

Abstract
Prostate cancer (PCa) progression to the lethal metastatic castration-resistant phenotype (mCRPC) is driven by αv integrins and is associated with Golgi disorganization and activation of the ATF6 branch of unfolded protein response (UPR). Overexpression of integrins requires N-acetylglucosaminyltransferase-V (MGAT5)-mediated glycosylation and subsequent cluster formation with Galectin-3 (Gal-3). However, the mechanism underlying this altered glycosylation is missing. For the first time, using HALO analysis of immunohistochemistry, we found a strong association of Integrin αv and Gal-3 at the plasma membrane (PM) in primary PCa and mCRPC samples. We discovered that MGAT5 activation is caused by Golgi fragmentation and mislocalization of its competitor, N-acetylglucosaminyltransferase-III, MGAT3, from Golgi to the endoplasmic reticulum (ER). This was validated in an ethanol-induced model of ER stress, where alcohol treatment in androgen-refractory PC-3 and DU145 cells or alcohol consumption in PCa patient samples aggravates Golgi scattering, activates MGAT5, and enhances integrin expression at PM. This explains known link between alcohol consumption and PCa mortality. ATF6 depletion significantly blocks UPR and reduces the number of Golgi fragments in both PC-3 and DU145 cells. Inhibition of autophagy by hydroxychloroquine (HCQ) restores compact Golgi, rescues MGAT3 intra-Golgi localization, blocks glycan modification via MGAT5, and abrogates delivery of Gal-3 to the cell surface. Importantly, the loss of Gal-3 leads to reduced integrins at PM and their accelerated internalization. ATF6 depletion and HCQ treatment synergistically decrease Integrin αv and Gal-3 expression and temper orthotopic tumor growth and metastasis.
Implications: Combined ablation of ATF6 and autophagy can serve as new mCRPC therapeutic.
Keywords: metastatic prostate cancer, integrins, Golgi disorganization, ER stress, alcohol.
INTRODUCTION
In the United States, prostate cancer (PCa) is the second-leading cause of cancer lethality in men. Prostate cancer death is attributed to metastatic castration-resistant prostate cancer (mCRPC) and multiple alterations in homeostatic signaling networks. Primary therapeutic approaches (androgen deprivation, prostatectomy, and radiation therapy) have failed, and normal signaling networks are altered, including integrins, the surface transmembrane αβ heterodimer receptors, which play a crucial role in prostate tumor progression. Integrins bind to various ligands in the extracellular matrix (ECM), including fibronectin (FN). The alteration of integrin signaling increases cell migration, invasion, proliferation, and cancer cell survival. It is well established that CRPC is associated with the atypical expression of different integrin proteins, especially those containing the αv subunit (1).
Multiple studies have clarified the link between endoplasmic reticulum stress (ER stress), unfolded protein response (UPR), and cancer (reviewed in (2)). Several reports indicate that cancer cell Golgi fragmentation is driven by prolonged, sub-lethal ER stress (3–5). One branch of UPR, mediated by the activating transcription factor 6 (ATF6), is mechanistically linked to the Golgi. Under normal conditions, cleavage of ATF6 in the Golgi by S1P and S2P proteases is necessary for the transactivation of ATF6-mediated ER stress response. Molecular studies in our laboratory confirm that the dimeric trans-Golgi matrix protein, GCC185, serves as a Golgi retention partner for S1P and S2P proteases. However, Golgi disorganization in advanced PCa cells results in the monomerization of GCC185 and its downregulation. This, in turn, leads to the translocation of S1P and S2P to the ER, where these proteins cleave ATF6 (6). This elegant escape mechanism employed by PCa cells accelerates ATF6-mediated UPR, thus maintaining the fragmented Golgi phenotype of prostate tumor cells. In previous studies, we introduced the “onco-Golgi” concept, postulating that Golgi structural dispersal is associated with specific pro-metastatic glycosyl epitopes (7). For instance, androgen-responsive LNCaP and 22Rv1 cells have a compact perinuclear Golgi, while androgen-refractory, docetaxel-resistant PC-3 and DU145 cells demonstrate disorganized Golgi, which is associated with de-dimerization of the key Golgi matrix protein, Giantin (4). This is associated with the mislocalization of critical O-glycosylation enzymes, which leads to the formation of pro-metastatic glycan epitopes (4,8). It was not known whether the same association exists between fragmented Golgi phenotype and activation of N-glycans that promote prostate tumor growth and metastases.
In cells with normal compact Golgi morphology, biantennary N-linked glycan chains can be modified by adding a bisecting GlcNAc. The reaction is catalyzed by N-acetylglucosaminyltransferase-III (MGAT3) (Fig. 1A, asterisk). This suppresses further elongation of the N-glycan by N-acetylglucosaminyltransferase-V (MGAT5) or N-acetylglucosaminyltransferase-IV (MGAT4) (9). Enhanced expression of MGAT3 has been found to block MGAT5-mediated glycosylation and suppress metastasis (10). In tumor cells, MGAT5-modified glycans are processed by beta-1,4-galactosyltransferase (β1,4-GalT) and elongated with poly-N-acetyllactosamine (LacNAc), followed by the addition of sialic acid and fucose. The αv integrins, especially αvβ3, αvβ5, and αvβ6, form an abnormal cell surface repertoire due to the high affinity of their LacNAc to Galectin-3 (Gal-3). At the PM, integrins and pentameric Gal-3 form clusters, termed lattices, which modulate tumor cell behavior, including the cardinal metastatic factors: adhesion to ECM and migration (11). Integrin αvβ3 and αvβ5 promote prostate tumor dissemination to lymph nodes and distant organs, including bones (12–14), suggesting their potential as prognostic markers. Overexpression of MGAT5 was also detected in other types of tumors (15–17). Importantly, enhanced glycosylation by MGAT5 was observed in mice metastatic xenografts derived from LNCaP and PC-3 cells, as well as in PCa patients (18). Overexpression of MGAT5 significantly enhanced LNCaP cell invasion (19). Bisecting GlcNAc structures in certain glycoproteins, including integrins, results in their altered expression levels and decreased sorting to the cell surface (20). Conversely, integrins and other ECM-binding proteins glycosylated by MGAT5 demonstrate enhanced representation at the plasma membrane (PM) via interaction with Gal-3, which is associated with a more aggressive tumor phenotype (21,22). It is unclear what mechanism dictates the downregulation of MGAT3 and subsequent upregulation of MGAT5 in advanced PCa.
Figure 1.
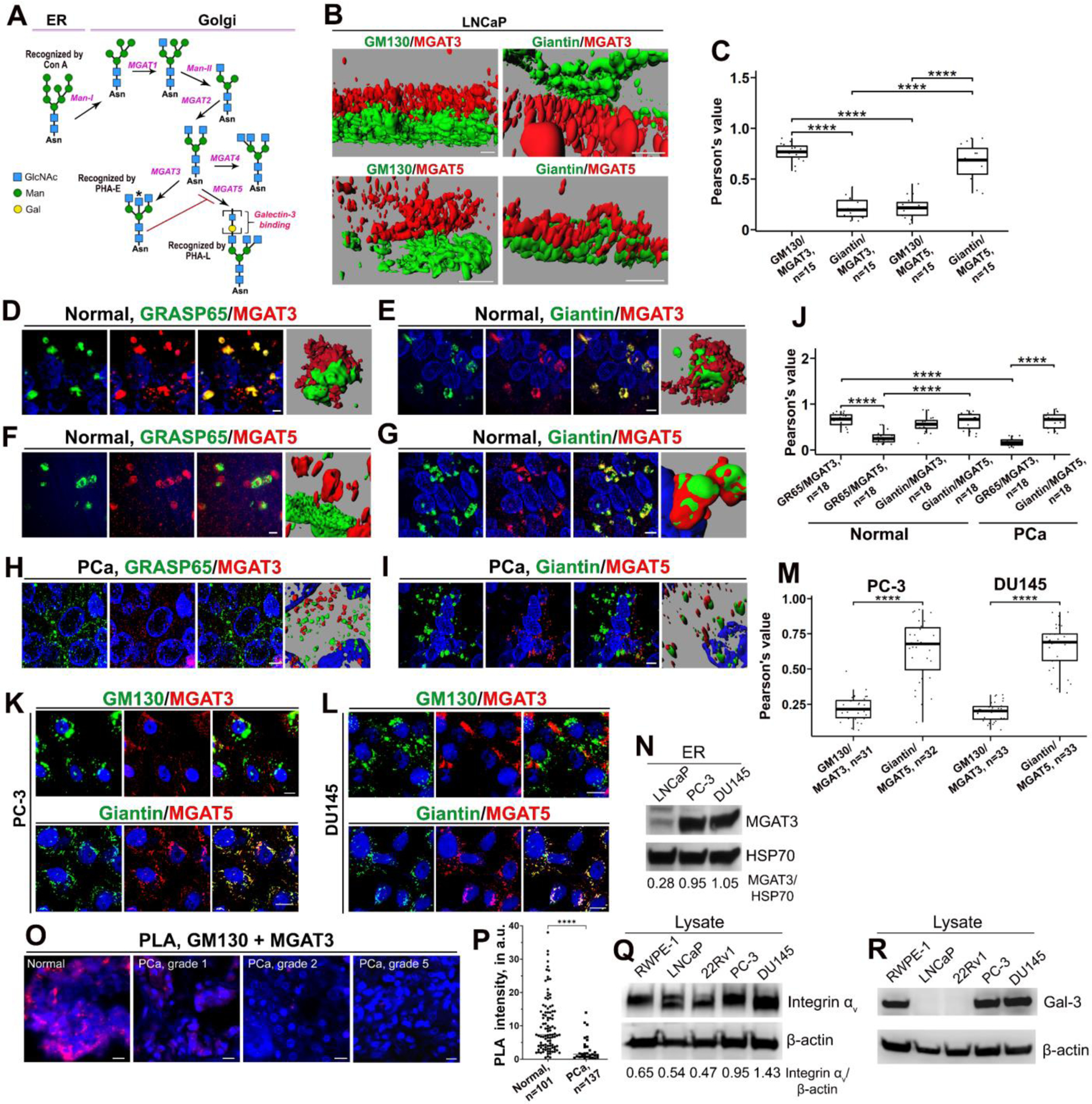
Differential distribution of MGAT3 and MGAT5 in PCa cells. (A) Schema of the N-glycosylation of proteins in ER and Golgi. High-Man-bearing proteins shift from ER to Golgi, where the (Man)5(GlcNAc)2 structure occurs after trimming by Mannosidase I (Man-I, α−1,2-specific). MGAT1 then adds an N-acetylglucosamine (GlcNAc) residue to the terminal Man residue, followed by the removal of two Man residues by Mannosidase II (Man-II, α−1,3/6-specific). Further processing by MGAT2 is followed by one of three glycosyltransferases: MGAT3, MGAT4, or MGAT5. Importantly, bisecting GlcNAc formed by MGAT3 (asterisk) blocks MGAT5-mediated glycan branching. Note that each variation will result in complex-type oligosaccharides (not shown). (B) Representative 3D SIM images of Golgi membranes in LNCaP cells stained for MGAT3 or MGAT5 and Golgi markers: GM130 (cis-Golgi) or Giantin (medial-Golgi); bars, 2 μm. (C) Quantification of Pearson’s correlation coefficient (PCC) for indicated proteins from B. Tukey Method, p-adjusted using Benjamini-Hochberg; n indicates the number of Golgi stacks counted, median ± SD. (D-I) 3D SIM images of normal prostate and PCa (grades 2–3) immunostained for MGAT3 or MGAT5 and Golgi markers: GRASP65 (cis-Golgi) or Giantin. Representative reconstruction by Imaris is presented to the right of each panel; bars, 10 μm. Panels H and I represent only reconstructed 3D images. (J) Quantification of PCC for indicated proteins from D-I. Tukey Method, p-adjusted using Benjamini-Hochberg; n indicates the number of Golgi counted, median ± SD. (K, L) Immunostaining of MGAT3 or MGAT5 with GM130 or Giantin in PC-3 (K) and DU145 (L) cells; bars, 10 μm. (M) Quantification of PCC for indicated proteins from K and L. Tukey Method, p-adjusted using Benjamini-Hochberg; n indicates the number of images analyzed, median ± SD. (N) MGAT3 W-B of ER fraction isolated from LNCaP, PC-3, and DU145 cells; samples were normalized by HSP70. (O) Proximity ligation assay (PLA) of normal prostate and PCa (grades 1–5) using Ms-anti-GM130 and Rb-anti-MGAT3 Abs; bars, 10 μm. (P) Quantification of PLA intensity (red) in normal and PCa tissue samples (all grades). Kruskal-Wallis test; medians with individual values. (Q, R) Integrin αv (Q) and Gal-3 (R) W-B of lysate samples from RWPE-1, LNCaP, 22Rv1, PC-3, and DU145 cells; β-actin is a loading control. All data presented are representative of at least three independent experiments. For all statistics: ****p<0.0001.
