

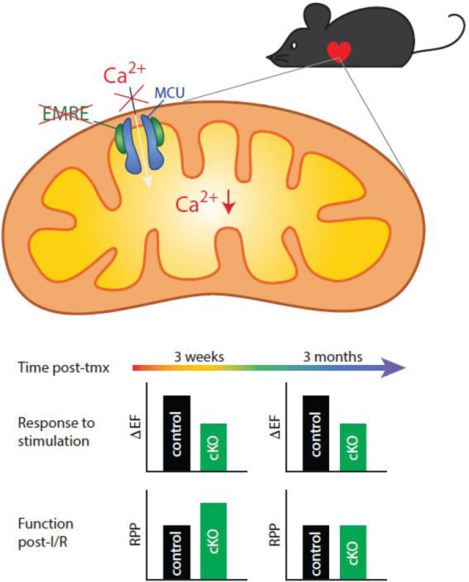
Abstract
Transport of Ca2+ into mitochondria is thought to stimulate the production of ATP, a critical process in the heart’s fight or flight response, but excess Ca2+ can trigger cell death. The mitochondrial Ca2+ uniporter complex is the primary route of Ca2+ transport into mitochondria, in which the channel-forming protein MCU and the regulatory protein EMRE are essential for activity. In previous studies, chronic Mcu or Emre deletion differ from acute cardiac Mcu deletion in response to adrenergic stimulation and ischemia/reperfusion (I/R) injury, despite equivalent inactivation of rapid mitochondrial Ca2+ uptake. To explore this discrepancy between chronic and acute loss of uniporter activity, we compared short-term and long-term Emre deletion using a novel conditional cardiac-specific, tamoxifen-inducible mouse model. After short-term Emre deletion (3 weeks post-tamoxifen) in adult mice, cardiac mitochondria were unable to take up Ca2+, had lower basal mitochondrial Ca2+ levels, and displayed attenuated Ca2+-induced ATP production and mPTP opening. Moreover, short-term EMRE loss blunted cardiac response to adrenergic stimulation and improved maintenance of cardiac function in an ex vivo I/R model. We then tested whether the long-term absence of EMRE (3 months post-tamoxifen) in adulthood would lead to distinct outcomes. After long-term Emre deletion, mitochondrial Ca2+ handling and function, as well as cardiac response to adrenergic stimulation, were similarly impaired as in short-term deletion. Interestingly, however, protection from I/R injury was lost in the long-term. These data suggest that several months without uniporter function are insufficient to restore bioenergetic response but are sufficient to restore susceptibility to I/R.
Keywords: mitochondrial calcium uniporter, mitochondria, calcium, permeability transition pore, ATP, ischemia/reperfusion
Graphical Abstract
1. Introduction
The heart, which requires large amounts of energy to continually pump blood, relies primarily on mitochondria to generate ATP [1]. Cardiomyocytes are thus among the most mitochondria-rich cells in the body; approximately 30% or more of the volume of a cardiomyocyte is comprised of mitochondria [2]. Among its many roles in vital cell processes such as gene expression, signal transduction and muscle contraction [3], calcium (Ca2+) has been shown to stimulate mitochondrial ATP production [4]. In the mitochondrial matrix, Ca2+ activates pyruvate dehydrogenase (PDH) as well as the tricarboxylic acid (TCA) cycle enzymes alpha-ketoglutarate dehydrogenase and isocitrate dehydrogenase [5,6]. This process is thought to be critical for matching energy supply to demand, such as during periods of increased workload [7]. However, overload of mitochondrial Ca2+ can induce opening of the mitochondrial permeability transition pore (mPTP) [8]. Mitochondrial Ca2+ overload has been implicated in a range of different pathological disorders [9–12], and Ca2+ induced mPTP opening has been linked to cell death in ischemia/reperfusion (I/R) injury in the heart [13,14]. Inhibition of mPTP opening or inhibition of the upstream event of mitochondrial Ca2+ uptake could therefore be beneficial therapeutically in myocardial infarction. Deeper understanding of the regulation of mitochondrial Ca2+ is thus critical for targeting mitochondrial dysfunction in pathology while not compromising physiological bioenergetics.
Mitochondrial Ca2+ uptake occurs through a highly selective multiprotein Ca2+ channel known as mitochondrial Ca2+ uniporter complex [15,16] (hereafter referred to as the “uniporter”), located in the inner mitochondrial membrane. The uniporter is made up of multiple auxiliary subunits, including MCU, the transmembrane protein that oligomerizes to form the Ca2+-permeant pore of the complex [17,18] and MCUb, a homologous protein that can act as a negative regulator [19]. Further, the uniporter is regulated by a family of intermembrane space-localized Ca2+-sensing proteins comprising three members, MICU1 [20], MICU2 [21], and MICU3 [22], and by a small transmembrane protein named Essential MCU Regulator (EMRE) [23]. EMRE has been shown to stabilize the uniporter serving as an anchor between MCU and MICU1 [23,24], and in a recently solved structure was shown to bind 1:1 with MCU to allow Ca2+ conductance through conformational changes in the tetrameric channel [25].
Recent research has continued to provide insight into the contribution of each subunit in regulating the physiological functions of various organ systems. Multiple mouse models of MCU deletion and inactivation have been used to study the role of mitochondrial Ca2+ in the heart. The first mouse model used a gene-trap method to globally delete Mcu in the germline in outbred CD-1 mice [26], as germline Mcu deletion was embryonic lethal in inbred C57Bl/6 mice. In these Mcu−/− mice, mitochondrial Ca2+ uptake was impaired across tissues [27] and intramitochondrial Ca2+ was decreased in the heart [28]. As expected, given these findings, sensitivity to Ca2+-induced mPTP opening was decreased; however, germline Mcu deletion did not confer protection in an ex vivo Langendorff model of I/R injury [26]. This counterintuitive result was supported by similar ex vivo I/R data in a study of mouse model expressing cardiac-specific dominant negative Mcu [29]. Subsequently, a cardiac-specific tamoxifen-inducible mouse model of Mcu deletion (hereafter referred to as McucKO mice) was developed by crossing floxed Mcufl/fl mice to the αMHC-MerCreMer (MCM) transgenic strain, enabling the acute deletion of Mcu in adulthood [30,31]. Though intramitochondrial Ca2+ levels were unchanged, upon in vivo I/R via left coronary artery ligation, infarct area was decreased in adult McucKO mice fed [30] or injected with [31] tamoxifen, showing that acute Mcu deletion did protect against I/R injury (see Table 1 in Discussion). Though protection was found in in vivo I/R and not found in ex vivo I/R, making the type of model used a possible source of this discrepancy, the more likely and interesting possibility would be that mice that have lost MCU acutely have the expected protection against I/R injury, but Mcu−/− and DN-Mcu mice that experience chronic loss of uniporter function do not retain protection. While activation of unknown compensatory pathways in the chronic absence of MCU could feasibly explain these findings, whether this could occur over long timescales even in adulthood or requires MCU loss during development is unclear.
Table 1.
Genetic mouse models of mitochondrial Ca2+ uniporter inactivation.
| Author and year | Knock out model | Tamoxifen route and dose | Timing of experiments | Response to β-adrenergic stimulation | Protection after ischemia/reperfusion |
|---|---|---|---|---|---|
| Pan et al, 2013 [27] | global MCU deletion | N/A | age 3–5 months | unchanged | not protected, ex vivo |
| Rasmussen et al, 2015 [29] | cardiac (αMHC) expression of a dominant-negative MCU | N/A | unspecified | diminished | not protected, ex vivo |
| Kwong et al, 2015 [30] | cardiac (αMHC-MerCreMer) MCU deletion | chow 4 weeks | 6 weeks post-tamoxifen | diminished | protected, in vivo |
| Luongo et al, 2015 [31] | cardiac (αMHC-MerCreMer) MCU deletion | injection, 40 mg/kg/day 5 days | 10 days post-tamoxifen (hemodynamics) 3 weeks post-tamoxifen (IR) | diminished | protected, in vivo |
| Liu et al, 2020 [32] | global EMRE deletion | N/A | age 3–6 months | unchanged | not protected, ex vivo |
In metazoan uniporters, EMRE is essential for the transport of Ca2+ through MCU [23]. The first mouse model of CRISPR-mediated deletion of Emre in the germline (Emre−/− mice) in outbred mice turned out to have substantial similarity to the Mcu−/− model [32]. The absence of mitochondrial Ca2+ uptake, decrease in intramitochondrial Ca2+ concentration, and decrease in sensitivity to Ca2+-induced mPTP opening in mitochondria from Emre−/− mice [32] recapitulated findings in Mcu−/− mice. Furthermore, like the Mcu−/− mice, Emre−/− mice did not exhibit protection in the Langendorff ex vivo I/R model [32]. Therefore, the potential compensatory response in chronic Mcu deletion models occurs as a result of loss of uniporter Ca2+ uptake activity and not loss of MCU per se. Here we sought to determine how short-term and long-term deletion of Emre in the heart would alter mitochondrial and tissue function. We developed a tamoxifeninducible cardiomyocyte-specific model of Emre deletion (EmrecKO mice) and characterized these mice 3 weeks (short-term) and 3 months (long-term) post-tamoxifen. As expected, we confirmed in the short and long term that EMRE is required for cardiac mitochondrial Ca2+ uptake. Like Emre−/− mice, EmrecKO mice exhibited a significant reduction in cardiac intramitochondrial Ca2+ levels and decreased Ca2+-induced mPTP opening, which persisted from short to long-term deletion. Unlike Emre−/− mice, however, in EmrecKO mice the short-term deletion of Emre led to blunted responses to adrenergic stimulation and protection from I/R injury in an ex vivo Langendorff model. In the long-term absence of EMRE, mitochondrial Ca2+ uptake was still absent in EmrecKO cardiac mitochondria, and adrenergic response continued to be attenuated. Strikingly, the protection from I/R injury seen upon short-term EMRE loss was no longer observed with long-term EMRE absence, suggesting that protection from I/R is lost in a time-dependent manner with chronic uniporter inactivity.
2. Methods
2.1. Development of cardiac-specific Emre knockout mouse model (EmrecKO)
Emre flox/flox mice (C57BL6/N) from the Mutant Mouse Resource & Research Centers (MMRRC) were crossed with cardiac-specific tamoxifen-inducible Cre mice (αMHC-Mer-Cre-Mer, abbreviated MCM+, C57BL6/J) to generate cardiac-specific EmrecKO mice (C57BL6/N and C57BL6/J hybrids). To induce Emre deletion, 9- to 11-week-old male and female mice were injected intraperitoneally (i.p.) with tamoxifen (30 mg/kg body weight) per day for 5 days, and experiments were conducted 3 weeks or 3 months post-tamoxifen as described in the text. All groups, including Emreflox/flox and MCM+ controls, received tamoxifen. The animal procedures were approved by the University of Minnesota Institutional Animal Care and Use Committee and performed in compliance with all relevant laws and regulations. Data are shown from female mice and from male mice where indicated.
2.2. Mitochondria heart isolation
Heart mitochondrial fractions were obtained as previously described [32]. Briefly, the heart was isolated and placed in an ice-cold isolation buffer, containing (mM): 225 mannitol, 75 sucrose, 5 MOPS, 0.5 EGTA, 2 taurine, pH 7.2. After being minced and homogenized with a T25 Ultra-Turrax disperser, heart tissue was digested for 5 min using trypsin in cold isolation buffer followed by addition of 0.2% BSA to stop digestion. The homogenates were centrifuged at 500 g for 5 min and then the resulting supernatant was centrifuged at 11,000 g for 5 min. The final mitochondria pellet was resuspended in isolation buffer and protein concentration was measured by Bradford assay.
2.3. Ca2+ retention capacity (CRC), membrane potential (Δψ), and swelling
The Ca2+ retention capacity (CRC) assay was performed by monitoring extramitochondrial Ca2+ levels via the fluorescence of the membrane-impermeable Ca2+ indicator Calcium Green-5N (Ca Green-5N, 1.0 μM). Isolated mitochondria were suspended (0.1 mg/mL) in respiration buffer containing (mM): 125 KCl, 2 K2HPO4, 20 HEPES, 1 MgCl2, 0.010 EGTA and pH 7.3. The mitochondria were energized with 10 mM succinate and 2 μM of rotenone or where indicated 10 mM glutamate and 5 mM malate. Successive boluses of Ca2+ (10 μM) were added to induce mitochondrial Ca2+ uptake. Loss of mitochondrial membrane potential (Δψ) upon addition of a large bolus of Ca2+ (400 μM) was measured via the fluorescence of the indicator safranin (2 μM) [33]. Carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP) was used as uncoupling agent. Mitochondrial swelling was measured in isolated mitochondria upon addition of a large bolus of Ca2+ (400 μM) as the reduction of absorbance at 540 nm. Fluorescence or absorbance was monitored using a Synergy H1 microplate reader (BioTek Instruments, Winooski, VT, USA).
