

Abstract
Lineage plasticity, a process whereby cells change their phenotype to take on a different molecular and/or histological identity, is a key driver of cancer progression and therapy resistance. While underlying genetic changes within the tumor can enhance lineage plasticity, it is predominantly a dynamic process controlled by transcriptional and epigenetic dysregulation. This review explores the transcriptional and epigenetic regulators of lineage plasticity and their interplay with other features of malignancy, such as dysregulated metabolism, the tumor microenvironment and immune evasion. We also discuss strategies for the detection and treatment of highly plastic tumors.
1. Introduction
During normal development, cells undergo specific differentiation and organisation programs to produce tissues with defined functions. More specifically, multipotent embryonic progenitor cells undergo non-genetic phenotypic changes in response to environmental cues to yield lineage-specific progenitors and, eventually, terminally differentiated cells (1). It was originally postulated by Conrad Waddington that mature, differentiated cells were unable to produce progeny distinct from their committed lineage (2). While some cell types and tissues exhibit a restricted capability for phenotypic plasticity, later studies have revealed a remarkable level of cell state flexibility in many adult organs (3), particularly in the context of tissue repair and pathologic stress (4). Lineage plasticity can manifest via dedifferentiation, the reversal of a differentiated phenotype back to a progenitor-like state which may then re-differentiate to another cell lineage, or transdifferentiation, whereby cells switch from one lineage to another without transit via an intermediate pluripotent state.
The unlocking of phenotypic plasticity during tumor evolution is now recognised as a critical hallmark of cancer (5). By exploiting dedifferentiation and transdifferentiation, cancer cells can acquire new molecular features to facilitate metastasis and evade systemic therapies. Although genetic alterations can promote cancer cell lineage plasticity, this phenomenon is primarily driven by epigenetic and transcriptional reprogramming. Epigenetic/transcriptional mechanisms are advantageous over genetic mechanisms since they enable tumors to respond more rapidly to volatile microenvironmental pressures and therapy (6) as well as being more permissive of reversible phenotype switching, whereas genetic changes can lock cells into an evolutionary trajectory that may become disadvantageous in the future. This review describes different types of cancer cell lineage plasticity, the epigenetic and transcriptional underpinnings of such plasticity, and how cancer plasticity might be leveraged therapeutically.
2. Manifestations of lineage plasticity in cancer
Cells exist in a certain configuration – termed a cell state – based on specific molecular (e.g., gene expression) and functional (e.g., self-renewal capacity) features. Multiple regulatory levels, including epigenetic and transcriptional, are fundamental for determining and maintaining cells in a given state. However, it is becoming increasing clear that cells are not locked in a particular state but rather can be rewired and transition along a continuum of phenotypes. A key feature of this rewiring is its complexity, with transitions being frequently characterized by uncertain outcomes and ambiguity in cancer cell identity. Nevertheless, common manifestations of lineage plasticity observed in solid tumors and with documented roles in cancer growth, metastasis and therapy resistance are summarized in Figure 1A and described in more detail below.
Figure 1. Lineage plasticity triggers phenotypic and histological diversity within a tumor.
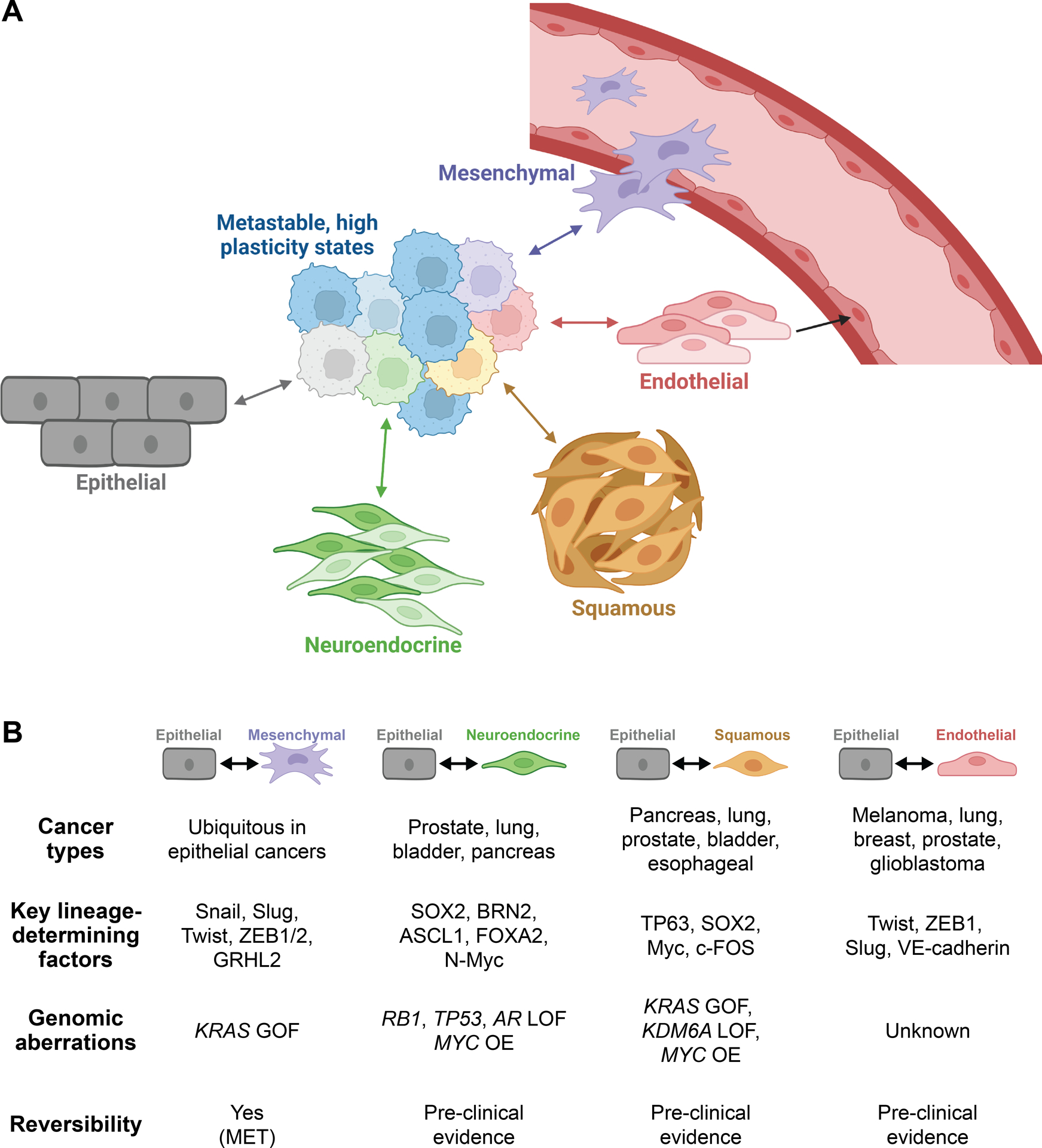
(A) During cancer progression, malignant cells can lose their epithelial identity and enter a hybrid state characterized by heightened plasticity that is primed for multi-lineage differentiation. Varying combinations of transcriptional and epigenetic changes within these hyper-plastic cells, as well as microenvironmental influences, drive transdifferentiation into divergent lineages. Active and bi-directional reprogramming supports tumor growth, metastasis and escape from lineage-directed therapies. (B) Major features of distinct manifestations of cancer cell lineage plasticity. GOF, gain of function; LOF, loss of function; MET, mesenchymal-epithelial transition; OE, overexpression. Created with BioRender.com.
Epithelial-mesenchymal plasticity
The loss of epithelial phenotype and acquisition of mesenchymal properties, a process referred to as epithelial-mesenchymal transition (EMT), was first described in the context of developmental genetics by Elizabeth Hay and colleagues when studying chick embryogenesis (7,8). In the intervening time, compounding evidence has pointed to EMT as a developmental program ubiquitously co-opted by cancer cells to shed epithelial cell polarity and manifest a mesenchymal, motile phenotype (9–11) (Figure 1B). This process is bi-directional; mesenchymal-like cells are capable of gaining epithelial features through a mesenchymal-epithelial transition (MET). The dynamic flexibility of these processes is collectively termed “epithelial-mesenchymal plasticity.”
The regulatory framework controlling epithelial-mesenchymal plasticity is well defined, incorporating both core intrinsic elements, namely the EMT transcription factors Slug, Snail, Twist, and Zeb1/2, as well as context-dependent extrinsic factors such as extracellular matrix stiffness and heterotypic signals originating from tumor-associated stromal cells (9) (see Section 6). A plethora of studies have demonstrated that EMT is associated with enhanced metastatic capacity, therapy resistance and stem-like features (11). Consistent with these findings, tumors derived from tissues of mesenchymal origin are frequently highly invasive (e.g., glioblastoma) or characterized by early metastasis (e.g., melanoma) (12). However, these observations contrast with a parallel body of work showing that stable mesenchymal features can abrogate metastatic outgrowth in pre-clinical models along with reports that tumor cells with an epithelial phenotype retain complete metastatic capacity. For example, recent findings from the Brisken lab and others have demonstrated that, in mouse models of estrogen receptor-positive breast cancer, cells in an epithelial differentiation state exhibit increased fitness, anti-apoptotic signalling, and metastatic potential (13,14).
The divergent and somewhat contradictory findings surrounding epithelial-mesenchymal plasticity in cancer progression may be a result of studying extreme epithelial and mesenchymal states in a process that is likely transient and episodic. More recent evidence has shown that cells existing in a hybrid EMT (hEMT) state – that is, co-expression of epithelial and mesenchymal markers in the same cells – have the greatest malignant and metastatic potential (15,16). Moreover, isolation and sequencing of circulating tumor cells (CTCs) from breast cancer patients revealed that the hEMT state is linked to elevated expression of genes that promote a stem cell-like state (17). Notably, this hybrid phenotype is very common, with a large-scale transcriptomic profiling effort of over 7,100 human epithelial tumors uncovering a hEMT state in ~40% of tumors (18). Whether the hEMT state represents a stable phenotype or is a snapshot of the fluctuating EMT status of individual cells remains an open question, although recent single cell data from multiple tumor types suggests the existence of a continuum of cell states (19).
Neuroendocrine transdifferentiation
Histological transitions from adenocarcinoma to neuroendocrine carcinoma are frequently observed in prostate, lung and bladder tumors (Figure 1B). However, a neuroendocrine gene signature was found to be associated with metastasis and poor outcome across 21 epithelial malignancies, suggesting that this phenomenon may be underappreciated (20). The concept that the neuroendocrine cell state arises via lineage plasticity rather than expansion of pre-existing alternative lineage clones is supported by the observation that prostate tumors frequently retain the genetic hallmarks of their precursor adenocarcinoma histology (21). Validating this hypothesis, murine lineage tracing studies revealed that neuroendocrine cancer cells can arise from alveolar type 2 and tracheobronchial basal cells in the lung and luminal epithelial cells in the prostate (22–24). Moreover, in a series of elegant multiomics and spatial profiling experiments of human lung adenocarcinomas, the Ciriello lab provided additional evidence for epithelial-neuroendocrine transdifferentiation as a result of epigenetic and transcriptional reprogramming (25). Epithelial-to-neuroendocrine lineage plasticity frequently manifests following therapies targeted against driver oncogenes: anti-EGFR therapy in EGFR-mutant lung adenocarcinoma, ALK inhibition in ALK-rearranged lung cancer, and anti-androgen receptor (AR) pathway therapy in prostate adenocarcinoma (26–30). However, evidence for such plasticity in untreated tumors suggests that it can be induced by other factors, such as microenvironmental stressors, hypoxia or oncogene dysregulation. For example, in genetically-engineered mouse models (GEMMs) of pancreatic ductal adenocarcinoma, ductal-neuroendocrine lineage plasticity is governed by deregulated MYC expression in the context of KRAS mutation (31). Similarly, normal epithelial cells of prostate and lung origin can be reprogrammed into neuroendocrine-like cancer cells by ectopic upregulation of AKT, c-MYC and BCL-2 concomitant with loss of the tumor suppressors TP53 and RB1 (32).
The recent observation of tumors with mixed adenocarcinoma/neuroendocrine features following targeted therapy is suggestive of epithelial-to-neuroendocrine plasticity occurring via transitory, metastable phenotypic state(s). Spatial profiling of a prostate tumor with distinct adenocarcinoma and neuroendocrine foci revealed cells along a continuum of plastic gene expression signatures between the two extreme differentiation states (33), a concept reinforced by the frequency of therapy-resistant prostate tumors exhibiting amphicrine phenotypes (i.e. co-expression of both AR and neuroendocrine markers) (30,34,35). Similarly, mixed/transitory high plasticity cell states emerge during lung cancer progression in GEMMs and human patient-derived xenografts (PDXs) and are postulated to underlie chemoresistance (36).
Squamous transdifferentiation
Squamous transdifferentiation represents an alternative lineage plasticity mechanism (Figure 1A) that often develops as a subpopulation within another primary histology. While adeno-squamous histology is most commonly observed to arise de novo in pancreatic ductal adenocarcinomas (37), squamous differentiation in the lung has been reported in the context of acquired resistance to EGFR tyrosine kinase inhibitor therapy as well as ALK inhibitors in the setting of EML4-ALK fusion positive non-small cell lung cancer (NSCLC) (38,39). Although limited in number, cases of squamous transformation with molecular profiling were found to retain the characteristic genetic features of the adenocarcinoma (e.g., EGFR mutation), supporting the hypothesis of lineage plasticity. This is reinforced by mouse models of Kras-driven lung adenocarcinoma whereby loss of a kinase, Lkb1, mediates epigenetic reprogramming that drives tumor cells into a plastic state to enable squamous transformation (40). More recently, squamous differentiation has been reported in human prostate tumors following AR pathway antagonism (35) and in a prostate cancer GEMM driven by Nmyc overexpression and Rb1 loss (41). The observation that prostate, lung, and pancreatic adenocarcinomas with squamous differentiation are enriched for mutations in oncogenes such as MYC and KRAS (KRASG12D) suggests that certain genotypes may be more permissive for squamous-like identity (42–44).