In PCa cells, autophagy is involved in the dysregulation of androgen receptor (AR) signaling and may interfere with other pathways, ultimately leading to castration resistance and dissemination of prostate tumors (reviewed in (23)). Autophagy inhibition by Chloroquine and Hydroxychloroquine (HCQ), alone or in combination with other agents, have been shown to overcome chemoresistance (24,25), reduce PCa cell proliferation, and induce apoptosis (26,27). Despite our increased understanding of the antitumor effect of HCQ, no mechanism is known that can clearly describe its potential effect on PCa cell metastasis and the expression of integrins.
The primary objective of this study was to examine the impact of fragmented Golgi phenotype and ATF6-mediated UPR on the activation of MGAT5-mediated glycosylation of integrins and their expression at the PM of prostate tumor cells. Using both in vitro and in vivo models of PCa, we discovered exciting links between cell surface expression of Integrin αv and Gal-3 and detected that the internalization of αv integrins does not require Gal-3. Finally, we found that inhibition of autophagy by HCQ with parallel ablation of ATF6 remarkably decreases the expression of αv integrins, suggesting a novel effective therapeutic strategy for prostate tumor metastases.
MATERIALS AND METHODS
In vivo Mouse Orthotopic Xenograft Model.
Male athymic nude mice (BALB/c nu/nu, 25–31 g, 5–6 weeks old, Jackson Laboratory) were housed in filter-top cages in the animal facility at the University of Nebraska Medical Center (UNMC) and provided food and water ad libitum. The mice underwent surgical implantation of control or ATF6 KD PC-3 cells as described in the supplemental methods. Two weeks after implantation, mice were randomly assigned to PBS or HCQ groups and received by i/p injection of 25 mg/kg HCQ or PBS twice per week (based on a weekly physiological dosage of 50 mg/kg) for four weeks. Body weights were recorded weekly, and mice were sacrificed six weeks after implantation. Tumors and serum were collected for IHC, IF, and ALT/AST analysis, and tumor volume was recorded. This protocol was approved by the Institutional Animal Care and Use Committee (IACUC) at UNMC.
Patient-Derived Tissues.
The tissue sections from normal prostate and PCa patients were obtained from US Biomax, Novus Biological, and Creative Bioarray. Also, sections were provided through the Department of Pathology and Microbiology (IRB protocol # 304–16-EP) at the University of Nebraska Medical Center and the Johns Hopkins University School of Medicine (Prostate Cancer Biorepository Network), and the Vernadsky Crimean Federal University (Russian Federation, IRB protocol # 98–2019-KFU). Prostate cancer was graded in accordance with the 2014 International Society of Urological Pathology (ISUP) Guideline. Alcohol consumption was stratified as follows: non-alcoholic – less than one drink per month; moderate-alcoholic – 5–6 beers per week, 3–5 glasses of wine per week, or 2–3 3.4 oz servings of hard liquor per week; heavy-alcoholic – 2 or more beers per day, one or more glasses of wine per day, or one or more 3.4 oz servings of hard liquor per day.
Quantification and Statistical Analysis.
Statistical analysis was performed using Microsoft Excel, Prism 9.0 (GraphPad), and R Software. Statistical parameters are described in each figure legend. Multi-parametric analysis was performed based on median values in R software as indicated using: a) Tukey method, p-adjusted by Benjamini-Hochberg, b) Dunn Test (1964) Kruskal-Wallis multiple comparisons, p-adjusted by Benjamini-Hochberg, c) Pairwise comparison using Wilcoxon rank sum exact test, p-adjusted by Benjamini-Hochberg, or d) Pairwise t-test, p-adjusted by Benjamini-Hochberg. Other analyses based on mean ± SD included the Mann-Whitney test, Student’s t-tests (unpaired, two-sided, or two-tailed), and one-way or two-way analysis of Variance (ANOVA). The specific test and parameters used, sample size, and p-values are reported in the figures and figure legends.
For IHC, the data of Integrin αv or Gal-3 from each microarray of tissue specimens were sorted by groups (normal or PCa grades), and correlation between all combinations of Abs (Pearson or Spearman r value) was evaluated. If the coefficient was greater than or equal to 0.4, those combinations were considered correlated and used to calculate the median value for each patient, which was then incorporated into the final analysis. After subtracting combinations that showed no correlation and samples with damaged tissues, we analyzed 19 donors and the following numbers for PCa samples: n=18 for grade 1, n=33 for grades 2–3, and n=56 for grades 4–5.
To evaluate lymphatic or organ metastases statistically in the orthotopic model of prostate tumor, the number of mice with and without lymphatic and/or organ metastasis was counted as “yes” or “no”, as in, yes metastasis or no metastasis. The results were inputted into a table, then, using R-statistical software, a chi-square analysis was done. If the p-value was less than or equal to 0.05, then a post hoc evaluation using Fisher’s Exact test was done to compare each group. The p-value was adjusted using the Benjamini-Hochberg method.
The remaining methods are detailed in the Supplemental Information,
including: Antibodies and Reagents; Cell Culture, EtOH and HCQ Treatment; LC3B Plasmid Transfection; Plasma Membrane Protein Isolation; RNA Isolation and RT-qPCR; Lectin-affinity chromatography, Fibronectin Adhesion Assay; Pulse-Chase Endocytosis Assay; Confocal Immunofluorescence Microscopy and Analysis; 3D SIM and Analysis; Proximity Ligation Assay; Electron Microscopy; Immunohistochemistry, Lectin Histochemistry and Analysis; ALT and AST Activity Assays; Detailed Orthotopic Implantation.
RESULTS
MGAT3 is situated in more proximal Golgi compartments than MGAT5 but is mislocalized in advanced PCa
Previously, we reported that expression of MGAT3 protein in high passage androgen-refractory LNCaP cells (c-86) is reduced compared with androgen-responsive, low passage LNCaP (c-26) cells (hereafter, LNCaP) (28). In contrast, the MGAT5 expression was higher. Additionally, ethanol (EtOH)-induced Golgi disorganization in LNCaP cells was accompanied by translocation of MGAT3 from the Golgi to the ER, while intra-Golgi localization of MGAT5 appeared unaffected (28). Previous studies indicated that MGAT3 and MGAT5 reside predominantly within medial-Golgi membranes (29,30). However, since MGAT3 and MGAT5 compete for N-glycan elongation (Fig. 1A), and activation of MGAT3 leads to the blockage of glycosylation via MGAT5 and reduction of metastasis (19), it is logical that MGAT3-mediated glycosylation has priority over MGAT5 under normal conditions, potentially due to more proximal intra-Golgi localization. To check this hypothesis, we performed a detailed analysis of the sub-Golgi position of these enzymes, using structured illumination superresolution microscopy (SIM) of Golgi stacks, which allows us to achieve two-color 3D imaging at ~110 nm resolution. In LNCaP cells, MGAT3 colocalization with the cis-Golgi marker, GM130, was higher than with the medial-Golgi marker, Giantin (Fig. 1B and C). The distribution of MGAT5 was opposite to that of MGAT3; more colocalization with Giantin than GM130. These data indicate that MGAT3 is localized in the earlier Golgi compartments than MGAT5, explaining the MGAT3 priority over MGAT5 for modification of proteins carrying N-glycans in Golgi. This cell line finding was further validated in human normal and neoplastic prostate tissues. Indeed, in normal prostate, MGAT3’s colocalization with cis-Golgi marker GRASP65 was higher than that for MGAT5. In contrast, MGAT5 exhibits stronger colocalization with Giantin than GRASP65 (Fig. 1D–G, J). In tumor tissues from PCa patients, the Golgi is fragmented, and we observed that MGAT3 loses cis-Golgi positioning while MGAT5 maintains its localization within the medial-Golgi (Fig. 1H–J). A similar tendency was observed in PC-3 and DU145 cells with fragmented Golgi: MGAT5 displayed strong colocalization with Giantin, but colocalization between MGAT3 and GM130 was weak (Fig. 1K–M). Western blotting of the ER fraction isolated from PC-3 and DU145 cells detected a significantly higher amount of MGAT3 protein than that in LNCaP cells (Fig. 1N). Finally, using mouse anti-GM130 and rabbit anti-MGAT3 antibodies (Abs), we performed Proximity Ligation Assay (PLA) in normal prostate and PCa tissue samples. Significant reduction of MGAT3 and GM130 closeness was documented in tumor cells, indicating non-Golgi positioning of MGAT3 (Fig. 1O and P).
In sum here, MGAT3 demonstrates cis-Golgi localization while MGAT5 is positioned in medial-Golgi, and in advanced PCa cells, MGAT5, but not MGAT3, maintains a strong intra-Golgi signal; this may justify the enhanced integrin expression in prostate tumors. Higher expression of Integrin αv in PC-3 and DU145 compared with non-malignant prostate epithelial (RWPE-1) cells, low aggressive malignant LNCaP, and 22Rv1 cells supports this observation (Fig. 1Q). Also, it is known that PC-3 and DU145 cells demonstrate higher expression of MGAT5 compared with androgen-responsive cells (19,31). Interestingly, LNCaP and 22Rv1 cells, which exhibit the lowest expression of Integrin αv, also express little, if any, Gal-3 (Fig. 1R). This aligns with previous observations indicating marginal expression of LGALS3 (Gal-3) mRNA in these cell lines (32,33).
Differential expression of αv integrins and Gal-3 in tumor tissue samples
It is well-known that expression of αv integrins and Gal-3 in primary PCa and patients with mCRPC is aberrant and positively correlated with Gleason grades (13,34–37). However, to our knowledge, no prior work convincingly analyzes Integrin αv and Gal-3 co-expression on the prostate tumor cell surface. Here, we performed comprehensive immunohistochemistry (IHC) analysis, staining Integrin αv and Gal-3, then measuring their PM signal using Multiplex IHC v3.1.4 algorithms by Halo v3.4 (Indica Labs, Inc.). To clearly distinguish the expression of these proteins at the cell surface, the PM was labeled with one of two different markers, E-cadherin or Na+/K+-ATPase. The PM stain was then segmented to match the visually observed PM cues most appropriately. Each microarray of prostate tissue from donors (normal, n=49) and primary PCa patients with varying grades of tumor (n=160) was stained with different combinations of anti-Integrin αv, anti-Gal-3, and anti-E-cadherin or anti-Na+/K+-ATPase Abs (see Supplemental Table S1). Representative images from one Ab combination are presented in Fig. 2A. The H-score of Integrin αv at PM demonstrated no difference between normal and PCa grade 1; however, a significant increase was detected in all other PCa grades (Fig. 2C). Also, cancer patient specimens with grades 4–5 exhibited higher Integrin αv PM H-score than their grades 2–3 counterparts. The data of PM-specific H-score for Gal-3 were slightly different. While we quantified a significant increase in PCa grades 2–3 compared with normal and grade 1, there was no substantial difference between grades 2–3 and 4–5 (Fig. 2D). Importantly, colocalization between Integrin αv and Gal-3 at the cell surface was higher in grade 1 and grades 2–3 compared with normal. However, patients with grades 4–5 demonstrated significantly lower colocalization than grades 2–3 (Fig. 2E).
Figure 2.

(A) IHC analysis of Integrin αv and Gal-3 expression in prostate tumor. Representative images of triple IHC staining of Integrin αv (red), Gal-3 (green), and E-cadherin (brown) in normal prostate and tumor tissues from PCa patients with different grades. (B) Representative images of triple IHC staining of Na+/K+-ATPase (red), Integrin αv (brown), and Gal-3 (green) in tumor tissues from PCa patients: non-metastatic (grade 4) and mCRPC patients with tissue or bone metastasis. Black boxes are highlighted at the right to present deconvoluted images of Gal-3 and Integrin αv. Bars for A and B, 100 μm. (C, D) Quantification of Integrin αv (C) and Gal-3 (D) H-scores at PM in patients from A; this includes only patients that demonstrate the correlation of parameters in different combinations of anti-Integrin αv and anti-Gal-3 Abs (see Supplemental Table S1 and supplemental methods). Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. (E) Quantification of Integrin αv and Gal-3 colocalization at PM within different grades of PCa from images in A. Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. (F, G) Quantification of Integrin αv (F) and Gal-3 (G) H-score at PM in patients from B. Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. For G, non-metastatic group includes patients with all grades. (H) Quantification of Integrin αv and Gal-3 colocalization at PM in patients from B. Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. (I) Representative images of triple IHC staining of Na+/K+-ATPase (red), Integrin αv (brown), and PHA-L lectin (green) in the tumor tissues from PCa patients: non-metastatic (grade 4) and CRPC patients with tissue or bone metastasis. Black boxes are presented at the right as deconvoluted images of Integrin αv and PHA-L lectin; bars, 100 μm. (J) Quantification of Integrin αv/PHA-L PM colocalization for samples from I; non-metastatic group includes patients with all grades Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. For all graphs, n indicates the number of tissue samples. For all statistics: ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05, median ± SD.