2.4. Intramitochondrial Ca2+ content (mCa2+)
Intramitochondrial free Ca2+ was measured as previously described [32]. Briefly, heart mitochondria were isolated in the isolation buffer supplemented with 2 μM Ru360 and 10 μM CGP-37157 to avoid calcium influx and efflux, respectively, during the isolation process. Isolated mitochondria were loaded with 20 μM of Fluo-4 AM at room temperature and incubated for 45 minutes in the dark. Afterwards, the pellet was washed twice with isolation buffer and then was washed again with isolation buffer without EGTA. Finally, the pellet was resuspended in buffer containing (mM): 137 KCl, 20 HEPES, 2 K2HPO4 at pH 7.2. After obtaining the fluorescence of the mitochondrial sample F, to calibrate, Fmax was obtained after addition of 10 μM of ionomycin and 15 μM of Ca2+, and Fmin was obtained after addition of 2 mM of EGTA. Fluorescence was monitored using a Synergy H1 microplate reader. Intramitochondrial Ca2+ was estimated using the formula: mCa2+ (nM)= Kd*((F – Fmin)/(Fmax – F)), where Kd is the dissociation constant for Fluo-4, 335 nM. The data were reported normalized to control groups performed in the same experiment.
2.5. ATP production rate
The ATP production rate was measured in fresh heart mitochondria using the ATP bioluminescence assay Kit CLS II (Roche, 11699695001) [34]. Briefly, 10 μg of protein was added to 75 μL of ATP buffer (mM): 125 KCl, 10 HEPES, 5 MgCl2 and 2 K2HPO4, pH: 7.4, along with 75 μL of luciferin/luciferase buffer. Luminescence was monitored using a Synergy H1 microplate reader. The baseline signal was recorded for 1 minute, then 10 mM of succinate or 10 mM glutamate/5 mM malate and 50 μM of ADP were added. The slope was analyzed, and the ATP concentration (nanomoles per minute per mg of protein) was calculated from a fresh ATP standard curve. Where indicated, the ADP buffer was supplemented with 20 μM of Ca2+.
2.6. Immunoblotting
Mitochondrial or cardiac tissue protein was resolved on NuPAGE Bis-Tris 4 to 12% gels (Invitrogen) and transferred to nitrocellulose membranes and incubated with following antibodies: MCU (1:2000, Cell Signaling, 14997S), EMRE (1:200, Santa Cruz Biotechnology, sc-86337), MICU1 (1:500, Sigma Aldrich, HPA037480), PDH (1:2000, Santa Cruz Biotechnology, sc-377092), p-PDH (Ser293) (1:2000, Cell Signaling, 31866S), CPT1B (1:1000, Proteintech, 22170-1-AP), p-CAMKII (1:1000, Cell Signaling, 12716S), CAMKIIα/β/γ/δ (1:1000, Santa Cruz Biotechnology, sc-5306), CypD (1:1000, abcam, ab110324), GAPDH (1:5000, Cell Signaling, 97166S, VDAC1 (1:1000, Santa Cruz Biotechnology, sc-390996) and then with LI-COR IRDye secondary antibodies. The membranes were scanned on the Licor Odyssey system.
2.7. Blue native PAGE (BN-PAGE)
Freshly isolated cardiac mitochondria were incubated with NativePAGE sample buffer (Invitrogen) containing 2% ReadyShield protease & phosphatase inhibitor cocktail (Sigma) and 2% digitonin [32]. After 20 minutes on ice the samples were centrifuged for 30 minutes at 18,000 g at 4 °C. Coomassie blue (5%) was added to the supernatants, then 40 μg of protein was loaded. The electrophoresis running buffers were prepared according to the manufacturer’s protocol for the NativePAGE Novex Bis-Tris Gel System (Invitrogen). After electrophoresis gels were transferred onto nitrocellulose membranes for 18 hours at 4 °C. After blocking with 5% milk for 6 hours at 4 °C, membranes were incubated with MCU antibody (1:1000, Sigma Aldrich, HPA016480) overnight at 4 °C and then with HPR-conjugated secondary antibody. The blots were developed with ECL Western Blotting Substrate (Pierce) and quantified using a ChemiDoc MP Imaging System (BioRad).
2.8. Real-Time Quantitative PCR (RT-qPCR)
Heart tissue RNA was isolated by RNA MiniPrep (Zymo Research, R2052). Reverse transcription (10 ng of RNA) and real-time PCR assay were performed by Luna® Universal One-step RT-qPCR kit (New England BioLabs, E3005L) on a Bio-Rad CFX96 System. The following primer sequences were used: MCU, forward primer: 5’-GTGCCCTCTGATGACGTGACGG-3’, reverse primer: 5’- ATGACAAGCTTAAAGTCATC-3’; EMRE, forward primer: 5’- CATTTTGCCCAAGCCGGTG-3’, reverse primer: 5’-CCTGTGCCCTGTTAATCGTCGT-3’. The following primer sequences were used for reference genes: eukaryotic translation initiation factor EIF35S, forward primer: 5’-CTGAGGATGTGCTGTCTGGGAA-3’, reverse primer: 5’-CCTTTGCCTCCACTTCGGTC-3’; ribosomal protein 36B4, forward primer: 5’-GGCCCTGCACTCTCGCTTTC-3’, reverse primer: 5’-TGCCAGGACGCGCTTGT-3’[35].
2.9. Oxygen consumption rate (OCR)
As previously described [36], mitochondria were loaded in a 96-well Seahorse plate (4 μg of protein per well) in mitochondrial assay solution (MAS) (mM): 220 mannitol, 70 sucrose, 10 KH2PO4, 5 MgCl2, 2 HEPES, 1 EGTA and 0.2% of fatty acid-free BSA, pH: 7.2 supplemented with: 5 mM pyruvate/0.5 mM malate or 40 μM palmitoylcarnitine/0.5 mM malate. Using an Agilent Seahorse XFe96 Analyzer, OCR was analyzed at baseline and following sequential injections of 4mM ADP (state III), 2.5 μg/mL oligomycin (state IVo), 6 μM FCCP (state IIIu), and 4 μM rotenone/4 μM antimycin A. Basal respiration was calculated as the difference between the baseline OCR and post-rotenone/antimycin A OCR (non-mitochondrial oxygen consumption). The respiratory control ratio (RCR) was calculated as the ratio of state IIIu to state IVo. OCR values were normalized to μg of protein.
2.10. Mitochondrial enzyme activity
As described in [37], Complex I (NADH: ubiquinone oxidoreductase) activity was measured by following the absorbance change of NADH at 340 nm (ε: 6.2 mM−1 cm−1 ) using potassium phosphate buffer (50 mM potassium phosphate dibasic with 50 mM of potassium phosphate monobasic, pH: 7.5), 0.100 mM NADH, 3 mg/mL BSA, 0.300 mM KCN and 0.060 mM ubiquinone. Rotenone (10μM) was added to inhibit Complex I to allow subtraction of inhibitor-insensitive activity from the data. Complex II (succinate dehydrogenase) activity was measured spectrophotometrically at 600 nm (ε: 19.1 mM−1 cm−1) in potassium phosphate buffer (25 mM potassium phosphate dibasic with 25 mM potassium phosphate monobasic, pH: 7.5), 20 mM succinate, 0.080 mM 2,6-dichlorophenolindophenol (DCPIP), 1 mg/mL BSA, 0.300 mM KCN and 0.050 mM decylubiquinone (DUB). Malonate (10 mM) was added to inhibit complex II. Complex I and II activity were normalized to citrate synthase activity (CS), measured at 412 nm (ε : 13.6 mM−1 cm−1) in Tris 100 mM (pH: 8.0) buffer containing 0.1% Triton X-100, 0.100 mM 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB), 0.300 mM acetyl CoA, and 0.5 mM oxaloacetic acid. All assays were performed in triplicate on a Synergy H1 microplate reader at 37 °C in volumes of 200 μl/ well at a protein concentration of 0.05 mg/ml. The enzymatic activities were calculated as: nmol min−1 mg−1 = (Δ Absorbance/min × 1,000)/[(extinction coefficient × volume of sample) × (sample protein concentration)].
2.11. H2O2 production
Mitochondrial ROS generation was measured spectrofluorometrically (560 nm excitation and 590 nm emission) on a Synergy H1 microplate reader with the H2O2-sensitive dye Amplex Red (50 μM) (Invitrogen, A1222) and horseradish peroxidase (5 U/mL). Mitochondria (100 μg protein) were incubated in a solution containing (in mM): 120 KCl, 10 Tris-HCl, 5 MOPS, 5 K2HPO4 and 10 μM EGTA, pH 7.4. Baseline fluorescence was monitored for 1 minute, then 10 mM glutamate/5 mM malate was added and monitoring continued for 6 or more minutes. After subtraction of values from a blank well containing no mitochondria, slopes were calculated for the first 5 minutes after addition of glutamate/malate, and converted to units of pmol H2O2/(min*mg protein) using a linear fit to a standard curve of Amplex Red fluorescence across H2O2 concentrations. As positive and negative controls, antimycin A (500 nM) or 2,4-dinitrophenol (10 nM) respectively were present with a mitochondria sample in separate wells.
2.12. Protein carbonylation assay
The levels of protein carbonyl groups were assessed using the protein carbonyl assay kit (Abcam) according to manufacturer’s instructions. Briefly, freshly isolated mitochondria (100 μg) were diluted with 12% SDS and incubated with DNPH solution for 15 minutes at room temperature, then the reaction was neutralized. Gel electrophoresis (3 μg of protein per sample) and immunoblotting was performed as described under immunoblotting above with the anti-DNP antibody provided in the kit.
2.13. Transthoracic echocardiography
To measure left ventricular (LV) response to beta-adrenergic stimulation, we used serial transthoracic echocardiography to evaluate LV ejection fraction as a surrogate for LV function. We used a Vevo 2100 (Fujifilm VisualSonics, Toronto, CA) ultrasound imaging system to perform echocardiography. Mice were anesthetized with 2% isoflurane and affixed in a supine position to an ECG measuring platform heated to 37C. Ultrasound measurements were performed with a MS400 linear array ultrasound probe operating between 22–55 MHz. The ultrasound probe was placed across the mouse’s thorax to obtain a parasternal long axis view of the heart, and to capture two-dimensional and M-mode cine at mid-LV level. After acquisition of baseline images, at a heart rate between 400 and 550 bpm, mice were injected with isoproterenol (0.4 μg/kg) and serial images were taken every minute for five minutes. Echocardiography images were then analyzed using the Vevo Lab software. LV ejection fraction was calculated using the modified Quinones method from M-mode measurements of at least 3 different cardiac cycles from the parasternal long axis view.
2.14. Langendorff ex vivo ischemia/reperfusion (I/R)
The mice were euthanized using 100U of heparin and 250 mg/kg of sodium pentobarbital. The hearts were then harvested and perfused on the Langendorff apparatus using modified Krebs buffer (in mM): 118 NaCl, 0.5 EDTA, 4.7 KCl, 1.2 MgSO4, 1.2 Na2HPO4, 15 glucose, 2.5 CaCl2, 25 NaHCO3, and 0.5 pyruvate. Oxygen probes (Microelectrodes, Inc.) were used throughout the protocol to monitor oxygen, sampling both the cardiac effluent and perfusate. The left ventricular pressure was measured using a balloon filled with water that was coupled to a solid-state pressure transducer (Millar Instruments). After 20 minutes of equilibration on the apparatus, the hearts were subjected to 20 minutes of no-flow global ischemia, followed by 90 minutes of reperfusion. Coronary flow was monitored by an ultrasonic flow probe (Transonic). At baseline the cannula oxygen levels were 524 ± 18 mmHg, while during hypoxia they were 175 ± 11 mmHg. At the start of reperfusion, effluent samples were collected to measure creatine kinase (CK) release, an indicator of heart damage. Heart rate and left ventricular developed pressure were also recorded to calculate the rate pressure product (RPP = HR × DP).
2.15. Data analysis
Data are expressed as mean ± standard deviation (SD). Unpaired Student’s t-test, one-way ANOVA or two-way ANOVA were performed when appropriate to compare experimental groups. A difference was considered statistically significant when p<0.05. Data processing and statistical tests were carried out with GraphPad Prism (v.9.3.1, San Diego, CA, USA).