Epithelial-endothelial transition
The concept of epithelial-to-endothelial transition (Figure 1A), more commonly referred to as vascular or vasculogenic mimicry, was first described by Maniotis and colleagues in 1999, who observed a neoteric blood supply in malignant melanoma formed by stem-like cancer cells transdifferentiating into an endothelial-like phenotype and forming vascular-like networks (45). Neo-vascularization via endothelial transdifferentiation has subsequently been reported in various other solid tumors, including breast, prostate, lung, and glioblastoma (46) (Figure 1B). Conceptually, the concept of vascular mimicry is akin to vasculogenesis during embryonic development, providing a conduit for increased plasma and blood cells to meet the metabolic demands necessitated by rapid growth. Not surprisingly, therefore, cancer cells undergoing vascular mimicry have been reported to hijack embryonic vasculogenesis programs including activation of Nodal signaling (47) and upregulation of VE-cadherin (48), which functions as an “adhesion zipper” between the endothelial-like cancer cells. Interestingly, the development of Kaposi’s sarcoma is characterized by transdifferentiation of endothelial cells into epithelial tumor cells (49), hinting that the epithelial-endothelial transdifferentiation axis is bidirectional.
Melanomas that are resistant to angiogenesis inhibitors display greater levels of vascular mimicry (46). This could be the result of the inability of these inhibitors to effectively suppress cancer cell-derived neo-vascular networks, a hypothesis supported by in vitro studies using human melanoma cell lines (50). Although definitive evidence of treatment-induced vascular mimicry has yet to be observed, it is tempting to speculate that the poor clinical efficacy of angiogenesis inhibitors could be due, at least in part, to the phenomenon of epithelial-to-endothelial transdifferentiation.
Hybrid cell states and cancer stem cells
Although there are clear distinctions between epithelial-mesenchymal, epithelial-neuroendocrine, epithelial-squamous and epithelial-endothelial plasticity, it must be noted that there is also substantial overlap between the features and mediators of these phenomena (Figure 1 and Box 1). An important concept is that these various transitions are generally associated with hybrid metastable states (Figure 1A) characterised by elevated plasticity and stem-like features. Notably, breakdown of lineage commitment and transition into a hyper-plastic state is not unique to cancer but is also observed during wound healing when stress-responsive enhancers become activated and override homeostatic enhancers that govern lineage specificity (51).
Box 1. Shared transcriptional and epigenetic features of lineage plasticity and implications for cancer treatment.
The diversity and breadth of oncogenic lineage plasticity – whereby tumors exhibit diverse manifestations of plasticity in response to the unique set of external pressures to which they are exposed, with the consequent possibility being almost limitless unique cellular states – creates a formidable barrier to fully understanding and effectively targeting this phenomenon to improve patient outcomes. Although the explosion of research in this field in the past 2 decades or so has exacerbated the complexity of this issue, it has also revealed convergent transcriptional and epigenetic mechanisms and principles that can guide the development of tools to monitor and target cancer cell lineage plasticity.
The Snail, Twist, and Zeb families of EMT-TFs represent one such convergent mechanism. As outlined in Section 3, these factors mediate a diverse array of cell state transitions beyond EMT. The mechanisms underlying the “pan-plasticity” activity of EMT-TFs remain to be fully elucidated, although one possibility is that these factors directly promote distinct oncogenic transdifferentiation events in a context-dependent manner: for example, Twist can repress the E-cadherin (CDH1) gene to drive EMT and can also activate vascular endothelial (VE)-cadherin (CDH5) expression during epithelial-endothelial transition (see Section 3). Another (non-mutually exclusive) mechanism is that EMT-TFs promote dedifferentiation and rely on other transcriptional and epigenetic regulators to endow cells with new lineage states, a concept supported by well-described roles for EMT-TFs in enhancing dedifferentiation and stemness in cancer, in some cases independent of their EMT-inducing capacity (see Section 3). Whatever the mechanism, “pan-plasticity” EMT-TF activity would enhance the adaptability of cancer cells and thereby maintain fitness in the face of environmental pressures such as therapies, nutrient deprivation and immune attack. It follows that suppressing the activity of these factors may be an effective approach to block multiple aggressive features of tumors; initially thought to be “undruggable”, it is now recognised that oncogenic TFs are bona fide targets (see Section 9).
Another molecular principle underpinning cancer cell plasticity is the ability to rapidly modulate gene expression programs by reprogramming chromatin states, with one prominent example being the exploitation of bivalent chromatin (see Section 3). Elevated activity of enzymes and other factors that regulate epigenetic topographies in cancer cells enables activation of pro-plasticity genes and repression of genes responsible for maintaining the original differentiated state, a phenomenon that is a prominent feature of malignancy-associated cell state transitions. The druggability of epigenetic enzymes, as well as the availability of molecular tools to monitor their activity, has facilitated the development of therapeutic strategies to counter plasticity-associated chromatin reprogramming (see Sections 8–9).
Lineage plasticity is also frequently associated with acquisition of immune-evasive phenotypes. In multiple cancer types and across distinct manifestations of cell state transitions, plasticity-promoting TFs and epigenetic enzymes have been found to suppress immune-activating genes and induce immune-inhibitory genes (see Section 7). This may have important implications for application of immunotherapies. More specifically, therapies that suppress lineage plasticity could induce anti-tumor immune responses and be fast-tracked into immunotherapy combination trials; additionally, markers of lineage switching and stemness could be used to identify patients unlikely to respond to immunotherapies and thereby guide treatment decision-making.
Another important concept when considering lineage plasticity is that a population of cells within a heterogenous tumor can be endowed with heightened stemness, plasticity and drug resistance, which is frequently referred to as the “cancer stem cell” or “therapy-tolerant persister” paradigm. When modelling acute responses to anti-cancer therapies across drug-sensitive human cancer cell lines, Settleman and colleagues made the initial discovery that a small population of “cancer stem-like cells (CSCs)” maintain viability (52) and serve as a reservoir from which heterogeneous drug-resistance mechanisms can evolve to enable tumor re-establishment. Notably, the persister state was found to be transiently acquired and relinquished by individual cells within a population (52), suggesting that tumor cells are constantly in flux along a continuum of “stem-ness.” The intimate relationship between stem-like states and lineage plasticity is an important principle when considering transcriptional and epigenetic mechanisms governing cancer cell state transitions.
3. Transcriptional and epigenetic regulators of lineage plasticity
Transcription factor control of lineage plasticity
The coordinated actions of lineage-specific transcription factors (TFs) are essential for normal development. In contrast, this tightly coordinated process goes awry in cancer, rerouting transcriptional pathways to enhance cell plasticity and drive disease progression. In this context, a major sequela of the malignant state is activation of aberrant lineage-specific TF activities. For example, therapy-mediated epithelial-neuroendocrine transdifferentiation in prostate cancer involves activation of BRN2, ASCL1, FOXA2 and N-Myc, TFs that mediate neural differentiation programs in normal development (53–56), whereas squamous transdifferentiation of pancreatic and lung adenocarcinomas is mediated by p63, a master transcriptional regulator of the normal squamous lineage (57,58).
Perhaps the most prominent example of lineage-specific TFs that are aberrantly activated in cancer to drive plasticity are the core EMT-TFs of the Snail, Twist and Zeb families (59). These TFs enable epithelial cancer cells to acquire features associated with a mesenchymal lineage by directly repressing epithelial genes (e.g. CDH1, encoding E-cadherin) and activating mesenchymal genes (e.g. CDH2, encoding N-cadherin, and VIM, encoding Vimentin). However, the terminology “EMT-TF” is perhaps unfortunate since this class of factors can also mediate epithelial-neuroendocrine (60–62), epithelial-endothelial (63) and endothelial-mesenchymal (64) cell state transitions as well as eliciting dedifferentiation, in some cases independent of their EMT-inducing capacity (65–67). Thus, EMT-TFs could be regarded as “pan-plasticity” factors, at least in some cancer contexts (Box 1).
Acquisition of new lineage-specific TF activity in cancer cells often occurs concomitantly with activation of one or more of the core embryonic stem cell pluripotency TFs, OCT4, SOX2, KLF4 and NANOG. These factors not only promote cancer cell dedifferentiation to a more stem-like state (68–71), as expected, but can also promote transdifferentiation, as illustrated by SOX2-mediated epithelial-neuroendocrine plasticity and EMT (70,72,73) and KLF4-mediated acinar-to-ductal phenotype switching in pancreatic cancer (74). Thus, pluripotency TFs can enable complex transdifferentiation/dedifferentiation coalescence that engenders highly plastic cell states.
Loss of developmental TFs that specify the original tumor lineage is another feature of cancer cell lineage plasticity, with some key examples being: loss of PDX1 in pancreatic cancer, which enables tumors to suppress the epithelial state and gain mesenchymal/squamous (pancreatic) features (75); loss of the luminal epithelial lineage factor AR in prostate cancer, which can lead to neuroendocrine transdifferentiation and/or dedifferentiation to a more stem-like state (35,76); loss of GATA3 in breast cancer, resulting in acquisition of basal/mesenchymal phenotypes (77); loss of the lung epithelial lineage factor NKX2–1 in lung cancer, which is associated with switching to squamous cell states and various gut lineages (78); loss of HOXA5 and SMAD4 in colon cancer, which enables dedifferentiation to a progenitor-like state (79,80); and loss of the master regulator of melanocyte differentiation, MITF, in melanoma, leading to reactivation of a neural crest progenitor state (81). A number of noteworthy elements related to lineage plasticity arising from reduced TF expression/activity are apparent. First, in some cases the TFs that are lost were the initial primary drivers of tumorigenesis and growth – such as AR in prostate cancer – highlighting the extreme adaptability of tumor progression. Second, targeted therapies are a significant selection pressure leading lead to TF loss and/or inactivation. Treatment of a GEMM model of melanoma with the BRAF inhibitor vemurafenib causes down-regulation of SOX10, a neural crest lineage-specifying transcription factor essential for melanocyte development, which provokes tumor dedifferentiation and therapy resistance (82). Other prominent examples are loss of the sex steroid hormone receptors AR and ERα in prostate and breast cancer, respectively, in response to hormonal therapies that directly inhibit their activity (35,83). Interestingly, the development of more potent hormonal therapies for prostate cancer has led to a substantial increase in the frequency of tumors exhibiting highly plastic phenotypes (84), a cautionary tale when applying therapies that inhibit lineage-specific TFs. Finally, paralleling other aspects of lineage plasticity (see below), epigenetic mechanisms frequently underpin loss of lineage-specific TFs. For example, hormone therapy-induced lineage switching in breast and prostate cancer has been linked to epigenetic silencing of the ESR1 and AR genes (85–87). Such epigenetic mechanisms could enable oscillating loss/gain of key epithelial TFs, in turn driving complex rounds of cell state switching, in response to microenvironmental cues and therapy.
The above examples imply scenarios whereby concomitant gain of TFs that enhance cell plasticity and loss of TFs that maintain differentiated epithelial states converge to yield unequivocal binary lineage switching. However, as mentioned above, such binary phenomena are likely to be rare and associated with reduced cellular flexibility. Instead, selection pressures enable the acquisition of high plasticity hybrid states; interplay between, and opposing actions of, TFs are important mediators of such states. In the hybrid EMT setting, TFs that counteract EMT-TFs to prevent full transition have been referred to as phenotypic stability factors (59); one such factor is GRHL2, which supports the proliferation of epithelial cancer cell growth (88,89). Similarly, hybrid states are also observed in cell line models of prostate cancer and patient tumors undergoing epithelial-neuroendocrine transdifferentiation, whereby TFs mediating the luminal epithelial state (i.e. AR, FOXA1) can be co-expressed with TFs driving neuronal and stemness transcriptional circuitry (i.e. N-Myc, SOX2, ASCL1, BRN2, etc) (34,35).
In addition to gain and loss of TF function, modulation of the activity of cancer regulatory TFs to support cancer cell lineage plasticity is frequently observed, a process referred to as TF “reprogramming” or “rewiring”. One notable example is altered function of two key prostate cancer-associated TFs, AR and FOXA1, in response to AR-targeted therapies. In the normal prostate and in early-stage prostate cancer, FOXA1 and AR play complementary and critical roles in maintaining the luminal epithelial lineage (90,91). However, both chronic and acute AR-targeted therapies can cause redistribution of AR/FOXA1 cistromes from epithelial-associated to neuroendocrine-associated regulatory elements in cell lines, PDXs and patient tumors (34,92,93), illustrating a paradigm whereby TF reprogramming is a driver of cell state switching. The mechanisms underlying TF reprogramming in cancer cell lineage plasticity are complex and multifactorial but include altered expression/activity of other TFs and chromatin-modifying factors, often in a combinatorial manner. Indeed, reprogramming of FOXA1 and AR activities in prostate cancer can be mediated via interplay with pluripotency TFs (OCT4 and NANOG) and the histone methyltransferase EZH2, leading to altered cistromes, interacting protein partners and gene targets (34,68,69).
