

SUMMARY
Oligodendrocytes are the sole myelin-producing cells in the central nervous system. Oligodendrocyte number is tightly controlled across diverse brain regions to match local axon type and number, yet the underlying mechanisms remain unclear. Here, we show that autophagy, an evolutionarily conserved cellular process that promotes cell survival under physiological conditions, elicits premyelinating oligodendrocyte apoptosis during development. Autophagy flux is increased in premyelinating oligodendrocytes, and its genetic blockage causes ectopic oligodendrocyte survival throughout the entire brain. Autophagy functions cell autonomously in the premyelinating oligodendrocyte to trigger cell apoptosis, and it genetically interacts with the TFEB pathway to limit oligodendrocyte number across diverse brain regions. Our results provide in vivo evidence showing that autophagy promotes apoptosis in mammalian cells under physiological conditions and reveal key intrinsic mechanisms governing oligodendrogenesis.
In brief
Autophagy cooperates with apoptosis to maintain cellular homeostasis. Zhang et al. discover that autophagy limits mature oligodendrocyte number by promoting apoptosis in subsets of premyelinating oligodendrocytes, thereby enabling the spatiotemporal specificity of CNS myelination during early brain development.
Graphical Abstract
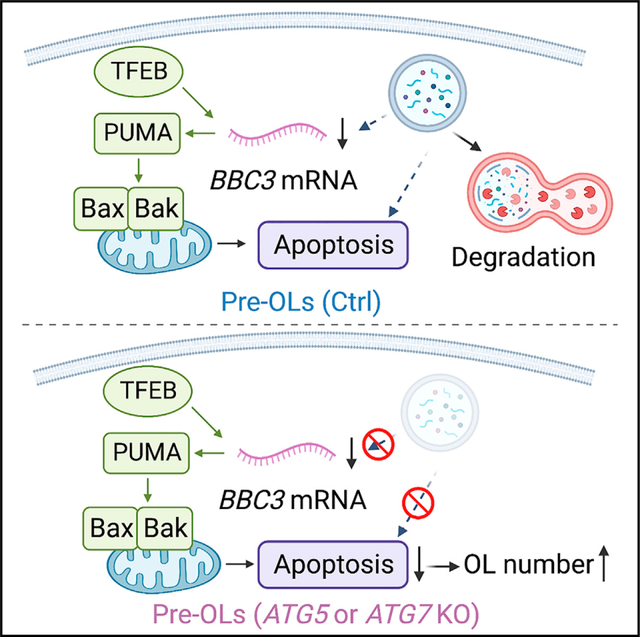
INTRODUCTION
Oligodendrocytes (OLs) are the sole myelin-producing cells in the central nervous system (CNS) and are critical for many aspects of neural function.1,2 OL number is tightly controlled across diverse brain regions to possibly match local axon type and number. However, the underlying cellular and molecular mechanisms remain elusive. Once OL precursor cells (OPCs) cease division, the control of OL number is primarily achieved at the premyelinating OL (pre-OL) stage. In rodents, pre-OLs are overproduced, and a significant portion of them undergo programmed cell death before committing to myelination, thereby contributing to the spatiotemporal specificity in myelination during development and throughout adulthood.3,4
The survival rate of pre-OLs and their ability to continue myelinating axons vary depending on the brain regions. For instance, about 20%–80% pre-OLs in the cortex die during early postnatal development and throughout adulthood.5,6 In the cerebellar molecular layer, nearly 100% pre-OLs undergo programmed cell death, resulting in a unique “unmyelinated” area that lacks mature OLs and myelin.7 Intriguingly, pre-OLs are at a “stressed” differentiation stage with high energy demands: after OPCs differentiate into pre-OLs, they begin expressing large quantities of mRNAs to encode myelin proteins, drastically expand their plasma membrane areas for myelination, and compete against each other for limited nutrients provided by axons and other cell types.8 Previous work showed that the TFEB-PUMA-Bax/Bak pathway powerfully promotes pre-OL apoptosis to control OL number.4 It remains unclear, however, whether the TFEB-PUMA-Bax/Bak pathway is the sole controller or if it interacts with other pathways to determine pre-OL fate.
Macroautophagy (hereafter referred to as autophagy) is an evolutionarily conserved cellular process that degrades unnecessary or dysfunctional cytoplasmic components, thereby promoting cell adaption under nutrient deprivation and stress.9–11 Under physiological settings, autophagy promotes cell survival, while in rare cases, it elicits cell death directly or indirectly through intimate crosstalk with apoptosis pathways.12,13 For instance, during Drosophila, midgut development autophagy functions independent of apoptosis to promote midgut cell death, known as autophagy-dependent cell death.14 In addition, the selective autophagy of anti-apoptotic proteins, such as dBruce, contributes to the induction of apoptosis during Drosophila oogenesis.15 In mammals, when apoptosis is genetically blocked, autophagy can act as a compensatory mechanism to facilitate the elimination of interdigit web cells, thereby contributing to tissue remodeling during early embryogenesis.16 The in vivo evidence demonstrating that autophagy promotes mammalian cell apoptosis under physiologically relevant circumstances, however, is still lacking.
Here, we discovered that autophagy collaborates with apoptosis pathways to eliminate subsets of pre-OLs during development, thereby limiting mature OL number and regulating the spatiotemporal specificity of CNS myelination. We showed that autophagy flux is elevated in the pre-OLs during differentiation and that genetic perturbation of autophagy within the pre-OLs leads to ectopic myelination in the unmyelinated brain regions and increased OL numbers across diverse brain areas, as well as to altered oligodendrogenesis onset in the forebrain. These phenotypes arise from autophagy’s cell-autonomous role in promoting apoptosis in subsets of pre-OLs. Finally, autophagy genetically interacts with the TFEB pathway to control pre-OL cell fate. Our findings demonstrate that autophagy plays an active role in promoting oligodendroglia apoptosis during brain development, shedding light on the cellular and molecular mechanisms governing oligodendrogenesis and CNS myelination.
RESULTS
Autophagy flux is elevated in pre-OLs
Recent work showed that autophagy plays critical roles in mature, myelinating OLs to prevent neurodegeneration.17 It remains unclear, however, if autophagy has a separate function at the immature, premyelinating stage during OL differentiation. To determine at which differentiation stage autophagy flux is elevated in OL lineage cells (Figure 1A), we first analyzed the in vivo bulk RNA sequencing (RNA-seq) dataset that contains the transcripts of 399 autophagy genes (Gene Ontology: 0006919).18 This dataset covers seven major brain cell types, including the OPCs, pre-OLs, and myelinating OLs (Table S1). We found that autophagy genes exhibited differentiation-stage-dependent expression patterns; many of them started mRNA expression at the OPC and pre-OL stages, whereas a subset of them exhibited the highest mRNA expression in the myelinating OL stage (Figure 1B). To validate the differentiation stage when autophagy flux is elevated, we acutely purified OPCs from early postnatal mouse brains and differentiated them in vitro (Figures 1C and S1A–S1G). We found that several autophagy proteins, including Lamp2, Beclin1, WIPI2, ATG5, and ATG7, exhibited elevated expression levels at differentiation day 4 and remained expressed at day 6 (Figures 1D and S1K–S1L, quantified in Figure 1F). To address if autophagy flux indeed increases at the pre-OL stage, we treated differentiating OLs with bafilomycin A1 (BafA1), a potent autophagosome-lysosome fusion blocker that allowed us to analyze transient autophagy flux events.19 We found that in the presence of BafA1, autophagosome adaptor protein p62/SQSTM1 and autophagosome marker LC3-II were significantly increased at the beginning of pre-OL differentiation and throughout its maturation (Figure 1E, quantified in Figures 1F and 1G). Finally, we performed transmission electron microscopy (TEM) analysis and found that upon BafA1 treatment, autophagosome density and total areas were significantly increased in cultured pre-OLs (Figures 1H, 1H′, S1I, and S1J, quantified in Figure 1I; see more examples in Figures 3 and S3). Therefore, autophagy flux increases during OL differentiation and is elevated in pre-OLs.
Figure 1. Autophagy flux increases in premyelinating and myelinating oligodendrocytes.
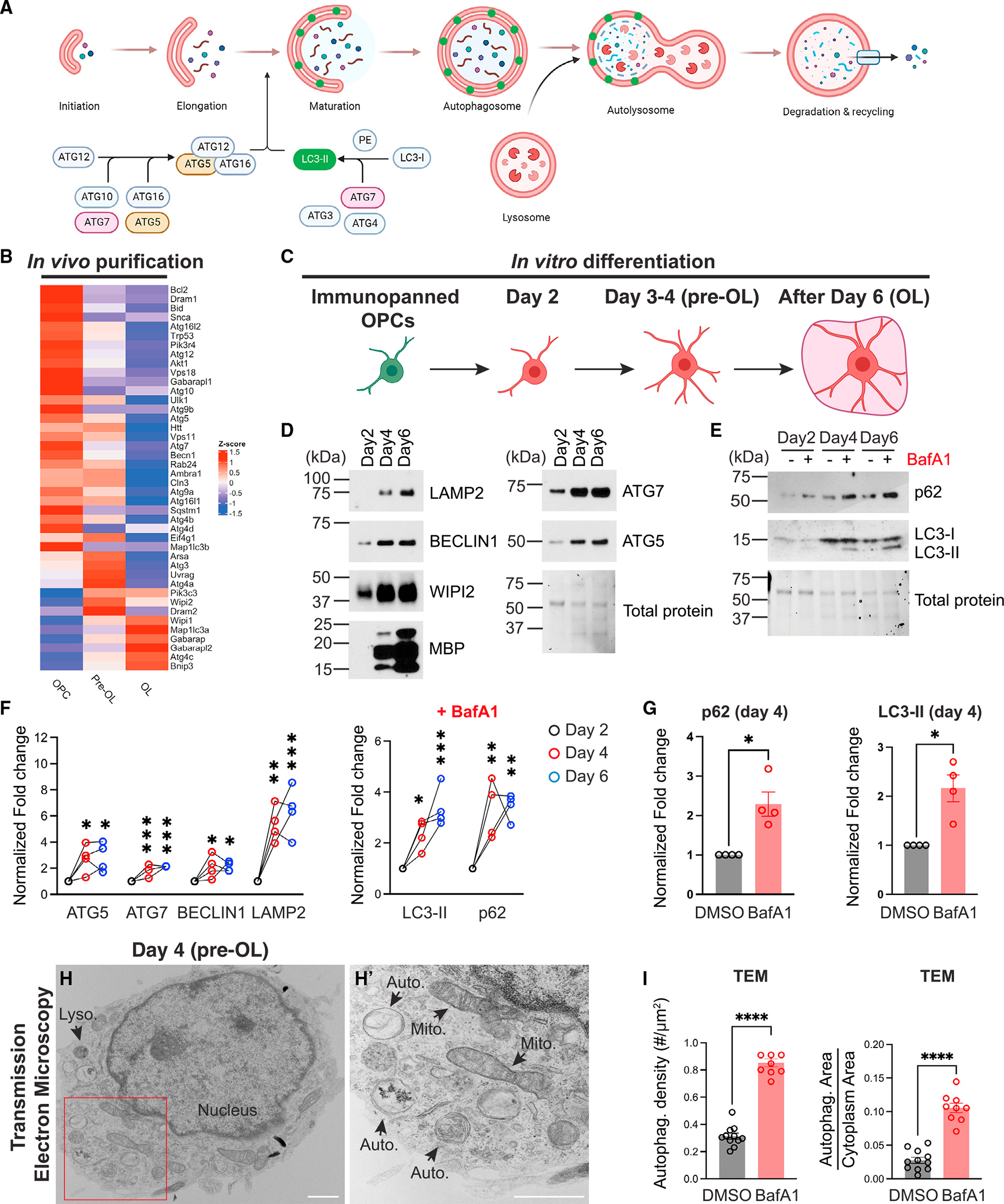
(A) Diagram showing the autophagy flux (modified from a template provided by BioRender).
(B) Heatmap showing major autophagy gene expression levels (in Z scores) in acutely isolated OPCs, pre-OLs, and myelinating OLs.18
(C) Diagram of OL in vitro differentiation (created by BioRender). See representative images of differentiating OLs in Figure S1.
(D and E) Western blot analysis of autophagy-related protein expression during OL in vitro differentiation and autophagy flux markers p62 and LC3-II with or without bafilomycin A1 (BafA1) treatment.
(F) Quantification of autophagy protein expression (left) and LC3-II and p62 levels with BafA1 treatment (right) during OL in vitro differentiation. Protein levels were normalized to the total protein level.
