

Abstract
Background and aims:
Overdose of acetaminophen (APAP) is the major cause of acute liver failure in the Western world. We report a novel signaling interaction between Hepatocyte Nuclear Factor 4 alpha (HNF4α) cMyc and Nrf2 during liver injury and regeneration after APAP overdose.
Approach and Results:
APAP-induced liver injury and regeneration were studied in male C57BL/6J (WT) mice, hepatocyte specific HNF4α knockout mice (HNF4α -KO) and HNF4α-cMyc double knockout mice (DKO). C57BL/6J mice treated with 300 mg/kg maintained nuclear HNF4α expression and exhibited liver regeneration, resulting in recovery. However, treatment with 600 mg/kg APAP, where liver regeneration was inhibited and recovery was delayed, showed rapid decline in HNF4α expression. HNF4α-KO mice developed significantly higher liver injury due to delayed GSH recovery after APAP overdose. HNF4α -KO mice also exhibited significant induction of cMyc, and deletion of cMyc in HNF4α -KO mice (DKO mice) reduced APAP-induced liver injury. The DKO mice had significantly faster GSH replenishment due to rapid induction in Gclc and Gclm genes. Co-IP and ChIP analysis revealed that HNF4α interacts with Nrf2 and affects its DNA binding. Further, DKO mice showed significantly faster initiation of cell proliferation resulting in rapid liver regeneration and recovery.
Conclusions:
These data show that HNF4α interacts with Nrf2 and promotes GSH replenishment aiding in recovery from APAP-induced liver injury, a process inhibited by cMyc. These studies indicate that maintaining HNF4α function is critical for regeneration and recovery after APAP overdose.
Keywords: DILI, cell proliferation, regeneration, HNF4α, cMyc, Nrf2
Lay summary:
Hepatocyte nuclear factor 4 alpha (HNF4α) is a master regulator of hepatic differentiation and plays important role in proliferation and disease progression. These studies show that maintaining HNF4α expression is essential for liver regeneration to take place after acetaminophen overdose induced acute liver injury. Loss of HNF4α induces cMyc, resulting in inhibition of regeneration and delayed recovery via Nrf2 suppression.
Introduction:
Acetaminophen (APAP), a widely used over the counter analgesic and antipyretic drug, is safe and effective at therapeutic doses. However, overdose of APAP results in acute liver injury, which can progress to acute liver failure. In fact, APAP overdose is the leading cause of liver failure in the Western world [1–3]. The mechanism of liver injury after APAP overdose involves a series of steps that begin with APAP bioactivation by CYP2E1 to its reactive metabolite N-acetyl benzoquinone imine (NAPQI). NAPQI is detoxified by glutathione (GSH) in hepatocytes but during APAP overdose GSH levels quickly deplete leaving extensive NAPQI for protein binding in the cells, which triggers a cascade of cellular events including oxidative stress, activation of c-Jun N terminal kinase (JNK), mitochondrial damage, and eventually hepatocyte necrosis [4–7]. Cell death is followed by compensatory liver regeneration, which is an essential step in recovery from APAP overdose [5, 8–10]. Identifying mechanisms that stimulate liver regeneration after APAP overdose has great potential to develop novel therapeutic strategies.
Hepatocyte nuclear factor 4 alpha (HNF4α) is a highly conserved orphan nuclear receptor that regulates the liver specific expression of genes involved in glycolysis, gluconeogenesis, ureagenesis, fatty acid metabolism, bile acid synthesis, drug metabolism, and blood coagulation [11–14]. It is considered the master regulator of hepatocyte differentiation because it regulates the majority of hepatocyte specific functions. Further, HNF4α functions as a tumor suppressor in the liver and is involved in the regulation of hepatocyte proliferation [15–17]. In resting hepatocytes, HNF4α regulates the genes involved in differentiation and suppresses expression of pro-mitogenic genes. Loss of HNF4α causes dedifferentiation and increased proliferation [18]. Deletion of HNF4α results in dysregulation of liver regeneration after partial hepatectomy due to failure of newly divided hepatocytes to re-differentiate into functional cells [19]. However, the role of HNF4α in drug-induced liver injury in general, and in APAP-induced acute liver failure is not known, which was investigated in these studies. Our studies have revealed a novel signaling network of HNF4α and cMyc, which is critical for recovery from APAP overdose.
Materials and Methods:
Animals, Treatment and Tissue Harvesting
All animals were housed in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care at the University of Kansas Medical Center under a standard 12 h light/dark cycle with free access to chow and water. All studies were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center. For all APAP experiments, mice were fasted overnight, and APAP (Sigma, St Louis, Missouri) dissolved in warm 0.9% saline was administered intraperitoneally (i.p.). Eight-week-old male C57BL/6J mice were purchased from Jackson Laboratories and used for incremental dose studies. Mice were treated with either 300 or 600 mg/kg APAP, and euthanized at 0, 1, 3, 6, 12, 24, 72, and 96 h following APAP treatment. Wild type (WT) and hepatocyte specific HNF4α -KO were generated as described before by injecting AAV8-TBG-eGFP or AAV8-TBG-CRE (Vector Biolabs), respectively to HNF4α -floxed mice [19]. To study the effect of both HNF4α and cMyc deletion on liver regeneration, HNF4α-cMyc double floxed mice were injected i.p. with AAV8-TBG-eGFP or AAV8-TBG-CRE, resulting in WT and hepatocyte specific HNF4α-cMyc double KO (DKO) animals, respectively. All knockout mouse experiments were performed one week after AAV8 injections. Mice were fasted overnight, treated with 300 mg/kg of APAP, and euthanized at 0, 1, 3, 6, 12, 24, and 72 h following APAP treatment to obtain blood and livers. Parts of liver tissue were processed separately to obtain paraffin sections, frozen sections, RNA samples, and nuclear, cytoplasmic, and total protein extracts, as described previously [20]. More details are located in the supplementary materials.
Statistical Analysis
Data presented in the form of bar graphs show mean ± standard error of mean. GraphPad Prism 9 was used to graph and calculate statistics. One and Two Way ANOVA and Student’s t test was applied to all analyses with p < 0.05 being considered significant.
Results:
Rapid HNF4α nuclear to cytoplasmic translocation after a non-regenerating dose of APAP
We used the incremental dose model developed in our laboratory [21] to investigate changes in HNF4α expression following an APAP overdose. In this experiment, mice were treated with either a 300 mg/kg (referred as regenerating dose), after which they experienced significant liver injury but recovered due to robust liver regeneration, or they were treated with a 600 mg/kg dose of APAP (referred as non-regenerating dose), which also induces significant liver injury but results in a considerable delay in regeneration. Serum ALT levels indicated that during the first 6 h after APAP administration, mice treated with either regenerating or non-regenerating doses developed significant but similar liver injury consistent with previous studies [21] (Fig. 1A). HNF4α mRNA remained unchanged at 1 h but then significantly declined at 3 and 6 h after administration of the regenerating dose. However, HNF4α mRNA declined by 50% within 1 h after treatment with the non-regenerating dose of APAP and remained low at subsequent time points (Fig. 1B). Consistent with these results, Western blot analysis showed rapid nuclear to cytoplasmic translocation of HNF4α following the non-regenerating dose but not after the regenerating dose (Fig. 1C–D, Supplementary Fig. 1A–B). The western blot results were corroborated by HNF4α immunohistochemistry (Fig. 1E).
Figure 1: Rapid HNF4α nuclear to cytoplasmic translocation after non-regenerating dose of APAP.