Epigenetic regulators of lineage plasticity
A critical epigenetic mechanism underlying cancer cell lineage plasticity is the addition and removal of post-translational modifications of histones, the so-called “histone code”. In particular, a histone code state characterized by the simultaneous enrichment of transcriptionally active (e.g. histone H3 lysine 4 monomethylation [H3K4me1] or trimethylation [H3K4me3]) and transcriptionally repressive (e.g. H3K27me3) post-translational modifications, termed bivalent chromatin, plays a major role in determining expression of plasticity-associated genes. Bivalently-marked genes tend to exhibit a low level of transcription that can be activated by loss of H3K27me3 or repressed by loss of H3K4me3, a status referred to as transcriptionally “poised” (94). In normal development, chromatin bivalency is enriched at elements regulating expression of lineage-specific TFs and other developmental factors, enabling dynamic and rapid control of cell state (95,96). A general feature of lineage plasticity is hijacked control of the bivalent chromatin state, resulting in activation of genes that promote plasticity and repression of genes that block plasticity, which collectively enable dedifferentiation and transdifferentiation processes (Box 1). For example, in prostate cancer, bivalent chromatin controls genes involved in epithelial and neuronal identity; stimuli that enhance cell plasticity (i.e. androgen withdrawal or over-expression of N-Myc) cause an increased H3K27me3:H3K4me3 ratio at epithelial genes and a decreased ratio at neuronal genes, leading to acquisition of neuronal features (97). Moreover, genes encoding EMT-TFs and other plasticity-associated TFs such as N-Myc and SOX9 are dynamically regulated by bivalent chromatin during metastatic progression of melanoma, enabling dynamic control of mesenchymal cell states (98).
Enzymes responsible for controlling histone post-translational modifications, and in particular the bivalent chromatin state, are key regulators of lineage plasticity. EZH2 is the enzymatic subunit of the Polycomb-repressive complex 2 (PRC2), which catalyzes the deposition of H3K27me3. Inhibition of EZH2 activity can block and, in some cases, reverse epithelial-neuroendocrine transdifferentiation in human and mouse models of prostate cancer and SCLC (34,56,72,99–102). By regulating deposition of H3K27me3, EZH2 also promotes EMT in pancreatic (103) and prostate cancer (104), the latter via direct repression of the CDH1 promoter. Another key mediator of chromatin bivalency and the histone code more generally is LSD1, an enzyme that demethylates H3K4me1, H3K4me2, H3K9me1 and H3K9me2. In embryonic stem cells, LSD1 regulates pluripotency and stemness by controlling chromatin bivalency at key developmental genes, including lineage-specific transcription factors such as FOXA2 (105). Given its overarching role in pluripotency, it is not surprising that dysregulation of LSD1 in cancer cells has been linked to EMT (106,107), epithelial-neuroendocrine plasticity (108,109) and maintenance of cancer stem cell states (110). EZH2 and LSD1 are increasingly recognised as therapeutic targets, a concept explored in more detail below.
Another key epigenetic factor regulating cancer cell lineage plasticity is DNA methylation, a modification that is deposited by DNA methyltransferases (DNMT1, DNMT3A and DNMT3B) and removed by ten-eleven translocation proteins (TET1, TET2 and TET3). DNMT1, in particular, has been identified as a major mediator of plasticity. In prostate cancer, upregulation of DNMT1 in NEPC tumors is associated with dramatically altered DNA methylation patterns; neuronal, cell-cell adhesion, developmental, EMT and stem cell transcriptional programs are enriched within genes exhibiting altered methylation, providing evidence that DNMT1 is a direct mediator of plasticity in this disease context (21,111). In support of this theory, DNMT1 inhibition dampens the neuroendocrine phenotype in cell line, organoid and xenograft models of prostate cancer (112). Paralleling these observations, SCLC is characterised by elevated DNMT expression and exhibits similar DNA methylation profiles to NEPC, including hypomethylation of neuronal TFs ASCL1, HES6 and ONECUT2 (111). Elevated DNMT1 activity and specific DNA methylation profiles have also been linked to EMT and stemness in breast cancer (113,114) and pancreatic cancer (115). Although the above examples reveal a robust link between elevated DNMT1 and lineage plasticity, this principle does not appear to apply to other DNA methylation-modifying enzymes. As examples, DNMT3A loss of function mutations can drive stemness and malignant progression of leukemia cells (116,117), while TET1 can enhance self-renewal and expansion of CSCs in triple-negative breast cancer cell lines (118). The precise mechanisms underlying these complex relationships between DNA methylation and cell plasticity states remain to be elucidated.
4. Insights into cancer cell lineage plasticity from single cell analyses
The unprecedented resolution afforded by single cell profiling has deepened our understanding of the origins and transitions between transcriptional states as well as the evolutionary paths that tumors follow. Indeed, such profiling has demonstrated that cells can display both developmental and unique evolutionary trajectories, which often involve a transient increase in cellular plasticity followed by adoption of increasingly metastatic states (119).
Pseudo-temporal approaches to evaluate single cell data have reinforced the notion that perturbation of tumor suppressors or therapeutic pressure accelerates plasticity-mediated progression by creating novel cellular trajectories as well as illustrating the complexity and heterogeneity of cell state changes. For example, single cell transcriptomic/epigenomic analysis of tumor progression in a GEMM expressing an oncogenic Kras-G12D mutant in combination with TP53 deletion, which recapitulates human lung adenocarcinoma progression, revealed a loss of fidelity of the lung lineage coincident with the acquisition of transcriptional features associated with lung progenitors, embryonic ectoderm and EMT (36,120). In contrast to embryogenesis, tumor progression in this model was characterized by persistence of earlier cell states coincident with acquisition of new cellular states, giving rise to intra-tumoral diversity. A similar phenomenon has been described in SCLC at the single cell level with multiple, concurrent plasticity-related resistance mechanisms and cell states (e.g. EMT, neuroendocrine-like phenotypes) co-existing within the same tumor (121,122). Likewise, in prostate cancer, single cell RNA and chromatin accessibility profiling of patient tumors, organoids and cell line models has demonstrated that AR-targeted hormonal therapies cause the onset of multi-lineage phenotypes comprised of epithelial, neuroendocrine, mesenchymal and tuft-like cell states, amongst others (41,123,124).
Single cell profiling has also provided new insight into the mediators of cancer cell lineage plasticity, uncovering evolutionary bottlenecks and vulnerabilities that could pave the way for new anti-plasticity therapies. Using single cell chromatin accessibility profiling, heterogenous self-renewing populations of stem-like cells were detected in human glioblastoma multiforme (GBM) tumors (125). Further work revealed that YAP/TAZ signalling is responsible for instilling stem-like properties; inhibition of this pathway irreversibly locked differentiated GBM cells in a non-tumorigenic state, preventing plasticity and regeneration of the stem-like cell compartment (126). Likewise, intratumoral subtype switching in SCLC has been reported as a mechanism of acquired resistance to platinum-based chemotherapy (127). Longitudinal single-cell transcriptome analysis of SCLC tumors uncovered a role for MYC-mediated Notch signalling activation in promoting a temporal shift from ASCL1-positive SCLC to a non-neuroendocrine YAP1-positive state, a subtype switch that could be blocked with a Notch inhibitor (128). Finally, single-cell studies in prostate and breast cancer have implicated JAK/STAT and FGFR signalling in lineage plasticity from luminal epithelial to neuroendocrine and mesenchymal states, revealing pharmacological inhibition of JAK/STAT as a viable therapeutic strategy (123,124,129).
Despite the increasing catalogue of single cell data, findings in relation to cellular trajectories inferred by pseudo-temporal approaches should be taken with caution as they make the assumption that transcriptional similarity denotes a developmental relationship. The next wave of single cell technologies that enable lineage barcoding (for review, see (130)) will provide more robust capture of plasticity, cell state transitions and clonal mosaicism. Indeed, application of a lineage recorder system that exploits CRISPR-Cas9-induced insertions and deletions (“indels”) to a mouse xenograft model of metastatic pancreatic cancer reinforced the notion – originally proposed from bulk transcriptomic data – that cells occupy a continuum of EMT states and that “late-hybrid” cells have the highest metastatic potential (131). More recently, an equivalent CRISPR-Cas9-based lineage tracing system was incorporated into the Kras-G12D mutant/TP53 deletion lung cancer GEMM, yielding an autochthonous mouse model that provided important insights into tumor evolution, plasticity and subclonal origins (119).
5. Interplay between cancer cell lineage plasticity and metabolism
The intimate relationship between metabolism and tumor cell plasticity is evident by the distinct metabolic profile between stem-like and differentiated cell populations within a heterogeneous tumor. In general, stem-like cells exhibit suppression of oxidative phosphorylation (OXPHOS), resulting in redox niches with low reactive oxygen species (ROS) levels. Multiple epigenetic and transcriptional drivers of lineage plasticity have been demonstrated to control this metabolic reprogramming (Figure 2). For example, in mouse models of liver cancer, Nanog directly represses OXPHOS genes and concomitantly activates fatty acid oxidation to support cancer cell self-renewal and drug resistance (132). Similarly, JAK/STAT pathway activation (described above as a feature of lineage plasticity) promotes fatty acid β-oxidation activity to sustain breast cancer stem-like cells (133). A switch from OXPHOS to glycolysis is also a common feature of lineage plasticity in tumors. The Snail-G9a-DNMT1 complex mediates promoter methylation and repression of fructose-1,6-biphosphatase (FBP1) in breast cancer cell lines, leading to a switch from mitochondrial OXPHOS to glycolysis that supports EMT, stemness and tumorigenicity (134). Acquisition of enhanced glycolysis is also associated with epithelial-neuroendocrine plasticity in prostate cancer, mediated at least in part by aberrant epigenetic/transcriptional activities of the histone lysine demethylase KDM8 and HIF1α (135).
Figure 2. Interplay between transcriptional and epigenetic mediators of lineage plasticity and cancer cell metabolism.
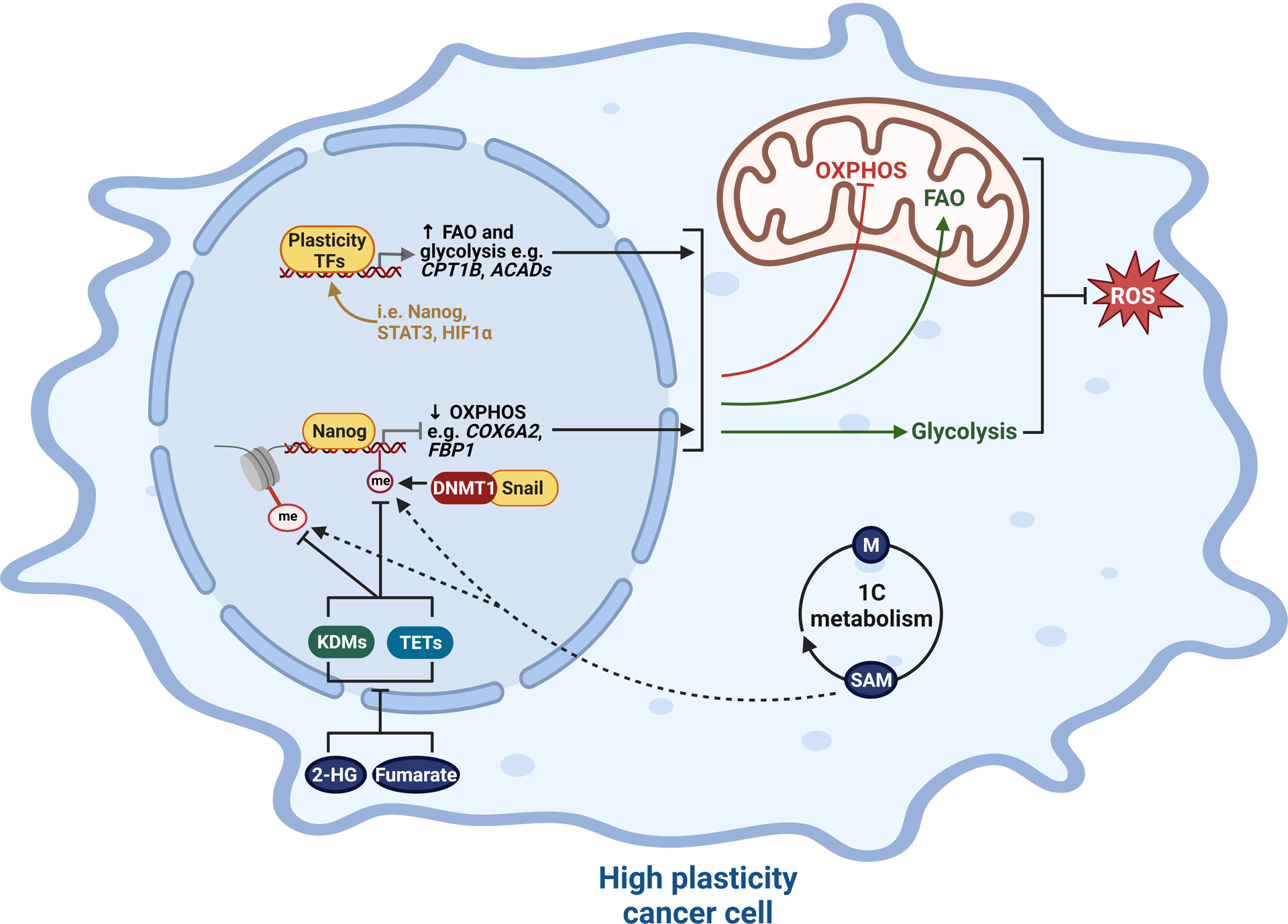
Plasticity-enhancing transcription factors (TFs) and epigenetic enzymes regulate a gene expression program that favors fatty acid oxidation (FAO) and glycolysis over oxidative phosphorylation (OSPHOS), one consequence of which is suppression of ROS production. Concomitantly, oncometabolites impinge on multiple transcriptional/epigenetic mechanisms, prominent examples being dysregulation of histone and DNA methylation by elevated levels of S-Adenosyl methionine (SAM), 2-Hydroxyglutarate (2-HG) and fumarate. 1C metabolism = one carbon metabolism; ACADs = acyl-CoA dehydrogenases; KDMs, lysine demethylases; me, methyl groups; TETs, ten-eleven translocation methylcytosine dioxygenases. Created with BioRender.com.
