
Fig. 1 |. Oestrogen receptor-α signalling and modes of inhibition.
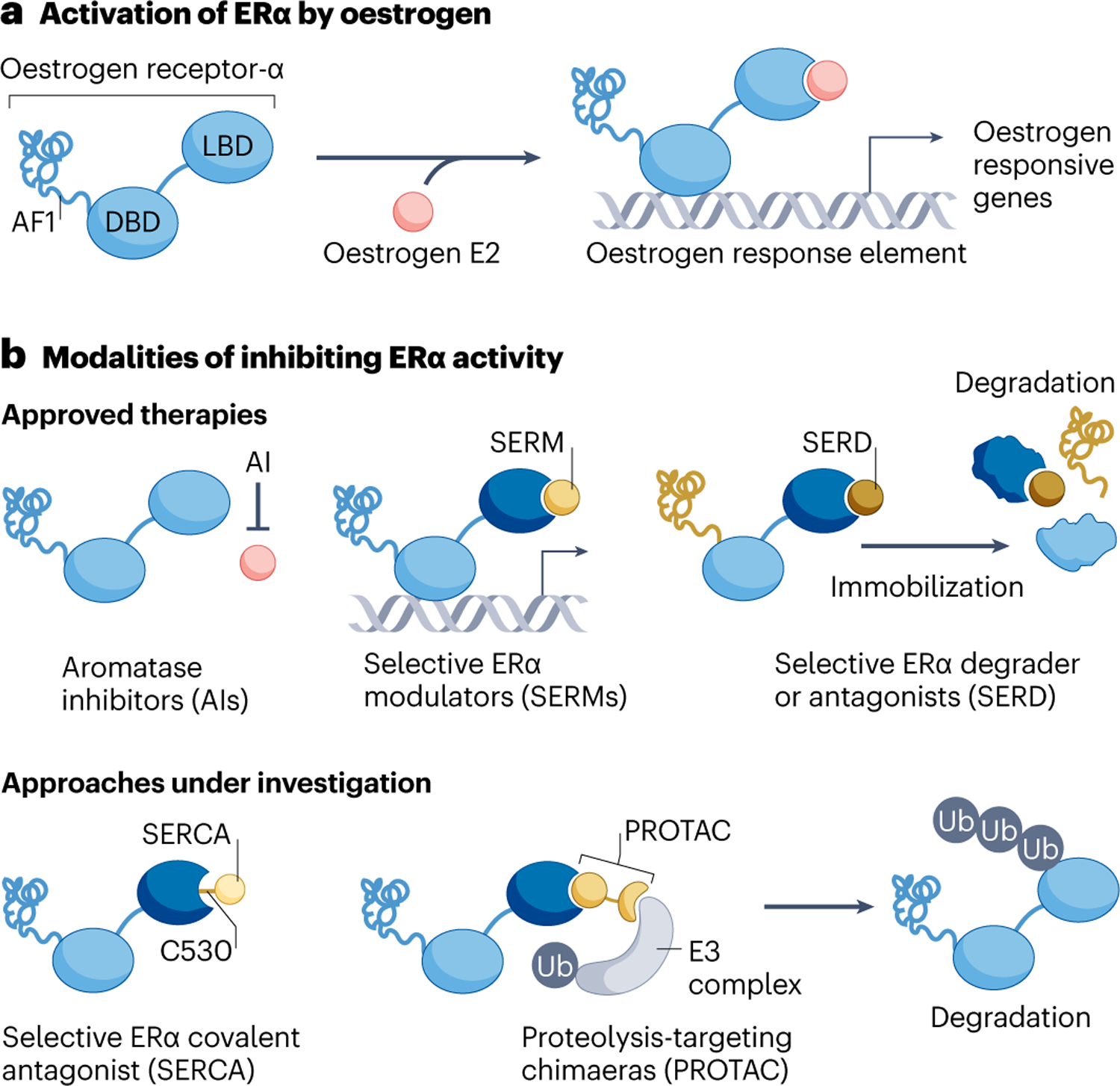
Oestrogen receptor-α (ER) signalling can be inhibited via multiple therapeutic strategies. a, ER is a multidomain ligand-inducible transcription factor that when activated drives a transcriptional programme that supports progression through the cell cycle. b, Strategies for ER targeting that are clinically approved are shown on the top. Aromatase inhibitors, which deplete oestrogen via inhibition of CYP19A1, are also known to select for oestrogen-independent ESR1 mutations. Selective ER modulators inhibit the ligand-binding domain (LBD) but not DNA-binding domain (DBD) or activation function-1 (AF1) domain of ER. They can also act as a partial ER agonist in some contexts. Selective ER degraders inhibit both LBD and AF1 domains by preventing recruitment of co-activators, immobilizing and destabilizing ER, leading to degradation. Approaches for inhibiting ER that are currently under investigation are shown on the bottom. Covalent ER antagonists react to cysteine C530 in the ER ligand-binding pocket and enforce an antagonist conformation in both wild-type and mutant ER. ER-specific PROTACs recruit E3 ligase to the ER, promoting poly-ubiquitination and proteasomal degradation. (Note: ER forms a homodimer but is depicted here as a monomer for visual clarity). Ub, ubiquitin.