

Abstract
NAFLD is the most common chronic liver disease worldwide, characterized by lipid accumulation in the liver, and usually evolves from steatohepatitis to fibrosis, cirrhosis, or even HCC. Its incidence is rapidly rising in parallel with the increasing prevalence of obesity and metabolic syndrome. Current therapies are limited to lifestyle changes including dietary intervention and exercise, in which dietary modification exerts an important part in losing weight and preventing NAFLD. In this review, we briefly discuss the roles and mechanisms of dietary components including fructose, non-nutritive sweeteners, fat, proteins, and vitamins in the progression or prevention of NAFLD. We also summarize several popular dietary patterns such as calorie-restricted diets, intermittent fasting, ketogenic diets, Mediterranean diets, and dietary approach to stop hypertension diets and compare the effects of low-fat and low-carbohydrate diets in preventing the development of NAFLD. Moreover, we summarize the potential drugs targeting metabolic-related targets in NAFLD.
INTRODUCTION
NAFLD, characterized as a spectrum of pathological stages including NAFL, NASH, and advanced fibrosis, can lead to cirrhosis and finally end-stage liver disease. Driven partially by rising obesity levels, the global prevalence of NAFLD among adults, children, and adolescents has increased in the last few decades,1 reaching 25% in 2016, with higher rates observed in South America, the Middle East, and Asia.2 The overall prevalence of NAFLD has reached 29.1% in Chinese people, and the proportion of young patients is rising, portending a heavier disease burden in the future.3 Though the majority of patients live with NAFL and early NASH without progression to later disease stages, the economic burden of NAFLD is getting heavier due to its huge affected population.1 NAFLD has been a primary cause of HCC in many countries.4 Being able to develop in patients without cirrhosis, NAFLD-related HCC is associated with a later stage at diagnosis and, conceivably, a poorer prognosis.5 It is also reported that NAFLD is an independent risk factor for cardiovascular diseases (CVDs), type 2 diabetes mellitus (T2DM), and chronic kidney diseases.6
So far, there is no licensed drug for NAFLD treatment. Lifestyle alternation, especially dietary intervention, is recommended to tackle NAFLD and its potential complications, with the primary principle of reducing calories intake and restricting the consumption of lipogenic nutrients. However, specific recommendations for diets are vague and inconsistent among guidelines.7 In this review, we briefly generalize the effects of common dietary components on NAFLD and their potential mechanisms, and provide an overview of the dietary patterns found beneficial to NAFLD patients, thereby promoting NAFLD prevention and management in aspects of modern lifestyle.
SWEETENERS AND NAFLD
Key points
High fructose consumption increases hepatic de novo lipogenesis (DNL) and reduces fatty acid β-oxidation (FAO), leading to fatty acid deposition. Increasing oxidative stress and low-grade hepatic inflammation contribute to further NAFLD progression. A series of animal studies suggest that non-nutritive sweeteners (NNSs) have adverse impairment on metabolism as well.
The global change in dietary habits, especially the introduction of sugar-sweetened beverages, has led to increased consumption of carbohydrates, mostly refined sugar, over the last decades. Consumption of added sugar has long been proved a risk factor for the development and progression of NAFLD.8 Habitual exposure to sugar-sweetened beverages leads to higher visceral adipose tissue volume, increased liver fat, impaired serum liver enzyme concentrations, and higher NAFLD odds in adults.9–12 Glucose, fructose, and sucrose are the 3 main carbohydrates consumed in modern diets, among which fructose has in particular gained research attention, not only for the evolution of sweeteners from sucrose to the “cheaper and sweeter” high-fructose corn syrup (HFCS), but also for its unique lipogenic potential.
Fructose and NAFLD
Once used as a sugar substitute for diabetic patients for its smaller influence on plasma glucose and serum insulin levels,13 fructose-containing diets have been confirmed in animal experiments as well as human studies to induce DNL and insulin resistance (IR) to a greater extent than normal or glucose-containing ones.14–17 Researchers have showed an average fructose:glucose ratio of about 60:40 in the 5 most popular HFCS-sweetened sodas in the United States,18 much higher than that of the traditional HFCS-55, which contains 55% of free fructose, and HFCS-42, containing 42% of free fructose. Thus, it is of significant importance to further demonstrate the mechanisms of fructose-induced NAFLD.
Fructose is mainly metabolized in the liver, since its transporter, glucose transporter type-5 (GLUT5), is poorly expressed in most cells.19,20 When it comes to hepatocytes, the membrane glycoprotein transmembrane 4 L 6 family member 5 (TM4SF5)–mediated GLUT8 relocalization to the plasma membrane helps transport extracellular fructose.21,22 The activity of ketohexokinase (KHK), the first enzyme of fructose metabolism, is greater than that of glucokinase, indicating a greater chance of fructose to enter the hepatic lipogenesis compared with glucose.23 Moreover, fructose metabolism bypasses phosphofructokinase-1, which acts as a rate-limiting enzyme of glycolysis, thereby escaping metabolic regulation.24 Small amounts of fructose are mostly converted by the small intestine to glucose, while large amounts of fructose, especially those consumed in liquid form, can be directly absorbed and access the liver via the portal vein, adding to liver stress and increasing the risk of NAFLD.19,25,26
High-carbohydrate diets stimulate the expression of carbohydrate-responsive element–binding protein (ChREBP) and sterol regulatory element–binding protein 1C (SREBP-1c) in rats,27–29 the 2 major transcription factors regulating hepatic lipogenesis.30,31 ChREBP and SREBP-1c induce lipogenic genes, including acetyl-CoA carboxylase (ACC), fatty acid synthase (FASN), ATP citrate lyase, and Stearoyl-CoA desaturase-1.30,32 Consistently, all of the abovementioned lipogenic genes have been reported to be significantly upregulated after high carbohydrate exposure,29,33,34 and several studies have demonstrated a greater change in mice fed with fructose-containing sugar.15,16
While both SREBP-1c and ChREBP are required for full induction of liver lipogenic genes in response to carbohydrates,31 ChREBP is the only one reported to modify monosaccharide transportation and glycolysis. Intestinal ChREBP stimulated by oral fructose ingestion upregulates Slc2a5, which encodes fructose transporter GLUT5, and the intestine-specific ChREBP-KO mice have been proved to be fructose-intolerant.35 Hepatic ChREBP induces liver enzyme KHK and pyruvate kinase (l-PK), both involved in fructose glycolysis.16,31,36 Increased levels of KHK16 and l-Pk,37 along with higher pyruvate levels34 after fructose ingestion, indicate thriving glycolysis, while in a cell model of human hepatocyte metabolism, intermediates of glycolysis decrease indeed,38 suggesting an increased consumption of glycolysis intermediates as the carbon source in hepatic DNL. It is worth mentioning that liver X receptors34 and mTORC130 are the potential upstream regulators of fructose-mediated ChREBP and SREBP-1c activation.
While increased hepatic DNL has been widely accepted as the main cause of fructose-mediated hepatic lipid deposition, there is mounting evidence that the reduction in FAO and increased production of reactive oxygen species (ROS) contribute as well. Hepatic malonyl-CoA, an intermediate from DNL, is increased after fructose ingestion,28 which inhibits carnitine palmitoyl transferase 1 (CPT1), thus disturbing the time-limiting process of FAO of acyl-CoAs transported across the mitochondrial membrane in the form of acylcarnitines.39 Impaired mitochondrial function is another reason for the diminished FAO, since the high-fructose-high-fat diet (HFD) led to a persistent decrease in mitochondrial size and DNA in animal models, which was not observed in high-glucose-high-fat groups.40,41 The tryptophan pathway, known as an NAD+ synthesis pathway, is significantly decreased in female juvenile rats after fructose consumption, and diminished supply of NAD+ is proved to impair mitochondrial fatty acid oxidation.42–44 Most HFD-induced NAFLD animal models show increased FAO in response to lipid deposition, indicating that sugar consumption may deteriorate the adverse effects of HFDs.45 The rise of antioxidant peptides including ascorbate and glutathione in response to HFCS-sweetened diets suggests increasing ROS production.43 HFCS-fed mice showed a lower diacylglycerol O-acyltransferase 2 expression, which is associated with impaired antioxidant ability and increased fibrosis.29,46 Furthermore, a sex-specific pattern can be seen in the HFCS-induced hepatic steatosis, with a greater reduction of FAO and higher antioxidant potential in female rats.43
Fructose consumption is linked to low-grade hepatic inflammation, which is a leading cause of NAFLD progression. After consuming 60% fructose solution for 9 weeks, both male and female Wistar rats showed increased hepatic TNF alpha (TNFα), one of the markers of inflammation.47,48 However, NFκB activation was only detected in male rats, which may be due to the protection effect of the enhanced glucocorticoid signaling restricted to female rats.48 Intriguingly, consuming fructose under stressful conditions, as performed by Kovačević et al49 in female Wistar rats, counteracted the effects of stress on hepatic lipid accumulation alone. Whether this is also a secondary effect of the elevated glucocorticoids remains unknown.
In addition to the lipogenic nature of the added sugar itself, gluttony induced by sweet food deserves concern as well. Scientists do describe a decreased ability toward moderate energy intake in mice fed with sugar-sweetened diets while provided an additional pre-meal.50 Though sugar-fed animals tend to reduce chow intake, they still consume more energy in total.50 It has been reported that fructose can induce leptin resistance and reduce satiety in animals by activating central AMP-activated protein kinase, thus inhibiting glucagon-like peptide-1 (GLP-1).51,52 fMRI studies in humans also showed that fructose ingestion failed to reduce the activity of the hypothalamus and striatum, the 2 brain regions associated with satiety.53 Interestingly, Levy et al54 found in mice that the fructose:glucose ratio was negatively correlated to sugar self-administration, resulting in fewer nose pokes and a lower caloric intake in the HFCS-fed group compared with the sucrose-fed one. One reasonable explanation for this conflicting observation is that fructose has a stronger reinforcing nature, which may be associated with reduced self-administration following higher substance concentration as observed in amphetamine and cocaine55 (Figure 1).
FIGURE 1.
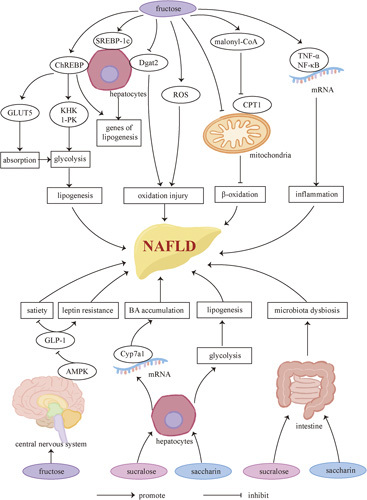
Sweeteners and NAFLD. Fructose induces ChREBP and SPEBR-1c, resulting in upregulation of glycolytic enzymes and lipogenic genes, both of which contribute to increasing hepatic DNL. The consequent accumulation of malonyl-CoA, an intermediate from DNL, along with the fructose-induced mitochondrial dysfunction, diminishes lipid metabolism and leads to further fatty acid deposition. Increasing oxidative stress and low-grade hepatic inflammation are found after high-fructose consumption, potentially leading to NAFLD progression. It should not be ignored that high-fructose diets can increase food intake by inhibiting satiety and inducing leptin resistance, since excessive energy intake is considered a crucial risk factor of NAFLD. As far as we know, some widely used NNSs such as sucralose and saccharin also exhibit adverse effects including bile acid accumulation, microbiota dysbiosis, and increasing hepatic DNL as well as oxidative stress, which may link them to the development and progression of NAFLD. Abbreviations: AMPK, AMP-activated protein kinase; BA, bile acid; ChREBP, carbohydrate-responsive element-binding protein; CPT, carnitine palmitoyl transferase; Dgat2, diacylglycerol O-aAcyltransferase 2; GLUT, glucose transporter type; KHK, ketohexokinase; l-PK, liver-pyruvate kinase; ROS, reactive oxygen species; SREBP-1c, sterol regulatory element–-binding protein 1C; TNFα, TNF alpha.
Non-nutritive Sweeteners and NAFLD
Although NNSs including sucralose, acesulfame, saccharin, and aspartame are often used as a control in fructose-related experiments to exclude the interference of sweet taste, there have been a series of animal studies showing that NNSs also have adverse impairment on metabolism. Ryuk et al56 reported the ability of non-nutritive aspartame and sucralose to induce IR in rats, indicating that the metabolic impairments may start with the sweet taste itself. Sucralose upregulates hepatic expression of Cyp7a1 mRNA, leading to bile acid (BA) accumulation,57 which is associated with the development and progression of NAFLD. Altered hepatic protein metabolism is also reported after sucralose consumption.58 Aspartame, saccharin, and aspartame may increase hepatic oxidative stress, deplete antioxidant levels, and elevate plasma aminotransferase and alkaline phosphatase activities.59–61 Saccharin consumption is also reported to potentially increase hepatic enzymes, including phosphofructokinase-1 (PFK-1), l-PK, and Fasn.62
Microbiota dysbiosis is another focused mechanism of NNS-associated hepatic disorders. Sucralose consumption significantly increases the Bacteroides and Clostridium population, resulting in higher deoxycholic acid serum concentration, a potential risk factor for NAFLD.57 Both sucralose and saccharin have been found to cause hepatic and systemic inflammation in mice through the alternation of gut microbiomes and their metabolites.63,64 Moreover, maternal sucralose intake can impair intestinal function and alter the gut microbiota of offspring65 (Figure 1).
However, current results are conflicting and sparse about whether NNSs impair hepatic lipogenesis, and whether the detected metabolism effects are sufficient to increase NAFLD risks.66–68 While some studies have mentioned a weight-loss effect of artificial sweeteners, there is also contrary evidence that aspartame treatment induces abdominal adiposity in rats.61 When consumed with a HFD, sucralose is observed by Pino-Seguel et al69 to prevent weight gain and alleviate glucose intolerance in mice. Contrast results were detected by Wu et al70 under a similar protocol wherein sucralose not only had no effect on the body weight of HFD-fed mice but also exacerbated HFD-induced fatty liver. Therefore, more studies are required to precisely evaluate the metabolism effect of NNSs.
LIPIDS AND NAFLD
Key point
Dietary lipids have a dualistic influence on NAFLD. On one hand, excess lipids, including free cholesterol (FCs) and free fatty acid (FFAs), especially saturated fatty acid (SFAs), promote the development of NAFLD. In addition, a higher dietary ratio of n-6 PUFAs to n-3 PUFAs is an independent promoter of NAFLD. On the other hand, increasing the dietary intake of n-3 can alleviate NAFLD.
A strong correlation between HFD and NAFLD is already known. A large amount of dietary lipid intake may cause the hepatocytes to be in an environment with high FFAs and high FCs, which may disrupt the homeostasis of liver lipid metabolism by lipotoxicity and lead to the development of NAFLD. In mouse NASH models induced by HFD or palmitic acid treatment, upregulation of vascular cell adhesion molecule 1 in liver sinusoidal endothelial cells has been observed, which promotes adhesion with liver sinusoidal endothelial cells and activation of macrophages.71,72 Another study showed that in cell and murine models, the activation of kinase receptor-interacting protein 1 (RIP1) of macrophages mediates inflammatory responses and apoptosis.73 In a high-fat environment, hepatocytes also play an important role in the activation of macrophages. Hepatocytes stimulate activation of macrophages as well as mediate inflammation and oxidative stress by secreting various inflammatory extracellular vesicles.74–77 Hepatocytes can induce apoptosis and HSC activation through autocrine and paracrine IL-11, respectively.78 In addition, the upregulation of lysophosphatidylcholine acyltransferase 2, cytoplasmic phospholipase A2, and 15-lipoxygenase in hepatocytes can cause the increase of arachidonic acid, a proinflammatory factor in the cell membrane,79 which results in ROS accumulation, oxidative stress, ER stress, mitochondrial damage, and hepatocyte apoptosis, thus promoting NAFLD. The activated macrophage stimulating 1-AMP-activated protein kinase pathway in macrophages, tank-binding protein kinase 1 (TBK1) in hepatocytes, and the inhibited Sirtuin 3 (SIRT3)-ERK-CREB-Bnip3 pathway all induce hepatocyte apoptosis through mitochondrial autophagy inhibition.80–82
Oxysterols, the oxidation products of excessive cholesterol, and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK)-mediated c-Jun N-terminal activation can cause mitochondrial damage, thereby exerting lipotoxicity on hepatocytes in vivo and vitro.83,84 Caspase-2–mediated hepatocyte apoptosis and SMS1-DAG-PKCδ-NLRC4 axis–mediated pyroptosis are also involved in lipotoxicity damage.85,86 In addition, Li et al87 found that the expression of 3-mercaptopyruvate sulfurtransferase, a key enzyme of hydrogen sulfide biosynthesis, is upregulated by high FFAs, which inhibits CSE/hydrogen sulfide in hepatocytes and leads to NAFLD. High FFAs may also promote NAFLD by BA accumulation.88 Besides, IR is one of the crucial factors for NAFLD development. Diabetic mice are more likely to develop NAFLD under HFD conditions than healthy wild-type mice, suggesting that HFD is more harmful to diabetic patients.89 In primate experiments conducted by McCurdy et al90, it was found that having HFD during pregnancy can increase the risk of developing NAFLD in offspring, whose vulnerability may persist into adulthood, regardless of maternal obesity and IR.
Notably, an equal-calorie high-SFA diet is more harmful to the liver than a high unsaturated fat diet, which may be caused by the enhanced activity of SIRT3 in hepatocytes. SIRT3 is a manganese superoxide dismutase located mainly in the mitochondria. The enhanced activity of SIRT3 leads to the activation of deacetylation-induced manganese superoxide dismutase, inhibition of AMPK, and mammalian target of rapamycin C1 (mTORC1)–related autophagy inhibition.91
Both omega-3 (n-3) and omega-6 (n-6) PUFAs are essentials for the composition of cell membranes. n-3 is rich in fish oil while n-6 is rich in vegetable oils. The dietary ratio of n-6 to n-3 (n-6/n-3) is another factor that affects lipid deposition in the liver. In mice fed with equal-calorie equal-fat (35%) high-n-3 and high-n-6 diets, respectively, the high-n-6 diet model showed higher levels of liver steatosis, increased cell apoptosis, and decreased hepatocyte proliferation, which may be related to the increased NF-κ–mediated inflammation in the liver.92 Bogl team compared the diets of 10 pairs of identical twins with significant differences in liver fat and found that the higher-liver-fat group has an average of 6.6:1 n-6/n-3, while the lower-liver-fat group has an average of 3.2:1 n-6/n-3.93 These studies suggest that the high proportion of n-6 intake may be a promoting factor of NAFLD independently. Similarly, Okada and colleagues found that ER stress and mitochondrial dysfunction caused by lipid deposition and oxidative stress can be significantly reduced after 6 months of treatment with n-3 in NAFLD patients, which is possibly due to the reduction of serum n-6/n-3. They also partially illustrated the mechanism that n-3 competes with n-6 for lipoxygenase and cyclooxygenase during the process of metabolism. Eicosapentaenoic acid and docosahexaenoic acid, metabolites of n-3, have anti-inflammatory effects, while arachidonic acid, the metabolite of n-6, has strong proinflammatory effects. In addition, this process involves the increased expression of protective genes related to lipid metabolism, including fatty acid binding protein – liver type and peroxiredoxin 6 (PRDX6).94
Šmíd et al95 reported that NAFLD patients treated with n-3 for 12 months undergo weight loss, liver fat reduction, and a significant decrease in serum liver enzymes including γ-glutamyltransferase (GGT), but liver fibrosis is not effectively improved.95 In mouse NAFLD models and in vitro experiments, the results suggested that fish oil may play a role in regulating lipid metabolism by inhibiting adipose cells, and inducing adipose-derived stromal cell and 3T3-L1 preadipocyte proliferation, which results in reduction of blood lipids, liver enzymes, and liver fat.96 In terms of mechanism, it may be the expression of neuregulin 4, Ucp1, and Pgc1a mRNA in NAFLD model adipose tissue upregulated by n-3 treatment. In vitro cell experiments have shown that n-3 induces browning of white adipose tissue by promoting peroxisome proliferator–activated receptor gamma pathway–dependent neuregulin 4 production.97 Besides, in the NAFLD rat model, supplementation of n-3 can regulate lipid metabolism by improving the bile components and regulating the biliary secretion rhythm.98
Hong et al99 compared the potential of various components of n-3, including α-linoleic acid, eicosapentaenoic acid, and docosahexaenoic acid, to alleviate NAFLD; the results showed that docosahexaenoic acid is the most effective in reducing liver fatty acid synthesis and has the potential to reverse liver fibrosis.
However, a small number of studies have disputed the role of n-3 in NAFLD treatment. Experimental results of fish oil supplementation for 12 weeks in overweight NAFLD patients showed that fish oil has no significant effect in the reduction of liver fat, liver enzymes, and other indicators.100 Another similar study found that although supplementation of n-3 for 12 weeks in NAFLD patients and overweight or obese people with hypertriglyceridemia can decrease serum TG levels, it also increased serum aspartate transaminase (AST) and alanine transferase (ALT), markers of liver damage, the mechanism of which remains unclear.101 Nevertheless, taking into account previous studies, these results may only suggest that short-term supplementation of n-3 has no significant therapeutic effect on NAFLD.
In addition, different meta-analyses have drawn positive conclusions on the therapeutic effect of n-3 supplementation in patients with NAFLD. Parker et al’s102 analysis showed that n-3 supplementation can effectively reduce liver fat and AST levels, and cause a downward trend of ALT. Yang et al103 found that the therapeutic effect of n-3 on NAFLD may be only effective in patients with early and mild NAFLD, but not in patients with severe NAFLD. According to the latest meta-analysis, n-3 can not only effectively reduce liver fat, but also significantly improve the levels of serum triglyceride, total cholesterol, HDL, body mass index (BMI), and other parameters, so it has a definite therapeutic effect on NAFLD.104 However, they also suggested that a more comprehensive analysis is needed due to the heterogeneity of the dose and n-3 treatment duration in the studies included (Figure 2).
FIGURE 2.