The above examples describe how epigenetic enzymes and TFs mediate metabolic reprogramming concomitant with increased cancer cell plasticity. However, the interplay between these processes is very much a two-way street, perhaps best epitomised by the fact that the lineage-shaping activities of chromatin modifying enzymes are dependent on metabolic pathways and oncometabolites that provide the “fuel” for epigenetic modifications, particularly DNA and histone methylation (Figure 2). For example, one-carbon metabolism is central to lineage plasticity due to its role in the synthesis of substrates for methylation reactions. The methionine cycle controls the conversion of methionine into S-Adenosyl methionine (SAM), the universal methyl donor for histone and DNA methyltransferase enzymes, such as EZH2. Accordingly, regulation of SAM metabolism dictates a cell’s ability to impart histone and DNA modifications to support lineage reprogramming (136,137). Indeed, methionine has been reported to be required for maintenance of human embryonic stem cells (138). Similarly, in malignancy, stem-like cells rely on methionine and SAM biosynthesis for their self-renewal and tumor-forming capacity (139,140). In triple-negative breast cancer, methionine deprivation is associated with H3K4me3 demethylation and suppression of the pro-plasticity/developmental transcription factor Sox9, effects that were partially rescued by SAM supplementation (140). Increased SAM levels have also been reported during prostate cancer epithelial-neuroendocrine transdifferentiation (112). Beyond SAM, other oncometabolites that support lineage plasticity via dysregulation of DNA and histone methylation include fumarate and 2-hydroxyglutarate, both of which can inhibit TET DNA demethylases and histone demethylases: by suppressing TET-mediated DNA demethylation of miR-200 promoters, fumarate was found to stabilise EMT-related transcription factors (141), whereas 2-hydroxyglutarate was recently demonstrated to mediate an altered chromatin landscape that drives breast cancer cell state transitions and heterogeneity (142).
6. Interplay between the tumor microenvironment and lineage plasticity
The microenvironment in which a tumor grows can have a profound impact on cancer cell lineage plasticity. These external cues have been referred to as extrinsic regulators of plasticity (143), as opposed to cell-intrinsic factors that are predominantly described above. Although in many cases the precise mechanisms underlying the cross-talk between the microenvironment and cancer cell plasticity remain to be uncovered, an emerging body of work is elucidating epigenetic and transcriptional mechanisms underlying this phenomenon.
Growth factor secretion by cancer-associated fibroblasts (CAFs) can have a profound influence on the transcriptional and epigenetic regulators of cancer cell plasticity. For example, Lau and colleagues demonstrated that hepatocyte growth factor (HGF) release by CAFs induced the AP-1 transcription factor FOSL1 (FRA1) in liver cancer cells; subsequent transcriptional changes mediated by FRA1, including induction of the NOTCH signalling factor HEY1, resulted in enhanced plasticity, stemness and cancer-initiating capacity (144). Acidic fibroblast growth factor (FGF1) derived from pancreatic cancer CAFs can signal through AKT and GSK-3β to increase Myc half-life in tumor cells, which increases Myc promoter occupancy and transcriptional activity (145). Another study found that NSCLC CAFs release various growth factors, most notably insulin-like growth factor 2, to induce the stem TF Nanog in lung cancer, which drives transcriptional programs associated with stemness and plasticity (146). Collectively, this body of evidence supports the concept that paracrine signals play a major role in establishing transcriptional and epigenetic programs required for cancer cell plasticity. Importantly, the composition of such signals and subsequent impact on cancer cell state will depend on the specific microenvironmental niche; lung cancer brain metastases exhibited epithelial-neuroendocrine lineage switching but this was reversed when the tumor cells were separated from brain stroma (147).
Rapid tumor growth frequently outstrips the process of vascularization, resulting in a hypoxic microenvironment. An emerging knowledge base has linked hypoxia to epigenetic and transcriptional reprogramming of plasticity in cancer cells. Arguably the most well-studied mechanism underlying this relationship is induction of the transcription factor HIF-1α, which occurs in response to hypoxia and can directly activate genes encoding mediators of EMT, such as Twist, ZEB1, Snail and lysyl oxidase and TGF-β (148–151). HIF-1α can also regulate other forms of cancer cell lineage plasticity: in hypoxic GEMM prostate tumors, HIF-1α protein is stabilised and can interact with the transcription factor FOXA2 to drive expression of a gene expression program that mediates epithelial-neuroendocrine plasticity and metastasis (152). Beyond cell-autonomous mechanisms, hypoxia can also influence stroma to influence plasticity of cancer cells. For example, hypoxia induces expression of VEGF by stromal cells, which can activate stem-related transcriptional programs in breast and lung cancer cell lines via Myc and SOX2 (153).
Inflammation is another important microenvironmental factor that is intimately connected to lineage plasticity. Early lineage tracing studies were key in establishing this relationship. For example, a murine model of bacteria-induced prostatic inflammation resulted in basal-luminal transdifferentiation that was linked to carcinogenesis (154). Although the epigenetic and transcriptional bases that link inflammation and cancer cell plasticity remain to be fully established, one emerging theme is activation of transcriptional regulators such as NF-κB, JAK-STAT3 and Notch by pro-inflammatory chemokines and cytokines (e.g., TNFα, IL-1β, IL-6, IL-8 and CXLC8). Indeed, IL-6 can elicit stemness by activating JAK-STAT3 and Notch in breast cancer models in vitro and in vivo (155,156). More recent studies have also highlighted the essential role of JAK-STAT inflammatory signalling in prostate cancer lineage plasticity, whereby this pathway is essential for establishing complex admixed cellular phenotypes (including neuroendocrine-like and stem-like populations) and resistance to AR-targeted therapies (123,124). Soluble pro-inflammatory factors are frequently produced at high levels in cancer cells but can also be derived from other tumor microenvironmental components, most notably myeloid cells (156,157) and CAFs (158).
Mechanical features of the TME can also regulate cancer cell transcriptional and epigenetic plasticity. Integrins in cancer cells sense TME stiffness and mediate activation of key plasticity TFs, such as Twist and YAP/TAZ, leading to EMT transcriptional programs (159,160). YAP/TAZ-mediated mechanotransduction was also reported to enhance stem-like traits in breast and pancreatic cancer cell lines via modulating autophagy (161).
Two other aspects of tumor:microenvironment interplay in relation to cancer cell plasticity are worth noting. First, although our discussion has had a unidirectional focus – i.e. microenvironmental cues that influence transcriptional and epigenetic regulators of plasticity – this is of course a bidirectional relationship. An elegant example of such bidirectionality was provided by Somerville and colleagues, who demonstrated that squamous transdifferentiation of human and mouse pancreatic cancer cells results in conversion of pancreatic stellate cells into inflammatory CAFs; mechanistically, this was mediated by p63-mediated transcription of genes encoding pro-inflammatory secreted factors in the cancer cells (37). Thus, p63 regulates a transcriptional program that simultaneously drives squamous transdifferentiation (57,58) and fibroblast reprogramming (37). Second, experimental conditions can dramatically influence cancer cell plasticity, which has significant implications for studying the influence of the microenvironment on this process. Indeed, cell states of pancreatic adenocarcinoma cell lines and organoids were highly sensitive to culture medium and artificial states altered anti-cancer drug responses (162). Additionally, non-malignant and neoplastic mammary epithelial cells can gain stem-like characteristics via epigenetic mechanisms when grown in 2-dimensional culture but this phenomenon is mitigated if cells are cultured within 3-dimensional matrices (163). Thus, approaches to study the impact of the tumor microenvironment on transcriptional and epigenetic mechanisms underlying cancer cell plasticity should use models and optimise culture conditions that faithfully recapitulate patient-relevant cell states (162,164).
7. Cancer cell plasticity as a driver of immune escape
The ability of cancer cells to evade attack and elimination by immune cells is a hallmark of cancer and emerging evidence points to a critical role for non-genetic cell tumor plasticity in promoting non-immunogenic cell states (Figure 3 and Box 1). This concept was first established via the identification of a relationship between epithelial-mesenchymal plasticity and an immunosuppressive phenotype (165,166). Surveying clinical data from lung cancer, a strong correlation between “tumor EMT score” (based on expression of 76 canonical EMT genes) and expression of the immune checkpoint PD-L1 was uncovered (165). This finding was validated in a subsequent large-scale analysis of eleven cancer types, which demonstrated that tumors with a high EMT score exhibited elevated expression of immune checkpoint proteins, including PD-1, PD-L1, PD-L2, and CTLA-4 (166). Subsequent work has begun to provide mechanistic insight into the link between EMT and immune evasion. For example, an elegant set of experiments from the Weinberg lab using a SNAIL-driven fluorescent reporter in the MMTV-PyMT breast cancer mouse model demonstrated the relevance of this EMT-TF in directly mediating resistance to immune checkpoint inhibitors (ICIs) (167,168). While SNAILlo epithelial cells were immunogenic, SNAILhi mesenchymal-like cells promoted an immunosuppressive tumor microenvironment characterized by cytotoxic T lymphocyte exclusion, M2 pro-tumor macrophage polarization, and decreased response to anti-CTLA-4 therapy. Interestingly, in mixed SNAILlo/SNAILhi tumors, only a small proportion (<10%) of mesenchymal-like cells were required to shield the surrounding epithelial cells from immune attack (167), likely due to secretion of immunosuppressive factors, such as TGFβ, by mesenchymal-like cells (169). SNAIL directly binds to the promoters of key immunosuppressive paracrine factors, including CD73 (Nt5e), macrophage colony-stimulating factor (Csf1), and Spp1, identifying a putative mechanism by which it inhibits anti-tumor immune activity (168). A similar immunosuppressive mechanism was identified in ovarian cancer whereby SNAIL directly binds to and upregulates the expression of genes encoding the CXCL1 and CXCL2 chemokines, leading to infiltration of myeloid-derived suppressor cells (MDSCs) (170). Direct transcriptional regulation by ZEB1 can activate expression of immunosuppressive factors (PD-L1 and CD47) (171) and repress expression of T cell-attracting chemokines, such as CXCL10 (172). Thus, direct transcriptional control of immunoregulatory factors by EMT-TFs could be a key contributor to tumor immune evasion and response to immunotherapy. Interestingly, positive feedback loops between tumor cells and immune cell populations can enhance this phenomenon: for example, MDSCs can reinforce a EMT/stemness phenotype and facilitate metastasis in syngeneic mouse models of breast cancer (173).
Figure 3. Cancer cell plasticity as a driver of immune escape.
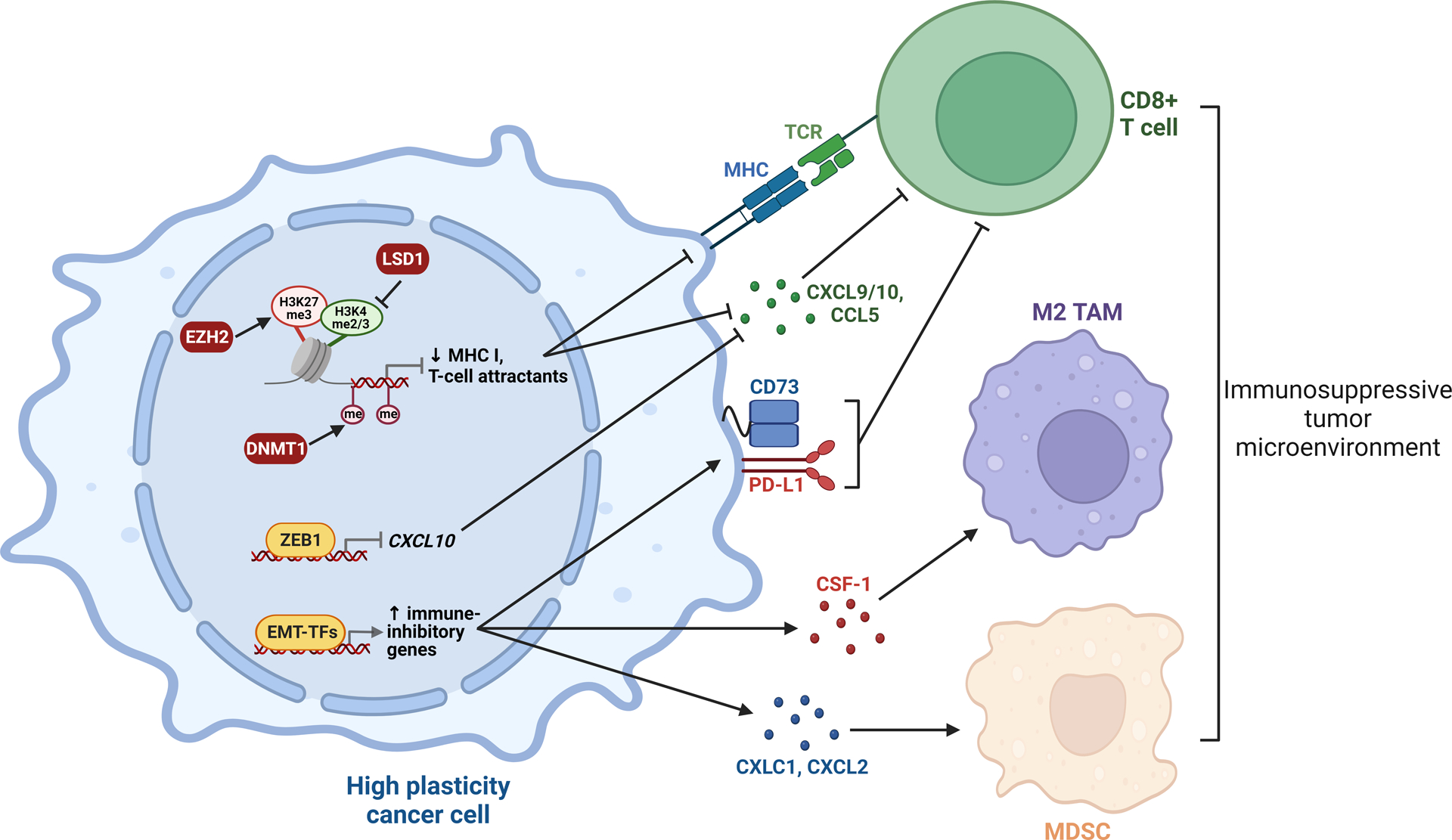
Plasticity-enhancing transcription factors and epigenetic enzymes downregulate immune-activating genes and upregulate immune-inhibitory genes, resulting in escape or exclusion of tumor-killing CD8+ T cells and recruitment of M2 tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs). Thus, the high plasticity state is associated with cancer cell evasion of the immune system. EMT-TFs, EMT transcription factors; MHC, major histocompatibility complex; me, methyl groups; TCR, T cell receptor. Created with BioRender.com.