(G) Comparison of p62 (left) and LC3-II (right) between vehicle treatment (DMSO) and BafA1 treatment at differentiation day 4.
(H and H′) Representative transmission electron microscopy (TEM) micrograph of a pre-OL at differentiation day 4 in vitro (H). (H′) represents the enlarged view of the red inset in (H). Mito., mitochondria; Auto., autophagosome; Lyso., lysosome.
(I) Quantification of autophagosome density (left) and the ratio of total autophagosome area over cytoplasm area (right) between vehicle treatment (DMSO) and BafA1 treatment at differentiation day 4. n ≥ 8 cells.
n = 4 separate cell purifications for (F) and (G). Error bars represent SEM. Scale bars: 1 μm. One-way ANOVA followed by Tukey’s multiple comparisons test for (F). Two-tailed t tests for (G) and (I). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
Figure 3. ATG5- and ATG7-deficient OLs exhibit disrupted autophagy flux and autophagosome abnormalities.
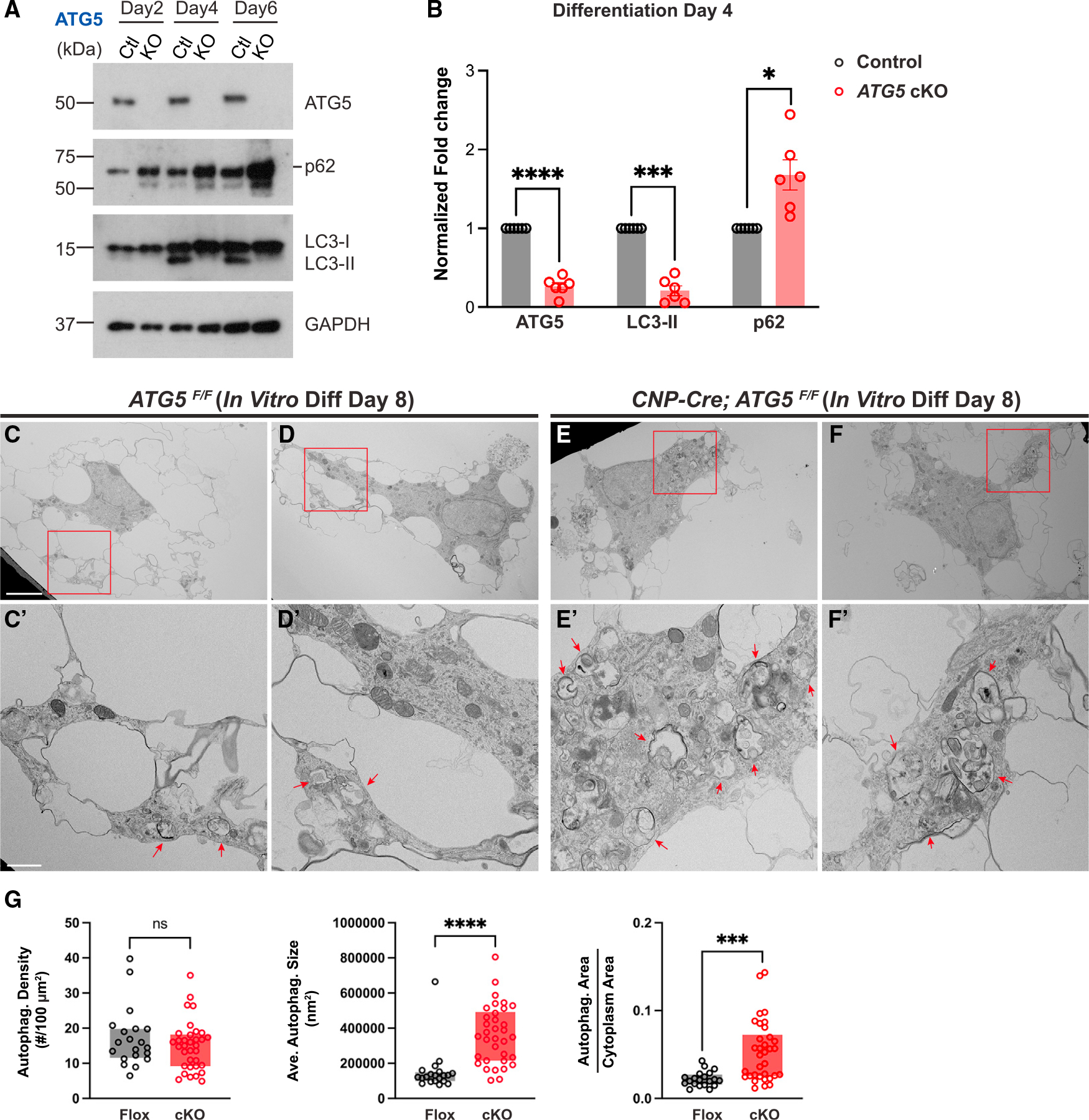
(A and B) Western blot analysis (A) and quantification (B) of ATG5, p62, and LC3-II in ATG5F/F (control) and CNP-Cre; ATG5F/F (ATG5 cKO) OLs during in vitro differentiation. Protein levels were normalized to GAPDH. The short line indicates the protein band of p62. n = 6 separate cell preparations.
(C–F′) Representative TEM images of ATG5F/F OLs (C–D′) and CNP-Cre; ATG5F/F OLs (E–F′) at differentiation day 8, showing that autophagosomes exhibited defective morphologies and enlarged sizes in CNP-Cre; ATG5F/F OLs (red arrows in E′ and F′). (C′)–(F′) represent red insets in (C)–(F), respectively.
(G) Quantifications (box and whisker, min to max) of autophagosome density, average autophagosome size, and the ratio of total autophagosome area over cytoplasm area in ATG5F/F (Flox) and CNP-Cre; ATG5F/F (cKO) OLs at differentiation day 8. n ≥ 20 cells per condition per genotype.
Error bars represent SEM. Scale bars: 5 μm in (C) for (C)–(F) and 1 mm in (C′) for (C′)–(F′). Two-tailed t tests for (B) and (G). *p < 0.05, ***p < 0.001, ****p < 0.0001, ns, not significant.
Genetic blockade of autophagy flux in OLs disrupts the spatiotemporal specificity of CNS myelination
We next characterized the mouse cerebellum, a brain region that harbors numerous pre-OLs during early brain development.4 We conditionally deleted ATG7, a critical gene mediating autophagosome formation, in OL lineage cells (CNP-Cre; ATG7F/F). Myelin basic protein (MBP) as well as myelination were restricted beneath the cerebellar molecular layer (ML) in control animals (Figures 2A, 2B, and S2J). In strong contrast, CNP-Cre; ATG7F/F mutants exhibited the fully penetrant, ectopic presence of myelin proteins and myelin wrapping in the ML across the entire cerebellum (Figures 2A′, 2B′, S2J′, and S2J″). To determine if the aberrant myelin was caused by ectopic OLs in the ML, we utilized in situ hybridization probes that recognize MBP mRNAs, a marker expressed by pre-OLs and myelinating OLs.20 We found that at postnatal day 56 (P56), both ATG7F/F and CNP-Cre; ATG7F/+ mice harbored very few MBP+ OLs in the cerebellar ML, likely representing newly formed pre-OLs that had not yet undergone programmed cell death (Figure 2C). Conversely, ATG7 conditional knockout (cKO) mice exhibited significantly increased numbers of MBP+ cells in the ML at P56 (Figure 2C′, quantified in Figure 2F). We found similar phenotypes in two separate mutant mouse strains including the CNP-Cre; ATG5F/F mutants and the Olig2-Cre; ATG5F/F mutants, where ATG5, another gene critical for autophagosome formation, was genetically deleted in OL lineage cells (Figures 2D–2E′, quantified in Figures 2F and S2I, and 2N and 2N′, quantified in Figure 2O). At P56, a majority of MBP+ cells in CNP-Cre; ATG5F/F ML co-expressed a mature OL marker, myelin OL glycoprotein (MOG), indicating that these ectopic cells were mature OLs in adulthood (Figures S2G and S2G′, quantified in Figure S2H).
Figure 2. Genetic deletion of ATG7 or ATG5 in OL lineage cells disrupts the spatiotemporal specificity of myelination.
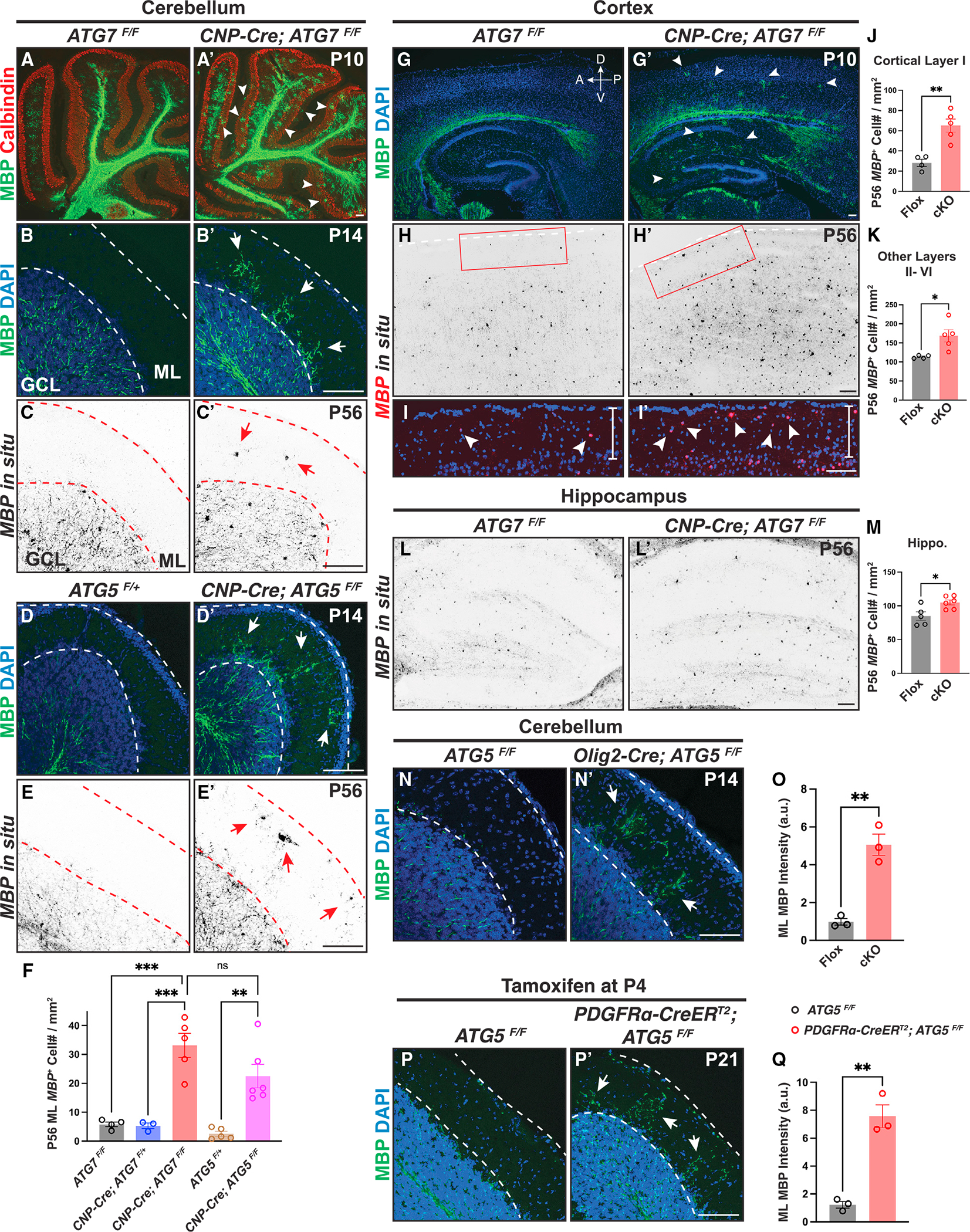
(A–B′) Characterization of ectopic OLs and aberrant myelination in CNP-Cre; ATG7F/F cerebellar molecular layer (ML) at P10 (A′) and P14 (B′) compared with the littermate controls (A and B). White arrowheads in (A′) and white arrows in (B′) indicate ectopic MBP immunolabeling. GCL, granule cell layer.
(C and C′) In situ hybridization using the MBP mRNA probes showing that CNP-Cre; ATG7F/F mutants exhibited ectopic MBP+ OLs in the cerebellar ML (red arrows in C′).