3. Results
3.1. Emre deletion eliminates mitochondrial Ca2+ uptake
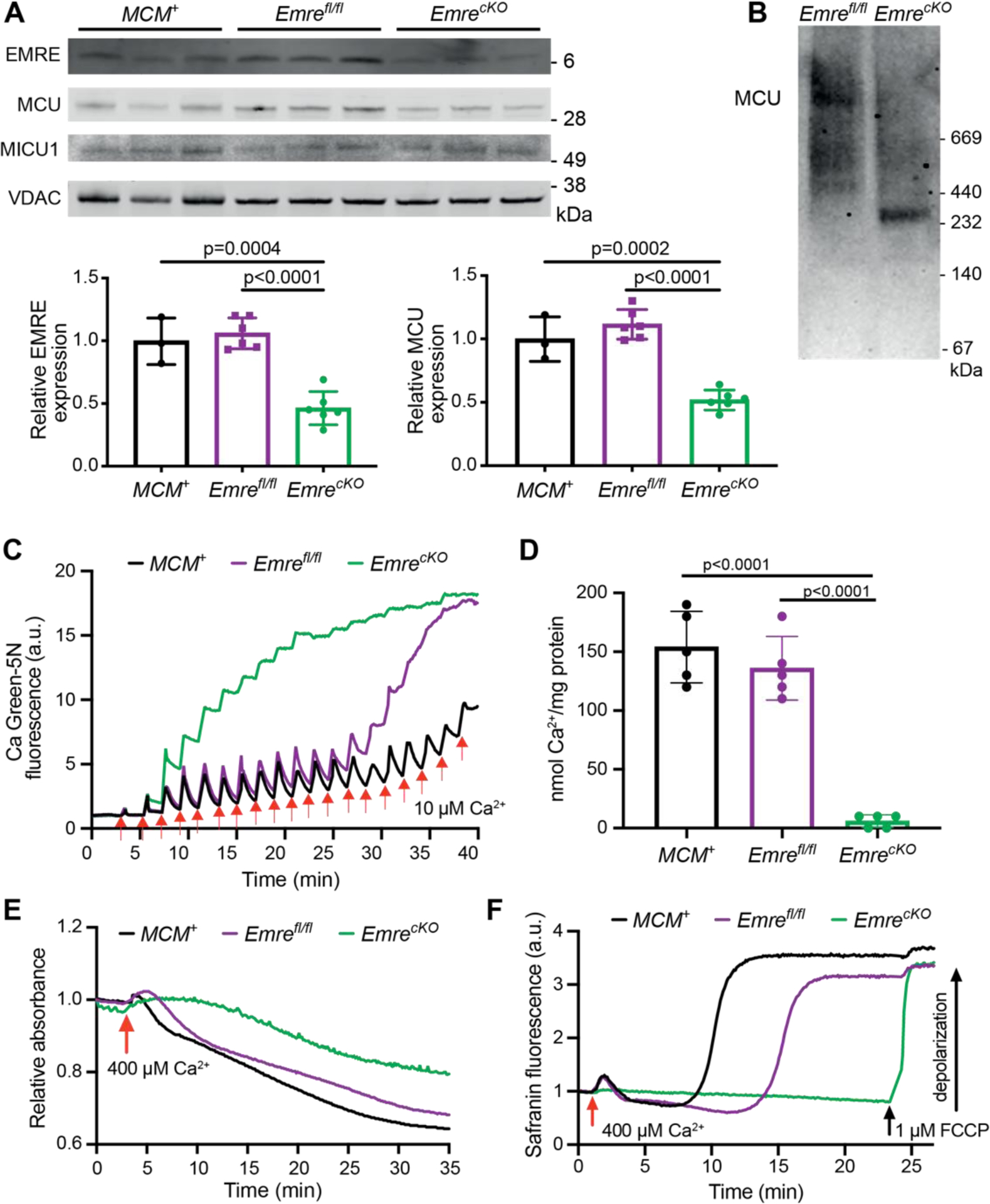
Emreflox/flox (Emrefl/fl) mice were crossed with the well-characterized αMHC-MerCreMer (MCM+) transgenic mouse line to generate a model of acute tamoxifen-inducible cardiomyocyte-specific Emre deletion in adulthood. We confirmed that 3 weeks post-tamoxifen injection, Emre mRNA expression decreased by 92.5% (Fig. S1A) and protein expression was reduced by 90% in EmrecKO mitochondria relative to MCM+ and Emrefl/fl controls (Fig. 1A). MCU protein expression also decreased but to a lesser extent (60% reduction), but no decrease was observed in Mcu mRNA expression (Fig. S1B), suggesting that in the absence of EMRE downregulation of MCU occurs not at the transcriptional level but potentially due to diminished protein stability. No differences were observed in the levels of other uniporter proteins, for instance MICU1 (Fig. 1A and Fig. S1C), nor proteins involved in mitochondrial Ca2+ efflux, including the Na+/Ca2+/Li+ exchanger (NCLX) and Leucine zipper EF-hand transmembrane protein 1 (Letm1) (Fig. S1D). Blue native polyacrylamide gel electrophoresis (BN-PAGE) analysis of cardiac mitochondria revealed that loss of EMRE decreased the molecular weight of the uniporter complex from over 440 kDa [23,38] to approximately 300 kDa, as reported previously [32]. To assess how Ca2+ handling is affected by EMRE deletion, we performed Ca2+ retention capacity (CRC), mitochondrial swelling, and membrane potential assays. EmrecKO cardiac mitochondria were unable to take up Ca2+ in CRC assays, unlike control MCM+ and Emrefl/fl mitochondria, in which normal mitochondrial Ca2+ uptake was observed (Fig. 1C–D, S1E–F). To determine whether EmrecKO mitochondria can undergo mitochondrial Ca2+ overload-induced mitochondrial permeability transition pore (mPTP) opening, we analyzed mitochondrial swelling after adding a large Ca2+ bolus. EmrecKO mitochondria were resistant to swelling relative to MCM+ and Emrefl/fl controls (Fig. 1E, S1G), presumably due to loss of mitochondrial Ca2+ uptake. Similarly, in an assay using the dye safranin to monitor depolarization of mitochondrial membrane potential, EmrecKO mitochondria were able to maintain mitochondrial membrane potential after addition of a large Ca2+ bolus, in contrast to MCM+ and Emrefl/fl controls (Fig. 1F).
Figure 1. Short-term Emre deletion impairs mitochondrial Ca2+ uptake.
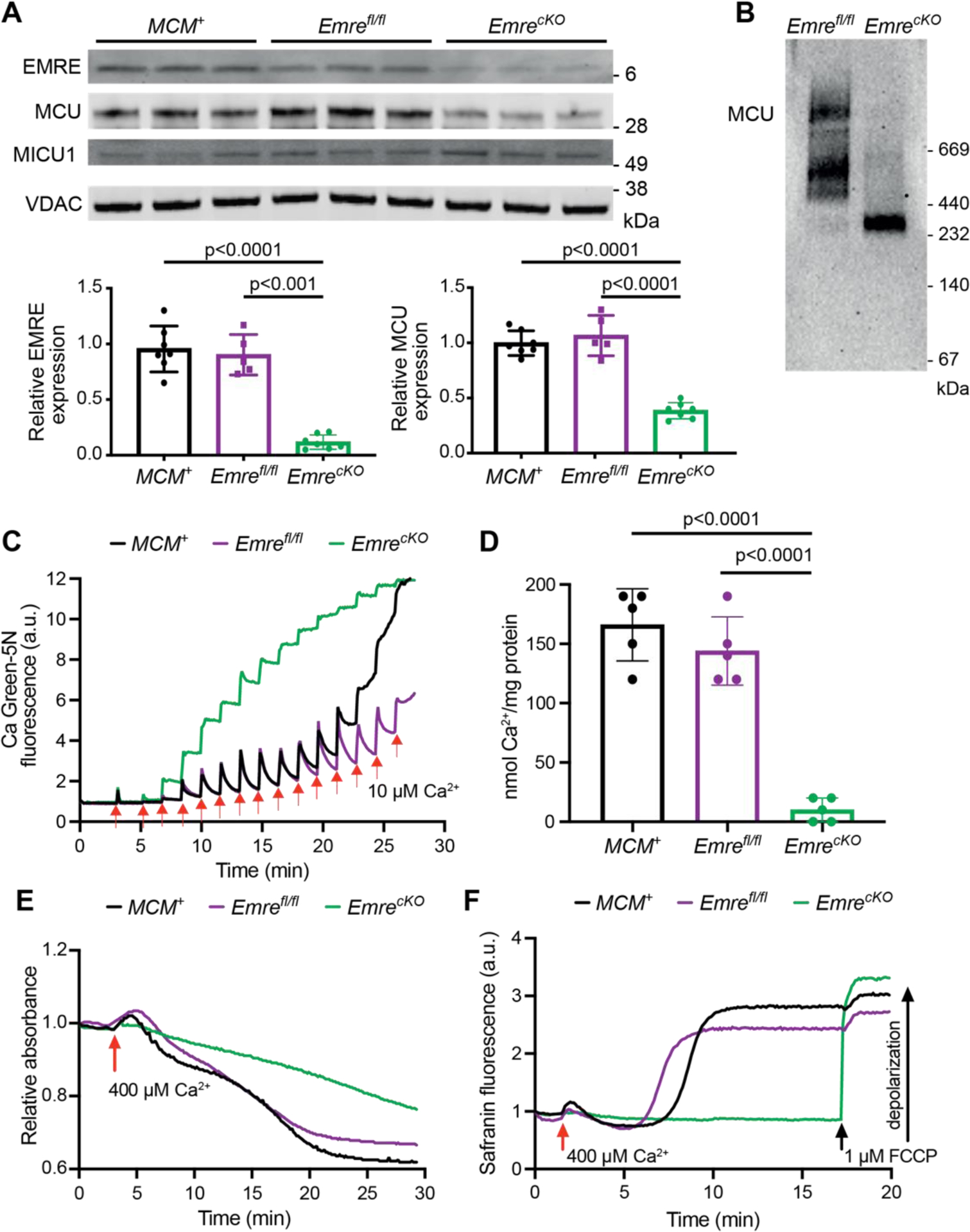
Analyses were performed 3 weeks post-tamoxifen. A) Representative Western blot of EMRE, MCU, and MICU1 in cardiac mitochondria from MCM+, Emrefl/fl and EmrecKO mice. VDAC was used as a loading control. Below, Western blot quantification shows a 90% reduction in EMRE and a 60% reduction in MCU in EmrecKO mitochondria compared to control groups. Values represent mean ± SD.**p<0.0001, n = 7 in MCM+ and EmrecKO, n = 5 in Emrefl/fl. One-way ANOVA test was used for statistical analysis. B) Blue Native gel of freshly isolated mitochondria from Emrefl/fl and EmrecKO hearts, immunoblotted with MCU antibody. Gel is representative of 3 mice (combined males and females) per group. C) Representative Ca2+ retention capacity (CRC) assay in isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice. The fluorescent Ca2+ indicator Calcium Green-5N was used to monitor extramitochondrial Ca2+. The arrows represent 10 μM Ca2+ additions. Traces are representative of n = 5 independent experiments. D) Ca2+ retention capacity calculated from independent traces as shown in (C). The estimated mean Ca2+/mg protein was for MCM+: 166 ± 30.49, for Emrefl/fl: 144 ± 28.89, and for EmrecKO: 10 ± 5 nmol of Ca2+/mg protein. Values represent mean ± SD.**p<0.0001, n = 5 per group. One-way ANOVA test was used for statistical analysis. E) Representative mitochondrial swelling assay monitoring absorbance of isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice after addition of 400 μM Ca2+. Traces are representative of n = 5 independent experiments. F) Representative traces of safranin fluorescence as an indicator of membrane potential (△Ψ) depolarization in isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice after addition of 400 μM Ca2+. As a positive control in EmrecKO mitochondria, depolarization was induced by addition of 1 μM FCCP. Traces are representative of n = 4 independent experiments. The mean time to Ca2+-induced depolarization, calculated as the time from Ca2+ addition to 50% of the rise in fluorescence, was for MCM+: 9.1 ± 2.8 minutes, for Emrefl/fl: 5.9 ± 1 minute, and could not be calculated for EmrecKO as FCCP was required for depolarization.