Epigenetic programs associated with high plasticity tumor states also play a critical role in regulating cancer cell immunogenicity. For example, bivalent chromatin or DNA methylation at the promoters of key immune regulators, including major histocompatibility complex (MHC) class I genes, the type III interferon receptor IFNLR1 and T cell-attracting chemokines (CCL5, CXCL9 and CXCL10), are associated with transcriptional repression and evasion of T cell-mediated immunity in leukaemia, SCLC, various types of squamous cell carcinoma and tripe-negative breast cancer (99,174–176). Collectively, these studies demonstrate that epigenetic reprogramming associated with lineage plasticity can mediate immune escape and reveal immunosensitization therapies for cancer, as discussed below.
8. Detection of cancer cell lineage plasticity for patient benefit
Clinically, lineage plasticity in cancer is not always recognizable, particularly since changes may be dynamic and occur at the cellular level. The most obvious recognition of lineage plasticity is when a primary tumor or metastatic biopsy shows mixed histology or a change in phenotype that differs from the original tumor histology. In the case of histologic transformation (e.g., SCLC or NEPC conversion), clinical genomic assays such as targeted exome sequencing may be performed to exclude a second primary cancer, which confirms similarities with the original cancer’s gene alterations (e.g., EGFR mutation in NSCLC) and sometimes acquisition of additional alterations associated with plasticity or treatment resistance (e.g., loss of RB1 and/or TP53) (21,177). Tumor protein assessment may be performed by immunohistochemistry to confirm loss of target expression (for example, loss of the luminal epithelial lineage factor AR in a subset of prostate tumors undergoing neuroendocrine transdifferentiation (30)) and acquisition of alternative lineage markers (e.g., neuroendocrine markers such as chromogranin, synaptophysin), although mixed luminal/neuroendocrine features are frequently observed. Often, the clinical detection of transdifferentiation is after it has already occurred; early detection strategies are important to identify susceptible individuals and for the development of novel approaches to target the lineage plasticity process.
Serial biopsies are not always feasible to perform in patients, and therefore a comprehensive understanding of the timing of activation of transcriptomic and epigenetic drivers of lineage plasticity in individual patients has been a major challenge. Non-invasive approaches such as liquid biopsies and molecular imaging are in early development and hold promise as tools for early detection and monitoring of lineage plasticity. For example, loss of luminal programs may be suspected in prostate cancer based on features such as progressive disease with low or non-rising serum prostate specific antigen (PSA) or loss of prostate specific membrane antigen, the latter evaluated using PET/CT imaging (178,179). Circulating tumor cell (CTC) analyses across varied cancer types have revealed heterogeneity in epithelial marker expression, pointing to EMT changes that may contribute to metastasis (180). Cell free DNA (cfDNA) also holds significant promise for detecting both genetic and epigenetic changes associated with lineage plasticity. Indeed, cfDNA methylation can distinguish NEPC from prostate adenocarcinoma (181), and other emerging approaches such as cfDNA nucleosome footprinting (182) and 5-hydroxymethylcytosine (5hmC) sequencing (183) may more robustly identify lineage plasticity changes as well as other epigenetic drivers of disease progression. Additional studies applying liquid biopsy platforms to evaluate lineage plasticity, with the aim of improving early detection strategies, disease monitoring and therapy selection, are warranted.
Distinguishing early facilitators, key drivers, and downstream consequences of lineage plasticity has important biomarker and therapeutic implications. For instance, an early facilitator such as RB1 loss may not be sufficient to drive a change in phenotype but might still be a useful biomarker for strategies geared toward the prevention of lineage plasticity (184). Identifying key drivers or markers of lineage plasticity such as TF activity, stem cell features, transcriptome and/or DNA methylation changes, may serve to guide strategies geared towards targeting or reversing the plasticity process. Detecting downstream consequences of lineage plasticity, such as INSM1 or DLL3 expression that only emerges when a neuroendocrine differentiated state occurs, may be leveraged to target the new emergent phenotype (185).
9. Targeting lineage plasticity to treat cancer
Since lineage plasticity most often emerges as a mechanism for cancers to metastasize and/or bypass therapy, patients developing lineage plasticity often have a poor prognosis. There are no current strategies specifically tailored toward targeting the lineage plasticity process. As new therapies are developed in the context of lineage plasticity, the goal of treatment may be to prevent, reverse, or treat the new lineage. For example, based on the hypothesis that RB1 and TP53 alterations occur “before” the onset of SCLC lineage plasticity, an ongoing trial is using these markers to intervene early in patients with NSCLC with SCLC-chemotherapy regimens to potentially prevent cell state conversions and improve outcomes (NCT03567642). Approaches to target EMT-TFs or other lineage determinant TFs has been particularly challenging but, with advances in drug development including protein degraders, are looking increasingly feasible (186,187).
Using epigenetic therapies to reverse cancer cell lineage plasticity and induce ‘re-differentiation’, which in some cases can re-sensitize tumors to standard-of-care therapies, is a compelling concept. Notably, this strategy would only be effective in the context of cancer cell states that are capable of transitioning and not ‘locked in’ to new lineages. One prominent example is prostate cancer, whereby acquired neuroendocrine-like states can be reversed by EZH2 inhibition, which in some cases results in re-expression of the AR and re-sensitization to AR-targeted therapies (34,72). This effect is in part due to the catalytic function of EZH2 (i.e. H3K27 trimethylation) but also via a non-canonical role of EZH2 in driving lineage plasticity by forming transcriptional complexes with N-Myc and AR (34,56). LSD1 inhibitors can reverse neuroendocrine phenotypes in prostate cancer and SCLC and are also emerging as a viable strategy to re-differentiate cancer cells for therapeutic benefit (109,188). Various epigenetic therapies including EZH2 inhibitors (eg., tazemetostat, valemetostat, PF-06821497, CPI- 0209) and LSD1 inhibitors (e.g., CC-90011, seclidemstat, JBI-802) are in clinical development across solid tumors but trials are currently in biomarker-unselected populations. Since these epigenetic therapies target factors with pleiotropic functions, understanding how they modulate lineage plasticity and/or act to ‘re-differentiate’ tumors, and in which context, will be critical. Co-targeting epigenetic pathways along with lineage-specific drivers in patients at high risk of developing lineage plasticity, or sequentially cycling between these distinct therapeutic approaches, are intriguing possibilities but are yet to be tested clinically.
Arguably the most exciting application of plasticity-suppressing epigenetic therapies is to prime tumors for immune rejection. As described above, key epigenetic mediators of lineage plasticity (i.e. EZH2, LSD1 and DNMT1) play a critical role in establishing chromatin states that result in repression of immune-activating genes and activation of immune-inhibitory genes (Figure 3). It is therefore unsurprising that targeting these enzymes potentiates response to ICIs (99,174–176,189,190). Furthermore, epigenetic therapies can also activate immune signaling through the viral defense pathway, a process referred to as “viral mimicry”. More specifically, inhibitors of EZH2, DNMT1 and LSD1 lead to de-repression of endogenous retroviruses, resulting in accumulation of dsRNA and anti-tumor type-I interferon (IFN) responses, a phenomenon that can be harnessed to improve tumor response to ICIs (191–197). Importantly, combining DNMT inhibitors with histone deacetylase inhibitors (HDACis) was found to enhance viral mimicry and immunotherapy responses in non-small cell lung cancer and ovarian cancer models (193,194). Collectively, these studies demonstrate that epigenetic reprogramming associated with lineage plasticity can mediate immune escape and uncover novel therapeutic strategies for immunosensitization.
Could lineage plasticity be harnessed to treat cancer? Historical and emerging evidence suggests that highly plastic malignant cells may be susceptible to enforced transdifferentiation into post-mitotic differentiated cells. This strategy has been applied for decades in acute promyelocytic leukemia by treatment with the retinoid all-trans retinoic acid, which induces terminal differentiation of leukaemic cells into granulocytes via activation of retinoic acid receptor-α (198). The application of differentiation therapy in solid tumors has been long studied with several agents including retinoids, histone deacetylase inhibitors, and PPARγ agonists inducing differentiation in solid tumor models (198). While this approach has been challenging to translate to clinical benefit, an increased understanding of differentiation and lineage plasticity processes has resulted in new opportunities. For example, in preclinical models of breast cancers developing EMT in vivo, forced transdifferentiation of cancer cells into post-mitotic adipocytes via the combination of MEK inhibition and the PPAR-gamma agonist rosiglitazone resulted in cell cycle arrest and repression of tumor growth and metastasis (199). Another breast cancer study demonstrated that retinoids can reverse EMT (i.e. promote MET) and thereby block metastasis of breast cancer, which occurs at least in part by causing a metabolic switch whereby fatty acids are directed towards lipid storage rather than β-oxidation (200). In IDH1-mutant glioma organoids or models of IDH1-mutant biliary cancer that exhibit dedifferentiation due to accumulation of 2-hydroxyglutarate, inhibition of IDH1 or DNA methylation using azacytidine enables tumor control at least in part by promoting differentiation (201,202). Chemotherapy-mediated lineage plasticity in urothelial carcinoma leads to semi-squamatization and targeting of cathepsin H promoted terminal squamous differentiation and cell death via pyroptosis (203).
Since lineage plasticity is often detected late after changes in phenotype manifest clinically, patients are typically treated based on their new histology extrapolated from other cancer types. Nonetheless, they still might harbor features of their ancestral tumor of origin, which should also be considered for disease management. The prognosis of lineage plasticity tends to be poor regardless of tumor type or mechanism of plasticity. An increased understanding of the biology of lineage plasticity and collaborative efforts across cancer types will hopefully lead to better biomarkers and therapies for this growing subgroup of cancer patients.
10. Conclusions and future perspectives
Lineage plasticity is a hallmark of cancer and a critical facilitator of other oncogenic features such as metastasis, therapy resistance, dysregulated metabolism, TME reprogramming and immune evasion. The centrality of phenotypic plasticity in cancer progression necessitates dissection of the molecular mediators and regulators of this phenomenon and development of strategies to effectively target them. Herein, we have focussed on the transcriptional and epigenetic mechanisms that mediate cell state transitions in cancer, in the process highlighting features of these phenomena that may be exploited for patient benefit.
Given the extreme epigenomic/transcriptomic heterogeneity in cell states that can be found in highly plastic tumors, the advent of high-throughput single cell and spatial profiling techniques alongside tools to trace individual cells are anticipated to drive progress in this field. Similarly, tumor models that accurately recapitulate the various manifestations of plasticity and which can be sampled at relevant time-points are critical. However, one major challenge will be to effectively unravel and exploit the cell atlases that will arise from these comprehensive large-scale approaches; high-throughput genetic perturbation strategies will assist in this respect.
Achieving a more complete understanding of the fundamental epigenetic/transcriptional mechanisms underlying cell state transitions in cancer will unlock new therapeutic strategies to target, and tools to monitor, lineage plasticity. In this respect, a key question that remains poorly understood is whether plasticity is a cause or consequence of therapy resistance. We propose that evaluation of rational combinatorial strategies will provide major insights into this question and are likely to be necessary to realise significant benefit for patients. Using epigenetic therapies to sensitize tumors to immunotherapies and/or re-sensitize tumors to drugs that block distinct oncogenic signalling pathways is a compelling possibility that is under clinical evaluation. Of course, context will be key for effective targeting of lineage plasticity for patient benefit. While a goal of precision cancer medicine is to treat patients with the right treatment at the right time, excess plasticity enables cancer cells to effectively deal with environmental stressors, signals and therapy by switching on the right phenotype(s) at the right time. Since epigenetics and TFs play a major role in this extreme adaptability, understanding their context-dependent activities will be crucial to improve precision cancer medicine.