(D–E′) CNP-Cre; ATG5F/F mutants showed ectopic MBP immunolabeling (white arrows in D′) and aberrant MBP+ OLs (red arrows in E′) in the cerebellar ML at P14 (D and D′) and P56 (E and E′).
(F) Quantification of MBP+ OL numbers in the cerebellar ML at P56.
(G–I′) CNP-Cre; ATG7F/F mutants exhibited MBP immunolabeling precociously in the cortex and hippocampus at P10 (white arrowheads in G′; n = 3 animals per genotype) and increased MBP+ OL numbers in cortical layer I at P56 (red inset in H′ and white arrowheads in I′; n ≥ 4 animals per genotype). (I) and (I′) represent the insets shown in (H) and (H′), respectively. A, anterior; P, posterior; D, dorsal; V, ventral.
(J and K) Quantification of MBP+ OLs in cortical layer I (J) and cortical layer II-VI (K) of ATG7F/F (Flox) and CNP-Cre; ATG7F/F (cKO) mice at P56.
(L, L′, and M) Representative images (L and L′) and quantification (M) of MBP+ OLs in the hippocampus.
(N, N′, and O) Representative images (N and N′) and quantification (O) showing that Olig2-Cre; ATG5F/F mice exhibited ectopic MBP immunolabeling in the cerebellar ML.
(P, P′, and Q) Representative confocal micrographs (P and P′) and quantification (Q) showing that inducible deletion of ATG5 from OL precursor cells at P4 led to ectopic MBP immunolabeling in P21 cerebellar ML.
Error bars represent SEM. Scale bars: 100 μm. Open circles in (F), (J), (K), (M), (O), and (Q) represent individual animals; n ≥ 3 animals per category. One-way ANOVA followed by Tukey’s multiple comparisons test for (F). Two-tailed t tests for (J), (K), (M), (O), and (Q). *p < 0.05, **p < 0.01, ***p < 0.001, ns, not significant.
Because CNP-Cre and Olig2-Cre are transiently expressed in neurons,21,22 the ectopic myelination phenotype observed in the CNP-Cre; ATG7F/F, CNP-Cre; ATG5F/F, and Olig2-Cre; ATG5F/F mutants could be due to neuronal disruption of autophagy.23 To rule out this possibility, we conditionally deleted ATG5 only in OPCs from P4 by administering tamoxifen to PDGFRα-CreERT2; ATG5F/F mice. We found similar phenotypes in these mutants (Figures 2P, 2P′, S2F, and S2F′, quantified in Figure 2Q), together showing that the ectopic myelination phenotype is indeed due to disrupted autophagy within OL lineage cells.
To determine if autophagy functions globally to control OL number, we characterized a variety of brain regions in ATG5 and ATG7 cKO mice throughout development. Oligodendrogenesis in the wild-type forebrain did not begin until P7, with only a few brain areas harboring differentiating MBP+ OLs (Figures 2G and S2K). In contrast, ATG7 cKO mice exhibited a fully penetrant, “precocious” oligodendrogenesis phenotype where numerous MBP+ OLs were present throughout the entire forebrain as early as P7 (Figure S2K′). By P10, MBP+ OLs readily appeared in the deep cortical layers and hippocampus in ATG7 cKO mice, whereas the littermate controls did not contain any MBP+ OLs in those brain regions (Figures 2G and 2G′). At P56, both ATG7 cKO and ATG5 cKO mice exhibited significantly increased MBP+ OL numbers in the cortex and hippocampus (Figures 2H–2M and S2A–S2C). These phenotypes were not caused by defects in OPC migration, patterning, or proliferation, as evidenced by the unchanged distribution, number, and proliferation rate of OPCs in ATG7 or ATG5 cKO mice (Figures S2L–S2N and S2D–S2E). Thus, genetic perturbation of autophagy in OL lineage cells generates ectopic OLs in the cerebellar ML and throughout the brain, disrupting the spatiotemporal specificity of oligodendrogenesis and myelination.
Autophagy functions cell autonomously to promote apoptosis in subsets of pre-OLs
To address the cellular mechanisms underlying autophagy-dependent pre-OL elimination, we analyzed autophagy flux in ATG5- and ATG7-deficient OLs. We found that the level of autophagy adaptor protein p62 was significantly increased and that autophagosome marker LC3-II displayed a significant decrease at differentiation days 4 (pre-OL stage) and 6 (mature OL stage) in ATG5 cKO OLs compared with control cells (Figure 3A, quantified in Figure 3B). To visualize autophagosome abnormalities in ATG5 cKO OLs in culture, we conducted ultra-resolution TEM analysis and found that ATG5 cKO OLs exhibited several deficits in autophagosome formation, including impaired autophagosome morphology and significantly enlarged autophagosome size and total area (Figures 3C–3F′, quantified in Figure 3G, and S3A–S3C). ATG7 cKO OLs exhibited similar defects on differentiation day 4 when they were treated with BafA1 to temporarily block autophagosome-lysosome fusion (Figures S3D–S3G″, quantified in Figures S3H and S3I). Notably, in the presence of BafA1, ATG7 cKO OLs harbored increased numbers of large degradative compartments (DGCs) that contained autophagosomes and amphisomes (compare Figures S3G–S3G″ with Figures S3F–S3F″). Thus, ATG5− and ATG7-deficient OLs exhibit disrupted autophagy flux and aberrant autophagosomes during differentiation.
To test if autophagy promotes pre-OL cell death, we first analyzed the cerebellar ML at P11, a stage at which wild-type pre-OLs undergo transient differentiation followed by rapid programmed cell death (Figures 4A and 4B).4 Because ATG5 and ATG7 cKO mice exhibit similar phenotypes (Figures 2 and 3), we focused on analyzing ATG5 cKO mice for the rest of our study. By using an antibody raised against MBP, which begins its expression in pre-OLs,20 we found that a fraction of MBP+ pre-OLs were cleaved caspase-3+ in ATG5F/F cerebellar ML (Figure 4B′ and 4B″, quantified in Figure 4E). Although MBP+ cleaved caspase-3+ double-positive cells were occasionally found in ATG5 cKO cerebellar ML (Figures 4D–4D″), the ratio of MBP+ cleaved caspase-3+ double-positive cells over total MBP+ cells in ATG5 cKO mice was significantly reduced compared with in littermate controls (Figures 4C–4C″, quantified in Figure 4E). Consistent with these results, ATG5 cKO mice exhibit increased numbers of ENPP6+ and Pcdh17it+ pre-OLs in the cerebellar ML and cortex (Figures 4G–4J and S4A–S4D). Importantly, these deficits were not due to increased OPC-to-pre-OL differentiation, as the percentages of MBP+ and galactocerebroside+ (GalC+) cells were not changed in ATG5 cKO OLs in vitro (Figures S4H–S4N″), and the density of EdU pulse-labeled OPCs that subsequently differentiated into pre-OLs (EdU+ Pcdh17it+) remained unchanged in vivo (Figures S4E–S4G). Therefore, ATG5 cKO mice exhibit reduced pre-OL cell death and transiently elevated pre-OL numbers.
Figure 4. Autophagy functions cell autonomously to promote pre-OL apoptosis.
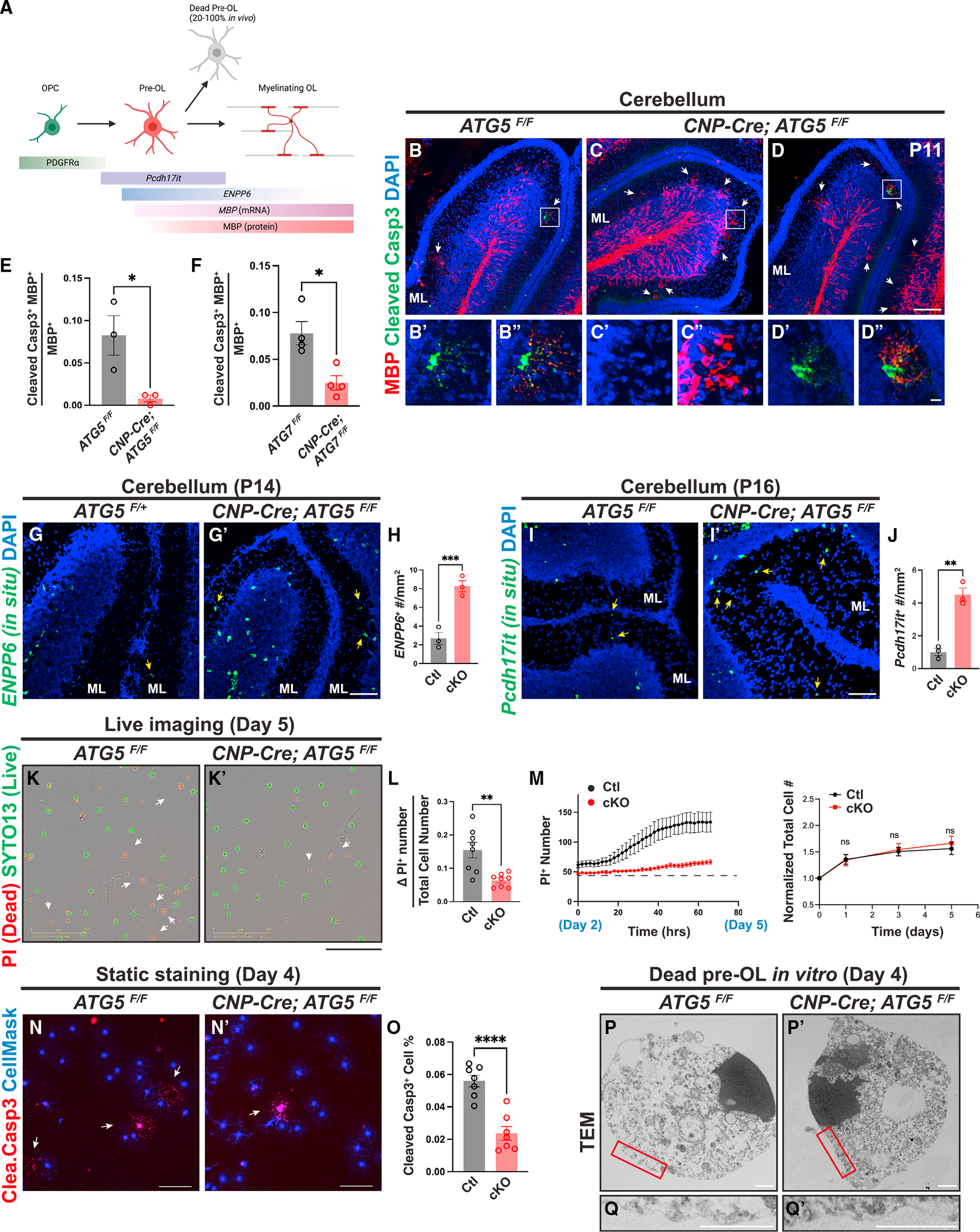
(A) Diagram showing OL differentiation and the markers that delineate OPC, pre-OLs, and myelinating OLs (modified from Kasuga et al.24 and created by BioRender).
(B–D″) Representative confocal micrographs of P11 cerebella from ATG5F/F (B–B″) and CNP-Cre; ATG5F/F (C–D″) stained by MBP (red) and cleaved caspase-3 (green) antibodies. (B′) and (B″), (C′) and (C″), and (D′) and (D″) represent the insets in (B), (C), and (D), respectively. White arrows indicate transiently differentiated pre-OLs in the cerebellar ML at P11.
(E and F) Quantification of the ratio of MBP+ cleaved caspase-3+ double-positive cells over total MBP+ cells in the cerebellar ML from P11 ATG5 cKO (E) and P10 ATG7 cKO (F) mice.
(G and H) In situ hybridization using the ENPP6 probes (G and G′) and quantification (H) showing that CNP-Cre; ATG5F/F mutants exhibited increased ENPP6+ cells in the cerebellar ML at P14 (yellow arrows in G and G′).
(I, I′, and J) In situ hybridization using the Pcdh17it probes (I and I′) and quantification (J) showing that CNP-Cre; ATG5F/F mutants exhibited increased Pcdh17it+ cells in the cerebellar ML at P16 (yellow arrows in I and I′).
(K and K′) Representative live-cell imaging micrographs of ATG5F/F (K) and CNP-Cre; ATG5F/F (K′) OLs at differentiation day 5 labeled by propidium iodide (PI; red for dead cells) and SYTO13 (green for live cells).
(L) Quantification of the increased PI+ cell number from day 2 to 5 (ΔPI+ number, reflecting pre-OL cell death) to the total cell number at day 5.
