

SUMMARY
Progenitor cells are critical in preserving organismal homeostasis, yet their diversity and dynamics in the aged brain remain underexplored. We introduced TrackerSci, a single-cell genomic method that combines newborn cell labeling and combinatorial indexing to characterize the transcriptome and chromatin landscape of proliferating progenitor cells in vivo. Using TrackerSci, we investigated the dynamics of newborn cells in mouse brains across various ages and in a mouse model of Alzheimer’s disease. Our dataset revealed diverse progenitor cell types in the brain and their epigenetic signatures. We further quantified aging-associated shifts in cell-type-specific proliferation and differentiation and deciphered the associated molecular programs. Extending our study to the progenitor cells in the aged human brain, we identified conserved genetic signatures across species and pinpointed region-specific cellular dynamics, such as the reduced oligodendrogenesis in the cerebellum. We anticipate that TrackerSci will be broadly applicable to unveil cell-type-specific temporal dynamics in diverse systems.
In brief
A comprehensive view of single-cell transcriptome and chromatin accessibility dynamics of brain newborn cells provides insights into aging-associated shifts in cell-type-specific proliferation and differentiation and associated molecular programs.
Graphical Abstract

INTRODUCTION
New neurons and glial cells are continuously produced in adult mammalian brains, a critical process associated with memory, learning, and stress.1,2 There is a consensus that adult neurogenesis and oligodendrogenesis decline with advancing ages and in neuropathological conditions,3,4 but to what extent is debated.5,6 The ambiguity stems partly from technical limitations—most studies rely upon the utilization of proxy markers, potentially introducing biases when quantifying the highly rare progenitor cells in aged tissues. Furthermore, the identity of progenitor cells is intricately defined through tightly controlled epigenetic programs. While prior works, such as Div-Seq,7,8 have profiled the gene expression signatures of progenitor cells within the adult brain, our understanding of their epigenetic landscape and how it changes during aging remain limited. Therefore, new approaches for quantitatively capturing newborn cells and tracking their transcriptome and chromatin state changes are critical to understanding cell population dynamics in development, aging, and disease states.
Single-cell combinatorial indexing has emerged as a powerful strategy to label the nucleic acid contents of individual cells or nuclei in a scalable manner.9–16 In this context, we introduce TrackerSci, a single-cell combinatorial indexing approach that integrates click chemistry to label newly synthesized DNA with a thymidine analog, 5-Ethynyl-2-deoxyuridine (EdU),8 along with single-cell combinatorial indexing sequencing for transcriptome (sci-RNA-seq14) or chromatin accessibility profiling (sci-ATAC-seq9). As a demonstration, we applied TrackerSci to profiling the single-cell transcriptome or chromatin accessibility dynamics of 14,689 newborn cells from entire mouse brains spanning three age stages and two genotypes. With the resulting datasets, we recovered rare progenitor cell populations missed in conventional single-cell analysis and tracked their cell-type-specific proliferation and differentiation dynamics across ages. Furthermore, we identified the genetic and epigenetic signatures associated with the alteration of cellular dynamics that occur in the aged mammalian brain. Finally, to compare rare progenitor cells across species, we profiled ~800,000 single-nucleus transcriptomes of the human brain across various regions. By integration with the TrackerSci dataset, we identified region- and cell-type-specific signatures of rare progenitor cells in the aged human brain and recovered conserved and divergent molecular signatures. The experimental and computational methods described here could be broadly applied to track cellular regenerative capacity and differentiation potential across mammalian organs and other biological systems.
RESULTS
A global view of newborn cells across the mammalian brain by TrackerSci
The TrackerSci method is delineated in the following steps (Figure 1A). (1) Mice are labeled with EdU, a thymidine analog that can be incorporated into replicating DNA for labeling in vivo cellular proliferation.8,17 (2) Brain nuclei are extracted, fixed, and then subjected to click chemistry-based in situ ligation18 to an azide-containing fluorophore, followed by fluorescence-activated cell sorting (FACS) to enrich the EdU+ cells (Figure S1A). (3) A first round of indexing is introduced through barcoded reverse transcription or transposition. Cells from all wells are pooled and then redistributed into multiple 96-well plates via FACS to further purify the EdU+ cells (Figure S1B). (4) We then follow library preparation protocols similar to sci-RNA-seq14 for transcriptome profiling or sci-ATAC-seq9 for chromatin accessibility analysis. Notably, the two sorting steps implemented in TrackerSci are essential for excluding contaminating cells and capturing rare proliferating cell populations, especially in the aged brain where EdU+ cells constitute less than 0.1% of the total. In addition, we extensively optimized the reaction conditions to ensure the methodology’s compatibility with newborn cell labeling and single-cell combinatorial indexing (Figures S1C–S1H). Notably, the transcriptome and chromatin profiles derived from TrackerSci closely matched standard profiling (Figure S1I), highlighting the minimal impact of TrackerSci on the molecular state of cells.
Figure 1. TrackerSci enables single-cell transcriptome and chromatin accessibility profiling of rare proliferating cells in the mammalian brain.
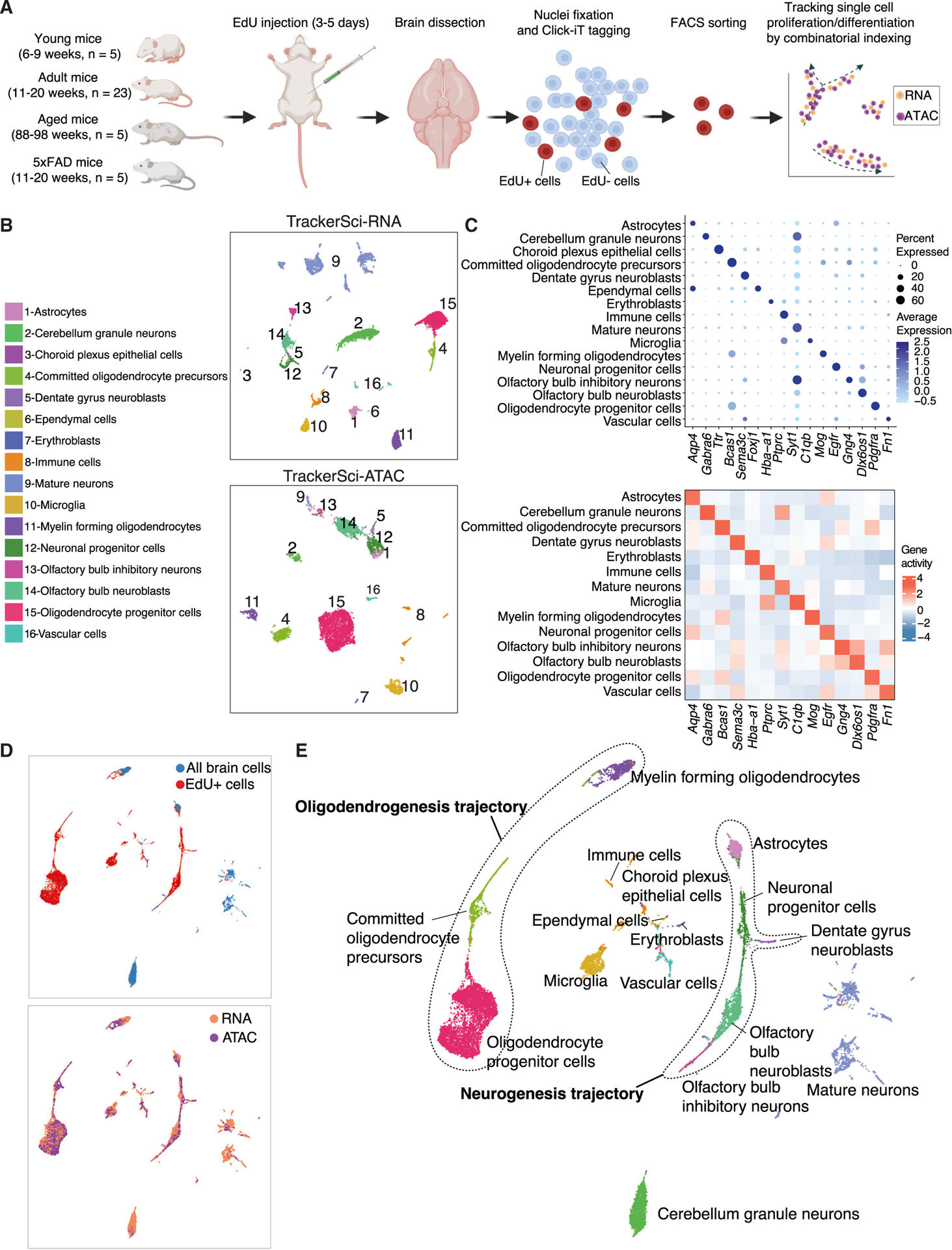
(A) TrackerSci workflow and experiment scheme. Key steps are outlined in the text.
(B) UMAP visualization of single-cell transcriptomes (top) and single-cell chromatin accessibility profiles (bottom), including EdU+ cells (profiled by TrackerSci) and all brain cells (without enrichment of EdU+ cells), colored by main cell types.
(C) Dot plot and heatmap showing gene expression and gene activity of marker genes for each cluster defined by TrackerSci-RNA (top) and TrackerSci-ATAC (bottom), respectively.
(D and E) UMAP visualization of mouse brain cells, integrating the single-cell transcriptome and chromatin accessibility profiles of EdU+ cells and DAPI singlets (representing the global brain cell population). Cells are colored by sources (D, top), molecular layers (D, bottom), and main cell types (D). The identified neurogenesis and oligodendrogenesis trajectories are both annotated in (E).
We next applied TrackerSci to capture rare newborn cells from mouse brains spanning three age stages and two genotypes. Briefly, following 3–5 days of continuous EdU labeling, we isolated nuclei from the whole brain of 38 sex-balanced C57BL/6 mice (Figure 1A; Table S1A), including 33 wild-type mice across multiple development stages (young: 6–9 weeks; adult: 11–20 weeks; and aged: 88–98 weeks), as well as five 5xFAD mutant mice (11–20 weeks) harboring multiple Alzheimer’s disease (AD) mutations.19 Following the TrackerSci protocol, we obtained transcriptomic profiles for 5,715 newborn cells (median 2,909 UMIs [unique molecular identifier]) (Figures S1J and S1K) and chromatin accessibility profiles for 8,974 newborn cells (median 50,225 unique reads) (Figures S1L and S1M). In addition, we included DAPI singlets representing “all” brain cells (i.e., without enrichment of the EdU+ cells) as a background control and obtained transcriptomic profiles for 8,380 nuclei (median 1,553 UMIs) and chromatin accessibility profiles for 342 nuclei (median 24,521 unique reads). The EdU+ nuclei and DAPI singlets were collected from the same set of samples and processed in parallel to minimize any batch effect.
We first subjected the 14,095 TrackerSci transcriptome profiles, including both EdU+ nuclei and DAPI singlets, to Louvain clustering20 and UMAP (uniform manifold approximation and projection) visualization21 (Figures 1B and S1N). Sixteen cell clusters were identified and annotated based on established markers (Figure 1C; Table S1B), ranging in size from 25 cells (choroid plexus epithelial cells) to 3,141 cells (mature neurons). We next performed clustering analysis of 9,316 TrackerSci chromatin accessibility profiles (8,974 EdU+ nuclei and 342 DAPI singlets) and identified 14 clusters (Figures 1B and S1O; STAR Methods), which mapped 1:1 to the main cell types identified in the transcriptome analysis. Two rare cell types (i.e., ependymal cells and choroid plexus epithelial cells) were only detected in the RNA dataset, mainly because of their low abundance. The corresponding cell types defined by the two molecular layers overlapped well, unveiling detailed trajectories of neurogenesis and oligodendrogenesis (Figures 1D and 1E).
We observed a significantly altered distribution of cell-type-specific fractions between all brain cells and the EdU+ cells (Figure 2A; p value < 2.2 × 10–26, chi-squared test). For example, in contrast to all brain cells that are dominated by mature neurons (e.g., cerebellum granule neurons: 32.7% in DAPI singlets vs. 2.85% in EdU+ cells) and differentiated glial cells (e.g., myelin-forming oligodendrocytes: 11.9% in DAPI singlets vs. 0.75% in EdU+ cells), the EdU+ population showed prominent enrichment of progenitor cells such as immature neurons (e.g., olfactory bulb neuroblasts: 0.14% in DAPI singlets vs. 13.4% in EdU+ cells) and glia progenitors (e.g., oligodendrocyte progenitor cells [OPCs]: 1.11% in DAPI singlets vs. 45.4% in EdU+ cells). Of note, the cell-type-specific distribution of newborn cells was highly correlated between TrackerSci transcriptome and chromatin accessibility datasets (Pearson’s correlation r = 0.9987, p value < 2.2 × 10–26; Figure 2B) and across conditions (Figure S1P).
Figure 2. TrackerSci captures rare newborn cells that are less represented in conventional single-cell studies.
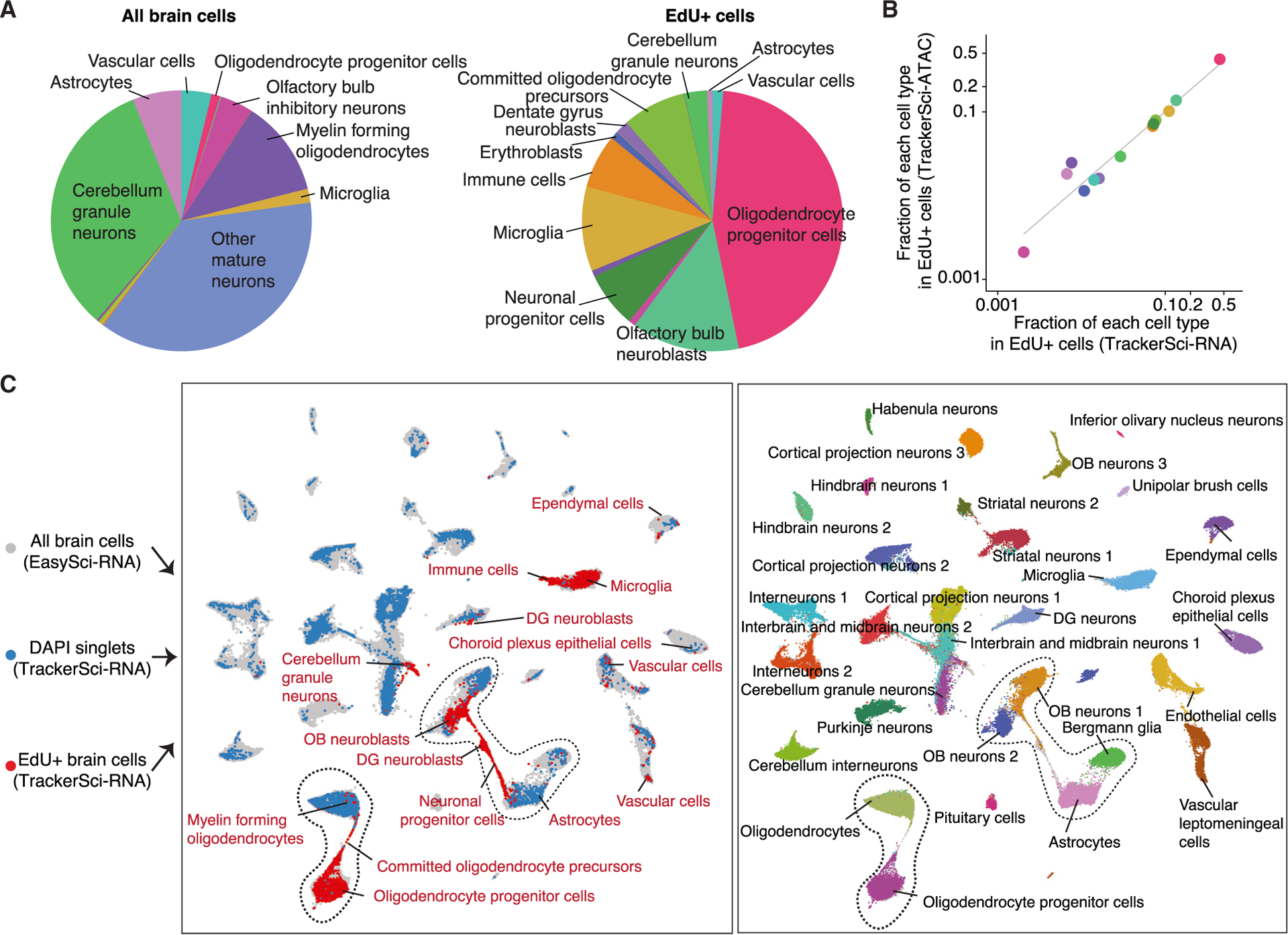
(A) Pie plots showing the proportion of main cell types identified in the global cell population (left) and the enriched EdU+ cell population (right) from single-cell transcriptome data.
(B) Scatterplot showing the fraction of each cell type in the enriched EdU+ cell population by single-cell transcriptome (x axis) or chromatin accessibility analysis (y-axis) in TrackerSci, together with a linear regression line.
(C) We integrated the TrackerSci dataset, including both EdU+ cells and DAPI singlets, with a large-scale brain cell atlas.15 The UMAP plots show the integrated cells, colored by assay types (left, cell types from TrackerSci are annotated) or cell annotations from the brain cell atlas (right, cells from TrackerSci are colored in gray).