Significance.
Lineage plasticity is a hallmark of cancer and a critical facilitator of other oncogenic features such as metastasis, therapy resistance, dysregulated metabolism and immune evasion. It is essential that the molecular mechanisms of lineage plasticity are elucidated to enable the development of strategies to effectively target this phenomenon. In this review, we describe key transcriptional and epigenetic regulators of cancer cell plasticity, in the process highlighting therapeutic approaches that may be harnessed for patient benefit.
Acknowledgements
This study was supported by: the Cancer Council of South Australia (2012127 to LAS); a joint grant from the Freemasons Centre for Male Health and Wellbeing and Flinders Foundation (LAS); Cancer Council New South Wales (23-04 to LAS); Terry Fox New Frontier Program Project (F22-00589 to AZ); the Canadian Institutes of Health Research (CIHR-169173 to AZ); U.S. Department of Defense (W81XWH-17-1-0653 to HB; W81XWH‐18‐1‐0690 to AZ); the Prostate Cancer Foundation; and the National Institutes of Health (NIH) / National Cancer Institute (NCI) (R37CA241486 to HB; P50-CA211024 to HB). LAS is supported by a Principal Cancer Research Fellowship awarded by Cancer Council’s Beat Cancer project on behalf of its donors, the state Government through the Department of Health, and the Australian Government through the Medical Research Future Fund. The authors apologize that, due to space limitations, many important studies in the field could not be cited.
Author disclosures
AD is employed by and holds stock in Pfizer, Inc. HB has served as consultant/advisory board member for Janssen, Astellas, Astra Zeneca, Merck, Pfizer, Foundation Medicine, Blue Earth Diagnostics, Amgen, Bayer, Oncorus, LOXO, Daicchi Sankyo, Curie Therapeutics, and has received research funding (to institution) from Janssen, AbbVie/Stemcentrx, Eli Lilly, Astellas, Millennium, Bristol Myers Squibb, Circle Pharma, Daicchi Sankyo and Novartis. No disclosures were reported by the other authors.
References
- 1.Tata PR, Rajagopal J. Cellular plasticity: 1712 to the present day. Curr Opin Cell Biol 2016;43:46–54 doi 10.1016/j.ceb.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waddington CH. The strategy of the genes; a discussion of some aspects of theoretical biology. London,: Allen & Unwin; 1957. ix, 262 p. p. [Google Scholar]
- 3.Blanpain C, Fuchs E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014;344(6189):1242281 doi 10.1126/science.1242281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jessen KR, Mirsky R, Arthur-Farraj P. The Role of Cell Plasticity in Tissue Repair: Adaptive Cellular Reprogramming. Dev Cell 2015;34(6):613–20 doi 10.1016/j.devcel.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Hanahan D Hallmarks of Cancer: New Dimensions. Cancer Discov 2022;12(1):31–46 doi 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 6.Zhang LT, Goodrich DW. RB1, Cancer Lineage Plasticity and Therapeutic Resistance. Annual Review of Cancer Biology 2022;6:201–21 doi 10.1146/annurev-cancerbio-070120-092840. [DOI] [Google Scholar]
- 7.Trelstad RL, Hay ED, Revel JD. Cell contact during early morphogenesis in the chick embryo. Dev Biol 1967;16(1):78–106 doi 10.1016/0012-1606(67)90018-8. [DOI] [PubMed] [Google Scholar]
- 8.Trelstad RL, Revel JP, Hay ED. Tight junctions between cells in the early chick embryo as visualized with the electron microscopy. J Cell Biol 1966;31(1):C6–10 doi 10.1083/jcb.31.1.c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009;119(6):1420–8 doi 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009;139(5):871–90 doi 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 11.Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 2019;20(2):69–84 doi 10.1038/s41580-018-0080-4. [DOI] [PubMed] [Google Scholar]
- 12.Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med 2014;6(10):1279–93 doi 10.15252/emmm.201404208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aouad P, Zhang Y, De Martino F, Stibolt C, Ali S, Ambrosini G, et al. Epithelial-mesenchymal plasticity determines estrogen receptor positive breast cancer dormancy and epithelial reconversion drives recurrence. Nat Commun 2022;13(1):4975 doi 10.1038/s41467-022-32523-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Padmanaban V, Krol I, Suhail Y, Szczerba BM, Aceto N, Bader JS, et al. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019;573(7774):439–44 doi 10.1038/s41586-019-1526-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature 2018;556(7702):463–8 doi 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 16.Kroger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A 2019;116(15):7353–62 doi 10.1073/pnas.1812876116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng YH, Chen YC, Lin E, Brien R, Jung S, Chen YT, et al. Hydro-Seq enables contamination-free high-throughput single-cell RNA-sequencing for circulating tumor cells. Nat Commun 2019;10(1):2163 doi 10.1038/s41467-019-10122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malagoli Tagliazucchi G, Wiecek AJ, Withnell E, Secrier M. Genomic and microenvironmental heterogeneity shaping epithelial-to-mesenchymal trajectories in cancer. Nat Commun 2023;14(1):789 doi 10.1038/s41467-023-36439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook DP, Vanderhyden BC. Context specificity of the EMT transcriptional response. Nat Commun 2020;11(1):2142 doi 10.1038/s41467-020-16066-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. Pan-cancer Convergence to a Small-Cell Neuroendocrine Phenotype that Shares Susceptibilities with Hematological Malignancies. Cancer Cell 2019;36(1):17–34 e7 doi 10.1016/j.ccell.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22(3):298–305 doi 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferone G, Song JY, Krijgsman O, van der Vliet J, Cozijnsen M, Semenova EA, et al. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/p53 Mice. Cell Rep 2020;30(11):3837–50 e3 doi 10.1016/j.celrep.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011;19(6):754–64 doi 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 24.Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov 2017;7(7):736–49 doi 10.1158/2159-8290.CD-16-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tavernari D, Battistello E, Dheilly E, Petruzzella AS, Mina M, Sordet-Dessimoz J, et al. Nongenetic Evolution Drives Lung Adenocarcinoma Spatial Heterogeneity and Progression. Cancer Discov 2021;11(6):1490–507 doi 10.1158/2159-8290.CD-20-1274. [DOI] [PubMed] [Google Scholar]
- 26.Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol 2018;15(5):271–86 doi 10.1038/nrurol.2018.22. [DOI] [PubMed] [Google Scholar]
- 27.Quintanal-Villalonga A, Chan JM, Yu HA, Pe’er D, Sawyers CL, Sen T, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol 2020;17(6):360–71 doi 10.1038/s41571-020-0340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubin MA, Bristow RG, Thienger PD, Dive C, Imielinski M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol Cell 2020;80(4):562–77 doi 10.1016/j.molcel.2020.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Offin M, Chan JM, Tenet M, Rizvi HA, Shen R, Riely GJ, et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J Thorac Oncol 2019;14(10):1784–93 doi 10.1016/j.jtho.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018;36(24):2492–503 doi 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrell AS, Joly MM, Allen-Petersen BL, Worth PJ, Lanciault C, Sauer D, et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat Commun 2017;8(1):1728 doi 10.1038/s41467-017-01967-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, et al. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018;362(6410):91–5 doi 10.1126/science.aat5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun 2021;12(1):1426 doi 10.1038/s41467-021-21615-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davies A, Nouruzi S, Ganguli D, Namekawa T, Thaper D, Linder S, et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat Cell Biol 2021;23(9):1023–34 doi 10.1038/s41556-021-00743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest 2019;129(10):4492–505 doi 10.1172/JCI128212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marjanovic ND, Hofree M, Chan JE, Canner D, Wu K, Trakala M, et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer cell 2020;38(2):229–46 e13 doi 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somerville TD, Biffi G, Dassler-Plenker J, Hur SK, He XY, Vance KE, et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife 2020;9 doi 10.7554/eLife.53381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park S, Han J, Sun JM. Histologic transformation of ALK-rearranged adenocarcinoma to squamous cell carcinoma after treatment with ALK inhibitor. Lung Cancer 2019;127:66–8 doi 10.1016/j.lungcan.2018.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Shukuya T, Takahashi T, Kaira R, Ono A, Nakamura Y, Tsuya A, et al. Efficacy of gefitinib for non-adenocarcinoma non-small-cell lung cancer patients harboring epidermal growth factor receptor mutations: a pooled analysis of published reports. Cancer Sci 2011;102(5):1032–7 doi 10.1111/j.1349-7006.2011.01887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun 2017;8:14922 doi 10.1038/ncomms14922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brady NJ, Bagadion AM, Singh R, Conteduca V, Van Emmenis L, Arceci E, et al. Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer. Nat Commun 2021;12(1):3372 doi 10.1038/s41467-021-23780-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andricovich J, Perkail S, Kai Y, Casasanta N, Peng W, Tzatsos A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018;33(3):512–26 e8 doi 10.1016/j.ccell.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531(7592):47–52 doi 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 44.Quintanal-Villalonga A, Taniguchi H, Zhan YA, Hasan MM, Chavan SS, Meng F, et al. Comprehensive molecular characterization of lung tumors implicates AKT and MYC signaling in adenocarcinoma to squamous cell transdifferentiation. J Hematol Oncol 2021;14(1):170 doi 10.1186/s13045-021-01186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe’er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol 1999;155(3):739–52 doi 10.1016/S0002-9440(10)65173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer 2003;3(6):411–21 doi 10.1038/nrc1092. [DOI] [PubMed] [Google Scholar]
- 47.Topczewska JM, Postovit LM, Margaryan NV, Sam A, Hess AR, Wheaton WW, et al. Embryonic and tumorigenic pathways converge via Nodal signaling: role in melanoma aggressiveness. Nat Med 2006;12(8):925–32 doi 10.1038/nm1448. [DOI] [PubMed] [Google Scholar]
- 48.Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, et al. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci U S A 2001;98(14):8018–23 doi 10.1073/pnas.131209798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Flore O, Rafii S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature 1998;394(6693):588–92 doi 10.1038/29093. [DOI] [PubMed] [Google Scholar]
- 50.van der Schaft DW, Seftor RE, Seftor EA, Hess AR, Gruman LM, Kirschmann DA, et al. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J Natl Cancer Inst 2004;96(19):1473–7 doi 10.1093/jnci/djh267. [DOI] [PubMed] [Google Scholar]
- 51.Ge Y, Gomez NC, Adam RC, Nikolova M, Yang H, Verma A, et al. Stem Cell Lineage Infidelity Drives Wound Repair and Cancer. Cell 2017;169(4):636–50 e14 doi 10.1016/j.cell.2017.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141(1):69–80 doi 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov 2017;7(1):54–71 doi 10.1158/2159-8290.CD-15-1263. [DOI] [PubMed] [Google Scholar]
- 54.Nouruzi S, Ganguli D, Tabrizian N, Kobelev M, Sivak O, Namekawa T, et al. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat Commun 2022;13(1):2282 doi 10.1038/s41467-022-29963-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han M, Li F, Zhang Y, Dai P, He J, Li Y, et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer Cell 2022;40(11):1306–23 e8 doi 10.1016/j.ccell.2022.10.011. [DOI] [PubMed] [Google Scholar]
- 56.Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016;30(4):563–77 doi 10.1016/j.ccell.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han X, Li F, Fang Z, Gao Y, Li F, Fang R, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun 2014;5:3261 doi 10.1038/ncomms4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Somerville TDD, Xu Y, Miyabayashi K, Tiriac H, Cleary CR, Maia-Silva D, et al. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep 2018;25(7):1741–55 e7 doi 10.1016/j.celrep.2018.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Migault M, Sapkota S, Bracken CP. Transcriptional and post-transcriptional control of epithelial-mesenchymal plasticity: why so many regulators? Cell Mol Life Sci 2022;79(3):182 doi 10.1007/s00018-022-04199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bery F, Cancel M, Gueguinou M, Potier-Cartereau M, Vandier C, Chantome A, et al. Zeb1 and SK3 Channel Are Up-Regulated in Castration-Resistant Prostate Cancer and Promote Neuroendocrine Differentiation. Cancers (Basel) 2021;13(12) doi 10.3390/cancers13122947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017;547(7664):453–7 doi 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKeithen D, Graham T, Chung LW, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate 2010;70(9):982–92 doi 10.1002/pros.21132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun T, Zhao N, Zhao XL, Gu Q, Zhang SW, Che N, et al. Expression and functional significance of Twist1 in hepatocellular carcinoma: its role in vasculogenic mimicry. Hepatology 2010;51(2):545–56 doi 10.1002/hep.23311. [DOI] [PubMed] [Google Scholar]
- 64.Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci 2008;121(Pt 20):3317–24 doi 10.1242/jcs.028282. [DOI] [PubMed] [Google Scholar]