(M) Left: representative curves of PI+ cell number from differentiation day 2 (before pre-OL stage) to 5 (after pre-OL stage). Right: normalized total cell number (including live and dead cells) between control and ATG5 cKO OLs during in vitro differentiation.
(N and N′) Representative micrographs of ATG5F/F (N) and CNP-Cre; ATG5F/F OLs (N′) showing cleaved caspase-3+ cells (red) at differentiation day 4.
(O) Quantification of cleaved caspase-3+ cell percentage at differentiation day 4.
(P–Q′) Representative TEM micrographs of ATG5F/F (P and Q; n = 31 cells) and CNP-Cre; ATG5F/F OLs (P′ and Q′; n = 15 cells) at differentiation day 4. (Q) and (Q′) represent the red insets in (P) and (P′), respectively.
Error bars represent SEM. Scale bars: 100 μm in (D) for (B)–(D); 10 μm in (D″) for (B′)–(D′); 100 μm in (G) for (G) and (G′); 100 μm in (I′) for (I) and (I′); 200 μm in (K′) for (K) and (K′); 100 μm in (N) and (N′); and 1 μm in (P)–(Q′). Open circles in (E), (F), (H), and (J) represent individual animals; n ≥ 3 animals per category. n = 8 wells (L and M) and n = 7 coverslips (O) from 3 separate OPC purifications. Two-tailed t tests for (E), (F), (H), (J), (L), (M), and (O). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
To address whether autophagy functions cell autonomously to promote pre-OL programmed cell death, we acutely purified OPCs from ATG5 cKO’s and littermate control’s brains and performed live-cell imaging assays from the onset of OPC differentiation (day 0) to day 5, when the cells fully differentiated to OLs. We utilized a fluorescent dye-conjugated Annexin V probe to monitor apoptosis during OL differentiation.4 As expected, we found that a subset of control OLs were Annexin V+ on day 3 when they first differentiated into the pre-OL stage (Figure S4O; see also Video S1). In contrast, ATG5 cKO OLs exhibited significantly reduced Annexin V+ areas (compare Figures S4O and S4O′, quantified in Figures S4P–S4Q; see also Video S2). This result was further confirmed by the live-cell imaging assay using another cell death marker, propidium iodide (PI), which labels dead cell nuclei (Figures 4K and 4K′, quantified in Figures 4L–4M), as well as by immunostaining of cleaved caspase-3 in pre-OLs (Figures 4N and 4N′, quantified in Figure 4O). Finally, to rule out the possibility that pre-OLs undergo a different form of programmed cell death other than apoptosis, we analyzed dead pre-OL’s ultra-structure by TEM. Wild-type dead pre-OLs exhibited apoptotic characteristics including chromatin condensation and maintenance of intact plasma membrane (Figures 4P, 4Q, S4R, and S4S). Similarly, unlike autosis, which exhibits swollen perinuclear space or necrosis where the plasma membrane erupts,25,26 dead ATG5 cKO pre-OLs still displayed apoptotic characteristics (Figures 4P′, 4Q′, S4R′, and S4S′). Taken together, our results showed that autophagy functions cell autonomously to promote apoptosis in subsets of pre-OLs.
Autophagy genetically interacts with the TFEB pathway to control pre-OL cell fate
To determine the molecular mechanisms through which autophagy promotes pre-OL apoptosis, we attempted to identify proteins that were misregulated when autophagy flux was disrupted in pre-OLs by quantitative liquid chromatography with tandem mass spectrometry (LC-MS-MS) (Figure S5A). ATG5 was among the most downregulated proteins and autophagy adaptor protein p62 and Gabarapl2/ATG8 were significantly upregulated in ATG5 cKO pre-OLs, showing that the assay was successful (Figure S5B). We found that proteins involved in ER-phagy (Calcoco1 and Retreg1), protein kinase A activity (Prkar1a), and lipid biosynthesis and metabolism were significantly upregulated in ATG5 cKO OLs (Figures S5B and S5C; Table S2). Due to its limited sensitivity, this assay did not provide a clear indication of the candidates directly involved in the apoptosis pathways. We then employed the bulk RNA-seq approach to quantitatively measure transcriptomic changes when autophagy flux was perturbed (Table S3). We focused on the apoptosis pathways and found that genes directly involved in apoptosis, including Pmaip1 (Noxa) and PUMA (Bbc3), exhibited significantly decreased expression levels in ATG5 cKO pre-OLs (Figure 5A). We further validated that mRNA and protein levels of PUMA, a pro-apoptotic gene belonging to the Bcl-2 family, were significantly reduced in ATG5 cKO pre-OLs (Figures 5B and 5C). Consistent with the RNA-seq results, major pro- and anti-apoptotic proteins including Bax, Bak, Bcl2, and XIAP remained unchanged in ATG5 cKO pre-OLs (Figures 5B, 5D, and S5F).
Figure 5. Autophagy genetically interacts with the TFEB pathway to control pre-OL cell fate.
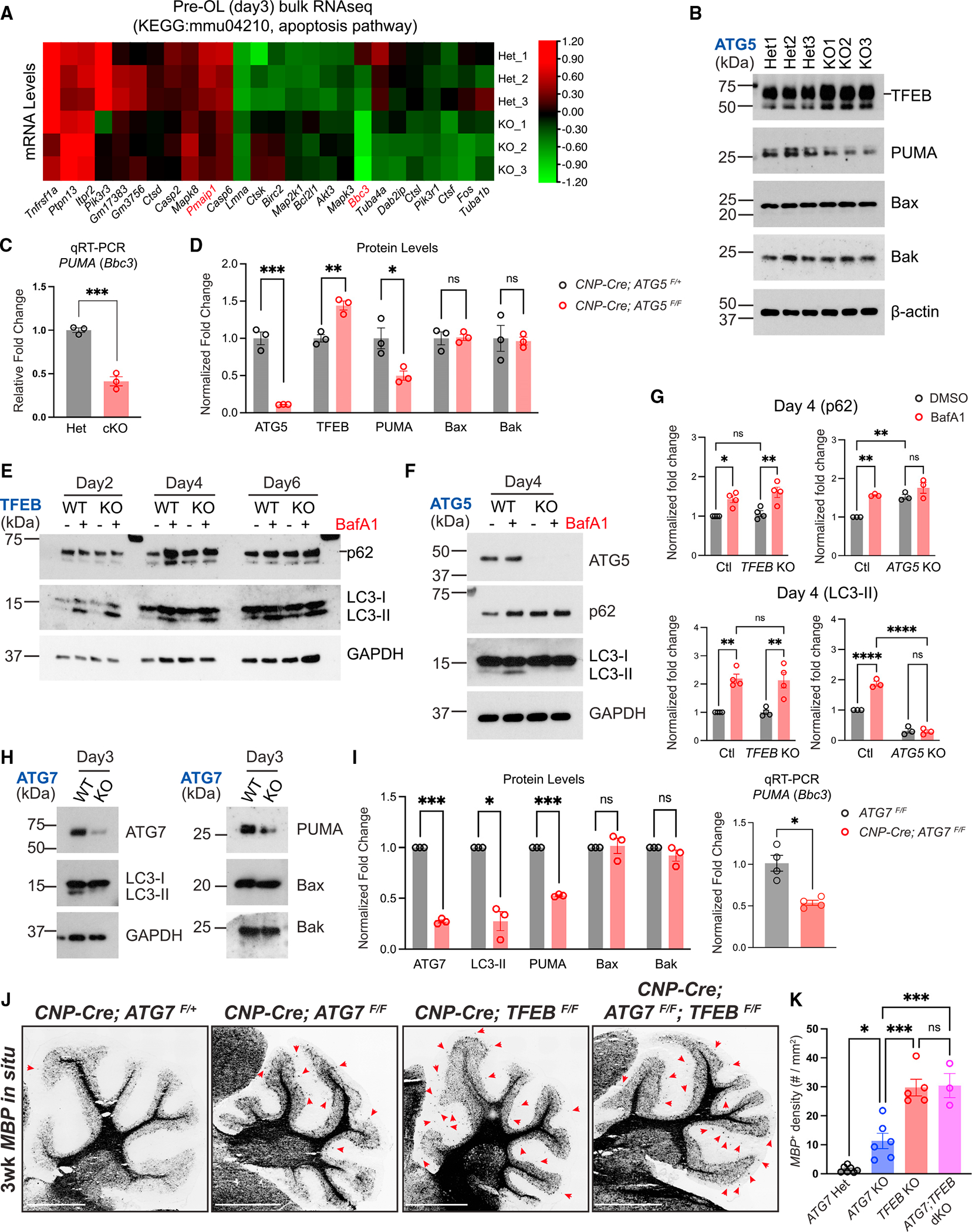
(A) Heatmap showing expression levels (in Z scores) of genes belonging to the KEGG mmu04210: apoptosis pathway that were significantly up- or downregulated (adjusted p value < 0.05) in ATG5 cKO pre-OLs at differentiation day 3 by bulk RNA-seq. Each row represents a biological repeat, and each column represents a gene. The heatmap was generated using the TBtools software.27
(B) Western blot analysis of ATG5 Het and ATG5 cKO pre-OLs at differentiation day 3. The short line indicates the protein band of TFEB.
(C) Quantitative RT-PCR showing PUMA mRNA levels in control and ATG5 cKO pre-OLs.
(D) Quantification of ATG5, TFEB, PUMA, Bax, and Bak protein levels in ATG5 Het and ATG5 cKO pre-OLs at differentiation day 3. n = 3 separate cell purifications.
(E) Biochemical analysis of autophagy flux in TFEB KO OLs with or without BafA1 compared with wild-type (WT) cells under the same conditions. The short line indicates the protein band of p62.
(F) Western blot analysis of p62 and LC3-I/II levels in the presence or absence of BafA1 in ATG5 WT (ATG5F/F) and KO (CNP-Cre; ATG5F/F) pre-OLs.
(G) Quantification of p62 and LC3-II protein levels in OLs at differentiation day 4 (pre-OL stage) in the presence or absence of BafA1. The protein levels were normalized to GAPDH. Two-way ANOVA followed by Sidak’s multiple comparisons test. n = 4 separate cell purifications for TFEB control and KO. n = 3 separate cell purifications for ATG5 control and KO.
(H) Western blot analysis of LC3-II, PUMA, Bax, and Bak protein levels in ATG7 WT and ATG7 cKO pre-OLs.
(I) Quantification of ATG7, LC3-II, PUMA, Bax, and Bak protein levels as well as PUMA mRNA levels in ATG7 WT and ATG7 cKO pre-OLs at differentiation day 3. n = 3 separate cell purifications for western blot analysis. n = 4 separate cell purifications for qRT-PCR analysis.
(J) Representative micrographs from 3-week-old control and mutant cerebella labeled by MBP in situ probes. Red arrows indicate ectopic OLs in the cerebellar ML.
(K) Quantification of MBP+ OL density in the cerebellar ML. Open circles represent individual animals; n ≥ 3 animals per category.
Error bars represent SEM. Scale bars: 1 mm in (J). Two-tailed t tests for (C), (D), and (I). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant.
Previous work showed that Foxo3, p53, and c-Jun can function as upstream transcriptional activators for PUMA, thereby promoting cell death under different cellular contexts.28,29 To test if autophagy regulates these upstream transcription activators to promote pre-OL apoptosis, we first analyzed an OL-specific Foxo3 KO mouse strain (CNP-Cre; Foxo3F/F, or Foxo3 cKO). We found that Foxo3 cKO mice exhibited normal myelination in the cerebellum (Figures S5G and S5G′). Moreover, ATG5 cKO pre-OLs exhibited unchanged protein levels of c-Jun and phosphorylated c-Jun (Ser73) (Figure S5F). Together with a previous study showing that genetic deletion of p53 in OLs does not affect their survival during development,4 our results suggest that autophagy regulates PUMA mRNA levels through an uncharacterized mechanism.