3.2. Emre deletion leads to lower intramitochondrial Ca2+ content and attenuated Ca2+-stimulated ATP production
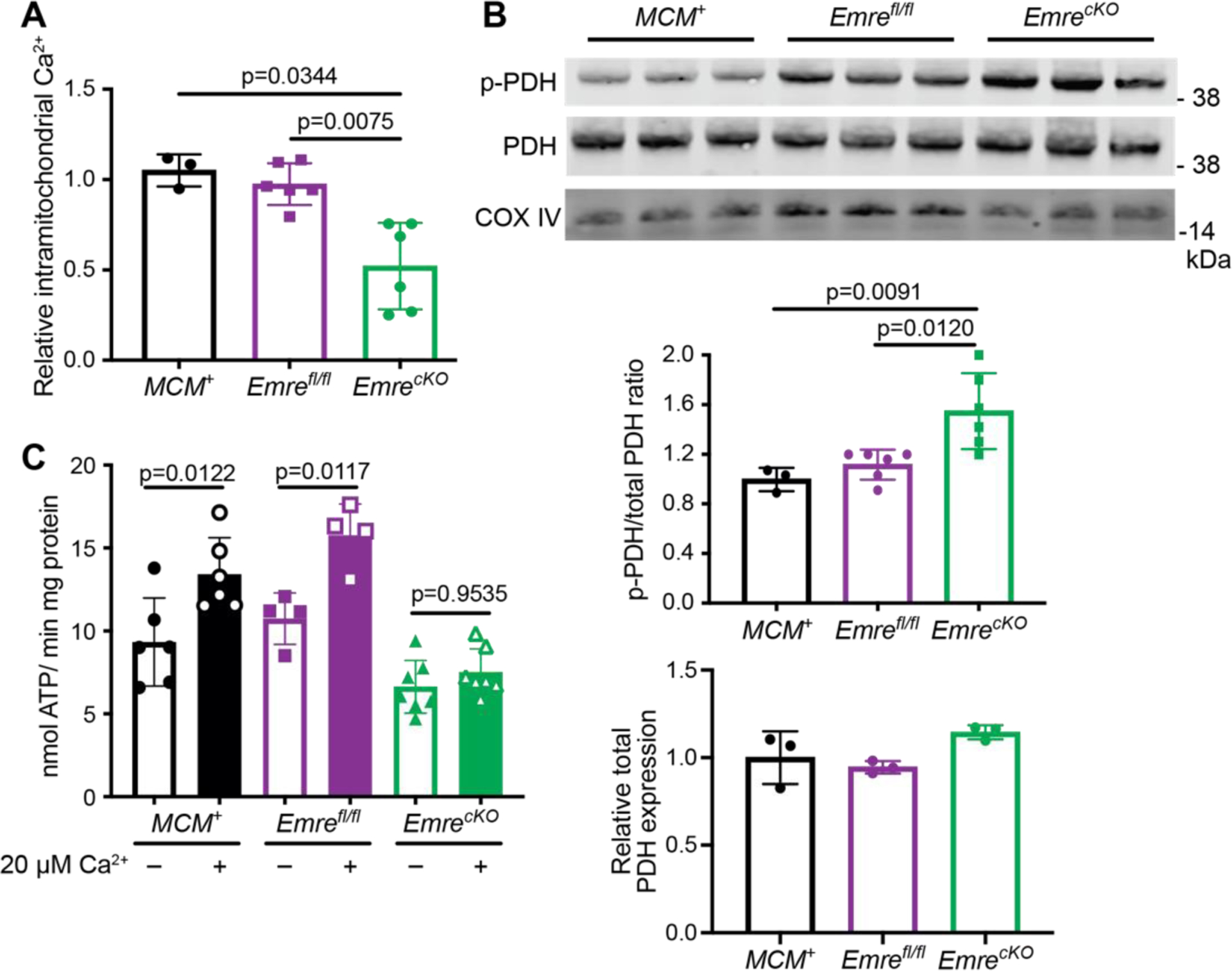
Once we confirmed that cardiac-specific EMRE deletion eliminated mitochondrial Ca2+ uptake in heart mitochondria as expected, our next question was whether this deletion affects basal intramitochondrial Ca2+ (mCa2+) levels. Relative to MCM+ and Emrefl/fl controls, EmrecKO mitochondria had lower mCa2+ levels, as might be expected in the absence of mitochondrial Ca2+ uptake activity (Fig. 2A). As the dephosphorylation of PDH is mCa2+-dependent [5], we next measured the ratio of phosphorylated pyruvate dehydrogenase (p-PDH) to total PDH in EmrecKO heart mitochondria and found a ~20% increase compared with control mitochondria (Fig. 2B). These data support that loss of EMRE leads to decreased mCa2+ levels. Because mCa2+ has been shown to activate mitochondrial energy production [39], we sought to assess whether mitochondrial respiration is affected by Emre deletion. Seahorse analysis of isolated mitochondria supplied with either pyruvate and malate or palmitoylcarnitine and malate did not reveal significant differences in oxygen consumption rate (OCR) at baseline, after ADP addition, or after FCCP treatment to decouple mitochondria and promote maximal respiration (Fig. S2A–B). Respiratory control ratios (RCR) likewise were similar between control and EmrecKO heart mitochondria for either substrate combination (Fig. S2C–D). These data suggest that, unlike Mcu deletion in heart and skeletal muscle [40–42], short-term Emre deletion did not lead to a shift toward for fatty acid oxidation in cardiac mitochondria. Consistently, protein levels of carnitine O-palmitoyl transferase 1b (CPT1) were similar across genotypes (Fig. S2E). Furthermore, though dysfunction of Complex I of the mitochondrial electron transport chain has been associated with increased uniporter stability [43,44], we did not find significant differences in the activity of either respiratory Complex I or Complex II in short-term EmrecKO and control heart mitochondria (Fig. S2F–G). We then asked whether directly measuring ATP production rate in the presence and absence of Ca2+ would reveal Ca2+-induced energetic differences in EmrecKO and control cardiac mitochondria. While the rate of ATP production upon providing ADP was similar in all groups, the addition of Ca2+ induced an increase in ATP production rate in control mitochondria but not in EmrecKO mitochondria (Fig. 2C, S2H). These results suggest that the loss of EMRE prevents stimulation of ATP production by mitochondrial Ca2+ uptake.
Figure 2. Short-term Emre deletion leads to lower intramitochondrial Ca2+ content and attenuated Ca2+-stimulated ATP production.
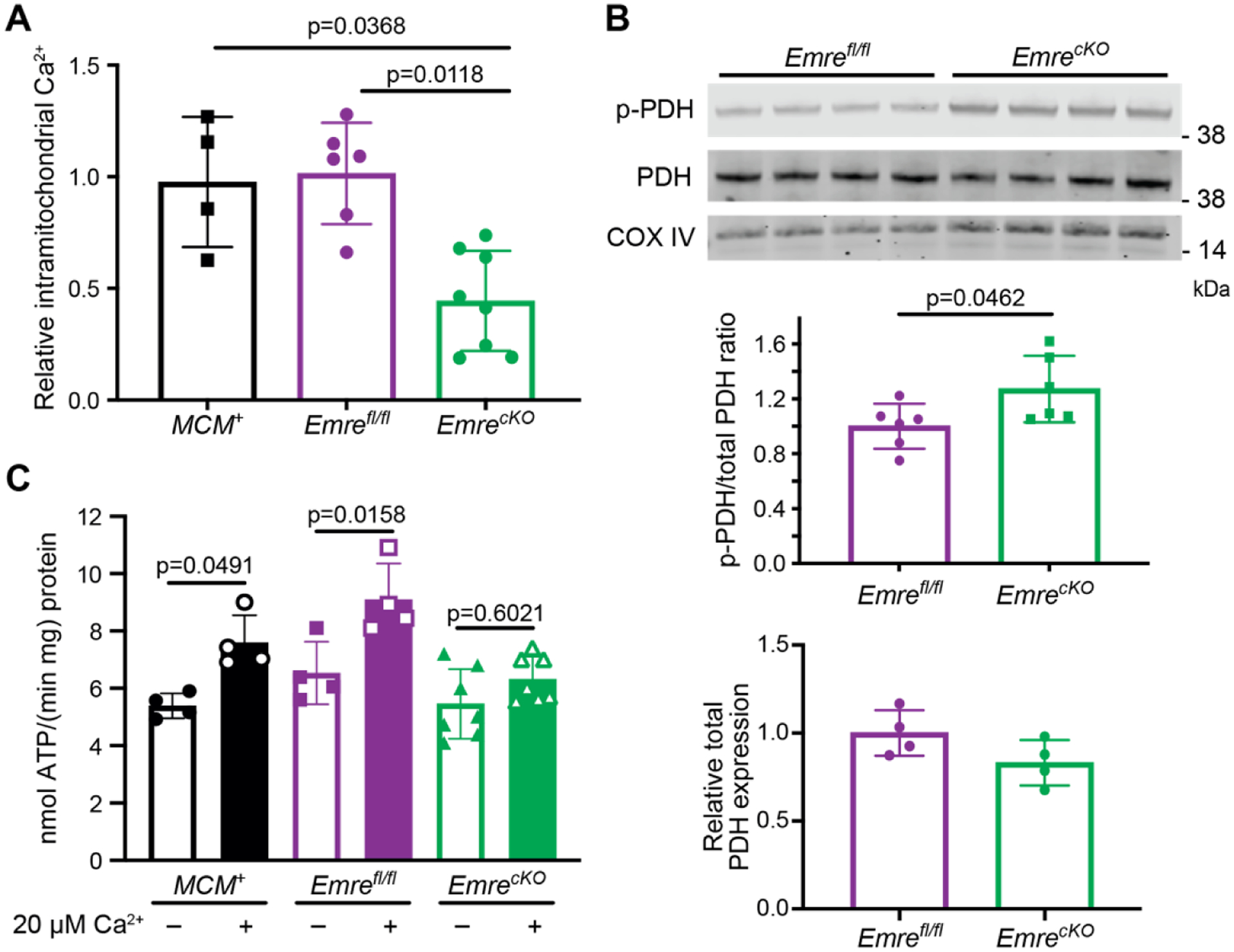
Analyses were performed 3 weeks post-tamoxifen. A) Quantification of relative free mCa2+. EmrecKO mitochondria show a reduction in mCa2+ by 56% relative to controls. n=4–8, *p<0.05. One-way ANOVA was used for statistical analysis. B) Representative Western blot of phosphorylated PDH (p-PDH) and total PDH from Emrefl/fl and EmrecKO mitochondria. COX IV was used as a loading control. Below, Western blot quantification shows a 25% increase in p-PDH to total PDH ratio in EmrecKO relative to Emrefl/fl mitochondria. n=6 per group. Student t-test was used for statistical analysis. C) Analysis of ATP production rate in isolated heart mitochondria stimulated with 50 μM ADP and 10 mM succinate ± 20 μM Ca2+. MCM+: 5.39 ± 0.43 vs MCM++ 20 μM Ca2+: 7.59 ± 0.96, Emrefl/fl: 6.35 ± 1.09 vs Emrefl/fl + 20 μM Ca2+: 9.01 ± 1.25, and EmrecKO: 5.46 ± 1.21 vs EmrecKO + 20 μM Ca2+: 6.32 ± 0.78 nmol ATP/(min*mg) protein. Values represent mean ± SD. *p<0.05, n=4–7. One-way ANOVA was used for statistical analysis.
3.3. Short-term Emre deletion leads to reduced response to β-adrenergic stimulation
We next sought to determine whether cardiac-specific Emre deletion in adult mice had physiological consequences in the short term. At 3 weeks post-tamoxifen, EmrecKO mice showed no overt phenotype and had similar body weights and heart weights as controls (Fig. S3A–B). Because the Ca2+-stimulated rate of ATP production was reduced in EmrecKO cardiac mitochondria, we reasoned that loss of EMRE might attenuate an increase in cardiac energy production upon increased workload. We thus evaluated the ability of EmrecKO mice to respond to β-adrenergic stimulation. Using echocardiography, we measured left ventricular ejection fraction (EF) and fractional shortening (FS) at baseline and then each minute up to 5 minutes after intraperitoneal injection of the β-adrenergic receptor agonist isoproterenol. In MCM+ and Emrefl/fl mice, EF (Fig. 3A) and FS (Fig. 3B) increased substantially upon stimulation, but in EmrecKO mice this response was more gradual. Although interplay between CaMKII and MCU in the context of adrenergic stimulation has been documented [45,46], we did not observe a difference in the phosphorylation status of CaMKII with Emre deletion (Fig. S3C). These results suggest that the absence of rapid mitochondrial Ca2+ uptake impairs the heart’s ability to respond to adrenergic stimulation, in agreement with previous studies investigating acute cardiac deletion of Mcu.
Figure 3. Short-term Emre deletion leads to reduced response to β-adrenergic stimulation.