- 65.Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013;154(1):61–74 doi 10.1016/j.cell.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X, Xu H, Cheng C, Ji Z, Zhao H, Sheng Y, et al. Identification of a Zeb1 expressing basal stem cell subpopulation in the prostate. Nat Commun 2020;11(1):706 doi 10.1038/s41467-020-14296-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beck B, Lapouge G, Rorive S, Drogat B, Desaedelaere K, Delafaille S, et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 2015;16(1):67–79 doi 10.1016/j.stem.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 68.Jeter CR, Liu B, Lu Y, Chao HP, Zhang D, Liu X, et al. NANOG reprograms prostate cancer cells to castration resistance via dynamically repressing and engaging the AR/FOXA1 signaling axis. Cell Discov 2016;2:16041 doi 10.1038/celldisc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takayama KI, Kosaka T, Suzuki T, Hongo H, Oya M, Fujimura T, et al. Subtype-specific collaborative transcription factor networks are promoted by OCT4 in the progression of prostate cancer. Nat Commun 2021;12(1):3766 doi 10.1038/s41467-021-23974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mamun MA, Mannoor K, Cao J, Qadri F, Song X. SOX2 in cancer stemness: tumor malignancy and therapeutic potentials. J Mol Cell Biol 2020;12(2):85–98 doi 10.1093/jmcb/mjy080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murgai M, Ju W, Eason M, Kline J, Beury DW, Kaczanowska S, et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat Med 2017;23(10):1176–90 doi 10.1038/nm.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017;355(6320):78–83 doi 10.1126/science.aah4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017;355(6320):84–8 doi 10.1126/science.aah4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z, et al. KLF4 Is Essential for Induction of Cellular Identity Change and Acinar-to-Ductal Reprogramming during Early Pancreatic Carcinogenesis. Cancer Cell 2016;29(3):324–38 doi 10.1016/j.ccell.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roy N, Takeuchi KK, Ruggeri JM, Bailey P, Chang D, Li J, et al. PDX1 dynamically regulates pancreatic ductal adenocarcinoma initiation and maintenance. Genes Dev 2016;30(24):2669–83 doi 10.1101/gad.291021.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res 2014;74(4):1227–37 doi 10.1158/0008-5472.CAN-13-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bi M, Zhang Z, Jiang YZ, Xue P, Wang H, Lai Z, et al. Enhancer reprogramming driven by high-order assemblies of transcription factors promotes phenotypic plasticity and breast cancer endocrine resistance. Nat Cell Biol 2020;22(6):701–15 doi 10.1038/s41556-020-0514-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tata PR, Chow RD, Saladi SV, Tata A, Konkimalla A, Bara A, et al. Developmental History Provides a Roadmap for the Emergence of Tumor Plasticity. Dev Cell 2018;44(6):679–93 e5 doi 10.1016/j.devcel.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perekatt AO, Shah PP, Cheung S, Jariwala N, Wu A, Gandhi V, et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res 2018;78(17):4878–90 doi 10.1158/0008-5472.CAN-18-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ordonez-Moran P, Dafflon C, Imajo M, Nishida E, Huelsken J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015;28(6):815–29 doi 10.1016/j.ccell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 81.Kohler C, Nittner D, Rambow F, Radaelli E, Stanchi F, Vandamme N, et al. Mouse Cutaneous Melanoma Induced by Mutant BRaf Arises from Expansion and Dedifferentiation of Mature Pigmented Melanocytes. Cell Stem Cell 2017;21(5):679–93 e6 doi 10.1016/j.stem.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 82.Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018;33(5):890–904 e5 doi 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aurilio G, Disalvatore D, Pruneri G, Bagnardi V, Viale G, Curigliano G, et al. A meta-analysis of oestrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 discordance between primary breast cancer and metastases. Eur J Cancer 2014;50(2):277–89 doi 10.1016/j.ejca.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 84.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017;32(4):474–89 e6 doi 10.1016/j.ccell.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Zhou C, Jiang H, Liang L, Shi W, Zhang Q, et al. ZEB1 induces ER-alpha promoter hypermethylation and confers antiestrogen resistance in breast cancer. Cell Death Dis 2017;8(4):e2732 doi 10.1038/cddis.2017.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nakayama T, Watanabe M, Suzuki H, Toyota M, Sekita N, Hirokawa Y, et al. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab Invest 2000;80(12):1789–96 doi 10.1038/labinvest.3780190. [DOI] [PubMed] [Google Scholar]
- 87.Kleb B, Estecio MR, Zhang J, Tzelepi V, Chung W, Jelinek J, et al. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics 2016;11(3):184–93 doi 10.1080/15592294.2016.1146851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Werner S, Frey S, Riethdorf S, Schulze C, Alawi M, Kling L, et al. Dual roles of the transcription factor grainyhead-like 2 (GRHL2) in breast cancer. J Biol Chem 2013;288(32):22993–3008 doi 10.1074/jbc.M113.456293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Paltoglou S, Das R, Townley SL, Hickey TE, Tarulli GA, Coutinho I, et al. Novel Androgen Receptor Coregulator GRHL2 Exerts Both Oncogenic and Antimetastatic Functions in Prostate Cancer. Cancer Res 2017;77(13):3417–30 doi 10.1158/0008-5472.CAN-16-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bernardo GM, Keri RA. FOXA1: a transcription factor with parallel functions in development and cancer. Biosci Rep 2012;32(2):113–30 doi 10.1042/BSR20110046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Coutinho I, Day TK, Tilley WD, Selth LA. Androgen receptor signaling in castration-resistant prostate cancer: a lesson in persistence. Endocr Relat Cancer 2016;23(12):T179–T97 doi 10.1530/ERC-16-0422. [DOI] [PubMed] [Google Scholar]
- 92.Baca SC, Takeda DY, Seo JH, Hwang J, Ku SY, Arafeh R, et al. Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat Commun 2021;12(1):1979 doi 10.1038/s41467-021-22139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Linder S, Hoogstraat M, Stelloo S, Eickhoff N, Schuurman K, de Barros H, et al. Drug-Induced Epigenomic Plasticity Reprograms Circadian Rhythm Regulation to Drive Prostate Cancer toward Androgen Independence. Cancer Discov 2022;12(9):2074–97 doi 10.1158/2159-8290.CD-21-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006;125(2):315–26 doi 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 95.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007;448(7153):553–60 doi 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Voigt P, Tee WW, Reinberg D. A double take on bivalent promoters. Genes Dev 2013;27(12):1318–38 doi 10.1101/gad.219626.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J Clin Invest 2019;129(9):3924–40 doi 10.1172/JCI127961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Terranova CJ, Tang M, Maitituoheti M, Raman AT, Ghosh AK, Schulz J, et al. Reprogramming of bivalent chromatin states in NRAS mutant melanoma suggests PRC2 inhibition as a therapeutic strategy. Cell Rep 2021;36(3):109410 doi 10.1016/j.celrep.2021.109410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigi M, Campisi M, et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov 2021;11(8):1952–69 doi 10.1158/2159-8290.CD-20-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N, et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun 2018;9(1):4080 doi 10.1038/s41467-018-06177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Puca L, Bareja R, Prandi D, Shaw R, Benelli M, Karthaus WR, et al. Patient derived organoids to model rare prostate cancer phenotypes. Nat Commun 2018;9(1):2404 doi 10.1038/s41467-018-04495-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murai F, Koinuma D, Shinozaki-Ushiku A, Fukayama M, Miyaozono K, Ehata S. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-beta-Smad-ASCL1 pathway. Cell Discov 2015;1:15026 doi 10.1038/celldisc.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ma J, Zhang J, Weng YC, Wang JC. EZH2-Mediated microRNA-139–5p Regulates Epithelial-Mesenchymal Transition and Lymph Node Metastasis of Pancreatic Cancer. Mol Cells 2018;41(9):868–80 doi 10.14348/molcells.2018.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem 2015;290(13):8271–82 doi 10.1074/jbc.M115.637496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adamo A, Sese B, Boue S, Castano J, Paramonov I, Barrero MJ, et al. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol 2011;13(6):652–9 doi 10.1038/ncb2246. [DOI] [PubMed] [Google Scholar]
- 106.Feng J, Xu G, Liu J, Zhang N, Li L, Ji J, et al. Phosphorylation of LSD1 at Ser112 is crucial for its function in induction of EMT and metastasis in breast cancer. Breast Cancer Res Treat 2016;159(3):443–56 doi 10.1007/s10549-016-3959-9. [DOI] [PubMed] [Google Scholar]
- 107.Lin T, Ponn A, Hu X, Law BK, Lu J. Requirement of the histone demethylase LSD1 in Snai1-mediated transcriptional repression during epithelial-mesenchymal transition. Oncogene 2010;29(35):4896–904 doi 10.1038/onc.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Augert A, Eastwood E, Ibrahim AH, Wu N, Grunblatt E, Basom R, et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci Signal 2019;12(567) doi 10.1126/scisignal.aau2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sehrawat A, Gao L, Wang Y, Bankhead A 3rd, McWeeney SK, King CJ, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A 2018;115(18):E4179–E88 doi 10.1073/pnas.1719168115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J, Lu F, Ren Q, Sun H, Xu Z, Lan R, et al. Novel histone demethylase LSD1 inhibitors selectively target cancer cells with pluripotent stem cell properties. Cancer Res 2011;71(23):7238–49 doi 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Smith BA, Balanis NG, Nanjundiah A, Sheu KM, Tsai BL, Zhang Q, et al. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep 2018;24(12):3353–66 e5 doi 10.1016/j.celrep.2018.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, et al. Increased Serine and One-Carbon Pathway Metabolism by PKClambda/iota Deficiency Promotes Neuroendocrine Prostate Cancer. Cancer Cell 2019;35(3):385–400 e9 doi 10.1016/j.ccell.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu X, Li C, Zhang R, Xiao W, Niu X, Ye X, et al. The EZH2- H3K27me3-DNMT1 complex orchestrates epigenetic silencing of the wwc1 gene, a Hippo/YAP pathway upstream effector, in breast cancer epithelial cells. Cell Signal 2018;51:243–56 doi 10.1016/j.cellsig.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 114.Pathania R, Ramachandran S, Elangovan S, Padia R, Yang P, Cinghu S, et al. DNMT1 is essential for mammary and cancer stem cell maintenance and tumorigenesis. Nat Commun 2015;6:6910 doi 10.1038/ncomms7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zagorac S, Alcala S, Fernandez Bayon G, Bou Kheir T, Schoenhals M, Gonzalez-Neira A, et al. DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17–92 Cluster. Cancer Res 2016;76(15):4546–58 doi 10.1158/0008-5472.CAN-15-3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lu R, Wang P, Parton T, Zhou Y, Chrysovergis K, Rockowitz S, et al. Epigenetic Perturbations by Arg882-Mutated DNMT3A Potentiate Aberrant Stem Cell Gene-Expression Program and Acute Leukemia Development. Cancer Cell 2016;30(1):92–107 doi 10.1016/j.ccell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mayle A, Yang L, Rodriguez B, Zhou T, Chang E, Curry CV, et al. Dnmt3a loss predisposes murine hematopoietic stem cells to malignant transformation. Blood 2015;125(4):629–38 doi 10.1182/blood-2014-08-594648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bao B, Teslow EA, Mitrea C, Boerner JL, Dyson G, Bollig-Fischer A. Role of TET1 and 5hmC in an Obesity-Linked Pathway Driving Cancer Stem Cells in Triple-Negative Breast Cancer. Mol Cancer Res 2020;18(12):1803–14 doi 10.1158/1541-7786.MCR-20-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang D, Jones MG, Naranjo S, Rideout WM 3rd, Min KHJ, Ho R, et al. Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution. Cell 2022;185(11):1905–23 e25 doi 10.1016/j.cell.2022.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.LaFave LM, Kartha VK, Ma S, Meli K, Del Priore I, Lareau C, et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020;38(2):212–28 e13 doi 10.1016/j.ccell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-Induced Evolution of Human Lung Cancer Revealed by Single-Cell RNA Sequencing. Cell 2020;182(5):1232–51 e22 doi 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stewart CA, Gay CM, Xi Y, Sivajothi S, Sivakamasundari V, Fujimoto J, et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer 2020;1:423–36 doi 10.1038/s43018-019-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chan JM, Zaidi S, Love JR, Zhao JL, Setty M, Wadosky KM, et al. Lineage plasticity in prostate cancer depends on JAK/STAT inflammatory signaling. Science 2022;377(6611):1180–91 doi 10.1126/science.abn0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Deng S, Wang C, Wang Y, Xu Y, Li X, Johnson NA, et al. Ectopic JAK-STAT activation enables the transition to a stem-like and multilineage state conferring AR-targeted therapy resistance. Nat Cancer 2022;3(9):1071–87 doi 10.1038/s43018-022-00431-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Guilhamon P, Chesnelong C, Kushida MM, Nikolic A, Singhal D, MacLeod G, et al. Single-cell chromatin accessibility profiling of glioblastoma identifies an invasive cancer stem cell population associated with lower survival. Elife 2021;10 doi 10.7554/eLife.64090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Castellan M, Guarnieri A, Fujimura A, Zanconato F, Battilana G, Panciera T, et al. Single-cell analyses reveal YAP/TAZ as regulators of stemness and cell plasticity in Glioblastoma. Nat Cancer 2021;2(2):174–88 doi 10.1038/s43018-020-00150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39(3):346–60 e7 doi 10.1016/j.ccell.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer cell 2020;38(1):60–78 e12 doi 10.1016/j.ccell.2020.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stevens LE, Peluffo G, Qiu X, Temko D, Fassl A, Li Z, et al. JAK-STAT Signaling in Inflammatory Breast Cancer Enables Chemotherapy-Resistant Cell States. Cancer Res 2023;83(2):264–84 doi 10.1158/0008-5472.CAN-22-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wagner DE, Klein AM. Lineage tracing meets single-cell omics: opportunities and challenges. Nat Rev Genet 2020;21(7):410–27 doi 10.1038/s41576-020-0223-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Simeonov KP, Byrns CN, Clark ML, Norgard RJ, Martin B, Stanger BZ, et al. Single-cell lineage tracing of metastatic cancer reveals selection of hybrid EMT states. Cancer Cell 2021;39(8):1150–62 e9 doi 10.1016/j.ccell.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen CL, Uthaya Kumar DB, Punj V, Xu J, Sher L, Tahara SM, et al. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab 2016;23(1):206–19 doi 10.1016/j.cmet.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang T, Fahrmann JF, Lee H, Li YJ, Tripathi SC, Yue C, et al. JAK/STAT3-Regulated Fatty Acid beta-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab 2018;27(6):1357 doi 10.1016/j.cmet.2018.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013;23(3):316–31 doi 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wang HJ, Pochampalli M, Wang LY, Zou JX, Li PS, Hsu SC, et al. KDM8/JMJD5 as a dual coactivator of AR and PKM2 integrates AR/EZH2 network and tumor metabolism in CRPC. Oncogene 2019;38(1):17–32 doi 10.1038/s41388-018-0414-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, et al. Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 2013;339(6116):222–6 doi 10.1126/science.1226603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ulanovskaya OA, Zuhl AM, Cravatt BF. NNMT promotes epigenetic remodeling in cancer by creating a metabolic methylation sink. Nat Chem Biol 2013;9(5):300–6 doi 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, et al. Methionine metabolism regulates maintenance and differentiation of human pluripotent stem cells. Cell Metab 2014;19(5):780–94 doi 10.1016/j.cmet.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 139.Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY, Chong PKW, et al. Methionine is a metabolic dependency of tumor-initiating cells. Nat Med 2019;25(5):825–37 doi 10.1038/s41591-019-0423-5. [DOI] [PubMed] [Google Scholar]