We next integrated TrackerSci datasets with a global brain cell atlas from our companion study,15 for which we profiled 1.5 million cells from entire mouse brains spanning three age groups and two mutants associated with AD. This integration included EdU+ brain cells (5,715 single-cell transcriptomes), all brain cells (8,380 DAPI singlets), and all brain cells from the global atlas (5,000 cells per main cell type) in the same UMAP space. As expected, all brain cells from the TrackerSci highly overlapped with cells from the global brain cell atlas in the integrated UMAP space (Figure 2C). Remarkably, the EdU+ cells (from TrackerSci) formed continuous cellular differentiation trajectories bridging several terminally differentiated cell types, including the oligodendrogenesis trajectory from the OPCs to differentiated oligodendrocytes and the neurogenesis trajectory connecting astrocytes and olfactory bulb (OB) neurons (Figure 2C). Notably, the bridge cells are validated by the expression of known progenitor markers, such as Bmp4 and Enpp6 for committed oligodendrocyte precursors (COPs)22,23 and Mki67 and Egfr for neuronal progenitor cells24 (Figure S1Q). These “bridge” cells were absent in the original analysis (Figure S1R), highlighting the application of TrackerSci in recovering continuous cellular differentiation trajectories in adult tissues.
Identifying cell-type-specific epigenetic signatures and TF regulators of newborn cells
We next performed differential expression (DE) and differential accessibility (DA) analysis, yielding 5,610 DE genes (false discover rate [FDR] of 5%, Figure 3A; Table S2A; STAR Methods) and 68,556 DA sites (FDR of 5%, Table S2B; STAR Methods) with significant changes across cell types. Notably, 1,744 (31.1%) of DE genes have DA promoters enriched in the same cell type (median Pearson r = 0.81, Figure 3A). While canonical gene markers were observed and used for the annotation (Figure S2A), we detected many less-characterized markers that are highly cell-type specific but have not been reported in prior research, including markers for neuronal progenitor cells (e.g., Adgrv1 and Rmi2) and dentate gyrus neuroblasts (DGNBs; e.g., Prdm8 and Marchf4) (Figure S2A). Notably, common neurogenesis markers like Sox2 and Dcx were identified in various cell types (e.g., OPCs; Figure S2B), potentially impacting their reliability for cell labeling in neurogenesis.25 In contrast, we identified two putative markers for neuronal progenitor cells, Tead2 and Esco2, which displayed high cell-type-specific expression in the same cell type in a published dataset26 (Figures S2B and S2C) and were specifically enriched in the neurogenesis subventricular zone, as evidenced by their overlap with the expression of Ascl1 and Mki6727 (Figure S2D).
Figure 3. Identifying epigenetic elements and TFs associated with heterogeneous cellular states of newborn cells in the mouse brain.
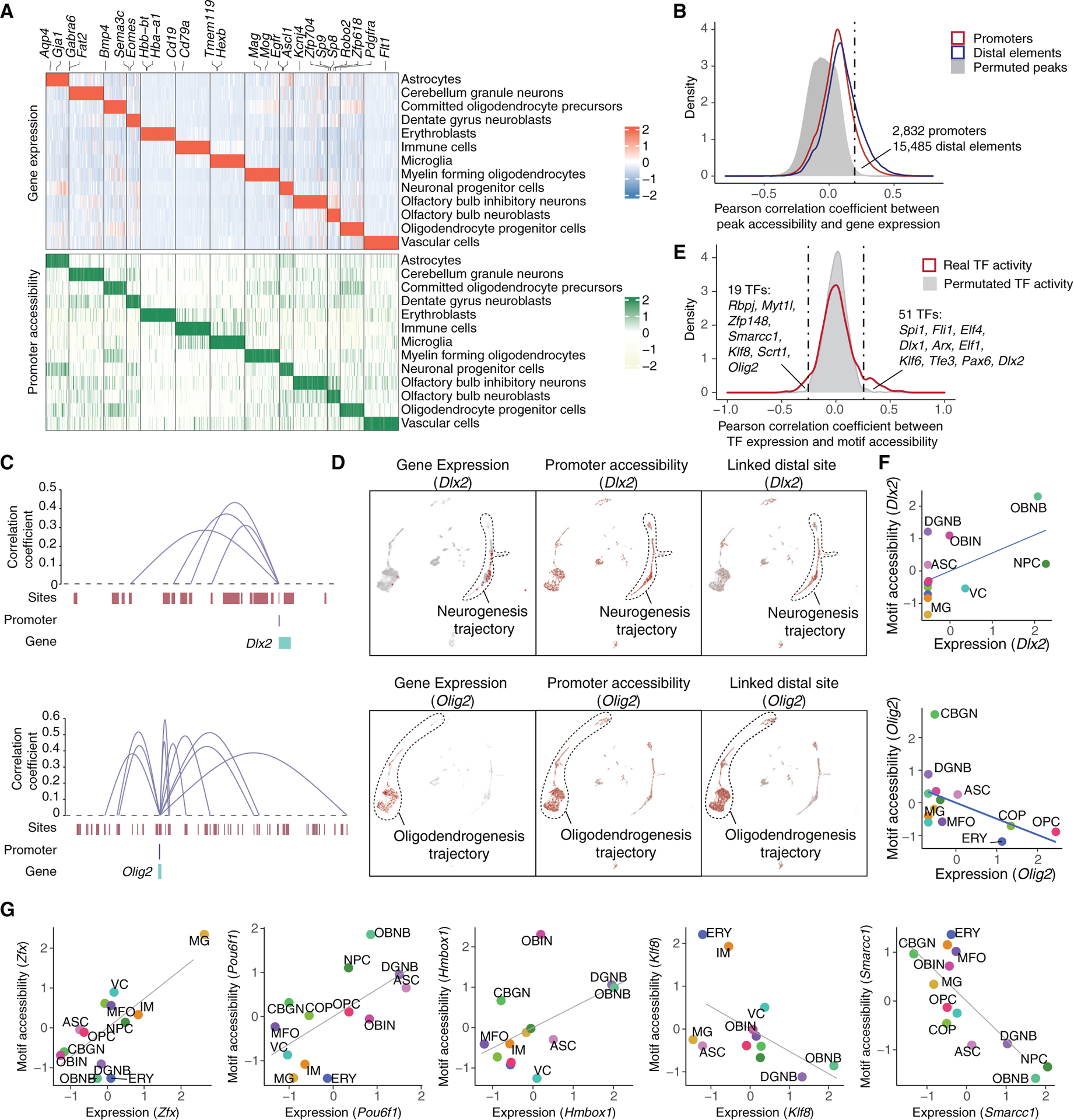
(A) Heatmap showing the relative expression (top) and chromatin accessibility (bottom) of cell-type-specific genes across cell types. Each row represents the aggregated gene expression or promoter accessibility for a specific cell type. All conditions are included into the calculation.
(B) Density plot showing the distribution of Pearson correlation coefficients between gene expression and the accessibility of promoter (red) or nearby accessible elements (±500 kb of the promoter, blue) across pseudo-cells. Background distribution by permuting pseudo-cells is colored in gray.
(C) Genome browser plot showing links between distal regulatory sites and genes for a neurogenesis marker (Dlx2, top) and an oligodendrogenesis marker (Olig2, bottom).
(D) UMAP plots showing the cell-type-specific expression (left), the accessibility of promoter (middle), and linked distal site (right) for genes Dlx2 (top) and Olig2 (bottom).
(E) Density plot showing the distribution of Pearson correlation coefficients between TF expression and their motif accessibility across pseudo-cells. Background distribution by permuting pseudo-cells is colored in gray.
(F) Scatterplots showing the correlation between the scaled gene expression and motif accessibility across cell types for Dlx2 (top) and Olig2 (bottom), together with a linear regression line. ASC, astrocyte; CBGN, cerebellum granule neuron; COP, committed oligodendrocyte precursor; DGNB, dentate gyrus neuroblast; ERY, erythroblast; MFO, myelin-forming oligodendrocyte; MG, microglia; NPC, neuronal progenitor cell; OBNB, olfactory bulb neuroblast; OBIN, olfactory bulb inhibitory neuron; OPC, oligodendrocyte progenitor cell; VC, vascular cell.
(G) Scatterplots showing the correlation between the scaled gene expression and motif accessibility of less-characterized TF regulators, together with a linear regression line.
To investigate the epigenetic landscape that shapes the transcriptome of newborn cells, we next sought to identify the cis-regulatory elements underlying the cell-type-specific expression of gene markers. We first computed the correlation between the expression of each gene and the accessibility of its nearby sites across 88 “pseudo-cells” (a subset of cells with adjacent integrative UMAP coordinates grouped by k-means clustering, Figures S3A–S3D; STAR Methods). To control for any potential artifacts of the analysis, we permuted the sample IDs of the data matrix followed by the same analysis pipeline. Altogether, we identified 15,485 positive links between genes and distal sites (plus 2,832 associations between genes and promoters) at an empirically defined significance threshold of FDR = 0.05 and based on their cell-type specificity (Figure 3B; Table S3A; STAR Methods).
The identified distal site-gene linkages were significantly closer than all possible pairs tested (median 159 kb for identified links vs. 251 kb for all pairs tested; p value < 5 × 10–5, unpaired permutation test based on 20,000 simulations, Figure S3E). Most genes were associated with a few links (median two distal sites per gene, out of a median of 94 distal sites within 500 kb of the transcription start site [TSS] tested, Figure S3E). For example, Dlx2, a canonical neurogenesis marker,28 was significantly linked to four distal peaks, all exhibiting remarkable cell-type specificity similar to its gene expression (Figures 3C, 3D, and S3F). By contrast, a small subset of genes (3.5%) were linked to many peaks (≥ 10 peaks). For instance, Olig2 was linked to 10 distal peaks (Figure 3C), all highly enriched in the OPCs and COPs (Figures 3D and S3G). Some genes (e.g., Dlx2) showed strong cell-type specificity in their linked distal sites compared to their promoters (Figure S3H, 8.8-fold vs. 3.2-fold enrichment), indicating the impact of long-range transcriptional regulation in shaping cell-type specificities.
We next computed the Pearson correlation coefficient between transcription factor (TF) expression and motif accessibility across all previously described pseudo-cells. The same analysis was performed with the permuted data as control. At an empirically defined significance threshold of FDR = 0.05, 70 cell-type-specific TF regulators were found (Figure 3E; Table S3B), including 19 potential repressors featured with negative correlations between gene expression and motif accessibility (e.g., Olig2, Figure 3F). Most cell-type-specific TFs are readily validated by previous studies.29–34 Notably, several less-characterized TFs were identified by the cell-type-specific enrichment of both gene expression and motif accessibility, such as Pou6f1 and Hmbox1 enriched in immature neurons and Zfx enriched in microglia, representing potential regulators of progenitor cells in the adult brain (Figures 3G and S3I).
A global view of cell-type-specific proliferation rates across the adult lifespan
We next compared the fraction of EdU+ cells across young, adult, and aged mice brains and observed a strongly decreased proliferation with age (Figure 4A). To investigate the cell-type-specific changes in proliferation rates, we quantified the relative fractions of each newborn cell type by their fractions in the EdU+ cell population, multiplied by the ratio of EdU+ cells in the global cell population. Interestingly, we detected highly heterogeneous responses to aging across various progenitor cell types, confirmed by single-cell transcriptome and chromatin accessibility profiles (Figure 4B). For example, dentate gyrus neuroblasts showed an 18-fold reduction in the aged brain (vs. the adult brain), while vascular cell proliferation was only mildly affected. In contrast, microglia and other immune cells showed a remarkable boost in producing newborn cells (Figures 4B–4D), possibly due to the elevated inflammatory signaling in the aged brain.35 Compared with the aged brain, we detected overall mild changes in cellular proliferation (except the microglia) in the AD-associated mouse model (5xFAD), probably because the mutant mice were profiled at a relatively early stage (before 3 months).
Figure 4. Deciphering the impact of aging on the proliferation status and differentiation dynamics of different cell types in the mammalian brain.
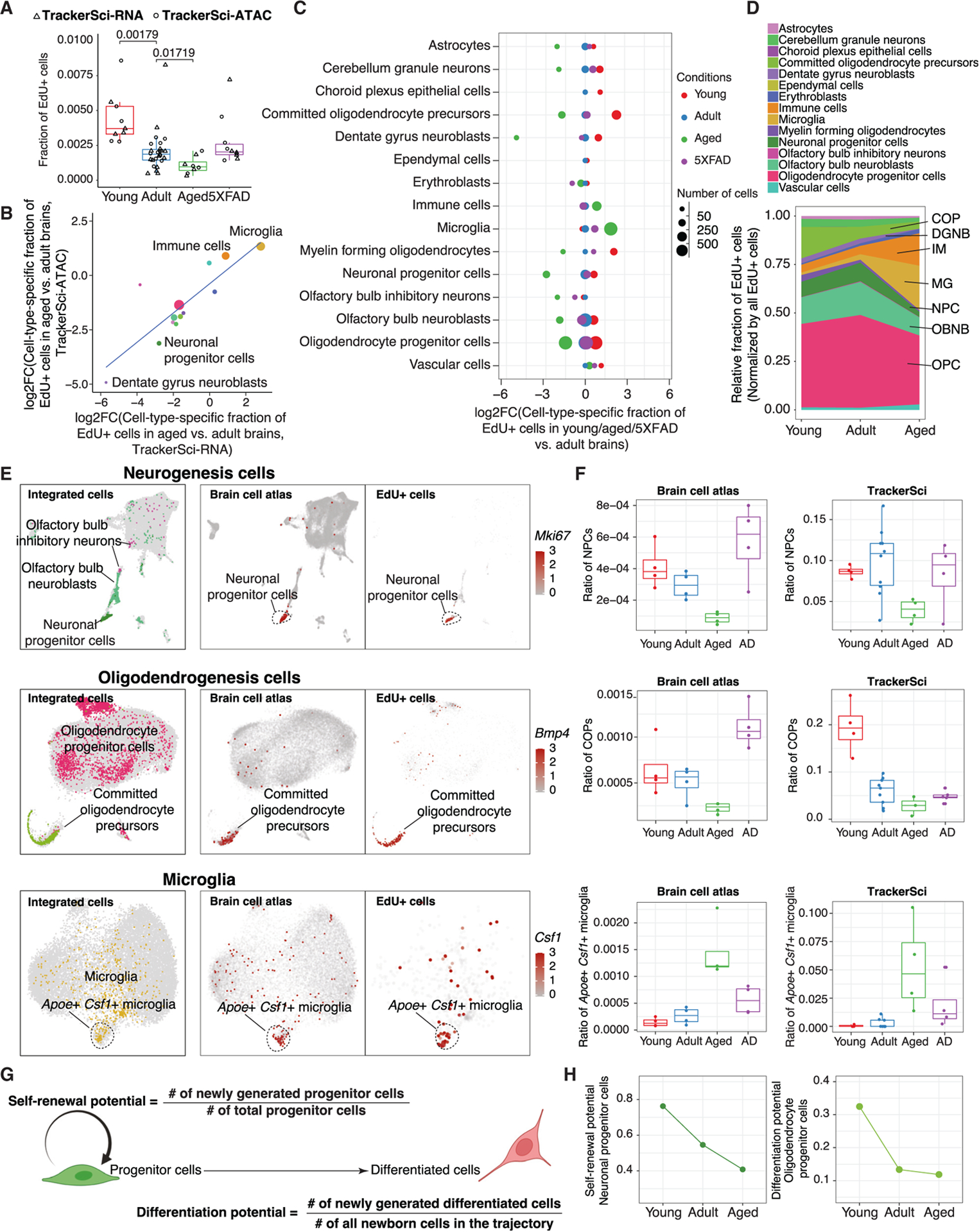
(A) Boxplot showing the fraction of EdU+ cells in the mouse brain after 5 days of EdU labeling from both single-cell transcriptome and chromatin accessibility experiments. Numbers represent the p values using the Wilcoxon rank-sum test.
(B) With the single-cell RNA (scRNA)-seq or ATAC-seq data of TrackerSci, we first calculated the cell-type-specific fractions among EdU+ cells and multiplied them by the EdU+ ratio from FACS for both aged and adult brains. We then quantified the fold changes of the normalized cell-type-specific fractions. The scatterplot shows logFC correlation between scRNA and scATAC analysis.
(C) Similar to the analysis in (B), the dot plot shows the log-transformed cell-type-specific fold changes between each condition and adult.
(D) Area plot showing the cell-type-specific proportions in EdU+ cells over time.
(E) UMAP showing integrated cells corresponding to OB neurogenesis (top), oligodendrogenesis (middle), and microglia (bottom) between TrackerSci and brain cell atlas,15 colored by cell type annotations in TrackerSci (left) and the expression of the NPC marker Mki67 (top), the COP marker Bmp4 (middle), and the aging/AD-associated microglia marker Csf1 (bottom).
(F) Boxplots showing the cell-type-specific fractions of NPCs (top), COPs (middle), and aging/AD-associated microglia (bottom) across different conditions in the brain cell atlas (left) or newborn cells from TrackerSci (right).
(G) Schematic showing the calculation of the self-renewal and differentiation potential of progenitor cells.
(H) Left: line plot showing the estimated self-renewal potential of NPCs over time. Right: line plot showing the estimated differentiation potential of OPCs across three age groups.
To further validate the cell-type-specific dynamics in brain aging, we integrated the newborn cells recovered from TrackerSci and a global mice brain cell atlas15 for subclustering analysis. Indeed, the integration facilitated the identification of rare progenitor cells, such as neuronal progenitor cells (NPCs; marked by Mki67, Top2a, and Egfr) and committed oligodendrocyte precursors (marked by Bmp4 and Enpp6) (Figure 4E), both significantly reduced in aging across datasets (Figure 4F). In addition, we identified a reactive microglia subtype (marked by Apoe and Csf1), which has been reported to be enriched in aged and AD mammalian brains.36 Consistent with these studies, the reactive microglia exhibited increased proliferation in both aged and 5xFAD brains when compared to adult brains (p value = 0.0045, 0.028, respectively, Wilcoxon rank-sum test), explaining their rapid expansion in aging and disease (Figure 4F).