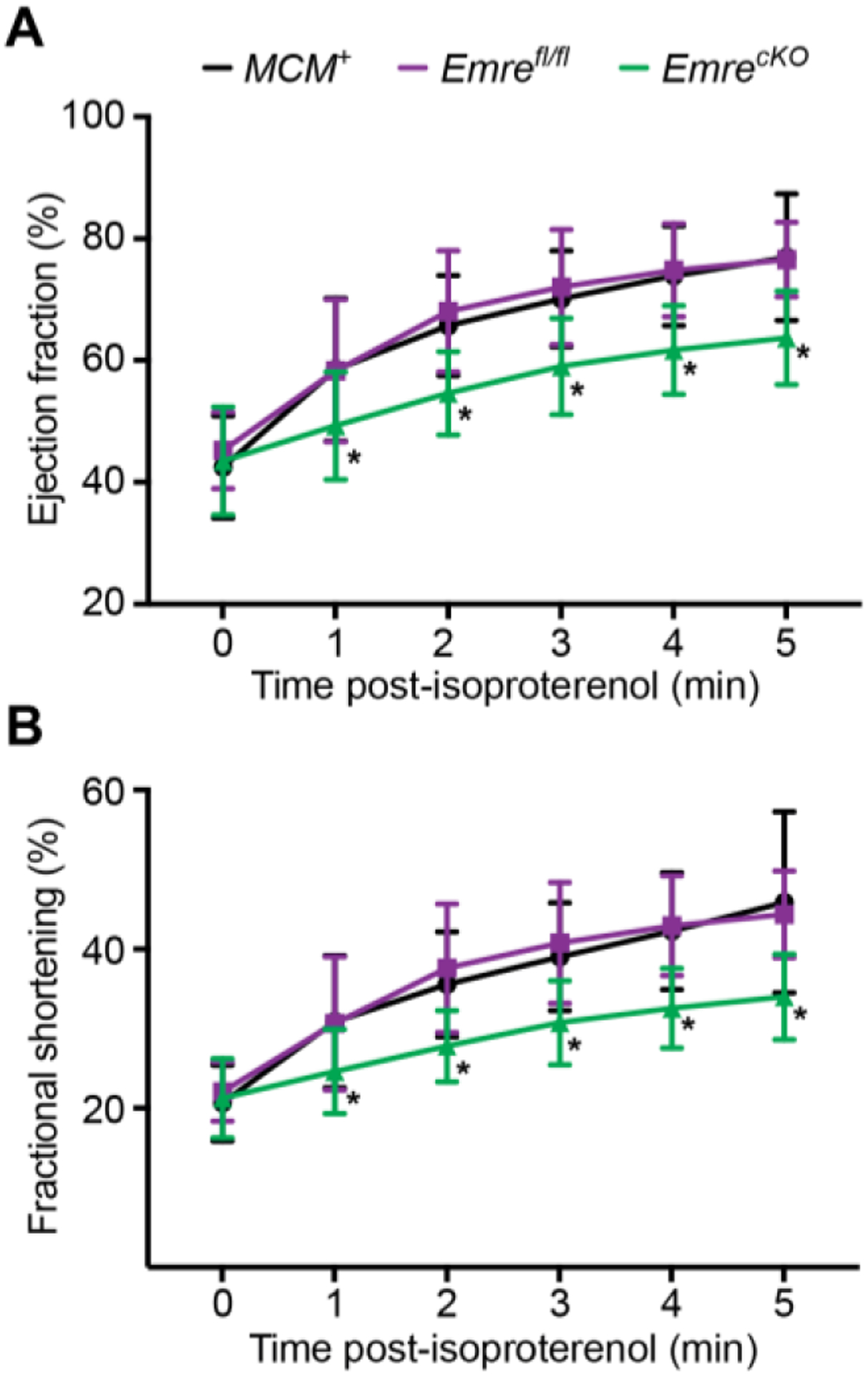
Three weeks post-tamoxifen, echocardiography at baseline (time 0), then 1, 2, 3, 4 and 5 minutes after isoproterenol injection (0.4 μg/kg) was used to analyze A) ejection fraction (EF) and B) fractional shortening (FS). A) EF at minute 5: MCM+: 76.95% ± 10.38, Emrefl/fl: 76.55% ± 6.11 and EmrecKO: 63.70% ± 7.65. B) FS at minute 5: MCM+: 45.90% ± 11.39, Emrefl/fl: 44.38% ± 5.45 and EmrecKO: 34.01% ± 5.38. Values represent mean ± SD. *p<0.05, n=14–16 per group. Two-way ANOVA test was used for statistical analysis.
3.4. Short-term Emre deletion preserves cardiac function in I/R
So far, we have shown that the short-term deletion of Emre in the adult mouse heart is sufficient to block Ca2+ uptake into mitochondria and decrease mCa2+ levels. Consistent with these effects, EmrecKO mitochondria are resistant to mPTP opening following mCa2+ overload. Cell death due to mPTP opening is thought to contribute to I/R injury; therefore, we evaluated whether EmrecKO mice would show preserved cardiac function in an ex vivo Langendorff perfusion model of I/R injury. Our results show that during reperfusion EmrecKO hearts maintained higher rate pressure product (RPP) relative to baseline pre-ischemic levels compared with control hearts, and this protection is even preserved until the end of the reperfusion period (Fig. 4A). Likewise, measures of systolic and diastolic pressure changes (dP/dtmax and dP/dtmin, respectively) were preserved to a greater degree during reperfusion in EmrecKO mice compared to control mice (Fig. 4B–C). To determine the degree of injury in the hearts, we measured the release of creatine kinase (CK), a marker of cardiomyocyte membrane injury, and found reduced CK release in EmrecKO mice relative to controls (Fig. 4D). Although Cyclophilin D (CypD) is a known activator of the mPTP, resistance to pore opening does not appear to be mediated by any decrease in CypD protein expression (Fig. S3D). Altogether, the data show that shortly after the loss of EMRE in adulthood, hearts are protected against I/R injury, consistent with the idea that preventing mitochondrial Ca2+ uptake during I/R prevents tissue damage.
Figure 4. Short-term Emre deletion preserves cardiac function in I/R.
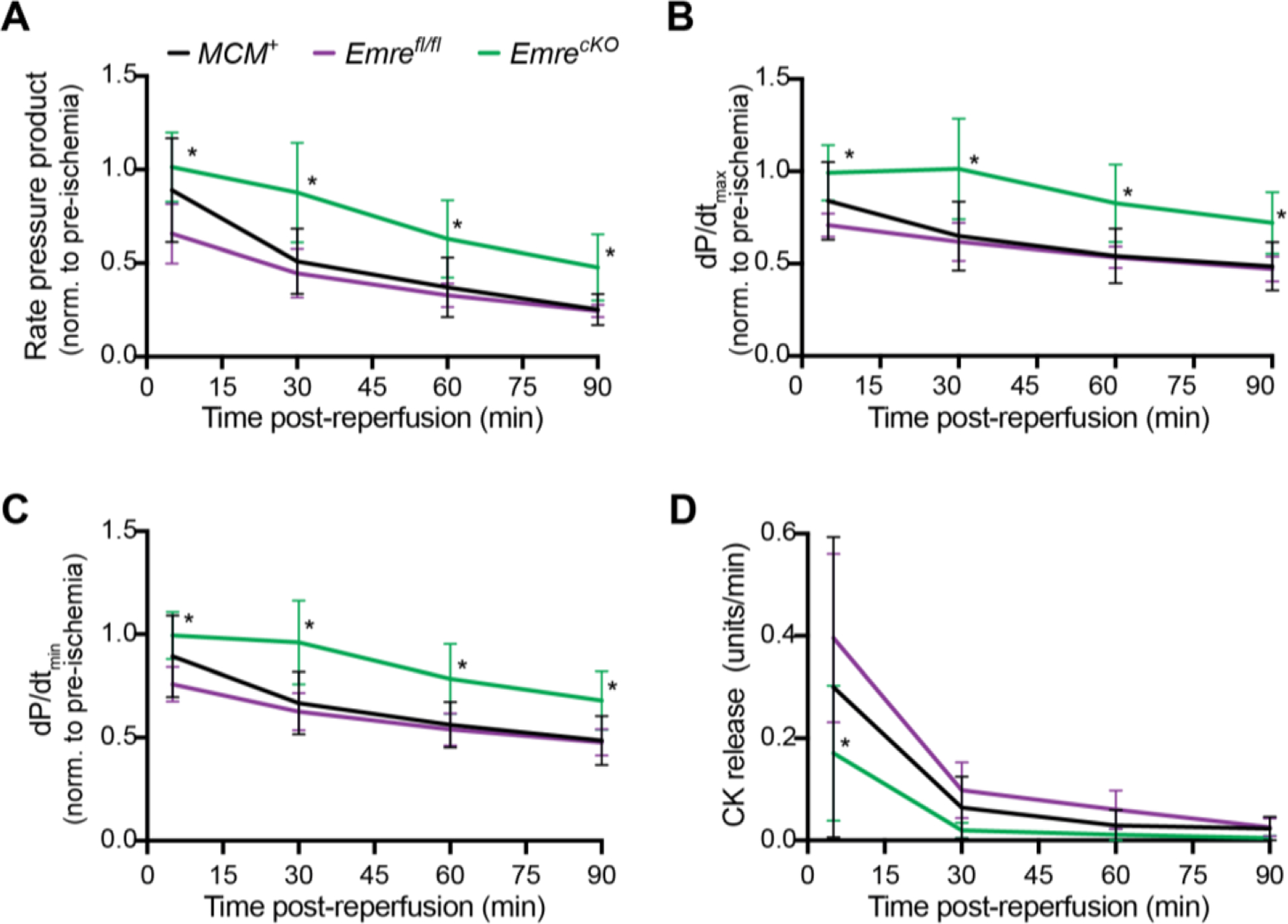
Three weeks post-tamoxifen, mouse hearts were subjected to 20 minutes of stabilization, 20 minutes of global ischemia and 90 minutes of reperfusion. At 5, 30, 60, and 90 minutes after the onset of reperfusion, relative to baseline pre-ischemia, A) rate pressure product (RPP) = heart rate (HR) × left ventricular pressure (LVP), B) dP/dtmax, C) dP/dtmin, and D) creatine kinase (CK) release were assessed. Values at 90 minutes: A) RPP: MCM+: 50.96% ± 17.39, Emrefl/fl: 44.55% ± 13.05 and EmrecKO: 87.64% ± 26.68. B) dP/dtmax: MCM+: 48.50% ± 13.10, Emrefl/fl: 47.14% ± 6.74 and EmrecKO: 72.04% ± 16.62. C) dP/dtmin: MCM+: 48.53% ± 11.95, Emrefl/fl: 47.64% ± 6.45 and EmrecKO: 67.88% ± 14.23. Values represent mean ± SD. *p<0.05, n=5–6 per group. Two-way ANOVA test was used for statistical analysis.
3.5. Mitochondrial Ca2+ handling remains impaired in EmrecKO mice over time
Thus far, we have confirmed that the short-term loss of EMRE in adulthood is consistent with many aspects of the McucKO phenotype, further solidifying that like MCU, EMRE is essential for uniporter activity. We next sought to explore why germline Mcu−/− and Emre−/− mice do not exhibit the impaired response to adrenergic stimulation and protection from I/R injury seen in the McucKO mice. This discrepancy has been attributed to remodeling in the chronic absence of uniporter activity, but whether long timescales alone are sufficient for remodeling is unclear. Therefore, we waited 3 months post-tamoxifen, which we will refer to as long-term Emre deletion, to reexamine the phenotype of EmrecKO mice. We first measured the mRNA and protein expression of EMRE and MCU in isolated cardiac mitochondria to confirm that protein levels relative to controls remained significantly decreased (Fig. 5A), although as before Mcu mRNA is expressed at control levels (Fig. S4A–B). Expression of MICU1 as well as the efflux proteins NCLX and LETM1 were still unchanged between EmrecKO and control mitochondria (Fig. 5A, S4C–D). Similarly, we verified that at 3 months post-tamoxifen the molecular weights of the uniporter complex with and without EMRE remained the same as at 3 weeks post-tamoxifen (Fig. 5B). Next, we conducted CRC, swelling, and membrane potential assays in EmrecKO mitochondria 3 months post-tamoxifen and found that the loss of mitochondrial Ca2+ uptake and resistance to mPTP opening were maintained, as observed in short-term deletion (Fig. 5C–F, S4E–G).
Figure 5. Mitochondrial Ca2+ handling remains impaired with long-term Emre deletion.
Analyses were performed 3 months post-tamoxifen. A) Representative Western blot of EMRE, MCU, and MICU1 in cardiac mitochondria from MCM+, Emrefl/fl and EmrecKO mice. VDAC was used as a loading control. Below, Western blot quantification of mitochondria from MCM+ (n = 3), Emrefl/fl (n = 6), and EmrecKO (n = 6) hearts. Values represent mean ± SD. One-way ANOVA was used for statistical analysis. B) Blue Native gel of freshly isolated mitochondria from Emrefl/fl and EmrecKO hearts, immunoblotted with MCU antibody. Gel is representative of 3 mice (combined males and females) per group. C) Representative Ca2+ retention capacity (CRC) assay in isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice. The fluorescent Ca2+ indicator Calcium Green-5N was used to monitor extramitochondrial Ca2+. The arrows represent 10 μM Ca2+ added. Traces are representative of n = 5 independent experiments. D) Ca2+ retention capacity calculated from independent traces as shown in (C). The estimated mean Ca2+/mg protein was for MCM+ : 154 ± 30.50, Emrefl/fl : 136 ± 27.01 and EmrecKO : 6 ± 5.47 nmol of Ca2+ / mg protein. Values represent mean ± SD.**p<0.0001, n = 5 in each group. One-way ANOVA test was used for statistical analysis. E) Representative mitochondrial swelling assay monitoring absorbance of isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice after addition of 400 μM Ca2+. Traces are representative of n = 5 independent experiments. F) Representative traces of membrane potential (△Ψ) depolarization in isolated heart mitochondria from MCM+ (black line), Emrefl/fl (purple line) and EmrecKO (green line) mice after the addition of 400 μM Ca2+. As a positive control in EmrecKO mitochondria, depolarization was induced by addition of 1 μM FCCP. Traces are representative of n = 4 independent experiments. The mean time to Ca2+-induced depolarization, calculated as the time from Ca2+ addition to 50% of the rise in fluorescence, was for MCM+: 10.5 ± 2 minutes, for Emrefl/fl: 12.7 ± 2.3 minutes, and could not be calculated for EmrecKO as FCCP was required for depolarization.