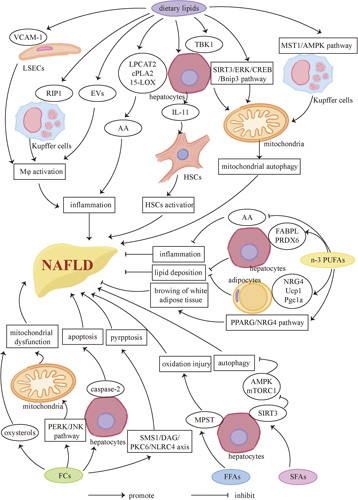
Lipids and NAFLD. Dietary lipids have a dualistic influence on NAFLD. On one hand, excess lipids, including FCs and FFAs and so forth, especially SFAs, promote the development of NAFLD. Inflammation can be promoted by activating KCs, liver macrophages (Mφ), and stimulating the release of the proinflammatory factor AA and IL-11. Mitochondrial autophagy is also associated with one of the promoting factors of NAFLD through the macrophage stimulating 1/AMPK pathway. Excess FCs increase the occurrence of mitochondrial dysfunction and cause caspase-2–mediated hepatocyte apoptosis and SMS1/DAG/PKC6/NLRC4 axis–mediated pyroptsis. Besides, inhibition of autophagy in hepatocytes may also be one of the factors that promote NAFLD. On the other hand, increasing dietary intake of n-3 PUFAs can alleviate NAFLD mainly by inhibiting proinflammatory factors and AA, and reducing lipid deposition. Besides, n-3 PUFAs contribute to browning of white adipose tissue. Abbreviations: AA, arachidonic acid; AMPK, AMP-activated protein kinase; EV, extracellular vesicle; FC, free cholesterol; FFA, free fatty acid; JNK, c-Jun N-terminal; LOX, lipoxygenase; LPCAT2, lysophosphatidylcholine acyltransferase 2; MPST, mercaptopyruvate sulfurtransferase; mTORC1, mammalian target of rapamycin complex 1; NRG4, neuregulin 4; PERK, PKR-like endoplasmic reticulum kinase; Pgc1α, peroxisome proliferators–-activated receptor γ coactivator lα; PPARG, peroxisome proliferator– activated receptor gamma; RIP1, receptor-interacting protein 1; SIRT3, sirtuin 3; SMS1, sphingomyelin synthaseUcp1, uncoupling protein 1; VCAM-1, vascular cell adhesion molecule 1.
Dietary changes are currently the most important measures to prevent the development of NAFLD. However, current studies on NAFLD mainly focus on animal models, and there is still a lack of research exploring the most appropriate lipid intake for NAFLD patients. Besides, few studies have explored the impact of an extreme low-fat diet (LFD) on liver metabolism and the specific mechanism promoting NAFLD, which are still necessary to find out through larger samples and more accurate experimental programs.
PROTEIN AND NAFLD
Key point
Dietary protein has been reported to exert beneficial effects on NAFLD patients, including reducing the intrahepatic lipid accumulation and improving IR. However, there is a controversy over the application of high-protein diets in NAFLD patients, which may be attributed to the different effects of animal and plant protein on NAFLD. High-protein diets also have some adverse effects.
The impact of protein and amino acids on NAFLD is little known since most studies focused on carbohydrates and fats. It has been reported that a high-protein diet can reduce hepatic lipid accumulation compared with a normal protein diet in a rat model, with an even greater decrease when combined with restriction of calories.105 Similar to the findings in rats, supplementing proteins without caloric restriction has been shown to lower intrahepatic fat in human studies, despite the stable BMI,106–108 while dietary protein insufficiency causes lipid accumulation.109 The additional metabolic advantages of high-protein diets have been displayed in obese and NAFLD patients,106,110 with improvement in serum lipid levels and hepatic enzymes. Moreover, sarcopenia, which is an independent risk factor of NAFLD, leads to NASH or even advanced fibrosis as well as CVDs, while high-protein diets can prevent muscle loss.111,112 A high-protein diet is also more suitable for lean individuals, accounting for 25% of the NAFLD patients.113 Since it is unnecessary and difficult for them to lose weight or be on a diet, changing the relative macronutrient consumption is more practicable. Notably, an increase in the proportion of protein in a diet usually leads to a decrease in carbohydrate intake. Despite patients with a HFD displaying less lipid deposition when increasing their dietary protein, with no differences in carbohydrate intake,107 whether the reduction of lipid accumulation is attributed to the high protein or low carbohydrate requires further investigation.
Dietary protein can promote more satiety than carbohydrates and fat, which contributes to more weight loss.114,115 Mechanically, elevated plasma amino acid contents and gut hormones may exert the satiety effects in NAFLD patients. The branched-chain amino acids have been reported to stimulate satiety signaling through receptors in the duodenums and intestines in rat models.116–118 Anorexigenic hormones such as GLP-1, cholecystokinin, and peptide YY (PYY) are increased in overweight individuals after high protein intake, which can stimulate the vagal activities controlling food intake.119,120 Glucagon secretion induced by dietary protein also suppresses DNL.121,122 What’s more, the catabolism of amino acids increases energy expenditure, which causes oxidation of hepatic lipid and depletion of liver fat in rat models consequently.123 A decrease in the expression of acetyl-CoA carboxylase and FAS, which are genes of hepatic lipogenesis, has also been observed in response to high-protein diets, with a lower serum insulin level and reduced hepatic glucose uptake in rat models as well.124 In addition, dietary protein deficiency is associated with mitochondrial dysfunction, which contributes to the pathogenesis of NAFLD. Rats with low-protein diets show decreased expression of antioxidant enzymes and peroxisomal membrane protein 70 (PMP70), with a consequent severe oxidative injury and lipid deposition.125 Peroxisomal acyl-CoA oxidase 1 and mitochondrial CPT2, which can promote peroxisomal and mitochondrial β-oxidation of fatty acids, are also increased after supplementing protein in diets126 (Figure 3).
FIGURE 3.
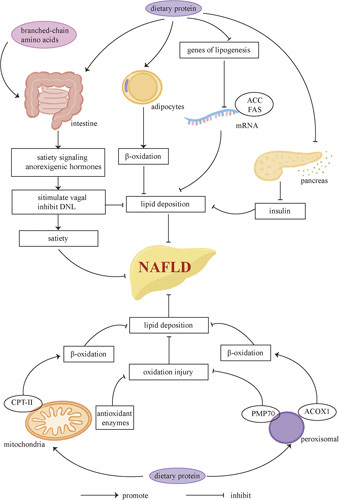
Protein and NAFLD. Dietary protein can help treat NAFLD by promoting satiety and inhibiting lipid deposition. Elevated branched-chain amino acids can stimulate satiety signaling. Anorexigenic hormones can also promote vagal activities and inhibit DNL. What’s more, dietary protein can increase β-oxidation of lipids in adipocytes, suppress the expression of lipogenesis genes, and decrease serum insulin levels, all of which inhibit hepatic lipid deposition. Proteins can also increase the expression of antioxidant enzymes in the mitochondria and PMP70 in the peroxisomes to decrease oxidation injury. The expression of peroxisomal ACOX1 and mitochondrial CPT2, which can increase β-oxidation of lipids, is also increased after supplementing dietary protein. Abbreviations: ACC, acetyl-CoA carboxylase; ACOX1, acyl-CoA oxidase 1; CPT, carnitine palmitoyl transferase; DNL, de novo lipogenesis; PMP70, peroxisomal membrane protein 70.
However, conflicting results regarding the effects of dietary protein on NAFLD have been exhibited. Individuals who consume high-protein diets show an increased prevalence of NAFLD127 and more severe histological manifestations.128 Another population study also reported a higher risk of NAFLD development among individuals consuming diets rich in animal protein, especially in overweight patients.129 The controversial findings can be attributed to different sources of protein, as plant protein seems to be beneficial to NAFLD while animal protein has an opposite effect.130 NAFLD patients consume more red meat than healthy controls,131 while red meat is reported to be associated with the development of NAFLD, IR, and CVDs,132,133 even without high intakes,134 mainly because red meat contains a high level of homocysteine.135 Bergeron et al136 reported that either white meat or red meat leads to higher levels of LDL-C and apoB than nonmeat protein sources, suggesting that vegetable-based food is a better choice for health (Figure 3). But Mariya reported that both animal and plant proteins can decrease intrahepatic lipid accumulation to a similar content in patients with NAFLD and T2DM.130 Therefore, the effect of different sources of protein on NAFLD requires further investigation. In addition, the results may be attributed to more than the effect of protein, as red meat and vegetables contain other compounds. Red meat contains cholesterol, heme iron, and nitrates, which are related to the increased mortality from chronic liver diseases,137,138 while vegetables are rich in phytonutrients and fibers.
Although dietary proteins may be beneficial in decreasing hepatic lipid accumulation, their adverse effects should be taken into consideration. High protein intake can lead to glomerular sclerosis as well as renal malfunction in patients vulnerable to kidney diseases,139 while it has been reported that the glomerular filtration rate does not decrease when consuming vegetable protein instead of animal protein.140,141 Furthermore, a higher proportion of protein intake is associated with an increased risk of CVDs as mentioned above.142 Current evidence supports that more plant protein can decrease the risks of CVD than animal-based protein, while meat that is unprocessed and low in saturated fat is beneficial as well.143 The increased levels of nitrosamine and heterocyclic amine in feces resulting from high-protein diets are harmful to the environment of the colon, leading to a higher risk of colorectal cancer.144 In addition, research in rats showed that a diet high in branched-chain amino acids and fat could lead to the accumulation of succinyl and propionyl-CoA, which can impair insulin sensitivity and is associated with a higher risk of diabetes consequently.145 A high intake of amino acids can inhibit insulin signaling through the activation of mTOR signaling as well.146,147 Hence, proper doses of daily protein intake and their sources need to be detected to balance the benefits and risks.
VITAMIN AND NAFLD
Key points
Both vitamin E (VE) and vitamin D (VD) can improve the biochemical and histological manifestations of NAFLD patients. The vitamin D receptor (VDR) is also a promising target for treating NAFLD. However, more research on the effects of vitamins on NAFLD is required to support their application.
Chronic liver diseases are reported to be associated with the deficiency of vitamins,148 in which context VE and VD are most investigated. VE, which has an antioxidant property, can improve both biochemical and histological manifestations, including aminotransferases, ballooning, inflammation, and steatosis, in NASH patients without T2DM.149 However, in NAFLD patients with T2DM, the results are conflicting. Bril et al150 reported that the application of VE alone did not improve liver fibrosis, while a combination of VE and pioglitazone exhibited improvement in histological outcomes. Eduardo reported that transplant-free survival and liver decompensation were improved by the application of VE in NASH patients both with and without T2DM.151 Studies on rats uncovered the potential mechanisms of VE on NAFLD treatment, showing that VE can alleviate oxidative stress and prevent NAFLD-driven HCC by downregulating inducible nitric oxide synthase and NADPH oxidase.152 Furthermore, VE lowers the expression of TGF-β, which plays a critical part in the progression of liver fibrosis.153 VE is also reported to improve lipid and glucose metabolism via activating the nuclear factor erythroid 2-related factor 2/carboxylesterase 1 pathway154 and enhance insulin sensitivity by inducing the expression of adiponectin via activation of PPAR.155 Regardless of its effects on NAFLD improvement, VE has been reported to be associated with an increasing risk of prostate cancer156 and CVDs.157 Considering the controversial results of VE and its side effects, more investigation is required before its clinical application.
What’s more, there is a deficiency of VD in NAFLD patients compared with healthy controls,158 associated with the severity of NAFLD’s histological manifestations.159 Supplementation of VD can improve IR and the levels of serum liver enzymes.160 In NAFLD rat models, deficiency of VD is associated with upregulation of TLR4-mediated inflammatory signaling in NAFLD rat models, while supplementing VD can reverse the activation of the TLR4 pathway161 and reduce the activation of HSCs as well, exerting an antifibrogenic effect.160 In addition, VD deficiency can downregulate defensins of intestinal Paneth cells, which damages the intestinal innate immunity and causes IR and NASH consequently.75
Although several studies have shown that supplementation of VD yielded no improvement in liver steatosis in NASH patients,162,163 the VDR is a promising target for treating NAFLD and is widely researched in rat models. Macrophages exhibit the highest expression of VDR mRNA among macrophages, hepatocytes, HSCs, and cholangiocytes.164 It has been reported that activation of VDRs on hepatic macrophages can improve insulin sensitivity and relieve liver steatosis, therefore exhibiting anti-inflammatory and antidiabetic effects.165 Activation of VDRs on HSCs can antagonize TGF-β/SMAD signaling, decreasing the expression of several profibrogenic genes consequently.166 However, although hepatocytes express low levels of VDRs, activation of VDRs on hepatocytes causes lipid accumulation, which suggests that drugs are required to target specific cells.
Furthermore, the impaired metabolism and increased vitamin A accumulation in the liver are associated with the progression of NAFLD, with elevated expression of inflammatory markers such as c-c motif chemokine ligand 2 and IL-6, as well as markers of liver fibrosis such as Timp1 and TGF-β.167 Supplementation of vitamin C also showed improvement in liver enzymes and glucose metabolism in NAFLD patients.168 Therefore, the roles of different types of vitamins in NAFLD progression deserve more investigation (Figure 4).
FIGURE 4.
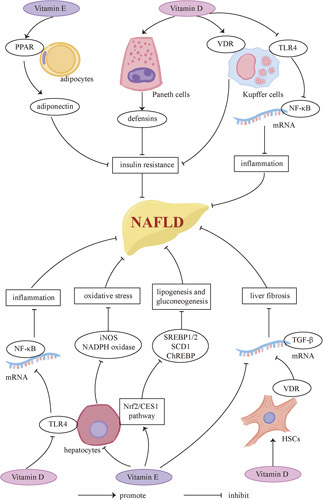
Vitamin and NAFLD. Both vitamin E and vitamin D contribute to the regression of NAFLD. VE can induce the expression of adiponectin by activation of PPAR, enhancing insulin sensitivity consequently. Moreover, VE can inhibit lipogenesis and gluconeogenesis through the Nrf2/CES1 pathway, and alleviate oxidative stress via downregulation of iNOS and NADPH oxidase. It can lower the expression of TGF-β as well, which prevents liver fibrosis. Vitamin D supplementation can upregulate defensins associated with intestinal innate immunity, preventing IR consequently. VD can also suppress TLR4 on KCs and hepatocytes, which can downregulate NF-κB and is associated with inflammation. Activation of the VDRs on KCs and HSCs can improve insulin sensitivity and alleviate liver fibrosis, respectively. Abbreviations: CES1, carboxylesterase 1; iNOS, inducible nitric oxide synthase; Nrf2, nuclear factor erythroid 2-related factor 2; PPAR, peroxisome proliferation–activated receptor; TLR4, Toll-like receptor 4; VDR, vitamin D receptor.
DIET AND NAFLD
Key points
Changes in dietary patterns are promising treatments for NAFLD. The Mediterranean diet (MD) has already been recommended by several guidelines. Effects of other popular diets, including intermittent fasting (IF), calorie-restricted diet (CRD), ketogenic diet (KD), the dietary approach to stop hypertension (DASH) diet, low-carbohydrate diet (LCD), and LFD, are also widely studied in NAFLD patients.
A large number of clinical trials have tested the effects of dietary modifications in the regression of NAFLD. Here we will briefly discuss several popular diets, including IF, CRDs, KD, MDs, and DASH diets, and compare the effects of low-fat and LCDs on NAFLD (detailed in Table 1). Other diets such as plant-based diets and paleolithic diets have also been widely investigated, but until now there is little high-quality evidence to support any diet. We also summarize macronutrients’ distribution in the following diets (Table 2).
TABLE 1.