Next, in the same patient cohort, we analyzed the PM expression of different αv integrins using specific Abs against both αv and different β subunits. The PM signal for Integrin αvβ3 in all grades of PCa was significantly higher than that in normal prostate. Interestingly, patients with grades 4–5 showed a slight, although still significant, decrease compared with grades 2–3 (Fig. S1A and S1B). Also, the colocalization between Integrin αvβ3 and Gal-3 at PM was higher in PCa samples than normal (Fig. S1C). The expression of Integrin αvβ5 was higher in all PCa groups compared with normal; however, no difference was detected between grades (Fig. S2A and S2B). Like αvβ3, αvβ5 colocalized with Gal-3 on the PM more in PCa than in normal donors (Fig. S2C). The results of the IHC signal for PM Integrin αvβ6 were most interesting: in addition to the difference between all grades and normal, there was a significant rise in grades 2–3 and 4–5 compared to grade 1 (Fig. S3A and S3B). Again, the association of Gal-3 with Integrin αvβ6 at the cell surface was more evident in PCa (Fig. S3C). These data indicate that most αv integrins are highly associated with Gal-3 on the PCa cell surfaces and increase with the cancer grade.
Then, we compared Integrin αv and Gal-3 PM expression in mCRPC patients (with tissue or bone metastases) and non-metastatic patients (Fig. 2B). We found that Integrin αv H-score at the cell surface of metastatic samples (both tissue and bone) was substantially higher than that in primary tumors (Fig. 2F). Importantly, patients with bone metastases demonstrated higher H-score than those with tissue metastases. A similar tendency was observed for Gal-3: patients with tissue metastases display less PM-specific Gal-3 than those with bone metastases. However, only bone metastasis cases had a significant increase compared with non-metastatic patients (Fig. 2B and G). The colocalization between Integrin αv and Gal-3 at PM in bone metastases mirrored results from primary tumors of patients with grades 2–3 and 4–5 (Fig. 2E). While this parameter was higher in tissue metastatic patients than in patients with the primary tumor, we detected a significant decrease in bone metastases compared with tissue and primary prostate tumors (Fig. 2H).
Evaluation of integrin glycosylation by lectin IHC
The data presented above suggest that the development of metastases is associated with enhanced expression of abnormally glycosylated integrins. We performed a series of lectin IHC staining of tumor samples mentioned above. To evaluate MGAT5-mediated glycosylation of Integrin αv at the cell surface, we co-stained Integrin αv with Na+/K+-ATPase and Phaseolus Vulgaris Leucoagglutinin (PHA-L) lectin, which binds preferentially to GlcNAc residues on β1–6 branches of tri- or tetra-antennary sugar chains (the product of MGAT5 glycosylation) (38,39) (Fig. 1A). As shown in Fig. 2I and J, colocalization of Integrin αv with PHA-L lectin at PM was significantly higher in both groups of mCRPC patients compared with non-metastatic patients. However, the difference between tissue and bone metastases was non-significant. Therefore, the development of mCRPC is linked to an increase of MGAT5-modified Integrin αv at the PM of tumor cells.
HCQ restores compact Golgi in advanced PCa cells and recovers MGAT3’s intra-Golgi localization
In previous studies, we reported that ablation of motor protein non-muscle Myosin IIA (NMIIA) restores compact Golgi in PC-3 and DU145 cells (4). Given that, broadly speaking, Golgi and autophagy are interconnected in many aspects (reviewed in (40)), including that Golgi membranes serve as a source for phagophores, we wandered whether Golgi disorganization in PCa cells could be attributed to enhanced autophagy. Thus, we hypothesized that autophagy blockage would restore Golgi morphology. Here, we found that treatment of PC-3 cells with HCQ (50 μM HCQ for 72 h) converts the Golgi phenotype from fragmented to more compact (Fig. 3A and B). A similar effect of HCQ was observed in DU145 cells, though it required a higher concentration (60 μM HCQ for 72 h) (Fig. S4A and B). The dosage of HCQ was determined based on the physiological concentration that, within 72 hours, induced a significant reduction in the number of Golgi membranes but did not induce apoptosis.
Figure 3.

The effect of HCQ on Golgi in PCa cells. (A) Representative images of Golgi in PC-3 cells stained by GM130 (green): control and ATF6 KD cells treated with 50 μM HCQ for 72 h or an appropriate amount of water (Ctrl); bars, 20 μm. (B) Quantification of Golgi spots per cell in samples from A. Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg; n represents the cells counted. (C) ATF6 W-B of the lysates of PC-3 cells transfected with control or ATF6α shRNA; β-actin is a loading control. (D-I) ERp72 (D), GRP78 (E), PERK (F), PERK-P (G), IRE1 (H), and IRE1-P (I) W-Bs of the lysates from control and ATF6 KD PC-3 cells. (J) Giantin W-B of the lysates from control and ATF6 KD cells. The samples were prepared under low (1%) concentrations of β-mercaptoethanol. Giantin-dimer and monomer are indicated by arrows. The Nesprin 2 band at 800 kDa in control PC-3 lysate corresponds to the size of the Giantin dimer. (K) Representative EM images of control and HCQ-treated PC-3 cells. Note multiple Golgi (G) fragments in control cells (insets a and b) and compact reorganized Golgi in HCQ-treated; N, nucleus. Orange squares indicate the areas of Golgi enlarged at the right; bars, 1 μm. (L) IF staining of cis-Golgi (GRASP65) and MGAT3 in PC-3 cells treated with HCQ. The right panels show representative Z-stack images collected by SIM and reconstructed using Imaris. Bar sizes are: 5 μm and 3 μm for Ctrl and Ctrl-Golgi 3D SIM, respectively, and 7 μm and 2 μm for HCQ and HCQ-Golgi 3D SIM, accordingly. (M) Quantification of GRASP65 and MGAT3 colocalization from the cells in L; only images of 3D SIM were counted. Unpaired t test; n indicates number of cells. (N) PC-3 cells were treated with HCQ and then transfected with the Premo™ Autophagy Tandem Sensor RFP-GFP-LC3B. The Golgi membranes were stained using Alexa Fluor 647 anti-GM130 Ab. White squares indicate an area of LC3B punctae around the Golgi membranes enlarged at the right. Bar size, 5 μm. (O) The autophagic index was counted as the ratio of the areas of autophagosomes (overlapped red and green) to autolysosomes (red); Mann Whitney test. (P) LC3B W-B of the lysates of cells from A. All data presented are representative of at least three independent experiments. (Q) Quantification of PCC between yellow punctae and GM130 (magenta). Unpaired t test. For O and Q, n indicates the number of cells. For all statistics: ****p<0.0001, ***p<0.001, **p<0.01, and *p<0.05, median ± SD.
Additionally, to determine the effect of ATF6-mediated ER stress response on the Golgi, we developed shRNA-mediated stable ATF6α knockdown (KD) in PC-3 cells (Fig. 3C). In ATF6 KD cells, we detected decreased expression of ATF6 target proteins, including ERp72 and GRP78 (Fig. 3D and E). Interestingly, such downregulation of ATF6 did not induce a compensatory response from the other two branches of UPR: PERK and IRE-1. Despite a slight increase in total PERK, the expression of its phosphorylated form was identical to control cells (Fig. 3F and G). Also, we observed a moderate reduction of both total and phosphorylated IRE-1 (Fig. 3H and I). These results support the conclusion that ATF6 KD cells lack excessive ER stress response, which is typically observed in control PC-3 cells and in advanced prostate tumor cells (6). Consequently, Giantin’s dimerization in ATF6 KD cells was higher than in control cells, implying the restoration of Golgi morphology (Fig. 3J) (4,41). Indeed, depletion of ATF6 has an effect close to HCQ: in both PC-3 (Fig. 3A and B) and DU145 cells (Fig. S4C-E) lacking ATF6α, Golgi appeared more compact and perinuclear compared with cells transfected with an appropriate control shRNA/siRNA. Remarkably, treating ATF6 KD cells with HCQ had the most noticeable impact on Golgi morphology, leading to a significantly smaller number of Golgi fragments than HCQ treatment or ATF6 KD alone (Fig. 3A and B).
To validate these results, we performed electron microscopy (EM) of PC-3 cells treated with HCQ. In control cells, multiple unstacked Golgi fragments were detected, consistent with our previous observations (Fig. 3K, top panel, indicated as G) (4). On the contrary, in HCQ-treated cells, we found stacks of Golgi cisternae fused in a continuous ribbon structure (Fig. 3K, bottom panel). Similarly, PC-3 and DU145 cells depleted of ATG5, a key protein for the extension of the phagophore membrane, demonstrate a more compact Golgi phenotype (Fig. S4F-K). Notably, treatment of PC-3 cells with Bafilomycin A1 (10 μM for 72 h), which also inhibits autophagosome-lysosome fusion and autolysosome acidification (42), results in a significant reduction of the number of Golgi membranes (Fig. S4L and M).
Our previous study found that, in PC-3 and DU145 cells, O-glycosylation enzyme core 2 N-acetylglucosaminyltransferase-L (C2GnT-L) mislocalized from Golgi to the ER; however, restoration of Golgi through KD or inhibition of NMIIA resulted in the recovery of its Golgi positioning (4). This stimulated us to examine whether HCQ-induced Golgi reconstruction will bring MGAT3 back to the Golgi. Using 3D SIM, we rigorously analyzed the colocalization of MGAT3 with GRASP65 before and after HCQ treatment of PC-3 cells. As shown in Fig. 3L and M, the colocalization of MGAT3 with GRASP65 was substantially enhanced in HCQ-treated cells, indicating the restoration of MGAT3 intra-Golgi localization (movies S1 and S2). Importantly, we noticed that recovery of compact perinuclear Golgi is associated with a reduction of Golgi area. This was confirmed by the expression of Golgi scaffold proteins, golgins, which represents the matrix of Golgi membranes. The protein level of cis-golgins, GRASP65 and GM130; medial-golgin, Giantin; and trans-golgins, Golgin-245 and TGN46, was substantially reduced after HCQ (Fig. S4N-R).
The next question of interest was whether fragmented Golgi membranes in PCa cells feed directly into autophagy, i.e., whether PCa cells demonstrate a high rate of Golgiphagy. To check this, we employed the Autophagy Tandem Sensor RFP-GFP-LC3B Kit (Thermo Fisher, see Supplementary methods for details) that allows for the detection of the maturation of the autophagosomes or autophagic vacuoles (AVs) to the autolysosome. Briefly, by combining an acid-sensitive GFP with an acid-insensitive RFP, the change from AV (neutral pH) to autolysosome (with an acidic pH) can be visualized by the specific loss of the GFP fluorescence, leaving only a red signal. To monitor Golgi morphology and its incorporation into AVs, PC-3 cells transfected with this LC3B-plasmid were also stained with Alexa Fluor® 647 anti-GM130 Ab (Abcam, ab195303). First, we verified the autophagy flux rate by taking the AV ratio (yellow-red and green colocalized) to autolysosome (red). Predictably, since HCQ blocks the fusion of AVs with lysosomes, the area of autolysosomes was significantly reduced in HCQ-treated cells (Fig. 3N and O). This was further validated by W-B analysis of LC3B-II protein in control and ATF6 KD cells treated with HCQ (Fig. 3P). HCQ treatment alone significantly enhanced LC3B-II expression, indicating the blockage of its degradation. Meanwhile, depletion of ATF6 alone had no impact on the level of LC3B-II compared to control cells. However, comparison of control and ATF6 KD cells treated with HCQ indicates even more accumulation of LC3B-II in cells lacking ATF6, suggesting that alleviation of the ER stress response may intensify the effect of HCQ. Next, we analyzed the colocalization of autophagosomes with Golgi membranes. In control PC-3 cells, some autophagosome (yellow) and Golgi membrane (magenta) punctae were merged, indicating that under normal conditions, PCa cells are experiencing steady-state Golgiphagy (Fig. 3N, Ctrl, and 3Q). Indeed, EM imaging revealed in both PC-3 and DU145 cells multiple phagophores nucleating from Golgi cisternae and AVs localized in the vicinity of Golgi (Fig. S5A and B). Moreover, Golgi membranes isolated from PC-3 and DU145 cells contain a substantially higher level of early autophagy marker WIPI2 compared to LNCaP cells (Fig. S5C). In cells treated with HCQ, Golgi compaction was associated with a significant decrease in its colocalization with autophagosomes (Fig. 3N, HCQ, and 3Q), implying the blockage of Golgiphagy.
