
Fig. 7.
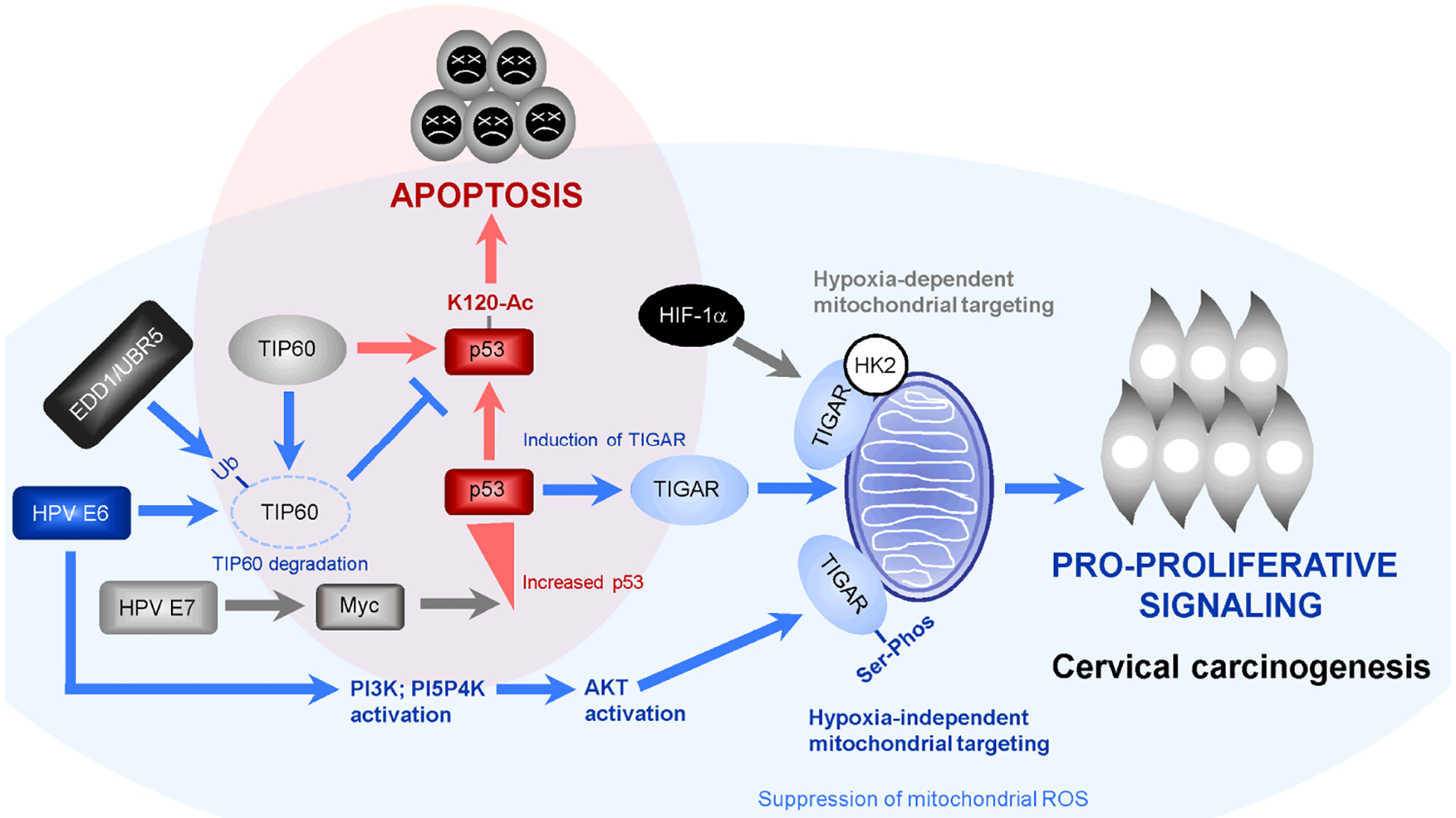
Model of the roles of p53-regulated pro-survival signals and hypoxia-independent mitochondrial targeting of TIGAR in the cooperation between hrHPV E6 and cellular oncogenes during viral carcinogenesis. The HPV E6/E7 oncoproteins have been shown to activate c-Myc-dependent transcription and cooperate with c-Myc to induce cellular proliferation and immortalization (Strickland and Vande Pol, 2016; Zhang et al., 2017; McMurray and McCance, 2003). However, aberrant oncogenic c-Myc expression can cause DNA-damage, activate p53, and induce p53-K120-acetylation and p53-dependent cellular apoptosis (shaded in red; Vafa et al., 2002; Hermeking and Eick, 1994; Romeo et al., 2018). The HPV E6 oncoprotein destabilizes the p53-cofactor TIP60 through recruitment of the E3 ubiquitin-ligase, EDD1/UBR5, and inhibits p53-K120-acetylation and p53-dependent apoptosis (Jha et al., 2010; Subbaiah et al., 2016). Whereas the TIGAR protein has been shown to target mitochondrial membranes and protect cells against the accumulation of damaging ROS under hypoxic conditions –dependent upon HIF-1α activation and interactions with hexokinase-2 (HK2) (Bensaad et al., 2006; Cheung et al., 2012), our studies demonstrate that the hrHPV E6 oncoproteins induce a pseudohypoxic response and activate PI3K/PI5P4K/AKT-signaling to promote the serine-phosphorylation and mitochondrial targeting of TIGAR which counters the oxidative stress/cytotoxicity associated with oncogenic proproliferative signaling during HPV-associated cervical carcinogenesis (shaded in blue).