Clinical trials testing effects of different dietary approaches in NAFLD
| References | Type of the study and duration | No. patients and their features | Dietary intervention | Mean weight loss | Changes in liver enzymes | Changes in liver fat, steatosis and stiffness | Other major outcomes |
|---|---|---|---|---|---|---|---|
| Clinical trials comparing diverse dietary approaches with control (usual diets or exercise) | |||||||
| Oshakbayev et al169 | RCT 24 wk |
N = 80 Patients with severe NASH and metabolic syndrome including T2DM |
Main group: fast weight loss (CRD and walking) vs. drug treatment | In the main group, the weight loss varied from 7–16 kg (p < 0.0001) and achieved in the first 8–10 wk. In the drug group, the weight loss was 2–5 kg and in the end of the treatment. | ALT (p = 0.02) and AST (p < 0.0001) significantly reduced in the main group compared with the drug group. These levels in the control group did not achieve normality. | In the histological scoring system in the main vs. control group there was significant improvement in liver inflammation (p = 0.0019), hepatocellular ballooning (p = 0.0025) and steatosis/fibrosis (p = 0.0056 and 0.0043, respectively). | Glucose metabolism (2-h OGTT: p = 0.012; HbA1c: p < 0.0001), insulin (p < 0.0001) and HOMA-IR (p = 0.002) all improved significantly in the main group compared with the drug group. |
| Asghari et al170 | RCT 12 wk |
N = 60 BMI:25–35 kg/m2 |
CRD vs. control | The weight reduced significantly in the CRD group (−4.06 kg) compared with the control. | ALT and AST significantly reduced in the CRD group compared with the control (p < 0.05). | No significant changes were observed in patients’ liver steatosis grades either within or between the 2 groups. | LDL-C and HDL-C levels did not change significantly between the 2 groups. Antioxidant levels did not improve significantly between the 2 groups. |
| Johari et al171 | RCT 8 wk |
N = 43 NAFLD patients |
ADF vs. control | There was a significant reduction in mean weight in the intervention group (p = 0.003) but not within the control group (p = 0.86). | There was a significant reduction of ALT (p = 0.001) and AST (p = 0.004) in the intervention group but not within the control group (p > 0.32). ALT reduced more significantly in the intervention group compared with the control (p = 0.02). But no difference was reported between the 2 groups with AST level (p = 0.34). |
There was significant reduction of liver steatosis grading (p = 0.001) and fibrosis (SWE scores) (p = 0.001) in the intervention group but not within the control group (p > 0.30). In between-group analysis, a statistically significant reduction was observed in the intervention vs. control group, for liver steatosis and SWE scores (p = 0.01). |
There was no reduction of all lipid parameters (total cholesterol, LDL, HDL and TG; all p > 0.28) in both groups. Fasting blood sugar reduced significantly in both groups (p = 0.006 vs. 0.08), but no significant changes were observed between the 2 groups (p = 0.34). |
| Cai et al172 | RCT 12 wk |
N = 271 NAFLD patients |
ADF vs. TRF vs. control | −4.04 kg vs. −3.25 kg vs. −1.85 kg (p-value not specified) The body weight decreased significantly in the ADF and TRF group compared with the control group. No differences between the ADF and CRF groups are observed. |
Not evaluated. | Liver stiffness did not improve after the intervention in 3 groups, | TC was significantly decreased in the ADF group compared with the TRF group and control (p-value not specified). Both the ADF and TRF group achieved a significant reduction in serum triglycerides (p < 0.001) after 12 wk of intervention. |
| Varkaneh et al173 | RCT 12 wk |
N = 52 Obese or overweight patients with NAFLD |
TRF (16 h fasting/8 h feeding daily) plus a low-sugar diet vs. control | There was a significant reduction of weight (p < 0.001) in the intervention group but not within the control group (p = 0.064). And significant between-group differences in body weight loss were observed (p = 0.029). | Significant decreases in serum levels of ALT (p = 0.013) and AST (p = 0.010) were observed for the intervention group compared with control. | Fibrosis score (p = 0.009) and steatosis score/CAP (p < 0.001) were significantly reduced in the intervention group compared with control. | The intervention group experienced significant reductions in TG (p<0.001), TC (p = 0.001), and LDL-C (p = 0.014) compared with the control. But no changes in HDL-C levels were observed (p = 0.159). |
| Ezpeleta et al174 | RCT 3 mo |
N = 80 With obesity and NAFLD |
ADF with exercise vs. ADF vs. exercise vs. control | The combination intervention produces superior reductions in body weight vs. exercise alone and control (p < 0.01), but not fasting alone (p = 0.43). | Change in serum ALT in the combination group was significantly different compared with the control group (p = 0.01), but not significantly different compared with the ADF group (p = 0.48) or exercise group (p = 0.09). Change in serum AST did not significantly differ among the 4 groups. | IHTG content was significantly reduced in the combination group compared with the exercise group (p = 0.02) and the control group (p < 0.01). However, the reduction in IHTG content in the combination group was not significantly different compared with the ADF group (p = 0.05). | The combination intervention improved insulin resistance and insulin sensitivity vs. controls, but not vs. exercise alone or fasting alone. |
| Misciagna et al175 | RCT 6 mo |
N = 98 With moderate or severe NAFLD |
MD vs. control | No significant changes of BMI were observed between the 2 groups. | Lower levels of GGT were observed in the MD group but not in the control. | Fatty liver index (FLI) and NAFLD score measure by liver ultrasonography reduced significantly in the MD group (p-value not specified). A negative interaction between time and MD on the NAFLD score was observed. | — |
| Zade et al176 | RCT 8 wk |
N = 60 overweight and obese patients aged with NAFLD diagnosed by ultrasonography scan |
DASH diet vs. control | −3.8 kg vs. −2.3 kg (p = 0.006) | ALT (p = 0.02) and ALP (p = 0.001) significantly reduced in the DASH diet group compared with the control. | Both groups showed significant reduction in percentage of NAFLD grades (p < 0.001) and the DASH group had more significant changes (p = 0.003). | Insulin levels (p = 0.01), homeostasis model of assessment-estimated insulin resistance (HOMA-IR) (p = 0.01) significantly decreased and quantitative insulin sensitivity check index (QUICKI) (p = 0.004) significantly increased. Compared with the control diet, the DASH diet has resulted in significant reductions in serum triglycerides (p = 0.04) and total-/HDL-cholesterol ratio (p = 0.01). |
| Schwimmer et al177 | RCT 8 wk |
N = 40 adolescent boys with histologically diagnosed NAFLD and evidence of active disease (hepatic steatosis > 10% and ALT level ≥45 U/L) |
Diets low in free sugar vs. control | Not evaluated | ALT (p < 0.001), AST (p = 0.005) and GGT (p < 0.001) were significantly lower in the intervention group than the control. | The mean decrease in hepatic steatosis was 8% vs. 1% (p < 0.001) | Adherence to the diet was higher in the intervention diet group. There were no significant differences in glucose, insulin, homeostasis model assessment for insulin resistance, TG, LDL-C, or HDL-C. |
| Domínguez-Coello et al178 | RCT 24 wk |
N = 239 BMI: 29-40.99 kg/m2 |
Low-fructose diet vs. control | Larger decreases in waist circumference, waist circumference/height ratio were seen in the low-fructose diet group compared with the control. | Not evaluated. | Not evaluated. | Larger decreases of fasting blood glucose were seen in the intervention group compared with the control. But the intervention did not reduce insulin resistance. |
| Cohen et al179 | RCT 8 wk |
N = 29 adolescent boys with NAFLD |
Diets low in free sugar vs. control | −1.4 kg vs. 0.6 kg (p-value not specified) | ALT, AST and GGT all significantly reduced in the intervention group compared with the control (all p < 0.05). | The intervention group experienced a greater decrease in hepatic fat measured by magnetic resonance imaging-proton density fat fraction (MRI-PDFF) compared with the control (p < 0.05). Hepatic DNL was significantly decreased in the intervention group (from 34.6% to 24.1%) vs. the control group (33.9% to 34.6%). |
Fasting insulin, fasting glucose, TGs, TC and LDL-C all significantly reduced in the intervention group compared with the control (all p < 0.05). Percentage change in DNL during the intervention correlated significantly with changes in free sugar intake (r = 0.48, p = 0.011), insulin (r = 0.40, p = 0.047), and alanine aminotransferase (ALT) (r = 0.39, p = 0.049), but not hepatic fat (r = 0.13, p = 0.532). |
| Schmidt et al180 | RCT 12 wk |
N = 105 Latino youth |
Diets low in free sugar intake vs. control | No significant changes in weight were observed in both groups over the intervention period (p = 0;48) | There were no differential changes in AST (p = 0.71) and ALT (p = 0.68) between the 2 groups. | Changes in liver fat fraction (p = 0.50) and liver fibrosis measured by MRE (p = 0.31) did not differ between the intervention group and the control. | Participants with whole-body fat mass (FM) reduction, irrespective of randomization, had significant reductions in liver fat compared with participants without FM reduction (p < 0.001). |
| Clinical trials comparing MD with LFD | |||||||
| Ryan et al181 | RCT 6 wk |
N = 12 nondiabetic individuals with biopsy-proven NAFLD |
MD vs. LFD | The weight loss in both diets (MD: −1.0 kg vs. LFD: 2.4 kg) was not significant and the difference in weight change between the groups was also not significant (p = 0.1). | ALT and AST did not change appreciably with either diet. | Intrahepatic lipid measured by MRS reduced significantly in MD group compared with the LFD (p = 0.012). | Both the HOMA-IR and the circulating insulin concentration declined significantly (p = 0.008 and p = 0.003) in the MD group but there was no significant improvement in the LFD group. |
| Properzi et al182 | RCT 12 wk |
N = 51 With NAFLD |
Two isocaloric diets: MD vs. LFD | −2.1 kg vs. −1.6 kg (p = 0.52) | ALT and GGT improved significantly in both groups. But no significant differences were observed between the 2 groups. | Hepatic steatosis measured by MRS had reduced significantly in both groups (p < 0.01), but there was no difference in liver fat reduction between the groups (p = 0.32). | Within-group improvements in Framingham Risk Score, TC, TG, and HbA1c were observed in MD (all p < 0.05), but not in LFD. Adherence was higher for MD compared with LFD (p = 0.048). |
| Biolato et al183 | Crossover clinical trail 48 wk |
N = 20 with biopsy-verified NAFLD and increased transaminases. |
MD vs. LFD: 16 wk of an MD (W1–W16), 16 wk of a free washout diet (W17–W32) and 16 wk of a LFD (W33–W48). | A significant reduction of weight loss was observed after 16 wk of MD (p = 0.003). There was a significant reduction in mean body weight after 16 wk of MD compared wit LFD (p = 0.004). |
A significant reduction of ALT (p = 0.0001) and AST (p = 0.001) was observed after 16 wk of MD. There was a significant reduction in ALT after 16 wk of MD compared with LFD (p = 0.04). |
Not evaluated. | No significant changes in intestinal permeability were observed at the end of the MD period or the LFD period. |
| Gepner et al184 | RCT 18 mo |
N = 278 With abdominal obesity or dyslipidemia |
Mediterranean and low-carbohydrate diet vs. LFD for 6 mo. Physical activity vs. no exercise for 12 mo. |
Not evaluated. | Decreased hepatic fat content was associated with reductions in serum GGT and ALT (p < 0.05). Differences between the 2 groups were not specified. | Mediterranean and low-carbohydrate diet induced a greater hepatic fat content decrease (p = 0.036) and greater improvements in cardiometabolic risk parameters (p < 0.05) than LFD. | — |
| Ristic-Medic et al185 | RCT 3 mo |
N = 27 BMI:25−35 kg/m2 |
MD vs. LFD | −9.23 kg vs. −9.71 kg (p = 0.342) | Both diets lowered liver enzymes, but no significant differences were observed between the 2 groups. | Both diets improved FLI and hepatic steatosis index. FLI in MD reduced more significantly (p = 0.022). | Participants on MD had higher levels of HDL-C and monounsaturated and n-3 docosahexaenoic acids in serum phospholipids and lower levels of saturated fatty acids, TG, and TG/HDL ratio when compared with participants on LFD. |
| Akbulut et al186 | RCT 12 wk |
N = 70 Children with NAFLD |
MD vs. LFD | 2.0 kg vs. 2.7 kg (p = 0.006) | ALT and AST levels both decreased significantly in both groups, but no differences between the 2 groups were observed. | Marked decreases in the degree of steatosis and liver stiffness were observed in both groups compared with the baseline. But there were no significant differences between the 2 groups. | Significant reductions in HOMA-IR were obtained in the MD group (18%) compared with the LFD group (12%) (p = 0.010). |
| George et al2022187 | RCT 12 wk |
N = 42 With NAFLD |
MD vs. LFD | The weight was not significantly different from preintervention to postintervention in both groups. | ALT, AST, and GGT were not statistically significant following the MD (p = 0.32, 0.29 and 0.26, respectively), but significant reductions were noted in the LFD group (p = 0.009, 0.040 and 0.029, respectively). Changes between groups were significant (p-value not specified). |
There was a significant improvement of intrahepatic lipids in the low-fat group (p = 0.02) but not in the MD group (p = 0.07). No significant differences for intrahepatic lipids (p = 0.865) and liver stiffness (p = 0.58) measurement were observed between the 2 groups. | No significant differences of HOMA-IR were observed between the 2 groups (p = 0.58). |
| Yurtdaş et al188 | RCT 12 wk |
N = 44 Adolescents with NAFLD |
MD vs. LFD | The mean body weight of participants significantly decreased in both groups (p < 0.001), but weight loss was not statistically different between groups (p = 0.532). | ALT, AST, and GGT decreased significantly in both groups with no significant differences between groups except for AST, which reduced more in the MD group than in the low-fat diet group (p = 0.039). | A statistically significant decrease was found in the grade of hepatic steatosis after the intervention in both diet groups (p < 0.05). There was no significant difference between the groups in terms of reduction in the degree of liver fat (p = 0.875). | Total antioxidant status, paraoxanase-1, and GSH-Px levels increased in the MD (p < 0.05) group but did not change in the LFD group (p > 0.05) compared with baseline. |
| Clinical trials comparing LCD with LFD | |||||||
| Ryan et al189 | RCT 16 wk |
N = 52 With obesity |
Two hypocaloric diets: LCD vs. LFD | Both groups experienced a significant decrease in weight (7.0 kg vs. 5.7 kg) (both p < 0.001). But it did not differ between the 2 groups. | ALT significantly reduced in the LCD group (p < 0.04). ALT changes correlated with improvement in insulin sensitivity (p = 0.04). Individuals with ALT concentrations above the proposed upper limits experienced a significant decline in ALT, unlike those with lower ALT levels. |
Not evaluated. | SSPG (p < 0.04) and circulating insulin (p < 0.01) significantly reduced in the LCD group but not in the LFD group. |
| Hernández et al190 | RCT 6 mo |
N = 59 With NAFLD |
LCD vs. LFD | −5.7% vs. −5.5% There was no significant difference in weight loss between the 2 groups. |
The LCD group showed more reduction in ALT (41 vs. 33.3%) and AST (31.7 vs. 22.4%) compared with the low-fat group, but there were no significant statistical differences. Changes in weight were positively related with changes in ALT and AST, irrespective of the type of diet. | Not evaluated. | — |
| Haufe et al191 | RCT 6 mo |
N = 170 Overweight and obese individuals |
LCD vs. LFD | The weight decreased significantly in the LCD group compared with the LFD group (p <0.001). | ALT and AST did not change significantly after the intervention in both groups. | Subjects with high baseline intrahepatic lipids (>5.56%) lost about 7-fold more intrahepatic lipids compared with those with low baseline values (<5.56%) irrespective of diet composition. | TC and LDL-C decreased more in the LFD group compared with the LCD group (p-value not specified). |
| Goss 2020192 | RCT 8 wk |
N = 32 Children/adolescents with obesity and NAFLD |
LCD vs. LFD | −2.4% vs. −0.4% (p = 0.06) | ALT and AST decreased significantly only in the LCD group (p < 0.05), but no significant differences were observed between groups (p = 0.15 and 0.43, respectively). | Changes in hepatic lipid accumulation did not differ between the 2 groups, but declined significantly (p < 0.001) only within the LCD group. | Significantly greater decreases in insulin resistance (HOMA-IR, p < 0.05), abdominal fat mass (p < 0.01), and body fat mass (p < 0 .01) were found in the LCD group compared with the LFD group. |
| Hansen et al193 | 6 mo | N = 165 T2DM |
LCD vs. LFD (Both are calorie-unrestricted.) |
Participants on the LCD lost more weight than LFD. | Not evaluated. | No statistically significant between-group changes were detected in the assessment of NAFLD by liver biopsies. | Participants on the LCD had greater improvements in HbA1c but less favorable changes in LDL-C. Both groups had higher HDL-C and lower TG at 6 mo but no significant changes were observed between the 2 groups. Changes were not sustained at the 9-month follow-up. |
| Clinical trials comparing other dietary approaches | |||||||
| Browning et al194 | RCT 2 wk |
N = 18 BMI: 35 ± 7 kg/m2 |
CRD vs. LCD | −4.0 kg vs. −4.6 kg (p = 0.0363) | ALT did not change significantly in either group. AST reduced significantly after 2 wk of weight loss (p < 0.001), but no differences were observed between the 2 groups. |
Not evaluated. | Liver triglycerides decreased significantly with weight loss (p < 0.001) but decreased significantly more (p = 0.008) in the LCD group than in the CRD group. Dietary fat (p = 0.004), carbohydrate (p = 0.008), post-treatment plasma ketones (p = 0.006), and respiratory quotient (p < 0.001) were related to the reduction in liver triglycerides. |
| Skytte et al195 | RCT 6 wk |
N = 30 T2DM |
Low-carbohydrate, high-protein diet vs. conventional diabetes diet | −1.4 kg vs. −0.8 kg (p = 0.071) | Not evaluated. | The low-carbohydrate, high-protein diet had more significant reduction in liver fat fraction (p < 0.01) and pancreatic fat fraction (p < 0.05) compared with the conventional diabetes diet. | The low-carbohydrate, high-protein diet had more significant reduction in HbA1c (p < 0.001) and fasting glucose (p < 0.05). TC reduced significantly in the low-carbohydrate, high-protein group compared with the conventional group (p < 0.05). |
| Thomsen et al196 | 6 wk | N = 72 T2DM, BMI >25 kg/m2 |
Low-carbohydrate, high-protein diet vs. conventional diabetes diet | −5.8 kg vs. −5.8 kg (p = 0.83) | Not evaluated. | Hepatic fat content was reduced significantly by 64% and 51% after the low-carbohydrate, high-protein diet, and conventional diabetes diet, respectively, with the difference between groups reaching borderline significance (p = 0.051). | The low-carbohydrate, high-protein diet had more significant reduction in HbA1c (p = 0.018) and diurnal mean glucose (p < 0.001). Fasting glucose, insulin, HOMA-IR, and cholesterol concentrations (total, LDL, and HDL) were reduced significantly and similarly by both diets. |
| Cunha et al197 | RCT 2 mo |
N = 39 BMI >30 kg/m2 |
low-calorie KD vs. CRD | −9.59 kg vs. −1.87 kg (p < 0.001) | AST decreased in the low-calorie KD group (p < 0.05) but not in the CRD group. | Liver fat fraction reduced more significantly in the low-calorie KD group (p < 0.005). No differences in mean liver stiffness values were seen between groups. |
Mean reductions in visceral adipose tissue were significantly more pronounced in the low-calorie KD group than CRD group (p < 0.05). |
| Lin et al198 | RCT 12 wk |
N = 132 BMI ≥ 30 kg/m2 |
CRD (450 kcal/d) vs. CRD (800 kcal/d) | −8.37 kg vs. −8.42 kg (p = 0.092) | Not evaluated. | The improvement rate of NAFLD was 41.5% vs. 50.0% (p-value not specified). | Fat mass, blood pressure, triglycerides, and blood glucose were statistically improved from baseline but not between the 2 groups. There is no additional benefit in prescribing the more restrictive diet intervention. |
| Crabtree et al199 | RCT 6 wk |
N = 37 Overweight and obese |
KD vs. KD + KS (KD with an exogenous ketone salt supplement) vs. KD + PL (KD with placebo) | Mean weight loss at 6 wk was significant (p < 0.001) in all 3 groups. There was no significant difference in weight loss among the KD + KS, KD + PL, and KD groups, representing decreases of 8.1%, 8.5%, and 6.7% of the initial body mass, respectively (p > 0.05). | There were no differences in AST and ALT, from baseline to postintervention. | There was a significant (p = 0.004) decrease in liver fat measured by MRI and hepatic steatosis index postintervention, but no differences among the 3 groups (p > 0.05). | The change in liver fat is highly associated with baseline liver fat rather than weight loss or other changes in anthropometric or circulating markers. |
| Parry et al200 | Crossover clinical trials 15 wk |
N = 16 BMI: 25–30 kg/m2 |
Two eucaloric diets enriched in saturated fat (SFA) vs. free sugars (SUGAR) (4 weeks of SFA or SUGAR, 7 wk of washout period and 4 wk of the alternate diet) | Body weight significantly increased after consumption of the SFA (p < 0.05) but not the SUGAR diet. | No significant changes of ALT were observed in both groups after the intervention. | IHTG significantly increased following the SFA diet (p < 0.05), while it remained unchanged in response to the SUGAR diet. | Significant decreases in plasma TC and HDL-C were observed in the SUGAR group compared with the SFA group (p < 0.05). |
Abbreviations: ADF, alternate-day fasting; ALP, alkaline phosphatase; ALT, alanine transferase; AST, aspartate transaminase; BMI, body mass index; CRD, calorie-restricted diet; DASH, dietary approach to stop hypertension; DNL, de novo lipogenesis; GGT, γ-glutamyltransferase; HbA1c, glycosylated hemoglobin type A1c; HOMA-IR, homeostasis model assessment – insulin resistance; IHTG, intrahepatic triglyceride; KD, ketogenic diet; LCD, low-carbohydrate diet; LFD, low-fat diet; MD, Mediterranean diet; MRE, magnetic resonance elastography; MRS, magnetic resonance spectroscopy; OGTT, oral glucose tolerance test; RCT, randomized controlled trial; SFA, saturated fatty acid; SWE, shear-wave elastography; T2DM, type 2 diabetes mellitus; TC, total cholesterol; TG, triglyceride; TRF, time-restricted fasting.
TABLE 2.
Macronutrients of diet patterns
| Diet | Carbohydrate (%) | Fat (%) | Protein (%) |
|---|---|---|---|
| KD | <10 | >70–90 | 10–15 |
| MD | 35–45 | 30–40 | 15–20 |
| DASH diet | 52–55 | 30 | 16–18 |
| LFD | 60 | 25 | 15 |
| LCD | 40 | 45 | 15 |
Abbreviations: DASH, dietary approach to stop hypertension; KD, ketogenic diet; LCD, low-carbohydrate diet; LFD, low-fat diet; MD, Mediterranean diet.
Calorie-restricted diet
A CRD, regardless of its macronutrient composition, can induce weight loss, which is associated with decreased total body fat and intrahepatic lipid depletion consequently.191,201 Indeed, Promrat et al202 reported an almost linear dose response between weight loss and resolution of hepatic steatosis, assessed by liver biopsy in NAFLD individuals. Moreover, the negative energy balance is related to reduced serum ALT and AST levels and metabolic benefits, including improved β-cell function and insulin sensitivity.203 NAFLD patients following CRD have also shown NASH and fibrosis regression histologically.204 However, Asghari et al170 reported that no changes were observed in the total antioxidant capacity, erythrocyte superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) activities between the hypocaloric group and the control group after 12 weeks of intervention, indicating that CRD cannot improve the antioxidant abilities of NAFLD patients, which is related to several chronic diseases. Further studies are warranted to target the effects of long-term intervention in NAFLD patients.
Unfortunately, significant weight loss can be difficult to maintain for many NAFLD patients in the long term. The average weight loss is only about 3%–5% from baseline weight after 2–4 years.205 Chronic nutrient deficiency, which is related to liver dysregulation, is another concern for the wide application of CRD in NAFLD patients. Individuals with eating disorders are more susceptible to both acute and chronic liver injury.206 Kwashiorkor, which is a severe consequence of protein deficiency, is also associated with fatty liver and hepatomegaly.207 Therefore, changing mealtimes or the composition of macronutrients in the diet rather than restricting calorie intake alone may be a better alternative.
Intermittent fasting and time-restricted fasting
IF has become increasingly popular in the past few decades. There are several patterns of IF, including alternate-day fasting, every-other-day fasting, and 5:2 fasting, which means consuming extremely low calories on 2 discontinuous days followed by normal eating patterns on 5 unrestricted days. Another widely used pattern of IF is time-restricted fasting (TRF), which refers to feeding within a certain period of time (usually 4–8 h) and fasting for the rest of the day. TRF does not restrict the caloric intake in the feeding time, which seems to be easier than other IF patterns for NAFLD patients to adopt. However, some researchers believe that TRF is not synonymous with IF.208
IF has been shown to be equally effective as traditional caloric restriction diets in weight loss and improving cardiometabolic outcomes.209 An observational cohort study found that the fatty liver index notably reduced after a mean period of 8.5 days of fasting, with a more significant decrease in individuals with diabetes. The improvement of fatty liver index correlated with the duration of fasting and the magnitude of weight loss.210 TRF has also been demonstrated to improve insulin sensitivity, blood pressure, and oxidative stress in prediabetes patients without weight loss. However, another study showed that no changes were observed in the fasting insulin level and liver stiffness between the fasting intervention group and the control.172 Moreover, Ezpeleta et al174 reported that a combination of IF and exercise can reduce intrahepatic triglyceride (IHTG) content, body weight, and fat mass and improve IR in NAFLD individuals. A combination intervention showed superior improvement versus exercise alone but not fasting alone, indicating that IF is the primary intervening factor benefiting NAFLD patients. Furthermore, an 8-week RCT showed that alternate fasting resulted in a greater improvement in steatosis and fibrosis as measured by ultrasound and elastography.13–107 However, current evidence concerning the effects of TRE on hepatic fat is still weak.
Research on mice provides more insight into the potential mechanisms of fasting on NAFLD treatment. When glycogen stored in the liver is depleted after fasting for over 12 hours, the metabolic switch, which refers to the shift from utilization of glucose from glycogenolysis to fatty acids and ketones, will occur.211 Consequently, the increase of adipose tissue lipolysis results in weight loss, improved insulin sensitivity, and decreased risks of CVDs due to lower contents of TC and LDL-C.212 Furthermore, IF can elevate the expression of a variety of genes involved in lipolysis and fatty acid oxidation in NAFLD mouse models induced by high-fat, high-sucrose diets (HFHSD), accompanied by decreased accumulation of lipid in the liver.213 Hatori et al214 also reported that TRF improved oscillations of the circadian clock, which is related to obesity and diabetes.
However, there are safety concerns about applying IF for NAFLD treatment. IF may aggravate starvation effects and even trigger binge eating in unrestricted periods. Some researchers are also worried that IF may lead to hypoglycemia in patients with diabetes despite only one clinical trial of IF in NAFLD patients reporting it until now.215 Although no significant adverse effects were observed in the current research, it is difficult to maintain the daily feeding window because of family and social life, which is another limitation of applying IF.208
The effects of IF in NAFLD prevention and treatment still require further investigation. In most of the clinical trials, the calorie intake of the control group is not the same as the IF group, so we cannot distinguish whether fasting or weight loss contributes to the beneficial effects. The duration of the research is also too short, so the long-term effects and adherence of IF cannot be observed. However, IF provides an easier way for some patients to lose weight than calorie-restricted diets.
Low and very LCD (KD)
KDs are dietary patterns that restrict carbohydrate intake to below 20–50 g per day, accounting for less than 10% of the daily caloric intake. KD has been proven to be effective in losing weight and reducing visceral fat.216 A few studies tested KD as a treatment for NAFLD patients. Tendler et al217 reported an improvement in hepatic inflammation, steatosis, and fibrosis in 5 NAFLD patients following KDs. Luukkonen et al218 also reported that KD could reduce accumulation of IHTGs and improve IR.