HCQ-mediated Golgi restoration leads to the reduction of Integrin αv and Gal-3 at the PM and their aggregation in the early endosome (EE)
The recovery of MGAT3’s intra-Golgi localization after HCQ suggests that the pool of integrins modified by MGAT5 will be reduced. To examine this, we performed lectin chromatography using immobilized PHA-L lectin. The lysates from control and HCQ-treated PC-3 cells were incubated with PHA-L-Separopore 4B beads, followed by extensive washing and elution by GlycoElute Elution Buffer. Integrin αv immunoblotting of eluates showed a significant reduction of integrins captured by PHA-L, indicating the blockage of glycan modification via MGAT5 (Fig. 4A).
Figure 4.
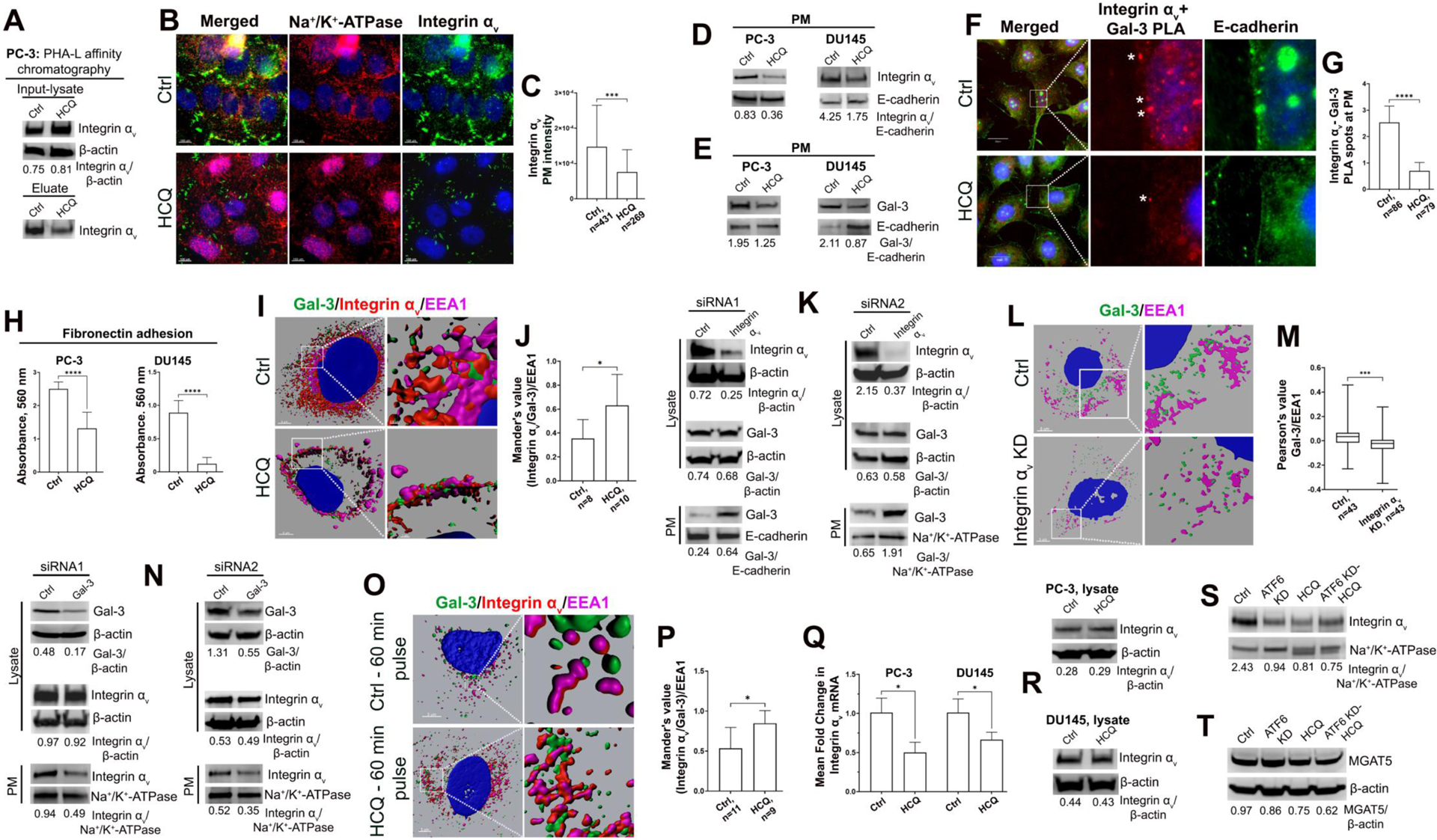
HCQ-induced redistribution of Integrin αv and Gal-3. (A) PHA-L lectin affinity chromatography of the lysate samples from control and HCQ-treated PC-3 cells. Integrin αv W-B of the input (top panel) and eluate (bottom panel). The input was normalized by β-actin and eluate by total protein concentration. (B) Representative images of PC-3 cells IF co-staining of Integrin αv (green) and Na+/K+-ATPase (red) in control and HCQ-treated cells; bars, 100 μm. (C) Quantification of Integrin αv IF intensity at PM in cells from B; Mann Whitney test. (D, E) Integrin αv (D) and Gal-3 (E) W-B of the PM samples from control and HCQ-treated PC-3 and DU145 cells. (F) PLA in control and HCQ-treated PC-3 cells, using Ms-anti-Gal-3 and Rb-anti-Integrin αv, and co-stained with Gt-anti-E-cadherin (green); bars, 20 μm. Asterisks indicate PLA-specific red spots at PM. (G) Quantification of PLA spots at PM per cell in samples from F; unpaired t test. (H) Adhesion of PC-3 and DU145 cells (control and HCQ-treated) to polystyrene microtiter plates coated with fibronectin; Mann Whitney test. (I) Representative reconstructions of the distribution of Gal-3 (green), Integrin αv (red), and early endosomal marker EEA1 (magenta) in control and HCQ-treated PC-3 cells. Z-stack images were collected, and a 3D image projection was created using Imaris for visualization; bars, 5 μm. (J) Mander’s coefficient of colocalization for Gal-3 and Integrin αv merged spots with EEA1. Unpaired t test. (K) W-B analysis of PC-3 cells treated with two different combinations of Integrin αv siRNAs. Lysate samples were tested for Integrin αv (top peanel) and Gal-3 (middle panel); PM samples were tested for Gal-3 (bottom panel). (L) Representative reconstructions of Gal-3 (green) and EEA1 (magenta) distribution in control and Integrin αv KD PC-3 cells; bars, 5 μm. (M) PCC between Gal-3 and EEA1; Mann Whitney test. (N) W-B analysis of PC-3 cells treated with two different combinations of Gal-3 siRNAs. Lysate samples were tested for Gal-3 (top panel) and Integrin αv (middle panel); PM samples were tested for Integrin αv (bottom panel). (O) Representative reconstructions of pulse-chase endocytosis experiment. Integrin αv Ab (red) was internalized by control or HCQ-treated PC-3 cells, which were then stained for Gal-3 (green) and EEA1 (magenta); bars, 5 μm. (P) Mander’s coefficient of colocalization for Gal-3 and Integrin αv merged spots with EEA1; Mann Whitney test. (Q) RT-qPCR analysis of Integrin αv mRNA in control and HCQ-treated PC-3 and DU145, calculated using the 2−ΔΔCT method with GAPDH as the reference gene. Results are the means of three independent experiments performed in triplicate. (R) Integrin αv W-B of the lysate samples from PC-3 and DU145 cells: control and treated with HCQ. (S, T) Integrin αv (S) and MGAT5 (T) W-B of the PM samples from PC-3 cells: control and ATF6 KD treated with HCQ. For W-B data: E-cadherin and Na+/K+-ATPase are the loading controls for PM samples, and lysates were normalized by β-actin. For all statistics: median ± SD, ***p<0.001, ****p<0.0001, *p<0.05; n indicates the number of cells. All data presented are representative of at least three independent experiments.
Abrogation of glycosylation via MGAT5 reduces the complex formed between Integrin αv and Gal-3. This, in turn, should limit integrins’ retention at the cell surface due to the inability to form clusters. To check this, we performed a series of immunofluorescence (IF) experiments, employing the same strategy as IHC, co-staining Integrin αv with PM marker, Na+/K+-ATPase. As demonstrated in Fig. 4B and C, the PM intensity of Integrin αv was significantly reduced in HCQ-treated PC-3 cells. These data were further verified by W-B analysis: in HCQ-treated PC-3 and DU145 cells, the level of Integrin αv protein was decreased in the PM fraction (Fig. 4D). Analogously to Integrin αv, we found that after HCQ, Gal-3 exhibits a reduction of IF signal on the cell surface (Fig. S6A and B). W-B analysis of PM fraction from PC-3 and DU145 cells validated this finding (Fig. 4E). Next, we examined whether the Integrin αv/Gal-3 association was decreased alongside the reduction in PM Integrin αv and Gal-3. In PC-3 cells, we performed PLA using mouse anti-Gal-3 and rabbit anti-Integrin αv Abs and co-stained PM with goat anti-E-cadherin Ab. In control cells, multiple PLA dots were observed at the cell surface; however, their numbers significantly declined in cells receiving HCQ (Fig. 4F and G).
Overall, these data prompt us to conclude that HCQ-induced abrogation of MGAT5-mediated glycosylation impacts the amount of both Gal-3 and Integrin αv at the cell surface. With all this information in hand, we proceeded to evaluate the adhesion potential of these cells to FN. Using polystyrene microtiter plates coated with FN, we measured the adhesion of PC-3 and DU145 cells. The average from three independent experiments strongly indicated a reduction in the adhesion of cells treated with HCQ (Fig. 4H).
It is known that cell surface expression and function of integrins are regulated by their internalization to early endosomes (EE), where they can be recycled back to the membrane to promote cell migration or routed to the lysosome for degradation (43). A study in breast carcinoma cells reported that Gal-3 mediates the endocytosis of beta-1 integrins (44). Thus, the next question we asked was: what is the role of Gal-3 in HCQ-induced redistribution of αv integrins?
To address this question, PC-3 cells were stained for Integrin αv, Gal-3, and EEA1, an EE marker. Three-color colocalization analysis of these images revealed more Integrin αv/Gal-3 complexes in EEs after HCQ treatment (Fig. 4I and J, movies S3 and S4). The simplest explanation of these results is that HCQ blocks the fusion of EE with AV (40). However, integrin aggregation in EE could also be caused by the intensification of their internalization. We hypothesize that HCQ-induced deficiency in Gal-3 at PM stimulates internalization of MGAT5-modified integrins, which were delivered to the cell surface before HCQ’s effect on Golgi and trafficking. To examine this assumption, we rigorously analyzed the impact of Integrin αv gene silencing on Gal-3 expression and vice versa. For depletion of each protein, we conducted two independent experiments using different siRNAs (see Supplemental Table S2). After Integrin αv KD, the total Gal-3 in lysates was unchanged (Fig. 4K, middle panels), but Gal-3 on the PM increased (Fig. 4K, bottom panels), indicating that Gal-3 cannot be internalized without integrins. Indeed, co-staining of Gal-3 and EEA1 in Integrin αv KD cells and quantification of colocalization between them indicates that lack of Integrin αv abrogates relocation of Gal-3 to EE (Fig. 4L and M). Therefore, to be transferred to EE, Gal-3 needs integrins. On the other hand, after Gal-3 KD, total Integrin αv was unchanged in lysate samples (Fig. 4N, middle panels). However, PM-localized Integrin αv was decreased (Fig. 4N, bottom panels), indicating that Gal-3 is required for the retention of Integrin αv on the PM.
We concluded that upon deficiency in Gal-3 (in this case, induced by HCQ), integrins are unable to form clusters and are quickly internalized. To prove this, we performed a proof-of-principle pulse-chase experiment. The detail of this experiment is described in the Supplementary methods. Briefly, cells were treated with HCQ for 72 hours and then incubated with anti-Integrin αv Ab. The Abs were allowed to internalize for 1 hour, and then cells were stained for Gal-3 and EEA1, followed by secondary antibodies for all three antibodies (Fig. 4O). Then, three-color colocalization (Integrin αv/Gal-3 with EEA1) was measured again. It is important to stress that the IF signal of Integrin αv presented here can be only ascribed to the portion of integrins internalized from the PM together with anti-Integrin αv Ab within the one-hour incubation. We found that in HCQ-treated cells, merged complexes αv-integrins/Gal-3 were increased in EE (Fig. 4P). These data suggest that abrogation of Gal-3 trafficking to the cell surface may stimulate the internalization of integrins and Gal-3 that form a complex at the cell surface. Hence, in terms of Gal-3 expression at PM, it appears that HCQ treatment mimics the effect of Gal-3 depletion by intensifying integrin internalization.