TFEB has been shown to be a master regulator to induce autophagy in a variety of cells upon starvation and stress.30 A previous study revealed that TFEB promotes PUMA/Bbc3 transcription and subsequently triggers Bax/Bak-dependent apoptosis in pre-OLs.4 To determine if TFEB induces autophagy in pre-OLs, we performed western blot analysis on control (TFEBF/F) and OL-specific TFEB KO pre-OLs (TFEB KO, Olig2-Cre; TFEBF/F) with a battery of autophagy markers (Figure S5E). As expected, in the presence of BafA1, p62 and LC3-II levels were significantly increased in control pre-OLs (Figure 5E, quantified in Figure 5G). However, TFEB KO pre-OLs exhibited similar levels of p62 and LC3-II compared with control cells in the presence or the absence of BafA1 (Figures 5E and S5H, quantified in Figure 5G), suggesting that autophagy flux was minimally affected in TFEB KO pre-OLs. These observations were in strong contrast to ATG5 cKO pre-OLs, as they showed diminished LC3-II in the presence or the absence of BafA1, and their p62 levels did not respond to BafA1 treatment (Figure 5F, quantified in Figure 5G). Finally, we found that TFEB mRNA and protein levels were not reduced in ATG5 cKO pre-OLs (Figures 5B and S5D, quantified in Figure 5D).
The TFEB-PUMA-Bax/Bak pathway strongly promotes pre-OL apoptosis in vitro and in vivo (Figure S5I).4 To determine if autophagy functions in this pathway or acts in a separate pathway, we conducted genetic interaction analysis and characterized MBP+ cells in the cerebellar ML among littermates with four genotypes, including the CNP-Cre; ATG7F/+ (ATG7 heterozygous [Het]), CNP-Cre; ATG7F/F (ATG7 KO), CNP-Cre; TFEBF/F (TFEB KO), and CNP-Cre; ATG7F/F; TFEBF/F (ATG7; TFEB dKO). Like ATG5-deficient OLs, ATG7-deficient OLs exhibited significantly reduced PUMA mRNA and protein levels, as well as decreased apoptosis in vitro (Figures 5H–5I and S5J–S5L). In vivo, as expected, ATG7 KO mice harbored significantly higher numbers of MBP+ cells in the cerebellar ML compared with ATG7 Het mice (left two panels in Figure 5J,. quantified in Figure 5K). TFEB KO mice exhibited enhanced phenotypes compared to ATG7 KO mice, but co-deletion of TFEB and ATG7 in OL lineage cells (CNP-Cre; ATG7F/F; TFEBF/F) did not further enhance the TFEB KO phenotype (right two panels in Figure 5J, quantified in Figure 5K). These results suggest that autophagy acts in the TFEB axis to limit OL number. Taken together, autophagy functions cell autonomously to promote apoptosis in subsets of pre-OLs, thereby controlling OL number and regulating myelination specificity during development.
DISCUSSION
The fine balance between cell construction and destruction governs organogenesis, tissue remodeling, cancer, and aging.31 Autophagy and apoptosis pathways intimately interact with each other to determine cell fate. Although autophagy can promote apoptosis in invertebrates, it remains controversial whether it plays similar functions in vertebrates under physiological conditions. In this study, we revealed that autophagy promotes apoptosis in subsets of immature myelinating glia (pre-OLs), thereby controlling OL number and regulating the spatiotemporal specificity of CNS myelination.
What are the molecular mechanisms underlying autophagy-mediated pre-OL apoptosis? Our data showed that autophagy genetically interacts with the TFEB pathway, which powerfully promotes pre-OL apoptosis.4 Intriguingly, both in vitro and in vivo phenotypes of autophagy-deficient pre-OLs are milder than those shown by TFEB cKO and PUMA−/− mutants (Figure S5).4 In addition, there is no enhancement of the phenotype in ATG7; TFEB double-KO mice compared with TFEB cKO mice (Figure 5). Together, these data suggest that the TFEB-PUMA-Bax/Bak apoptotic pathway is the main driver for pre-OL cell death, while autophagy plays a modulatory role. Moreover, our data showed that autophagy is not induced by TFEB under basal conditions during OL differentiation (Figure 5). This is consistent with recent work showing that Tfeb and Tfe3 are dispensable for basal levels of autophagy and lysosomal gene expression under homeostatic conditions in zebrafish microglia.32 Therefore, further work is needed to fully understand TFEB’s roles and underlying mechanisms in glial cells.
Although ATG5 and ATG7 are long considered elongation factors for autophagosomes, ATG5 and ATG7 cKO OLs exhibit enlarged autophagosome areas. One possible explanation is that ATG5 and ATG7 might be critical for autophagosome closure in OLs, disruption of which can lead to defective autophagosome morphology and increased size.33,34 Furthermore, even though ATG5 and ATG7 are essential for autophagy flux, we cannot rule out the possibility that they may function as direct mediators of apoptosis. For instance, in various cell lines under apoptotic stimuli, ATG5 undergoes cleavage by the enzyme calpain to generate a fragment that is capable of translocating to mitochondria. There it deactivates BCL-xL and activates Bax, ultimately leading to apoptosis.35 Moreover, ATG5 and ATG7 might participate in the formation of other vesicle-like structures including endosomes and exosomes.36,37 Therefore, genetic deletion of ATG5 or ATG7 in pre-OLs, a sensitive stage during OL differentiation and membrane expansion, could elicit pre-OL metabolic stress and contribute to pre-OL cell death. It will be of great interest to determine the precise molecular mechanisms governing ATG5/7-mediated pre-OL apoptosis.
A previous study found that conditionally deleting ATG5 in OPCs via an inducible manner causes OL cell death, hypomyelination, and lethality early postnatally.38 However, our work showed that four cKO mouse strains with genetic perturbation of autophagy in the OL lineage cells are viable. In fact, we found opposite phenotypes in these mutants where all of them exhibit increased OL numbers across diverse brain regions. Our results are consistent with a recent finding showing that ATG7 cKO mice are viable until at least 6 months of age and that they exhibit hypermyelination in adulthood.17 Our study, which was primarily focused on developmental myelination, uncovered a new ectopic myelination phenotype that has not been previously reported. Given the complexity of autophagy during OL differentiation and myelination, further studies are necessary to elucidate the differentiation-stage-specific functions of autophagy and the underlying molecular mechanisms.
In conclusion, our findings show that autophagy promotes pre-OL apoptosis to limit OL number during development. Our observations provide a mechanistic link connecting autophagy and apoptosis, two evolutionarily conserved cellular processes that intimately interact with each other to determine cell fate.
Limitations of the study
In this study, we show that ATG5 and ATG7 function cell autonomously to eliminate subsets of pre-OLs in vitro and in vivo. Although our work supports a model where autophagy acts in the TFEB axis to control pre-OL cell fate, the precise molecular mechanisms governing autophagy-mediated apoptosis remain elusive and require further investigation. Furthermore, we do not know if the reduced levels of PUMA in ATG5 and ATG7 KO OLs are responsible for the decreased pre-OL apoptosis in those mutants. Finally, our study is limited by the usage of ATG5 and ATG7 cKO mice, and we have not exhaustively tested whether deleting other autophagy genes in OLs will lead to similar phenotypes.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lu O. Sun (Lu.Sun@UTSouthwestern.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Proteomic datasets related to Figure S5 in the paper are available in Table S2. The gene expression file of bulk RNA-seq related to Figure 5 are available in Table S3. The raw data of proteomics have been deposited to MassIVE (accession number: MSV000092434). The raw data of bulk RNA-seq have been deposited at NCBI BioProject (accession number: PRJNA986522).
No original codes were generated in this study.
Any additional information related to this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All experiments related to animals complied with the Guide for Care and Use of Laboratory Animals and followed protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Texas Southwestern Medical Center. Mice were housed on normal light-dark cycles (12:12) with food and water ad libitum. The day of birth in this study was designated postnatal day 0 (P0) and all the developmental stages were specified in figures and figure legends. Both male and female mice were used for the experiments and analyses. Transgenic animals include the ATG7Flox (ATG7F, RIKEN RBRC02759), ATG5Flox (ATG5F, RIKEN RBRC02975), Olig2-Cre (Jax#025567), CNP-Cre,40 PDGFRα-CreERT2 (Jax#018280), TFEBFlox (TFEBF),4 PUMA− (Jax#011067),4 and Foxo3Flox (Foxo3F, Jax#024668).
Immunopanning purification of mouse oligodendrocyte precursor cells (OPCs), in vitro differentiation, and bafilomycin A1 treatment
The immunopanning purification procedure was following the protocol previously described.4 In brief, postnatal day 6 to day 10 pups were quickly decapitated, and the whole brains except olfactory bulbs were dissected out and diced into ~1mm3 chunks. The tissues were immediately subjected to digestion with a buffer containing 1× Earle’s Balanced Salt Solution (EBSS 10×; Sigma-Aldrich, Cat#E7501) supplemented with 1 mM MgSO4, 0.46% glucose, 2 mM EGTA, 26 mM NaHCO3, 20 units/ml of papain (Worthington Biochemical, Cat#LS003126), and 250 units/ml of DNase I (Worthington Biochemical, Cat#LS002007) under 5% CO2/95% O2 gas flow at 34°C for 90 min. The tissues were then triturated with 1 mL pipettes to form a single-cell suspension. The single-cell suspension was incubated on three BSL-1-coated Petri dishes (Vector Laboratories, Cat#L1100) for 10 min each to get rid of endothelial cells and microglia. Finally, the suspension was incubated on a rat anti-PDGFRα antibody (rat anti-mouse CD140a, BD Biosciences, Cat#558774) coated Petri dish for harvesting OPCs.
The purified OPCs were plated in the proliferation medium containing DMEM-SATO Base Growth Medium39 supplemented with 4.2 μg/mL forskolin (Sigma-Aldrich, Cat#F6886), 10 ng/mL PDGF (Peprotech, Cat#100–13A), 10 ng/mL CNTF (Peprotech, Cat#450–02), and 1 ng/mL neurotrophin-3 (NT-3; Peprotech, Cat#450–03) in a 37°C 10% CO2 humidified incubator. To differentiate OPCs in vitro, the proliferation medium was replaced with the differentiation medium containing DMEM-SATO Base Growth Medium supplemented with 4.2 μg/mL forskolin, 10 ng/mL CNTF, and 40 ng/mL thyroid hormone (T3; Sigma-Aldrich, Cat#T6397). Half of the culture medium was replaced with fresh medium every 2 days.
To treat cultured oligodendrocytes with bafilomycin A1 (BafA1), 50 nM BafA1 (Sigma-Aldrich, Cat#196000) diluted in DMSO or the same amount of DMSO (vehicle) was added into cell culture medium and cells were incubated at 37°C for 2–3 h.
METHOD DETAILS
In situ hybridization and quantification
In situ hybridization was performed on fresh frozen sagittal brain sections with 10-μm or 12-mm thickness using the RNAscope Fluorescent Multiplex Reagent Kit (ACDbio, Cat#320850) or RNAscope Multiplex Fluorescent Reagent Kit v2 with TSA Vivid Dyes (ACDbio, Cat#323270). MBP in situ probes (Cat#451491 and Cat#451491-C2), PDGFRα probes (Cat#480661 and Cat#480661-C2), MOG probes (Cat#492981-C2), ENPP6 probes (Cat#511021-C2), and Pcdh17it probes (Cat#526741-C3) were used per manufacturer’s instructions. Fluorescent images were taken using the Keyence BZ-810 all-in-one fluorescence microscope or Zeiss LSM700 inverted confocal microscope. For the quantification of oligodendrocyte lineage cell numbers in different brain regions, the entire brain of each brain section was captured using the automated image-stitching function of Keyence BZ-810. The stitched images were analyzed by the ImageJ software. Briefly, the area of different brain regions was selected by the “Freehand Selections” tool followed by the area measurement. The cell number was counted, and the density was calculated afterward. The cell density from 1 or 2 brain sections per animal was quantified and averaged, and at least 3 animals per genotype were analyzed.
Immunohistochemistry
For mouse brain staining, mice were euthanized with CO2 and immediately perfused with chilled PBS and 4% PFA in PBS solutions. The brains were post-fixed in 4% PFA/PBS solution for 3 h at 4°C and then cryopreserved with 30% sucrose in PBS at 4°C for 24 h. Sagittal brain sections with 18-μm thickness were used for immunostaining. The sections were incubated with 10% normal goat serum (Jackson ImmunoResearch, Cat# 005-000-121) or 10% normal donkey serum (Jackson ImmunoResearch, Cat#017-000-121) in PBS supplemented with 0.1% Triton X-100 (PBST; Sigma-Aldrich, Cat#T8787) for 10–15 min at room temperature. The sections were then incubated with the diluted primary antibodies in the staining solution (PBST supplemented with 2% normal goat or donkey serum) at 4°C overnight. The brain sections were rinsed 20 min for 4 times in PBST at room temperature and incubated with appropriate AlexaFluor 488- or 594-conjugated secondary antibodies (Thermo Fisher Scientific, 1:1000) and DAPI (Thermo Fisher Scientific, 5 μg/mL, Cat#D1306) for 1 h at room temperature. The sections were rinsed 20 min for 4 times in PBST at room temperature and mounted with coverslips in the VectaShield Hardset Antifade mounting medium (Vector Laboratories, Cat#H-1400-10). Images were taken by a Keyence BZ-810 all-in-one fluorescence microscope or a Zeiss LSM700 inverted confocal microscope. For the quantification of MBP fluorescent intensity in the cerebellum molecular layer, the entire cerebellum of each brain section was captured using the automated image-stitching function of Keyence BZ-810. The stitched images were analyzed by the ImageJ software. Briefly, the images were converted to a binary image with the “Make Binary” tool in ImageJ, then the molecular layer area of cerebellum was then selected on the binary image by the “Freehand Selections” tool. The readout of “mean gray value” was used for MBP fluorescent intensity. The fluorescence density from 2 brain sections per animal was quantified and averaged, and at least 3 animals per genotype were analyzed.