We next investigated the impact of aging on the self-renewal and differential potential of progenitor cells. The self-renewal potential was determined by the number of newly generated progenitor cells divided by total progenitor cells in the brain (i.e., the number of new cells generated per progenitor cell in a fixed time, Figure 4G). For instance, the NPCs exhibited downregulated self-renewal potential over age (Figure 4H), which readily explained the depleted neural stem cell pool in the aged brain. Meanwhile, the differentiation potential of a cell type can be defined by the fraction of newly generated differentiated cells divided by all newborn cells in the same lineage (Figure 4G). For example, we observed a substantially reduced differentiation potential in OPCs across the adult lifespan, especially during early growth (Figure 4H). This demonstrates an application of TrackerSci in quantifying cell-type-specific self-renewal and differentiation capacities in vivo.
The impact of aging on adult neurogenesis
We next sought to interrogate the impact of aging on adult neurogenesis and oligodendrogenesis and delineate the underlying transcriptional and epigenetic controls. For adult neurogenesis, we identified three main trajectories that differentiated into DG neuroblasts, OB neuroblasts, and astrocytes, consistent with the cell-state transition directions inferred by the RNA velocity analysis37 and prior report38 (Figure 5A). The “root cells” coincided with activated neuronal stem cells, validated through integrated analysis with a published dataset39 (Figures S4A and S4B). The trajectory was further validated through a pulse-chase experiment, where we harvested cells for TrackerSci profiling at different time points (i.e., 1 day, 3 days, and 9 days post-labeling). Indeed, we observed a gradual accumulation of more differentiated cell states with longer chasing time (Figure 5B). Through DE gene analysis, we identified 2,072 and 6,473 DE genes along the DG neurogenesis and OB neurogenesis trajectories, respectively (Table S4A). Of all DE genes, 1,799 genes were shared between the two trajectories, including upregulated genes (e.g., Dcx) enriched in neuron development40 and downregulated genes (e.g., Notum) enriched in negative Wnt signaling regulation40 (Figure S4E). In addition, putative trajectory- and region-specific neurogenesis programs were identified, such as Neurod1 and Neurod2 enriched in the DG trajectory (Figure S4F), aligning with their known roles.41–43
Figure 5. Characterizing the impact of aging on neurogenesis.
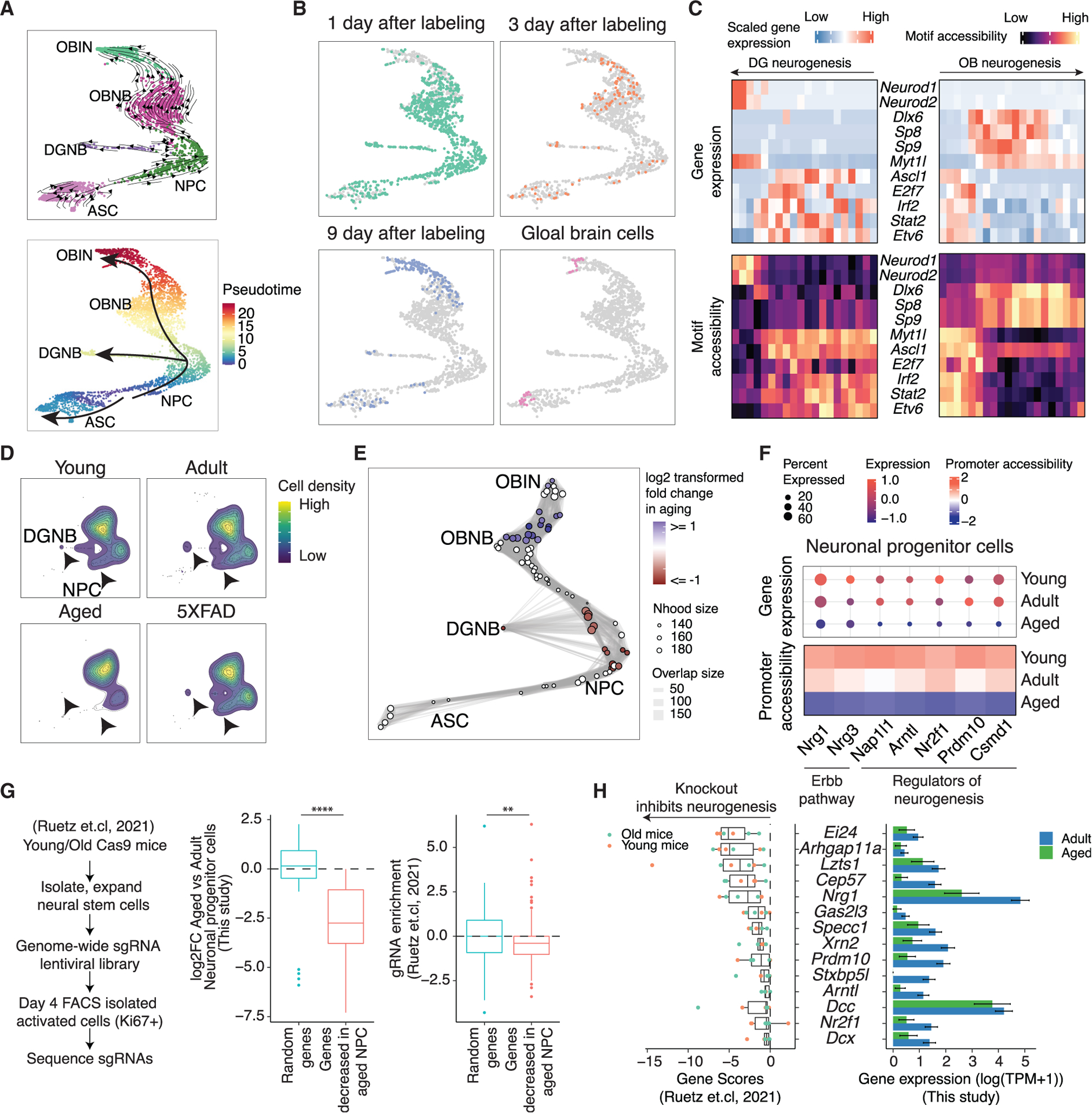
(A) UMAP plots showing the differentiation trajectory of neurogenesis, colored by main cell types (top) or pseudotime (bottom), inferred by RNA velocity analysis (top).
(B) Scatterplots show the distribution of EdU+ cells harvested at different time points after 5-day EdU labeling and cells without EdU+ enrichment along neurogenesis.
(C) Heatmap showing the dynamics of gene expression and motif accessibility of cell-type-specific TFs across the pseudotime of neurogenesis trajectories. Each bin along the x axis represents a collection of cells stratified based on their respective positions along the pseudotime trajectory.
(D) Contour plots showing the distribution of EdU+ cells from TrackerSci-RNA in the neurogenesis trajectory across conditions. The arrows point to the significantly reduced cell states.
(E) A neighborhood graph from Milo differential abundance analysis on the neurogenesis trajectory. Nodes represent cellular neighborhoods from the k-nearest neighbor (KNN) graph. Differential abundance neighborhoods are colored by the log-transformed fold change across ages. Graph edges depict the number of cells shared between neighborhoods.
(F) The dot plots and heatmaps show the scaled gene expression and promoter accessibility of top differentially expressed genes in the NPCs.
(G) Left: summary of the study design used to validate the knockout effects of aging-decreased genes in the NPCs. Right: boxplots showing the expression changes of aging-decreased genes from current study and the gRNA enrichment of these genes compared to randomly selected genes from the knockout study. Stars indicate p values using the Wilcoxon rank-sum test. Left: p = 3.94e−7; right: p = 0.00285.
(H) Left: boxplot showing the top gene candidates that impair neurogenesis after CRISPR knockout from the published study. Right: bar plot showing their decreased expression in NPCs comparing aged to adult in our current study. Error bars represent standard error of mean (SEM).
With the chromatin accessibility profiling, we identified 3,095 and 13,790 sites showing dynamics patterns along the DG and OB neurogenesis trajectories, respectively (Table S4B), from which we further identified 20 TFs exhibiting significantly changed motif accessibility in the DG neurogenesis trajectory and 283 TFs in OB neurogenesis (FDR of 0.05, Table S4C). Key TFs were further validated by strong correlations between their expression and motif accessibility dynamics (Figure 5C). For example, the expression of the neurogenesis regulators Neurod1 and Neurod2 positively correlates with their motif accessibility, while Myt1l, a known repressor of neural differentiation,44 ex hibited a negative correlation. This approach identified TFs shared between two neurogenesis trajectories (e.g., Myt1l and Ascl1) and TFs that regulate the specification of different neuron types (e.g., Dlx6 and Sp8 uniquely enriched in OB neurogenesis45,46). Meanwhile, several TFs (e.g., Irf2 and Stat2) exhibited strong enrichment of gene expression and motif accessibility in NPCs. While their functions in neurogenesis were less characterized, some have been reported as essential regulators of other stem cell types.47–49
To investigate the impact of aging on adult neurogenesis, we next compared the cellular density recovered from TrackerSci transcriptome profiling across different conditions along the neurogenesis trajectory. Consistent with the cell-type level analysis (Figure 4C), we observed a dramatic age-dependent reduction in the cellular density of NPCs and DGNBs, but not in OB neuroblasts (Figure 5D). The finding was consistent with the chromatin accessibility profiles, where we applied a differential abundance testing algorithm, Milo,50 to identify the cellular neighborhoods that are significantly altered upon aging. We identified 14 cellular neighborhoods that exhibited a differential decrease (Figure 5E; 5% FDR, log2FC < 0), predominantly originating from NPCs. This analysis further validated that aging affects neurogenesis by downregulating the proliferation rate of its progenitor cells.
To further decipher the molecular mechanisms underlying the age-dependent changes in NPCs, we then performed differential gene expression analysis across young, adult, and aged conditions, yielding 30 genes showing concordant changes over time, supported by both gene expression and the accessibility of promoters or linked distal sites (Figure 5F; Table S5; STAR Methods). For example, two neurotrophic factors involved in the Erbb pathway, Nrg1 and Nrg3, exhibited strongly reduced expression and promoter accessibility upon aging. Indeed, Nrg1 has been reported to increase neurogenesis upon in vivo administration.51 In addition, we identified several other known regulators of neurogenesis, such as Nr2f1 and Nap1l1,52,53 that were significantly downregulated upon aging, which serve as potential targets for restoring neurogenesis in aged brains.
For validation, we utilized a recently published dataset54 (Figure 5G, left) that employed a genome-wide CRISPR screen to systematically dissect the roles of various genes in neurogenesis by quantifying the enrichment of gene-specific single-guide RNAs (sgRNAs) within proliferation-active (Ki67+) primary neural stem cells in vitro. We examined the downregulated genes in aged NPCs (Figure 5G, middle) and detected a significant reduction in their sgRNA enrichment compared to a set of randomly chosen genes (Figure 5G, right). This observation suggests that the knockout of these genes impedes neural stem cell proliferation, aligning with our observation of decreased NPC proliferation in aged mice. Top candidate drivers were identified based on the negative ranking of gene scores, including genes related to cell proliferation (e.g., Ei24, Arhgap11a, and Cep5755–58) and aforementioned neurogenesis regulators, such as Prdm10, Nrg1, and Nr2f1.53,59–62 Notably, these candidate genes were validated in CRISPR screens using NPCs derived from young and aged mouse brains (Figure 5H).
Leveraging the TrackerSci dataset, we identified a significant correlation between the expression changes of Terf2, a critical telomere protector, and cellular proliferation rates across various cell types in aged vs. adult mice (Pearson correlation r = 0.71, p value = 0.047, Figure S4G). To functionally examine whether perturbing telomere-maintenance-related pathways influence neurogenesis, we treated 8-week-old mice with a known telomerase inhibitor, azidothymidine (AZT),63 followed by TrackerSci analysis (Figure S4H). We sequenced and annotated a total of 17,916 EdU+ cells (9,902 treated, 8,014 control) and 3,222 DAPI singlets (1,709 treated and 1,513 control) based on established cell markers (Figures S4I and S4J). The augmented recovery of EdU+ cells from young brains facilitated the identification of rare proliferating cell types previously overlooked, such as arachnoid barrier cells located within the blood- cerebrospinal fluid (CSF) barrier64 and pituitary stem cells from the pituitary gland65 (Figure S5). Notably, the telomerase inhibitor resulted in enriched intermediate neuroblasts, associated with reduced fraction of early progenitor cells and late-stage differentiated cells along the neurogenesis trajectory (Figures S4K and S4L), consistent with previous studies54,66,67 (Figure S4M). This analysis demonstrates the potential of TrackerSci to aid in characterizing cell proliferation and differentiation dynamics upon in vivo perturbations.
The impact of aging on adult oligodendrogenesis
We next in silico isolated cell types that span multiple stages of oligodendrogenesis for pseudotime analysis, yielding a simple trajectory defined by integrated transcriptome and chromatin accessibility profiles (Figure 6A). The oligodendrogenesis trajectory was further validated by the RNA velocity analysis and the time-dependent labeling experiment mentioned above (Figure 6B). Through DE and DA analysis, we identified 8,443 DE genes and 15,164 DA sites that were significantly changed along the trajectory (5% FDR, Tables S4A and S4B). This analysis identified known oligodendrogenesis regulators (e.g., Zfp27668 and Myrf68,69), associated pathways (e.g., cholesterol biosynthesis70), and less-studied gene markers (e.g., Snx10 and Rfbox2) along the trajectory of oligodendrogenesis (Figure S6A).
Figure 6. Characterizing the impact of aging on oligodendrogenesis.
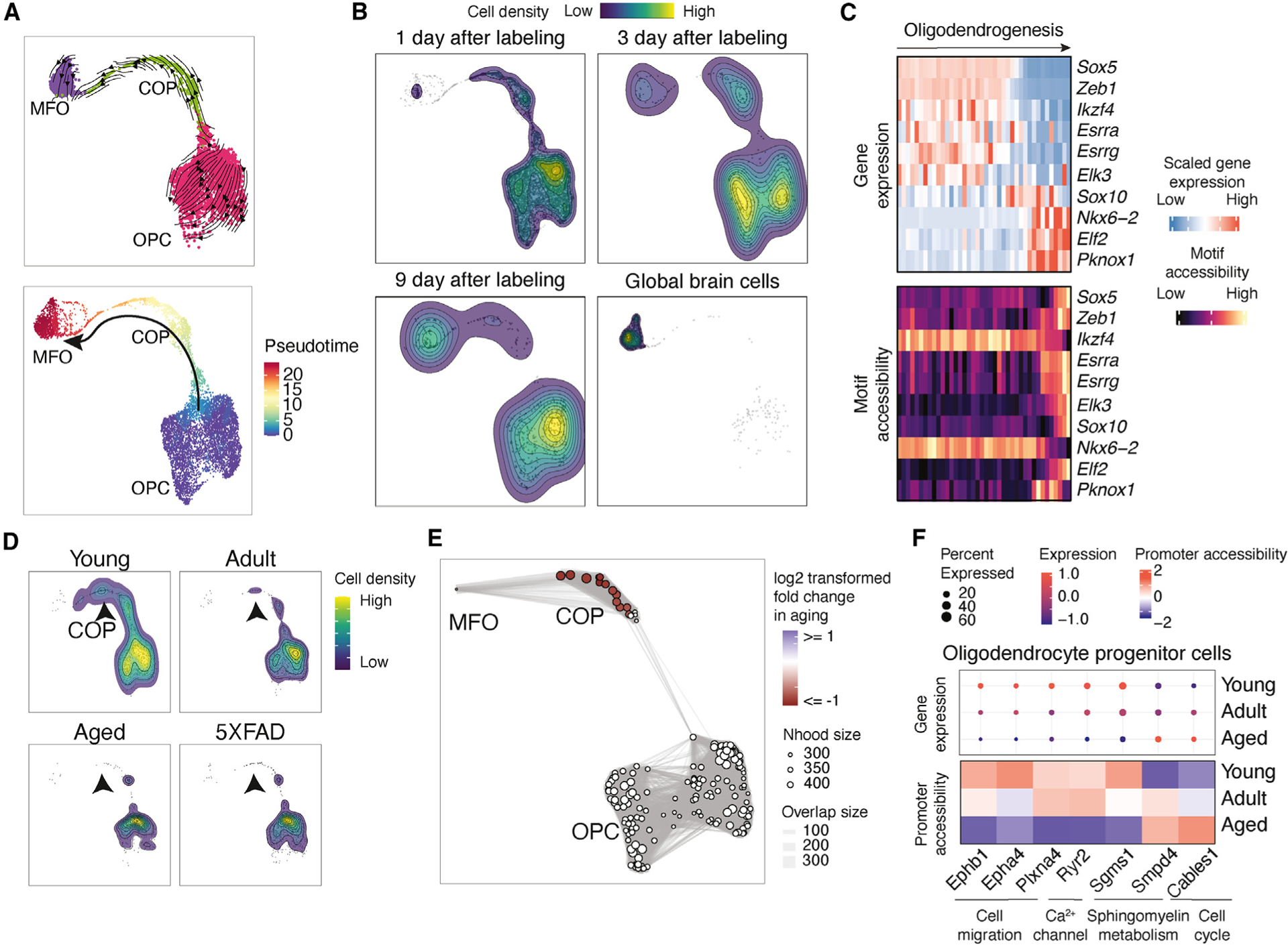
(A) UMAP plots showing the differentiation trajectory of oligodendrogenesis, colored by main cell types (top) or pseudotime (bottom), inferred by RNA velocity analysis (top).