These data raise the logical question of whether the decrease of integrins at the cell surface after HCQ treatment is due to altered gene transcription or altered trafficking to and stability at the PM. In both HCQ-treated PC-3 and DU145 cells, we found a significant reduction of Integrin αv mRNA expression by qRT-PCR analysis (Fig. 4Q). However, the total protein level of Integrin αv was identical to the control (Fig. 4R). This suggests a multi-faceted effect. While transcription of Integrin αv mRNA decreases after HCQ treatment, so does degradation as HCQ blocks lysosomal cleavage of autophagosome contents. Thus, the total protein levels remain constant, as less protein is produced, but less protein is degraded. Meanwhile, we see lower overall levels on the PM caused by increased internalization of Integrin αv. Therefore, the reduction of Integrin αv in the PM fraction cannot be ascribed solely to its gene downregulation but rather also to intensified internalization.
Interestingly, the effect of HCQ on PM expression of Integrin αv was close to that in ATF6 KD cells. Still, more reduction was observed in HCQ-treated ATF6 KD cells (Fig. 4S). Similarly, these cells demonstrated the most noticeable decrease of MGAT5 protein amount compared with ATF6 KD or HCQ-treated cells (Fig. 4T).
Examination of Integrin αv and Gal-3 suborganellar distribution by immunogold EM in HCQ-treated cells
To further define the subcellular distribution of Integrin αv and Gal-3 after HCQ treatment, we performed double pre-embedding immunogold EM of Integrin αv and Gal-3 using Abs conjugated to 1.4-nm and 10-nm gold particles, respectively, in PC-3 cells. In control cells, we detected the association of both proteins at the PM as both single merged spots and clusters (Fig. 5A, Ctrl-PM). Also, Integrin αv and Gal-3 joined spots were observed in the cytoplasm, late endosomes (LE), EE, and Golgi, which is surprising because Gal-3 is suggested to be delivered to the cell surface via non-canonical trafficking (45). After HCQ treatment, we could not detect joined Integrin αv and Gal-3 spots in cytoplasm or Golgi. Nevertheless, many merged spots were found in EE. Quantification of Integrin αv and Gal-3 association indicated a significant decrease at the PM but a robust increase in EE (Fig. 5B and C), echoing the results presented above. Thus, enhanced internalization of αv-integrins and Gal-3 is associated with their segregation at the cell surface.
Figure 5.
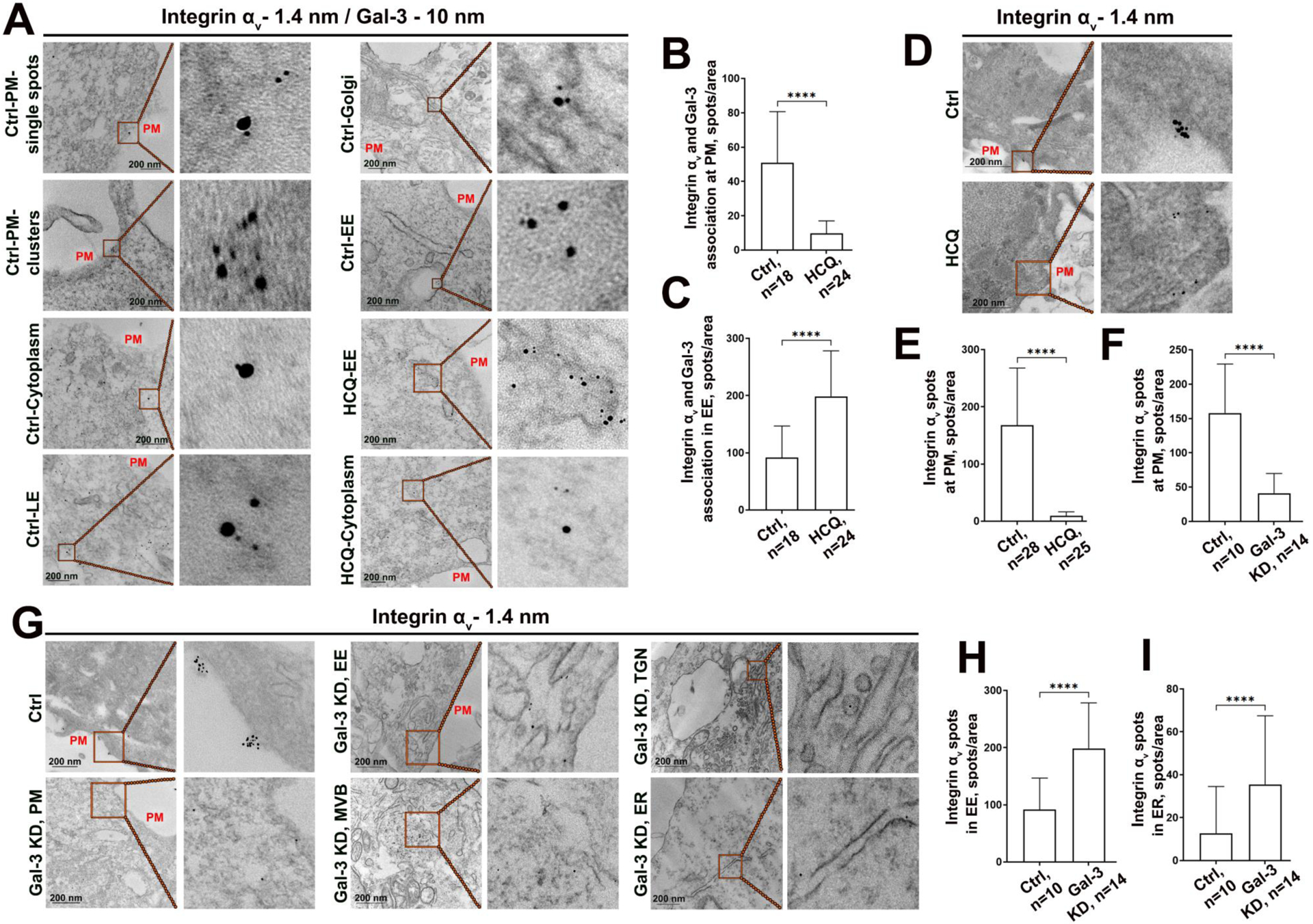
Analysis of Integrin αv and Gal-3 distribution using immunogold EM. (A) Representative micrographs of double pre-embedding immunogold EM analyses of Integrin αv and Gal-3 in PC-3 cells using Abs conjugated to 1.4-nm and 10-nm gold particles, respectively. Cells were treated with 50 μM HCQ or a proper amount of water (Ctrl). The two upper left panels (Ctrl-PM) demonstrate the merged Integrin αv-Gal-3 spots at PM: single or clusters. Others indicate their distribution in the cytoplasm, late endosomes (LE), Golgi, and early endosomes (EE). (B, C) Quantification of merged Integrin αv-Gal-3 spots at PM (B) and in EE (C) in cells from A per area in μm2. (D) Representative micrographs of single pre-embedding immunogold EM analyses of Integrin αv in PC-3 cells treated with 50 μM HCQ for 72 h or an appropriate amount of water. (E) Quantification of Integrin αv spots at PM from cells in D. (F) Quantification of Integrin αv spots at PM from control and Gal-3 KD PC-3 cells. (G) Representative micrographs of single pre-embedding immunogold EM analyses of Integrin αv in PC-3 cells treated with control or Gal-3 siRNAs; MVB, multivesicular bodies; TGN, trans-Golgi network; ER, endoplasmic reticulum. (H, I) Quantification of Integrin αv spots in EE and ER, respectively, in cells from G. For all graphs, n is the number of cells, bars 200 nm. Mann Whitney test for all graphs; ****p<0.0001, median ± SD.
In another series of single Integrin αv immunogold staining, we validated that treatment of PC-3 cells with HCQ substantially reduces the content of Integrin αv particles at PM (Fig. 5D and E). Then, we corroborated that Gal-3 depletion decreases Integrin αv at the PM, as presented above. In cells lacking Gal-3, the number of Integrin αv-positive particles was reduced at the cell surface (Fig. 5F and G). Importantly, in terms of Integrin αv distribution, we found two different aspects of Gal-3 depletion. On the one hand, Gal-3 KD leads to accumulation of Integrin αv in EE, confirming its enhanced internalization (Fig. 5G and H). Thus, the internalization of αv integrins is independent of Gal-3. On the other hand, deficiency in Gal-3 impacts Integrin αv trafficking as we detected aggregation of Integrin αv in ER (Fig. 5G and I). Also, we found more Integrin αv in the trans-Golgi network (TGN) and multivesicular bodies (MVBs) (Fig. 5G), indicating its elevated recycling. In sum here, Gal-3 is required not only for the retention of αv integrins at the cell surface but also for their anterograde trafficking. Conversely, the restoration of MGAT3-mediated glycosylation of integrins may impact the trafficking of Gal-3 to the cell surface because integrins carrying these glycans do not form a complex with Gal-3.
The impact of alcohol-induced Golgi disorganization on the MGAT5-Integrin αv axis
The link between alcohol consumption and the progression of PCa is well-documented, but the mechanism is not fully understood (46). Our laboratory revealed how EtOH and its metabolites could interfere with the growth of prostate tumors. EtOH-induced Golgi disorganization in androgen-sensitive LNCaP and 22Rv1 cells (a) relocates GSK3β from Golgi to ER, which in turn, activates the HDAC6-HSP90-AR axis (8), and (b) accelerates ATF6-mediated ER stress response via induction of Golgi dispersal and redistribution of S1P and S2P to the ER (6). Importantly, we have shown that the frequency of alcohol consumption positively correlates with Golgi fragmentation and activation of ATF6 (6,28).
Considering this information, the next question to address was whether EtOH administration aggravates Golgi disorganization in PCa cells and accelerates Integrin αv expression, thus potentially promoting prostate tumor expansion. Notably, both PC-3 and DU145 cells abundantly express the main EtOH-metabolizing enzymes, alcohol dehydrogenase (ADH1A) and aldehyde dehydrogenase (ALDH2) (Fig. S7). First, we found that the number of Golgi fragments was significantly increased in both PC-3 and DU145 cells after 72 h of EtOH treatment (Fig. 6A, B and D, E). This effect was detected at the dosage of 11.5 μM EtOH, which is within the physiological dosage of alcohol administration that ranges from 1 to 100 mM (47). Thus, this in vitro model falls at a very moderate to low-level dose, yet still elicits Golgi fragmentation, avoiding the cytotoxic effects of higher doses in vitro. Second, the intensity of Integrin αv IF was also enhanced at the PM following EtOH treatment (Fig. 6A, C and D, F). Interestingly, we detected a strong positive correlation between the number of Golgi fragments and the PM Integrin αv IF signal, demonstrating a nexus between Golgi disorganization and the expression of integrins (Fig. 6G). W-B analysis of PM fraction from EtOH-treated PC-3 and DU145 cells confirmed enhanced retention of Integrin αv at the cell surface (Fig. 6H and I). This was further validated by immunogold staining of Integrin αv and EM imaging. In DU145 cells, clusters formed by Integrin αv-positive punctae were significantly enhanced in EtOH-treated cells (Fig. 6J and K, arrowheads, Fig. 6L). Notably, in both cell lines, EtOH also upregulates MGAT5 (Fig. 6M and N). We further examined whether this in vitro data could be replicated in primary PCa patients with the same tumor grade, consuming alcohol at different levels. We sorted PCa patients with grades 2–3 into three groups: non-alcoholic, moderate-alcoholic, and heavy-alcoholic, based on the criteria of alcohol consumption history published by our group previously and described in the methods (6,28). First, we measured Integrin αv H-score at the PM of the prostate tumor cells. We found that this parameter was significantly higher in patients heavily consuming alcohol compared to their non-alcoholic counterparts (Fig. 6O and P). Analysis of Integrin αv and Gal-3 colocalization at PM revealed a significant increase in both alcohol-consuming groups compared to nondrinkers; however, no difference was detected between drinkers (Fig. 6Q). Next, we examined MGAT5 immunoreactivity. Again, we did not observe a substantial difference between patients drinking alcohol at moderate or heavy levels; nevertheless, heavy drinkers demonstrated a higher level of MGAT5 compared to nondrinkers (Fig. 6R and S).
Figure 6.