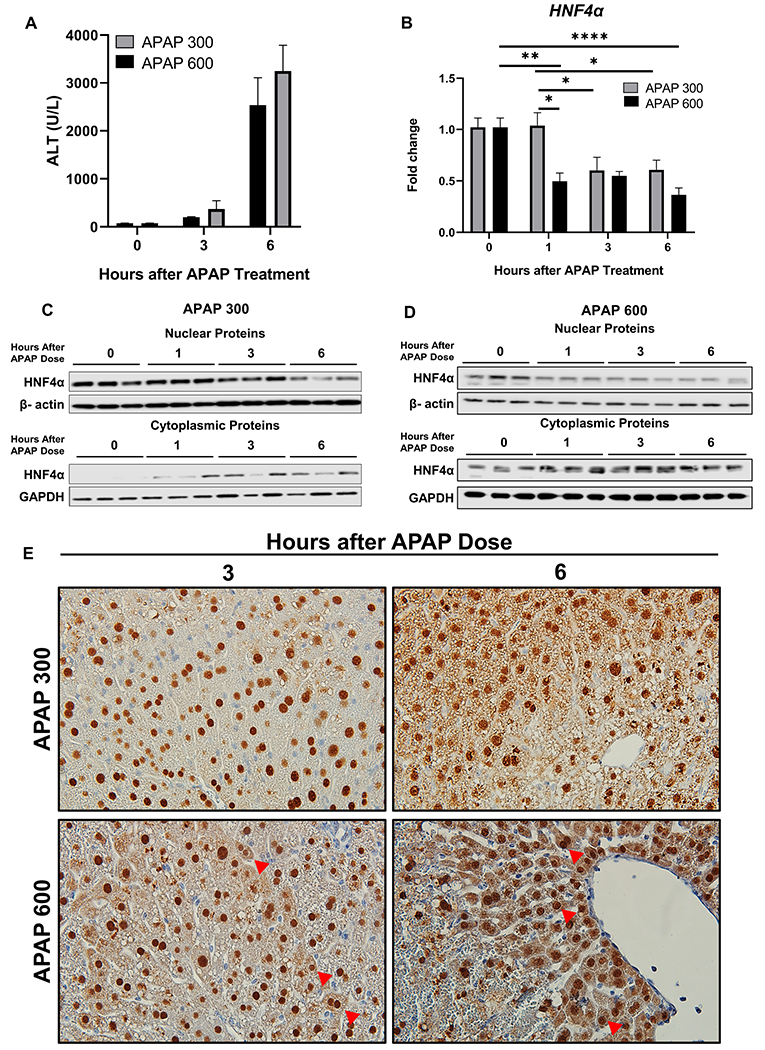
(A) Serum ALT levels (B) qPCR analysis of HNF4α mRNA at various time points after administration of 300 mg/kg and 600 mg/kg APAP in C57BL/6J mice. (C) Western blot analysis of HNF4α using nuclear and cytoplasmic extracts showing nuclear to cytoplasmic translocation of HNF4α after 300 mg/kg dose of APAP and (D) 600 mg/kg dose of APAP. (E) Representative photomicrographs of HNF4α immunohistochemistry staining at 3 and 6 h after 300 mg/kg and 600 mg/kg dose of APAP. Arrows pointing to cytoplasmic HNF4α staining. Original magnification, 600X; * Indicates significant difference at, *=P<0.05, **=P<0.01, ****=P<0.0001
HNF4α-KO mice show complete recovery following APAP overdose despite higher initial liver injury
Hepatocyte specific HNF4α-KO mice were successfully generated by injecting HNF4αfl/fl mice with AAV8-TBG-CRE as previously described [19]. HNF4αfl/fl mice treated with AAV8-TBG-GFP were used as controls (referred to as WT). Western blot analysis of microsomes isolated from WT and HNF4α -KO mice showed similar basal CYP2E1 levels, which plays an important role in the bioactivation of APAP (Fig 2A). Further, hepatic GSH levels, which detoxifies NAPQI via conjugation, were similar in the liver sections of both WT and HNF4α-KO mice (Fig. 2B). Liver injury was studied in WT and HNF4α -KO mice after 300 mg/kg dose of APAP over a time course of 0 to 96 h. Serum ALT (Fig. 2C) and histopathological analysis of H&E-stained paraffin sections (Fig. 2D, Supplementary Fig. 2A) showed significantly higher liver injury in HNF4α -KO mice as compared to the WT mice at 6 h after APAP overdose. Despite this higher injury, HNF4α-KO mice recovered at the same time as the WT mice i.e. 48 h post-APAP administration.
Figure 2: Kinetics of APAP-induced liver injury in HNF4α-KO mice.
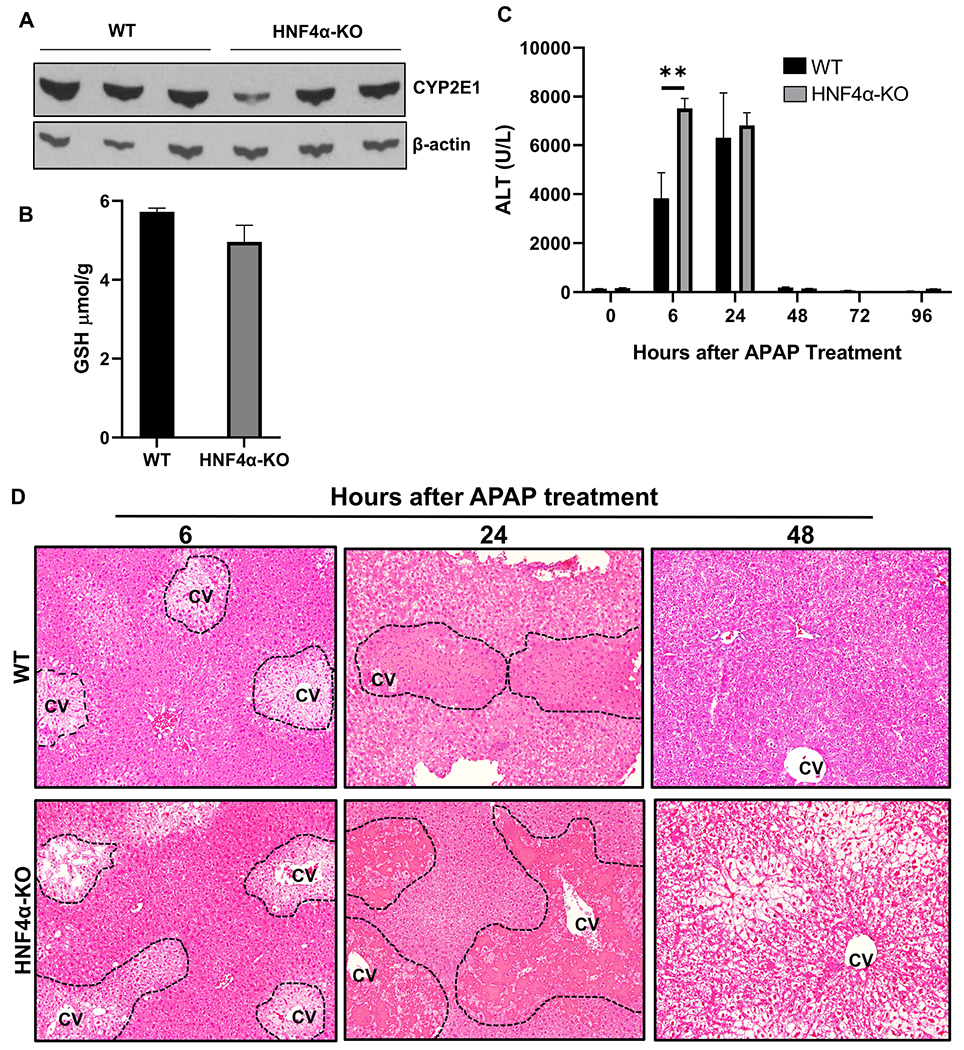
(A) Western blot analysis of hepatic CYP2E1 and (B) Hepatic GSH levels in WT and HNF4α KO mice at 0 h time point (C) Serum ALT levels and (D) representative photomicrographs of H&E-stained liver sections of WT and HNF4α-KO mice treated with 300 mg/kg APAP. Dotted lines demark area of necrosis. CV, central vein; Original magnification, 200X; *=P<0.05
Mechanisms of APAP-induced liver injury in HNF4α-KO mice
Rapid GSH depletion followed by gradual replenishment is an important part of the mechanism of APAP-induced liver injury. GSH depletion at 1 h after APAP treatment was similar between WT and HNF4α-KO mice. WT mice exhibited a rapid GSH replenishment and GSH levels returned to pre-APAP treatment levels within 6 h after APAP treatment. However, GSH replenishment was significantly lower in HNF4α -KO mice at 6 h following APAP administration. GSH levels were again equal at 24 h post-APAP treatment in WT and HNF4α -KO mice (Fig. 3A). APAP-induced necrotic cell death involves a cascade of molecular events including APAP-protein adduct formation, reactive oxygen species (ROS) generation because of the excessive amount of free NAPQI, and activation of JNK, RIP, and GSK3β Kinases, which ultimately leads to mitochondrial oxidative stress and injury [22]. Surprisingly, HNF4α-KO mice did not show increased JNK, RIP, or GSK3β activation (Fig. 3–D, Supplementary Fig. 2B–C). Loss of mitochondrial integrity due to mitochondrial permeability transition and release of endonucleases is critical part of APAP-induced cell necrosis [22]. We investigated changes in mitochondrial integrity in WT and HNF4α-KO mice after APAP treatment. While Apoptosis-inducing factor (AIF), Cytochrome C changes were similar between WT and HNF4α-KO mice, significantly higher cytoplasmic SMAC and endonuclease G was observed in HNF4α-KO mice at 6h after APAP (Fig. 3E–F).
