
FIGURE 4. The basic steps and principle of working of molecular dynamics (MD) simulations used in new drug development.
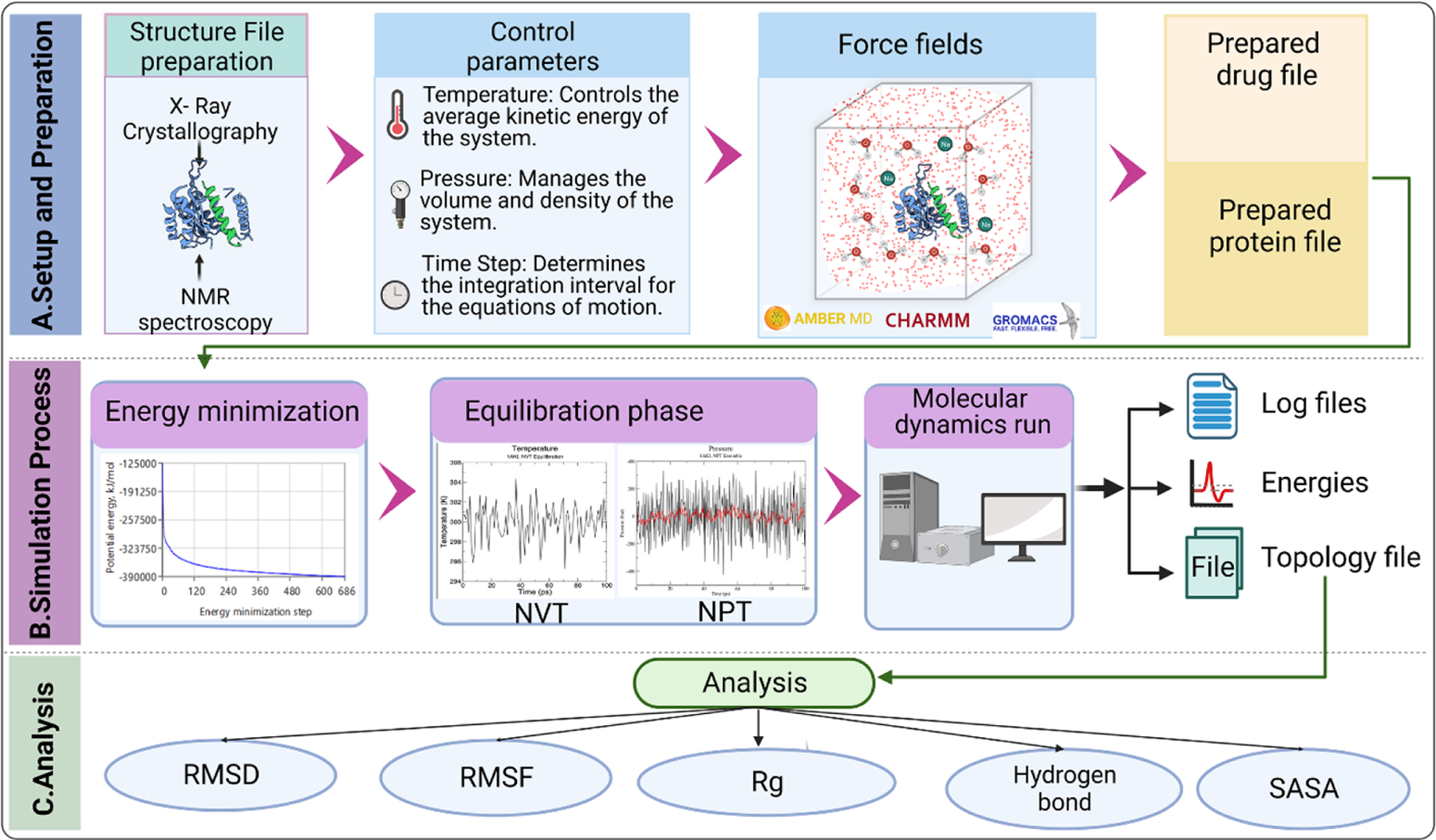
(a) Schematic representation of the steps involved in MD simulation for drug research. This figure illustrates the sequential steps in conducting MD simulations for drug research. The setup and preparation phase involves obtaining and converting the three-dimensional structures of protein and drug molecules into appropriate file formats. Essential simulation parameters, such as temperature, pressure, and time step, are defined. Additionally, an appropriate force field is chosen based on the system and research objectives. It is important to select simulation software that is compatible with the chosen force field. (b) During the simulation process, energy minimization is performed to alleviate steric clashes and minimize potential energy in the system. The equilibration phase allows the system to gradually relax by restraining specific atoms and allowing solvent molecules to adjust around the protein–drug complex. Subsequently, a time-dependent molecular dynamics run is carried out, where the equations of motion are numerically integrated to generate atom trajectories. (c) The analysis of the topology file includes various metrics. Root-mean-square deviation (RMSD) is used to measure the average deviation between different structures at different time points and a reference structure. Root-mean-square fluctuation (RMSF) determines the atomic fluctuations of the protein and drug, providing insights into their flexibility and stability. The radius of gyration (Rg) quantifies the compactness or spatial extent of the protein–drug complex. Hydrogen bonds are assessed to understand their formation and dynamics, indicating potential interactions. Finally, the solvent accessible surface area (SASA) is calculated to analyze the interactions of the protein and drug with the surrounding solvent molecules.