Similarly, measurements of free mCa2+ levels in EmrecKO mitochondria 3 months post-tamoxifen suggested that mCa2+ remained decreased (Fig. 6A), consistent with significantly elevated p-PDH to total PDH ratio in EmrecKO mitochondria (Fig. 6B). As found in the short-term, long-term Emre deletion did not alter OCR or RCR in either pyruvate/malate or palmitoylcarnitine/malate substrate conditions (Fig. S5A–D). Hence, long-term absence of Emre deletion did not appear to lead to a shift toward preferential fatty acid oxidation, and correspondingly CPT1 protein levels were unchanged (Fig. S5E). Complex II activity remained similar between groups 3 months post-tamoxifen; Complex I activity in EmrecKO mitochondria exhibited a slight downward trend that however did not reach significance (p = 0.0735) (Fig. S5F–G). Additionally, though baseline ATP production rates were similar in EmrecKO and control mitochondria, the increase in ATP production rate in the presence of Ca2+ was still absent in EmrecKO mitochondria (Fig. 6C, S5H). Taken together, these data suggest that, at the level of isolated mitochondria, long-term absence of EMRE does not lead to substantially different effects compared to those of the short-term.
Figure 6. Lower intramitochondrial Ca2+ content and attenuated Ca2+-stimulated ATP production are maintained over long-term Emre deletion.
Analyses were performed 3 months post-tamoxifen. A) Quantification of relative free mCa2+ in mitochondria from MCM+ (n = 3), Emrefl/fl (n = 6), and EmrecKO (n = 6) hearts. EmrecKO mitochondria show a reduction in mCa2+ by 40% ± 15.01 relative to controls. Values represent mean ± SD. One-way ANOVA was used for statistical analysis. B) Representative Western blot of phosphorylated PDH (p-PDH) and total PDH from Emrefl/fl and EmrecKO mitochondria. COX IV was used as a loading control. Below, Western blot quantification of mitochondria from MCM+ (n = 3), Emrefl/fl (n = 6), and EmrecKO (n = 6) hearts. Values represent mean ± SD. One-way ANOVA was used for statistical analysis. C) ATP production rate in isolated heart mitochondria stimulated with 50 μM ADP and 10 mM succinate ± 20 μM Ca2+. MCM+: 9.33 ± 2.65 vs MCM++ 20 μM Ca2+: 13.41 ± 2.21, Emrefl/fl: 10.73 ± 1.55 vs Emrefl/fl + 20 μM Ca2+: 15.76 ± 1.90, and EmrecKO: 6.63 ± 1.59 vs EmrecKO + 20 μM Ca2+: 7.52 ± 1.40 nmol ATP/min*mg protein. Values represent mean ± SD. *p<0.05, n=4–7. One-way ANOVA was used for statistical analysis.
3.6. β-adrenergic response remains diminished in long-term Emre deletion, but protection against I/R injury is lost
After confirming that long-term Emre deletion revealed similar mitochondrial Ca2+ handling as in short-term Emre deletion, we sought to assess at 3 months post-tamoxifen how EmrecKO mice responded to β-adrenergic stimulation. We first verified that long-term Emre deletion did not lead to any significant changes in body weight or heart weight (Fig. S6A–B). We then repeated the echocardiography experiment measuring ejection fraction and fractional shortening at baseline and after isoproterenol injection. Notably, in contrast to germline Emre or Mcu deletion, long-term cardiac-specific Emre deletion in adulthood still resulted in diminished adrenergic response (Fig. 7A–B). The phosphorylation status of CaMKII also remained similar between groups (Fig. S6C). This result suggests that the fight or flight response cannot be restored in a time-dependent manner in the chronic absence of rapid mitochondrial Ca2+ uptake ability. After confirming that β-adrenergic response remained blunted in mice with long-term cardiac Emre deletion, we next sought to determine whether protection from I/R injury is similarly preserved. We repeated the Langendorff ex vivo I/R experiment in 3-month post-tamoxifen EmrecKO and control hearts. Surprisingly, and in contrast to short-term Emre deletion, we observed that long-term Emre deletion did not have a significant effect relative to controls on RPP, dP/dtmax, dP/dtmin, or CK release following reperfusion (Fig. 7C–F). This lack of protection in long-term Emre deletion could not be attributed to increased CypD expression, as CypD protein levels did not vary significantly between EmrecKO and control mitochondria (Fig. S6D).
Figure 7. β-adrenergic response remains diminished in long-term Emre deletion, but protection against I/R injury is lost.
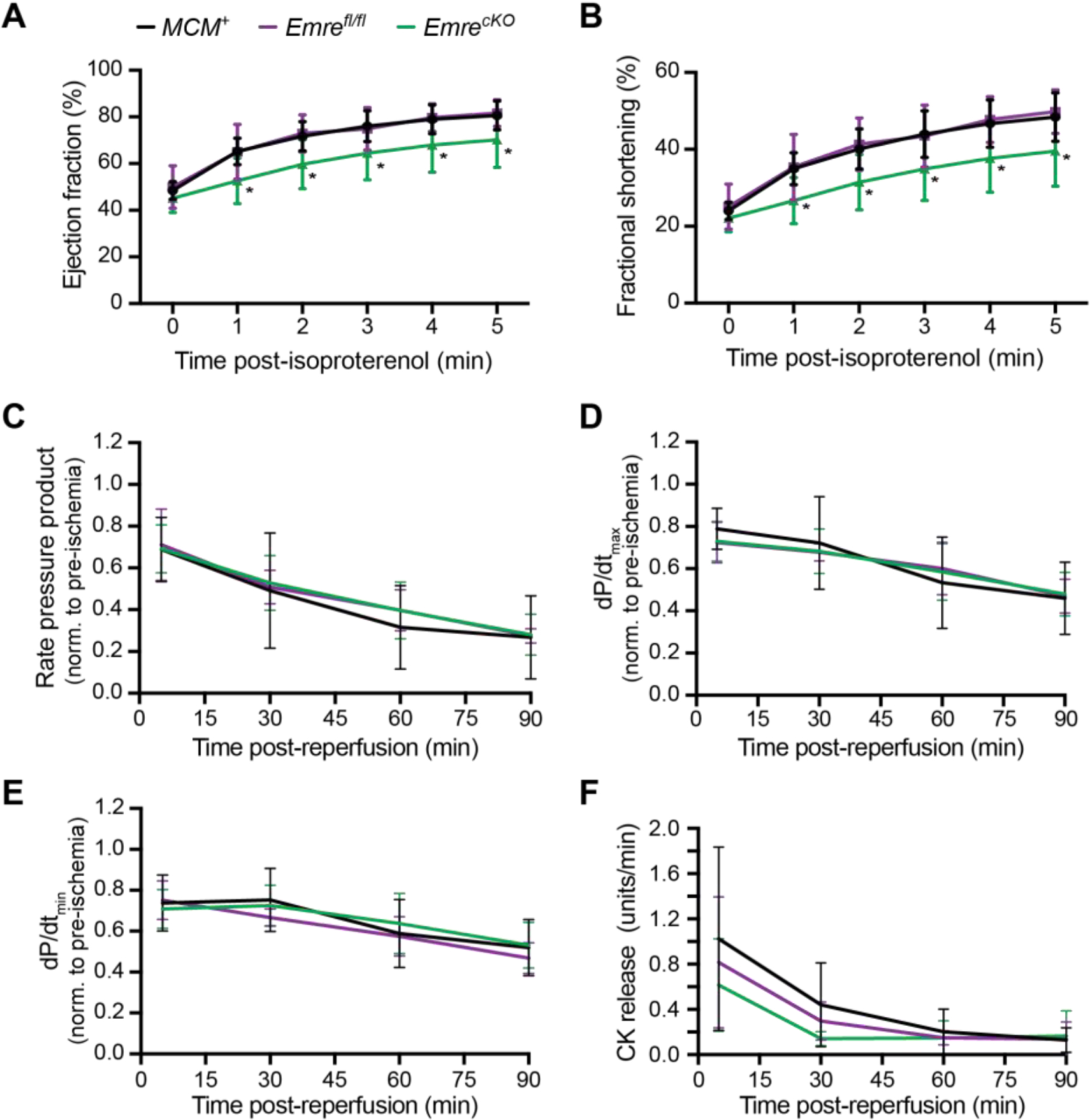
Three months post-tamoxifen, echocardiography at baseline (time 0), then 1, 2, 3, 4 and 5 minutes after isoproterenol injection (0.4 μg/kg) was used to analyze A) ejection fraction (EF) and B) fractional shortening (FS). A) EF at minute 5: MCM+(black line): 80.58% ± 6.19, Emrefl/fl (purple line): 81.62% ± 5.69 and EmrecKO (green line): 70.20% ± 11.08. B) FS at minute 5: MCM+: 48.42% ± 6.32, Emrefl/fl: 49.80% ± 5.67 and EmrecKO: 39.58% ± 9.14. Values represent mean ± SD. *p<0.05, n=10–12 per group. Two-way ANOVA was used for statistical analysis. Three months post-tamoxifen, mouse hearts were subjected to 20 minutes of stabilization, 20 minutes of global ischemia and 90 minutes of reperfusion. At 5, 30, 60, and 90 minutes after the onset of reperfusion, relative to baseline pre-ischemia, C) rate pressure product (RPP) = heart rate (HR) × left ventricular pressure (LVP), D) dP/dtmax, E) dP/dtmin, and F) creatine kinase (CK) release were assessed. Values represent mean ± SD. *p<0.05, n=5–6 per group. Two-way ANOVA was used for statistical analysis.
We then considered that apart from excessive mitochondrial Ca2+, reactive oxygen species (ROS) can also act as a trigger for mPTP opening [47]. Despite equivalent rates of ROS production in isolated EmrecKO and control mitochondria in the short and long term as measured by Amplex Red (Fig. S7A–C), assessment of protein carbonylation revealed that EmrecKO mitochondria have significantly less oxidative stress in the short-term than in the long-term (Fig. 8A–B). Furthermore, although oxidative stress levels between EmrecKO and control mitochondria in the long-term are similar, in the short-term EmrecKO mitochondria show a trend toward decreased oxidative stress relative to control mitochondria that approaches significance (p = 0.0548). These data suggest that a transient decline in oxidative stress in short-term but not long-term Emre deletion is associated with protection against I/R injury; over time, oxidative stress returns to control levels, and concurrently protection against I/R injury conferred by acute loss of mitochondrial Ca2+ uniporter activity is lost.
Figure 8. Short-term Emre deletion leads to lower oxidative stress levels than long-term Emre deletion.
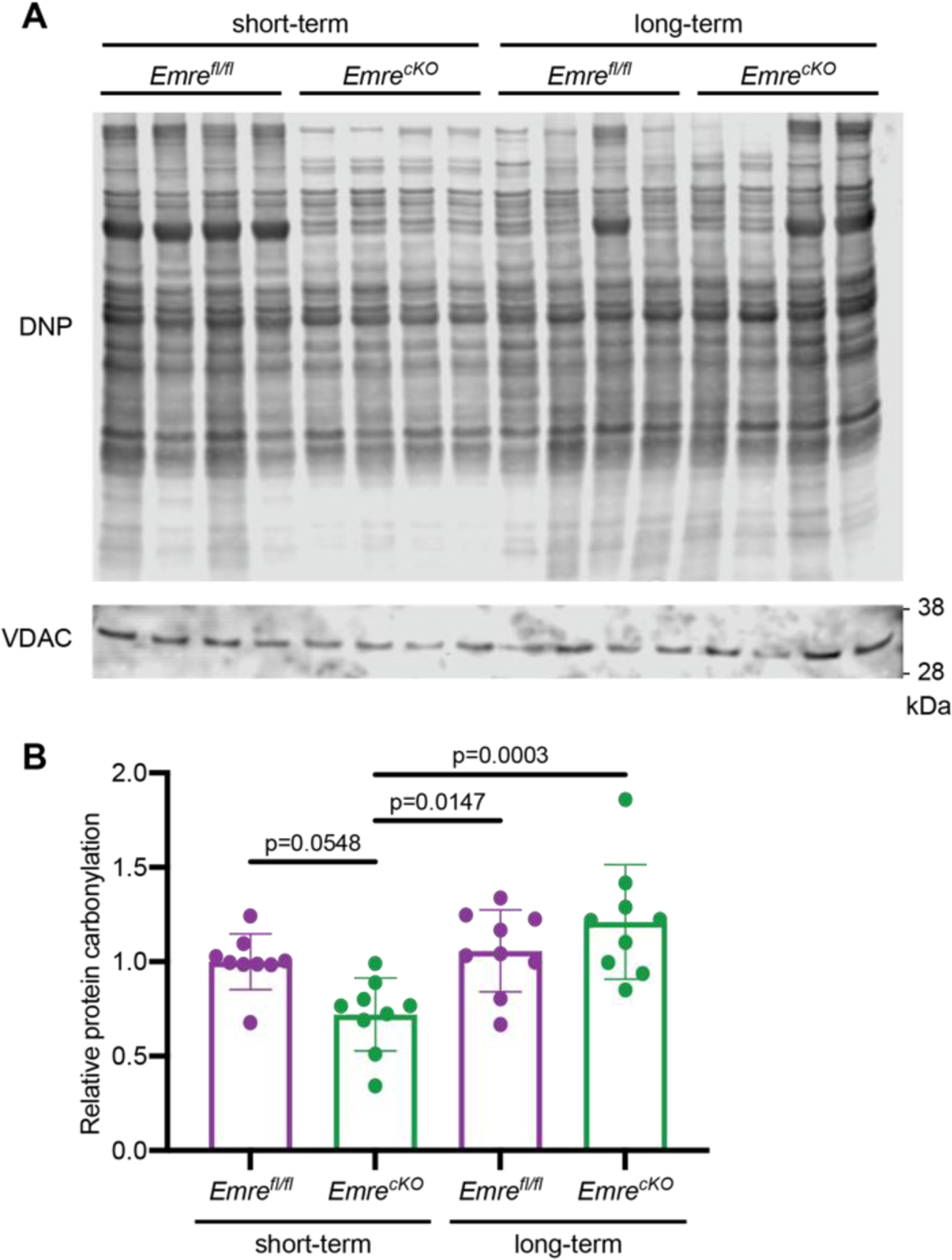
A) Representative Western blot using anti-DNP antibody to detect carbonyl groups modified by DNPH as a marker of oxidized proteins in cardiac mitochondria from short-term and long-term Emrefl/fl and EmrecKO mice. VDAC was used as a loading control. B) Western blot quantification of mitochondria from short-term Emrefl/fl: 1.00 ± 0.14 (n = 9), short-term EmrecKO: 0.72 ± 0.19 (n = 9), long-term Emrefl/fl: 1.05 ± 0.21 (n = 9), and long-term EmrecKO: 1.21 ± 0.30 (n = 9). Values represent mean ± SD, *p<0.05. One-way ANOVA was used for statistical analysis.