Figure 3: Mechanisms of liver regeneration in HNF4α-KO mice.
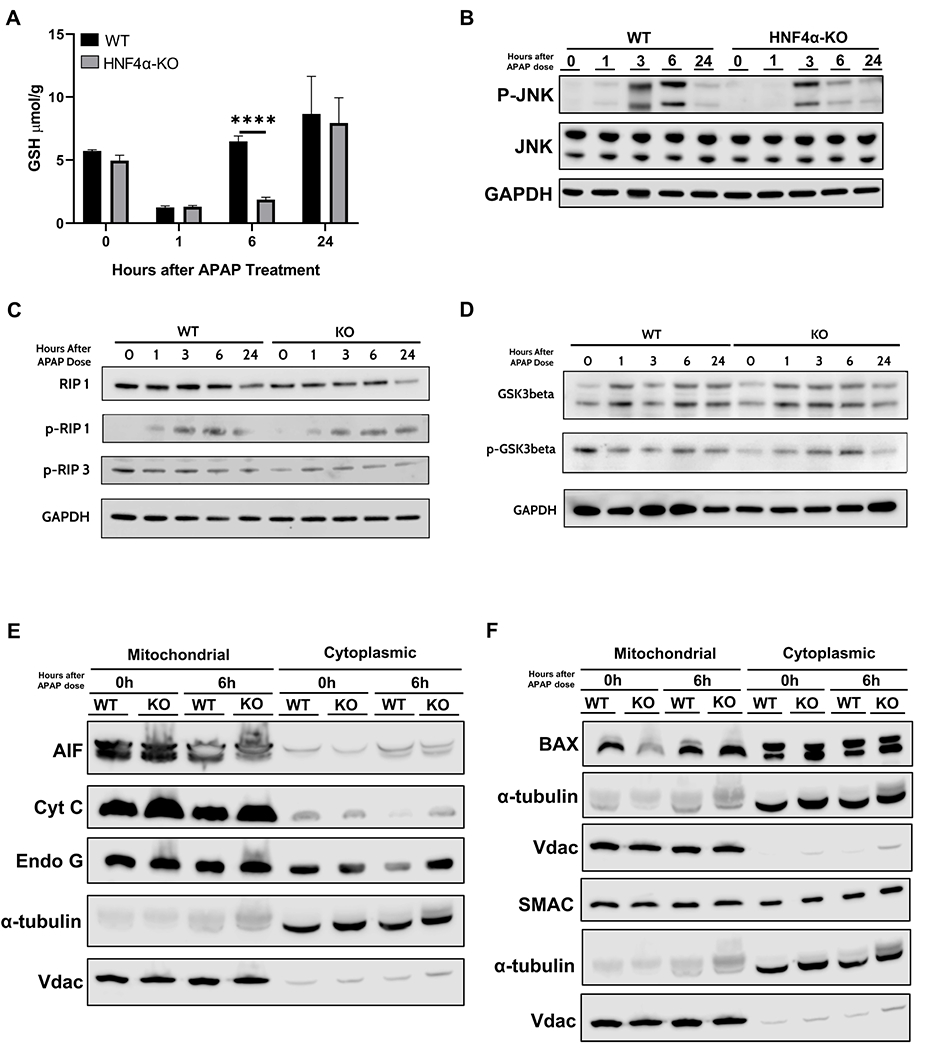
(A) Total glutathione levels in liver lysates of WT and HNF4α-KO mice at various time points after APAP 300 mg/kg dose. Western blot analysis of (B) total and phospho-JNK, (C) total and phospho-RIP1 and phospho-RIP3, and (D) total and phospho-GSK3β performed using pooled (n= 3-5) liver lysates and (E) AIF, Cyt C, Endo G (F) BAX and SMAC performed using pooled (n=3-5) liver mitochondrial and cytoplasmic fractions of WT and HNF4α-KO mice treated with APAP 300 mg/kg. *=P<0.05
Sustained cell proliferation in HNF4α-KO mice after APAP overdose accompanied by cMyc and CylinD1 induction
Surprisingly, all HNF4α mice recovered despite high initial liver injury. We investigated compensatory cell proliferation and core cell cycle machinery using Western blot analysis. HNF4α-KO mice had significant ongoing proliferation as demonstrated by a significant increase in the hepatic protein levels of the proliferation marker PCNA at 0 h (Fig. 4A). This finding was consistent with previous reports by our group [15]. PCNA levels remained higher throughout the time course. (Fig. 4B). Consistent with previous findings, HNF4α -KO mice also showed significant induction in protein and mRNA of cMyc and CyclinD1, both major drivers of hepatocyte proliferation (Fig. 4C and D).
Figure 4: Sustained cell proliferation in HNF4α-KO mice after APAP overdose accompanied by cMyc and CylinD1 induction.
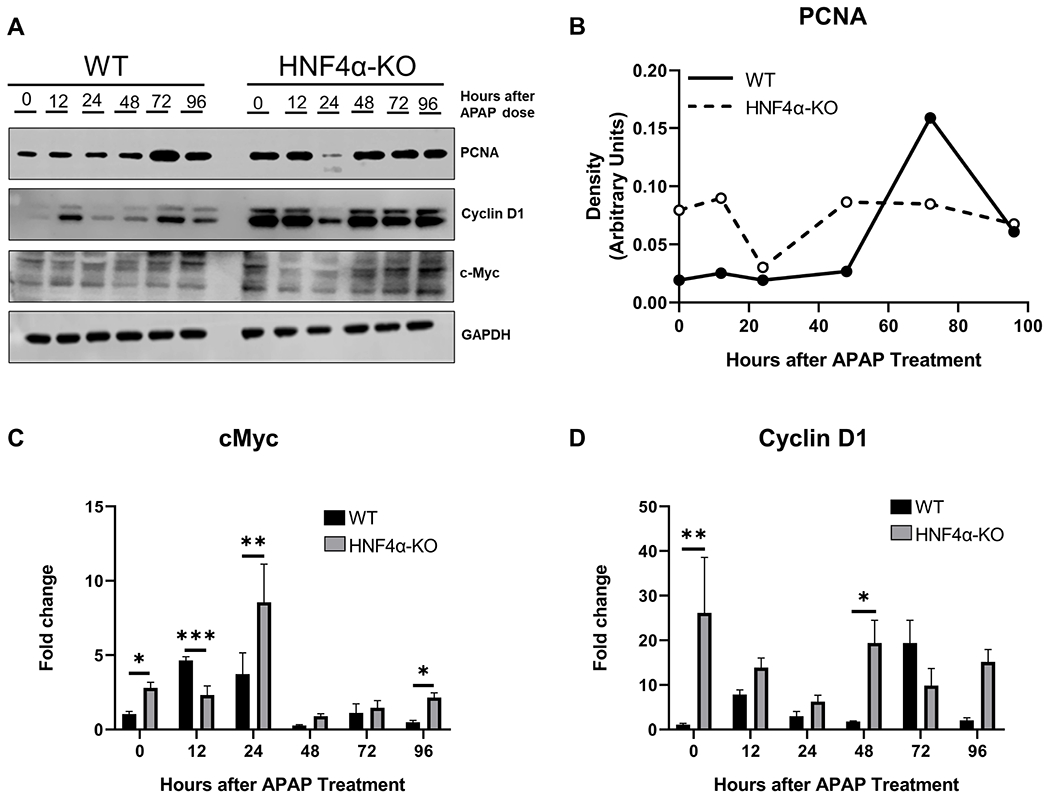
(A) Western blot analysis of proliferating cell nuclear antigen (PCNA), cMyc and Cyclin D1 performed using pooled (n=3-5) liver lysates of WT and HNF4α-KO mice treated with APAP 300 mg/kg. (B) Densitometric analysis of protein levels of PCNA in WT and HNF4α-KO mice. (C) qPCR analysis of cMyc and (D) Cyclin D1. *=P<0.05, **=P<0.01, ***=P<0.001
cMyc deletion in HNF4α -KO mice reduced APAP induced liver injury
To determine whether the response of HNF4α-KO mice to an APAP overdose was due to induction of cMyc, we generated hepatocyte specific HNF4α-cMyc double knock out (DKO) mice as described in the methods section (Fig. 5A, Supplementary Fig. 3A). Western blot analysis showed that basal CYP2E1 levels (Fig. 5B), and hepatic GSH levels (Fig. 5C) were similar in both WT and DKO mice. APAP-induced liver injury and recovery were studied in WT and DKO mice after a 300 mg/kg dose of APAP. Serum ALT and histopathological analysis of H&E-stained paraffin sections revealed equal liver injury in WT and DKO mice 6 h after APAP. However, DKO mice exhibited significantly lower liver injury as compared to WT mice at 24 h post-APAP, indicating a significantly faster recovery (Fig. 5D, 5E Supplementary Fig.3B, 4A–B).