Carbohydrate restriction can improve insulin sensitivity, which results in more oxidation of fatty acids and less lipogenesis in overweight and obese individuals.201 What’s more, KDs can induce the production of ketone bodies, which can increase hepatic glutathione peroxidase 1 and decrease oxidized glutathione, as well as elevating mitochondrial transcription factor A (TFAM) in NAFLD rat models, which is associated with mitochondrial biogenesis.219 Furthermore, NAFLD patients have been reported to display impaired capacity of ketogenesis and elevated acetyl-CoA oxidation in the TCA cycle alternatively, associated with increased oxidative metabolism and impaired glucose homeostasis.220 Therefore, stimulation of ketogenesis is believed to be a potential target in NAFLD.
However, some researchers argue that it is hard to determine whether weight loss or carbohydrate restriction contributes to the beneficial effects. Pérez-Guisado et al221 reported an improvement in serum liver enzyme levels and a reduction in steatosis degrees in 14 overweight NAFLD individuals who applied KDs, with no restriction in caloric intake. Moreover, patients who apply carbohydrate-restricted diets exhibit a more significant decrease in liver triglyceride compared with calorie-restricted diets.194 However, in 22 obese individuals who received a calorie-restricted diet, the reduction of hepatic triglyceride was similar in both the low-carbohydrate group and the high-carbohydrate group.222 A meta-analysis suggested that both macronutrient distribution and calorie restriction are crucial in the regression of NAFLD.223 Conflicting results indicate that more large-scale research is required to clarify the roles of carbohydrate restriction in NAFLD.
Furthermore, it is controversial to apply KDs in NAFLD patients since they are also high in fat. TC and LDL-C values change unfavorably in patients with low carbohydrate intake, which is associated with a higher risk of CVDs.224 It is also reported that KD can lead to hypotension, electrolyte disturbance, and symptomatic gallstones.225,226 Moreover, individuals following KDs show numerous discomforts such as vomiting, constipation, fatigue, and irritability,212 so it is difficult for them to adhere for a long time. Hence, the side effects of KDs should be taken into consideration and more long-term investigation is required before the wide range of applications. However, considering that KD restricts sugary foods in the diet and helps lose weight, it can provide an opportunity for obese individuals to achieve normal BMIs.223
Mediterranean diet
The MD, which has been considered as a healthy diet worldwide, is recommended by European Society for Parenteral and Enteral Nutrition guidelines and European Association for the Study of the Liver–European Association for the Study of Diabetes–European Association for the Study of Obesity (EASL–EASD–EASO) Clinical Practice Guidelines to treat NAFLD.227,228 It is characterized by a high intake of olive oil, which is a major source of fat in the diet, and a low intake of added sugars and refined carbohydrates. Actually, it is also a high-fat, LCD. What’s more, MD contains a large number of vegetables, fruits, and whole grains, as well as moderate wine and dairy food in the diet, while red meat, especially processed meat, intake is low.
MD has been reported to show a more significant reduction of liver steatosis and improvement of insulin sensitivity compared with LFD (39% vs. 7%), although there is no difference in weight loss between the 2 groups.181 Tsaban et al229 also reported a decrease of serum LDL-C levels and Framingham Risk Score, which is associated with cardiovascular risks, in 293 patients consuming green MD.
Moreover, MD is a dietary pattern rich in antioxidants, mainly because the major source of fat is monounsaturated and polyunsaturated fats instead of saturated fats. The appropriate ratio of n-3 to n-6 fatty acids in MD provides an opportunity to benefit from n-3 polyunsaturated fats,230 which can prevent intrahepatic lipid accumulation and decrease the risks of steatosis.231 What’s more, patients following MD have decreased serum hypersensitive-C-reactive-protein and inflammatory cytokines such as IL-6, IL-7, and IL-18 compared with the control groups. Endothelial function and IR are also improved in the MD group, associated with decreased rates of metabolic syndromes and risks of CVDs.232 Another clinical trial, in which patients were divided into 3 groups (MD, MD with supplementation of antioxidants, control), also reported that individuals consuming MD with or without antioxidants showed decreased liver lipid accumulation and fibrosis. However, groups with extra antioxidant intake exhibited more significantly reduced BMI, waist, hip circumference, and improved glucose metabolism compared with patients with MD alone, indicating the role of antioxidants in the prevention of NAFLD.233
Furthermore, another characteristic of MD is the consumption of coffee, which is considered to reduce the risk of liver fibrosis in NAFLD.234 Coffee intake can lower the risks of HCC as well, but interestingly, caffeine may not contribute to this beneficial effect.235 Paola reported that gut permeability and the gut-liver axis modulated by coffee participate in the prevention of NAFLD in HFD-induced mouse models, with reduced liver lipid accumulation and improved energy metabolism.236 Coffee is also reported to regulate the expression of long noncoding RNA in pathways of NAFLD progression, such as inhibiting lipogenesis through the long noncoding RNA Gm16551/Srebf1 pathway as well as long noncoding RNA H19 associated with fibrosis.237 Although the mechanisms of coffee in preventing NAFLD are unclear, coffee consumption provides a hopeful option for patients.
However, MD recommends moderately drinking wine. It is controversial whether NAFLD patients should accept it since current studies in humans show conflicting results. Long et al238 reported an association between alcohol and hepatic steatosis, indicating that alcohol is a risk factor for NAFLD and should be avoided, while Mitchell et al239 reported that modest wine consumption(1–70 g/wk) can lower the rates of liver fibrosis. What’s more, it has been reported that any amount of alcohol consumption increases the risk of NASH-induced HCC,240 but specific components of wine such as resveratrol show antioxidant properties and therefore can improve endothelial function and hypertension.241 Therefore, NAFLD patients should balance between the risks and benefits. More studies concerning the mechanisms of how wine modulates NAFLD and longitudinal clinical trials are required before clinical recommendation.
DASH diet
The DASH diet, which can decrease cardiovascular risks,242 was originally used to regulate blood pressure, while NAFLD patients have been found to benefit from it as well.176 The DASH diet is similar to MD but characterized by reduced sodium, added sugars, and saturated fats.
Mohsen et al reported that NAFLD patients following the DASH diet had significantly decreased weight, ALT, alkaline phosphatase, and serum triglyceride levels, as well as improved insulin sensitivity and markers of inflammation.176 Hekmatdoost et al243 also reported an inverse relationship between adherence to DASH diets and risks of NAFLD. Moreover, a meta-analysis suggested that the DASH diet is more suitable for obese individuals to lose weight than a low-calorie diet.244 However, currently, studies on the effects of DASH diet in NAFLD patients are limited. More clinical trials concerning the influence and side effects of DASH diets in NAFLD are needed.
Comparison between LFD and LCD
It is controversial whether a LFD or a LCD is a better choice for NAFLD patients, although both of them are effective in losing weight. Ryan et al189 reported that in 2 groups with hypocaloric diets, individuals following LCDs showed more significantly improved ALT levels and insulin sensitivity than those following LFDs, although body weight loss was similar in both groups. Likewise, there was a more significant reduction of IHTG in the LCD group than in the low-fat, high-carbohydrate group after 48 hours. However, over a long period of 11 weeks, both groups exhibited similar weight loss and no differences in the decrease of IHTG.222 On the contrary, Jukka et al(245) reported that liver fat decreased in the LFD group but increased in the high-fat, low-carbohydrate group, in parallel with changes in serum insulin levels [245]. A meta-analysis in 2019 also suggested that the LCD and LFD groups had similar improvements in hepatic fat and serum liver enzymes, indicating that current evidence to support either diet is limited.246
There has been no conclusion on which diet pattern is superior to the other for NAFLD patients. The conflicting results may be attributed to the different sources of carbohydrates and fat, as well as adherence of participants to the diets, which should be taken into consideration in the future investigation.
METABOLIC TARGETS IN NAFLD
Key points
Various metabolic drugs have been approved for clinical trials in the treatment of NAFLD/NASH. farnesoid X receptor (FXR), THRβ, and hepatic DNL enzymes have been identified as promising drug targets. Plenty of FDA-approved antidiabetic and lipid-modifying agents are undergoing phase 4 clinical trials for NAFLD/NASH treatment.
Though there has been no licensed drug for NAFLD treatment so far, various metabolic targets have been discovered and plenty of potential drugs have been approved for clinical trials in recent years. Here, we summarized the major metabolic targets and potential monotherapy drugs under development for the treatment of NAFLD/NASH (Table 3).
TABLE 3.
Update of major metabolic targets and potential monotherapy drugs under development for NAFLD/NASH
| Clinical improvements | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Target(s) | Agent | Current phase | Condition | Weight loss | ALT | LFC/steatosis | fibrosis | Sponsor | References |
| FXR | Nidufexor (LMB763)a | Phase 2 (terminated) | NASH | √ | √ | √ | — | Novartis | Chianelli et al247 |
| TERN-101 | Phase 2a | NASH | NA | √ | √ | √ | Terns | Wang et al248 | |
| Cilofexor (GS-9674)a | Phase 2 | NASH | — | — | √ | — | Gilead | Patel et al249 | |
| MET642a | Phase 2 | NASH | NA | NA | √ | NA | Metacrine | — | |
| Px-104 | phase 2 | NAFLD | — | √ | — | TBD | Phenex | Traussnigg et al250 | |
| EDP-297a | Phase 1 | NASH | NA | NA | NA | NA | Enanta | — | |
| EDP-305a | phase 2b | NAFLD | — | √ | √ | — | Enanta | Ratziu et al251 | |
| HPG1860 | Phase 2a | NASH | TBD | √ | TBD | TBD | Hepagene | — | |
| HEC96719 | Phase 2 | NASH | TBD | TBD | TBD | TBD | HEC Pharm | — | |
| Tropifexor (LJN452)a | Phase 2 (terminated) | NASH | √ | √ | √ | — | Novartis | Sanyal et al252 | |
| Vonafexor (EYP001a) | Phase 2 | NASH | √ | √ | √ | √ | Enyo Pharma | Ratziu et al253 | |
| XZP-5610 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Sihuan pharm | — | |
| MET409a | Phase 2 | NASH | √ | — | √ | NA | Metacrine | Harrison et al254 | |
| ASC42 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Ascletis Pharma | — | |
| Obeticholic acid | Phase 3 | NAFLD/NASH with or without T2D | √ | √ | √ | √ | Intercept | Younossi et al255 | |
| iBAT/ASBT/SLC10A2 | Elobixibat (A3309)a | Phase 2 | NAFLD NASH |
NA | — | — | NA | Albireo | — |
| Volixibat (SHP626)a | Phase 2 (terminated) | NASH | NA | — | — | — | Mirum | Newsome et al (2020)256 | |
| SGLT2 | Empagliflozin (Jardiance) | Phase 4 | NAFLD with or without T2D | √ | √ | √ | — | — | Kuchay et al257 Taheri et al258 |
| Ertugliflozin | Phase 4 | NAFLD with T2D | TBD | TBD | TBD | TBD | Getz Pharma | — | |
| Dapagliflozin | Phase 4 | NAFLD with T2D | √ | √ | √ | — | — | Shimizu et al259 Phrueksotsai et al260 | |
| Canagliflozin | Phase 4 | NAFLD with T2D | √ | √ | √ | — | — | Inoue et al261 | |
| SGLT1&2 | Licogliflozin (LIK066) | Phase 2 | NASH | √ | √ | √ | √ | Novartis | Harrison et al262 |
| PPARα | Pemafibrate (K-877) | Phase 2 | NAFLD | — | √ | — | — | Kowa Company | Nakajima et al263 |
| Fenofibrate | Phase 4 | NAFLD | √ | √ | — | — | — | Fernández-Miranda et al264 Oscarsson et al101 | |
| PPARγ | Rosiglitazone | Phase 4 | — | √ | √ | — | — | Ratziu V, et al. (2008)265 | |
| Lobeglitazone | Phase 4 | — | — | √ | √ | — | — | Lee et al266 | |
| Pioglitazone | Phase 4 | NAFLD/NASH with T2D | — | √ | √ | √ | — | Aithal et al267 Cusi et al268 | |
| PXL065 | Phase 2 | NASH | — | — | √ | — | Poxel | Harrison et al269 | |
| PPARδ(PPARβ) | Seladelpar | Phase 2 | NASH | NA | √ | — | NA | CymaBay | — |
| PPARα/δ | Elafibranora | Phase 3 (terminated) | NASH | — | √ | √ | √ | Genfit | Ratziu et al270 |
| PPARα/γ | Saroglitazar | Phase 2 | NAFLD/NASH | — | √ | √ | — | Zydus Therapeutics | Siddiqui et al271 Gawrieh et al272 |
| pan-PPAR | Lanifibranor (IVA337) | Phase 3 | NAFLD/NASH | — | √ | NA | √ | — | Sven et al273 |
| Chiglitazar | Phase 2 | NASH | TBD | TBD | TBD | TBD | Chipscreen Biosciences | — | |
| DPP-4 | Sitagliptin | Phase 4 (terminated) | NAFLD/NASH | — | — | — | — | — | Cui et al274 Joy et al275 |
| Evogliptin | Phase 4 | NAFLD With T2D | — | √ | √ | TBD | Han E, et al. (2022)276 | ||
| GLP-1R | Liraglutide | Phase 4 | NAFLD /NASH | √ | √ | √ | TBD | — | Armstrong et al277 Petit et al278 |
| Semaglutide | Phase 4 | NASH | √ | √ | √ | — | — | Flint et al279 Newsome et al280 | |
| Dulaglutide | Phase 4 | NAFLD/NASH with T2D | √ | — | √ | NA | Kuchay et al281 | ||
| Ecnoglutide (XW003) | Phase 1 | NASH, T2D and obesity | TBD | TBD | TBD | TBD | Sciwind Biosciences | — | |
| Exenatide | Phase 4 | — | √ | √ | √ | √ | — | Shao et al282 Liu et al283 | |
| ECC5004 | Phase 1 | NASH T2D |
TBD | TBD | TBD | TBD | Eccogene | — | |
| GLP-1R& GIPR | VK2735 | Phase 1 | NASH | NA | NA | NA | NA | Viking Therapeutics | — |
| Tirzepatide(LY3298176) | Phase 2 | NASH | √ | √ | √ | NA | Eli Lilly and Company | — | |
| GLP-1R& GCGR | Efinopegdutide | Phase 2a | NASH | TBD | TBD | TBD | TBD | Merck Sharp & Dohme | — |
| Cotadutide (MEDI0382)a | Phase 3 | NASH | — | — | √ | NA | AstraZeneca | — | |
| ALT-801 (Pemvidutide) | Phase 1 | NAFLD with or without T2D | √ | √ | √ | TBD | Altimmune | — | |
| BI 456906 | Phase 2 | NASH | √ | TBD | TBD | TBD | Boehringer Ingelheim | Yazawa et al284 Jungnik et al285 | |
| SAR425899 (Bamadutide) | Phase 2 (withdrawn) | NASH | √ | NA | NA | NA | Sanofi | — | |
| DD01 | Phase 1 | NASH with T2D | TBD | TBD | TBD | TBD | D&D Pharmatech | — | |
| GLP-1R & GIPR & GCGR | Efocipegtrutide (HM15211) | Phase 2b | NASH | TBD | TBD | TBD | TBD | Hanmi | — |
| FGF21 | pegozafermin (BIO89-100) | Phase 2 | NASH | — | √ | √ | TBD | 89bio | Loomba et al286 |
| B1344 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Tasly Biopharma | — | |
| NNC0194 0499 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Novo Nordisk A/S | — | |
| Pegbelfermin (BMS-986036) | Phase 2 | NASH | — | √ | √ | √ | Bristol-Myers Squibb | Sanyal et al287 Brown et al288 | |
| BOS-580 | Phase 2 | NASH | TBD | TBD | TBD | TBD | Boston Pharmaceuticals | — | |
| Efruxifermin (EFX) | Phase 2 | NASH | √ | √ | √ | √ | Akero Therapeutics | Harrison et al289 | |
| GLP-1&FGF21 | HEC88473 | Phase 1 | NASH | TBD | TBD | TBD | TBD | HEC Pharm | — |
| FGFR1&KLB | BFKB8488A | Phase 2 | NASH | √ | √ | √ | TBD | Genentech | Baruch et al290 Wong et al291 |
| MK-3655 (NGM313)a | Phase 2 | NASH | NA | NA | NA | NA | Merck Sharp & Dohme LLC | — | |
| FGF19 | Aldafermin (NGM282)a | Phase 2 | NASH | — | √ | √ | — | NGM Bio | Harrison et al292 |
| GR | miricorilant | Phase 2 (suspended) | NASH | TBD | TBD | TBD | TBD | Corcept Therapeutics | — |
| FASN | Denifanstat (TVB-2640) | Phase 2 | NASH | — | √ | √ | √ | Sagimet Biosciences | Syed-Abdulet al293 Loomba et al294 |
| FT-4101a | Phase 2 (terminated) | NASH | — | NA | √ | NA | Forma Therapeutics | Beysen et al295 | |
| KHK | PF-06835919a | Phase 2 | NAFLD | — | — | √ | NA | Pfizer | Kazierad et al296 Saxena et al297 |
| XZP-6019 | Phase 1 | NAFLD | TBD | TBD | TBD | TBD | Sihuan Pharmaceutical Holdings Group Ltd. | — | |
| DGAT2 | PF-07202954 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Pfizer | — |
| Ervogastat (PF-06865571) | Phase 2 | NASH | NA | NA | √ | NA | Pfizer | — | |
| Pradigstat (LCQ908)a | Phase 2 | NASH | NA | — | √ | NA | Novartis | — | |
| SNP-612a | Phase 2 (terminated) | NASH | NA | NA | NA | NA | Sinew Pharma | — | |
| SNP-610 | Phase 2 | NASH | TBD | TBD | TBD | TBD | Sinew Pharma | — | |
| ION224 | Phase 2 | NASH | TBD | TBD | TBD | TBD | Ionis Pharmaceuticals | — | |
| SCD-1 | Aramchol | Phase 3 (suspended) | NASH | — | √ | √ | — | Galmed Pharmaceuticals | Ratziu et al298 |
| ACC | Clesacostat (PF-05221304)a | Phase 2 | NAFLD NASH |
NA | √ | √ | NA | Pfizer | Bergman et al299 Calle et al300 |
| Firsocostat (GS-0976)a | Phase 2 | NASH | NA | — | √ | √ | Gilead Sciences | Loomba et al301 | |
| MK-4074a | Phase 1 | NAFLD | NA | NA | √ | NA | Merck Sharp & Dohme LLC | Kim et al302 | |
| THR-β | ALG-055009 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Aligos Therapeutics | — |
| Resmetirom (MGL-3196) | Phase 3 | NASH | — | √ | √ | √ | Madrigal Pharmaceuticals | Harrison et al303 Harrison et al304 | |
| ASC41 | Phase 2 | NASH | TBD | TBD | TBD | TBD | Ascletis Pharma | — | |
| TERN-501 | Phase 2 | NASH | TBD | TBD | TBD | TBD | Terns Pharmaceuticals | — | |
| ECC4703 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Eccogene | — | |
| VK2809 | Phase 2 | NASH | TBD | √ | √ | TBD | Viking Therapeutics | — | |
| PNPLA3 | AMG 609 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Amgen | — |
| ALN-PNP | Phase 1 | NASH | TBD | TBD | TBD | TBD | Alnylam | — | |
| AZD2693 | Phase 2 | NASH | TBD | TBD | TBD | TBD | AstraZeneca | — | |
| JNJ-75220795 (ARO-PNPLA3) | Phase 1 | NASH | TBD | TBD | TBD | TBD | Arrowhead | — | |
| ANGPTL3 | Vupanorsen (ISIS 703802)a | Phase 2 | NAFLD with T2D | NA | — | — | NA | Ionis Pharmaceuticals | Gaudet D, et al.305 |
| MPC | MSDC-0602K | Phase 2 | NASH | — | √ | — | — | Cirius Therapeutics | Harrison et al306 |
| MOTS-c | CB4211 | Phase 1 | NASH | — | √ | — | TBD | CohBar | — |
| HTR2A | GM-60106 | Phase 1 | NASH | TBD | TBD | TBD | TBD | JD Bioscience | — |
| GDF15 | NGM395a | Phase 1 | NAFLD | NA | NA | NA | NA | NGM Biopharmaceuticals | — |
| HSD17B13 | ARO-HSD (GSK4532990) | Phase 2 | NASH | — | √ | — | — | Arrowhead Pharmaceuticals (Licensed to GlaxoSmithKline) | Mak et al307 |
| AZD7503 | Phase 1 | NASH | TBD | TBD | TBD | TBD | AstraZeneca | — | |
| ALN-HSD | Phase 2 | NASH | TBD | TBD | TBD | TBD | Alnylam | — | |
| HSD11B1 | RO5093151a | Phase 1 | NASH | √ | √ | √ | NA | Roche | Stefan et al308 |
| miR-103/107 | RG-125 (AZD4076)a | Phase 1 | NAFLD with T2D | NA | NA | NA | NA | AstraZeneca | — |
| RORα | TB-840 | Phase 1 | NASH | TBD | TBD | TBD | TBD | Therasid Bioscience | — |
| CB1 | Namacizumab (RYI-018)a | Phase 1 | NASH | TBD | TBD | TBD | TBD | Bird Rock Bio (Johnson & Johnson) | — |
| Otenabant (CP-945598)a | Phase 1 | NASH | √ | NA | NA | NA | Pfizer | Aronne et al309 | |
| Rimonabanta | Phase 3 (terminated) | NAFLD with or without T2D | NA | NA | NA | NA | Sanofi | — | |
| AMPK | PXL770 | Phase 2 | NASH | — | √ | √ | TBD | Poxel | Cusi et al310 |
| Androgen | Spironolactone | Phase 4 | NAFLD | √ | — | √ | NA | — | Polyzos et al311 |
Future development for NAFLD/NASH monotherapy is not pursued.
