically significant. Symbols of significance used: ns p > 0.05, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-NeuN | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Donkey Anti-Mouse IgG NL557 Affinity Purified | R&D Systems | Cat# NL007; RRID: AB_663768 |
| Anti-beta3-tubulin | Synaptic Systems | Cat# 302-302; RRID: AB_10637424 |
| Goat Anti-Rabbit IgG H&L (Alexa Fluor® 405) | Abcam | Cat# ab175652; RRID: AB_2687498 |
| Bacterial and virus strains | ||
| AAV1/2 hSyn:2mtPF | Ben Gurion University | This paper; Kugler et al. (2003) |
| AAV1/2 hSyn:2mtGCaMP6 | Ben Gurion University | This paper; Kugler et al. (2003) |
| AAV1/2 hSyn:mito-DsRed2 | Ben Gurion University | Stavsky et al. 2021 |
| Biological samples | ||
| Primary Hippocampi of 0–3 days old mouse pups | Ben Gurion University | This paper |
| Chemicals, peptides, and recombinant proteins | ||
| Protease inhibitor | Sigma Aldrich | Cat# P2714 |
| PhosSTOP | Roches | Cat# 04 906 845 001 |
| Forskolin | Cayman | Cat# 11018 |
| DL-threo-beta-benzyloxyaspartate (TBOA) | Tocris | Cat# 1223 |
| Bay 60-7550 | Santa Cruz, | Cat# sc-396772A |
| H89 | Sigma Aldrich | Cat# B1427 |
| KT 5720 | Cayman chemical | Cat# 10011011 |
| ATP | Amresco | Cat# 0220 |
| Digitonin | Sigma Aldrich | Cat# D141 |
| Mito-View 405 | Biotium | Cat# 70070-T |
| Vasopressin | Santa Cruz | Cat# sc-356188 |
| Norepinephrine | Levophed | Cat# NDC 0409-3375-04 |
| Fura2-AM | Sigma Aldrich | Cat# F0888 |
| Rhod2-AM | Biotium | Cat# 50024 |
| Magnesium Green | Invitrogen | Cat# M3735 |
| MitoTracker Deep red | Invitrogen | Cat# M22426 |
| Sytox green | Invitrogen | Cat# S7020 |
| CEPIA2-mt | Addgene | Cat# 58218; Kostic et al. (2015) |
| Kolliphor | Sigma Aldrich | Cat# C5135-500G |
| DAPI | Sigma-Aldrich | Cat# D9542 |
| Experimental models: Cell lines | ||
| Primary culture of hippocampal neurons | Ben Gurion University | For this paper |
| SH-SY5Y | ATCC | Cat# CRL-2266 |
| HEK-293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| C57BL6 Wild-Type | Envigo | Cat# 6JRCCH5B043 |
| NCLX KO (C57BL6) | Jackson Laboratories | Cat# 026242 |
| Oligonucleotides | ||
| ATGCGAATTCCACCATGTCCGTCCTGAG | HyLabs | For this paper |
| GCTAGGATCCAGAACCAAGCTTCCCCTCCG | HyLabs | For this paper |
| Recombinant DNA | ||
| mtPF | De Stefani Diego, Department of Biomedical Sciences, University of Padua | Patron et al. 2014 |
| PF Vector | Addgene; Harada et al. (2017) | Cat# 102356 |
| 2mtGCaMP6 | De Stefani Diego, Department of Biomedical Sciences, University of Padua | Patron et al. 2014 |
| Human synapsin 1 promoter (hSyn) | Kugler et al. 2003 | N/A |
| pD1 and pD2 helper plasmids | Groh et al. 2008 | N/A |
| Software and algorithms | ||
| Ethovision© | Noldus, The Netherlands | https://www.noldus.com/ethovision-xt |
| KaleidaGraph | https://www.synergy.com | N/A |
| GraphPad Prism 6 | www.graphpad.com | N/A |
| Adobe Illustrator | https://www.adobe.com/products/illustrator | N/A |
Highlights.