4. Discussion
Here we present the novel finding that cardiac-specific Emre deletion in adult mice has distinct effects in the short and long term. While a blunted response to isoproterenol is preserved from 3 weeks to 3 months after Emre deletion, an initial protection against I/R injury is no longer observed after 3 months. This dichotomy exists despite our data showing that in isolated mitochondria at both time points Emre deletion eliminates mitochondrial Ca2+ uptake and reduces sensitivity to Ca2+-induced mPTP opening.
Lack of rapid mitochondrial Ca2+ uptake was also seen in mitochondria from mice with germline Mcu or Emre deletion as well as mice undergoing cardiac-specific Mcu deletion in adulthood [30,31]. Our results in a new cardiac-specific Emre knockout mouse model support the multiple studies showing that EMRE is essential to uniporter function [23,24,32]. Furthermore, like Mcu−/− and Emre−/− mice [28,32] but unlike McucKO mice [30,31], EmrecKO mice have reduced cardiac intramitochondrial Ca2+ concentration, with lasting effects even 3 months after tamoxifeninduced deletion. In EmrecKO mice, we also observe a large decrease in MCU expression, to a greater extent than observed in the germline Emre−/− mice in which MCU decreased only slightly [32]. Our findings in these mice are thus not attributable solely to the absence of EMRE alone; rather, EmrecKO mice represent another model in which the uniporter is nonfunctional. Nonetheless, in both EmrecKO and germline Emre−/− mice, and in the germline Mcu−/− mice where EMRE was significantly decreased [28], the data suggest that a coordinated downregulation of MCU in the absence of EMRE and vice versa leads to lower intramitochondrial Ca2+ levels. As EMRE expression was not reported in McucKO mice, it is possible that lesser EMRE downregulation with MCU in these circumstances might explain why intramitochondrial Ca2+ was unchanged in McucKO mice [30,31].
Despite reduced basal intramitochondrial Ca2+ levels after short-term Emre deletion, EmrecKO cardiac mitochondria produced ATP at the same rate as control mitochondria when supplied with ADP without Ca2+. This result echoes previous measurements of oxygen consumption or ATP production in Mcu−/−, Emre−/−, or McucKO cardiac mitochondria [28,30,32]. However, Ca2+-induced stimulation of ATP production was observed in control but not EmrecKO and other uniporter-deficient cardiac mitochondria [28,30], consistent with the uniporter’s purported role in delivering Ca2+ to the matrix to provide a boost in ATP. Thus, in the absence of a functional uniporter, matching of energy supply to demand, for instance in the fight or flight response, might be expected to be attenuated. Consistent with this hypothesis, here we show with echocardiography that short-term deletion of Emre in adulthood, as was shown for Mcu deletion in in vivo hemodynamics experiments [30,31], resulted in impaired cardiac contractile response to adrenergic stimulation (see Table 1). In contrast, the hearts of global Mcu−/− and Emre−/− mice responded indistinguishably from wild-type mice to isoproterenol, measured in the former by cardiac catheterization [28] and in the latter by echocardiography [32]. Recent work has shown that Langendorff-perfused global Mcu−/− hearts show no increase in mitochondrial Ca2+ following isoproterenol addition, yet contractility increases similarly in Mcu−/− and wild-type hearts [48]. These results suggest that in the Mcu−/− and Emre−/− models, the chronic absence of uniporter activity beginning in the germline may have led to compensatory mechanisms that act to meet increased energy demand. However, what form this compensation takes or when it occurs are unclear. To address the latter question, we assessed EmrecKO mice at a long time interval (3 months) after tamoxifen administration in adulthood and found that impaired adrenergic response persisted even after long-term EMRE deletion. Although it cannot be excluded that recovery of the rapid response to adrenergic stimulation could gradually take place at timescales longer than 3 months after the loss of uniporter activity starting in adulthood, one possibility is that restoration of the adrenergic response in global Mcu−/− and Emre−/− mice does not occur merely in a time-dependent manner and instead may require the absence of uniporter function during embryogenesis. Notably, the expression of DN-Mcu driven by the αMHC promoter, which is expressed in the myocardium shortly after birth [49], also results in reduced response to isoproterenol [29]. Taken together, one might speculate that the onset of uniporter deficiency at any time after birth stably attenuates the ability of the mitochondria to respond to a sudden increase in energy demand in the fight or flight response. It remains to be tested whether compensation for uniporter loss in adulthood can occur gradually over timescales longer than 3 months or whether compensation is only possible when uniporter loss is experienced early in development in conditions of germline Mcu or Emre deletion.
The inactivation of mitochondrial Ca2+ uptake due to Emre deletion would be predicted to protect against I/R injury, and in line with this reasoning, at 3 weeks post-tamoxifen, EmrecKO mice were able to better maintain cardiac function compared with controls in an ex vivo Langendorff I/R model. These results are consistent with studies in McucKO mice undergoing in vivo I/R [30,31], demonstrating that protection from I/R can manifest in both in vivo and ex vivo models (see Table 1). Hence, this experimental distinction is not the cause of divergent results in McucKO mice (shown in vivo to be protected from I/R) compared with Mcu−/−, Emre−/−, and DN-Mcu mice (shown ex vivo to be unprotected). Moreover, it was previously unclear whether the lack of protection against I/R injury in Mcu−/−, Emre−/−, and DN-Mcu mice arose from compensatory cellular pathways in response to chronic uniporter loss, and if so whether these pathways are only active when the uniporter is absent in development or are activated as function of time. Our data support the latter hypothesis, as by 3 months post-tamoxifen, EmrecKO hearts exhibited a similar decrease in cardiac function during I/R as control hearts, suggesting that the protection from I/R injury evident at 3 weeks post-tamoxifen was lost in the intervening time. We conclude that although mice gain protection from I/R injury after acute deletion of Mcu or Emre in adulthood, this effect appears to be transient. Within a few months, EmrecKO hearts, from which isolated mitochondria are still unable to rapidly take up Ca2+, experience I/R-induced damage to roughly the same extent as control hearts. Furthermore, concurrently with I/R protection in short-term Emre deletion, protein carbonylation potentially decreases but later recovers to control levels in the long term. This interesting phenomenon argues that time-dependent compensation for uniporter loss, potentially due to a return to baseline oxidative stress levels, is sufficient to restore susceptibility to I/R, but the nature of this compensation remains incompletely resolved.
We examined multiple cellular pathways known to be linked to MCU activity to determine whether or not in the short- or long-term absence of EMRE these pathways would be altered. Loss of MCU has been shown to alter metabolic substrate preference in the heart and skeletal muscle, in which glucose oxidation is impaired or unchanged while fatty acid oxidation is favored [40–42]. Our data suggest that OCR in the presence of substrates for either glucose oxidation or fatty acid oxidation is similar between control and EmrecKO heart mitochondria in both the short-and long-term. However, it was noted within one study that although OCR measured in Langendorff-perfused and paced hearts increased in DN-Mcu mice, Seahorse analysis of control and DN-Mcu isolated mitochondria showed no differences [29]. Hence, though we were unable to detect intrinsic mitochondrial differences, further study is needed to uncover metabolic alterations, including potentially fuel preference, in animals with short- and long-term Emre deletion.
Additionally, it has been shown that during chronic β-adrenergic stimulation, CaMKIIδB activation upregulates Mcu expression at the transcriptional level [45]. However, in the context of both short- and long-term EMRE loss our data show that while MCU protein decreases, CaMKII phosphorylation as well as Mcu mRNA levels remain unchanged. Indeed, the modulation of uniporter activity was shown to be downstream of the CaMKII pathway, and not necessarily vice versa. Similarly, decreased Complex I activity in multiple contexts was shown to lead to compensatory increase in MCU levels and activity [43,44]; whether alterations in MCU activity can reciprocally impact Complex I is less clear. Mild decreases in Complex I and II protein levels were previously observed following acute isoproterenol challenge in McucKO hearts 3 months post-tamoxifen [40]. In our study, though no statistically significant decrease in Complex II activity occurred in short- or long-term Emre deletion, and short-term EmrecKO and control Complex I activity were also similar, at the long-term timepoint Complex I activity trended downwards in EmrecKO mitochondria relative to controls, nearly achieving significance. Since it is thought that Complex I and MCU interact such that mild ROS leak from Complex I oxidizes MCU [44], one might speculate that gradually over time, a prolonged absence of this interaction in EmrecKO mice could result in even slightly increased oxidative damage to Complex I itself that may decrease its activity. The interaction of Complex I and the uniporter complex, and how perturbation thereof might directly and indirectly affect sensitivity to I/R injury, is amply deserving of future study.
Furthermore, we observed that protein carbonylation is transiently decreased in the short-term absence of EMRE relative to long-term EMRE absence. These data, which temporally correlate with the transient protection from I/R injury, are notable given the well-documented involvement of ROS in opening the mPTP [47]. A gradual decline in Complex I activity and potential uptick in ROS leakage may play a part in the resurgence of oxidative stress in the long-term absence of EMRE, yet what mediates the transient decrease in oxidative damage is still an open question. Which proteins undergo less or more oxidation in the short or long term is also important: for instance, oxidation of CypD is reported to target it to the mitochondrial inner membrane where it can activate mPTP [50]. The germline loss of MCU has been suggested to lead to alterations in the post-translational modification status of CypD that prime mitochondria toward mPTP opening [51]. More detailed study in EmrecKO mice of time-dependent CypD expression and modifications, particularly oxidation, is warranted. Finally, cardiomyocytes that chronically lack a functional uniporter may undergo a cell death pathway in I/R distinct from mPTP-triggered necroptosis, given evidence that the CypD inhibitor Cyclosporine A is protective in I/R in wild-type hearts but not Mcu−/− or Emre−/− hearts [26,32]. What this cell death pathway or pathways might be or what might lead to its activation during the chronic absence of uniporter activity is still unclear.
In summary, we have reported the characterization of a novel tamoxifen-inducible cardiac-specific mouse model of Emre deletion, with which we have examined mitochondrial Ca2+ handling, response to adrenergic stimulation, and susceptibility to I/R injury at short (3 week) and long (3 month) post-tamoxifen time points. In most respects long-term Emre deletion closely resembled short-term Emre deletion. In isolated mitochondria, the differences between EmrecKO and controls at 3 weeks post-tamoxifen – lack of mitochondrial Ca2+ uptake, increased p-PDH to total PDH ratio, reduced Ca2+ levels, resistance to Ca2+-induced swelling, and attenuated Ca2+-stimulated ATP production – were still present at 3 months post-tamoxifen. We found that Emre deletion in both the short- and long-term impaired isoproterenol-induced increase in contractility; however, only short-term Emre deletion was protective against reduced cardiac function during I/R, potentially due to reduced oxidative stress that appears restricted to the short-term time point. This discrepancy underscores that the compensatory mechanism that allows only mice lacking a functional uniporter starting in the germline to retain their adrenergic response is fundamentally different from the mechanism that in the span of a few months following uniporter loss in adult mice restores sensitivity to I/R. The nature of these uniporter deficiency-induced pathways and the cellular changes they elicit remain a complex problem to be addressed in future research.