Abbreviations: ACC, acetyl-CoA carboxylase; ALT, alanine transferase; AMPK, AMP-activated protein kinase; ANGPTL3, angiopoietin like protein 3; CB1, cannabinoid; DPP-4, dipeptidyl peptidase-4; FASN, FGFR1&KLB, β-Klotho/FGFR1c receptor complex; FXR, farnesoid X receptor; GIPR, glucose-dependent insulinotropic polypeptide receptor; GLP-1R, glucagon-like peptide-1 receptor; HSD11B1, 11β-hydroxysteroid dehydrogenase type 1; HSD17B13, hydroxysteroid 17β-dehydrogenase 13; HTR2A, 5-hydroxytryptamine receptor 2A; iBAT, ileal bile acid transporter; LFC, liver fat content; MOTS-c, mitochondrial open-reading frame of the 12 SrRNA type-c; MPC, mitochondrial pyruvate carrier; NA, not available; PNPLA3, patatin-like phospholipase domain–-containing protein 3; PPAR, peroxisome proliferation– activated receptor; PPARG, peroxisome proliferator– activated receptor gamma; SCD-1, stearoyl-CoA desaturase-1; SGLT, sodium-glucose cotransporter; SSPG, steady- state plasma glucose; SWE, shear-wave elastography; TBD, to be determined.
FXR has been one of the most extensively studied targets for NAFLD/NASH since 2015, with the release of the remarkable interim data from a 72-week randomized trial of a FXR agonist, obeticholic acid, in biopsy-proven NASH patients.312 FXR is a nuclear receptor expressed in both hepatocytes and intestinal enterocytes. Activation of FXR alleviates hepatic lipid accumulation potentially by 2 mechanisms: (1) the downregulation of hepatic DNL via an FXR-SHP-SREBP-1c pathway, and (2) the reduction of intestinal lipid absorption.313 Improvement in steatosis, inflammation, and fibrosis, as well as a weight-loss effect, was observed in the obeticholic acid therapy.312 Despite its efficacy, obeticholic acid was not approved for NASH because of its common adverse events of pruritus and transient increase in LDL-C. Though it remains unclear whether pruritus and LDL-C increases are drug-specific or target-related, they are also observed in most FXR agonists, including Tropifexor, Cilofexor, Vonafexor, EDP-297, EDP-305, and MET-409.253,314,315 In the recently released data of 2 phase 1 trials of a nonsteroidal FXR agonist, TERN-101, no pruritus and lipid profile change was reported.248 Further studies are required to evaluate its safety and efficacy in patients with longer treatment duration.
As a direct target of FXR, a FGF19 analog, Aldafermin, was also investigated for NAFLD/NASH treatment. However, although improvements in LFC, liver enzymes, and fibrosis markers were observed after the 24-week phase 2b trial, Aldafermin failed to reach the primary end point of fibrosis improvement.292 Another potential target in BA metabolism is the ileal BA transporter, the inhibition of which interrupts enterohepatic recirculation and promotes the hepatic de novo production of BAs. Nevertheless, 2 ileal BA transporter inhibitors, Elobixibat and Volixibat, both failed to improve ALT levels and steatosis in NASH patients, indicating that ileal BA transporter may not be the best choice for NAFLD/NASH therapy.256
Compared with FGF19, FGF21 has attracted much more interest in terms of its multiple effects on obesity and metabolism.316 Several FGF21 analogues are going through early clinical trials for NASH patients in various fibrosis stages. Among them, pegozafermin, pegbelfermin, and efruxifermin have shown promising results in phase 2 studies, where all of these drugs significantly improved ALT levels, the hepatic fat fraction, and metabolic biomarkers.286–289
Resmetirom, a thyroid hormone receptor-β (THRβ) agonist, is perhaps the most promising drug under development for NAFLD/NASH. Significant reduction of hepatic fat measured by MRI-PDFF, reduction of liver enzymes, and improvement of inflammation and fibrosis biomarkers were observed in a multicenter, randomized 36-week phase 2 study.303 Positive phase 3 results have been released recently, announcing that the dual primary end points of NASH resolution and fibrosis improvement are achieved by resmetirom. Other THRβ agonists under investigation include ALG-055009, ASC41, TERN-501, ECC4703, and VK2809.
Several drugs targeting hepatic DNL have been approved for clinical trials in recent years, including FASN inhibitors (denifanstat, FT-4101), DGAT2 inhibitors (PF-07202954, ervogastat, pradigstat, SNP-610, ION224), the stearoyl-CoA desaturase-1 inhibitor (aramchol), and acetyl-CoA carboxylase inhibitors (clesacostat, firsocostat, MK-4074), most of which are still in early-phase clinical trials and further data are required before a proper review. So far, all 3 acetyl-CoA carboxylase inhibitors have been excluded from monotherapy development because of the side effect of hypertriglyceridemia. KHK, the key enzyme for fructose metabolism, was also investigated as a potential target. The KHK inhibitor PF-06835919 has been proved to be safe and well-tolerated, and reduces liver fat in patients with NAFLD and T2D. Yet, the clinical improvements were not significant enough to support further development.297 The phase 1 clinical trial of another KHK inhibitor, XZP-6019, is currently ongoing.
Since NAFLD/NASH is related to obesity and T2D, plenty of FDA-approved antidiabetic and lipid-modifying agents have been going through phase 4 clinical trials to evaluate the possibility for NAFLD/NASH treatment. Pioglitazone has been recommended by KASL clinical practice guidelines for NAFLD management.317 While the side effects of weight gain, edema, and bone loss limit the use of TZDs,318 PXL065, a deuterium-stabilized stereoisomer of pioglitazone, is expected to avoid the safety concerns and improve efficacy through non–PPARγ-mediated mechanisms.269,319
CONCLUSIONS AND FUTURE PERSPECTIVES
There are no drugs approved for the treatment of NAFLD until now, and therapies are limited to weight loss and exercise. The nutrient compositions of diets and dietary patterns have significant impacts on weight loss and progression of NAFLD. In this review, we briefly summarize the effects of 3 macronutrients, vitamins, and several dietary patterns, including CRD, IF, KD, MD, and the DASH diet, in the development of NAFLD. We also compare the effects of LCD and LFD. What’s more, we summarize the potential metabolic targets that seem to be promising in the NAFLD treatments.
However, the current findings are still incomplete. In studies on the effects of sugar on NAFLD, fructose consumption in current mouse models far exceeds the human daily intake, which may overestimate the harm of fructose. There are also significant sex differences in fructose metabolism in the liver, so only using male mouse models in some research may lead to incomplete results. Moreover, the current research is limited to the effects of fructose on NAFLD, but other sugars such as sucrose are widely used in daily cooking as well. The effects of NNSs, which are becoming increasingly popular, on IR, promotion of NAFLD, and microbiota dysbiosis are still uncertain. Furthermore, fructose-induced leptin resistance is likely to increase the intake of other foods. The impacts of fructose on other dietary elements’ metabolic effects require further studies since fructose may exacerbate the adverse effects of glucose, fat, or alcohol320 on NAFLD. In studies on the effect of fat on NAFLD, research on the effects of low-fat diets is limited to clinical settings, while the mechanism is uncertain. The exact dose of n-3 and its duration are also under investigation before recommending it for the clinical use. In studies on the effects of protein on NAFLD, there is little research concerning the effects of specific amino acids. What’s more, whether the advantages and disadvantages of plant and animal proteins are attributed to the proteins itself or other elements in food and their mechanism requires further research. The side effects of high-protein diets should also be considered before using them on a large scale.
The major problems of studies on dietary patterns are the small sample sizes, short observation time, and insufficiency of means to assess patients’ adherence to the diets. So far, only MD is recommended by some NAFLD guidelines, and a tool to assess adherence to MD—Mediterranean adequacy index—has already been designed.321 However, more clinical trials are needed to standardize the characteristics of MD. More large-scale, long-term studies should be designed to gain further evidence about other dietary patterns, and specific tools should be developed to assess individuals’ adherence.
Notably, there is a lack of research on the most appropriate intake of the 3 macronutrients in the diets. The intake of carbohydrates, fat, and proteins in current clinical trials is inconsistent, and more studies are required to standardize the recommended doses. Another factor that needs to be considered is that although weight loss is traditionally thought to alleviate NAFLD, some individuals with normal BMIs develop NAFLD after losing too much weight in some cases. Therefore, the specific rate of weight loss and whether weight loss is applicable to lean NAFLD patients need further studies.
The development of drugs for the treatment of NAFLD is even more difficult. Most drugs fail to enter the phase III clinical trials, probably due to insufficient sample sizes or the short duration. As reversal of liver histology may be a slow process, it is unlikely that efficacy will be seen in a short period of time. In addition, the mechanisms of these metabolic targets, as well as the pathogenesis of NAFLD, still require more research.
Footnotes
Abbreviations: AA, arachidonic acid; ACC, acetyl-CoA carboxylase; ACOX1, acyl-CoA oxidase 1; ADF, alternate-day fasting; ALP, alkaline phosphatase; ALT, alanine transferase; AMPK, AMP-activated protein kinase; ANGPTL3, angiopoietin like protein 3; apoB, apolipoprotein B; AST, aspartate transaminase; BA, bile acid; BMI, body mass index; Bnip3, BCL2 adenovirus E1B 19kDa interacting protein 3; CB1, cannabinoid receptors type 1; CES1, carboxylesterase 1; ChREBP, carbohydrate-responsive element–binding protein; CPT, carnitine palmitoyl transferase; CRD, calorie-restricted diet; CREB, cAMP response element binding; CSE, cystathionine-γ-lyase; CVD, cardiovascular disease; DAG, diacylglycerol; DASH, dietary approach to stop hypertension; Dgat2, diacylglycerol O-acyltransferase 2; DNL, de novo lipogenesis; DPP-4, dipeptidyl peptidase-4; EASD, European Association for the Study of Diabetes; EASL, European Association for the Study of the Liver; EASO, European Association for the Study of Obesity; ER, endoplasmic reticulum; ERK, extracellular regulated protein kinase; EV, extracellular vesicle; FAO, fatty acid β-oxidation; FASN, fatty acid synthase; FC, free cholesterol; FFA, free fatty acid; FGFR1&KLB, β-Klotho/FGFR1c receptor complex; FLI, fatty liver index; FM, fat mass; FXR, farnesoid X receptor; GCGR, glucagon receptor; GDF15, growth differentiation factor 15; GGT, γ-glutamyltransferase; GIPR, glucose-dependent insulinotropic polypeptide receptor; GLP-1, glucagon-like peptide-1; GLP-1R, glucagon-like peptide-1 receptor; GLUT, glucose transporter type; GPBAR1, G protein–coupled bile acid receptor 1; GR, glucocorticoid receptor; GSH-Px, glutathione peroxidase; H2S, hydrogen sulfide; HbA1c, glycosylated hemoglobin type A1c; HFCS, high-fructose corn syrup; HFD, high-fat diet; HFHSD, high-fat high-sucrose diet; HOMA-IR, homeostasis model assessment – insulin resistance; HSD11B1, 11β-hydroxysteroid dehydrogenase type 1; HSD17B13, hydroxysteroid 17β-dehydrogenase 13; HTR2A, 5-hydroxytryptamine receptor 2A; iBAT, ileal bile acid transporter; IF, intermittent fasting; IHTG, intrahepatic triglyceride; iNOS, inducible nitric oxide synthase; IR, insulin resistance; JNK, c-Jun N-terminal; KD, ketogenic diet; KHK, ketohexokinase; LDL-C, LDL-cholesterol; LCD, low-carbohydrate diet; LFC, liver fat content; LFD, low-fat diet; LOX, lipoxygenase; LPCAT2, lysophosphatidylcholine acyltransferase 2; l-PK, liver-pyruvate kinase; MD, Mediterranean diet; MOTS-c, mitochondrial open-reading frame of the 12 SrRNA type-c; MPC, mitochondrial pyruvate carrier; MPST, mercaptopyruvate sulfurtransferase; MRE, magnetic resonance elastography; MRI-PDFF, magnetic resonance imaging-proton density fat fraction; MRS, magnetic resonance spectroscopy; mTORC1, mammalian target of rapamycin complex 1; n-3, omega-3; n-6, omega-6; NLRC4, nucleotide-binding oligomerization domain, leucine-rich repeat and caspase recruitment domain–containing 4; NNS, non-nutritive sweetener; Nrf2, nuclear factor erythroid 2-related factor 2; NRG4, neuregulin 4; OGTT, oral glucose tolerance test; PERK, PKR-like endoplasmic reticulum kinase; PFK-1, phosphofructokinase-1; Pgc1α, peroxisome proliferator–activated receptor γ coactivator lα; PKCδ, protein kinase C-δ; PKR, protein kinase R; PMP70, peroxisomal membrane protein 70; PNPLA3, patatin-like phospholipase domain–containing protein 3; PPAR, peroxisome proliferation–activated receptor; PPARG, peroxisome proliferator–activated receptor gamma; PRDX6, peroxiredoxin 6; PYY, peptide YY; QUICKI, quantitative insulin sensitivity check index; RCT, randomized controlled trial; RIP1, receptor-interacting protein 1; RORα, retinoic acid receptor–related orphan receptor α; ROS, reactive oxygen species; SCD-1, stearoyl-CoA desaturase-1; SFA, saturated fatty acid; SGLT, sodium-glucose cotransporter; SIRT3, sirtuin 3; SMAD, Drosophila mothers against decapentaplegic; SMS1, sphingomyelin synthase; SOD, superoxide dismutase; Srebf1, sterol regulatory element–binding transcription factor 1; SREBP-1c, sterol regulatory element–binding protein 1C; SSPG, steady-state plasma glucose; SWE, shear-wave elastography; T2DM, type 2 diabetes mellitus; TBK1, tank-binding protein kinase 1; TC, total cholesterol; TCA, tricarboxylic acid; TFAM, transcription factor A; TG, triglyceride; THR-β, thyroid hormone receptor-β; Timp1, tissue inhibitors of metalloproteinase 1; TLR4, Toll-like receptor 4; TM4SF5, transmembrane 4 L 6 family member 5; TNFα, TNF alpha; TRF, time-restricted fasting; TZDs, thiazolidinediones; Ucp1, uncoupling protein 1; VCAM-1, vascular cell adhesion molecule 1; VD, vitamin D; VDR, vitamin D receptor; VE, vitamin E.
Contributor Information
Tianyu Mao, Email: susanmao1@sjtu.edu.cn.
Yiwen Sun, Email: sun_777@sjtu.edu.cn.
Xinyi Xu, Email: xxy1219@sjtu.edu.cn.
Kang He, Email: hekang929@163.com.
FUNDING INFORMATION
This study was supported by the Project of the Shanghai Municipal Health Commission (20204Y0012), Seed Fund of Renji Hospital (RJZZ18-010), Shenkang 3-year action plan (SHDC2020CR2003A, SHDC2020CR5012), Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZDCX20180802), National Natural Science Foundation of China (81972205), and the Project of Shanghai key clinical specialties (shslczdzk05801).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
REFERENCES
- 1. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. 2022;19:60–78. [DOI] [PubMed] [Google Scholar]
- 2. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64:73–84. [DOI] [PubMed] [Google Scholar]
- 3. Zhou J, Zhou F, Wang W, Zhang X-J, Ji Y-X, Zhang P, et al. Epidemiological features of NAFLD From 1999 to 2018 in China. Hepatology. 2020;71:1851–64. [DOI] [PubMed] [Google Scholar]
- 4. Ioannou GN. Epidemiology and risk-stratification of NAFLD-associated HCC. J Hepatol. 2021;75:1476–84. [DOI] [PubMed] [Google Scholar]
- 5. Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2021;18:223–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab. 2020;42:101092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Semmler G, Datz C, Reiberger T, Trauner M. Diet and exercise in NAFLD/NASH: Beyond the obvious. Liver Int. 2021;41:2249–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sekkarie A, Welsh JA, Vos MB. Carbohydrates and diet patterns in nonalcoholic fatty liver disease in children and adolescents. Curr Opin Clin Nutr Metab Care. 2018;21:283–8. [DOI] [PubMed] [Google Scholar]
- 9. Shimony MK, Schliep KC, Schisterman EF, Ahrens KA, Sjaarda LA, Rotman Y, et al. The relationship between sugar-sweetened beverages and liver enzymes among healthy premenopausal women: a prospective cohort study. Eur J Nutr. 2016;55:569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park WY, Yiannakou I, Petersen JM, Hoffmann U, Ma J, Long MT. Sugar-sweetened beverage, diet soda, and nonalcoholic fatty liver disease over 6 years: The Framingham Heart Study. Clin Gastroenterol Hepatol. 2022;20:2524–32.e2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ma J, Sloan M, Fox CS, Hoffmann U, Smith CE, Saltzman E, et al. Sugar-sweetened beverage consumption is associated with abdominal fat partitioning in healthy adults. J Nutr. 2014;144:1283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Assy N, Nasser G, Kamayse I, Nseir W, Beniashvili Z, Djibre A, et al. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can J Gastroenterol. 2008;22:811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond). 2005;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Janevski M, Ratnayake S, Siljanovski S, McGlynn MA, Cameron-Smith D, Lewandowski P. Fructose containing sugars modulate mRNA of lipogenic genes ACC and FAS and protein levels of transcription factors ChREBP and SREBP1c with no effect on body weight or liver fat. Food Funct. 2012;3:141–9. [DOI] [PubMed] [Google Scholar]
- 16. Softic S, Gupta MK, Wang G-X, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127:4059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Geidl-Flueck B, Hochuli M, Németh Á, Eberl A, Derron N, Köfeler HC, et al. Fructose- and sucrose- but not glucose-sweetened beverages promote hepatic de novo lipogenesis: A randomized controlled trial. J Hepatol. 2021;75:46–54. [DOI] [PubMed] [Google Scholar]
- 18. Rw W, Ka D, Mi G. Fructose content in popular beverages made with and without high-fructose corn syrup. Nutrition. 2014:30. [DOI] [PubMed] [Google Scholar]
- 19. Basaranoglu M, Basaranoglu G, Bugianesi E. Carbohydrate intake and nonalcoholic fatty liver disease: Fructose as a weapon of mass destruction. Hepatobiliary Surg Nutr. 2015;4:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab. 2008;295:E227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee H, Kim E, Shin E-A, Shon JC, Sun H, Kim JE, et al. Crosstalk between TM4SF5 and GLUT8 regulates fructose metabolism in hepatic steatosis. Mol Metab. 2022;58:101451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Novelle MG, Bravo SB, Deshons M, Iglesias C, García-Vence M, Annells R, et al. Impact of liver-specific GLUT8 silencing on fructose-induced inflammation and omega oxidation. iScience. 2021;24:102071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zakim D, Pardini RS, Herman RH, Sauberlich HE. Mechanism for the differential effects of high carbohydrate diets on lipogenesis in rat liver. Biochim Biophys Acta. 1967;144:242–51. [DOI] [PubMed] [Google Scholar]
- 24. Sun SZ, Empie MW. Fructose metabolism in humans—what isotopic tracer studies tell us. Nutr Metab (Lond). 2012;9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bantle JP. Dietary fructose and metabolic syndrome and diabetes. J Nutr. 2009;139:1263S–8S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68:1063–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim M-S, Krawczyk SA, Doridot L, Fowler AJ, Wang JX, Trauger SA, et al. ChREBP regulates fructose-induced glucose production independently of insulin signaling. J Clin Invest. 2016;126:4372–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koo H-Y, Wallig MA, Chung BH, Nara TY, Cho BHS, Nakamura MT. Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta. 2008;1782:341–8. [DOI] [PubMed] [Google Scholar]
- 29. Mock K, Lateef S, Benedito VA, Tou JC. High-fructose corn syrup-55 consumption alters hepatic lipid metabolism and promotes triglyceride accumulation. J Nutr Biochem. 2017;39:32–9. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Viscarra J, Kim S-J, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol. 2015;16:678–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Linden AG, Li S, Choi HY, Fang F, Fukasawa M, Uyeda K, et al. Interplay between ChREBP and SREBP-1c coordinates postprandial glycolysis and lipogenesis in livers of mice. J Lipid Res. 2018;59:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Iizuka K, Takao K, Yabe D. ChREBP-mediated regulation of lipid metabolism: involvement of the gut microbiota, liver, and adipose tissue. Front Endocrinol (Lausanne). 2020;11:587189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ma L, Robinson LN, Towle HC. ChREBP*Mlx is the principal mediator of glucose-induced gene expression in the liver. J Biol Chem. 2006;281:28721–30. [DOI] [PubMed] [Google Scholar]
- 34. F Q, N Rc, B C, L C, D Kt, B E, et al. LXRα regulates hepatic ChREBPα activity and lipogenesis upon glucose, but not fructose feeding in mice. Nutrients. 2017;9:678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight. 2017;2:e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci USA. 2004;101:7281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hu Y, Semova I, Sun X, Kang H, Chahar S, Hollenberg AN, et al. Fructose and glucose can regulate mammalian target of rapamycin complex 1 and lipogenic gene expression via distinct pathways. J Biol Chem. 2018;293:2006–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Meissen JK, Hirahatake KM, Adams SH, Fiehn O. Temporal metabolomic responses of cultured HepG2 liver cells to high fructose and high glucose exposures. Metabolomics. 2015;11:707–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bonnefont J-P, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: Biochemical, molecular and medical aspects. Mol Aspects Med. 2004;25:495–520. [DOI] [PubMed] [Google Scholar]