(B) Contour plots show the distribution of EdU+ cells harvested at different time points after 5-day EdU labeling and cells without enrichment of EdU+ cells along oligodendrogenesis.
(C) Heatmap showing the dynamics of gene expression and motif accessibility of cell-type-specific TFs across the pseudotime of the oligodendrogenesis trajectory.
(D) Contour plots showing the distribution of EdU+ cells from TrackerSci-RNA in the oligodendrogenesis trajectory across conditions. The arrows point to the significantly reduced cell states.
(E) A neighborhood graph from Milo differential abundance analysis on the oligodendrogenesis trajectory. Nodes represent cellular neighborhoods from the KNN graph. Differential abundance neighborhoods are colored by the log-transformed fold change across ages. Graph edges depict the number of cells shared between neighborhoods.
(F) The dot plots and heatmaps show the scaled gene expression and promoter accessibility of top differentially expressed genes in the OPCs.
Moreover, we identified 97 TFs that exhibited highly correlated gene expression and motif accessibility in oligodendrogenesis (FDR of 5%, Table S4C), including known regulators of oligodendrocyte differentiation, such as Sox5, Sox10, Pknox1, and Nkx6-2.71,72 In addition, several less-characterized TF markers were recovered, including Ikzf4, a known regulator of Müller glia differentiation in the retina,73 and several potential transcriptional “repressors” (e.g., Esrra, Esrrg, and Elk3) with negative correlation between their expression and motif accessibility along the trajectory of oligodendrogenesis (Figure 6C).
We further investigated the impact of aging on adult oligodendrogenesis by examining cellular density along the cellular differentiation trajectory across different conditions. Unlike adult neurogenesis, we observed a remarkable reduction in COPs rather than the early progenitor cells in single-cell transcriptome analysis (Figure 6D). The result is further validated through the Milo50 analysis of chromatin accessibility profiles, where significantly decreased cellular neighborhoods exclusively overlapped with the COPs (Figure 6E, 5% FDR). This observation is in accordance with the aging-associated depletion of newly formed oligodendrocytes in our companion study15 and previous reports.74
To delineate the mechanism contributing to reduced oligodendrogenesis upon aging, we examined the significantly dysregulated genes in OPCs and identified 242 DE genes (FDR of 10%, Table S5). Many top DE genes are cross validated by gene expression and promoter accessibility (Figure 6F) and were involved in molecular processes critical for oligodendrocyte differentiations, such as cell cycle (e.g., Cables175) or cell migration pathway (e.g., Ephb1, Epha4, and Plxna4)76,77 (Figure 6F). Intriguingly, two sphingomyelin metabolism-related genes exhibited opposite dynamics between young and aged OPCs: Sgms1, a gene encoding a sphingomyelin synthase critical for sphingomyelin production,78,79 was substantially downregulated in the aged OPCs. By contrast, Smpd4, encoding a sphingomyelin phosphodiesterase that catalyzes the reverse reaction,80 was significantly upregulated in OPCs upon aging (Figure 6F). The age-dependent changes of both Sgms1 and Smpd4 could lead to ceramide accumulation and depletion of sphingomyelin in OPCs, which could increase cellular susceptibility to senescence and cell death.81–83 Furthermore, the downregulated differentiation of oligodendrocytes is associated with dysregulated immune responses during aging, such as the accelerated proliferation of the reaction microglia subtype (Figure 4F) and an increased C4b expression in OPCs from both the EdU+ population and the global pool (Figure S6B). Further investigation could be critical for deciphering the regulatory links between the elevated inflammation signaling and the dysregulated oligodendrocyte differentiation in the aged brain.
TrackerSci facilitates the identification of rare progenitor cells in the aged human brain
We next sought to investigate whether the TrackerSci dataset can be applied to facilitate the identification of rare progenitor cell types in the aged human brain. We conducted single-nucleus RNA-seq (snRNA-seq) on 29 human brain samples from six individuals ranging from 70 to 94 in age at death (Table S6). Up to five regions (cerebellum, hippocampus, inferior parietal, motor cortex, and superior and middle temporal lobe [SMTG]) for each individual were included to characterize the region-specific effect of cellular dynamics.
After removing low-signal cells and potential doublets, we recovered gene expression profiles in 798,434 cells for downstream analysis (a median of 23,504 nuclei per brain sample, with a median of 1,013 UMIs per nucleus, Figures S7A and S7B). Because of the rarity of proliferating cells in the aged human brain, it was challenging to recover cycling or differentiating cells in the initial unsupervised clustering analysis (Figure S7C). We next integrated the TrackerSci dataset (including 5,715 EdU+ mouse brain cells and 8,380 mouse brain cells without EdU enrichment) with the human brain dataset, followed by UMAP visualization (Figure 7A, left). Despite the species differences, the integration identified extremely rare proliferating cell populations in the aged human brain (Figures 7A, right, S7D, and S7E). For example, we identified a rare human cycling cell population overlapping with cycling progenitor cells from mice (Figure 7A). Further subclustering analysis separated the population into cycling microglia (569 cells, 0.07% of the total), cycling OPCs (56 cells, 0.007% of the total), and cycling erythroblasts (51 cells, 0.006% of the total) (Figures 7B and 7C). Interestingly, while the cycling microglia expressed typical cell-cycle-related genes and exhibited a similar ratio to the non-cycling microglia across regions (Figure S7H), we identified gene expression signatures unique to each region, suggesting a local control of microglia proliferation (Figure S7I). The proliferating microglia in the human brain were further validated by immunostaining (Figure S7J).
Figure 7. TrackerSci facilitates the identification of proliferating and differentiating cells in the human brain.
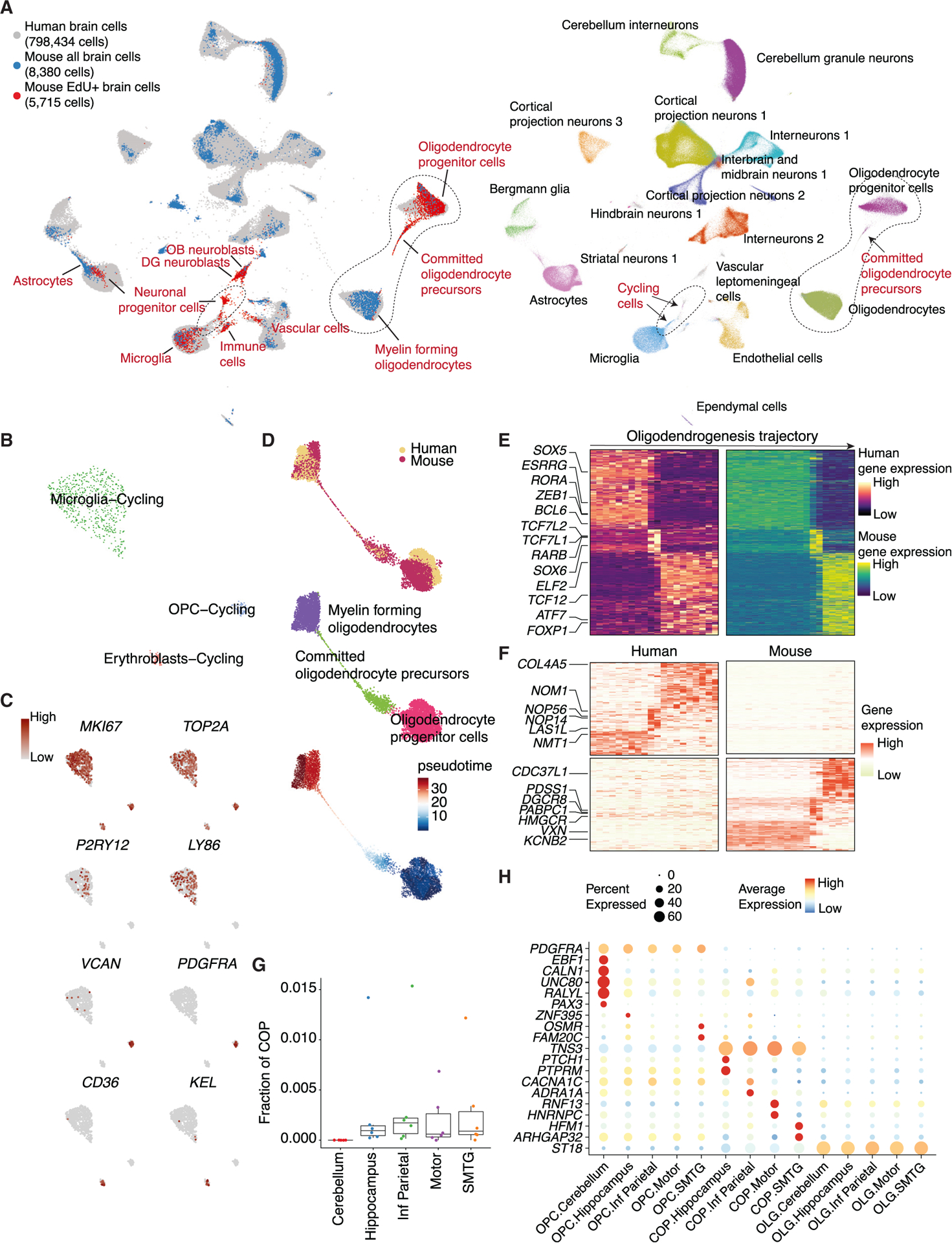
(A) UMAP plots showing integrated cells between TrackerSci and the human brain dataset, colored by assay types (left, cell types from TrackerSci are annotated) or cell annotations from the human brain dataset (right, cells from TrackerSci in gray).
(B) UMAP plots showing the subclustering analysis of cycling cells from the human dataset, colored by cell annotation.
(C) UMAP plots same as (B), colored by the expression of markers for proliferation (MKI67 and TOP2A), microglia (P2RY12 and LY86), OPCs (VCAN and PDGFRA), and erythroblasts (CD36 and KEL).
(D) UMAP plots showing the integrated trajectory of oligodendrogenesis-related cells between TrackerSci and the human dataset, colored by species (top), cell type annotations (middle), and pseudotime (bottom).
(E) Heatmaps showing conserved gene expression dynamics along the oligodendrogenesis trajectory for human (left) and mouse (right), with key TF regulators annotated on the left.
(F) Heatmaps showing divergent gene expression dynamics along the oligodendrogenesis trajectory enriched only in human (top) and mouse (bottom), with key genes annotated on the left.
(G) Boxplot showing the fraction of COPs among oligodendrogenesis-related cells across different brain regions in each sample.
(H) Dot plot showing examples of commonly changed and region-specific gene expression signatures across three differentiation stages along oligodendrogenesis.
Furthermore, integration analysis with the TrackerSci dataset facilitates the recovery of a stereotypical cell differentiation trajectory. For example, 188 COPs were identified in the aged human brain (0.02% of the total cell population), connecting the OPCs to mature oligodendrocytes (Figure 7A). To decipher the conserved gene dynamics underlying oligodendrogenesis across species, we integrated oligodendrogenesis-related cells from both datasets and detected a smooth cell transition trajectory from progenitors to differentiated cell state (Figure 7D). We identified 5,680 genes that significantly changed along the human oligodendrogenesis trajectory (FDR of 5%), out of which 1,162 genes (48 TFs) were shared between human and mouse (Figure 7E). While most of the conserved TFs have been previously reported as key regulators of oligodendrocyte differentiation (e.g., TCF7L1 and TCF7L284), several TFs (e.g., ZEB1 and ESRRG) have not been well characterized. Some less-characterized TFs were also nominated in previous motif analysis (Figure 6C). We also identified gene signatures contributing to interspecies differences in oligodendrogenesis (Figure 7F). For example, the human-specific genes are enriched in ribosome biogenesis (e.g., NOM1 and NOP56), while mouse-specific genes are involved in multiple pathways such as primary miRNA processing (e.g., DGCR8) and mRNA 3′ end processing (e.g., PABPN1).
We next investigated the differences in oligodendrogenesis across brain regions. Interestingly, we observed a depletion of the COPs in all cerebellum samples compared with other brain regions (Figures 7G and S7K; p value = 0.001, Fisher’s exact test), suggesting reduced oligodendrogenesis in the cerebellum. To delve into the detailed molecular programs, we performed DE analysis across regions and identified 45, 32, and 25 region-specific DE genes in OPCs, COPs, and oligodendrocytes, respectively (Figure 7H). Strikingly, 40 out of the 45 region-associated genes of OPCs (e.g., EBF1, PAX3,85 and CALN1) were highly enriched in the cerebellum (Figure 7H), indicating a unique molecular state of OPCs in the cerebellum compared with other regions. This is reinforced by the cerebellum’s higher OPC fraction and reduced mature oligodendrocytes compared with other regions (Figure S7L).
Very few neurogenesis cells were detected in the aged human brains by integrating with the TrackerSci dataset. As a further investigation, we integrated the TrackerSci dataset, our aged human brain dataset, with another published adult human brain dataset.86 Notably, a rare cell population (n = 388) from the published dataset demonstrated significant overlap with immature neurons from the TrackerSci dataset (Figure S8A) and was almost exclusively identified in hippocampus-derived samples (n = 357, 92%) with enrichment of gene markers associated with adult neurogenesis (e.g., SOX4 and SOX1187,88) (Figure S8B). This observation aligns with recent studies89 that suggest ongoing neurogenesis in the adult human hippocampus and reinforces the capacity of our mouse dataset in facilitating the detection of such rare immature populations in humans.
To further showcase the potential of the TrackerSci dataset, we integrated data from our TrackerSci murine dataset, our human brain atlas (a subsample of 5,000 cells per cell type), and a published snRNA-seq dataset90 that examines both AD brains and control brains not affected by dementia. Similar cell types across all three datasets demonstrated a remarkable overlap (Figure S8C). Notably, this integration allowed us to identify rare cell clusters indicative of committed oligodendrocyte precursors (17 cells, marked by GPR1791 and SIRT292; Figure S8D) and cycling microglia (176 cells, marked by MKI67 CSF1R; Figure S8E). Subsequent examination of cycling microglia revealed an over 4.7-fold increase in the occipital cortex (p value = 0.053, Wilcoxon rank-sum test) and an 8.5-fold increase in the occipitotemporal cortex (p value = 0.071, Wilcoxon rank-sum test), two regions examined in AD patients (Figures S8F and S8G). This observation aligns with our mouse data (Figure 4C) and the previous report linking AD with neuroinflammation,93 further demonstrating that our TrackerSci mouse dataset could facilitate the detection of proliferative populations in both normal and pathological brain samples.
DISCUSSION
Technological advancements in genomics have facilitated the exploration of gene expression and epigenetic landscape at a single-cell level.16,26,64,94–101 Nevertheless, analysis of single cells from adult or aged brains predominantly focuses on the most common cell types (e.g., differentiated neurons or glia), thus unable to precisely characterize the dynamics of rare progenitor cell types in aged tissues because of restrictions in throughput and resolution. Although prior research established molecular profiles of these rare progenitor cells by using distinct enrichment strategies,102 such as reporter mice for labeling specific progenitor cell types103–105 or the physical dissection of the stem cell niche in conjunction with single-cell sequencing,106–108 these methodologies predominantly target micro-dissected regions or depend on the availability of known cellular markers. Hence, they fail to identify lesser-known progenitor cells in non-canonical regions or to quantify cellular proliferation dynamics across the entire organ.
To address these constraints, here we described TrackerSci, a single-cell genomic sequencing strategy designed to quantitatively depict the temporal dynamics of progenitor cells in vivo. Compared with prior studies,7 TrackerSci can be effectively scaled up to profiling millions of cells using three levels of molecular barcoding14,15 and is able to monitor transcriptional and epigenetic dynamics of proliferating cells, providing additional insights into gene regulatory controls. We used TrackerSci to examine single-cell transcriptome and chromatin accessibility dynamics across 14,689 newborn cells from entire mouse brains, covering three age stages and two genotypes. Given the scarcity of progenitor cells, especially in aged brains, it requires deep sequencing of up to 15 million brain cells to recover the same amount of progenitor cells by conventional single-cell techniques. Through a “global view” of proliferating progenitor cells, TrackerSci enabled us to identify several previously overlooked proliferating cell types, such as vascular cells, arachnoid barrier cells, and pituitary stem cells. We have also identified molecular signatures of various newborn cell types, uncovering markers that show greater specificity over traditional markers for these cells. For example, our analysis pinpointed two neurogenesis markers, Tead2 and Esco2, that exhibited higher cell-type specificity than well-known neurogenesis markers (e.g., Sox2 and Dcx109). In contrast to previous studies that primarily emphasized gene expression,7 TrackerSci offers a comprehensive view of both gene expression and epigenetic landscapes in newborn cells, allowing us to explore the epigenetic foundation (e.g., cis-regulatory elements, TFs) underlying the gene expression signatures.
There is a consensus that the self-renewal and regeneration capacity of progenitor cells reduce as we age. However, we observed heterogeneous cellular responses across progenitor cell types. Notably, aging led to a marked reduction in proliferation potential in neuronal progenitors and increased proliferation capacity in reactive microglia, both in line with global cell population shifts.15 Although oligodendrocyte progenitors’ proliferation was mildly affected, the intermediate differentiation precursors significantly declined at an early stage (before 6 months), suggesting that aging primarily blocks its differentiation process, aligning with prior reports.110,111 Our study further proposes candidates that could be manipulated to counteract aging-related cellular renewal decline. For example, we identified genes involved in cell proliferation (e.g., Ei24, Arhgap11a, Lzts1, and Cep5755–58) and neurogenesis regulation (e.g., Prdm10, Nrg1, Nrg3, and Nr2f153,59–62), which were further confirmed in CRISPR screens using NPCs derived from both young and aged brains.54 In addition, we detected an age-dependent increase of Smpd4 (sphingomyelin phosphodiesterase) and a decrease of Sgms1 (sphingomyelin synthase) expression in the OPCs, suggesting that a high cellular ceramide level was associated with the aging-induced inhibition of oligodendrocyte differentiation.