Caffeine-activated mitochondrial Ca2+ efflux by NCLX in hippocampal neurons
Caffeine is acting by inhibiting PDE2
Inhibition of PDE2 triggers NCLX phosphorylating, thereby accelerating NCLX activity
PDE2-dependent activation of NCLX protects against excitotoxicity and enhances learning
ACKNOWLEDGMENTS
This study was funded by ISF 1424/17, ISF 1763/21, and DIP SE 2372/1-1 to I.S. and NIH grant (FR1Ag065628-O1A1) to G.S. and I.S.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111772.
REFERENCES
- Acin-Perez R, Russwurm M, Gunnewig K, Gertz M, Zoidl G, Ramos L, Buck J, Levin LR, Rassow J, Manfredi G, and Steegborn C (2011). A phosphodiesterase 2A isoform localized to mitochondria regulates respiration. J. Biol. Chem 286, 30423–30432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, and Manfredi G (2009). Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 9, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assali EA, Jones AE, Veliova M, Acín-Pérez R, Taha M, Miller N, Shum M, Oliveira MF, Las G, Liesa M, et al. (2020). NCLX prevents cell death during adrenergic activation of the brown adipose tissue. Nat. Commun 11, 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatti SK, O’Keefe JH, and Lavie CJ (2013). Coffee and tea: perks for health and longevity? Curr. Opin. Clin. Nutr. Metab. Care 16, 688–697. [DOI] [PubMed] [Google Scholar]
- Boess FG, Hendrix M, van der Staay FJ, Erb C, Schreiber R, van Staveren W, de Vente J, Prickaerts J, Blokland A, and Koenig G (2004). Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 47, 1081–1092. [DOI] [PubMed] [Google Scholar]
- Bubb KJ, Trinder SL, Baliga RS, Patel J, Clapp LH, MacAllister RJ, and Hobbs AJ (2014). Inhibition of phosphodiesterase 2 augments cGMP and cAMP signaling to ameliorate pulmonary hypertension. Circulation 130, 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin JD (2004). Mechanisms of action of PDE5 inhibition in erectile dysfunction. Int. J. Impot. Res 16 (Suppl 1), S4–S7. [DOI] [PubMed] [Google Scholar]
- Corsini S,Tortora M, and Nistri A (2016). Nicotinic receptor activation contrasts pathophysiological bursting and neurodegeneration evoked by glutamate uptake block on rat hypoglossal motoneurons. J. Physiol 594, 6777–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, and Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw CC, Ramachandran K, Enslow BT, Maity S, Bursic B, Novello MJ, Rubannelsonkumar CS, Mashal AH, Ravichandran J, Bakewell TM, et al. (2020). Lactate elicits ER-mitochondrial Mg(2+) dynamics to integrate cellular metabolism. Cell 183, 474–489.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, and Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Zhang C, Masood A, Li J, Sun J, Nadeem A, Zhang HT, O’ Donnell JM, and Xu Y (2014). Protective effects of phosphodiesterase 2 inhibitor on depression- and anxiety-like behaviors: involvement of antioxidant and anti-apoptotic mechanisms. Behav. Brain Res 268, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Takagishi Y, Feng J, Ren Y, Rodriguiz RM, Wetsel WC, Greengard P, and Augustine GJ (2004a). Different presynaptic roles of synapsins at excitatory and inhibitory synapses. J. Neurosci 24, 11368–11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Xu Y, Kao HT, Lin D, Lim S, Feng J, Greengard P, and Augustine GJ (2004b). Molecular determinants of synapsin targeting to presynaptic terminals. J. Neurosci 24, 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez L, and Breitenbucher JG (2013). PDE2 inhibition: potential for the treatment of cognitive disorders. Bioorg. Med. Chem. Lett 23, 6522–6527. [DOI] [PubMed] [Google Scholar]
- Gooderham M, and Papp K (2015). Selective phosphodiesterase inhibitors for psoriasis: focus on apremilast. BioDrugs 29, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh A, de Kock CPJ, Wimmer VC, Sakmann B, and Kuner T (2008). Driver or coincidence detector: modal switch of a corticothalamic giant synapse controlled by spontaneous activity and short-term depression. J. Neurosci 28, 9652–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada K, Ito M, Wang X, Tanaka M, Wongso D, Konno A, Hirai H, Hirase H, Tsuboi T, and Kitaguchi T (2017). Red fluorescent protein-based cAMP indicator applicable to optogenetics and in vivo imaging. Sci. Rep 7, 7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemnes AR, and Champion HC (2006). Sildenafil, a PDE5 inhibitor, in the treatment of pulmonary hypertension. Expert Rev. Cardiovasc Ther 4, 293–300. [DOI] [PubMed] [Google Scholar]
- Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D, and Elrod JW (2019). Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun 10, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Kostic M, Katoshevski T, and Sekler I (2018). Allosteric regulation of NCLX by mitochondrial membrane potential links the metabolic state and Ca(2+) signaling in mitochondria. Cell Rep. 25, 3465–3475.e4. [DOI] [PubMed] [Google Scholar]
- Kostic M, Ludtmann MHR, Bading H, Hershfinkel M, Steer E, Chu CT, Abramov AY, and Sekler I (2015). PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 13, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku BM, Lee YK, Jeong JY, Ryu J, Choi J, Kim JS, Cho YW, Roh GS, Kim HJ, Cho GJ, et al. (2011). Caffeine inhibits cell proliferation and regulates PKA/GSK3beta pathways in U87MG human glioma cells. Mol. Cells 31, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kügler S, Kilic E, and Bähr M (2003). Human synapsin 1 gene promoter confers highly neuron-specific long-term transgene expression from an adenoviral vector in the adult rat brain depending on the transduced area. Gene Ther. 10, 337–347. [DOI] [PubMed] [Google Scholar]
- Leyton-Jaimes MF, Kahn J, and Israelson A (2019). AAV2/9-mediated overexpression of MIF inhibits SOD1 misfolding, delays disease onset, and extends survival in mouse models of ALS. Proc. Natl. Acad. Sci. USA 116, 14755–14760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logrip ML (2015). Phosphodiesterase regulation of alcohol drinking in rodents. Alcohol 49, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lueptow LM, Zhan CG, and O’Donnell JM (2016). Cyclic GMP-mediated memory enhancement in the object recognition test by inhibitors of phosphodiesterase-2 in mice. Psychopharmacology 233, 447–456. [DOI] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. (2017). The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature 545, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP (2007). Mitochondrial regulation of neuronal plasticity. Neurochem. Res 32, 707–715. [DOI] [PubMed] [Google Scholar]
- Monterisi S, Lobo MJ, Livie C, Castle JC, Weinberger M, Baillie G, Surdo NC, Musheshe N, Stangherlin A, Gottlieb E, et al. (2017). PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. Elife 6, e21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Furuta T, Nagai T, and Horikawa K (2018). Red fluorescent cAMP indicator with increased affinity and expanded dynamic range. Sci. Rep 8, 1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Hershfinkel M, and Sekler I (2012). Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J. Biol. Chem 287, 31650–31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, Silverman WF, Argaman M, and Sekler I (2004). Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem 279, 25234–25240. [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 107, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannaccione A, Piccialli I, Secondo A, Ciccone R, Molinaro P, Boscia F, and Annunziato L (2020). The Na(+)/Ca(2+)exchanger in Alzheimer’s disease. Cell Calcium 87, 102190. [DOI] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, and Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poburko D, Liao CH, van Breemen C, and Demaurex N (2009). Mitochondrial regulation of sarcoplasmic reticulum Ca2+ content in vascular smooth muscle cells. Circ. Res 104, 104–112. [DOI] [PubMed] [Google Scholar]
- Rasola A, and Bernardi P (2011). Mitochondrial permeability transition in Ca(2+)-dependent apoptosis and necrosis. Cell Calcium 50, 222–233. [DOI] [PubMed] [Google Scholar]
- Ribeiro JA, and Sebastião AM (2010). Caffeine and adenosine. J. Alzheimers Dis 20, S3–S15. [DOI] [PubMed] [Google Scholar]