- 40. Bramlage KS, Bhattacharjee J, Kirby M, Myronovych A, Gupta R, Gonzalez R-MS, et al. A diet high in fat and fructose induces early hepatic mitochondrial aging. J Pediatr Gastroenterol Nutr. 2021;73:99–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Softic S, Meyer JG, Wang G-X, Gupta MK, Batista TM, Lauritzen HPMM, Fujisaka S, et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. 2019;30:735–53.e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hwang JH, Kim DW, Jo EJ, Kim YK, Jo YS, Park JH, et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes. 2009;58:965–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bhat SF, Pinney SE, Kennedy KM, McCourt CR, Mundy MA, Surette MG, et al. Exposure to high fructose corn syrup during adolescence in the mouse alters hepatic metabolism and the microbiome in a sex-specific manner. J Physiol. 2021;599:1487–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elhassan YS, Philp AA, Lavery GG. Targeting NAD + in metabolic disease: new insights into an old molecule. J Endocr Soc. 2017;1:816–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med. 2020;152:116–41. [DOI] [PubMed] [Google Scholar]
- 46. Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology (Baltimore, Md). 2007;45:1366–74. [DOI] [PubMed] [Google Scholar]
- 47. Vasiljević A, Bursać B, Djordjevic A, Milutinović DV, Nikolić M, Matić G, et al. Hepatic inflammation induced by high-fructose diet is associated with altered 11βHSD1 expression in the liver of Wistar rats. Eur J Nutr. 2014;53:1393–402. [DOI] [PubMed] [Google Scholar]
- 48. Elaković I, Kovačević S, Vojnović Milutinović D, Nikolić-Kokić A, Glban AM, Spasić M, et al. Fructose consumption affects glucocorticoid signaling in the liver of young female rats. Nutrients. 2020;12:3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kovačević S, Elaković I, Vojnović Milutinović D, Nikolić-Kokić A, Blagojević D, Matić G, et al. Fructose-rich diet attenuates stress-induced metabolic disturbances in the liver of adult female rats. J Nutr. 2021;151:3661–70. [DOI] [PubMed] [Google Scholar]
- 50. Kendig MD, Martire SI, Boakes RA, Rooney KB. Comparable metabolic effects of isocaloric sucrose and glucose solutions in rats. Physiol Behav. 2021;229:113239. [DOI] [PubMed] [Google Scholar]
- 51. Burmeister MA, Ayala J, Drucker DJ, Ayala JE. Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am J Physiol Endocrinol Metab. 2013;304:E677–85. [DOI] [PubMed] [Google Scholar]
- 52. S A, M W, R C, C Ky, J Rj, S Pj. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Page KA, Chan O, Arora J, Belfort-Deaguiar R, Dzuira J, Roehmholdt B, et al. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA. 2013;309:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Levy A, Marshall P, Zhou Y, Kreek MJ, Kent K, Daniels S, et al. Fructose:glucose ratios—A study of sugar self-administration and associated neural and physiological responses in the rat. Nutrients. 2015;7:3869–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Carroll ME, Lac ST. Acquisition of i.v. amphetamine and cocaine self-administration in rats as a function of dose. Psychopharmacology (Berl). 1997;129:206–14. [DOI] [PubMed] [Google Scholar]
- 56. Ryuk JA, Kang S, Daily JW, Ko B-S, Park S. Moderate intake of aspartame and sucralose with meals, but not fructose, does not exacerbate energy and glucose metabolism in estrogen-deficient rats. J Clin Biochem Nutr. 2019;65:223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shi Z, Chen G, Cao Z, Wu F, Lei H, Chen C, et al. Gut microbiota and its metabolite deoxycholic acid contribute to sucralose consumption-induced nonalcoholic fatty liver disease. J Agric Food Chem. 2021;69:3982–91. [DOI] [PubMed] [Google Scholar]
- 58. Liu CW, Chi L, Tu P, Xue J, Ru H, Lu K. Quantitative proteomics reveals systematic dysregulations of liver protein metabolism in sucralose-treated mice. J Proteomics. 2019;196:1–10. [DOI] [PubMed] [Google Scholar]
- 59. Alkafafy Mel S, Ibrahim ZS, Ahmed MM, El-Shazly SA. Impact of aspartame and saccharin on the rat liver: Biochemical, molecular, and histological approach. Int J Immunopathol Pharmacol. 2015;28:247–55. [DOI] [PubMed] [Google Scholar]
- 60. Finamor I, Pérez S, Bressan CA, Brenner CE, Rius-Pérez S, Brittes PC, et al. Chronic aspartame intake causes changes in the trans-sulphuration pathway, glutathione depletion and liver damage in mice. Redox Biol. 2017;11:701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lebda MA, Tohamy HG, El-Sayed YS. Long-term soft drink and aspartame intake induces hepatic damage via dysregulation of adipocytokines and alteration of the lipid profile and antioxidant status. Nutr Res. 2017;41:47–55. [DOI] [PubMed] [Google Scholar]
- 62. Mendoza-Pérez S, García-Gómez RS, Durán-Domínguez-de-Bazúa MD. Chronic intake of nutritive sweeteners and saccharin increases levels of glycolytic and lipogenic enzymes in rat liver. Int J Food Sci Nutr. 2022;73:927–39. [DOI] [PubMed] [Google Scholar]
- 63. Bian X, Chi L, Gao B, Tu P, Ru H, Lu K. Gut microbiome response to sucralose and its potential role in inducing liver inflammation in mice. Front Physiol. 2017;8:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bian X, Tu P, Chi L, Gao B, Ru H, Lu K. Saccharin induced liver inflammation in mice by altering the gut microbiota and its metabolic functions. Food Chem Toxicol. 2017;107:530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dai X, Guo Z, Chen D, Li L, Song X, Liu T, et al. Maternal sucralose intake alters gut microbiota of offspring and exacerbates hepatic steatosis in adulthood. Gut Microbes. 2020;11:1043–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Green CH, Syn WK. Non-nutritive sweeteners and their association with the metabolic syndrome and non-alcoholic fatty liver disease: a review of the literature. Eur J Nutr. 2019;58:1785–800. [DOI] [PubMed] [Google Scholar]
- 67. Sylvetsky AC, Rother KI. Nonnutritive sweeteners in weight management and chronic disease: A review. Obesity (Silver Spring). 2018;26:635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abbas T, Murad W. Do artificial sweeteners increase the risk of non-alcoholic fatty liver disease (NAFLD) ? EXCLI J. 2020;19:1158–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Pino-Seguel P, Moya O, Borquez JC, Pino-de la Fuente F, Díaz-Castro F, Donoso-Barraza C, et al. Sucralose consumption ameliorates high-fat diet-induced glucose intolerance and liver weight gain in mice. Front Nutr. 2022;9:979624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu H-T, Lin C-H, Pai H-L, Chen Y-C, Cheng K-P, Kuo H-Y, et al. Sucralose, a non-nutritive artificial sweetener exacerbates high fat diet-induced hepatic steatosis through taste receptor type 1 member 3. Front Nutr. 2022;9:823723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Furuta K, Guo Q, Pavelko KD, Lee JH, Robertson KD, Nakao Y, et al. Lipid-induced endothelial vascular cell adhesion molecule 1 promotes nonalcoholic steatohepatitis pathogenesis. J Clin Invest. 2021;131:e143690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Carr RM. VCAM-1: closing the gap between lipotoxicity and endothelial dysfunction in nonalcoholic steatohepatitis. J Clin Invest. 2021;131:e147556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ. 2021;28:1418–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology. 2016;150:956–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zeng Y, Luo M, Pan L, Chen Y, Guo S, Luo D, et al. Vitamin D signaling maintains intestinal innate immunity and gut microbiota: Potential intervention for metabolic syndrome and NAFLD. Am J Physiol Gastrointest Liver Physiol. 2020;318:G542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Liu H, Niu Q, Wang T, Dong H, Bian C. Lipotoxic hepatocytes promote nonalcoholic fatty liver disease progression by delivering microRNA-9-5p and activating macrophages. Int J Biol Sci. 2021;17:3745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, et al. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology. 2016;63:731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dong J, Viswanathan S, Adami E, Singh BK, Chothani SP, Ng B, et al. Hepatocyte-specific IL11 cis-signaling drives lipotoxicity and underlies the transition from NAFLD to NASH. Nat Commun. 2021;12:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology. 2017;65:1165–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. 2019;21:101120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018;18:229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cho CS, Park HW, Ho A, Semple IA, Kim B, Jang I, et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018;68:1331–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bellanti F, Villani R, Tamborra R, Blonda M, Iannelli G, di Bello G, et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2018;15:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol. 2015;62:1367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Machado MV, Michelotti GA, Pereira Tde A, Boursier J, Kruger L, Swiderska-Syn M, et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut. 2015;64:1148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Koh EH, Yoon JE, Ko MS, Leem J, Yun JY, Hong CH, et al. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut. 2021;70:1954–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li M, Xu C, Shi J, Ding J, Wan X, Chen D, et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut. 2018;67:2169–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bechmann LP, Kocabayoglu P, Sowa JP, Sydor S, Best J, Schlattjan M, et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57:1394–406. [DOI] [PubMed] [Google Scholar]
- 89. Arteel GE. Beyond reasonable doubt: who is the culprit in lipotoxicity in NAFLD/NASH ? Hepatology. 2012;55:2030–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest. 2009;119:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li S, Dou X, Ning H, Song Q, Wei W, Zhang X, et al. Sirtuin 3 acts as a negative regulator of autophagy dictating hepatocyte susceptibility to lipotoxicity. Hepatology. 2017;66:936–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Khadge S, Sharp JG, Thiele GM, McGuire TR, Klassen LW, Duryee MJ, et al. Dietary omega-3 and omega-6 polyunsaturated fatty acids modulate hepatic pathology. J Nutr Biochem. 2018;52:92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bogl LH, Kaprio J, Pietiläinen KH. Dietary n-6 to n-3 fatty acid ratio is related to liver fat content independent of genetic effects: evidence from the monozygotic co-twin control design. Clin Nutr. 2020;39:2311–14. [DOI] [PubMed] [Google Scholar]
- 94. Okada L, Oliveira CP, Stefano JT, Nogueira MA, Silva I, Cordeiro FB, et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH—proteomic and lipidomic insight. Clin Nutr. 2018;37:1474–84. [DOI] [PubMed] [Google Scholar]
- 95. Šmíd V, Dvořák K, Šedivý P, Kosek V, Leníček M, Dezortová M, et al. Effect of omega-3 polyunsaturated fatty acids on lipid metabolism in patients with metabolic syndrome and NAFLD. Hepatol Commun. 2022;6:1336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Antraco VJ, Hirata BKS, de Jesus Simão J, Cruz MM, da Silva VS, da Cunha de Sá RDC, et al. Omega-3 polyunsaturated fatty acids prevent nonalcoholic steatohepatitis (NASH) and stimulate adipogenesis. Nutrients. 2021;13:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yang F, Zhou N, Zhu X, Min C, Zhou W, Li X. n-3 PUFAs protect against adiposity and fatty liver by promoting browning in postnatally overfed male rats: a role for NRG4. J Nutr Biochem. 2021;93:108628. [DOI] [PubMed] [Google Scholar]
- 98. Liu Y, Li Q, Wang H, Zhao X, Li N, Zhang H, et al. Fish oil alleviates circadian bile composition dysregulation in male mice with NAFLD. J Nutr Biochem. 2019;69:53–62. [DOI] [PubMed] [Google Scholar]
- 99. Hong L, Zahradka P, Cordero-Monroy L, Wright B, Taylor CG. Dietary docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) operate by different mechanisms to modulate hepatic steatosis and hyperinsulemia in fa/fa Zucker rats. Nutrients. 2019;11:917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Parker HM, Cohn JS, O’Connor HT, Garg ML, Caterson ID, George J, et al. Effect of fish oil supplementation on hepatic and visceral fat in overweight men: a randomized controlled trial. Nutrients. 2019;11:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Oscarsson J, Önnerhag K, Risérus U, Sundén M, Johansson L, Jansson P-A, et al. Effects of free omega-3 carboxylic acids and fenofibrate on liver fat content in patients with hypertriglyceridemia and non-alcoholic fatty liver disease: a double-blind, randomized, placebo-controlled study. J Clin Lipidol. 2018;12:1390–403.e1394. [DOI] [PubMed] [Google Scholar]
- 102. Parker HM, Johnson NA, Burdon CA, Cohn JS, O’Connor HT, George J. Omega-3 supplementation and non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol. 2012;56:944–51. [DOI] [PubMed] [Google Scholar]
- 103. Yang J, Fernández-Galilea M, Martínez-Fernández L, González-Muniesa P, Pérez-Chávez A, Martínez JA, et al. Oxidative stress and non-alcoholic fatty liver disease: effects of omega-3 fatty acid supplementation. Nutrients. 2019;11:872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lee CH, Fu Y, Yang SJ, Chi CC. Effects of omega-3 polyunsaturated fatty acid supplementation on non-alcoholic fatty liver: a systematic review and meta-analysis. Nutrients. 2020;12:2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Margolis LM, Rivas DA, Ezzyat Y, Gaffney-Stomberg E, Young AJ, McClung JP, et al. Calorie restricted high protein diets downregulate lipogenesis and lower intrahepatic triglyceride concentrations in male rats. Nutrients. 2016;8:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bortolotti M, Maiolo E, Corazza M, Van Dijke E, Schneiter P, Boss A, et al. Effects of a whey protein supplementation on intrahepatocellular lipids in obese female patients. Clin Nutr. 2011;30:494–8. [DOI] [PubMed] [Google Scholar]
- 107. Bortolotti M, Kreis R, Debard C, Cariou B, Faeh D, Chetiveaux M, et al. High protein intake reduces intrahepatocellular lipid deposition in humans. Am J Clin Nutr. 2009;90:1002–1010. [DOI] [PubMed] [Google Scholar]
- 108. Theytaz F, Noguchi Y, Egli L, Campos V, Buehler T, Hodson L, et al. Effects of supplementation with essential amino acids on intrahepatic lipid concentrations during fructose overfeeding in humans. Am J Clin Nutr. 2012;96:1008–16. [DOI] [PubMed] [Google Scholar]
- 109. Ghosh TS, Gupta SS, Bhattacharya T, Yadav D, Barik A, Chowdhury A, et al. Gut microbiomes of Indian children of varying nutritional status. PLoS One. 2014;9:e95547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bezerra Duarte SM, Faintuch J, Stefano JT, Sobral de Oliveira MB, de Campos Mazo DF, Rabelo F, et al. Hypocaloric high-protein diet improves clinical and biochemical markers in patients with nonalcoholic fatty liver disease (NAFLD). Nutr Hosp. 2014;29:94–101. [DOI] [PubMed] [Google Scholar]
- 111. Hong HC, Hwang SY, Choi HY, Yoo HJ, Seo JA, Kim SG, et al. Relationship between sarcopenia and nonalcoholic fatty liver disease: the Korean Sarcopenic Obesity Study. Hepatology. 2014;59:1772–8. [DOI] [PubMed] [Google Scholar]
- 112. Lee YH, Jung KS, Kim SU, Yoon HJ, Yun YJ, Lee BW, et al. Sarcopaenia is associated with NAFLD independently of obesity and insulin resistance: nationwide surveys (KNHANES 2008-2011). J Hepatol. 2015;63:486–93. [DOI] [PubMed] [Google Scholar]
- 113. Young S, Tariq R, Provenza J, Satapathy SK, Faisal K, Choudhry A, et al. Prevalence and profile of nonalcoholic fatty liver disease in lean adults: systematic review and meta-analysis. Hepatol Commun. 2020;4:953–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cuenca-Sánchez M, Navas-Carrillo D, Orenes-Piñero E. Controversies surrounding high-protein diet intake: satiating effect and kidney and bone health. Adv Nutr. 2015;6:260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Halkjær J, Olsen A, Overvad K, Jakobsen MU, Boeing H, Buijsse B, et al. Intake of total, animal and plant protein and subsequent changes in weight or waist circumference in European men and women: the Diogenes project. Int J Obes (Lond). 2011;35:1104–13. [DOI] [PubMed] [Google Scholar]
- 116. Niijima A, Torii K, Uneyama H. Role played by vagal chemical sensors in the hepato-portal region and duodeno-intestinal canal: an electrophysiological study. Chem Senses. 2005;30(suppl 1):i178–9. [DOI] [PubMed] [Google Scholar]
- 117. Veldhorst MA, Nieuwenhuizen AG, Hochstenbach-Waelen A, Westerterp KR, Engelen MP, Brummer RJ, et al. Comparison of the effects of a high- and normal-casein breakfast on satiety, ‘satiety’ hormones, plasma amino acids and subsequent energy intake. Br J Nutr. 2009;101:295–303. [DOI] [PubMed] [Google Scholar]
- 118. Fromentin G, Darcel N, Chaumontet C, Marsset-Baglieri A, Nadkarni N, Tomé D. Peripheral and central mechanisms involved in the control of food intake by dietary amino acids and proteins. Nutr Res Rev. 2012;25:29–39. [DOI] [PubMed] [Google Scholar]
- 119. Belza A, Ritz C, Sørensen MQ, Holst JJ, Rehfeld JF, Astrup A. Contribution of gastroenteropancreatic appetite hormones to protein-induced satiety. Am J Clin Nutr. 2013;97:980–989. [DOI] [PubMed] [Google Scholar]
- 120. Diepvens K, Häberer D, Westerterp-Plantenga M. Different proteins and biopeptides differently affect satiety and anorexigenic/orexigenic hormones in healthy humans. Int J Obes (Lond). 2008;32:510–8. [DOI] [PubMed] [Google Scholar]
- 121. Gannon MC, Nuttall JA, Damberg G, Gupta V, Nuttall FQ. Effect of protein ingestion on the glucose appearance rate in people with type 2 diabetes. J Clin Endocrinol Metab. 2001;86:1040–7. [DOI] [PubMed] [Google Scholar]
- 122. Torres N, Tovar AR. The role of dietary protein on lipotoxicity. Nutr Rev. 2007;65:S64–8. [DOI] [PubMed] [Google Scholar]
- 123. Pichon L, Huneau JF, Fromentin G, Tomé D. A high-protein, high-fat, carbohydrate-free diet reduces energy intake, hepatic lipogenesis, and adiposity in rats. J Nutr. 2006;136:1256–60. [DOI] [PubMed] [Google Scholar]
- 124. Stepien M, Gaudichon C, Fromentin G, Even P, Tomé D, Azzout-Marniche D. Increasing protein at the expense of carbohydrate in the diet down-regulates glucose utilization as glucose sparing effect in rats. PLoS One. 2011;6:e14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. van Zutphen T, Ciapaite J, Bloks VW, Ackereley C, Gerding A, Jurdzinski A, et al. Malnutrition-associated liver steatosis and ATP depletion is caused by peroxisomal and mitochondrial dysfunction. J Hepatol. 2016;65:1198–208. [DOI] [PubMed] [Google Scholar]
- 126. Bjørndal B, Berge C, Ramsvik MS, Svardal A, Bohov P, Skorve J, et al. A fish protein hydrolysate alters fatty acid composition in liver and adipose tissue and increases plasma carnitine levels in a mouse model of chronic inflammation. Lipids Health Dis. 2013;12:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Alferink LJ, Kiefte-de Jong JC, Erler NS, Veldt BJ, Schoufour JD, de Knegt RJ, et al. Association of dietary macronutrient composition and non-alcoholic fatty liver disease in an ageing population: the Rotterdam Study. Gut. 2019;68:1088–98. [DOI] [PubMed] [Google Scholar]
- 128. Lang S, Martin A, Farowski F, Wisplinghoff H, Vehreschild M, Liu J, et al. High protein intake is associated with histological disease activity in patients with NAFLD. Hepatol Commun. 2020;4:681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zelber-Sagi S, Nitzan-Kaluski D, Goldsmith R, Webb M, Blendis L, Halpern Z, et al. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): a population based study. J Hepatol. 2007;47:711–717. [DOI] [PubMed] [Google Scholar]
- 130. Markova M, Pivovarova O, Hornemann S, Sucher S, Frahnow T, Wegner K, et al. Isocaloric diets high in animal or plant protein reduce liver fat and inflammation in individuals with type d Diabetes. Gastroenterology. 2017;152:571–85.e578. [DOI] [PubMed] [Google Scholar]
- 131. Fakhoury-Sayegh N, Younes H, Heraoui G, Sayegh R. Nutritional profile and dietary patterns of lebanese non-alcoholic fatty liver disease patients: a case-control study. Nutrients. 2017;9:1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Turner KM, Keogh JB, Clifton PM. Red meat, dairy, and insulin sensitivity: a randomized crossover intervention study. Am J Clin Nutr. 2015;101:1173–79. [DOI] [PubMed] [Google Scholar]
- 133. Medeiros G, Azevedo KPM, Mesquita GXB, Lima S, Silva DFO, Pimenta I, et al. Red meat consumption, risk of incidence of cardiovascular disease and cardiovascular mortality, and the dose-response effect: protocol for a systematic review and meta-analysis of longitudinal cohort studies. Medicine (Baltimore). 2019;98:e17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hashemian M, Merat S, Poustchi H, Jafari E, Radmard AR, Kamangar F, et al. Red meat consumption and risk of nonalcoholic fatty liver disease in a population with low meat consumption: the Golestan Cohort Study. Am J Gastroenterol. 2021;116:1667–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Dai Y, Zhu J, Meng D, Yu C, Li Y. Association of homocysteine level with biopsy-proven non-alcoholic fatty liver disease: a meta-analysis. J Clin Biochem Nutr. 2016;58:76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bergeron N, Chiu S, Williams PT, S MK, Krauss RM. Effects of red meat, white meat, and nonmeat protein sources on atherogenic lipoprotein measures in the context of low compared with high saturated fat intake: a randomized controlled trial. Am J Clin Nutr. 2019;110:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Etemadi A, Sinha R, Ward MH, Graubard BI, Inoue-Choi M, Dawsey SM, et al. Mortality from different causes associated with meat, heme iron, nitrates, and nitrites in the NIH-AARP Diet and Health Study: population based cohort study. BMJ. 2017;357:j1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Tricò D, Biancalana E, Solini A. Protein and amino acids in nonalcoholic fatty liver disease. Curr Opin Clin Nutr Metab Care. 2021;24:96–101. [DOI] [PubMed] [Google Scholar]