For cellular immunostaining, oligodendrocytes were cultured on Poly-D-lysine (Sigma-Aldrich, Cat# P6407) coated 12-mm plastic coverslips at the density of 10,000 cells/coverslip. Cells were rinsed with PBS and fixed with 4% PFA for 10–15 min and rinsed twice with PBS. The cells were then permeabilized and blocked with 10% normal donkey serum in PBST for 10–15 min. The cells were incubated with the diluted primary antibodies in PBS supplemented with 2% normal donkey serum solution overnight. The cells were washed with PBS 20 min for 4 times and incubated with AlexaFluor 488- or 594-conjugated secondary antibodies (Thermo Fisher Scientific, 1:1000) and DAPI (Thermo Fisher Scientific, 5 μg/mL) or HCS CellMask Blue Stain (Thermo Fisher Scientific, 1:1000, Cat#H32720) for 1 h at room temperature. After 4 times of PBS wash, cells were mounted in the Prolong Gold Antifade mounting medium (Thermofisher, Cat#P10144). Images were taken with the Keyence BZ-810 all-in-one fluorescence microscope.
Primary antibodies used in this study include: rat anti-MBP (Abcam, 1:100 for tissue staining and 1:500 for cell staining, Cat#ab7349), rabbit anti-cleaved caspase-3 (Asp175) (Cell Signaling Technology, 1:2000 for tissue staining and 1:500 for cell staining, Cat#9661S), mouse anti-calbindin (Swant,1:200 for tissue staining,Cat#CB38), rabbit anti-PDGFRα (Santa Cruz Biotechnology, 1:200 for tissue staining, Cat#sc-338), and mouse anti-galactocerebroside (Galc) hybridoma (1:50 for cell staining).43
Tamoxifen injections
Tamoxifen was dissolved in sunflower oil at the concentration of 10 mg/mL and was injected at postnatal day 4 intraperitoneally with the 100 mg/kg dosage.
Live-cell imaging
OPCs were seeded into PDL-coated 24-well plates at the density of 5,000 cells/well. Cells were maintained in the proliferation medium for 1–2 days before subjecting to the differentiation medium. For Annexin V imaging, cells were incubated with the differentiation medium supplemented with Alexa 594-conjugated Annexin V (Thermo Fisher Scientific, 1:500, Cat# A13203) or with IncuCyte Annexin V red dye for apoptosis (Sartorius, 1:200, Cat#4641). The plates were placed into IncuCuyte live cell imaging system (Sartorius) and imaged continuously for 5 days with 2-h intervals at 37°C and 10% CO2. Half of the differentiation medium was replaced with fresh differentiation medium supplemented with the indicated dye every 2 days. The Annexin V+ area was quantified by the IncuCyte software provided by the vendor. For propidium iodide (PI) staining, the cells were incubated with the differentiation medium for 2 days before the live-cell imaging. At the end of differentiation day 2, half of the medium was replaced with fresh differentiation medium supplemented with PI (Thermo Fisher Scientific, 1:2000, P1304MP). The plates were placed into IncuCyte live cell imaging system and imaged continuously for 3 days with a 2-h frame. PI+ cell numbers were quantified by the IncuCyte software. At the end of living imaging, Calcein AM (Thermo Fisher Scientific, 1:1000, Cat#C3100MP) or SYTO13 green dye (Thermo Fisher Scientific, 1:10000, Cat# S7575) was added into the medium to label live cells.
Western immunoblotting and quantification of protein expression
Oligodendrocytes cultured on 6-well plates (100,000 OPCs per plate as the plating density), 12-well plates (50,000 OPCs per plate as the plating density), or 6-cm dishes (250,000 OPCs per plate as the plating density), were rinsed with chilled PBS and lysed in RIPA Buffer (Thermo Fisher Scientific, Cat#89900) supplemented with 1× cOmplete protease inhibitor cocktail (Sigma-Aldrich, Cat#5892791001) and 1× PhosSTOP phosphatase inhibitors (Sigma-Aldrich, Cat#4906845001) for 1 h on a rocker at 4°C. Cell lysates were collected and centrifuged for 10 min at 14,000 rpm. The supernatants were collected for use. Protein concentration was determined by a BCA assay (Thermo Fisher Scientific, Cat#23225), and the samples were denatured with 4×LDS sample buffer (Thermo Fisher Scientific, Cat#NP0007) supplemented 10% 2-Mercaptoethanol (Sigma-Aldrich, Cat#M6250) for 10 min at 95°C. 1–5 μg total proteins were loaded into stain-free 4–15% or 4%–20% gradient precast polyacrylamide gel (Bio-Rad, Cat#4568086 or 4568096) for separation. The total protein images were captured by the Bio-Rad ChemiDoc imaging system with the stain-free gel imaging protocol. Then proteins were then transferred to 0.45 μM PVDF membranes (Thermo Fisher Scientific, Cat#88518) at 100V for 1 h on ice. Blots were blocked with 5% non-fat milk in TBS buffer with 0.1% Tween 20 (TBST) at room temperature for 1 h and then incubated with primary antibodies diluted in TBST with 3% BSA overnight at 4°C on a rocker. Blots were washed with TBST for 3 × 10 min and incubated with HRP-conjugated secondary antibodies in 5% non-fat milk at room temperature for 1 h. Blots were washed with TBST for 6 × 5 min. Blots were developed with Amersham ECLprime western blotting detection reagent (Cytiva, Cat#RPN2232) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermofisher, Cat#34095) for 3 min and imaged with X-ray films. For protein quantification, films were scanned, quantified with ImageJ and normalized to total proteins, GAPDH, or β-actin as indicated in the figure legends. For the experiment conducted with each cell purification carried out separately (Figures 1 and 3, 5E–I), we normalized the protein levels for each replicate separately. For the experiment performed with multiple cell purifications at the same time (Figures 5B and 5D), we normalized the protein levels to the average level of loading controls.
Primary antibodies used include: rabbit anti-TFEB (Bthyl Laboratories, 1:2000, Cat# A303–673A), mouse anti-β-actin (Santa Cruz Biotechnology, 1:500, Cat#sc-47778), rat anti-MBP (Abcam, 1:500, Cat#ab7349), rabbit anti-Beclin1 (Cell Signaling Technology, 1:1000, Cat#3495S), rabbit anti-ATG5 (Cell Signaling Technology, 1:1000, Cat#12994S), rabbit anti-ATG7 (Cell Signaling Technology, 1:1000, Cat#8558S), rabbit anti-PUMA (Cell Signaling Technology, 1:1000, Cat#24633S), rat anti-LAMP2 (Abcam, 1:1000, Cat#ab13524), rabbit anti-p62 (Cell Signaling Technology, 1:1000, Cat# 5114S), rabbit anti-Bak (Cell Signaling Technology, 1:1000, Cat#12105T), rabbit anti-Bax (Cell Signaling Technology, 1:1000, Cat#2772T), rabbit anti-XIAP (Cell Signaling Technology, 1:200, Cat#14334S), mouse anti-Bcl2 (Santa Cruz Biotechnology, 1:200, Cat# sc-7382), mouse anti-WIPI2 (Bio-Rad, 1:1000, Cat#mca5780GA), mouse anti-LC3 (MBL, 1:500, Cat#M186–3), rabbit anti-c-Jun (Cell Signaling Technology, 1:1000, Cat#9165T), rabbit anti-phospho-c-Jun (Ser73) (Cell Signaling Technology, 1:1000, Cat#3270T), and mouse anti-GAPDH (Santa Cruz Biotechnology, 1:500, Cat#sc-32233).
EdU pulse labeling
EdU (5-ethynyl-2′-deoxyuridine) was dissolved in DMSO at a concentration of 10 μg/mL as the stock solution.
For OPC proliferation assays, EdU was administered into P13 CNP-Cre; ATG7F/F and littermate control animals to pulse-label OPCs via intraperitoneal injections at a dose of 50 μg/g body weight. Mice were perfused with the chilled 4% PFA/PBS solution 24 h post-injection, and brains were collected and cryopreserved. Sagittal brain sections with 16-mm thickness were incubated with the primary antibody (rat anti-PDGFRα, Thermo Fisher Scientific, 1:100, Cat#14140182) at 4°C overnight followed by the incubation with Alexa Fluor 488 secondary antibodies (Thermo Fisher Scientific, 1:1000). The Click-iT Plus reaction cocktail for EdU detection was then made and applied as described by vendor’s protocol (Thermo Fisher Scientific, Cat#C10639).
For OPC differentiation assays, EdU was administered into P11 CNP-Cre; ATG5F/F and littermate control animals via intraperitoneal injections at a dose of 50 μg/g body weight. Brains were collected 5 days post-injection. Sagittal fresh-frozen brain sections with 12-μm thickness were subjected to in situ hybridization using Pcdh17it probes followed by EdU detection. Fluorescent images were taken using the Keyence BZ-810 all-in-one fluorescence microscope or Zeiss LSM700 inverted confocal microscope, and the whole brain region of each brain section was captured using the automated image stitching function of Keyence BZ-810.
Transmission electron microscopy and quantification of autophagosomes
Oligodendrocytes growing on plastic coverslips (Nunc Thermanox Plastic Coverslips, Cat# 174950 Lot#1023669) were fixed with 2.5% (v/v) glutaraldehyde in 0.1 M sodium cacodylate buffer with 2 mM CaCl2 at room temperature for 5 min. The samples were then submitted to the UT Southwestern electron microscopy core for further processing. Briefly, after five rinses in 0.1 M sodium cacodylate buffer, the samples were post-fixed in 1% osmium tetroxide plus 0.8% K3[Fe(CN6)] in 0.1 M sodium cacodylate buffer for 1 h at room temperature. Cells were rinsed with distilled water and en bloc stained with 2% aqueous uranyl acetate for 1 h. After five rinses with distilled water, specimens were dehydrated with increasing concentration of ethanol, infiltrated with Embed-812 resin. Beem capsules were overfilled with resin and coverslips were placed on top, cell side down, and polymerized in a 60°C oven overnight. Epoxy discs were removed by placing coverslips in liquid nitrogen. Beem capsule blocks were sectioned with a diamond knife (Diatome) on a Leica Ultracut UCT (7) ultramicrotome (Leica Microsystems) and collected onto copper grids. The sections were post-stained with 2% uranyl acetate in water and lead citrate. Images were acquired on a JEOL 1400+ transmission electron microscope (FEI) equipped with a LaB6 source using a voltage of 120 kV and an AMT camera system.
Imaging was performed at low magnifications to image the whole cell. The low-magnification images were used to calculate the whole cell area and nuclear area by tracing their outlines using ImageJ. High-magnification images of each cell were taken to quantify autophagosomes and lysosomes. Autophagosomes and autophagosome-like vesicles were identified by their stereotypical double-membrane enclosed structure with various subcellular contents. To prevent the inclusion of small vesicles that are likely not autophagosomes in the quantification, all structures below 5000 nm2 area were not taken for quantification.44 Elongated structures were not classified as autophagosomes because these structures could be swollen endoplasmic reticulum (ER).