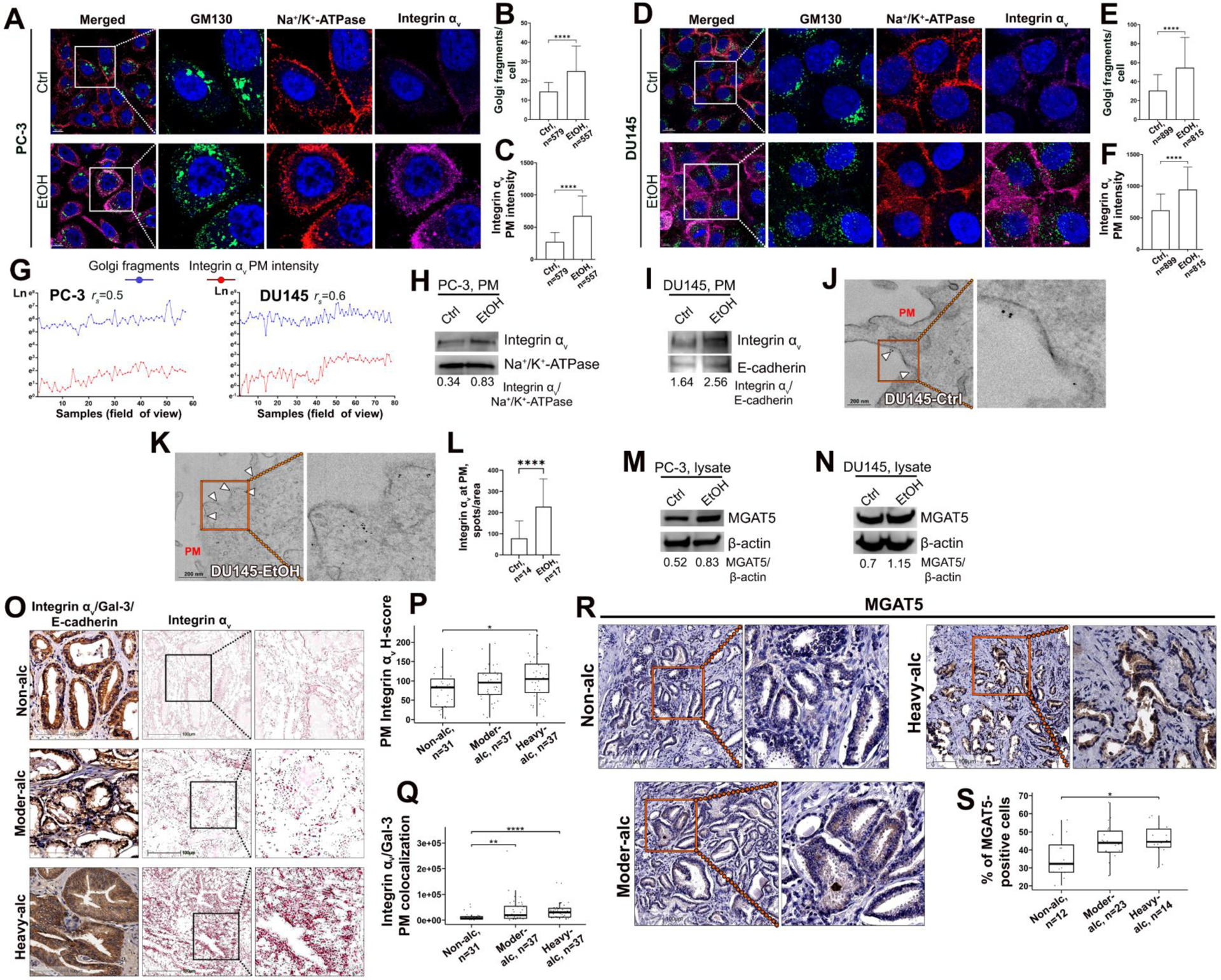
The impact of alcohol on Golgi morphology and expression of Integrin αv and MGAT5. (A-F) Representative IF images of Golgi (GM130, green), PM (Na+/K+-ATPase, red), and Integrin αv (magenta) in PC-3 (A) and DU145 (D) cells treated with 11.5 μM ethanol (EtOH) or isocaloric amount of media (Ctrl) for 72 hours; bars, 10 μm. B and E: quantification of the number of Golgi fragments per cell from images in A and D, respectively; C and F: quantification of Integrin αv integrated fluorescence intensity at PM from images in A and D, respectively. Mann Whitney test for B, E, and F, unpaired t test for C. For B, C, E, and F, n indicates the number of cells. (G) Correlation analysis between the number of Golgi spots (blue) and PM Integrin αv (red) counted as the average for each parameter in the cells from one field of view. Spearman Rank Correlation Coefficient, rs; p<0.0001. (H, I) Integrin αv W-B of the PM samples from the cells in A and D, respectively; Na+/K+-ATPase and E-cadherin are loading controls. (J, K) Representative micrographs of single pre-embedding immunogold EM analyses of Integrin αv in control (J) and EtOH-treated (K) DU145 cells. Arrowheads indicate Integrin αv-specific spots on PM. (L) Quantification of Integrin αv spots per area of PM (in μm2) in cells from J and K; Mann Whitney test. (M, N) MGAT5 W-B of the lysate samples from the cells in A and D, respectively; β-actin is a loading control. (O) Triple IHC staining of Integrin αv (red, deconvoluted in the middle), Gal-3 (green), and E-cadherin (brown) in tumor tissues from PCa patients: non-alcoholic or consuming alcohol at a moderate or high level; bars, 100 μm. (P, Q) Quantification of Integrin αv PM H-score (P) and colocalization of Integrin αv with Gal-3 at the PM (Q) from the samples in O. For P: Tukey Method, p-adjusted using Benjamini-Hochberg. For Q: Dunn Test (1964) Kruskal-Wallis multiple comparisons; n indicates the number of samples. (R) MGAT5 IHC (brown) in tumor tissues from PCa patients: non-alcoholic or consuming alcohol at the moderate or high level; bars, 100 μm. (S) Quantification of the percent of MGAT5-positive cells from the samples in R. Tukey Method, p-adjusted using Benjamini-Hochberg. For all statistics: median ± SD, ****p<0.0001, **p<0.005, *p<0.05. All data presented are representative of at least three independent experiments.
The implication of HCQ and ATF6 depletion in vivo
To investigate the clinical significance of our findings, we examined the effect of ATF6 depletion in combination with HCQ treatment using a mouse model of PCa. In nude mice, we performed the orthotopic implantation of control or ATF6 KD PC-3 cells, followed by treatment with PBS (control) or HCQ. Treatment was administered by i/p injection of 25 mg/kg HCQ in PBS or an equivalent volume of PBS twice per week, based on a weekly physiological dosage of 50 mg/kg (48). After 40 days of tumor growth, mice were sacrificed. Importantly, at this dosage, HCQ did not show hepatotoxicity, as the serum level of both aspartate aminotransferase (AST) and alanine aminotransferase (ALT) did not differ significantly from the control (Fig. S8). As shown in Fig. 7A and B, tumor size was substantially reduced after HCQ treatment in mice that received PC-3 control cells. Similarly, the tumors derived from ATF6 KD cells were smaller than the control and even smaller than in the HCQ group. However, mice injected with ATF6 KD cells and treated with HCQ demonstrated the smallest average tumor size, significantly lower than the control, HCQ, or ATF6 KD alone groups. Moreover, in control mice, various macro- and micro-metastases were detected in regional and distant lymph nodes, as well as in the liver and lung (Fig. 7A, Ctrl, arrowheads). In the HCQ-treated or ATF6 KD groups, we could not detect organ metastases; however, regional lymph node metastases were found (Fig. 7A, HCQ and ATF6 KD, arrowheads). Finally, the tumors derived from ATF6 KD cells in mice receiving HCQ did not demonstrate signs of metastasis. Detailed information about the tumor size and the number of metastases is presented in Supplemental Table S3. A significant difference in the occurrence of lymphatic metastases was detected between (a) Ctrl and ATF6 KD-HCQ, (b) HCQ and ATF6 KD-HCQ, and (c) ATF6 KD and ATD6 KD-HCQ groups. Additionally, a significant difference in the occurrence of organ metastases was detected between (a) Ctrl and HCQ and (b) Ctrl and ATF6 KD-HCQ groups. This Pearson’s Chi squared and Fisher’s Exact test statistical analyses are summarized in Supplemental Table S4.
Figure 7.

In vivo implication of ATF6 ablation and HCQ treatment, and the working model of HCQ treatment and ATF6 depletion on glycosylation and distribution of Integrin αv and Gal-3. (A) Representative images of orthotopic tumors in nude mice derived from control and ATF6 KD PC-3 cells. In the control group, images at the right indicate metastases (arrowheads) in regional (top panel) and mediastinal (middle panel) lymph nodes and in the liver (bottom panel). In HCQ and ATF6 KD groups, insets show metastases in regional lymph nodes. (B) Quantification of tumor size from mice in A. Pairwise comparisons using Wilcoxon rank sum exact test, p-adjusted using Benjamini-Hochberg. (C) Golgi disorganization in advanced PCa cells results in the mislocalization of MGAT3 from Golgi to ER; however, it does not affect Golgi localization for MGAT5. (1) Integrins are delivered to the cell surface via canonical (ER→Golgi→PM) trafficking and modified in Golgi by MGAT5, followed by interaction with pentameric Gal-3 and the formation of clusters at the PM. (2) The magnifying glass indicates the details of Golgi-associated autophagy at the left. Under normal conditions, the Golgi fragments in PCa cells can serve as a source of phagophores. The complex of Integrin αv and Gal-3 is internalized into early endosomes (EE), which, in turn, can fuse with autophagosome (AV). Most EEs mature into late endosomes (LE) or multivesicular bodies (MVB), followed by recycling or formation of an amphisome. The latter fuses with lysosomes to form autophagolysosomes. Hydroxychloroquine (HCQ) does not inhibit the maturation of EE, but it accelerates the internalization of Integrin αv/Gal-3 (3) due to a deficiency of Gal-3 at the cell surface. This, in addition to HCQ blocking the fusion of EE with AV, results in the aggregation of integrins in EE. Also, HCQ prevents the fusion of lysosomes with amphisomes and the formation of phagophores from Golgi membranes. Reconstitution of compact Golgi by ATF6 depletion and HCQ treatment (4) is associated with the recovery of intra-Golgi localization of MGAT3, which significantly reduces the retention of integrins at the cell surface. (D) Flow chart of HCQ’s effect on trafficking of Gal-3 and Integrin αv. Abrogation of MGAT5-mediated glycosylation and Gal-3 delivery to the cell surface destabilize the retention of integrins at PM, which accelerates their internalization.
Next, using both IHC and IF of the orthotopic tumor samples, we were able to reproduce our in vitro data. First, we demonstrated the restoration of Golgi after HCQ or ATF6 KD and significant compaction of Golgi in the ATF6 KD-HCQ group compared with HCQ or ATF6 KD groups (Fig. S9A and B). Second, we validated the lack of ATF6 in ATF6 KD cell-derived tumors (Fig. S9C and D). Third, PM expression of Integrin αv was reduced substantially in all groups of mice compared with the control (Fig. S9E and F). Moreover, a robust reduction of this parameter was found in ATF6 KD-HCQ relative to HCQ or ATF6 KD alone. Additionally, the PM expression of Gal-3 was significantly lower in ATF6 KD-HCQ than in the control group or ATF6 KD (Fig. S9E and G).
The recovery of MGAT3’s intra-Golgi localization after HCQ (Fig. 3L and M) suggests that integrins will not be modified by MGAT5 after HCQ treatment. To evaluate MGAT5-mediated glycosylation of Integrin αv in orthotopic tumor samples, we co-stained Integrin αv with Cy3-labeled PHA-L lectin. To obtain more detailed information about PHA-L and Integrin αv colocalization, we captured high-resolution Z-stack images. We reconstructed 3D image projection of images using Imaris and quantified the Mander’s overlap coefficient B, which reflects the proportion of the red signal (PHA-L) coincident with a signal in the green channel (Integrin αv) over the total red intensity (Fig. S10A and B). In all groups of mice, Mander’s coefficient was substantially reduced compared to the control. Also, ATF6 KD-HCQ samples demonstrated the lowest level of colocalization compared with ATF6 KD or HCQ.
A similar experiment was performed using Phaseolus Vulgaris Erythroagglutinin (PHA-E) lectin, which specifically recognizes complex type N-glycans containing bisecting GlcNAc, the product of MGAT3 (49) (Fig. 1A). Co-staining of the orthotopic tumor samples with PHA-E and Integrin αv revealed significantly higher colocalization in ATF6 KD and HCQ samples. Again, the number of merged PHA-E/Integrin αv spots was the highest in ATF6 KD-HCQ samples (Fig. S10C and D). Overall, these results mirror those from PHA-L, proving that restoration of MGAT3 glycosylation associates with the reduction of glycan modification via MGAT5.