Supplementary Material
Highlights.
A tamoxifen-inducible, cardiac-specific mouse model of Emre deletion was generated
Mitochondrial Ca2+ uptake is eliminated after short and long-term EMRE loss
Response to adrenergic stimulation is blunted after short and long-term EMRE loss
Short-term EMRE loss is protective against ischemia/reperfusion (I/R) injury
Protection against I/R injury is lost in the long-term absence of EMRE
Funding
This work was supported by the National Institutes of Health [1K22HL137901 and 1R01HL164491 to J.C.L].
Acknowledgements
We are grateful to Dr. Elizabeth Murphy and members of the Liu and Murphy labs for thoughtful discussions and feedback. We thank Lynn Hartweck and Dr. Madjda Bellamri for their help in optimizing methods to measure respiratory complex activity. We would also like to thank Maximiliano and Dorotea for their everlasting support.
Footnotes
Conflict of Interest: none declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Doenst T, Nguyen TD, Abel ED, Cardiac metabolism in heart failure: implications beyond ATP production, Circ Res. 113 (2013) 709–724. 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Barth E, Stämmler G, Speiser B, Schaper J, Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man, J. Mol. Cell. Cardiol 24 (1992) 669–681. 10.1016/0022-2828(92)93381-s. [DOI] [PubMed] [Google Scholar]
- [3].Duchen MR, Verkhratsky A, Muallem S, Mitochondria and calcium in health and disease, Cell Calcium. 44 (2008) 1–5. 10.1016/j.ceca.2008.02.001. [DOI] [PubMed] [Google Scholar]
- [4].Glancy B, Balaban RS, Role of mitochondrial Ca2+ in the regulation of cellular energetics, Biochemistry. 51 (2012) 2959–2973. 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Denton RM, Randle PJ, Martin BR, Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase, Biochem J 128 (1972) 161–163. 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Denton RM, McCormack JG, The role of calcium in the regulation of mitochondrial metabolism, Biochem. Soc. Trans 8 (1980) 266–268. [DOI] [PubMed] [Google Scholar]
- [7].Kwong JQ, The mitochondrial calcium uniporter in the heart: energetics and beyond, J Physiol. 595 (2017) 3743–3751. 10.1113/JP273059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kwong JQ, Molkentin JD, Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart, Cell Metabolism. 21 (2015) 206–214. 10.1016/j.cmet.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Santulli G, Xie W, Reiken SR, Marks AR, Mitochondrial calcium overload is a key determinant in heart failure, Proc. Natl. Acad. Sci. U.S.A 112 (2015) 11389–11394. 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vallejo-Illarramendi A, Toral-Ojeda I, Aldanondo G, López de Munain A, Dysregulation of calcium homeostasis in muscular dystrophies, Expert Rev Mol Med. 16 (2014) e16. 10.1017/erm.2014.17. [DOI] [PubMed] [Google Scholar]
- [11].and P.P. Giampaolo Morciano, Massimo Bonora, Gianluca Campo, Giorgio Aquila, Paola Rizzo, Carlotta Giorgi, Mariusz R. Wieckowski, Mitochondrial Dynamics in Cardiovascular Medicine, 2017. 10.1007/978-3-319-55330-6_9. [DOI] [Google Scholar]
- [12].Martin SD, McGee SL, The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes, Biochim Biophys Acta. 1840 (2014) 1303–1312. 10.1016/j.bbagen.2013.09.019. [DOI] [PubMed] [Google Scholar]
- [13].García-Rivas GDJ, Carvajal K, Correa F, Zazueta C, Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo., British Journal of Pharmacology. 149 (2006) 829–837. 10.1038/sj.bjp.0706932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].García-Rivas GDJ, Guerrero-Hernández A, Guerrero-Serna G, Rodríguez-Zavala JS, Zazueta C, Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart, FEBS Journal. 272 (2005) 3477–3488. 10.1111/j.1742-4658.2005.04771.x. [DOI] [PubMed] [Google Scholar]
- [15].Patron M, Raffaello A, Granatiero V, Tosatto A, Merli G, De Stefani D, Wright L, Pallafacchina G, Terrin A, Mammucari C, Rizzuto R, The mitochondrial calcium uniporter (MCU): molecular identity and physiological roles, J. Biol. Chem 288 (2013) 10750–10758. 10.1074/jbc.R112.420752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu JC, Parks RJ, Liu J, Stares J, Rovira II, Murphy E, Finkel T, The In Vivo Biology of the Mitochondrial Calcium Uniporter, Adv. Exp. Med. Biol 982 (2017) 49–63. 10.1007/978-3-319-55330-6_3. [DOI] [PubMed] [Google Scholar]
- [17].Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter, Nature. 476 (2011) 341–345. 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R, A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter, Nature. 476 (2011) 336–340. 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R, The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit, EMBO J 32 (2013) 2362–2376. 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, Rothberg BS, Mak D-OD, Foskett JK, Madesh M, MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival, Cell. 151 (2012) 630–644. 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK, MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling, PLoS ONE. 8 (2013) e55785. 10.1371/journal.pone.0055785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D, MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake, Cell Death & Differentiation. 26 (2019) 179–195. 10.1038/s41418-018-0113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK, EMRE is an essential component of the mitochondrial calcium uniporter complex, Science. 342 (2013) 1379–1382. 10.1126/science.1242993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tsai M-F, Phillips CB, Ranaghan M, Tsai C-W, Wu Y, Willliams C, Miller C, Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex, Elife. 5 (2016). 10.7554/eLife.15545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang Y, Nguyen NX, She J, Zeng W, Yang Y, Bai X, Jiang Y, Structural Mechanism of EMRE-Dependent Gating of the Human Mitochondrial Calcium Uniporter, Cell. 177 (2019) 1252–1261.e13. 10.1016/j.cell.2019.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nat. Cell Biol 15 (2013) 1464–1472. 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T, The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter, Nat. Cell Biol 15 (2013) 1464–1472. 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, Pan H, Parks RJ, Anderson S, Noguchi A, Springer D, Murphy E, Finkel T, Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter, J. Mol. Cell. Cardiol 85 (2015) 178–182. 10.1016/j.yjmcc.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rasmussen TP, Wu Y, Joiner MA, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Z, Wagner BA, Soto J, McCormick ML, Kutschke W, Weiss RM, Yu L, Boudreau RL, Abel ED, Zhan F, Spitz DR, Buettner GR, Song L-S, Zingman LV, Anderson ME, Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart, Proc. Natl. Acad. Sci. U.S.A 112 (2015) 9129–9134. 10.1073/pnas.1504705112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, Molkentin JD, The Mitochondrial Calcium Uniporter Selectively Matches Metabolic Output to Acute Contractile Stress in the Heart, Cell Rep. 12 (2015) 15–22. 10.1016/j.celrep.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW, The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition, Cell Rep. 12 (2015) 23–34. 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu JC, Syder NC, Ghorashi NS, Willingham TB, Parks RJ, Sun J, Fergusson MM, Liu J, Holmström KM, Menazza S, Springer DA, Liu C, Glancy B, Finkel T, Murphy E, EMRE is essential for mitochondrial calcium uniporter activity in a mouse model, JCI Insight. 5 (2020) 134063. 10.1172/jci.insight.134063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chapoy-Villanueva H, Silva-Platas C, Gutiérrez-Rodríguez AK, García N, Acuña-Morin E, Elizondo-Montemayor L, Oropeza-Almazán Y, Aguilar-Saenz A, García-Rivas G, Changes in the Stoichiometry of Uniplex Decrease Mitochondrial Calcium Overload and Contribute to Tolerance of Cardiac Ischemia/Reperfusion Injury in Hypothyroidism, Thyroid. 29 (2019) 1755–1764. 10.1089/thy.2018.0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sverdlov AL, Elezaby A, Qin F, Behring JB, Luptak I, Calamaras TD, Siwik DA, Miller EJ, Liesa M, Shirihai OS, Pimentel DR, Cohen RA, Bachschmid MM, Colucci WS, Mitochondrial Reactive Oxygen Species Mediate Cardiac Structural, Functional, and Mitochondrial Consequences of Diet-Induced Metabolic Heart Disease, JAHA. 5 (2016). 10.1161/JAHA.115.002555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, Yu Z-X, Springer DA, Halsey C, Liu C, Murphy E, Finkel T, MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload, Cell Rep. 16 (2016) 1561–1573. 10.1016/j.celrep.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang K, Doan MT, Stiles L, Divakaruni AS, Measuring CPT-1-mediated respiration in permeabilized cells and isolated mitochondria, STAR Protocols. 2 (2021) 100687. 10.1016/j.xpro.2021.100687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C, Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells, Nat Protoc. 7 (2012) 1235–1246. 10.1038/nprot.2012.058. [DOI] [PubMed] [Google Scholar]
- [38].Tsai C-W, Wu Y, Pao P-C, Phillips CB, Williams C, Miller C, Ranaghan M, Tsai M-F, Proteolytic control of the mitochondrial calcium uniporter complex, Proc. Natl. Acad. Sci. U.S.A 114 (2017) 4388–4393. 10.1073/pnas.1702938114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Glancy B, Balaban RS, Role of mitochondrial Ca2+ in the regulation of cellular energetics, Biochemistry. 51 (2012) 2959–2973. 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Altamimi TR, Karwi QG, Uddin GM, Fukushima A, Kwong JQ, Molkentin JD, Lopaschuk GD, Cardiac-specific deficiency of the mitochondrial calcium uniporter augments fatty acid oxidation and functional reserve, J. Mol. Cell. Cardiol 127 (2019) 223–231. 10.1016/j.yjmcc.2018.12.019. [DOI] [PubMed] [Google Scholar]
- [41].Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, Bonaldo P, De Stefani D, Rizzuto R, Mammucari C, Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference, Cell Death Differ. 26 (2019) 362–381. 10.1038/s41418-018-0191-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kwong JQ, Huo J, Bround MJ, Boyer JG, Schwanekamp JA, Ghazal N, Maxwell JT, Jang YC, Khuchua Z, Shi K, Bers DM, Davis J, Molkentin JD, The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle, JCI Insight. 3 (2018). 10.1172/jci.insight.121689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sommakia S, Houlihan PR, Deane SS, Simcox JA, Torres NS, Jeong M-Y, Winge DR, Villanueva CJ, Chaudhuri D, Mitochondrial cardiomyopathies feature increased uptake and diminished efflux of mitochondrial calcium, J Mol Cell Cardiol. 113 (2017) 22–32. 10.1016/j.yjmcc.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Balderas E, Eberhardt DR, Lee S, Pleinis JM, Sommakia S, Balynas AM, Yin X, Parker MC, Maguire CT, Cho S, Szulik MW, Bakhtina A, Bia RD, Friederich MW, Locke TM, Van Hove JLK, Drakos SG, Sancak Y, Tristani-Firouzi M, Franklin S, Rodan AR, Chaudhuri D, Mitochondrial calcium uniporter stabilization preserves energetic homeostasis during Complex I impairment, Nat Commun. 13 (2022) 2769. 10.1038/s41467-022-30236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang P, Xu S, Xu J, Xin Y, Lu Y, Zhang H, Zhou B, Xu H, Sheu S-S, Tian R, Wang W, Elevated MCU Expression by CaMKIIδB Limits Pathological Cardiac Remodeling, Circulation. 145 (2022) 1067–1083. 10.1161/CIRCULATIONAHA.121.055841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koval OM, Nguyen EK, Santhana V, Fidler TP, Sebag SC, Rasmussen TP, Mittauer DJ, Strack S, Goswami PC, Abel ED, Grumbach IM, Loss of MCU prevents mitochondrial fusion in G1-S phase and blocks cell cycle progression and proliferation, Sci Signal. 12 (2019). 10.1126/scisignal.aav1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bernardi P, Rasola A, Forte M, Lippe G, The Mitochondrial Permeability Transition Pore: Channel Formation by F-ATP Synthase, Integration in Signal Transduction, and Role in Pathophysiology, Physiological Reviews. 95 (2015) 1111–1155. 10.1152/physrev.00001.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kosmach A, Roman B, Sun J, Femnou A, Zhang F, Liu C, Combs CA, Balaban RS, Murphy E, Monitoring mitochondrial calcium and metabolism in the beating MCU-KO heart, Cell Reports. 37 (2021) 109846. 10.1016/j.celrep.2021.109846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Davis J, Maillet M, Miano JM, Molkentin JD, Lost in transgenesis: a user’s guide for genetically manipulating the mouse in cardiac research, Circ. Res 111 (2012) 761–777. 10.1161/CIRCRESAHA.111.262717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Connern CP, Halestrap AP, Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel, Biochem J 302 ( Pt 2) (1994) 321–324. 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Parks RJ, Menazza S, Holmström KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E, Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter, Cardiovascular Research. 115 (2019) 385–394. 10.1093/cvr/cvy218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.