Figure 5: cMyc deletion in HNF4α-KO mice reduced APAP induced liver injury.
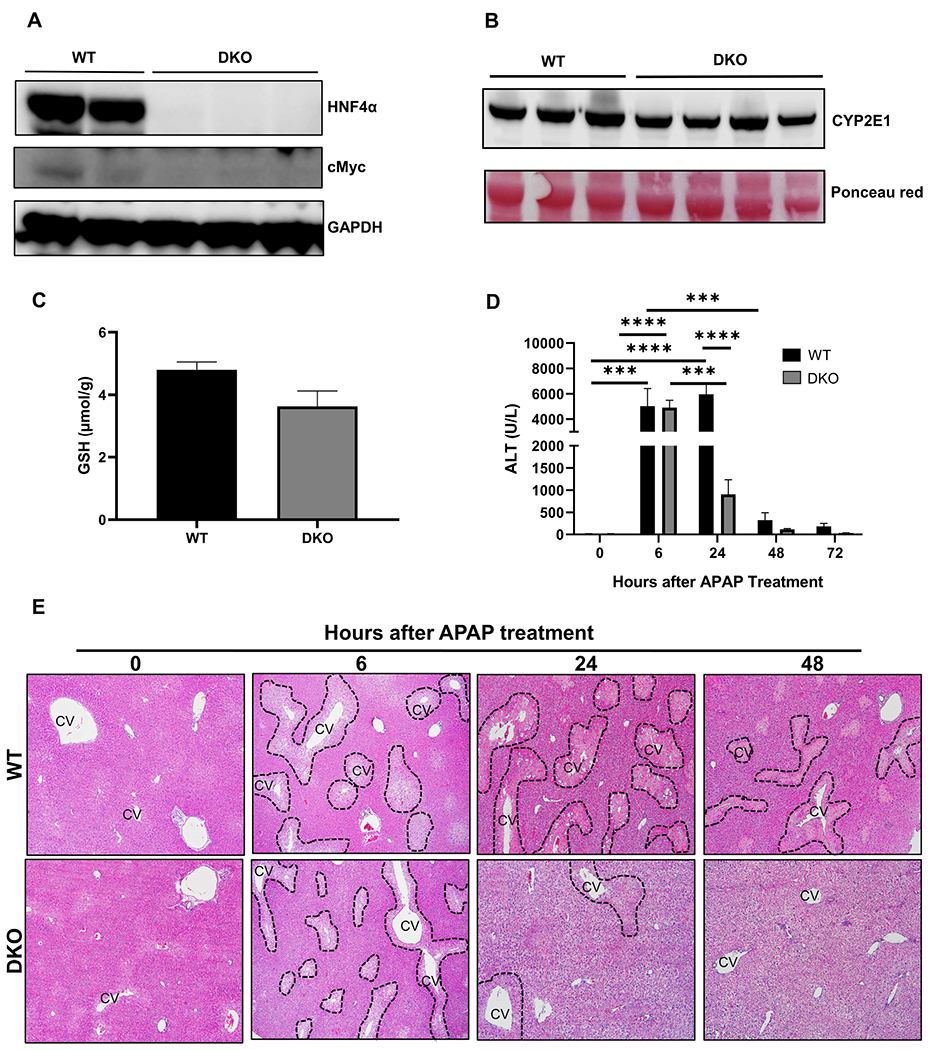
(A) Western blot analysis of HNF4α and cMyc showing deletion of HNF4α and cMyc in DKO mice (B) Western blot analysis of hepatic CYP2E1 and (C) Hepatic GSH levels in WT and DKO mice at 0 h time point. (D) Serum ALT levels of WT and DKO mice treated with APAP 300 mg/kg dose at various time points. (E) Representative photomicrographs of H&E-stained liver sections. Dotted lines demark area of necrosis. CV, central vein; Original magnification, 200X; * Indicates significant difference at, **=P<0.01, ***=P<0.001, ****=P<0.0001.
Faster initiation of cell proliferation in HNF4α-cMyc DKO mice after an APAP overdose
To investigate the mechanisms of faster recovery from APAP-induced liver injury in the DKO mice, we performed Western blot analysis of the key drivers of cell proliferation. DKO mice showed a significantly faster initiation of cell proliferation following APAP overdose as demonstrated by increased PCNA and CyclinD1 as compared to WT mice (Fig. 6A–B). The Western blot data were further corroborated by Ki67 staining, which confirmed a significantly faster compensatory proliferation in DKO mice after APAP overdose (Fig. 6C, Supplementary Fig. 5).
Figure 6: Faster initiation of cell proliferation in HNF4α-cMyc DKO mice after APAP overdose.