- 139. Zivkovic AM, German JB, Sanyal AJ. Comparative review of diets for the metabolic syndrome: implications for nonalcoholic fatty liver disease. Am J Clin Nutr. 2007;86:285–300. [DOI] [PubMed] [Google Scholar]
- 140. Haring B, Selvin E, Liang M, Coresh J, Grams ME, Petruski-Ivleva N, et al. Dietary protein sources and risk for incident chronic kidney disease: results from the Atherosclerosis Risk in Communities (ARIC) Study. J Ren Nutr. 2017;27:233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lew QJ, Jafar TH, Koh HW, Jin A, Chow KY, Yuan JM, et al. Red meat intake and risk of ESRD. J Am Soc Nephrol. 2017;28:304–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sinha R, Cross AJ, Graubard BI, Leitzmann MF, Schatzkin A. Meat intake and mortality: a prospective study of over half a million people. Arch Intern Med. 2009;169:562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Richter CK, Skulas-Ray AC, Champagne CM, Kris-Etherton PM. Plant protein and animal proteins: do they differentially affect cardiovascular disease risk? Adv Nutr. 2015;6:712–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Hughes R, Cross AJ, Pollock JR, Bingham S. Dose-dependent effect of dietary meat on endogenous colonic N-nitrosation. Carcinogenesis. 2001;22:199–202. [DOI] [PubMed] [Google Scholar]
- 145. Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15:606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. White PJ, Lapworth AL, An J, Wang L, McGarrah RW, Stevens RD, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab. 2016;5:538–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. [DOI] [PubMed] [Google Scholar]
- 148. Mahamid M, Mahroum N, Bragazzi NL, Shalaata K, Yavne Y, Adawi M, et al. Folate and B12 levels correlate with histological severity in NASH patients. Nutrients. 2018;10:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362:1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bril F, Biernacki DM, Kalavalapalli S, Lomonaco R, Subbarayan SK, Lai J, et al. Role of vitamin E for nonalcoholic steatohepatitis in patients with type 2 diabetes: a randomized controlled trial. Diabetes Care. 2019;42:1481–88. [DOI] [PubMed] [Google Scholar]
- 151. Vilar-Gomez E, Vuppalanchi R, Gawrieh S, Ghabril M, Saxena R, Cummings OW, et al. Vitamin E improves transplant-free survival and hepatic decompensation among patients with nonalcoholic steatohepatitis and advanced fibrosis. Hepatology. 2020;71:495–509. [DOI] [PubMed] [Google Scholar]
- 152. Calvisi DF, Ladu S, Hironaka K, Factor VM, Thorgeirsson SS. Vitamin E down-modulates iNOS and NADPH oxidase in c-Myc/TGF-alpha transgenic mouse model of liver cancer. J Hepatol. 2004;41:815–22. [DOI] [PubMed] [Google Scholar]
- 153. Nan YM, Wu WJ, Fu N, Liang BL, Wang RQ, Li LX, et al. Antioxidants vitamin E and 1-aminobenzotriazole prevent experimental non-alcoholic steatohepatitis in mice. Scand J Gastroenterol. 2009;44:1121–31. [DOI] [PubMed] [Google Scholar]
- 154. He W, Xu Y, Ren X, Xiang D, Lei K, Zhang C, et al. Vitamin E ameliorates lipid metabolism in mice with nonalcoholic fatty liver fisease via Nrf2/CES1 signaling pathway. Dig Dis Sci. 2019;64:3182–91. [DOI] [PubMed] [Google Scholar]
- 155. Landrier JF, Gouranton E, El Yazidi C, Malezet C, Balaguer P, Borel P, et al. Adiponectin expression is induced by vitamin E via a peroxisome proliferator-activated receptor gamma-dependent mechanism. Endocrinology. 2009;150:5318–25. [DOI] [PubMed] [Google Scholar]
- 156. Klein EA, Thompson IM, Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306:1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Schürks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010;341:c5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Eliades M, Spyrou E, Agrawal N, Lazo M, Brancati FL, Potter JJ, et al. Meta-analysis: vitamin D and non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2013;38:246–54. [DOI] [PubMed] [Google Scholar]
- 159. Elangovan H, Chahal S, Gunton JE. Vitamin D in liver disease: current evidence and potential directions. Biochim Biophys Acta Mol Basis Dis. 2017;1863:907–16. [DOI] [PubMed] [Google Scholar]
- 160. Della Corte C, Carpino G, De Vito R, De Stefanis C, Alisi A, Cianfarani S, et al. Docosahexanoic acid plus vitamin D treatment improves features of NAFLD in children with serum Vitamin v deficiency: results from a single centre trial. PLoS One. 2016;11:e0168216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Wang H, Zhang Q, Chai Y, Liu Y, Li F, Wang B, et al. 1,25(OH)2D3 downregulates the Toll-like receptor 4-mediated inflammatory pathway and ameliorates liver injury in diabetic rats. J Endocrinol Invest. 2015;38:1083–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Geier A, Eichinger M, Stirnimann G, Semela D, Tay F, Seifert B, et al. Treatment of non-alcoholic steatohepatitis patients with vitamin D: a double-blinded, randomized, placebo-controlled pilot study. Scand J Gastroenterol. 2018;53:1114–20. [DOI] [PubMed] [Google Scholar]
- 163. Kitson MT, Pham A, Gordon A, Kemp W, Roberts SK. High-dose vitamin D supplementation and liver histology in NASH. Gut. 2016;65:717–8. [DOI] [PubMed] [Google Scholar]
- 164. Zhou Y, Dong B, Kim KH, Choi S, Sun Z, Wu N, et al. Vitamin D receptor activation in liver macrophages protects against hepatic endoplasmic reticulum stress in mice. Hepatology. 2020;71:1453–66. [DOI] [PubMed] [Google Scholar]
- 165. Dong B, Zhou Y, Wang W, Scott J, Kim K, Sun Z, et al. Vitamin D receptor activation in liver macrophages ameliorates hepatic inflammation, steatosis, and insulin resistance in mice. Hepatology. 2020;71:1559–74. [DOI] [PubMed] [Google Scholar]
- 166. Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Saeed A, Bartuzi P, Heegsma J, Dekker D, Kloosterhuis N, de Bruin A, et al. Impaired hepatic vitamin A metabolism in NAFLD mice leading to vitamin A accumulation in hepatocytes. Cell Mol Gastroenterol Hepatol. 2021;11:309–25.e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. He Z, Li X, Yang H, Wu P, Wang S, Cao D, et al. Effects of oral vitamin C supplementation on liver health and associated parameters in patients with non-alcoholic fatty liver disease: a randomized clinical trial. Front Nutr. 2021;8:745609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Oshakbayev K, Bimbetov B, Manekenova K, Bedelbayeva G, Mustafin K, Dukenbayeva B. Severe nonalcoholic steatohepatitis and type 2 diabetes: liver histology after weight loss therapy in a randomized clinical trial. Curr Med Res Opin. 2019;35:157–65. [DOI] [PubMed] [Google Scholar]
- 170. Asghari S, Rezaei M, Rafraf M, Taghizadeh M, Asghari-Jafarabadi M, Ebadi M. Effects of calorie restricted diet on oxidative/antioxidative status biomarkers and serum fibroblast growth factor 21 levels in nonalcoholic fatty liver disease patients: a randomized, controlled clinical trial. Nutrients. 2022;14:2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Johari MI, Yusoff K, Haron J, Nadarajan C, Ibrahim KN, Wong MS, et al. A randomised controlled trial on the effectiveness and adherence of modified alternate-day calorie restriction in improving activity of non-alcoholic fatty liver disease. Sci Rep. 2019;9:11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Cai H, Qin YL, Shi ZY, Chen JH, Zeng MJ, Zhou W, et al. Effects of alternate-day fasting on body weight and dyslipidaemia in patients with non-alcoholic fatty liver disease: a randomised controlled trial. BMC Gastroenterol. 2019;19:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kord-Varkaneh H, Salehi-Sahlabadi A, Tinsley GM, Santos HO, Hekmatdoost A. Effects of time-restricted feeding (16/8) combined with a low-sugar diet on the management of non-alcoholic fatty liver disease: a randomized controlled trial. Nutrition. 2023;105:111847. [DOI] [PubMed] [Google Scholar]
- 174. Ezpeleta M, Gabel K, Cienfuegos S, Kalam F, Lin S, Pavlou V, et al. Effect of alternate day fasting combined with aerobic exercise on non-alcoholic fatty liver disease: a randomized controlled trial. Cell Metab. 2023;35:56–70.e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Misciagna G, Del Pilar Díaz M, Caramia DV, Bonfiglio C, Franco I, Noviello MR, et al. Effect of a low glycemic index mediterranean diet on non-alcoholic fatty liver disease. a randomized controlled clinici trial. J Nutr Health Aging. 2017;21:404–12. [DOI] [PubMed] [Google Scholar]
- 176. Zade MR, Telkabadi MH, Bahmani F, Salehi B, Farshbaf S, Asemi Z. The effects of DASH diet on weight loss and metabolic status in adults with non-alcoholic fatty liver disease: a randomized clinical trial. Liver Int. 2016;36:563–71. [DOI] [PubMed] [Google Scholar]
- 177. Schwimmer JB, Ugalde-Nicalo P, Welsh JA, Angeles JE, Cordero M, Harlow KE, et al. Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: a randomized clinical trial. JAMA. 2019;321:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Domínguez-Coello S, Carrillo-Fernández L, Gobierno-Hernández J, Méndez-Abad M, Borges-Álamo C, García-Dopico JA, et al. Decreased consumption of added fructose reduces waist circumference and blood glucose concentration in patients with overweight and obesity. The DISFRUTE Study: a randomised trial in primary care. Nutrients. 2020;12:1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Cohen CC, Li KW, Alazraki AL, Beysen C, Carrier CA, Cleeton RL, et al. Dietary sugar restriction reduces hepatic de novo lipogenesis in adolescent boys with fatty liver disease. J Clin Invest. 2021;131:e150996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Schmidt KA, Jones RB, Rios C, Corona Y, Berger PK, Plows JF, et al. Clinical intervention to reduce dietary sugar does not affect liver fat in Latino youth, regardless of PNPLA3 genotype: a randomized controlled trial. J Nutr. 2022;152:1655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Ryan MC, Itsiopoulos C, Thodis T, Ward G, Trost N, Hofferberth S, et al. The Mediterranean diet improves hepatic steatosis and insulin sensitivity in individuals with non-alcoholic fatty liver disease. J Hepatol. 2013;59:138–43. [DOI] [PubMed] [Google Scholar]
- 182. Properzi C, O’Sullivan TA, Sherriff JL, Ching HL, Jeffrey GP, Buckley RF, et al. Ad libitum mediterranean and low-fat diets both significantly reduce hepatic steatosis: a randomized controlled trial. Hepatology. 2018;68:1741–54. [DOI] [PubMed] [Google Scholar]
- 183. Biolato M, Manca F, Marrone G, Cefalo C, Racco S, Miggiano GA, et al. Intestinal permeability after Mediterranean diet and low-fat diet in non-alcoholic fatty liver disease. World J Gastroenterol. 2019;25:509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Gepner Y, Shelef I, Komy O, Cohen N, Schwarzfuchs D, Bril N, et al. The beneficial effects of Mediterranean diet over low-fat diet may be mediated by decreasing hepatic fat content. J Hepatol. 2019;71:379–88. [DOI] [PubMed] [Google Scholar]
- 185. Ristic-Medic D, Kovacic M, Takic M, Arsic A, Petrovic S, Paunovic M, et al. Calorie-restricted mediterranean and low-fat diets affect fatty acid status in individuals with nonalcoholic fatty liver disease. Nutrients. 2020;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Akbulut UE, Isik IA, Atalay A, Eraslan A, Durmus E, Turkmen S, et al. The effect of a Mediterranean diet vs. a low-fat diet on non-alcoholic fatty liver disease in children: a randomized trial. Int J Food Sci Nutr. 2022;73:357–66. [DOI] [PubMed] [Google Scholar]
- 187. George ES, Reddy A, Nicoll AJ, Ryan MC, Itsiopoulos C, Abbott G, et al. Impact of a Mediterranean diet on hepatic and metabolic outcomes in non-alcoholic fatty liver disease: the MEDINA randomised controlled trial. Liver Int. 2022;42:1308–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Yurtdaş G, Akbulut G, Baran M, Yilmaz C. The effects of Mediterranean diet on hepatic steatosis, oxidative stress, and inflammation in adolescents with non-alcoholic fatty liver disease: a randomized controlled trial. Pediatr Obes. 2022;17:e12872. [DOI] [PubMed] [Google Scholar]
- 189. Ryan MC, Abbasi F, Lamendola C, Carter S, McLaughlin TL. Serum alanine aminotransferase levels decrease further with carbohydrate than fat restriction in insulin-resistant adults. Diabetes Care. 2007;30:1075–80. [DOI] [PubMed] [Google Scholar]
- 190. Rodríguez-Hernández H, Cervantes-Huerta M, Rodríguez-Moran M, Guerrero-Romero F. Decrease of aminotransferase levels in obese women is related to body weight reduction, irrespective of type of diet. Ann Hepatol. 2011;10:486–92. [PubMed] [Google Scholar]
- 191. Haufe S, Haas V, Utz W, Birkenfeld AL, Jeran S, Böhnke J, et al. Long-lasting improvements in liver fat and metabolism despite body weight regain after dietary weight loss. Diabetes Care. 2013;36:3786–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Goss AM, Dowla S, Pendergrass M, Ashraf A, Bolding M, Morrison S, et al. Effects of a carbohydrate-restricted diet on hepatic lipid content in adolescents with non-alcoholic fatty liver disease: a pilot, randomized trial. Pediatr Obes. 2020;15:e12630. [DOI] [PubMed] [Google Scholar]
- 193. Hansen CD, Gram-Kampmann EM, Hansen JK, Hugger MB, Madsen BS, Jensen JM, et al. Effect of calorie-unrestricted low-carbohydrate, high-fat diet versus high-carbohydrate, low-fat diet on type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial. Ann Intern Med. 2023;176:10–21. [DOI] [PubMed] [Google Scholar]
- 194. Browning JD, Baker JA, Rogers T, Davis J, Satapati S, Burgess SC. Short-term weight loss and hepatic triglyceride reduction: evidence of a metabolic advantage with dietary carbohydrate restriction. Am J Clin Nutr. 2011;93:1048–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Skytte MJ, Samkani A, Petersen AD, Thomsen MN, Astrup A, Chabanova E, et al. A carbohydrate-reduced high-protein diet improves HbA(1c) and liver fat content in weight stable participants with type 2 diabetes: a randomised controlled trial. Diabetologia. 2019;62:2066–78. [DOI] [PubMed] [Google Scholar]
- 196. Thomsen MN, Skytte MJ, Samkani A, Carl MH, Weber P, Astrup A, et al. Dietary carbohydrate restriction augments weight loss-induced improvements in glycaemic control and liver fat in individuals with type 2 diabetes: a randomised controlled trial. Diabetologia. 2022;65:506–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Cunha GM, Guzman G, Correa De Mello LL, Trein B, Spina L, Bussade I, et al. Efficacy of a 2-month very low-calorie ketogenic diet (VLCKD) compared to a standard low-calorie diet in reducing visceral and liver fat accumulation in patients with obesity. Front Endocrinol (Lausanne). 2020;11:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Lin WY, Wu CH, Chu NF, Chang CJ. Efficacy and safety of very-low-calorie diet in Taiwanese: a multicenter randomized, controlled trial. Nutrition. 2009;25:1129–36. [DOI] [PubMed] [Google Scholar]
- 199. Crabtree CD, Kackley ML, Buga A, Fell B, LaFountain RA, Hyde PN, et al. Comparison of ketogenic diets with and without ketone salts versus a low-fat diet: liver fat responses in overweight adults. Nutrients. 2021;13:966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Parry SA, Rosqvist F, Mozes FE, Cornfield T, Hutchinson M, Piche ME, et al. Intrahepatic fat and postprandial glycemia increase after consumption of a diet enriched in saturated fat compared with free sugars. Diabetes Care. 2020;43:1134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Haufe S, Engeli S, Kast P, Böhnke J, Utz W, Haas V, et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology. 2011;53:1504–14. [DOI] [PubMed] [Google Scholar]
- 202. Promrat K, Kleiner DE, Niemeier HM, Jackvony E, Kearns M, Wands JR, et al. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology. 2010;51:121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Magkos F, Fraterrigo G, Yoshino J, Luecking C, Kirbach K, Kelly SC, et al. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016;23:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015;149:367–78.e365. [DOI] [PubMed] [Google Scholar]
- 205. Magkos F. The role of dietary protein in obesity. Rev Endocr Metab Disord. 2020;21:329–40. [DOI] [PubMed] [Google Scholar]
- 206. Rosen E, Bakshi N, Watters A, Rosen HR, Mehler PS. Hepatic complications of anorexia nervosa. Dig Dis Sci. 2017;62:2977–81. [DOI] [PubMed] [Google Scholar]
- 207. Risi R, Tuccinardi D, Mariani S, Lubrano C, Manfrini S, Donini LM, et al. Liver disease in obesity and underweight: the two sides of the coin. A narrative review. Eat Weight Disord. 2021;26:2097–107. [DOI] [PubMed] [Google Scholar]
- 208. Perez-Diaz-Del-Campo N, Castelnuovo G, Caviglia GP, Armandi A, Rosso C, Bugianesi E. Role of circadian clock on the pathogenesis and lifestyle management in non-alcoholic fatty liver disease. Nutrients. 2022;14:5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Barnosky AR, Hoddy KK, Unterman TG, Varady KA. Intermittent fasting vs daily calorie restriction for type 2 diabetes prevention: a review of human findings. Transl Res. 2014;164:302–11. [DOI] [PubMed] [Google Scholar]
- 210. Drinda S, Grundler F, Neumann T, Lehmann T, Steckhan N, Michalsen A, et al. Effects of periodic fasting on fatty liver index—a prospective observational study. Nutrients. 2019;11:2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Harvie M, Wright C, Pegington M, McMullan D, Mitchell E, Martin B, et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Br J Nutr. 2013;110:1534–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Pugliese N, Plaz Torres MC, Petta S, Valenti L, Giannini EG, Aghemo A. Is there an ‘ideal’ diet for patients with NAFLD ? Eur J Clin Invest. 2022;52:e13659. [DOI] [PubMed] [Google Scholar]
- 213. Zhao J, Bai M, Wei S, Li C, Lv Q, Chen Y. Improvement of non-alcoholic fatty liver disease in mice by intermittent use of a fasting-mimicking diet. Mol Nutr Food Res. 2021;65:e2100381. [DOI] [PubMed] [Google Scholar]
- 214. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012;15:848–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Holmer M, Lindqvist C, Petersson S, Moshtaghi-Svensson J, Tillander V, Brismar TB, et al. Treatment of NAFLD with intermittent calorie restriction or low-carb high-fat diet—a randomised controlled trial. JHEP Rep. 2021;3:100256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Moreno B, Crujeiras AB, Bellido D, Sajoux I, Casanueva FF. Obesity treatment by very low-calorie-ketogenic diet at two years: reduction in visceral fat and on the burden of disease. Endocrine. 2016;54:681–90. [DOI] [PubMed] [Google Scholar]
- 217. Tendler D, Lin S, Yancy WS, Jr, Mavropoulos J, Sylvestre P, Rockey DC, et al. The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: a pilot study. Dig Dis Sci. 2007;52:589–93. [DOI] [PubMed] [Google Scholar]
- 218. Luukkonen PK, Dufour S, Lyu K, Zhang XM, Hakkarainen A, Lehtimäki TE, et al. Effect of a ketogenic diet on hepatic steatosis and hepatic mitochondrial metabolism in nonalcoholic fatty liver disease. Proc Natl Acad Sci USA. 2020;117:7347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Moore MP, Cunningham RP, Kelty TJ, Boccardi LR, Nguyen NY, Booth FW, et al. Ketogenic diet in combination with voluntary exercise impacts markers of hepatic metabolism and oxidative stress in male and female Wistar rats. Appl Physiol Nutr Metab. 2020;45:35–44. [DOI] [PubMed] [Google Scholar]
- 220. Fletcher JA, Deja S, Satapati S, Fu X, Burgess SC, Browning JD. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight. 2019;5:e127737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Pérez-Guisado J, Muñoz-Serrano A. The effect of the Spanish Ketogenic Mediterranean Diet on nonalcoholic fatty liver disease: a pilot study. J Med Food. 2011;14:677–80. [DOI] [PubMed] [Google Scholar]
- 222. Kirk E, Reeds DN, Finck BN, Mayurranjan SM, Patterson BW, Klein S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology. 2009;136:1552–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Watanabe M, Tozzi R, Risi R, Tuccinardi D, Mariani S, Basciani S, et al. Beneficial effects of the ketogenic diet on nonalcoholic fatty liver disease: a comprehensive review of the literature. Obes Rev. 2020;21:e13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Nordmann AJ, Nordmann A, Briel M, Keller U, Yancy WS, Jr, Brehm BJ, et al. Effects of low-carbohydrate vs low-fat diets on weight loss and cardiovascular risk factors: a meta-analysis of randomized controlled trials. Arch Intern Med. 2006;166:285–93. [DOI] [PubMed] [Google Scholar]
- 225. Fock KM, Khoo J. Diet and exercise in management of obesity and overweight. J Gastroenterol Hepatol. 2013;28(suppl 4):59–63. [DOI] [PubMed] [Google Scholar]
- 226. Johansson K, Sundström J, Marcus C, Hemmingsson E, Neovius M. Risk of symptomatic gallstones and cholecystectomy after a very-low-calorie diet or low-calorie diet in a commercial weight loss program: 1-year matched cohort study. Int J Obes (Lond). 2014;38:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64:1388–402. [DOI] [PubMed] [Google Scholar]