Quantitative mass spectrometry
Oligodendrocytes (~6,000,000/dish) in two 15-cm dishes were briefly rinsed by DPBS and lysed in 500 μL RIPA buffer supplemented with 1×cOmplete protease inhibitor cocktail and 1× PhosSTOP phosphatase inhibitors. Protein concentration was measured by a BCA assay, and 500 mg proteins were used for quantitative mass spectrometry (MS). The samples were processed at the UT Southwestern Proteomics Core. Briefly, samples were reduced with tris (2-carboxyethyl) phosphine (TCEP), alkylated with iodoacetamide in the dark, and digested overnight with trypsin at 37°C using an S-Trap (Protifi). Following digestion, the peptide eluate was dried and reconstituted in 100 mM triethylammonium bicarbonate (TEAB) buffer. The samples were labeled with tandem mass tag (TMT) reagent, quenched with 5% hydroxylamine, and combined. The reverse-phase fractionation spin columns (Thermo Fisher Scientific, Cat#84868) were used according to the manufacturer’s directions to fractionate each sample into 8 fractions. The fractions were dried in a SpeedVac and reconstituted in a 2% acetonitrile, 0.1% trifluoroacetic acid (TFA) buffer. Fractions were injected into an Orbitrap Fusion Lumos mass spectrometer coupled to an Ultimate 3000 RSLC-Nano liquid chromatography system. Samples were injected onto a 75 μm i.d., 75-cm long EasySpray column (Thermo Fisher Scientific) and eluted with a gradient of 0–28% buffer B over 180 min. Buffer A contained 2% (v/v) acetonitrile (ACN) and 0.1% formic acid in water, and buffer B contained 80% (v/v) ACN, 10% (v/v) trifluoroethanol, and 0.1% formic acid in water. The mass spectrometer operated in positive ion mode with a source voltage of 2.0 kV and an ion transfer tube temperature of 275°C. MS scans were acquired at the 120,000 resolution in the Orbitrap and top speed mode was used for SPS-MS3 analysis with a cycle time of 2.5 s. MS2 was performed with CID with a collision energy of 35%. The top 10 fragments were selected for MS3 fragmentation using HCD, with a collision energy of 55%. Dynamic exclusion was set for 25 s after an ion was selected for fragmentation.
Raw MS data files were analyzed using Proteome Discoverer v2.4 (Thermo Fisher Scientific), with peptide identification performed using Sequest HT searching against the mouse protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.6 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of cysteine and TMT labeling of N-terminals and lysine side chains were set as a fixed modification, with oxidation of methionine set as a variable modification. The false-discovery rate (FDR) cutoff was 1% for all peptides.
RNA extraction, reverse-transcription, and quantitative real-time PCR
Oligodendrocytes (~1,000,000/dish) on 10-cm dishes were rinsed with DPBS and the total RNAs were isolated using the RNeasy Micro Kit (Qiagen, Cat#74004). The total RNAs were reverse-transcribed with the RT SuperMix Kit (New England BioLabs, Cat#E3010) according to vendors’ manuals. The cDNAs were used as the template for quantitative real-time PCR in 20 μL reactions using Fast SYBR Green Master Mix (Thermo Fisher Scientific, Cat# 4385612) in a QuantStudio 3 qPCR machine (Thermo Fisher Scientific). The primer efficiency was validated by plotting the template quantity vs. the Ct value and the individual primer pair’s specificity was confirmed with the single-peak melt curves. Primer sequences for mouse genes were shown in key resource table. The quantification was performed with the 2−ΔΔCt method and normalized to GAPDH.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-ATG5 (clone: D5F5U) | Cell Signaling Technology | Cat#12994; RRID: AB_2630393 |
| Rabbit monoclonal anti-ATG7 (clone: D12B11) | Cell Signaling Technology | Cat#8558; RRID: AB_10831194 |
| Rabbit monoclonal anti-Beclin-1 (clone: D40C5) | Cell Signaling Technology | Cat#3495; RRID: AB_1903911 |
| Rabbit polyclonal anti-SQSTM1/p62 | Cell Signaling Technology | Cat#5114; RRID: AB_10624872 |
| Rabbit polyclonal anti-TFEB | Bthyl Laboratories | Cat#A303-673A; RRID: AB_11204751 |
| Rabbit monoclonal anti-PUMA (clone: D7L9L) (Rodent Specific) | Cell Signaling Technology | Cat#24633, RRID: AB_2798879 |
| Rabbit polyclonal anti-Bax | Cell Signaling Technology | Cat#2772; RRID: AB_10695870 |
| Rabbit monoclonal anti-Bak (clone: D4E4) | Cell Signaling Technology | Cat#12105; RRID: AB_2716685 |
| Rabbit monoclonal anti-XIAP (clone: D2Z8W) | Cell Signaling Technology | Cat#14334; RRID: AB_2784533 |
| Rabbit monoclonal anti-phospho-c-Jun (Ser73) (clone: D47G9) | Cell Signaling Technology | Cat#3270; RRID: AB_2129575 |
| Rabbit monoclonal anti-c-Jun (clone:60A8) | Cell Signaling Technology | Cat#9165; RRID: AB_2130165 |
| Rabbit polyclonal anti-cleaved caspase-3 (Asp175) | Cell Signaling Technology | Cat#9661; RRID: AB_2341188 |
| Mouse monoclonal anti-WIPI2 | Bio-Rad | Cat#MCA5780GA; RRID: AB_10845951 |
| Mouse monoclonal anti-LC3 | MBL International | Cat#M186-3; RRID: AB_10897859 |
| Mouse monoclonal anti-Bcl2 (clone: C-2) | Santa Cruz Biotechnology | Cat#sc-7382; RRID: AB_626736 |
| Mouse monoclonal anti-GAPDH (clone:6C5) | Santa Cruz Biotechnology | Cat#sc-32233; RRID: AB_627679 |
| Mouse monoclonal anti-β-actin (clone:C4) | Santa Cruz Biotechnology | Cat#sc-47778; RRID: AB_626632 |
| Rat monoclonal anti-CD140a (PDGFRα) | Thermo Fisher Scientific | Cat#14-1401-82; RRID: AB_467491 |
| Rat monoclonal anti-CD140a (PDGFRα) (Clone: APA5) | BD Biosciences | Cat#558774; RRID: AB_397117 |
| Rat monoclonal anti-LAMP2 (clone: GL2A7) | Abcam | Cat#ab13524; RRID: AB_2134736 |
| Rat monoclonal anti-MBP | Abcam | Cat#ab7349; RRID: AB_305869 |
| Mouse anti-galactocerebroside (Galc) hybridoma | Emery and Dugas39 | N/A |
| Mouse monoclonal anti-Calbindin | Swant | Cat#CB38; RRID: AB_10000340 |
| AffiniPure goat anti-rat IgG (H + L) | Jackson ImmunoResearch Labs | Cat#112-005-003; RRID: AB_2338090 |
| Griffonia (Bandeiraea) simplicifolia lectin I (BSL I) | Vector Laboratories | Cat#L-1100; RRID: AB_2336491 |
| Peroxidase-AffiniPure goat anti-mouse IgG (H + L) | Jackson ImmunoResearch Labs | Cat#115-035-003, RRID: AB_10015289 |
| Peroxidase-AffiniPure goat anti-rat IgG (H + L) | Jackson ImmunoResearch Labs | Cat#112-035-167; RRID: AB_2338139 |
| Peroxidase-AffiniPure goat anti-rabbit IgG (H + L) | Jackson ImmunoResearch Labs | Cat#111-035-003; RRID: AB_2313567 |
| Donkey anti-rat IgG (H + L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21208; RRID: AB_2535794 |
| Donkey anti-rat IgG (H + L), Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21209; RRID: AB_2535795 |
| Goat anti-rat IgG (H + L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11006; RRID: AB_2534074 |
| Goat anti-rat IgG (H + L), Alexa Fluor 555 | Thermo Fisher Scientific | Cat#A-21434; RRID: AB_2535855 |
| Donkey anti-rabbit IgG (H + L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21206; RRID: AB_2535792 |
| Goat anti-rabbit IgG (H + L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11008, RRID: AB_143165 |
| Donkey anti-rabbit IgG (H + L), Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21207; RRID: AB_141637 |
| Goat anti-mouse IgG (H + L), Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11001; RRID: AB_2534069 |
| Donkey anti-mouse IgG (H + L), Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21203; RRID: AB_141633 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| CellMask Blue Stain | Thermo Fisher Scientific | Cat#H32720 |
| Calcein AM | Thermo Fisher Scientific | Cat#C3100MP |
| Annexin V, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A13203 |
| Incucyte Annexin V Dye, Red | Sartorius | Cat#4641 |
| Propidium Iodide | Thermo Fisher Scientific | Cat#P3566 |
| SYTO13 Green Fluorescent Nucleic Acid Stain | Thermo Fisher Scientific | Cat#S7575 |
| L-Cysteine hydrochloride | Sigma-Aldrich | Cat#C7477 |
| cOmplete protease inhibitor | Sigma-Aldrich | Cat#5892791001 |
| PhosSTOP phosphatase inhibitor | Sigma-Aldrich | Cat#4906845001 |
| RIPA | Thermo Fisher Scientific | Cat#89900 |
| Amersham ECL Prime Western Blotting Detection Reagent | Cytiva | Cat#RPN2232 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher Scientific | Cat#PI34095 |
| 0.05% Trypsin-EDTA | Thermo Fisher Scientific | Cat#25300–054 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat#M6250 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15710 |
| Glutaraldehyde | Electron Microscopy Sciences | Cat#16320 |
| Poly-D-lysine hydrobromide | Sigma-Aldrich | Cat#P6407 |
| Normal Goat Serum | Jackson ImmunoResearch Labs | Cat#005-000-121; RRID: AB_2336990 |
| Normal Donkey Serum | Jackson ImmunoResearch Labs | Cat#017-000-121; RRID: AB_2337258 |
| Dimethyl Sulfoxide | Sigma-Aldrich | Cat#D2650 |
| Bafilomycin A1 | Sigma-Aldrich | Cat#196000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Click-iT Plus EdU Alexa Fluor 594 Imaging Kit | Thermo Fisher Scientific | Cat#C10639 |
| RNAscope Fluorescent Multiplex Reagent Kit | ACDbio | Cat#320850 |
| RNAscope Multiplex Fluorescent Reagent Kit v2 with TSA Vivid Dyes | ACDbio | Cat#323270 |
| RNeasy Plus Micro Kit | QIAGEN | Cat#74034 |
| LunaScript RT SuperMix Kit | New England Biolabs | Cat#E3010S |
| Fast SYBR Green Master Mix | Thermo Fisher Scientific | Cat#4385612 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23227 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq raw reads | This paper | NCBI BioProject: PRJNA986522 |
| The raw data of proteomics | This paper | MassIVE: MSV000092434 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Primary murine oligodendrocyte precursor cells | This paper | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | Charles River | Strain #632; RRID: IMSR_JAX:000664 |
| Mouse: ATG5Flox: B6.129S-Atg5<tm1Myok> | RIKEN | Strain #RBRC02975; RRID: IMSR_RBRC02975 |
| Mouse: ATG7Flox: B6.Cg-Atg7<tm1Tchi> | RIKEN | Strain #RBRC02759; RRID: IMSR_RBRC02759 |
| Mouse: TFEBFlox | Sun et al.4 | N/A |
| Mouse: Foxo3Flox: STOCK Foxo3tm1Rdp/J | The Jackson Laboratory | Strain #024668; RRID: IMSR_JAX:024668 |
| Mouse: CNP-Cre | Lappe-Siefke et al.40 | N/A |
| Mouse: Olig2-Cre: B6.129-Olig2tm1.1(cre)Wdr/J | The Jackson Laboratory | Stock #025567; RRID: IMSR_JAX:025567 |
| Mouse: PDGFRα-CreERT2: B6N.Cg-Tg (Pdgfra-cre/ERT)467Dbe/J | The Jackson Laboratory | Stock #018280; RRID: IMSR_JAX:018280 |
| Mouse: PUMA−: C57BL/6-Bbc3tm1Ast/J | The Jackson Laboratory | Stock #011067; RRID: IMSR_JAX:011067 |
|
| ||
| Oligonucleotides | ||
|
| ||
| MBP in situ probes | ACDbio | Cat#451491 and Cat#451491-C2 |
| PDGFRα in situ probes | ACDbio | Cat#480661 and Cat#480661-C2 |
| MOG in situ probes | ACDbio | Cat#492981-C2 |
| ENPP6 in situ probes | ACDbio | Cat#511021-C2 |
| Pcdh17it in situ probes | ACDbio | Cat#526741-C3 |
|
TFEB qPCR Forward: CCACCCCAGCCATCAACAC Reverse: CAGACAGATACTCCCGAACCTT |
This paper | N/A |
|
Bbc3 (PUMA) qPCR Forward: ACCTCAACGCGCAGTACG Reverse: CACCTAGTTGGGCTCCATTT |
This paper | N/A |
|
Pmaip1(NOXA) qPCR Forward: GCAGAGCTACCACCTGAGTTC Reverse: CTTTTGCGACTTCCCAGGCA |
This paper | N/A |
|
GAPDH qPCR Forward: AGGTCGGTGTGAACGGATTTG, Reverse: TGTAGACCATGTAGTTGAGGTCA |
This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Fiji (ImageJ) | National Institutes of Health | RRID: SCR_002285 |
| Incucyte ZOOM System | Sartorius | N/A |
| Bio-Rad ChemiDoc MP Imaging System | Bio-Rad | RRID:SCR_019037 |
| DESeq2 v 1.20.0 | Love et al.41 | RRID: SCR_015687 |
| Proteome Discoverer v2.4 | Thermo Fisher Scientific | RRID: SCR_014477 |
| TBtools | Chen et al.27 | RRID: SCR_023018 |
| Gorilla | Eden et al.42 | RRID: SCR_006848 |
| GraphPad Prism 9 | GraphPad | RRID: SCR_002798 |
Bulk RNA sequencing (RNA-seq) and analysis
Total RNAs from differentiating oligodendrocytes in 6-well plates (Seeding density: 100,000 OPCs per well) were isolated with the RNeasy Micro Kit (Qiagen, Cat#74004). RNA quality was accessed by Bioanalyzer and samples with high RNA integrity number (>7.5) were used for library construction. Library preparation and messenger RNA (mRNA) sequencing were performed by Novogene Corporation Inc. (Sacramento, California). In brief, the libraries were constructed with NEBNext Ultra II RNA Prep for Illumina (New England BioLabs, Cat#E7770) and subject to 150-bp double-end sequencing using the Illumina NovaSeq 6000 sequencer with 30 million reads. The raw data were filtered and aligned to the mouse genome (mm10) by STAR Aligner. The gene expression was calculated by HTSeq. Differential gene expression analysis was performed using the DESeq2 R package (1.20.0). Genes with an adjusted P-value ≤ 0.05 found by DESeq2 were assigned as differentially expressed genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses except for the RNA-seq analyses (see details in Methods and Materials) were performed using GraphPad Prism 9. Data are shown as mean ± SEM. The “n” numbers represent biological replicates and are specified as open or closed circles in the figures. For the comparison between two groups, statistical significance was determined using the two-tailed Student’s t-tests. For multiple comparisons, one-way or two-way ANOVA followed by multiple comparisons tests were performed (Tukey’s multiple comparisons test for one-way ANOVA and Sidak’s multiple comparisons test for two-way ANOVA). The criterion for statistical difference was set at p < 0.05. p values are indicated in the figures and figure legends.