DISCUSSION
Here, for the first time, we explain the phenomenon of excessive MGAT5-mediated glycosylation in PCa tumor cells. We detected that the progression of prostate malignancy is associated with the mislocalization of MGAT3, which, otherwise, in cells with unaffected Golgi, blocks MGAT5-mediated sugar modification. Histochemical clinical data indicate that the increased Integrin αv PM expression in high-grade PCa and mCRPC patients is commensurate with enhanced MGAT5-catalyzed glycosylation. Analogously, patients with high alcohol consumption had significantly higher immunoreactivity for both MGAT5 and PM-specific Integrin αv. Downregulation of MGAT5 and recovery of MGAT3 localization to the Golgi following HCQ treatment caused a reduction of PM-specific Integrin αv in the PC-3 and DU145 cellular models and a substantial decline in FN adhesion. The in vivo data indicate that this expression is attributed to a decrease in glycosylation of αv integrins via MGAT5 and restoration of MGAT3 glycosylation.
For the first time, we observed that inhibition of autophagy converts fragmented Golgi phenotype to compact. This was accompanied by a reduction of Golgi size, suggesting that Golgi fragmentation in prostate tumor cells is also associated with a significant increase in its volume, probably driven by the intensification of Golgi membrane biogenesis. However, these nascent membranes are unable to fuse due to the ER stress and impaired dimerization of several critical golgins, such as Giantin (4).
So, how does HCQ restore Golgi morphology exactly? Currently, there is no consensus regarding the precise involvement of autophagy in carcinogenesis since its activation and inhibition have each been observed in different cancers (reviewed in (50)). However, emerging evidence indicates the critical role of autophagy during metastasis in mCRPC (reviewed in (23)). In this study, we observed the close association between Golgi membranes and autophagosomes, which was mitigated by HCQ treatment. Fragmented Golgi membranes serve as a steady-state source of phagophores, so by inhibiting the fusion of AV with EE or lysosomes, HCQ prevents Golgi disorganization (Fig. 7C).
Additionally, it is known that Golgi luminal pH in normal cells is slightly acidic (6.0–6.5); however, moderate alkalinization of Golgi has been shown in different cancers and is associated with the fragmentation of Golgi (51,52). Thus, it is unlikely that Chloroquine and HCQ significantly de-acidify the lumen of fragmented Golgi membranes in cancer cells and impair the function of Golgi. In fact, the restoration of Golgi by HCQ not only brings MGAT3 back to the Golgi but also rescues its function because the number of αv integrins colocalizing with PHA-E lectin was significantly increased.
We showed that in advanced PCa cells, steady-state activation of the ATF6 branch of UPR is critical for the maintenance of dispersed Golgi. Indeed, depletion of ATF6 restores dimerization of Giantin, recovers compact Golgi, and reduces Integrin αv in the PM fraction in both in vitro and in vivo experiments. The effect of ATF6 KD on Golgi is not mediated by autophagy inhibition, as we could not detect changes in LC3BII expression in PC-3 cells lacking ATF6. However, dimerization of Giantin requires PDIA3 chaperone delivered to the nascent Golgi membranes via COPII vesicles (53). At this point, we speculate that ATF6 may transcriptionally downregulate critical proteins required for the formation of the COPII complex. However, this hypothesis requires further investigation.
Importantly, we observed a synergistic effect of HCQ and ATF6 depletion on autophagy inhibition and a more prominent effect on Golgi morphology and expression of Integrin αv at the cell surface (Fig. 7C). Finally, in the mice orthotopic tumor model, the combination of ATF6 KD with HCQ had the most noticeable impact on MGAT5-mediated glycosylation and halted metastasis. It is essential to note here that we could not find any strong correlation between the orthotopic tumor size and the presence or absence of metastases. In some cases, large tumor volume was not associated with any sign of dissemination; in others, small tumors were found to be metastasized. Thus, the lack of metastases in the ATF6 KD-HCQ group cannot be ascribed to the smallest tumor size. Also, in clinical settings, small prostate tumors appear to be more aggressive than large (54).
Intriguingly, we found that in primary prostate tumors with grades 4–5, the expression of αv integrins at PM was higher than that in patients with grades 2–3. Also, these groups demonstrate a similar expression of Gal-3 at the cell surface. Nevertheless, colocalization of integrins and Gal-3 at PM was significantly lower in grades 4–5 tumors than that with grades 2–3. This implies that in high-grade PCa, some fraction of integrin molecules can be expressed at PM independently of Gal-3. Also, in mCRPC patients, the PM expression of Integrin αv and Gal-3 was substantially higher in samples from bone metastases compared with those from tissue metastases. However, the association of Integrin αv and Gal-3 was lower in bone metastases samples than in visceral. These data suggest that the expression of integrins during the growth and dissemination of prostate tumors may also involve Gal-3-independent mechanisms, for instance, the delivery of integrins to the cell surface via an alternative, non-canonical trafficking. Recently, we reported that alcohol-induced ER stress and Golgi dispersal stimulate the trafficking of one of the hepatic proteins, the asialoglycoprotein receptor, to the cell surface via ER-PM junctions, bypassing canonical (ER→Golgi→PM) anterograde transportation (55). Our preliminary data indicate that in PCa cells, some pool of αv integrins that bear high-mannose glycans can be detected at the cell surface. Thus, we cannot exclude the non-canonical trafficking of integrins and the contribution of their high-mannose glycans to the metastatic potential of cancer cells.
It was suggested that galectins are non-glycoproteins, which can be exported to the cell surface and excreted by a non-classical pathway, bypassing the classical biosynthetic ER→Golgi→PM pathway (45). Indeed, our multiple attempts to check the sensitivity of Gal-3 to different glycosidases confirmed that Gal-3 does not bear any N- or O-glycans. Paradoxically, Integrin αv-Gal-3 spots were observed in the medial and terminal Golgi compartments, indicating Gal-3 is integrated into canonical trafficking. IHC and W-B data revealed an interesting bidirectional relationship between Integrin αv and Gal-3. Gal-3 depletion did not affect the total level of Integrin αv but reduced PM content via deposition in EE and ER, substantiating Gal-3 is involved in αv integrins trafficking, but it is not required for their internalization.
Our unexpected EM finding of the association of Integrin αv and Gal-3 in the Golgi membranes of control PC-3 cells raises the possibility that a complex between these proteins can be formed en route to the cell surface (right after MGAT5-mediated glycosylation of integrin in Golgi) rather than at the cell surface as was previously suggested (11,56). Importantly, we could not find merged Integrin αv-Gal-3 immunogold spots in Golgi after HCQ treatment, which may be caused by the restoration of MGAT3-mediated Integrin αv glycosylation.
The results of Integrin αv depletion were surprising to us. While cells lacking Integrin αv showed no difference in the total level of Gal-3, they demonstrated an increase of Gal-3 at the PM. Thus, without Integrin αv, Gal-3 is not internalized and, instead, aggregates at the cell surface. Intriguingly, the expression of Gal-3 in androgen-responsive LNCaP and 22Rv1 cells is negligible, but they still express Integrin αv. However, we would like to stress again that in LNCaP and 22Rv1 cells, Golgi is intact, contrary to PC-3 and DU145 cells. Therefore, the chance of MGAT5-mediated glycosylation of integrins and their potential need for Gal-3 is low. Thus, it appears that the function of Gal-3 and trafficking to the cell surface are directly determined by Golgi disorganization and MGAT5-modified integrins. Also, we cannot rule out that Gal-3 binding to LacNAc-carrying Integrin αv is required not only for its retention at PM but also for the transition from the inactive to the active conformation and subsequent binding to ECM ligands, such as FN (22,57). These possibilities also await further verification.
Our data allow us to disentangle the effect of HCQ on integrin expression at the PM. On the one hand, Golgi recovery reduces MGAT5-mediated glycosylation, thus blocking the formation of integrin clusters with Gal-3. On the other hand, restoration of MGAT3 in Golgi and subsequent glycosylation of integrins by this enzyme reduce their association with Gal-3, which leads to abrogation of Gal-3 trafficking to the cell surface. Altogether, this results in instability of the Integrin αv-Gal-3 complex and subsequent acceleration of their internalization (Fig. 7D). Thus, we believe that HCQ-induced lack of Gal-3 at the PM is a direct consequence of Golgi reorganization, and it is a major contributing factor leading to a reduction of integrins at the PM.
In sum, we envisage that the results from this study can help to establish several new combinational therapeutic strategies. First, HCQ may be used in parallel with ceapins, a class of small molecules that selectively block the activity of ATF6 without effecting the other UPR pathways (58). Another selective ATF6 inhibitor, Melatonin (59), has shown therapeutic benefits in combination with conventional PCa therapy (reviewed in (60)). Additionally, given that the major integrin-binding ligand, FN, is heavily glycosylated in Golgi (61–63), it would be interesting to check whether fibroblasts in advanced PCa cells exhibit fragmented Golgi phenotype and whether they employ the same alteration of FN glycosylation as described here for integrins. Then, the critical question to address in the future is whether HCQ and ATF6 ablation can reduce the production of FN by cancer-associated fibroblasts. These approaches may also impact ECM and other microenvironmental factors that mediate PCa progression.
Supplementary Material
Acknowledgments:
We thank the UNMC Advanced Microscopy Core Facility and Tissue Sciences Facility for help with imaging and sample preparation. Portions of Fig. 7 were created using BioRender.com; subscription provided by the UNMC Center for Heart and Vascular Research.
Funding.
A.P. is supported by NIH grant NIAAA R01AA027242. J.R. is supported by NIGMS 1P20GM113126. A.K. is funded by the Russian Federation State program FZEG-2020–0060.
Abbreviations:
- PCa
prostate cancer
- mCRPC
metastatic castration-resistant prostate cancer
- AR
androgen receptor
- FN
fibronectin
- Gal-3
Galectin-3
- UPR
unfolded protein response
- PM
plasma membrane
- ER
endoplasmic reticulum
- MVB
multivesicular bodies
- TGN
trans-Golgi network
- EE
early endosome
- LE
late endosome
- AV
autophagosome
- ATF6
activating transcription factor 6
- MGAT3
N-acetylglucosaminyltransferase-III
- MGAT5
N-acetylglucosaminyltransferase-V
- PLA
Proximity Ligation Assay
- NMIIA
non-muscle Myosin IIA
- SIM
structured illumination superresolution microscopy
- EM
electron microscopy
- HCQ
Hydroxychloroquine
- EtOH
ethanol
- Ab
antibody
- PHA-L
Phaseolus Vulgaris Leucoagglutinin
- PHA-E
Phaseolus Vulgaris Erythroagglutinin lectin
Footnotes
Competing interests. The authors declare that they have no competing interests.
Ethics approval and consent to participate. The tissue sections from normal prostate were obtained from US Biomax and Novus Biological. Also, sections were provided through the Department of Pathology and Microbiology (IRB protocol # 304–16-EP) at the University of Nebraska Medical Center and the Johns Hopkins University School of Medicine (Prostate Cancer Biorepository Network), and the Vernadsky Crimean Federal University (Russia, IRB protocol # 98–2019-KFU). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study. The animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at UNMC.
Consent for publication. N/A – The manuscript has no individual person’s data in any form.
Availability of data and materials.