- 228. Plauth M, Bernal W, Dasarathy S, Merli M, Plank LD, Schütz T, et al. ESPEN guideline on clinical nutrition in liver disease. Clin Nutr. 2019;38:485–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Tsaban G, Yaskolka Meir A, Rinott E, Zelicha H, Kaplan A, Shalev A, et al. The effect of green Mediterranean diet on cardiometabolic risk; a randomised controlled trial. Heart. 2020. doi: 10.1136/heartjnl-2020-317802 [DOI] [PubMed] [Google Scholar]
- 230. Van Name MA, Savoye M, Chick JM, Galuppo BT, Feldstein AE, Pierpont B, et al. A low ω-6 to ω-3 PUFA ratio (n-6:n-3 PUFA) diet to treat fatty liver disease in obese youth. J Nutr. 2020;150:2314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Lu W, Li S, Li J, Wang J, Zhang R, Zhou Y, et al. Effects of omega-3 fatty acid in nonalcoholic fatty liver disease: a meta-analysis. Gastroenterol Res Pract. 2016;2016:1459790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, et al. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292:1440–6. [DOI] [PubMed] [Google Scholar]
- 233. Abenavoli L, Greco M, Milic N, Accattato F, Foti D, Gulletta E, et al. Effect of Mediterranean diet and antioxidant formulation in non-alcoholic fatty liver disease: a randomized study. Nutrients. 2017;9:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Hayat U, Siddiqui AA, Okut H, Afroz S, Tasleem S, Haris A. The effect of coffee consumption on the non-alcoholic fatty liver disease and liver fibrosis: a meta-analysis of 11 epidemiological studies. Ann Hepatol. 2021;20:100254. [DOI] [PubMed] [Google Scholar]
- 235. Tamura T, Wada K, Konishi K, Goto Y, Mizuta F, Koda S, et al. Coffee, green tea, and caffeine intake and liver cancer risk: a prospective cohort study. Nutr Cancer. 2018;70:1210–16. [DOI] [PubMed] [Google Scholar]
- 236. Vitaglione P, Mazzone G, Lembo V, D’Argenio G, Rossi A, Guido M, et al. Coffee prevents fatty liver disease induced by a high-fat diet by modulating pathways of the gut-liver axis. J Nutr Sci. 2019;8:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Di Mauro S, Salomone F, Scamporrino A, Filippello A, Morisco F, Guido M, et al. Coffee restores expression of lncRNAs involved in steatosis and fibrosis in a mouse model of NAFLD. Nutrients. 2021;13:2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Long MT, Massaro JM, Hoffmann U, Benjamin EJ, Naimi TS. Alcohol use is associated with hepatic steatosis among persons with presumed nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2020;18:1831–41.e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Mitchell T, Jeffrey GP, de Boer B, MacQuillan G, Garas G, Ching H, et al. Type and pattern of alcohol consumption is associated with liver fibrosis in patients with non-alcoholic fatty liver disease. Am J Gastroenterol. 2018;113:1484–93. [DOI] [PubMed] [Google Scholar]
- 240. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–8. [DOI] [PubMed] [Google Scholar]
- 241. Liberale L, Bonaventura A, Montecucco F, Dallegri F, Carbone F. Impact of red wine consumption on cardiovascular health. Curr Med Chem. 2019;26:3542–66. [DOI] [PubMed] [Google Scholar]
- 242. Vollmer WM, Sacks FM, Ard J, Appel LJ, Bray GA, Simons-Morton DG, et al. Effects of diet and sodium intake on blood pressure: subgroup analysis of the DASH-sodium trial. Ann Intern Med. 2001;135:1019–28. [DOI] [PubMed] [Google Scholar]
- 243. Hekmatdoost A, Shamsipour A, Meibodi M, Gheibizadeh N, Eslamparast T, Poustchi H. Adherence to the dietary approaches to stop hypertension (DASH) and risk of nonalcoholic fatty liver disease. Int J Food Sci Nutr. 2016;67:1024–9. [DOI] [PubMed] [Google Scholar]
- 244. Soltani S, Shirani F, Chitsazi MJ, Salehi-Abargouei A. The effect of dietary approaches to stop hypertension (DASH) diet on weight and body composition in adults: a systematic review and meta-analysis of randomized controlled clinical trials. Obes Rev. 2016;17:442–54. [DOI] [PubMed] [Google Scholar]
- 245. Westerbacka J, Lammi K, Häkkinen AM, Rissanen A, Salminen I, Aro A, et al. Dietary fat content modifies liver fat in overweight nondiabetic subjects. J Clin Endocrinol Metab. 2005;90:2804–9. [DOI] [PubMed] [Google Scholar]
- 246. Ahn J, Jun DW, Lee HY, Moon JH. Critical appraisal for low-carbohydrate diet in nonalcoholic fatty liver disease: review and meta-analyses. Clin Nutr. 2019;38:2023–30. [DOI] [PubMed] [Google Scholar]
- 247. Chianelli D, Rucker PV, Roland J, Tully DC, Nelson J, Liu X, et al. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. J Med Chem. 2020;63:3868–80. [DOI] [PubMed] [Google Scholar]
- 248. Wang Y, Crittenden DB, Eng C, Zhang Q, Guo P, Chung D, et al. Safety, pharmacokinetics, pharmacodynamics, and formulation of liver-distributed farnesoid X-receptor agonist TERN-101 in healthy volunteers. Clin Pharmacol Drug Dev. 2021;10:1198–208. [DOI] [PubMed] [Google Scholar]
- 249. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, et al. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology (Baltimore, Md). 2020;72:58–71. [DOI] [PubMed] [Google Scholar]
- 250. Traussnigg S, Halilbasic E, Hofer H, Munda P, Stojakovic T, Fauler G, et al. Open-label phase II study evaluating safety and efficacy of the non-steroidal farnesoid X receptor agonist PX-104 in non-alcoholic fatty liver disease. Wien Klin Wochenschr. 2021;133:441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Ratziu V, Rinella ME, Neuschwander-Tetri BA, Lawitz E, Denham D, Kayali Z, et al. EDP-305 in patients with NASH: a phase II double-blind placebo-controlled dose-ranging study. J Hepatol. 2022;76:506–517. [DOI] [PubMed] [Google Scholar]
- 252. Sanyal AJ, Lopez P, Lawitz EJ, Lucas KJ, Loeffler J, Kim W, et al. Tropifexor for nonalcoholic steatohepatitis: an adaptive, randomized, placebo-controlled phase 2a/b trial. Nat Med. 2023;29:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Ratziu V, Harrison SA, Loustaud-Ratti V, Bureau C, Lawitz E, Abdelmalek M, et al. Hepatic and renal improvements with FXR agonist vonafexor in individuals with suspected fibrotic NASH. J Hepatol. 2023;78:479–92. [DOI] [PubMed] [Google Scholar]
- 254. Harrison SA, Bashir MR, Lee K-J, Shim-Lopez J, Lee J, Wagner B, et al. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J Hepatol. 2021;75:25–33. [DOI] [PubMed] [Google Scholar]
- 255. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet (London, England). 2019;394:2184–96. [DOI] [PubMed] [Google Scholar]
- 256. Newsome PN, Palmer M, Freilich B, Sheikh MY, Sheikh A, Sarles H, et al. Volixibat in adults with non-alcoholic steatohepatitis: 24-week interim analysis from a randomized, phase II study. J Hepatol. 2020;73:231–40. [DOI] [PubMed] [Google Scholar]
- 257. Kuchay MS, Krishan S, Mishra SK, Farooqui KJ, Singh MK, Wasir JS, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT Trial). Diabetes Care. 2018;41:1801–8. [DOI] [PubMed] [Google Scholar]
- 258. Taheri H, Malek M, Ismail-Beigi F, Zamani F, Sohrabi M, Reza Babaei M, et al. Effect of empagliflozin on liver steatosis and fibrosis in patients with non-alcoholic fatty liver disease without diabetes: a randomized, double-blind, placebo-controlled trial. Adv Ther. 2020;37:4697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab. 2019;21:285–92. [DOI] [PubMed] [Google Scholar]
- 260. Phrueksotsai S, Pinyopornpanish K, Euathrongchit J, Leerapun A, Phrommintikul A, Buranapin S, et al. The effects of dapagliflozin on hepatic and visceral fat in type 2 diabetes patients with non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2021;36:2952–9. [DOI] [PubMed] [Google Scholar]
- 261. Inoue M, Hayashi A, Taguchi T, Arai R, Sasaki S, Takano K, et al. Effects of canagliflozin on body composition and hepatic fat content in type 2 diabetes patients with non-alcoholic fatty liver disease. J Diabetes Investig. 2019;10:1004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Harrison SA, Manghi FP, Smith WB, Alpenidze D, Aizenberg D, Klarenbeek N, et al. Licogliflozin for nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a study. Nat Med. 2022;28:1432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Nakajima A, Eguchi Y, Yoneda M, Imajo K, Tamaki N, Suganami H, et al. Randomised clinical trial: pemafibrate, a novel selective peroxisome proliferator-activated receptor α modulator (SPPARMα), versus placebo in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2021;54:1263–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Fernández-Miranda C, Pérez-Carreras M, Colina F, López-Alonso G, Vargas C, Solís-Herruzo JA. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig Liver Dis. 2008;40:200–5. [DOI] [PubMed] [Google Scholar]
- 265. Ratziu V, Giral P, Jacqueminet S, Charlotte F, Hartemann-Heurtier A, Serfaty L, et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology. 2008;135:100–110. [DOI] [PubMed] [Google Scholar]
- 266. Lee YH, Kim JH, Kim SR, Jin HY, Rhee EJ, Cho YM, et al. Lobeglitazone, a novel thiazolidinedione, improves non-alcoholic fatty liver disease in type 2 diabetes: its efficacy and predictive factors related to responsiveness. J Korean Med Sci. 2017;32:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–84. [DOI] [PubMed] [Google Scholar]
- 268. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- 269. Harrison SA, Thang C, Bolze S, Dewitt S, Hallakou-Bozec S, Dubourg J, et al. Evaluation of PXL065 - deuterium-stabilized (R)-pioglitazone in patients with NASH: a phase II randomized placebo-controlled trial (DESTINY-1). J Hepatol. 2023;78:914–25. [DOI] [PubMed] [Google Scholar]
- 270. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–59.e1145. [DOI] [PubMed] [Google Scholar]
- 271. Siddiqui MS, Idowu MO, Parmar D, Borg BB, Denham D, Loo NM, et al. A phase 2 double blinded, randomized controlled trial of saroglitazar in patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2021;19:2670–72. [DOI] [PubMed] [Google Scholar]
- 272. Gawrieh S, Noureddin M, Loo N, Mohseni R, Awasty V, Cusi K, et al. Saroglitazar, a PPAR-α/γ agonist, for treatment of NAFLD: a randomized controlled double-blind phase 2 trial. Hepatology (Baltimore, Md). 2021;74:1809–24. [DOI] [PubMed] [Google Scholar]
- 273. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pn-PPAR agonist lanifibranor in NASH. N Engl J Med. 2021;385:1547–58. [DOI] [PubMed] [Google Scholar]
- 274. Cui J, Philo L, Nguyen P, Hofflich H, Hernandez C, Bettencourt R, et al. Sitagliptin vs. placebo for non-alcoholic fatty liver disease: a randomized controlled trial. J Hepatol. 2016;65:369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Joy TR, McKenzie CA, Tirona RG, Summers K, Seney S, Chakrabarti S, et al. Sitagliptin in patients with non-alcoholic steatohepatitis: a randomized, placebo-controlled trial. World J Gastroenterol. 2017;23:141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Han E, Huh JH, Lee EY, Bae JC, Chun SW, Yu SH, et al. Efficacy and safety of evogliptin in patients with type 2 diabetes and non-alcoholic fatty liver disease: a multicentre, double-blind, randomized, comparative trial. Diabetes Obes Metab. 2022;24:752–6. [DOI] [PubMed] [Google Scholar]
- 277. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet (London, England). 2016;387:679–90. [DOI] [PubMed] [Google Scholar]
- 278. Petit J-M, Cercueil J-P, Loffroy R, Denimal D, Bouillet B, Fourmont C, et al. Effect of liraglutide therapy on liver fat content in patients with inadequately controlled type 2 diabetes: the Lira-NAFLD study. J Clin Endocrinol Metab. 2017;102:407–15. [DOI] [PubMed] [Google Scholar]
- 279. Flint A, Andersen G, Hockings P, Johansson L, Morsing A, Sundby Palle M, et al. Randomised clinical trial: semaglutide versus placebo reduced liver steatosis but not liver stiffness in subjects with non-alcoholic fatty liver disease assessed by magnetic resonance imaging. Aliment Pharmacol Ther. 2021;54:1150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A Placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2021;384:1113–24. [DOI] [PubMed] [Google Scholar]
- 281. Kuchay MS, Krishan S, Mishra SK, Choudhary NS, Singh MK, Wasir JS, et al. Effect of dulaglutide on liver fat in patients with type 2 diabetes and NAFLD: randomised controlled trial (D-LIFT trial). Diabetologia. 2020;63:2434–45. [DOI] [PubMed] [Google Scholar]
- 282. Shao N, Kuang HY, Hao M, Gao XY, Lin WJ, Zou W. Benefits of exenatide on obesity and non-alcoholic fatty liver disease with elevated liver enzymes in patients with type 2 diabetes. Diabetes Metab Res Rev. 2014;30:521–9. [DOI] [PubMed] [Google Scholar]
- 283. Liu L, Yan H, Xia M, Zhao L, Lv M, Zhao N, et al. Efficacy of exenatide and insulin glargine on nonalcoholic fatty liver disease in patients with type 2 diabetes. Diabetes Metab Res Rev. 2020;36:e3292. [DOI] [PubMed] [Google Scholar]
- 284. Yazawa R, Ishida M, Balavarca Y, Hennige AM. A randomised phase I study of the safety, tolerability, pharmacokinetics and pharmacodynamics of BI 456906, a dual glucagon receptor/glucagon-like peptide-1 receptor agonist, in healthy Japanese men with overweight/obesity. Diabetes Obes Metab. 2023;25:1973–84. [DOI] [PubMed] [Google Scholar]
- 285. Jungnik A, Arrubla Martinez J, Plum-Mörschel L, Kapitza C, Lamers D, Thamer C, et al. Phase I studies of the safety, tolerability, pharmacokinetics and pharmacodynamics of the dual glucagon receptor/glucagon-like peptide-1 receptor agonist BI 456906. Diabetes Obes Metab. 2023;25:1011–23. [DOI] [PubMed] [Google Scholar]
- 286. Loomba R, Lawitz EJ, Frias JP, Ortiz-Lasanta G, Johansson L, Franey BB, et al. Safety, pharmacokinetics, and pharmacodynamics of pegozafermin in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 1b/2a multiple-ascending-dose study. Lancet Gastroenterol Hepatol. 2023;8:120–32. [DOI] [PubMed] [Google Scholar]
- 287. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet (London, England). 2019;392:2705–17. [DOI] [PubMed] [Google Scholar]
- 288. Brown EA, Minnich A, Sanyal AJ, Loomba R, Du S, Schwarz J, et al. Effect of pegbelfermin on NASH and fibrosis-related biomarkers and correlation with histological response in the FALCON 1 trial. JHEP Rep. 2023;5:100661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Harrison SA, Ruane PJ, Freilich BL, Neff G, Patil R, Behling CA, et al. Efruxifermin in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled, phase 2a trial. Nat Med. 2021;27:1262–71. [DOI] [PubMed] [Google Scholar]
- 290. Baruch A, Wong C, Chinn LW, Vaze A, Sonoda J, Gelzleichter T, et al. Antibody-mediated activation of the FGFR1/Klothoβ complex corrects metabolic dysfunction and alters food preference in obese humans. Proc Natl Acad Sci USA. 2020;117:28992–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Wong C, Dash A, Fredrickson J, Lewin-Koh N, Chen S, Yoshida K, et al. Fibroblast growth factor receptor 1/Klothoβ agonist BFKB8488A improves lipids and liver health markers in patients with diabetes or NAFLD: a phase 1b randomized trial. Hepatology. 2022. doi: 10.1002/hep.32742 [DOI] [PubMed] [Google Scholar]
- 292. Harrison SA, Abdelmalek MF, Neff G, Gunn N, Guy CD, Alkhouri N, et al. Aldafermin in patients with non-alcoholic steatohepatitis (ALPINE 2/3): a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Gastroenterol Hepatol. 2022;7:603–16. [DOI] [PubMed] [Google Scholar]
- 293. Syed-Abdul MM, Parks EJ, Gaballah AH, Bingham K, Hammoud GM, Kemble G, et al. Fatty aAcid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology. 2020;72:103–18. [DOI] [PubMed] [Google Scholar]
- 294. Loomba R, Mohseni R, Lucas KJ, Gutierrez JA, Perry RG, Trotter JF, et al. TVB-2640 (FASN Inhibitor) for the treatment of nonalcoholic steatohepatitis: FASCINATE-1, a randomized, placebo-controlled phase 2a trial. Gastroenterology. 2021;161:1475–86. [DOI] [PubMed] [Google Scholar]
- 295. Beysen C, Schroeder P, Wu E, Brevard J, Ribadeneira M, Lu W, et al. Inhibition of fatty acid synthase with FT-4101 safely reduces hepatic de novo lipogenesis and steatosis in obese subjects with non-alcoholic fatty liver disease: results from two early-phase randomized trials. Diabetes Obes Metab. 2021;23:700–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Kazierad DJ, Chidsey K, Somayaji VR, Bergman AJ, Birnbaum MJ, Calle RA. Inhibition of ketohexokinase in adults with NAFLD reduces liver fat and inflammatory markers: a randomized phase 2 trial. Med. 2021;2:800–13.e803. [DOI] [PubMed] [Google Scholar]
- 297. Saxena AR, Lyle S-A, Khavandi K, Qiu R, Whitlock M, Esler WP, et al. A phase 2a, randomized, double-blind, placebo-controlled, three-arm, parallel-group study to assess the efficacy, safety, tolerability and pharmacodynamics of PF-06835919 in patients with non-alcoholic fatty liver disease and type 2 diabetes. Diabetes Obes Metab. 2023;25:992–1001. [DOI] [PubMed] [Google Scholar]
- 298. Ratziu V, de Guevara L, Safadi R, Poordad F, Fuster F, Flores-Figueroa J, et al. Aramchol in patients with nonalcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase 2b trial. Nat Med. 2021;27:1825–35. [DOI] [PubMed] [Google Scholar]
- 299. Bergman A, Carvajal-Gonzalez S, Tarabar S, Saxena AR, Esler WP, Amin NB. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a liver-targeting acetyl-CoA carboxylase inhibitor (PF-05221304): a three-part randomized phase 1 study. Clin Pharmacol Drug Dev. 2020;9:514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Calle RA, Amin NB, Carvajal-Gonzalez S, Ross TT, Bergman A, Aggarwal S, et al. ACC inhibitor alone or co-administered with a DGAT2 inhibitor in patients with non-alcoholic fatty liver disease: two parallel, placebo-controlled, randomized phase 2a trials. Nat Med. 2021;27:1836–48. [DOI] [PubMed] [Google Scholar]
- 301. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, et al. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018;155:1463–73.e1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Kim C-W, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26:394–406.e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019;394:2012–24. [DOI] [PubMed] [Google Scholar]
- 304. Harrison SA, Bashir M, Moussa SE, McCarty K, Pablo Frias J, Taub R, et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH.. Hepatol Commun. 2021;5:573–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Gaudet D, Karwatowska-Prokopczuk E, Baum SJ, et al., Vupanorsen, an N-acetyl galactosamine-conjugated antisense drug to ANGPTL3 mRNA, lowers triglycerides and atherogenic lipoproteins in patients with diabetes, hepatic steatosis, and hypertriglyceridaemia. Eur Heart J, 2020;41:3936–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, et al. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. 2020;72:613–26. [DOI] [PubMed] [Google Scholar]
- 307. Mak L-Y, Gane E, Schwabe C, Yoon KT, Heo J, Scott R, et al. A phase I/II study of ARO-HSD, an RNA interference therapeutic, for the treatment of non-alcoholic steatohepatitis. J Hepatol. 2023;78:684–92. [DOI] [PubMed] [Google Scholar]
- 308. Stefan N, Ramsauer M, Jordan P, Nowotny B, Kantartzis K, Machann J, et al. Inhibition of 11β-HSD1 with RO5093151 for non-alcoholic fatty liver disease: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014;2:406–16. [DOI] [PubMed] [Google Scholar]
- 309. Aronne LJ, Finer N, Hollander PA, England RD, Klioze SS, Chew RD, et al. Efficacy and safety of CP-945,598, a selective cannabinoid CB1 receptor antagonist, on weight loss and maintenance. Obesity (Silver Spring). 2011;19:1404–114. [DOI] [PubMed] [Google Scholar]
- 310. Cusi K, Alkhouri N, Harrison SA, Fouqueray P, Moller DE, Hallakou-Bozec S, et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol Hepatol. 2021;6:889–902. [DOI] [PubMed] [Google Scholar]
- 311. Polyzos SA, Kountouras J, Mantzoros CS, Polymerou V, Katsinelos P. Effects of combined low-dose spironolactone plus vitamin E vs vitamin E monotherapy on insulin resistance, non-invasive indices of steatosis and fibrosis, and adipokine levels in non-alcoholic fatty liver disease: a randomized controlled trial. Diabetes Obes Metab. 2017;19:1805–9. [DOI] [PubMed] [Google Scholar]
- 312. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Clifford BL, Sedgeman LR, Williams KJ, Morand P, Cheng A, Jarrett KE, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33:1671–84.e1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Kremoser C. FXR agonists for NASH: how are they different and what difference do they make? J Hepatol. 2021;75:12–5. [DOI] [PubMed] [Google Scholar]
- 315. Marotta C, Ahmad A, Luo E, Oosterhaven J, van Marle S, Adda N. EDP-297: A novel, farnesoid X receptor agonist—results of a phase I study in healthy subjects. Clin Transl Sci. 2023;16:338–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. 2020;16:654–67. [DOI] [PubMed] [Google Scholar]
- 317. Kang SH, Lee HW, Yoo JJ, Cho Y, Kim SU, Lee TH, et al. KASL clinical practice guidelines: management of nonalcoholic fatty liver disease. Clin Mol Hepatol. 2021;27:363–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Jacques V, Bolze S, Hallakou-Bozec S, Czarnik AW, Divakaruni AS, Fouqueray P, et al. Deuterium-stabilized (R)-pioglitazone (PXL065) is responsible for pioglitazone efficacy in NASH yet exhibits little to no PPARgamma activity. Hepatol Commun. 2021;5:1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Thomes PG, Benbow JH, Brandon-Warner E, Thompson KJ, Jacobs C, Donohue TM, Jr, et al. Dietary fructose augments ethanol-induced liver pathology. J Nutr Biochem. 2017;43:141–50. [DOI] [PubMed] [Google Scholar]
- 321. Vancells Lujan P, Viñas Esmel E, Sacanella Meseguer E. Overview of non-alcoholic fatty liver disease (NAFLD) and the role of sugary food consumption and other dietary components in its development. Nutrients. 2021;13:1442. [DOI] [PMC free article] [PubMed] [Google Scholar]