Lastly, we showcased the potential of the TrackerSci dataset as an anchor for detecting rare proliferating or differentiating cells in the human brain. By profiling nearly 800,000 single nuclei from five anatomical regions of the aged human brains and them integrating with the TrackerSci dataset, we discerned the region-specific and cell-type-specific signatures of rare progenitor cells in the aged human brain and observed both conserved and differing molecular signatures of oligodendrogenesis across species. We identified a decrease in oligodendrogenesis in the cerebellum relative to other brain areas and uncovered region-specific gene control mechanisms. Although our initial analysis did not reveal a clear neurogenesis-related population in aged human brain samples—possibly due to the inherent scarcity of neurogenesis cells or non-targeted sampling of neurogenic regions (e.g., the dentate gyrus)—integration with a larger-scale human dataset86 focused on adult brain samples enabled us to identify a rare population of immature neurons within the adult human hippocampus, which aligns with earlier published findings89,112 and further emphasizes our technique’s potential in identifying proliferative events in human samples.
In summary, our study represents a key step toward understanding the impact of aging on the proliferation and differentiation potential of progenitor cells in the mammalian brain. We anticipate that TrackerSci will be broadly used to identify and quantify cell-genesis processes across diverse systems, including other mammalian organs and humanized organoids. Additionally, we envision that similar strategies (i.e., coupling the sci-seq platform with in vivo cellular labeling) can be expanded to study other critical aspects, such as the cell-type-specific survival, apoptosis, and senescent states. This will facilitate a global view of molecular programs regulating cell-type-specific dynamics during aging, thereby informing potential pathways to restore tissue homeostasis for patients with aging-related diseases.
Limitations of the study
While the TrackerSci method provides several advances, there are certain limitations inherent to this study. First, the cellular differentiation trajectories inferred from TrackerSci are primarily based on gene expression and chromatin accessibility and do not directly map out cellular lineage relationships. The depth of our analysis could be further enhanced by concurrently assaying lineage history38,113,114 and different molecular dimensions in the same cell.99,115–119 In addition, the short interval of metabolic labeling in our study may have led to a lesser representation of the quiescent stem cell population. To address this, future iterations of TrackerSci could include label-retention experiments120 for assessing stem cells that have divided and entered quiescence over a more extended period. While TrackerSci might not be readily applicable for profiling progenitor cells in human tissues, this limitation can be partially addressed through our interspecies integration analysis. Additionally, alternative strategies to identify molecular markers of cell differentiation lineages across species can be pursued by leveraging computational methodologies,89,121,122 which will facilitate detection of human stem cells based on protein marker expression123 and allow tar geted characterizations of rare cell genesis in humans.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information should be directed to and will be fulfilled by the lead contact, Junyue Cao (jcao@rockefeller.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq and ATAC-seq data have been deposited at Gene Expression Omnibus. Accession numbers are listed in the key resources table. Interactive website for data exploration is available at UCSC cell browser138: https://adult-mouse-brain-newborn.cells.ucsc.edu.
Computational pipeline scripts for processing data are available at this github repository: https://github.com/ZiyuLu041/TrackerSci_analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Iba1 | Novus Biologicals | Cat#NB100–1028; RRID:AB_521594 |
| Mouse anti-Ki67 | Invitrogen | Cat#14-5699-82; RRID:AB_2016711 |
| Biological samples | ||
| Whole mouse brains | This study | N/A |
| Aged human brain tissues | University of Kentucky AD Center Tissue Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 5-Ethynyl-2′-deoxyuridine | Lumiprobe | Cat#20540 |
| SUPERase•In™ RNase Inhibitor | Thermo Fisher Scientific | Cat#AM2696 |
| BSA | NEB | Cat#B90000S |
| TRIS 1.0M Sterile Solution, pH 7.5 | VWR | Cat#97062–936 |
| 16% methanol-free formaldehyde | Thermo Fisher Scientific | Cat#28906 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | Cat#18090200 |
| mRNA Second Strand Synthesis buffer and enzyme | NEB | Cat#E6111L |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat#M0541L |
| EDTA-free Protease Inhibitor Cocktail | Sigma | Cat#11873580001 |
| 3′-Azido-3′-deoxythymidine | Thermo Fisher Scientific | Cat#J65127–06 |
| Critical commercial assays | ||
| Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat#C10634 |
| Nuclei Isolation Kit: Nuclei EZ Prep Millipore Sigma | Millipore Sigma | Cat#NUC101–1KT |
| Deposited data | ||
| Raw and analyzed data | This study | GEO: GSE212251 |
| Single-nucleus RNA-seq data of whole mouse brain across three age groups and two Alzheimer’s disease associated mutants | Sziraki et al.15 | GEO: GSE212606 |
| Single-cell RNA-seq data of the adolescent mouse nervous system | Zeisel et al.26 | http://mousebrain.org/ |
| Spatial transcriptomic Slide-seq data of adult mouse brain | Langlieb et al.27 | https://www.braincelldata.org/ |
| Single-cell RNA-seq data of adult mouse brain subventricular zone | Zywitza et al.39 | GEO: GSE111527 |
| Gene scores and effect sizes from in vitro genome-wide CRISPR screen in primary neural stem cells | Ruetz et al.54 | https://www.biorxiv.org/content/biorxiv/early/2021/11/23/2021.11.23.469762/DC1/embed/media-1.xlsx?download=true |
| Single-nucleus RNA-seq data of human brain tissues from patients with Alzheimer’s disease and healthy controls | Gerrits et al.90 | GEO: GSE148822 |
| Single-cell RNA-seq data from pituitary glands of 7-week-old male C57BL/6 mice | Cheung et al.65 | GEO: GSE120410 |
| Experimental models: Cell lines | ||
| HEK293T | Gift from J. Shendure, University of Washington | N/A |
| NIH/3T3 | Gift from J. Shendure, University of Washington | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: 5XFAD | The Jackson Laboratory | RRID:MMRRC_034840-JAX |
| Software and algorithms | ||
| R | R Core | https://www.r-project.org/ |
| Python | Python Software Foundation | https://www.python.org/ |
| bcl2fastq | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Trim Galore | Babraham Institute | https://github.com/FelixKrueger/TrimGalore |
| STAR | Dobin et al.127 | https://github.com/alexdobin/STAR |
| Samtools | Li et al.128 | http://www.htslib.org/download/ |
| Picard MarkDuplicates | Broad Institute | https://broadinstitute.github.io/picard/ |
| Scanpy | Wolf et al.129 | https://github.com/scverse/scanpy |
| Scrublet | Wolock et al.130 | https://github.com/swolock/scrublet |
| Seurat | Stuart et al.131 | https://satijalab.org/seurat/ |
| Monocle | Trapnell et al.132 | http://cole-trapnell-lab.github.io/monocle-release/ |
| SnapTools | Fang et al.133 | https://github.com/r3fang/SnapTools |
| SnapATAC | Fang et al.133 | https://github.com/r3fang/SnapTools |
| MACS2 | Zhang et al.134 | https://github.com/macs3-project/MACS |
| bedtools | Quinlan and Hall135 | https://bedtools.readthedocs.io/en/latest/ |
| chromVar | Schep et al.136 | https://github.com/GreenleafLab/chromVAR |
| scVelo | Bergen et al.37 | https://github.com/theislab/scvelo |
| ComplexHeatmap | Gu et al.137 | https://github.com/jokergoo/ComplexHeatmap |
| miloR | Dann et al.50 | https://github.com/MarioniLab/miloR |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture
HEK293T and NIH/3T3 cells (gift from J. Shendure, University of Washington) were cultured in 10 cm dishes at 37°C with 5% CO2 in high glucose DMEM (Gibco, 11965–118) supplemented with 10% Fetal Bovine Serum (Sigma-Aldrich, F4135) and 1X penicillin-streptomycin (Gibco, 15140–122).
Animals
The C57BL/6 wild-type mice and 5XFAD transgenic mice (heterozygous, JAX stock #034840) were obtained from The Jackson Laboratory. The ages of these mice ranged from 48 to 685 days. Both male and female mice were included in each condition. Detailed information of animal individuals in this study is provided in Table S1A. Mice were housed socially. All animal procedures were in accordance with institutional, state, and government regulations and approved under the IACUC protocol 21049.
Human brain sample
Twenty-nine postmortem human brain samples across five regions and six individuals (three male and three female, all cognitively normal proximal to death) ranging from 70 to 94 years of age at death, were collected from the University of Kentucky AD Center Tissue Bank.139,140 Each surveyed sample underwent rigorous quality control including short PMI (<4 h). Detailed information of human individuals in this study is provided in Table S6.
METHOD DETAILS
EdU labeling of mammalian cell culture
EdU (5-ethynyI-2′-deoxyuridine) (Thermo Fisher Scientific, A10044) was added to culture media at 10 μM final concentration for 1 h. After labeling, cells were harvested with 0.25% trypsin-EDTA. HEK293T and NIH/3T3 cells were combined at a 1:1 ratio, washed with ice-cold PBS, and lysed in 1 mL ice-cold EZ lysis buffer (Millipore Sigma, NUC101). The nuclei were then fixed on ice with 1% formaldehyde (Thermo Fisher Scientific, 28906) for 10 min and washed with EZ lysis buffer, filtered with 40 μm cell strainers (Ward’s Science, 470236–276), and resuspended in Nuclei Suspension Buffer (NSB) (10 mM Tris-HCl pH 7.5 (VWR, 97062–936), 10 mM NaCl (VWR, 97062–858), 3 mM MgCl2 (VWR, 97062–848) supplemented with 0.1% SUPERase•In RNase Inhibitor (Thermo Fisher Scientific, AM2696) and 1% BSA for TrackerSci-RNA or supplemented with 0.1% Tween 20 (Sigma, P9416–100ML), 1x cOmplete, EDTA-free Protease Inhibitor Cocktail (Sigma, 11873580001) and 0.1% IGEPAL CA-630 (VWR, IC0219859650) for TrackerSci-ATAC experiments).
EdU labeling of mouse tissues
C57BL/6J mice of different age groups were obtained from The Jackson Laboratory. The 5xFAD mice were obtained from The Jackson Laboratory (NIH Mutant Mouse Resource & Research Centers) and were hemizygous. All the mice lived socially. Mice were injected intraperitoneally with 50 mg/kg of EdU in PBS at 24-h intervals for five days, and mouse brains were harvested 24 h after the final injection.
C57BL/6J mice obtained from The Jackson Laboratory were labeled and harvested for pulse-chase labeling at various time points. Specifically, four mice (two male and two female) were injected intraperitoneally with 50 mg/kg of EdU in PBS for 3 days at 24-h intervals, and brains were harvested 24 h after the final injection. 12 mice were injected intraperitoneally with 50 mg/kg of EdU in PBS for 5 days at 24-h intervals. In addition, for 5-day injections, four mice (two male and two female) were harvested 1 day, 3 days, and 9 days after the final injection.
Tissue collection and nuclei isolation
Mice were euthanized utilizing inhalation of carbon dioxide (CO2), following IACUC protocol 20047. Whole brains were extracted from mice, immediately snap-frozen in liquid nitrogen, and stored at −80°C upon further usage. For nuclei isolations, thawed brains were cut into small pieces with fine scissors (Fine Science Tools, 14060–09) in 1 mL ice-cold PBS with 1% SUPERase•In RNase Inhibitor and 1% BSA, pelleted, resuspended in 1.5 mL Nuclei Isolation Buffer (EZ Lysis Buffer supplemented with 1% SUPERase•In RNase Inhibitor, 1% BSA and 1X cOmplete EDTA-free Protease Inhibitor Cocktail) for 5 min on ice, and homogenized through 40 μm cell strainers (VWR, 470236–276) with the rubber tips of syringes. Then, extracted nuclei were pelleted, fixed in 1% formaldehyde on ice for 10 min, washed twice with NSB, and divided into two aliquots for both sci-RNA-seq and sci-ATAC-seq profiling. Nuclei subjected to sci-RNA-seq were briefly sonicated (Diagenode, low power mode for 12 s) to reduce clumping. Finally, nuclei were filtered through pluriStrainer Mini 20 μm filters (Pluriselect, 43-10020-70), resuspended in 100 μL NSB, snap frozen in liquid nitrogen, and stored at −80°C until further usage.
Human brain sample
Established strategies were used to extract high-quality nuclei from frozen postmortem brain samples.15 Extracted nuclei were then fixed with formaldehyde, diluted, and flash-frozen for storage. For EasySci transcriptome profiling, nuclei from all samples were thawed and deposited into different wells for barcoded reverse transcription (RT), such that the first index identifies the source of each cell. The library was sequenced across two Illumina NovaSeq 6000 sequencer runs, altogether yielding 12 billion reads for ~900,000 cells (~13,000 sequencing reads per cell).
In vivo pharmacological inhibition
Twelve 8-week-old mice (six male and six female) were treated with 3-azido-3-deoxythymidine (AZT, 100 mg/kg/day67 in saline, n = 6) or saline (n = 6) for twelve consecutive days through intraperitoneal injection. EdU labeling was performed starting from the sixth day of drug treatment through intraperitoneal injection (50 mg/kg/day) and drinking water administration (0.3 mg/mL). On the day of harvest, mice were anesthetized with isoflurane and perfused with 20 mL cold PBS to remove peripheral blood cells. Brains were then collected, and divided sagittally. Left hemispheres were snap-frozen, stored at −80°C, and used for nuclei extraction and TrackerSci-RNA profiling.
TrackerSci-RNA
Briefly, EdU staining was performed on thawed nuclei using Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry assay Kit (Thermo Fisher Scientific, 10634). A 500 μL reaction buffer (prepared following the manufacturer’s protocol) supplemented with 1% SUPERase•In RNase Inhibitor was added directly to the nuclei suspension, mixed well and left in RT for 30 min. Then, nuclei were spun down for 5 min at 500g (4°C), washed once with 500 μL of 1X Click-iT saponin-based permeabilization and wash reagent, resuspended in 1 mL NSB with 1:20 dilution of 0.25 mg/mL 4′,6-diamidino-2-phenylindole (DAPI, Invitrogen D1306) and FACS sorted. Alexa 647 and DAPI positive nuclei were sorted into 96-well plates with each well (250~500 nuclei/well) containing 4 μL of NSB. Sorted plates were briefly centrifuged, mixed with 1 μL of 50 μM oligo-dT primer (5ʹ-ACGACGCTCTTCCGATCTNNNNNNNN [10bp-index]TTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3ʹ, where “N” is any base and “V” is either “A”, “C” or “G”, IDT) and 0.5 μL 10 mM dNTP mix (Thermo Fisher Scientific, R0194) and denatured at 55°C for 5 min and immediately placed on ice. 3.5 μL of first-strand reaction mix, containing 2 μL 5X SuperScript IV Reverse Transcriptase Buffer (Invitrogen, 18090200), 0.5 μL 100 mM DTT (Invitrogen, P2325), 0.5 μL SuperScript IV Reverse Transcriptase (Invitrogen, 18090200), 0.5 μL RNaseOUT Recombinant Ribonuclease Inhibitor (Invitrogen, 10777019) was then added to each well. Reverse transcription was carried out by incubating plates at the following temperature gradient: 4°C 2 min, 10°C 2 min, 20°C 2 min, 30°C 2 min, 40°C 2 min, 50°C 2 min and 55°C 10 min, and was stopped by adding 1 μL of 18 mM EDTA (VWR, 97062–656) to each well. All nuclei were then pooled, stained with DAPI at a final concentration of 3 μM, and sorted at 25 nuclei per well into 5 μL EB buffer. Cells were gated based on DAPI and Alexa 647 such that singlets were discriminated from doublets and EdU+ cells were purified. 0.66 μL mRNA Second Strand Synthesis buffer and 0.34 μL mRNA Second Strand Synthesis enzyme (NEB, E6111L) were then added to each well. Second strand synthesis was carried out at 16°C for 1 h. 6 μL tagmentation reaction mix (made by mixing 0.5 μL self-loaded Tn5 with 200 μL Tagmentation buffer containing 20 mM Tris-HCl pH 7.5, 20 mM MgCl2, 20% Dimethylformamide (Fisher, AC327175000)) was added to each well and tagmentation was performed at 55°C for 5 min. After tagmentation, each well was mixed with 0.4 μL 1% SDS, 0.4 μL BSA (NEB, B90000S), and 2 μL of 10 μM P5 primer (5′-AATGATACGGCGACCACCGAGATCTACA[i5]CCCTACACGACGCTCTTCCGATCT-3′, IDT), and incubated at 55°C for 15 min. Then, 2 μL 10% Tween 20, 1.2 μL nuclease-free water and 2 μL of 10 μM indexed P7 primer (5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3′, IDT), and 20 μL NEBNext High-Fidelity 2X PCR Master Mix (NEB, M0541L) were added to each well. Amplification was carried out using the following program: 72°C for 5 min, 98°C for 30 s, 18–22 cycles of (98°C for 10 s, 66°C for 30 s, 72°C for 1 min), and a final 72°C for 5 min. After PCR, samples were pooled and purified using 0.8 volumes of AMPure XP beads (Beckman Coulter, A63882) twice. Library concentrations were determined by Qubit (Invitrogen, Q33231), and the libraries were visualized by electrophoresis on a 2% E-Gel EX Agarose Gels (Invitrogen, G402022). All RNA-seq libraries were sequenced on the NextSeq 1000 platform (Illumina) using a 100 cycle kit (Read 1: 58 cycles, Read 2: 60 cycles, Index 1: 10 cycles, Index 2: 10 cycles). The TrackerSci-RNA libraries were sequenced to 70,000 reads per cell.