- Roy S, Dey K, Hershfinkel M, Ohana E, and Sekler I (2017). Identification of residues that control Li(+) versus Na(+) dependent Ca(2+) exchange at the transport site of the mitochondrial NCLX. Biochim. Biophys. Acta Mol. Cell Res 1864, 997–1008. [DOI] [PubMed] [Google Scholar]
- Rutter GA (1990). Ca2(+)-binding to citrate cycle dehydrogenases. Int. J. Biochem 22, 1081–1088. [DOI] [PubMed] [Google Scholar]
- Samanta K, Mirams GR, and Parekh AB (2018). Sequential forward and reverse transport of the Na(+) Ca(2+) exchanger generates Ca(2+) oscillations within mitochondria. Nat. Commun 9, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang L, Dick IE, and Yue DT (2016). Protein kinase A modulation of CaV1.4 calcium channels. Nat. Commun 7, 12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savai R, Pullamsetti SS, Banat GA, Weissmann N, Ghofrani HA, Grimminger F, and Schermuly RT (2010). Targeting cancer with phosphodiesterase inhibitors. Expert Opin. Investig. Drugs 19, 117–131. [DOI] [PubMed] [Google Scholar]
- Sharifullina E, and Nistri A (2006). Glutamate uptake block triggers deadly rhythmic bursting of neonatal rat hypoglossal motoneurons. J. Physiol 572, 407–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LM, Meyer E, Milani H, Steinbusch HWM, Prickaerts J, and de Oliveira RMW (2017). The phosphodiesterase type 2 inhibitor BAY 60-7550 reverses functional impairments induced by brain ischemia by decreasing hippocampal neurodegeneration and enhancing hippocampal neuronal plasticity. Eur. J. Neurosci 45, 510–520. [DOI] [PubMed] [Google Scholar]
- Stavsky A, Stoler O, Kostic M, Katoshevsky T, Assali EA, Savic I, Amitai Y, Prokisch H, Leiz S, Daumer-Haas C, et al. (2021). Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol 4, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Technikova-Dobrova Z, Sardanelli AM, Speranza F, Scacco S, Signorile A, Lorusso V, and Papa S (2001). Cyclic adenosine monophosphate-dependent phosphorylation of mammalian mitochondrial proteins: enzyme and substrate characterization and functional role. Biochemistry 40, 13941–13947. [DOI] [PubMed] [Google Scholar]
- Tevet Y, and Gitler D (2016). Using FRAP or FRAPA to visualize the movement of fluorescently labeled proteins or cellular organelles in live cultured neurons transformed with adeno-associated viruses. Methods Mol. Biol 1474, 125–151. [DOI] [PubMed] [Google Scholar]
- Traaseth N, Elfering S, Solien J, Haynes V, and Giulivi C (2004). Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta 1658, 64–71. [DOI] [PubMed] [Google Scholar]
- Vercesi AE, Bernardes CF, Hoffmann ME, Gadelha FR, and Docampo R (1991). Digitonin permeabilization does not affect mitochondrial function and allows the determination of the mitochondrial membrane potential of Trypanosoma cruzi in situ. J. Biol. Chem 266, 14431–14434. [PubMed] [Google Scholar]
- Vettel C, Lindner M, Dewenter M, Lorenz K, Schanbacher C, Riedel M, Lämmle S, Meinecke S, Mason FE, Sossalla S, et al. (2017). Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ. Res 120, 120–132. [DOI] [PubMed] [Google Scholar]
- Walz B, Baumann O, Zimmermann B, and Ciriacy-Wantrup EV (1995). Caffeine- and ryanodine-sensitive Ca(2+)-induced Ca2+ release from the endoplasmic reticulum in honeybee photoreceptors. J. Gen. Physiol. 105, 537–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witt M, Jaruzelska J, Kuczora I, Matuszak R, Cichy W, and Borski K (1993). A simplified method for detection of the mutations predominantly causing cystic fibrosis and phenylketonuria in Polish families. Clin. Genet 44, 44–45. [DOI] [PubMed] [Google Scholar]
- Yeh CY, Bulas AM, Moutal A, Saloman JL, Hartnett KA, Anderson CT, Tzounopoulos T, Sun D, Khanna R, and Aizenman E (2017). Targeting a potassium channel/syntaxin interaction ameliorates cell death in ischemic stroke. J. Neurosci 37, 5648–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, and Movsesian MA (2007). cAMP and cGMP signaling crosstalk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res 100, 1569–1578. [DOI] [PubMed] [Google Scholar]
- Zeitlin R, Patel S, Burgess S, Arendash GW, and Echeverria V (2011). Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res. 1417, 127–136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.