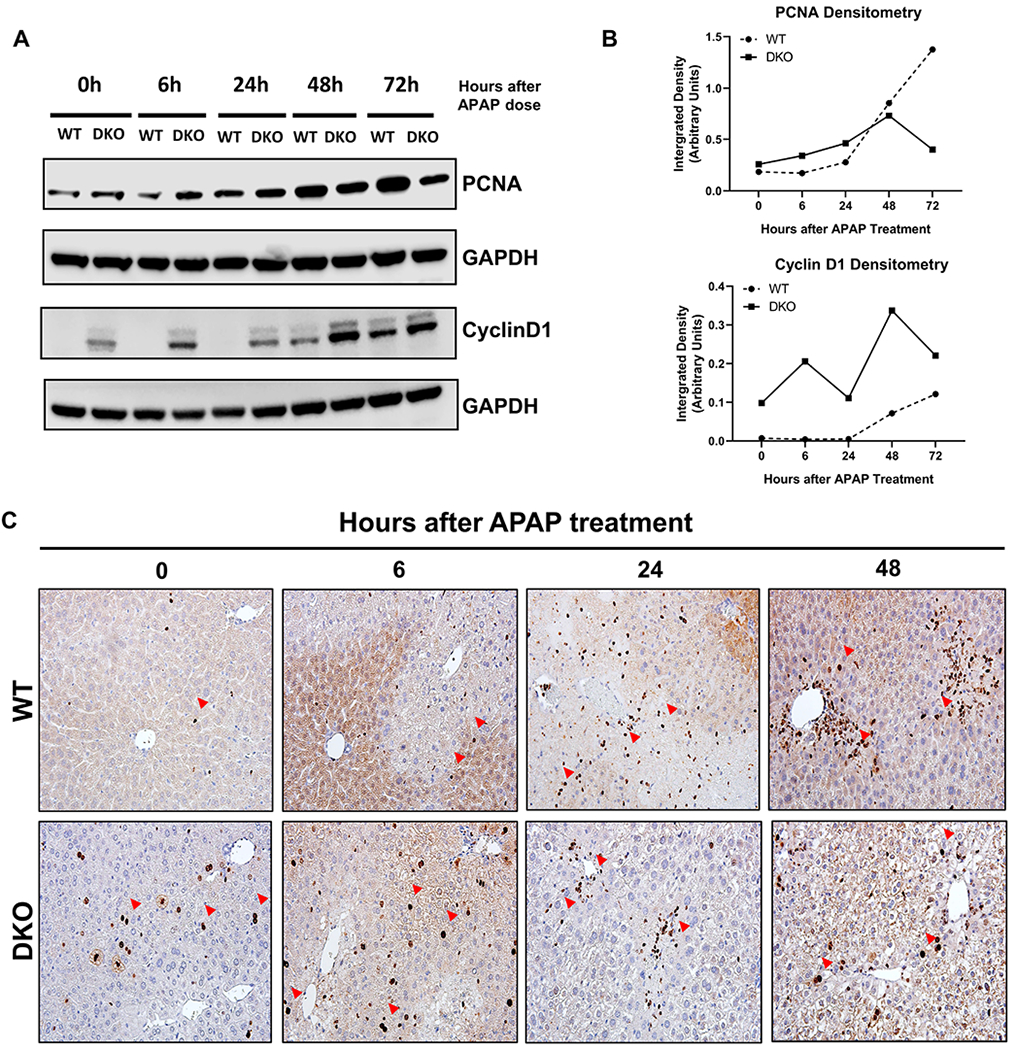
(A) Western blot analysis of PCNA and CyclinD1 performed using pooled (n=3-5) liver lysates from WT and DKO mice at various time points.(B) Densitometric analysis of PCNA and CyclinD1 Western blots. (C) Ki67 immunohistochemistry of WT and DKO mice at 0, 6, 24, 48 h after APAP overdose. Red arrows pointing to Ki67 positive cells. Original magnification, 400X
Rapid GSH replenishment in DKO mice after APAP treatment
We investigated the major intracellular mechanisms involved in APAP-induced hepatocyte necrosis in the WT and DKO mice after APAP overdose. Hepatic JNK activation was lower in DKO mice than in WT mice at 1 and 6 h (Fig. 7A). WT and DKO mice had similar GSH depletion after APAP treatment. However, DKO mice showed a significantly higher GSH replenishment 6 h after APAP administration (Fig. 7B). The recovery of hepatic GSH levels is regulated by Nrf2-dependent activation of genes involved in GSH synthesis including Gclc and Gclm. qPCR analysis showed significantly faster and higher induction of Gclc and Gclm in DKO mice (Fig. 7C, 7D). Consistent with these findings, expression of another Nrf2 target gene, Nqo1, was also significantly increased in DKO mice, suggesting an overall higher Nrf2 activation in DKO mice (Fig. 7E). Next, we performed co-immunoprecipitation (IP) experiments to determine interaction between Nrf2, HNF4α, and cMyc. Total liver lysates from WT mice were used to IP Nrf2 and then Western blotting was conducted to detect the binding between proteins. The data indicated that in WT livers, Nrf2 binds to both HNF4α and cMyc (Supplementary Fig. 6). Further, we performed Nrf2 ChIP analysis which showed moderate decrease in Nrf2 DNA binding in HFN4α-KO mice which was significantly increased in the DKO mice as both compared to the WT and HNF4α-KO mice (Fig. 7F).
Figure 7: Rapid GSH replenishment in DKO mice after APAP treatment.
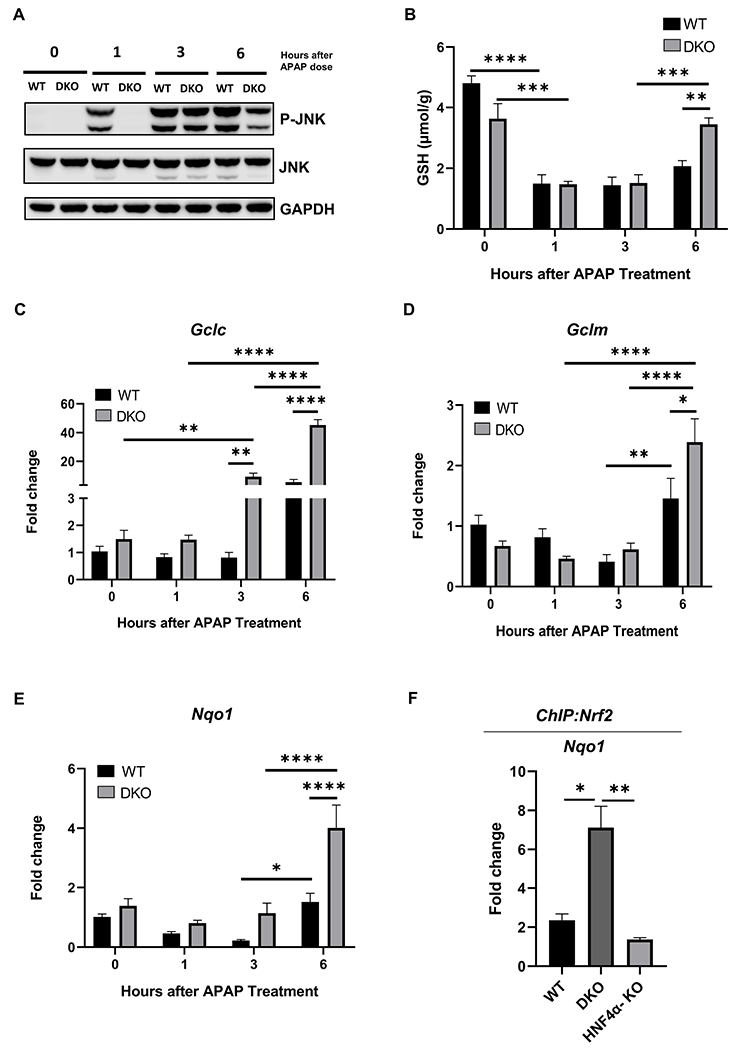
(A) Western blot analysis of JNK and phospho- JNK performed using pooled (n=3-5) liver lysates from WT and DKO mice after 300 mg/kg dose of APAP. (B) Total glutathione levels in liver lysates of WT and DKO mice. qPCR analysis of (C) Gclc, (D) Gclm and, (E) Nqo1 genes. (F) ChIP of Nrf2 in control WT, DKO and HNF4α-KO mice using the frozen liver tissues. Nqo1 qPCR was performed to confirm the pulldown of Nrf2. * Indicates significant difference at, *=P<0.05, **=P<0.01, ***=P<0.001, ****=P<0.0001.
Discussion and Conclusion:
HNF4α plays a key role in early embryonic liver development and in hepatocyte differentiation after birth [23]. Previous studies have shown that HNF4α is involved in the inhibition of hepatocyte proliferation [15–17]. Decreased HNF4α expression leads to loss of quiescence which leads to dedifferentiation and increased proliferation [15, 18, 24, 25]. Decreased HNF4α mRNA and especially protein expression has been associated with liver disease progression [26]. Recent studies from our laboratory indicated that loss of HNF4α activity is a major feature of all chronic liver diseases [27]. However, very little is known about the role of HNF4α in liver regeneration after drug-induced acute liver injury (DILI). The goal of this study was to investigate the role of HNF4α in liver regeneration after APAP induced acute liver injury.