TrackerSci-ATAC
EdU staining was performed on thawed nuclei using Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry assay Kit (Thermo Fisher Scientific, 10634). A 500 μL reaction buffer (prepared following the manufacturer’s protocol) supplemented with 1X cOmplete EDTA-free Protease Inhibitor Cocktail was added directly to the nuclei suspension, mixed well, and left in RT for 30 min. Then, nuclei were spun down for 5 min at 500g (4°C), washed once with 500 μL of 1X Click-iT saponin-based permeabilization and wash reagent, resuspended in 1 mL NSB with 1:20 dilution of 0.25 mg/mL 4′,6-diamidino-2-phenylindole (DAPI) and FACS sorted. Alexa 647 and DAPI positive nuclei were sorted into 96-well plates with each well (250~500 nuclei/well) containing 4 μL of NSB. Sorted plates were briefly centrifuged, mixed with 5 μL 2x TD buffer (20 mM Tris-HCl Ph 7.5, 20 mM MgCl2, 20% Dimethylformamide) and 1 μL barcoded Tn5. Tagmentation reaction was performed at 55°C for 30 min and stopped by adding 11 μL 2X Stop buffer (40 mM EDTA, 1 mM Spermidine (Sigma, S0266)) to each well. All nuclei were then pooled, stained with DAPI at a final concentration of 3 μM, and sorted at 25 nuclei per well into 5 μL EB buffer. Cells were gated based on DAPI and Alexa 647 such that singlets were discriminated from doublets and EdU+ cells were purified. After sorting, each well was mixed with 0.25 μL 18.9 mg / mL proteinase K (Sigma, 3115828001), 0.25 μL 1% SDS and 0.5 μL nuclease-free water, and reverse crosslinking was performed at 65°C for 16 h. Then, 2 μL 10% Tween 20 was added to each well to quench the SDS. Following on, 1 μL of 10 μM indexed P5 primer (5′-AATGATA CGGCGACCACCGAGATCTACA[i5]CCCTACACGACGCTCTTCCGATCT-3′, IDT), 1 μL of 10 μM indexed P7 primer (5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3′, IDT) and 10 μL NEBNext High-Fidelity 2X PCR Master Mix were added into each well. Amplification was carried out using the following program: 72°C for 5 min, 98°C for 30 s, 15–16 cycles of (98°C for 10 s, 66°C for 30 s, 72°C for 1 min), and a final 72°C for 5 min. Final PCR products were pooled and purified by a Zymoclean DNA clean and concentration kit (Zymoresearch, D4014). Library concentrations were determined by Qubit, and the libraries were visualized by electrophoresis on a 2% E-Gel EX Agarose Gels. All ATAC-seq libraries were sequenced on the NextSeq 1000 platform (Illumina) using a 100 cycle kit (Read 1: 58 cycles, Read 2: 60 cycles, Index 1: 10 cycles, Index 2: 10 cycles). The TrackerSci-ATAC libraries were sequenced to ~120,000 reads per cell.
TrackerSci-RNA data processing
Read alignment and gene count matrix generation for the scRNA-seq were performed using the pipeline we developed before.13 Briefly, base calls were converted to fastq format and demultiplexed using Illumina’s bcl2fastq/v2.19.0.316, tolerating one mismatched base in barcodes (edit distance (ED) < 2). The RT barcode for each read was corrected to its nearest barcode (edit distance (ED) < 2), and reads with uncorrected barcodes (ED ≥ 2) were removed. Demultiplexed reads were then adaptor clipped using trim_galore/v0.4.1 (https://github.com/FelixKrueger/TrimGalore) with default settings. Trimmed reads were mapped to a chimeric reference genome of human and mouse (hg39/mm39) for the species-mixing experiment and to the mouse only (mm39) for mouse brain experiments, using STAR/v2.5.2b127 with default settings. Uniquely mapping reads were extracted, and duplicates were removed using the unique molecular identifier (UMI) sequence, reverse transcription (RT) index, and read 2 end-coordinate (i.e., reads with identical UMI, RT index, and tagmentation site were considered duplicates). Finally, mapped reads were split into constituent cellular indices by further demultiplexing reads using the RT index.
To generate digital expression matrices, we calculated the number of strand-specific UMIs for each cell mapping to the exonic and intronic regions of each gene with python/v2.7.18 HTseq package.141 For multi-mapped reads, reads were assigned to the closest gene, except in cases where another intersected gene fell within 100 bp to the end of the closest gene, in which case the read was discarded. For most analyses, we included both expected-strand intronic and exonic UMIs in per-gene single-cell expression matrices. Exonic and intronic gene count matrices were used in RNA velocity analysis.
For the species-mixing experiment, RNA barcodes with more than 200 UMIs and 100 unique genes were identified as real cells, and those with fewer than that were discarded. The percentage of uniquely mapping reads for genomes of each species was calculated. Cells with over 90% of UMIs assigned to one species were regarded as species-specific cells, with the remaining cells classified as mixed cells or “collisions”. The collision rate was calculated as the ratio of mixed cells.
TrackerSci-ATAC data processing
Single-cell ATAC-seq data was performed using a published pipeline9,115 with mild modifications. Base calls were converted to fastq format and demultiplexed using Illumina’s bcl2fastq/v2.19.0.316 tolerating one mismatched base in barcodes (edit distance (ED) < 2). The indexed Tn5 barcode for each read was corrected to its nearest barcode (edit distance (ED) < 2), and reads with uncorrected barcodes (ED ≥ 2) were removed. Demultiplexed reads were then adaptor-clipped using trim_galore/0.4.1 with default settings. Trimmed reads were mapped to a chimeric reference genome of human and mouse (hg38/mm39) for the species-mixing experiment and to the mouse only (mm39) for mouse brain experiments, using STAR/v2.5.2b127 with default settings. Duplicates were removed by picard MarkDuplicates/v2.25.2142 per PCR sample. Deduplicated reads were split into constituent cellular indices by further demultiplexing reads using the Tn5 index.
A snap-format (Single-Nucleus Accessibility Profiles) file was generated from deduplicated bam files using SnapTools/v1.4.8 with default settings (https://github.com/r3fang/SnapTools).133 A cell-by-bin count matrix with 5kb bin size was created from the resulting snapfile. The promoter ratio for each cell was calculated as the number of fragments mapping to genomic bins overlapping with promoter regions (defined as 2kb upstream of the gene body).
For the species-mixing experiment, ATAC barcodes with more than 1000 fragments and more than 0.2 promoter ratio were identified as real cells, and those with fewer than that were discarded. The percentage of uniquely mapping reads for genomes of each species was calculated. Cells with over 90% of reads assigned to one species were considered species-specific cells, with the remaining cells classified as mixed cells or “collisions”. The collision rate was calculated as the ratio of mixed cells.
Cell filtering, clustering, and annotation for TrackerSci-RNA
A digital gene expression matrix was constructed from the raw sequencing data as described above. EdU+ cells and global cells were combined and analyzed together. Cells with less than 200 UMIs and 100 unique genes were discarded. Potential doublet cells and doublet-derived subclusters were detected using an iterative clustering strategy similar to before.143 Cells labeled as doublets(by scanpy/v1.6.0 and scrublet/v0.2.3)129,130 or from doublet-derived sub-clusters were filtered. The downstream dimension reduction and clustering analysis were done by Seurat/v4.0.2.144 Briefly, the dimensionality of the data was reduced by PCA (30 components) first and then with UMAP, followed by Louvain clustering. Clusters were assigned to known cell types based on cell type-specific markers (Table S2).
Differentially expressed genes across different cell types were identified using monocle/v2.22.0145 with the differentialGeneTest() function. Genes detected in less than 10 cells were filtered out before the analysis. To identify cell type-specific gene markers, we selected genes that were differentially expressed across different cell types (5% FDR, likelihood ratio test), with FC > 2 between the target cell type and the second highest expressed cell type, and with maximum transcripts per million (TPM) > 10 in the target cell types.
Cell filtering, clustering, and annotation for TrackerSci-ATAC
Single-cell ATAC-seq profiles were generated as described above. EdU+ cells and global cells are combined and analyzed together. Cells with less than 1000 fragments and less than 0.2 promoter ratio were discarded. Dimensionality reduction for ATAC-seq data was performed using the snapATAC/v1.0.0.133 A cell-by-bin matrix at 5-kb resolution was used. We focused on bins on chromosomes 1–19, X and Y. High-coverage bins (top 5% bins that overlap with invariant features) or low-coverage bins (bottom 5% bins that represent general inaccessible regions) were filtered out before the analysis. Diffusion maps dimensionality reduction was performed on the filtered cell-by-bin matrix after binarization. UMAP analyses were performed on the top 20 eigenvectors, followed by unsupervised clustering via the densityPeak algorithm implemented in R package densityClust/v0.3.146
We performed integration analysis between the TrackerSci-RNA dataset and TrackerSci-ATAC dataset to annotate the ATAC dataset. The gene activity score for ATAC cells was computed using the snapATAC function createGmatFromMat() by summing up the counts of bins overlapping with the gene body. A Seurat object was generated using the gene activity matrix and previously calculated diffusion map embeddings for single cell ATAC-seq. Then, variable genes were identified from TrackerSci-RNA data and used for identifying anchors between these two modalities. Next, we co-embedded the RNA-seq and ATAC-seq profiles in the same low-dimensional space to visualize all the cells together. We then used overlapped RNA clusters to annotate ATAC cells in the integrated UMAP space. ATAC cells without overlapped RNA cells were removed with careful inspection since they usually represent potential doublets or low-quality cells. Finally, single-cell ATAC dimension reduction, clustering, and integration analysis were rerun on the remaining dataset following the same procedure.
Peak calling and identifications of cell-type-specific peaks
To define peaks of accessibility across all sites, we used MACS2/v2.1.1.134 Nonduplicate ATAC-seq reads of cells from each main cell type were aggregated, and peaks were called on each group separately with these parameters: –nomodel –extsize 200 –shift −100 –q 0.1. Peak summits were extended by 250bp on either side and then merged with bedtools/v2.30.0,134,135 together with gene promoter regions (annotated transcription start site (TSS) in GENCODE VM27 minus/plus 1000 base pairs in a strand-specific manner). Each read alignment was extended by 100 bp upstream and downstream of the insertion site of tagmentation. Cells were determined to be accessible at a given peak if a read from a cell overlapped with the peak. The peak count matrix was generated by a custom python script with the HTseq package.134,135,141 Differentially accessible peaks across cell types were identified using monocle/v2.22.0145 with the differentialGeneTest() function. Peaks detected in less than 10 cells were filtered out before the analysis. When multiple peaks existed within the same promoter, each peak was considered independently. To determine cell-type-specific peak markers, we selected peaks that were differentially accessible across different cell types (5% FDR, likelihood ratio test), with FC > 2 between the target cell type and the second highest expressed cell type, and with reads per million > 10 in the target cell types.
Analysis for linking cis-regulatory elements (CRE) to regulated genes
We aim to identify links between chromatin-accessible sites and regulated genes based on their covariance. Only EdU+ cells were kept in this analysis. We first constructed pseudo-cells by aggregating the RNA-seq and ATAC-seq profiles of highly similar cells through k-means clustering the integrative UMAP coordinates using the kmeans function from R package stats/v4.1.2. The k was selected so that the average cell number per subcluster is 150. Subclusters overrepresented by one molecular layer (the percentage of cells from either RNA-seq or ATAC-seq profile greater than 90%) were merged with a nearby subcluster. After aggregating cells within each sub-cluster, we obtained a total of 88 pseudo-cells, with a median of 54 cells from RNA-seq profile and 93 cells from ATAC-seq profile. Aggregated count matrices for RNA-seq and ATAC-seq were normalized to transcripts per million (TPM) and log-transformed after adding one pseudocount. We only retained genes and peaks with TPM value greater than 10 in the maximum expressed pseudo-cells. Then, for each gene, we calculated the Pearson Correlation Coefficient (PCC) between its gene expression and the chromatin accessibility of its nearby accessible sites (minus/plus 500 kb from the TSS) across pseudo-cells. Sites overlapping with minus/plus 1kb from the TSS were considered promoters, while the rest were considered distal regions. To define a threshold at PCC score, we also generated a set of background pairs by permuting the pseudo cell id of the ATAC-seq matrix and with an empirically defined significance threshold of FDR < 0.05, to select significant positively correlated cCRE-gene pairs. We further filtered the linkage by requiring that either the maximum expressed cell types in the RNA profile and the ATAC profile were the same or the top two or top three highest expressed cell types were in the same cell trajectory (Oligodendrogenesis trajectory: OPC, COP, OLG; Astrocytes trajectory: ASC, NPC; DG neurogenesis trajectory: NPC, DGNB; OB neurogenesis trajectory: NPC, OBNB, OBIN). Finally, we only keep the one top linked gene with the highest PCC for each peak.
Transcription factor analysis
To identify key TF regulators of each main cell type, we searched for TF that can be validated in two molecular layers by correlating gene expression and motif accessibility. First, using the TrackerSci-ATAC dataset, we selected the top 300 sites per main cell type (from the differential peak analysis described above, filtered by q value < 0.05, maximum expressed TPM > 10 and ranked by FC between the highest and the second expressed cell type) to a combined peak set. We then resized the peaks to a fixed length of 500 bp (± 250 bp around the center) and generated a binarized peak-by-motif matrix using the R package motifmatchr/v1.16.0147 with the matchMotifs() function to identify the occurrences of motifs in each peak from a filtered collection of the cisBP motif database curated by chromVARmotifs/v0.2.0.136,148 A matrix of motif-by-cell counts was obtained by multiplying the peak-by-cell matrix with the peak-by-motif matrix, and was aggregated into pseudo-cells based on the k-means clustering described before. We then computed the PCC between the scaled TF motif accessibility and the scaled TF gene expression across pseudo-cells. To select significantly positive and negative correlations of TF gene expression and motif accessibility pairs, we permuted the pseudo cell id of the motif-by-cell matrix to compute a background PCC distribution and selected the TF pairs with an empirically defined significance threshold of FDR < 0.05. In addition, we only keep TF with TPM > 10 in the maximum expressed cell type.
Trajectory analysis
Cells corresponding to the neurogenesis trajectory (ASC, NPC, DGNB, OBNB and OBIN) or the oligodendrogenesis trajectory (OPC, COP and OLG) from both RNA-seq data and ATAC-seq data were selected for detailed investigation. We next performed UMAP dimension reduction at the trajectory level with the integration function from Seurat,144 using the top 3,000 highly variable genes and top 50 PCs. Each cell was assigned a pseudotime value based on its position along the trajectory using monocle3/v1.0.0 function order_cells().132 RNA velocity analyses were performed using scVelo/v0.2.337 using the exonic and intronic gene count matrix generated from sci-RNA-seq pipeline to validate the cell differentiation direction and estimate the position of the progenitor cell state. For the two neurogenesis trajectories (DG neurogenesis and OB neurogenesis), pseudotime assignment was calculated separately and scaled so that the cells shared between two trajectories received the same pseudotime value. Specifically, we first used the pseudotime value calculated from the OB trajectory for common progenitor cells in both DG and OB trajectories. We then fitted a linear regression line using R function lm() to predict the OB-pseudotime based on the DG-pseudotime. Then, for cells unique to the DG neurogenesis, we adjusted their pseudotime using the predict() function using DG-pseudotime as input. Gene expression and peak accessibility dynamics along pseudotime were identified using monocle/v2.22.0145 with the differentialGeneTest() function with pseudotime values and their main cluster identity as variables. Genes or peaks that passed a significant test (FDR of 5%) were considered as dynamically regulated genes or sites. Furthermore, differential accessible sites along pseudotime were used to infer TF motif accessibility dynamics. We computed a motif deviation score for each single cell using chromVar/v1.4.1136 with the dynamic peak set (resized to 500 bp) as input. Then, the motif deviation scores of each single cell were rescaled to (0, 10) using R function rescale() and differential accessible motifs were identified using monocle/v2.22.0 with the differentialGeneTest() function. TF motifs that passed a significant test (FDR of 5%) were considered as dynamically regulated motifs. For gene enrichment analysis we used the enrichR40 and the following pathways collections were considered: Panther_2016, Reactome_2016, KEGG_2019_Mouse, GO_Biological_Process_2018, GO_Molecular_Function_2018. For visualizing the dynamics of gene expression, peak accessibility and motif accessibility, we used R package ComplexHeatmap/v2.10.0.137
Cell proportion analysis
To quantify the cell-type-specific changes in the proliferation dynamics across conditions, we calculated the fraction of each cell type within EdU+ population from each condition for RNA-seq data and ATAC-seq data separately, which was further multiplied by the median of EdU+ ratio for each group obtained from FACS sorting. For adult WT mice, we only included those that were harvested 24h after five-day labeling to avoid artifacts introduced by the labeling time.
To quantify the effects of aging on cell differentiation dynamics along neurogenesis and oligodendrogenesis trajectories, we applied miloR/v1.3.1,50 a single-cell differential abundance testing framework using k-nearest neighbor (KNN) graphs. We first constructed the KNN graph on the UMAP space for each trajectory using the buildGraph() function with k = 120 for the neurogenesis trajectory and k = 250 for the oligodendrogenesis trajectory. Cell neighborhoods were then defined using the makeNhoods() function and the number of cells from each experiment sample were counted for each neighborhood using the countCells() function. Testing for differential abundance in neighborhoods was performed using the testNhoods() function and significance levels for Spatial FDR of 0.05 were used. Visualization of differential abundance neighborhoods was done using the plotNhoodGraphDA() function.