- 140.Strekalova E, Malin D, Weisenhorn EMM, Russell JD, Hoelper D, Jain A, et al. S-adenosylmethionine biosynthesis is a targetable metabolic vulnerability of cancer stem cells. Breast Cancer Res Treat 2019;175(1):39–50 doi 10.1007/s10549-019-05146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016;537(7621):544–7 doi 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kusi M, Zand M, Lin LL, Chen M, Lopez A, Lin CL, et al. 2-Hydroxyglutarate destabilizes chromatin regulatory landscape and lineage fidelity to promote cellular heterogeneity. Cell reports 2022;38(2):110220 doi 10.1016/j.celrep.2021.110220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bonfanti P, Barrandon Y, Cossu G. ‘Hearts and bones’: the ups and downs of ‘plasticity’ in stem cell biology. EMBO Mol Med 2012;4(5):353–61 doi 10.1002/emmm.201200220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK, et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep 2016;15(6):1175–89 doi 10.1016/j.celrep.2016.04.019. [DOI] [PubMed] [Google Scholar]
- 145.Bhattacharyya S, Oon C, Kothari A, Horton W, Link J, Sears RC, et al. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J Exp Med 2020;217(8) doi 10.1084/jem.20191805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun 2014;5:3472 doi 10.1038/ncomms4472. [DOI] [PubMed] [Google Scholar]
- 147.Wingrove E, Liu ZZ, Patel KD, Arnal-Estape A, Cai WL, Melnick MA, et al. Transcriptomic Hallmarks of Tumor Plasticity and Stromal Interactions in Brain Metastasis. Cell Rep 2019;27(4):1277–92 e7 doi 10.1016/j.celrep.2019.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A 2008;105(17):6392–7 doi 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol 2008;10(3):295–305 doi 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 150.Zhang W, Shi X, Peng Y, Wu M, Zhang P, Xie R, et al. HIF-1alpha Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS One 2015;10(6):e0129603 doi 10.1371/journal.pone.0129603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor −1alpha in hepatocellular carcinoma. BMC Cancer 2013;13:108 doi 10.1186/1471-2407-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell 2010;18(1):23–38 doi 10.1016/j.ccr.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015;34(24):3107–19 doi 10.1038/onc.2014.257. [DOI] [PubMed] [Google Scholar]
- 154.Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A 2014;111(5):E592–600 doi 10.1073/pnas.1318157111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal 2013;25(4):961–9 doi 10.1016/j.cellsig.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res 2016;76(11):3156–65 doi 10.1158/0008-5472.CAN-15-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wang N, Liu W, Zheng Y, Wang S, Yang B, Li M, et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis 2018;9(9):880 doi 10.1038/s41419-018-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kennel KB, Bozlar M, De Valk AF, Greten FR. Cancer-Associated Fibroblasts in Inflammation and Antitumor Immunity. Clin Cancer Res 2023;29(6):1009–16 doi 10.1158/1078-0432.CCR-22-1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wei SC, Fattet L, Tsai JH, Guo Y, Pai VH, Majeski HE, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol 2015;17(5):678–88 doi 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474(7350):179–83 doi 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 161.Totaro A, Zhuang Q, Panciera T, Battilana G, Azzolin L, Brumana G, et al. Cell phenotypic plasticity requires autophagic flux driven by YAP/TAZ mechanotransduction. Proc Natl Acad Sci U S A 2019;116(36):17848–57 doi 10.1073/pnas.1908228116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Raghavan S, Winter PS, Navia AW, Williams HL, DenAdel A, Lowder KE, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021;184(25):6119–37 e26 doi 10.1016/j.cell.2021.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gupta PB, Pastushenko I, Skibinski A, Blanpain C, Kuperwasser C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019;24(1):65–78 doi 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mosquera MJ, Kim S, Bareja R, Fang Z, Cai S, Pan H, et al. Extracellular Matrix in Synthetic Hydrogel-Based Prostate Cancer Organoids Regulate Therapeutic Response to EZH2 and DRD2 Inhibitors. Adv Mater 2022;34(2):e2100096 doi 10.1002/adma.202100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 2014;5:5241 doi 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mak MP, Tong P, Diao L, Cardnell RJ, Gibbons DL, William WN, et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin Cancer Res 2016;22(3):609–20 doi 10.1158/1078-0432.CCR-15-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL, et al. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res 2017;77(15):3982–9 doi 10.1158/0008-5472.CAN-16-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dongre A, Rashidian M, Eaton EN, Reinhardt F, Thiru P, Zagorulya M, et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov 2021;11(5):1286–305 doi 10.1158/2159-8290.CD-20-0603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8 doi 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Taki M, Abiko K, Baba T, Hamanishi J, Yamaguchi K, Murakami R, et al. Snail promotes ovarian cancer progression by recruiting myeloid-derived suppressor cells via CXCR2 ligand upregulation. Nat Commun 2018;9(1):1685 doi 10.1038/s41467-018-03966-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Guo Y, Lu X, Chen Y, Rendon B, Mitchell RA, Cuatrecasas M, et al. Zeb1 induces immune checkpoints to form an immunosuppressive envelope around invading cancer cells. Sci Adv 2021;7(21) doi 10.1126/sciadv.abd7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Plaschka M, Benboubker V, Grimont M, Berthet J, Tonon L, Lopez J, et al. ZEB1 transcription factor promotes immune escape in melanoma. J Immunother Cancer 2022;10(3) doi 10.1136/jitc-2021-003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ouzounova M, Lee E, Piranlioglu R, El Andaloussi A, Kolhe R, Demirci MF, et al. Monocytic and granulocytic myeloid derived suppressor cells differentially regulate spatiotemporal tumour plasticity during metastatic cascade. Nat Commun 2017;8:14979 doi 10.1038/ncomms14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Burr ML, Sparbier CE, Chan KL, Chan YC, Kersbergen A, Lam EYN, et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019;36(4):385–401 e8 doi 10.1016/j.ccell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhou L, Mudianto T, Ma X, Riley R, Uppaluri R. Targeting EZH2 Enhances Antigen Presentation, Antitumor Immunity, and Circumvents Anti-PD-1 Resistance in Head and Neck Cancer. Clin Cancer Res 2020;26(1):290–300 doi 10.1158/1078-0432.CCR-19-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Li Y, Goldberg EM, Chen X, Xu X, McGuire JT, Leuzzi G, et al. Histone methylation antagonism drives tumor immune evasion in squamous cell carcinomas. Mol Cell 2022;82(20):3901–18 e7 doi 10.1016/j.molcel.2022.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 2015;16(4):e165–72 doi 10.1016/S1470-2045(14)71180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Bakht MK, Yamada Y, Ku SY, Venkadakrishnan VB, Korsen JA, Kalidindi TM, et al. Landscape of prostate-specific membrane antigen heterogeneity and regulation in AR-positive and AR-negative metastatic prostate cancer. Nat Cancer 2023;4(5):699–715 doi 10.1038/s43018-023-00539-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tosoian JJ, Gorin MA, Rowe SP, Andreas D, Szabo Z, Pienta KJ, et al. Correlation of PSMA-Targeted (18)F-DCFPyL PET/CT Findings With Immunohistochemical and Genomic Data in a Patient With Metastatic Neuroendocrine Prostate Cancer. Clin Genitourin Cancer 2017;15(1):e65–e8 doi 10.1016/j.clgc.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Jie XX, Zhang XY, Xu CJ. Epithelial-to-mesenchymal transition, circulating tumor cells and cancer metastasis: Mechanisms and clinical applications. Oncotarget 2017;8(46):81558–71 doi 10.18632/oncotarget.18277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest 2020;130(4):1653–68 doi 10.1172/JCI131041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.De Sarkar N, Patton RD, Doebley AL, Hanratty B, Adil M, Kreitzman AJ, et al. Nucleosome Patterns in Circulating Tumor DNA Reveal Transcriptional Regulation of Advanced Prostate Cancer Phenotypes. Cancer Discov 2023;13(3):632–53 doi 10.1158/2159-8290.CD-22-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sjostrom M, Zhao SG, Levy S, Zhang M, Ning Y, Shrestha R, et al. The 5-Hydroxymethylcytosine Landscape of Prostate Cancer. Cancer Res 2022;82(21):3888–902 doi 10.1158/0008-5472.CAN-22-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J Clin Oncol 2017;35(26):3065–74 doi 10.1200/JCO.2016.71.9096. [DOI] [PubMed] [Google Scholar]
- 185.Puca L, Gavyert K, Sailer V, Conteduca V, Dardenne E, Sigouros M, et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci Transl Med 2019;11(484) doi 10.1126/scitranslmed.aav0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 2022;21(3):181–200 doi 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bushweller JH. Targeting transcription factors in cancer - from undruggable to reality. Nat Rev Cancer 2019;19(11):611–24 doi 10.1038/s41568-019-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen HY, Durmaz YT, Li Y, Sabet AH, Vajdi A, Denize T, et al. Regulation of neuroendocrine plasticity by the RNA-binding protein ZFP36L1. Nat Commun 2022;13(1):4998 doi 10.1038/s41467-022-31998-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019;38(3):390–405 doi 10.1038/s41388-018-0451-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Luo N, Nixon MJ, Gonzalez-Ericsson PI, Sanchez V, Opalenik SR, Li H, et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun 2018;9(1):248 doi 10.1038/s41467-017-02630-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Hirukawa A, Singh S, Wang J, Rennhack JP, Swiatnicki M, Sanguin-Gendreau V, et al. Reduction of Global H3K27me(3) Enhances HER2/ErbB2 Targeted Therapy. Cell Rep 2019;29(2):249–57 e8 doi 10.1016/j.celrep.2019.08.105. [DOI] [PubMed] [Google Scholar]
- 192.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162(5):974–86 doi 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Topper MJ, Vaz M, Chiappinelli KB, DeStefano Shields CE, Niknafs N, Yen RC, et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017;171(6):1284–300 e21 doi 10.1016/j.cell.2017.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A 2017;114(51):E10981–E90 doi 10.1073/pnas.1712514114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24(8):1143–50 doi 10.1038/s41591-018-0116-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Morel KL, Sheahan AV, Burkhart DL, Baca SC, Boufaied N, Liu Y, et al. EZH2 inhibition activates a dsRNA-STING-interferon stress axis that potentiates response to PD-1 checkpoint blockade in prostate cancer. Nat Cancer 2021;2(4):444–56 doi 10.1038/s43018-021-00185-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174(3):549–63 e19 doi 10.1016/j.cell.2018.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.de The H Differentiation therapy revisited. Nat Rev Cancer 2018;18(2):117–27 doi 10.1038/nrc.2017.103. [DOI] [PubMed] [Google Scholar]
- 199.Ishay-Ronen D, Diepenbruck M, Kalathur RKR, Sugiyama N, Tiede S, Ivanek R, et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019;35(1):17–32 e6 doi 10.1016/j.ccell.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 200.Loo SY, Toh LP, Xie WH, Pathak E, Tan W, Ma S, et al. Fatty acid oxidation is a druggable gateway regulating cellular plasticity for driving metastasis in breast cancer. Sci Adv 2021;7(41):eabh2443 doi 10.1126/sciadv.abh2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Turcan S, Fabius AW, Borodovsky A, Pedraza A, Brennan C, Huse J, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013;4(10):1729–36 doi 10.18632/oncotarget.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4alpha to block hepatocyte differentiation and promote biliary cancer. Nature 2014;513(7516):110–4 doi 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wang M, Chen X, Tan P, Wang Y, Pan X, Lin T, et al. Acquired semi-squamatization during chemotherapy suggests differentiation as a therapeutic strategy for bladder cancer. Cancer Cell 2022;40(9):1044–59 e8 doi 10.1016/j.ccell.2022.08.010. [DOI] [PubMed] [Google Scholar]