In our initial experiments using the incremental dose model in C57BL/6J mice, we observed a rapid nuclear to cytoplasmic translocation of HNF4α and decreased HNF4α expression within 1 h following administration of the non-regenerating dose (600 mg/kg) of APAP. Mice exhibited significant liver injury followed by a delay in liver regeneration and up to 30% mortality after the non-regenerating dose [21]. In contrast, mice given the regenerating dose (300 mg/kg) maintained HNF4α expression until 3 h after APAP treatment. These data indicate that maintaining hepatic HNF4α during the initial stages after APAP overdose is critical for long term survival. It should be noted that liver injury is similar following administration of either 300 mg/kg or 600 mg/kg APAP for the first 6 h. However, significant differences in HNF4α expression were observed within 1 h after APAP administration between these doses. The mechanisms of the rapid decline in HNF4α expression following the non-regenerating dose are not known, but the rapid posttranslational modification (PTM) of HNF4α is a plausible cause given the role of PTM in regulating HNF4α protein levels [20, 28, 29]. Interestingly, previous studies from our laboratory have shown a xdecline in HNF4α during the initial hours after 70% partial hepatectomy (PHX) [19]. However, in those studies, HNF4α expression was rapidly regained within 12 h and HNF4α decrease was not associated with delayed regeneration. In fact, the initial decline was associated with rapid entry of hepatocytes into the cell cycle. These data highlight the different roles played by HNF4α following PHX and DILI.
Studies on hepatocyte specific HNF4α-KO mice supported our observation with the incremental dose model. HNF4α-KO mice experienced significantly higher initial liver injury at 6 h after 300 mg/kg APAP administration. The basal CYP2E1 and GSH levels were similar in both the WT and HNF4α-KO mice. However, we observed a significant delay in GSH recovery following an APAP overdose in HNF4α-KO. These data suggested a link between loss of hepatocyte HNF4α and GSH replenishment. Further, these data are consistent with previous observations that GSH replenishment is significantly delayed after 600 mg/kg APAP as compared to 300 mg/kg APAP [30]. Studies on mitochondrial integrity and cellular injury revealed that HNF4α-KO mice had a moderate increase in cytoplasmic release of SMAC and Endonuclease G, both of which contribute to liver injury. However, these were not enough to explain the significantly higher liver injury. This further highlighted the role of failed GSH replenishment in significantly higher initial liver injury in HNF4α-KO mice. Surprisingly, despite the initial higher liver injury, HNF4α-KO mice (and the WT controls) recovered completely 48 h after a 300 mg/kg APAP. This observation is in striking contrast with our previous studies using PHX model where we observed a significant mortality in HNF4α-KO mice [19]. In that study, we observed that HNF4α-KO mice die several days after PHX, not because of lack of proliferation, but due to lack of redifferentiation of newly divided hepatocytes. These data further stress that the role of HNF4α in liver regeneration after PHX and DILI are different. It is possible that midzonal and periportal hepatocytes, which survive the centrilobular necrosis induced by APAP, can compensate for the loss of HNF4α and are able to proliferate and re-differentiate.
One of the consistent observations with hepatocyte specific deletion of HNF4α is the induced expression of cMyc mRNA and protein in normal, regenerating, and cancerous livers [15, 19, 31]. cMyc is a transcription factor that regulates cell growth, cell proliferation, differentiation, metabolism as well as cell growth arrest and apoptosis [32, 33], [34]. cMyc is a proto-oncogene and is often upregulated in various cancers. cMyc overexpression in hepatic cells leads to the development of Hepatocellular Carcinoma (HCC) [35]. Low expression of cMyc protein is correlated with poor outcomes in patients with HCC after hepatectomy [36]. After PHX, a transient increase in the expression of cMyc results in the transition of hepatocytes from G0/G1 phase to S phase [37]. However, published studies on the role of cMyc in liver regeneration are not fully consistent. One study showed that deletion of cMyc resulted in a decreased proliferative response after PHX [38]. In contrast, some studies showed that conditional deletion of cMyc did not affect restoration of liver mass during liver regeneration [39], [40]. We hypothesized that cMyc may compensate for HNF4α deletion after APAP overdose and investigated the role of cMyc-HNF4α interaction using double knockout mice.
The DKO studies led to two major observations. Deletion of cMyc in the HNF4α-KO mice resulted in normalizing liver injury. WT and DKO mice had similar injuries, which were lower than the HNF4α-KO mice. The decrease in injury was accompanied by rapid GSH replenishment in the DKO mice after APAP treatment. GSH replenishment is regulated by the transcription factor nuclear factor erythroid 2–related factor 2 (Nrf2), which controls the expression of Gclc and Gclm genes involved in the de novo synthesis of GSH. Consistently, we observed increased expression of Gclc, Gclm, and Nqo1, another Nrf2 target gene, in DKO mice 6 h after APAP overdose. Further, co-IP analysis showed that HNF4α and cMyc both can directly interaction with Nrf2. Furthermore, ChIP analysis confirmed that HNF4α can directly affect Nrf2 DNA binding and this process is significantly affected by cMyc. These data indicate that HNF4α supports Nrf2 activation, while cMyc inhibits Nrf2 function. In HNF4α-KO conditions, cMyc is induced, which in turn inhibits Nrf2, leading to higher liver injury. Inhibition of cMyc in HNF4α-KO, i.e. in the DKO mice, Nrf2 activation is restored resulting in rapid GSH replenishment and decreased liver injury.
Secondly, the DKO mice showed significantly faster recovery as compared to the WT mice following APAP overdose despite equal initial injury. This faster recovery was consistent with rapid cell proliferation after APAP treatment. We detected cell proliferation as early as 6 h after APAP treatment in the DKO mice. Previous studies have shown that faster replenishment of GSH after APAP overdose helps to initiate rapid regeneration [41]. Our studies are consistent with these observations. The DKO mice have significantly faster GSH replenishment, which may help in the rapid entry of hepatocytes into the cell cycle.
In summary, our studies have uncovered a novel signaling mechanism involved in recovery from an APAP overdose. cMyc inhibits GSH replenishment by suppressing Nrf2 function and promotes liver injury, a process inhibited by HNF4α. Our studies show that maintaining HNF4α function following an APAP overdose is critical for recovery because it suppresses cMyc activation, which can delay GSH recovery via Nrf2 inhibition (Fig. 8). This novel mechanism involving HNF4α-cMyc interaction links mechanisms of liver injury with the mechanisms that trigger liver regeneration after APAP overdose and could be a novel therapeutic target in DILI.
Figure 8: Graphical Abstract.
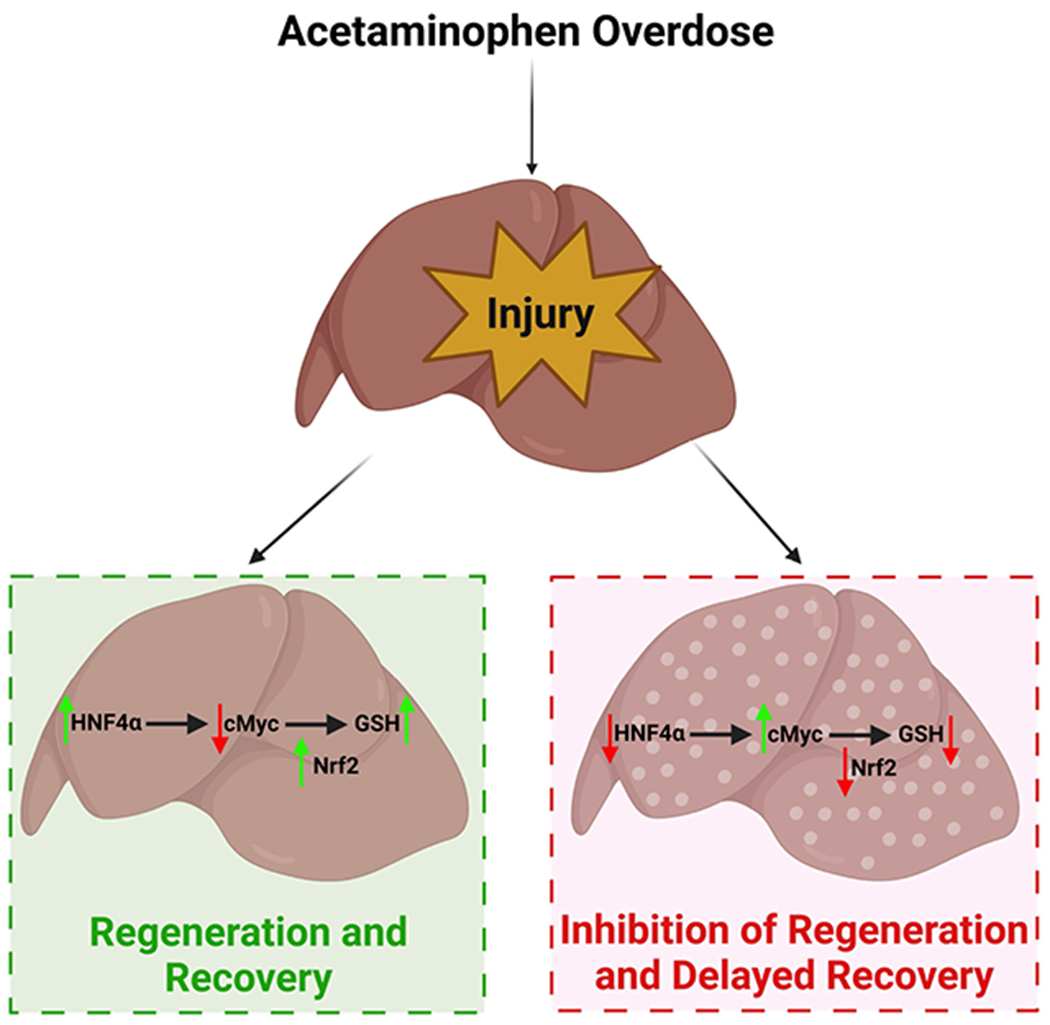
Schematic representation of final conclusions.