Supplementary Material
Highlights.
Autophagic flux is elevated in premyelinating oligodendrocytes
Genetic perturbation of autophagy in oligodendroglia causes ectopic myelination
Autophagy cell autonomously promotes premyelinating oligodendrocyte apoptosis
Autophagy interacts with the TFEB-PUMA pathway to limit oligodendrocyte number
ACKNOWLEDGMENTS
We thank Dr. Klaus-Armin Nave for sharing the CNP-Cre mice. We thank Drs. John Abrams, Yang Liu, and Helmut Kramer for helpful comments on the manuscript. We thank UT Southwestern Proteomics Core for the quantitative proteomic analysis. We also thank Dr. David J. Simon and members of Sun laboratory for their assistance and discussions. This work was supported by a UT Southwestern Endowed Scholarship (L.O.S.); NIH R00EY029330 (L.O.S.); the Texas Alzheimer’s Research and Care Consortium (L.O.S.); the Brain & Behavior Research Foundation (L.O.S.); the Welch Foundation (L.O.S.); DoD W81XWH-21-1-0830 (L.O.S.); NIH 1DP2MH129988 (L.O.S.); 1S10OD021685-01A1 (UT Southwestern electron microscopy core facility); and NIH R01DC03157 (J.H.K.). L.O.S. is a Southwestern Medical Foundation Scholar in Biomedical Research and the John and Polly Sparks Foundation Investigator.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112943.
REFERENCES
- 1.Nave KA, and Werner HB (2021). Ensheathment and Myelination of Axons: Evolution of Glial Functions. Annu. Rev. Neurosci. 44, 197–219. 10.1146/annurev-neuro-100120-122621. [DOI] [PubMed] [Google Scholar]
- 2.Elbaz B, and Popko B (2019). Molecular Control of Oligodendrocyte Development. Trends Neurosci. 42, 263–277. 10.1016/j.tins.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, and Raff MC (1992). Cell death and control of cell survival in the oligodendrocyte lineage. Cell 70, 31–46. 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- 4.Sun LO, Mulinyawe SB, Collins HY, Ibrahim A, Li Q, Simon DJ, Tessier-Lavigne M, and Barres BA (2018). Spatiotemporal Control of CNS Myelination by Oligodendrocyte Programmed Cell Death through the TFEB-PUMA Axis. Cell 175, 1811–1826.e21. 10.1016/j.cell.2018.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trapp BD, Nishiyama A, Cheng D, and Macklin W (1997). Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J. Cell Biol. 137, 459–468. 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes EG, Orthmann-Murphy JL, Langseth AJ, and Bergles DE (2018). Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 21, 696–706. 10.1038/s41593-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, et al. (2017). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat. Neurosci. 20, 10–15. 10.1038/nn.4425. [DOI] [PubMed] [Google Scholar]
- 8.Hughes EG, and Stockton ME (2021). Premyelinating Oligodendrocytes: Mechanisms Underlying Cell Survival and Integration . Front. Cell Dev. Biol. 9, 714169. 10.3389/fcell.2021.714169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Green DR, and Levine B (2014). To be or not to be? How selective autophagy and cell death govern cell fate. Cell 157, 65–75. 10.1016/j.cell.2014.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamamoto A, and Yue Z (2014). Autophagy and its normal and pathogenic states in the brain. Annu. Rev. Neurosci. 37, 55–78. 10.1146/annurev-neuro-071013-014149. [DOI] [PubMed] [Google Scholar]
- 11.Griffey CJ, and Yamamoto A (2022). Macroautophagy in CNS health and disease. Nat. Rev. Neurosci. 23, 411–427. 10.1038/s41583-022-00588-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doherty J, and Baehrecke EH (2018). Life, death and autophagy. Nat. Cell Biol. 20, 1110–1117. 10.1038/s41556-018-0201-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Denton D, and Kumar S (2019). Autophagy-dependent cell death. Cell Death Differ. 26, 605–616. 10.1038/s41418-018-0252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, and Kumar S (2009). Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 19, 1741–1746. 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjørkøy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, and Stenmark H (2010). Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J. Cell Biol. 190, 523–531. 10.1083/jcb.201002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arakawa S, Tsujioka M, Yoshida T, Tajima-Sakurai H, Nishida Y, Matsuoka Y, Yoshino I, Tsujimoto Y, and Shimizu S (2017). Role of Atg5-dependent cell death in the embryonic development of Bax/Bak double-knockout mice. Cell Death Differ. 24, 1598–1608. 10.1038/cdd.2017.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aber ER, Griffey CJ, Davies T, Li AM, Yang YJ, Croce KR, Goldman JE, Grutzendler J, Canman JC, and Yamamoto A (2022). Oligodendroglial macroautophagy is essential for myelin sheath turnover to prevent neurodegeneration and death. Cell Rep. 41, 111480. 10.1016/j.celrep.2022.111480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947. 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, and Reggiori F (2018). Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455. 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao L, Ohayon D, McKenzie IA, Sinclair-Wilson A, Wright JL, Fudge AD, Emery B, Li H, and Richardson WD (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat. Neurosci. 19, 1210–1217. 10.1038/nn.4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zawadzka M, Rivers LE, Fancy SPJ, Zhao C, Tripathi R, Jamen F, Young K, Goncharevich A, Pohl H, Rizzi M, et al. (2010). CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 6, 578–590. 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tognatta R, Sun W, Goebbels S, Nave KA, Nishiyama A, Schoch S, Dimou L, and Dietrich D (2017). Transient Cnp expression by early progenitors causes Cre-Lox-based reporter lines to map profoundly different fates. Glia 65, 342–359. 10.1002/glia.23095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo YR, Kim HR, Jang SY, Go H, Song MY, Park DK, Oh Y, Jo J, Shin YK, Lee SJ, et al. (2021). Potential neuron-autonomous Purkinje cell degeneration by 2’,3’-cyclic nucleotide 3’-phosphodiesterase promoter/Cre-mediated autophagy impairments. Faseb. J. 35, e21225. 10.1096/fj.202001366RR. [DOI] [PubMed] [Google Scholar]
- 24.Kasuga Y, Fudge AD, Zhang Y, and Li H (2019). Characterization of a long noncoding RNA Pcdh17it as a novel marker for immature premyelinating oligodendrocytes. Glia 67, 2166–2177. 10.1002/glia.23684. [DOI] [PubMed] [Google Scholar]
- 25.Koenig U, Robenek H, Barresi C, Brandstetter M, Resch GP, Gröger M, Pap T, and Hartmann C (2020). Cell death induced autophagy contributes to terminal differentiation of skin and skin appendages. Autophagy 16, 932–945. 10.1080/15548627.2019.1646552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, Posner B, Tran KA, Green DR, Xavier RJ, et al. (2013). Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 110, 20364–20371. 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, and Xia R (2020). TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data . Mol. Plant 13, 1194–1202. 10.1016/j.molp.2020.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Fitzwalter BE, Towers CG, Sullivan KD, Andrysik Z, Hoh M, Ludwig M, O’Prey J, Ryan KM, Espinosa JM, Morgan MJ, and Thorburn A (2018). Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 44, 555–565.e3. 10.1016/j.devcel.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tešić Mark M, Molina H, and Tessier-Lavigne M (2016). Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell 164, 1031–1045. 10.1016/j.cell.2016.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao YG, Codogno P, and Zhang H (2021). Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat. Rev. Mol. Cell Biol. 22, 733–750. 10.1038/s41580-021-00392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green DR (2019). The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107. 10.1016/j.cell.2019.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer H, Shen K, Meireles AM, and Talbot WS (2022). A lysosomal regulatory circuit essential for the development and function of microglia. Sci. Adv. 8, eabp8321. 10.1126/sciadv.abp8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kishi-Itakura C, Koyama-Honda I, Itakura E, and Mizushima N (2014). Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell Sci. 127, 4089–4102. 10.1242/jcs.156034. [DOI] [PubMed] [Google Scholar]
- 34.Melia TJ, Lystad AH, and Simonsen A (2020). Autophagosome biogenesis: From membrane growth to closure. J. Cell Biol. 219, e202002085. 10.1083/jcb.202002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, and Simon HU (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132. 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 36.Peng J, Zhang R, Cui Y, Liu H, Zhao X, Huang L, Hu M, Yuan X, Ma B, Ma X, et al. (2014). Atg5 regulates late endosome and lysosome biogenesis. Sci. China Life Sci. 57, 59–68. 10.1007/s11427-013-4588-8. [DOI] [PubMed] [Google Scholar]
- 37.Guo H, Chitiprolu M, Roncevic L, Javalet C, Hemming FJ, Trung MT, Meng L, Latreille E, Tanese de Souza C, McCulloch D, et al. (2017). Atg5 Disassociates the V(1)V(0)-ATPase to Promote Exosome Production and Tumor Metastasis Independent of Canonical Macroautophagy. Dev. Cell 43, 716–730.e7. 10.1016/j.devcel.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 38.Bankston AN, Forston MD, Howard RM, Andres KR, Smith AE, Ohri SS, Bates ML, Bunge MB, and Whittemore SR (2019). Autophagy is essential for oligodendrocyte differentiation, survival, and proper myelination. Glia 67, 1745–1759. 10.1002/glia.23646. [DOI] [PubMed] [Google Scholar]
- 39.Emery B, and Dugas JC (2013). Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harb. Protoc. 2013, 854–868. 10.1101/pdb.prot073973. [DOI] [PubMed] [Google Scholar]
- 40.Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, and Nave KA (2003). Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33, 366–374. 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 41.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 10, 48. 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, and Barres BA (2009). Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell 138, 172–185. 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith CM, Mayer JA, and Duncan ID (2013). Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J. Neurosci. 33, 8088–8100. 10.1523/JNEUROSCI.0233-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomic datasets related to Figure S5 in the paper are available in Table S2. The gene expression file of bulk RNA-seq related to Figure 5 are available in Table S3. The raw data of proteomics have been deposited to MassIVE (accession number: MSV000092434). The raw data of bulk RNA-seq have been deposited at NCBI BioProject (accession number: PRJNA986522).
No original codes were generated in this study.
Any additional information related to this paper is available from the lead contact upon request.