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Goel HL, Li J, Kogan S, Languino LR. Integrins in prostate cancer progression. Endocr Relat Cancer 2008;15:657–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yadav RK, Chae SW, Kim HR, Chae HJ. Endoplasmic reticulum stress and cancer. J Cancer Prev 2014;19:75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kellokumpu S, Sormunen R, Kellokumpu I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: dependence on intra-Golgi pH. FEBS Lett 2002;516:217–24 [DOI] [PubMed] [Google Scholar]
- 4.Petrosyan A, Holzapfel MS, Muirhead DE, Cheng PW. Restoration of compact Golgi morphology in advanced prostate cancer enhances susceptibility to galectin-1-induced apoptosis by modifying mucin O-glycan synthesis. Mol Cancer Res 2014;12:1704–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McKinnon CM, Mellor H. The tumor suppressor RhoBTB1 controls Golgi integrity and breast cancer cell invasion through METTL7B. BMC Cancer 2017;17:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pachikov AN, Gough RR, Christy CE, Morris ME, Casey CA, LaGrange CA, et al. The non-canonical mechanism of ER stress-mediated progression of prostate cancer. J Exp Clin Cancer Res 2021;40:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrosyan A Onco-Golgi: Is Fragmentation a Gate to Cancer Progression? Biochem Mol Biol J 2015;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manca S, Frisbie CP, LaGrange CA, Casey CA, Riethoven JM, Petrosyan A. The Role of Alcohol-Induced Golgi Fragmentation for Androgen Receptor Signaling in Prostate Cancer. Mol Cancer Res 2019;17:225–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakano M, Mishra SK, Tokoro Y, Sato K, Nakajima K, Yamaguchi Y, et al. Bisecting GlcNAc Is a General Suppressor of Terminal Modification of N-glycan. Mol Cell Proteomics 2019;18:2044–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi M, Kuroki Y, Ohtsubo K, Taniguchi N. Core fucose and bisecting GlcNAc, the direct modifiers of the N-glycan core: their functions and target proteins. Carbohydr Res 2009;344:1387–90 [DOI] [PubMed] [Google Scholar]
- 11.Fortuna-Costa A, Gomes AM, Kozlowski EO, Stelling MP, Pavao MS. Extracellular galectin-3 in tumor progression and metastasis. Front Oncol 2014;4:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisanz K, Yu J, Edlund M, Spohn B, Hung MC, Chung LW, et al. Targeting ECM-integrin interaction with liposome-encapsulated small interfering RNAs inhibits the growth of human prostate cancer in a bone xenograft imaging model. Mol Ther 2005;12:634–43 [DOI] [PubMed] [Google Scholar]
- 13.Connell B, Kopach P, Ren W, Joshi R, Naber S, Zhou M, et al. Aberrant integrin alphav and alpha5 expression in prostate adenocarcinomas and bone-metastases is consistent with a bone-colonizing phenotype. Transl Androl Urol 2020;9:1630–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutta A, Li J, Lu H, Akech J, Pratap J, Wang T, et al. Integrin alphavbeta6 promotes an osteolytic program in cancer cells by upregulating MMP2. Cancer Res 2014;74:1598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li D, Li Y, Wu X, Li Q, Yu J, Gen J, et al. Knockdown of Mgat5 inhibits breast cancer cell growth with activation of CD4+ T cells and macrophages. J Immunol 2008;180:3158–65 [DOI] [PubMed] [Google Scholar]
- 16.Murata K, Miyoshi E, Kameyama M, Ishikawa O, Kabuto T, Sasaki Y, et al. Expression of N-acetylglucosaminyltransferase V in colorectal cancer correlates with metastasis and poor prognosis. Clin Cancer Res 2000;6:1772–7 [PubMed] [Google Scholar]
- 17.Kang R, Saito H, Ihara Y, Miyoshi E, Koyama N, Sheng Y, et al. Transcriptional regulation of the N-acetylglucosaminyltransferase V gene in human bile duct carcinoma cells (HuCC-T1) is mediated by Ets-1. J Biol Chem 1996;271:26706–12 [DOI] [PubMed] [Google Scholar]
- 18.Lange T, Ullrich S, Muller I, Nentwich MF, Stubke K, Feldhaus S, et al. Human prostate cancer in a clinically relevant xenograft mouse model: identification of beta(1,6)-branched oligosaccharides as a marker of tumor progression. Clin Cancer Res 2012;18:1364–73 [DOI] [PubMed] [Google Scholar]
- 19.Tsui KH, Chang PL, Feng TH, Chung LC, Sung HC, Juang HH. Evaluating the function of matriptase and N-acetylglucosaminyltransferase V in prostate cancer metastasis. Anticancer Res 2008;28:1993–9 [PubMed] [Google Scholar]
- 20.Zhao Y, Nakagawa T, Itoh S, Inamori K, Isaji T, Kariya Y, et al. N-acetylglucosaminyltransferase III antagonizes the effect of N-acetylglucosaminyltransferase V on alpha3beta1 integrin-mediated cell migration. J Biol Chem 2006;281:32122–30 [DOI] [PubMed] [Google Scholar]
- 21.Pochec E, Litynska A, Amoresano A, Casbarra A. Glycosylation profile of integrin alpha 3 beta 1 changes with melanoma progression. Biochim Biophys Acta 2003;1643:113–23 [DOI] [PubMed] [Google Scholar]
- 22.Lagana A, Goetz JG, Cheung P, Raz A, Dennis JW, Nabi IR. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol Cell Biol 2006;26:3181–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loizzo D, Pandolfo SD, Rogers D, Cerrato C, di Meo NA, Autorino R, et al. Novel Insights into Autophagy and Prostate Cancer: A Comprehensive Review. Int J Mol Sci 2022;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamprou I, Tsolou A, Kakouratos C, Mitrakas AG, Xanthopoulou ET, Kassela K, et al. Suppressed PLIN3 frequently occurs in prostate cancer, promoting docetaxel resistance via intensified autophagy, an event reversed by chloroquine. Med Oncol 2021;38:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kranzbuhler B, Salemi S, Mortezavi A, Sulser T, Eberli D. Combined N-terminal androgen receptor and autophagy inhibition increases the antitumor effect in enzalutamide sensitive and enzalutamide resistant prostate cancer cells. Prostate 2019;79:206–14 [DOI] [PubMed] [Google Scholar]
- 26.Erkisa M, Aydinlik S, Cevatemre B, Aztopal N, Akar RO, Celikler S, et al. A promising therapeutic combination for metastatic prostate cancer: Chloroquine as autophagy inhibitor and palladium(II) barbiturate complex. Biochimie 2020;175:159–72 [DOI] [PubMed] [Google Scholar]
- 27.Lin C, Blessing AM, Pulliam TL, Shi Y, Wilkenfeld SR, Han JJ, et al. Inhibition of CAMKK2 impairs autophagy and castration-resistant prostate cancer via suppression of AMPK-ULK1 signaling. Oncogene 2021;40:1690–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kubyshkin AV, Fomochkina II, Petrosyan AM The Impact of Alcohol on Pro-Metastatic N-Glycosylation in Prostate Cancer. Krim Z Eksp Klin Med 2018;8:11–20 [PMC free article] [PubMed] [Google Scholar]
- 29.The Schachter H. ‘yellow brick road’ to branched complex N-glycans. Glycobiology 1991;1:453–61 [DOI] [PubMed] [Google Scholar]
- 30.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 1985;54:631–64 [DOI] [PubMed] [Google Scholar]
- 31.Gao Y, Chachadi VB, Cheng PW, Brockhausen I. Glycosylation potential of human prostate cancer cell lines. Glycoconj J 2012;29:525–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laderach DJ, Gentilini LD, Giribaldi L, Delgado VC, Nugnes L, Croci DO, et al. A unique galectin signature in human prostate cancer progression suggests galectin-1 as a key target for treatment of advanced disease. Cancer Res 2013;73:86–96 [DOI] [PubMed] [Google Scholar]
- 33.Dondoo TO, Fukumori T, Daizumoto K, Fukawa T, Kohzuki M, Kowada M, et al. Galectin-3 Is Implicated in Tumor Progression and Resistance to Anti-androgen Drug Through Regulation of Androgen Receptor Signaling in Prostate Cancer. Anticancer Res 2017;37:125–34 [DOI] [PubMed] [Google Scholar]
- 34.Ciardiello C, Leone A, Lanuti P, Roca MS, Moccia T, Minciacchi VR, et al. Large oncosomes overexpressing integrin alpha-V promote prostate cancer adhesion and invasion via AKT activation. J Exp Clin Cancer Res 2019;38:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tiraboschi C, Gentilini L, Velazquez C, Corapi E, Jaworski FM, Garcia Garcia JD, et al. Combining inhibition of galectin-3 with and before a therapeutic vaccination is critical for the prostate-tumor-free outcome. J Immunother Cancer 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gao J, Li T, Mo Z, Hu Y, Yi Q, He R, et al. Overexpression of the galectin-3 during tumor progression in prostate cancer and its clinical implications. Int J Clin Exp Pathol 2018;11:839–46 [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma S, Cwiklinski K, Sykes DE, Mahajan SD, Chevli K, Schwartz SA, et al. Use of Glycoproteins-Prostate-Specific Membrane Antigen and Galectin-3 as Primary Tumor Markers and Therapeutic Targets in the Management of Metastatic Prostate Cancer. Cancers (Basel) 2022;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cummings RD, Kornfeld S. Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J Biol Chem 1982;257:11230–4 [PubMed] [Google Scholar]
- 39.Petrosian AM, Britan AV. [Lectin-enzyme assay as a method of estimation of immunoglobulins’ glycosylation]. Ukr Biokhim Zh (1999) 2006;78:151–9 [PubMed] [Google Scholar]
- 40.Chen X, Geiger JD. Janus sword actions of chloroquine and hydroxychloroquine against COVID-19. Cell Signal 2020;73:109706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frisbie CP, Lushnikov AY, Krasnoslobodtsev AV, Riethoven JM, Clarke JL, Stepchenkova EI, et al. Post-ER Stress Biogenesis of Golgi Is Governed by Giantin. Cells 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015;11:1437–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bridgewater RE, Norman JC, Caswell PT. Integrin trafficking at a glance. J Cell Sci 2012;125:3695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furtak V, Hatcher F, Ochieng J. Galectin-3 mediates the endocytosis of beta-1 integrins by breast carcinoma cells. Biochem Biophys Res Commun 2001;289:845–50 [DOI] [PubMed] [Google Scholar]
- 45.Delacour D, Koch A, Jacob R. The role of galectins in protein trafficking. Traffic 2009;10:1405–13 [DOI] [PubMed] [Google Scholar]
- 46.Macke AJ, Petrosyan A. Alcohol and Prostate Cancer: Time to Draw Conclusions. Biomolecules 2022;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dolganiuc A, Szabo G. In vitro and in vivo models of acute alcohol exposure. World J Gastroenterol 2009;15:1168–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masson JD, Blanchet B, Periou B, Authier FJ, Mograbi B, Gherardi RK, et al. Long Term Pharmacological Perturbation of Autophagy in Mice: Are HCQ Injections a Relevant Choice? Biomedicines 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaneda Y, Whittier RF, Yamanaka H, Carredano E, Gotoh M, Sota H, et al. The high specificities of Phaseolus vulgaris erythro- and leukoagglutinating lectins for bisecting GlcNAc or beta 1–6-linked branch structures, respectively, are attributable to loop B. J Biol Chem 2002;277:16928–35 [DOI] [PubMed] [Google Scholar]
- 50.Ashrafizadeh M, Paskeh MDA, Mirzaei S, Gholami MH, Zarrabi A, Hashemi F, et al. Targeting autophagy in prostate cancer: preclinical and clinical evidence for therapeutic response. J Exp Clin Cancer Res 2022;41:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altan N, Chen Y, Schindler M, Simon SM. Defective acidification in human breast tumor cells and implications for chemotherapy. J Exp Med 1998;187:1583–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rivinoja A, Kokkonen N, Kellokumpu I, Kellokumpu S. Elevated Golgi pH in breast and colorectal cancer cells correlates with the expression of oncofetal carbohydrate T-antigen. J Cell Physiol 2006;208:167–74 [DOI] [PubMed] [Google Scholar]
- 53.Petrosyan A, Cheng PW, Clemens DL, Casey CA. Downregulation of the small GTPase SAR1A: a key event underlying alcohol-induced Golgi fragmentation in hepatocytes. Sci Rep 2015;5:17127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou Z, Yue F, Jin L, Liu X, Zhai TS, Zhang JX, et al. Characteristics and risk differences of different tumor size on localized prostate cancer: A retrospective cohort study in the SEER database. Cancer Med 2021;10:2763–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Casey CA, Macke AJ, Gough RR, Pachikov AN, Morris ME, Thomes PG, et al. Alcohol-Induced Liver Injury: Down-regulation and Redistribution of Rab3D Results in Atypical Protein Trafficking. Hepatol Commun 2022;6:374–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Honig E, Ringer K, Dewes J, von Mach T, Kamm N, Kreitzer G, et al. Galectin-3 modulates the polarized surface delivery of beta1-integrin in epithelial cells. J Cell Sci 2018;131. [DOI] [PubMed] [Google Scholar]
- 57.Saravanan C, Liu FT, Gipson IK, Panjwani N. Galectin-3 promotes lamellipodia formation in epithelial cells by interacting with complex N-glycans on alpha3beta1 integrin. J Cell Sci 2009;122:3684–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S, et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bu LJ, Yu HQ, Fan LL, Li XQ, Wang F, Liu JT, et al. Melatonin, a novel selective ATF-6 inhibitor, induces human hepatoma cell apoptosis through COX-2 downregulation. World J Gastroenterol 2017;23:986–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anim-Koranteng C, Shah HE, Bhawnani N, Ethirajulu A, Alkasabera A, Onyali CB, et al. Melatonin-A New Prospect in Prostate and Breast Cancer Management. Cureus 2021;13:e18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Akiyama SK, Yamada SS, Yamada KM. Analysis of the role of glycosylation of the human fibronectin receptor. J Biol Chem 1989;264:18011–8 [PubMed] [Google Scholar]
- 62.Wood AJ, Lin CH, Li M, Nishtala K, Alaei S, Rossello F, et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat Commun 2021;12:2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.da Costa Santos MAR, Dos Reis JS, do Nascimento Santos CA, da Costa KM, Barcelos PM, de Oliveira Francisco KQ, et al. Expression of O-glycosylated oncofetal fibronectin in alternatively activated human macrophages. Immunol Res 2022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.