Similarly, we applied miloR/v1.3.1 to examine the effects of telomerase inhibition on cell differentiation dynamics along neurogenesis. Of note, we observed a decrease of EdU intensity per cell during the FACS sorting in the AZT-treated group, which could due to its side effects on EdU incorporation considering previous report about its effects of pyrimidine metabolism,149 or the label dilution caused by differences in replication speed and lack of cell cycle reentry upon treatment.150 This resulted in two subpopulations with high and low EdU intensity, where the EdUhigh population contains more control cells, while the EdUlow population contains more AZT-treated cells. To minimize the artifacts introduced by the variations in EdU intensity, we focused on the cell state transition instead of the absolute quantifications of proliferation events. We achieved this by considering all the newborn cells from EdU+ population regardless of the intensity. Specifically, in some experiments where we gated EdU+ population further into EdUhigh and EdUlow and followed up with profiling separately, we combined those cells at a same fraction obtained from sorting for each animal into the analysis.
Differential analysis of NPC and OPC across aged groups
Differential gene expression analysis across young, adult, and aged groups of NPC and OPC was performed using monocle/v2.22.0145 function differentialGeneTest() with the number of genes detected per cell included as a covariant. For adult WT mice, only cells from the animals harvested at 24 h after 5-day labeling were included to avoid artifacts introduced by the labeling time. In addition, only differentially expressed genes (> expressed in more than 10 cells) along the neurogenesis or the oligodendrogenesis trajectory were included in the differential gene test. Differentially expressed genes were selected by a q value cutoff of 0.1, a TPM cutoff of 50 in the maximum expressed group, and with at least 1.5 FC between the maximum expressed group and the minimum expressed group. Next, differentially expressed genes were grouped to aged-depleted genes and aged-enriched genes by the following criteria: for aging-depleted genes, we first selected the genes with minimum expression in aged mice, and only kept those with either maximum expression in young mice or within less than 2 FC between the young group and the adult group. For aging-enriched genes, we first selected the genes with maximum expression in aged mice, and only kept those with either minimum expression in young mice or with less than 2 FC between the young group and the adult group. We then further filtered the DE genes based on the consistency on their promoters or linked sites. For aging-depleted genes, we required that the mean of promoter accessibility or linked site accessibility was at the minimum level in the aged group compared to young and adults. For aging-enriched genes, we required that the mean of promoter accessibility or the linked site accessibility was at the maximum level in the aged group compared to young and adults. Genes that were lowly detected in both promoter accessibility and linked sites (represented by the mean of reads per million < 10 in all conditions) were also discarded.
Integration analysis between TrackerSci-RNA and EasySci-RNA
Integration analysis of scRNA-seq dataset profiled using TrackerSci and EasySci was performed using Seurat/v4.0.2.144 We first in tegrated 14,095 TrackerSci-RNA cells (including 5,715 EdU+ cells and 8,380 all brain cells without EdU enrichment) with 126,285 EasySci-RNA cells (up to 5,000 cells randomly sampled from each of 31 cell types) in our companion study.15 Shared variable genes, selected by SelectIntegrationFeatures() function, were used for identifying anchors using FindIntegrationAnchors(). The two datasets were then integrated together with the IntegrateData() function. To visualize all the cells together, we co-embedded all the cells in the same low-dimensional space. We further applied the same integrative analysis strategy to cells matching the same cellular state from both datasets. Specifically, for the neurogenesis trajectory, we integrated 1,214 EdU+ cells from TrackerSci-RNA (NPC, OBNB, and OBIN) with 37,258 OB neurons 1 cells from EasySci-RNA. For the oligodendrogenesis trajectory, we integrated 3,044 EdU+ cells from TrackerSci-RNA (OPC and COP) to 22,718 oligodendrocyte progenitor cells from EasySci-RNA. For the microglia, we integrated 600 EdU+ microglia from TrackerSci-RNA to 15,754 microglia from EasySci-RNA. Microglia subclusters corresponding to peripheral immune cells were excluded before the analysis.
Quantifications of the self-renewal potential and the differentiation potential
The self-renewal potential was defined as the ratio of newly generated progenitor cells within 5 days of EdU labeling divided by the ratio of total progenitor cells detected from the global population. To account for potential variations due to slight differences of animal ages between TrackerSci and the brain cell atlas, we first fitted a linear model between the ages and the ratio of progenitor cells using the EasySci data for the following cell type: neuronal progenitor cells, oligodendrocyte progenitor cells, and microglia. We used that to predict the ratio of progenitor cells for each individual mice profiled by TrackerSci. We then divided the ratio of newly generated progenitor cells from each 5-day labeled mice by the predicted cellular fraction of the global progenitor pool for the same cell type. A line plot was generated using the median values of proliferation potential for each aged group normalized to the young mice. RNA and ATAC cells were both included, and samples with less than 50 cells were excluded from the calculation.
The differentiation potential was quantified by the ratio of differentiated cells divided by all EdU+ cells in the same trajectory. We calculated such a ratio only for oligodendrogenesis trajectory since it’s a unidirectional route. For this analysis, we divided the ratio of committed oligodendrocytes and myelin-forming oligodendrocytes to the ratio of oligodendrocyte progenitor cells for each sample and median values of each age group were used to generate the line plot. RNA and ATAC cells were included, and samples with less than 50 cells were excluded from the calculation.
Cell filtering, clustering, and annotation for the human dataset
A digital gene expression matrix was constructed from the raw sequencing data as described in our companion study.15 Potential doublet cells and doublet-derived subclusters were detected using an iterative clustering strategy similar to before.143 Cells labeled as doublets(by scanpy/v1.6.0 and scrublet/v0.2.3)129,130 or from doublet-derived sub-clusters were filtered. To identify distinct clusters of cells corresponding to different cell types in the human data, we performed the downstream dimension reduction and clustering analysis using Seurat/v4.0.2.144 Briefly, the dimensionality of the data was reduced by PCA (50 components) first and then with UMAP, followed by Louvain clustering. We then co-embedded the human data with the mouse brain atlas from profiled in our companion study15 through Seurat,131 and clusters were annotated based on overlapped cell types. The annotations were manually verified and refined based on marker genes.
Integration analysis between human and mouse
Integration analysis of scRNA-seq dataset of human and mouse was performed using Seurat/v4.0.2.144 Similar to the integration of mouse dataset profiled between TrackerSci-RNA and EasySci-RNA, we first integrated 14,095 mouse cells (including 5,715 EdU+ cells and 8,380 all brain cells without EdU enrichment) with 71,743 human cells (up to 5,000 cells randomly sampled from each of 18 cell types) to construct a coembedding UMAP space. We then project the rest of human cells into this UMAP structure using Map-Query() and TransferData() function. Cycling cells and committed oligodendrocytes from the human dataset were extracted based on the UMAP coordinates overlapping with mouse cells. Cycling cells were subjected to sub-clustering analysis for identifying their cell types. Markers for cycling cells were identified by comparing them to the rest of all cells using the Seurat function FindMarkers().
Identifications of shared and unique features between human and mouse oligodendrogenesis
To construct a continuous oligodendrogenesis trajectory shared between human and mouse, we subjected all 4,194 oligodendrogenesis-related cells (OPC, COP and OLG) from mouse data and took 2,188 oligodendrogenesis-related cells from human data (including all of 188 cells from COP and randomly sampled 1,000 cells from OPC and OLG) to integration analysis using Seurat/v4.0.2. Each cell was assigned a pseudotime value based on its position along the trajectory using monocle3 function order_cells(). For human cells, gene expression dynamics along pseudotime were identified using monocle/v2.22.0 (Qiu et al., 2017) with the differentialGeneTest() function with pseudotime values and their main cluster identity (i.e., OPC, COP and OLG) as variables. For mouse cells, we used the results from DE gene analysis along pseudotime calculated before. Conserved gene expression dynamics were selected by a q value cutoff of 0.05, a TPM (transcripts per million) cutoff of 50 in the same maximum expressed stage in both species. This reveals 1,162 DE genes along oligodendrogenesis shared between human and mouse. To select genes with species-unique expression dynamics, we filtered the DE genes with the following criteria: significantly changed along pseudotime (q value <0.05) and TPM of the maximum expressed stage larger than 50 in one species, while no significantly changed (q value >0.05) and TPM of the maximum expressed stage less than 50 in the other species. This reveals 458 and 361 DE genes along oligodendrogenesis unique to human and mouse respectively. For visualizations of gene expression dynamics, we use R package ComplexHeatmap/v2.10.0 and the genes were ordered by the hierarchical clustering implemented in the function Heatmap().
Analysis of region-specific oligodendrogenesis
To study region-specific effects of oligodendrogenesis, we quantified the ratio of each stage (OPC, COP and OLG) within all the cells along the oligodendrogenesis trajectory for each region. Cycling Oligodendrocyte progenitor cells were not included in the calculation. Statistical analysis was performed by comparing the ratio of COP to OPC in cerebellum vs. non-cerebellum cells using Fisher exact test. To study the region-specific transcriptional controls of each stage along oligodendrogenesis, we performed differential expression analysis across regions using monocle/v2.22.0 with the differentialGeneTest() function. Region-specific gene expression signatures were selected by the following cutoffs: q value < 0.05, with FC > 2 between the maximum expressed region and the second highest expressed region, and with maximum transcripts per million (TPM) > 50 in the highest expressed region.
Immunohistochemistry
For double label (Iba-1/Ki-67) immunohistochemistry, human brain sections (middle frontal gyrus, Brodmann area 9) were cut at 10 mm from a formalin-fixed, paraffin-embedded tissue block. Sections were deparaffinized prior to microwave antigen retrieval for 6 min (power 8) using Target Retrieval Solution High pH-TRS Hi (pH9) (Dako, GV800/GV804; Agilent Technologies; Santa Clara, CA, USA). Sections were next incubated for 45 s at room temperature in a 1X solution of TrueBlack (Cat # 23007, Biotium, Fremont, CA) prepared in 70% ethanol, to reduce auto-fluorescence. Sections were blocked in 10% normal goat serum in TRIS buffered saline (10% S + TBS) for 1 h at room temperature, then incubated in primary antibodies. Anti-Iba-1 (Novus Biologicals, NB100–1028, 1:1000 dilution) and anti-Ki67 (Invitrogen, 14-5699-82, 1:500 dilution ratio) were incubated overnight at 4C. Secondary antibodies were conjugated to Alexa Fluor probes 594 and 488 (Alexa 488 Goat-anti-Rabbit 1:1000, Cat # A32731 and Alexa 594 Goat-anti-Mouse 1:200, Cat # A11006, Life Technologies; Carlsbad, CA) diluted in 10% S + TBS for 1 h at room temperature. Slides were cover slipped using Invitrogen ProLong Gold mounting medium with DAPI (cat #P36935, Fisher Scientific, Waltham, MA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses used in this study included Wilcoxon test and permutation test and were reported in the corresponding figure legends. Correlations analyses were estimated by Pearson correlation. All statistical analyses were performed in R.
Supplementary Material
Highlights.
TrackerSci for characterizing the diversity and dynamics of progenitor cells in vivo
The epigenetic and genetic signatures of brain progenitor cell types
Aging-associated cell-type-specific alterations in proliferation and differentiation
Region-specific changes of oligodendrogenesis in the aged human brain
ACKNOWLEDGMENTS
We thank members of the Cao lab, R. Satija (New York Genome Center), and J. Shendure (University of Washington) for helpful discussions and feedback. We also thank members from the Rockefeller University Flow Cytometry Resource Center and Comparative Bioscience Center for their help on FACS experiments and animal maintenance. This work was funded by grants from the NIH (1DP2HG012522, 1R01AG076932, and RM1HG011014 to J.C; P30AG072946 and P01AG078116 to P.T.N.; and R01AG066912 to S.G.). This research was conducted while J.C. was a Sagol Network GerOmic Award for Junior Faculty awardee.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
J.C., W.Z., Z.L., and M.Z. are inventors of patent applications related to TrackerSci.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.08.042.
REFERENCES
- 1.Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Götz M, Haas CA, Kempermann G, Taylor V, and Giachino C (2010). Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6, 445–456. [DOI] [PubMed] [Google Scholar]
- 2.Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollina EA, and Brunet A (2011). Epigenetic regulation of aging stem cells. Oncogene 30, 3105–3126. [DOI] [PubMed] [Google Scholar]
- 4.Galvan V, and Jin K (2007). Neurogenesis in the aging brain. Clin. Interv. Aging 2, 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, et al. (2018). Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mathews KJ, Allen KM, Boerrigter D, Ball H, Shannon Weickert C, and Double KL (2017). Evidence for reduced neurogenesis in the aging human hippocampus despite stable stem cell markers. Aging Cell 16, 1195–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, Hession C, Zhang F, and Regev A (2016). Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salic A, and Mitchison TJ (2008). A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 105, 2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, and Shendure J (2015). Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vitak SA, Torkenczy KA, Rosenkrantz JL, Fields AJ, Christiansen L, Wong MH, Carbone L, Steemers FJ, and Adey A (2017). Sequencing thousands of single-cell genomes with combinatorial indexing. Nat. Methods 14, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, and Shendure J (2017). Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, Sun D, Sinnamon JR, Shendure J, Trapnell C, O’Roak BJ, et al. (2018). Highly scalable generation of DNA methylation profiles in single cells. Nat. Biotechnol 36, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ, et al. (2017). Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 357, 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ, et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sziraki A, Lu Z, Lee J, Banyai G, Anderson S, Abdulraouf A, Metzner E, Liao A, Epstein A, Xu Z, et al. (2022). A global view of aging and Alzheimer’s pathogenesis-associated cell population dynamics and molecular signatures in the human and mouse brains. Preprint at bioRxiv. 10.1101/2022.09.28.509825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 360, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin G, Huang Y-C, Shindel AW, Banie L, Wang G, Lue TF, and Lin C-S (2009). Labeling and tracking of mesenchymal stromal cells with EdU. Cytotherapy 11, 864–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke ST, Calderon V, and Bradford JA (2017). Click Chemistry for Analysis of Cell Proliferation in Flow Cytometry. Curr. Protoc. Cytom 82, 7–49. [DOI] [PubMed] [Google Scholar]
- 19.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech 2008, P10008. 10.1088/1742-5468/2008/10/p10008. [DOI] [Google Scholar]
- 21.McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 3, 861. 10.21105/joss.00861. [DOI] [Google Scholar]
- 22.Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Väremo L, Giacomello S, Falcão AM, Meijer M, Björklund ÅK, et al. (2018). Transcriptional Convergence of Oligodendrocyte Lineage Progenitors during Development. Dev. Cell 46, 504–517.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pastrana E, Cheng L-C, and Doetsch F (2009). Simultaneous prospective purification of adult subventricular zone neural stem cells and their progeny. Proc. Natl. Acad. Sci. USA 106, 6387–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hodge RD, and Hevner RF (2011). Expression and actions of transcription factors in adult hippocampal neurogenesis. Dev. Neurobiol 71, 680–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeisel A, Hochgerner H, Lönnerberg P, Johnsson A, Memic F, van der Zwan J, Häring M, Braun E, Borm LE, La Manno G, et al. (2018). Molecular Architecture of the Mouse Nervous System. Cell 174, 999–1014.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langlieb J, Sachdev N, Balderrama K, Nadaf N, Raj M, Murray E, Webber J, Vanderburg C, Gazestani V, Tward D, et al. (2023). The cell type composition of the adult mouse brain revealed by single cell and spatial genomics. Preprint at bioRxiv. 10.1101/2023.03.06.531307. [DOI] [Google Scholar]
- 28.Petryniak MA, Potter GB, Rowitch DH, and Rubenstein JLR (2007). Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron 55, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang K, Chen S, Yang Q, Guo S, Chen Q, Liu Z, Li L, Jiang M, Li H, Hu J, et al. (2022). The Oligodendrocyte Transcription Factor 2 OLIG2 regulates transcriptional repression during myelinogenesis in rodents. Nat. Commun 13, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yeh H, and Ikezu T (2019). Transcriptional and Epigenetic Regulation of Microglia in Health and Disease. Trends Mol. Med 25, 96–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwasaki H, and Akashi K (2007). Myeloid lineage commitment from the hematopoietic stem cell. Immunity 26, 726–740. [DOI] [PubMed] [Google Scholar]