Supplementary Material
Supplementary Figure 1:
(A) Densitometry analysis of HNF4α Western blot using nuclear and cytoplasmic extracts after 300 mg/kg dose of APAP and (B) 600 mg/kg dose of APAP normalized to the loading control.
Supplementary Figure 2:
(A) Quantification of necrotic area in the liver sections of WT and HNF4α-KO mice treated with 300 mg/kg APAP. (B) Densitometry analysis of phosphor- RIP1 and (C) phosphor GSK3β Western blot performed using liver lysates of WT and HNF4α-KO mice treated with APAP 300 mg/kg normalized to the loading control.
Supplementary Figure 3:
(A) HNF4α and cMyc genotyping data for DKO mice. (B) Quantification of necrotic area in the liver sections of WT and DKO mice treated with 300 mg/kg APAP.
Supplementary Figure 4:
(A) Serum ALT levels of WT, HNF4α-KO and DKO mice treated with APAP 300 mg/kg dose at various time points. (B) Representative photomicrographs of H&E-stained liver sections. Dotted lines demark area of necrosis. CV, central vein; Original magnification, 200X; * Indicates significant difference at *=P<0.05, **=P<0.01, ***=P<0.001, ****=P<0.0001
Supplementary Figure 5:
(A) Quantification of Ki67 positive cells in WT and DKO mice at 0, 6, 24, 48 h after APAP overdose.
Supplementary Figure 6:
(A) IP pull down of Nrf2 from liver tissues of WT mice. Western blot analysis was performed on Nrf2 to show successful pulldown and HNF4α and cMyc.
Table 1:
Antibodies used in this study
| Name | Manufacturer | Catalog number |
|---|---|---|
| HNF4α | Perseus Proteomics | PP-H1415-00 |
| CYP2E1 | Abcam | 28146 |
| JNK | Cell Signaling Technology | 4672S |
| Phospho-JNK | Cell Signaling Technology | 4668S |
| RIP | Cell Signaling Technology | 13526S |
| Phospho- RIP1 | Cell Signaling Technology | 53286 |
| Phospho- RIP3 | Cell Signaling Technology | 57220S |
| GSK3β | Cell Signaling Technology | 9315 |
| Phospho- GSK3β | Cell Signaling Technology | 9323 |
| AIF | Cell Signaling Technology | 5318 |
| Cyt C | BD Pharmigen | 556433 |
| Endonuclease G | Cell Signaling Technology | 4969 |
| BAX | Cell Signaling Technology | 2772 |
| SMAC | Cell Signaling Technology | 15108 |
| α-tubulin | Sigma-Aldrich | T6074 |
| Vdac | Cell Signaling Technology | 4661 |
| cMyc | Cell Signaling Technology | 56055 |
| Phospho- RB | Cell Signaling Technology | 8516S |
| Cyclin E | Cell Signaling Technology | 4129S |
| Cyclin D1 | Cell Signaling Technology | 2978 |
| PCNA | Cell Signaling Technology | 2586S |
| Nrf2 | Cell Signaling Technology | 12721 |
| GAPDH | Cell Signaling Technology | 2118S |
| β- actin | Cell Signaling Technology | 4970 |
Table 2:
Primers used in this study
| Gene | Forward (5′–3′) | Reverse (3′-5′) |
|---|---|---|
| HNF4α | AGAATGACCCTGAAGCACCAGG | GCCAGAGGTCTGTGAAACAAG |
| CyclinD1 | GAATTCTATGACCCCTTGACCCC | TGGTGTTGGGTAAGAGGTTG |
| cMyc | GGTGTTTGAAGGCTGGATTTC | GATGAAATAGGGCTGTACGGAG |
| GCLC | GGGGTGACGAGGTGGAGTA | GTTGGGGTTTGTCCTCTCCC |
| GCLM | AGGAGCTTCGGGACTGTATCC | GGGACATGGTGCATTCCAAAA |
| Nqo1 | AGGATGGGAGGTACTCGAATC | AGGCGTCCTTCCTTATATGCTA |
| Nqo1 (ChIP) | AGCAGAACGCAGCACGAAT | CACTCAGCCGTGGGAAGT |
| 18S | ACGGAAGGGCACCACCAGGA | TTTAGTATTGGACGCTGCCC |
Highlights:
Maintaining HNF4α expression is critical for regeneration after APAP overdose
HNF4α inhibits cMyc and promotes recovery by GSH replenishment via Nrf2 activation
Funding information:
These studies were supported by NIH-COBRE (P20 RR021940-03, P30 GM118247), NIEHS Toxicology Training Grant (T32 ES007079-34), NIH R01 DK0198414 and NIH R56 DK112768
Abbreviations:
- APAP
Acetaminophen
- AAV8-TBG-CRE
AA8 vector containing Cre recombinase coding plasmid under a TBG promoter
- AAV8-TBG-eGFP
AA8 vector containing enhanced green fluorescent protein coding plasmid under a TBG promoter
- AIF
Apoptosis-inducing Factor
- ALI
Acute liver injury
- ALT
Alanine aminotransferase
- BAX
Bcl-2-associated X protein
- CYP2E1
Cytochrome p45 family 2 subfamily E member 1
- Cyt C
Cytochrome C
- DAMPs
Damage associated molecular patterns
- DKO
Hepatocyte specific HNF4α-cMyc double knockout
- Endo G
Endonuclease G
- Gclc
Glutamate-Cysteine Ligase Catalytic Subunit
- Gclm
Glutamate-Cysteine Ligase Modifier Subunit
- GSH
Glutathione
- GSK3β
Glycogen Synthase Kinase 3 Beta
- HNF4α
Hepatocyte nuclear factor 4 alpha
- HNF4α-KO
Hepatocyte specific HNF4α knockout
- H&E
Hematoxylin and eosin
- i.p.
Intraperitoneal
- IP
Immunoprecipitation
- JNK
c-Jun N-terminal kinase
- NAPQI
N-acetyl benzoquinone imine
- Nqo1
NAD(P)H Quinone Dehydrogenase 1
- Nrf2
Nuclear factor-erythroid factor 2-related factor 2
- PCNA
Proliferating cell nuclear antigen
- PHX
Partial hepatectomy
- RIP
Receptor-interacting protein
- ROS
Reactive oxygen species
- WT
Wild type
Footnotes
Conflict of interest:
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References:
- 1.Bernal W and Wendon J, Acute liver failure. N Engl J Med, 2013. 369(26): p. 2525–34. [DOI] [PubMed] [Google Scholar]
- 2.Larson AM, et al. , Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology, 2005. 42(6): p. 1364–72. [DOI] [PubMed] [Google Scholar]
- 3.Shehab N, et al. , US Emergency Department Visits for Outpatient Adverse Drug Events, 2013-2014. JAMA, 2016. 316(20): p. 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaeschke H and Bajt ML, Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci, 2006. 89(1): p. 31–41. [DOI] [PubMed] [Google Scholar]
- 5.Bhushan B and Apte U, Acetaminophen Test Battery (ATB): A Comprehensive Method to Study Acetaminophen-Induced Acute Liver Injury. Gene Expr, 2020. 20(2): p. 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han D, et al. , Regulation of drug-induced liver injury by signal transduction pathways: critical role of mitochondria. Trends Pharmacol Sci, 2013. 34(4): p. 243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunawan BK, et al. , c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology, 2006. 131(1): p. 165–78. [DOI] [PubMed] [Google Scholar]
- 8.Mehendale HM, Tissue repair: an important determinant of final outcome of toxicant-induced injury. Toxicol Pathol, 2005. 33(1): p. 41–51. [DOI] [PubMed] [Google Scholar]
- 9.James LP, et al. , Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem Biophys Res Commun, 2003. 309(4): p. 857–63. [DOI] [PubMed] [Google Scholar]
- 10.Donahower BC, et al. , Human recombinant vascular endothelial growth factor reduces necrosis and enhances hepatocyte regeneration in a mouse model of acetaminophen toxicity. J Pharmacol Exp Ther, 2010. 334(1): p. 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inoue Y, et al. , Role of hepatocyte nuclear factor 4alpha in control of blood coagulation factor gene expression. J Mol Med (Berl), 2006. 84(4): p. 334–44. [DOI] [PubMed] [Google Scholar]
- 12.Inoue Y, et al. , Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem, 2004. 279(4): p. 2480–9. [DOI] [PubMed] [Google Scholar]
- 13.Inoue Y, et al. , Regulation of bile acid biosynthesis by hepatocyte nuclear factor 4alpha. J Lipid Res, 2006. 47(1): p. 215–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayhurst GP, et al. , Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol, 2001. 21(4): p. 1393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walesky C, et al. , Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology, 2013. 57(6): p. 2480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin C, et al. , Differentiation therapy of hepatocellular carcinoma in mice with recombinant adenovirus carrying hepatocyte nuclear factor-4alpha gene. Hepatology, 2008. 48(5): p. 1528–39. [DOI] [PubMed] [Google Scholar]
- 17.Ning BF, et al. , Hepatocyte nuclear factor 4 alpha suppresses the development of hepatocellular carcinoma. Cancer Res, 2010. 70(19): p. 7640–51. [DOI] [PubMed] [Google Scholar]
- 18.Walesky C and Apte U, Role of hepatocyte nuclear factor 4alpha (HNF4alpha) in cell proliferation and cancer. Gene Expr, 2015. 16(3): p. 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huck I, et al. , Hepatocyte Nuclear Factor 4 Alpha Activation Is Essential for Termination of Liver Regeneration in Mice. Hepatology, 2019. 70(2): p. 666–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robarts DR, et al. , Regulation of Liver Regeneration by Hepatocyte O-GlcNAcylation in Mice. Cell Mol Gastroenterol Hepatol, 2022. 13(5): p. 1510–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhushan B, et al. , Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am J Pathol, 2014. 184(11): p. 3013–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaeschke H, McGill MR, and Ramachandran A, Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev, 2012. 44(1): p. 88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duncan SA, et al. , Expression of transcription factor HNF-4 in the extraembryonic endoderm, gut, and nephrogenic tissue of the developing mouse embryo: HNF-4 is a marker for primary endoderm in the implanting blastocyst. Proc Natl Acad Sci U S A, 1994. 91(16): p. 7598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatziapostolou M, et al. , An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell, 2011. 147(6): p. 1233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai WY, et al. , Yes-associated protein/TEA domain family member and hepatocyte nuclear factor 4-alpha (HNF4alpha) repress reciprocally to regulate hepatocarcinogenesis in rats and mice. Hepatology, 2017. 65(4): p. 1206–1221. [DOI] [PubMed] [Google Scholar]
- 26.Yeh MM, Bosch DE, and Daoud SS, Role of hepatocyte nuclear factor 4-alpha in gastrointestinal and liver diseases. World J Gastroenterol, 2019. 25(30): p. 4074–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunewardena S, et al. , Progressive loss of hepatocyte nuclear factor 4 alpha activity in chronic liver diseases in humans. Hepatology, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokoyama A, et al. , Multiple post-translational modifications in hepatocyte nuclear factor 4alpha. Biochem Biophys Res Commun, 2011. 410(4): p. 749–53. [DOI] [PubMed] [Google Scholar]
- 29.Veto B, et al. , The transcriptional activity of hepatocyte nuclear factor 4 alpha is inhibited via phosphorylation by ERK1/2. PLoS One, 2017. 12(2): p. e0172020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGill MR, et al. , Plasma and liver acetaminophen-protein adduct levels in mice after acetaminophen treatment: dose-response, mechanisms, and clinical implications. Toxicol Appl Pharmacol, 2013. 269(3): p. 240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walesky C, et al. , Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol, 2013. 304(1): p. G26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adhikary S and Eilers M, Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol, 2005. 6(8): p. 635–45. [DOI] [PubMed] [Google Scholar]
- 33.Cole MD and Henriksson M, 25 years of the c-Myc oncogene. Semin Cancer Biol, 2006. 16(4): p. 241. [DOI] [PubMed] [Google Scholar]
- 34.Sodir NM and Evan GI, Nursing some sense out of Myc. J Biol, 2009. 8(8): p. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin CP, et al. , Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J Hepatol, 2010. 2(1): p. 16–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji F, et al. , Low expression of c-Myc protein predicts poor outcomes in patients with hepatocellular carcinoma after resection. BMC Cancer, 2018. 18(1): p. 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng K, Cubero FJ, and Nevzorova YA, c-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes (Basel), 2017. 8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baena E, et al. , c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci U S A, 2005. 102(20): p. 7286–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li F, et al. , Conditional deletion of c-myc does not impair liver regeneration. Cancer Res, 2006. 66(11): p. 5608–12. [DOI] [PubMed] [Google Scholar]
- 40.Sanders JA, et al. , Postnatal liver growth and regeneration are independent of c-myc in a mouse model of conditional hepatic c-myc deletion. BMC Physiol, 2012. 12: p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bajt ML, et al. , Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther, 2003. 307(1): p. 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1:
(A) Densitometry analysis of HNF4α Western blot using nuclear and cytoplasmic extracts after 300 mg/kg dose of APAP and (B) 600 mg/kg dose of APAP normalized to the loading control.
Supplementary Figure 2:
(A) Quantification of necrotic area in the liver sections of WT and HNF4α-KO mice treated with 300 mg/kg APAP. (B) Densitometry analysis of phosphor- RIP1 and (C) phosphor GSK3β Western blot performed using liver lysates of WT and HNF4α-KO mice treated with APAP 300 mg/kg normalized to the loading control.
Supplementary Figure 3:
(A) HNF4α and cMyc genotyping data for DKO mice. (B) Quantification of necrotic area in the liver sections of WT and DKO mice treated with 300 mg/kg APAP.
Supplementary Figure 4:
(A) Serum ALT levels of WT, HNF4α-KO and DKO mice treated with APAP 300 mg/kg dose at various time points. (B) Representative photomicrographs of H&E-stained liver sections. Dotted lines demark area of necrosis. CV, central vein; Original magnification, 200X; * Indicates significant difference at *=P<0.05, **=P<0.01, ***=P<0.001, ****=P<0.0001
Supplementary Figure 5:
(A) Quantification of Ki67 positive cells in WT and DKO mice at 0, 6, 24, 48 h after APAP overdose.
Supplementary Figure 6:
(A) IP pull down of Nrf2 from liver tissues of WT mice. Western blot analysis was performed on Nrf2 to show successful pulldown and HNF4α and cMyc.