- 32.Osumi N, Shinohara H, Numayama-Tsuruta K, and Maekawa M (2008). Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cell. 26, 1663–1672. [DOI] [PubMed] [Google Scholar]
- 33.Ninkovic J, Steiner-Mezzadri A, Jawerka M, Akinci U, Masserdotti G, Petricca S, Fischer J, von Holst A, Beckers J, Lie CD, et al. (2013). The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell 13, 403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colombo E, Collombat P, Colasante G, Bianchi M, Long J, Mansouri A, Rubenstein JLR, and Broccoli V (2007). Inactivation of Arx, the Murine Ortholog of the X-Linked Lissencephaly with Ambiguous Genitalia Gene, Leads to Severe Disorganization of the Ventral Telencephalon with Impaired Neuronal Migration and Differentiation. J. Neurosci 27, 4786–4798. 10.1523/jneurosci.0417-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corlier F, Hafzalla G, Faskowitz J, Kuller LH, Becker JT, Lopez OL, Thompson PM, and Braskie MN (2018). Systemic inflammation as a predictor of brain aging: Contributions of physical activity, metabolic risk, and genetic risk. Neuroimage 172, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Bergen V, Lange M, Peidli S, Wolf FA, and Theis FJ (2020). Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414. [DOI] [PubMed] [Google Scholar]
- 38.Ratz M, von Berlin L, Larsson L, Martin M, Westholm JO, La Manno G, Lundeberg J, and Frisén J (2022). Clonal relations in the mouse brain revealed by single-cell and spatial transcriptomics. Nat. Neurosci 25, 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zywitza V, Misios A, Bunatyan L, Willnow TE, and Rajewsky N (2018). Single-Cell Transcriptomics Characterizes Cell Types in the Subventricular Zone and Uncovers Molecular Defects Impairing Adult Neurogenesis. Cell Rep. 25, 2457–2469.e8. [DOI] [PubMed] [Google Scholar]
- 40.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brulet R, Zhu J, Aktar M, Hsieh J, and Cho K-O (2017). Mice with conditional NeuroD1 knockout display reduced aberrant hippocampal neurogenesis but no change in epileptic seizures. Exp. Neurol 293, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong SM, Liu Z, Fan Y, Neumann M, Won SJ, Lac D, Lum X, Weinstein PR, and Liu J (2007). Reduced hippocampal neurogenesis and skill reaching performance in adult Emx1 mutant mice. Exp. Neurol 206, 24–32. [DOI] [PubMed] [Google Scholar]
- 43.Micheli L, Ceccarelli M, Gioia R, D’Andrea G, Farioli-Vecchioli S, Costanzi M, Saraulli D, Cestari V, and Tirone F (2017). Terminal Differentiation of Adult Hippocampal Progenitor Cells Is a Step Functionally Dissociable from Proliferation and Is Controlled by Tis21, Id3 and NeuroD2. Front. Cell. Neurosci 11, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mall M, Kareta MS, Chanda S, Ahlenius H, Perotti N, Zhou B, Grieder SD, Ge X, Drake S, Euong Ang C, et al. (2017). Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J, Wang C, Zhang Z, Wen Y, An L, Liang Q, Xu Z, Wei S, Li W, Guo T, et al. (2018). Transcription Factors Sp8 and Sp9 Coordinately Regulate Olfactory Bulb Interneuron Development. Cereb. Cortex 28, 3278–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Díaz-Guerra E, Pignatelli J, Nieto-Estévez V, and Vicario-Abejón C (2013). Transcriptional regulation of olfactory bulb neurogenesis. Anat. Rec 296, 1364–1382. [DOI] [PubMed] [Google Scholar]
- 47.Minamide K, Sato T, Nakanishi Y, Ohno H, Kato T, Asano J, and Ohteki T (2020). IRF2 maintains the stemness of colonic stem cells by limiting physiological stress from interferon. Sci. Rep 10, 14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yi T, Lee D-S, Jeon M-S, Kwon SW, and Song SU (2012). Gene expression profile reveals that STAT2 is involved in the immunosuppressive function of human bone marrow-derived mesenchymal stem cells. Gene 497, 131–139. [DOI] [PubMed] [Google Scholar]
- 49.Hock H, Meade E, Medeiros S, Schindler JW, Valk PJM, Fujiwara Y, and Orkin SH (2004). Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 18, 2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dann E, Henderson NC, Teichmann SA, Morgan MD, and Marioni JC (2022). Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol 40, 245–253. 10.1038/s41587-021-01033-z. [DOI] [PubMed] [Google Scholar]
- 51.Mahar I, MacIsaac A, Kim JJ, Qiang C, Davoli MA, Turecki G, and Mechawar N (2016). Effects of neuregulin-1 administration on neurogenesis in the adult mouse hippocampus, and characterization of immature neurons along the septotemporal axis. Sci. Rep 6, 30467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiao H, Li Y, Feng C, Duo S, Ji F, and Jiao J (2018). Nap1l1 Controls Embryonic Neural Progenitor Cell Proliferation and Differentiation in the Developing Brain. Cell Rep. 22, 2279–2293. [DOI] [PubMed] [Google Scholar]
- 53.Bertacchi M, Romano AL, Loubat A, Tran Mau-Them F, Willems M, Faivre L, Khau van Kien P, Perrin L, Devillard F, Sorlin A, et al. (2020). NR2F1 regulates regional progenitor dynamics in the mouse neocortex and cortical gyrification in BBSOAS patients. EMBO J. 39, e104163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruetz TJ, Kashiwagi CM, Morton B, Yeo RW, Leeman DS, Morgens DW, Kimberly Tsui C, Li A, Bassik MC, and Brunet A (2021). In vitro and in vivo CRISPR-Cas9 screens reveal drivers of aging in neural stem cells of the brain. Preprint at bioRxiv. 10.1101/2021.11.23.469762. [DOI] [Google Scholar]
- 55.Momotani K, Khromov AS, Miyake T, Stukenberg PT, and Somlyo AV (2008). Cep57, a multidomain protein with unique microtubule and centrosomal localization domains. Biochem. J 412, 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hwang M, Jun DW, Kang EH, Yoon K-A, Cheong H, Kim Y-H, Lee C-H, and Kim S (2019). EI24, as a Component of Autophagy, Is Involved in Pancreatic Cell Proliferation. Front. Oncol 9, 652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vecchione A, Baldassarre G, Ishii H, Nicoloso MS, Belletti B, Petrocca F, Zanesi N, Fong LYY, Battista S, Guarnieri D, et al. (2007). Fez1/Lzts1 absence impairs Cdk1/Cdc25C interaction during mitosis and predisposes mice to cancer development. Cancer Cell 11, 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kagawa Y, Matsumoto S, Kamioka Y, Mimori K, Naito Y, Ishii T, Okuzaki D, Nishida N, Maeda S, Naito A, et al. (2013). Cell cycle-dependent Rho GTPase activity dynamically regulates cancer cell motility and invasion in vivo. PLoS One 8, e83629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ou G-Y, Lin W-W, and Zhao W-J (2021). Neuregulins in Neurodegenerative Diseases. Front. Aging Neurosci 13, 662474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paterson C, Wang Y, Hyde TM, Weinberger DR, Kleinman JE, and Law AJ (2017). Temporal, Diagnostic, and Tissue-Specific Regulation of NRG3 Isoform Expression in Human Brain Development and Affective Disorders. Am. J. Psychiatry 174, 256–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mahar I, Tan S, Davoli MA, Dominguez-Lopez S, Qiang C, Rachalski A, Turecki G, and Mechawar N (2011). Subchronic peripheral neuregulin-1 increases ventral hippocampal neurogenesis and induces antidepressant-like effects. PLoS One 6, e26610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinameri E, Inoue T, Aruga J, Imayoshi I, Kageyama R, Shimogori T, and Moore AW (2008). Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS One 3, e3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, Bolognesi D, Barry DW, and Broder S (1985). 3’-Azido-3’-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymph-adenopathy-associated virus in vitro. Proc. Natl. Acad. Sci. USA 82, 7096–7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, Mayweather BA, Nguyen L, Niziolek Z, Ozek C, et al. (2019). Single-cell transcriptomic profiling of the aging mouse brain. Nat. Neurosci 22, 1696–1708. [DOI] [PubMed] [Google Scholar]
- 65.Cheung LYM, George AS, McGee SR, Daly AZ, Brinkmeier ML, Ellsworth BS, and Camper SA (2018). Single-Cell RNA Sequencing Reveals Novel Markers of Male Pituitary Stem Cells and Hormone-Producing Cell Types. Endocrinology 159, 3910–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu Y, Fang K-H, Shen L-P, Cao S-Y, Yuan F, Su Y, Xu M, Pan Y, Chen Y, and Liu Y (2018). The telomerase inhibitor AZT enhances differentiation and prevents overgrowth of human pluripotent stem cell-derived neural progenitors. J. Biol. Chem 293, 8722–8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou Q-G, Hu Y, Wu D-L, Zhu L-J, Chen C, Jin X, Luo C-X, Wu H-Y, Zhang J, and Zhu D-Y (2011). Hippocampal telomerase is involved in the modulation of depressive behaviors. J. Neurosci 31, 12258–12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aberle T, Piefke S, Hillgärtner S, Tamm ER, Wegner M, and Küspert M (2022). Transcription factor Zfp276 drives oligodendroglial differentiation and myelination by switching off the progenitor cell program. Nucleic Acids Res. 50, 1951–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fletcher JL, Makowiecki K, Cullen CL, and Young KM (2021). Oligodendrogenesis and myelination regulate cortical development, plasticity and circuit function. Semin. Cell Dev. Biol 118, 14–23. [DOI] [PubMed] [Google Scholar]
- 70.Mathews ES, and Appel B (2016). Cholesterol Biosynthesis Supports Myelin Gene Expression and Axon Ensheathment through Modulation of P13K/Akt/mTor Signaling. J. Neurosci 36, 7628–7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Emery B, and Lu QR (2015). Transcriptional and Epigenetic Regulation of Oligodendrocyte Development and Myelination in the Central Nervous System. Cold Spring Harb. Perspect. Biol 7, a020461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kato K, Konno D, Berry M, Matsuzaki F, Logan A, and Hidalgo A (2015). Prox1 Inhibits Proliferation and Is Required for Differentiation of the Oligodendrocyte Cell Lineage in the Mouse. PLoS One 10, e0145334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Javed A, Mattar P, Cui A, and Cayouette M (2021). Ikaros family proteins regulate developmental windows in the mouse retina through convergent and divergent transcriptional programs. Preprint at bioRxiv. 10.1101/2021.12.01.470829. [DOI] [Google Scholar]
- 74.Sim FJ, Zhao C, Penderis J, and Franklin RJM (2002). The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J. Neurosci 22, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He L, Beghi F, Baral V, Dépond M, Zhang Y, Joulin V, Rueda BR, Gonin P, Foudi A, Wittner M, and Louache F (2019). CABLES1 Deficiency Impairs Quiescence and Stress Responses of Hematopoietic Stem Cells in Intrinsic and Extrinsic Manners. Stem Cell Rep. 13, 274–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Linneberg C, Harboe M, and Laursen LS (2015). Axo-Glia Interaction Preceding CNS Myelination Is Regulated by Bidirectional Eph-Ephrin Signaling. ASN Neuro 7, 1759091415602859. 10.1177/1759091415602859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smith A, Robinson V, Patel K, and Wilkinson DG (1997). The EphA4 and EphB1 receptor tyrosine kinases and ephrin-B2 ligand regulate targeted migration of branchial neural crest cells. Curr. Biol 7, 561–570. [DOI] [PubMed] [Google Scholar]
- 78.Tafesse FG, Huitema K, Hermansson M, van der Poel S, van den Dikkenberg J, Uphoff A, Somerharju P, and Holthuis JCM (2007). Both sphingomyelin synthases SMS1 and SMS2 are required for sphingomyelin homeostasis and growth in human HeLa cells. J. Biol. Chem 282, 17537–17547. [DOI] [PubMed] [Google Scholar]
- 79.Huitema K, van den Dikkenberg J, Brouwers JFHM, and Holthuis JCM (2004). Identification of a family of animal sphingomyelin synthases. EMBO J. 23, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krut O, Wiegmann K, Kashkar H, Yazdanpanah B, and Krönke M (2006). Novel tumor necrosis factor-responsive mammalian neutral sphingomyelinase-3 is a C-tail-anchored protein. J. Biol. Chem 281, 13784–13793. [DOI] [PubMed] [Google Scholar]
- 81.Hannun YA, and Obeid LM (2008). Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol 9, 139–150. [DOI] [PubMed] [Google Scholar]
- 82.Jana A, Hogan EL, and Pahan K (2009). Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J. Neurol. Sci 278, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoo S-W, Agarwal A, Smith MD, Khuder SS, Baxi EG, Thomas AG, Rojas C, Moniruzzaman M, Slusher BS, Bergles DE, et al. (2020). Inhibition of neutral sphingomyelinase 2 promotes remyelination. Sci. Adv 6, eaba5210. 10.1126/sciadv.aba5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Weng C, Ding M, Fan S, Cao Q, and Lu Z (2017). Transcription factor 7 like 2 promotes oligodendrocyte differentiation and remyelination. Mol. Med. Rep 16, 1864–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kioussi C, Gross MK, and Gruss P (1995). Pax3: a paired domain gene as a regulator in PNS myelination. Neuron 15, 553–562. [DOI] [PubMed] [Google Scholar]
- 86.Siletti K, Hodge R, Albiach AM, Hu L, Lee KW, Lönnerberg P, Bakken T, Ding S-L, Clark M, Casper T, et al. (2022). Transcriptomic diversity of cell types across the adult human brain. Preprint at bioRxiv. 10.1101/2022.10.12.511898. [DOI] [PubMed] [Google Scholar]
- 87.Bergsland M, Werme M, Malewicz M, Perlmann T, and Muhr J (2006). The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 20, 3475–3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Y, Lin L, Lai H, Parada LF, and Lei L (2013). Transcription factor Sox11 is essential for both embryonic and adult neurogenesis. Dev. Dyn 242, 638–653. [DOI] [PubMed] [Google Scholar]
- 89.Zhou Y, Su Y, Li S, Kennedy BC, Zhang DY, Bond AM, Sun Y, Jacob F, Lu L, Hu P, et al. (2022). Molecular landscapes of human hippocampal immature neurons across lifespan. Nature 607, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gerrits E, Brouwer N, Kooistra SM, Woodbury ME, Vermeiren Y, Lambourne M, Mulder J, Kummer M, Möller T, Biber K, et al. (2021). Distinct amyloid-βand tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, et al. (2009). The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat. Neurosci 12, 1398–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen R, Wu X, Jiang L, and Zhang Y (2017). Single-Cell RNA-Seq Reveals Hypothalamic Cell Diversity. Cell Rep. 18, 3227–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thakur S, Dhapola R, Sarma P, Medhi B, and Reddy DH (2023). Neuroinflammation in Alzheimer’s Disease: Current Progress in Molecular Signaling and Therapeutics. Inflammation 46, 1–17. [DOI] [PubMed] [Google Scholar]
- 94.Allen WE, Blosser TR, Sullivan ZA, Dulac C, and Zhuang X (2023). Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 186, 194–208.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F, and Macosko EZ (2019). Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li YE, Preissl S, Hou X, Zhang Z, Zhang K, Qiu Y, Poirion OB, Li B, Chiou J, Liu H, et al. (2021). An atlas of gene regulatory elements in adult mouse cerebrum. Nature 598, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xu W, Yang W, Zhang Y, Chen Y, Hong N, Zhang Q, Wang X, Hu Y, Song K, Jin W, and Chen X (2022). ISSAAC-seq enables sensitive and flexible multimodal profiling of chromatin accessibility and gene expression in single cells. Nat. Methods 19, 1243–1249. [DOI] [PubMed] [Google Scholar]
- 100.Zhu C, Zhang Y, Li YE, Lucero J, Behrens MM, and Ren B (2021). Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat. Methods 18, 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, Filippova GN, Huang X, Christiansen L, DeWitt WS, et al. (2018). A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kalinina A, and Lagace D (2022). Single-Cell and Single-Nucleus RNA-seq Analysis of Adult Neurogenesis. Cells 11, 1633. 10.3390/cells11101633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Basak O, Krieger TG, Muraro MJ, Wiebrands K, Stange DE, Frias-Aldeguer J, Rivron NC, van de Wetering M, van Es JH, van Oudenaarden A, et al. (2018). Troy+ brain stem cells cycle through quiescence and regulate their number by sensing niche occupancy. Proc. Natl. Acad. Sci. USA 115, E610–E619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dulken BW, Leeman DS, Boutet SC, Hebestreit K, and Brunet A (2017). Single-Cell Transcriptomic Analysis Defines Heterogeneity and Transcriptional Dynamics in the Adult Neural Stem Cell Lineage. Cell Rep. 18, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shah PT, Stratton JA, Stykel MG, Abbasi S, Sharma S, Mayr KA, Koblinger K, Whelan PJ, and Biernaskie J (2018). Single-Cell Transcriptomics and Fate Mapping of Ependymal Cells Reveals an Absence of Neural Stem Cell Function. Cell 173, 1045–1057.e9. [DOI] [PubMed] [Google Scholar]
- 106.Kalamakis G, Brüne D, Ravichandran S, Bolz J, Fan W, Ziebell F, Stiehl T, Catalá-Martinez F, Kupke J, Zhao S, et al. (2019). Quiescence Modulates Stem Cell Maintenance and Regenerative Capacity in the Aging Brain. Cell 176, 1407–1419.e14. [DOI] [PubMed] [Google Scholar]
- 107.Mizrak D, Levitin HM, Delgado AC, Crotet V, Yuan J, Chaker Z, Silva-Vargas V, Sims PA, and Doetsch F (2019). Single-Cell Analysis of Regional Differences in Adult V-SVZ Neural Stem Cell Lineages. Cell Rep. 26, 394–406.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cebrian-Silla A, Nascimento MA, Redmond SA, Mansky B, Wu D, Obernier K, Romero Rodriguez R, Gonzalez-Granero S, García-Verdugo JM, Lim DA, and Álvarez-Buylla A (2021). Single-cell analysis of the ventricular-subventricular zone reveals signatures of dorsal and ventral adult neurogenesis. Elife 10, e67436. 10.7554/eL-ife.67436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kuhn HG, Eisch AJ, Spalding K, and Peterson DA (2016). Detection and Phenotypic Characterization of Adult Neurogenesis. Cold Spring Harb. Perspect. Biol 8, a025981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang F, Ren S-Y, Chen J-F, Liu K, Li R-X, Li Z-F, Hu B, Niu J-Q, Xiao L, Chan JR, and Mei F (2020). Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat. Neurosci 23, 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang X, Huang N, Xiao L, Wang F, and Li T (2021). Replenishing the Aged Brains: Targeting Oligodendrocytes and Myelination? Front. Aging Neurosci 13, 760200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N,Ávila J, and Llorens-Martín M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- 113.Oren Y, Tsabar M, Cuoco MS, Amir-Zilberstein L, Cabanos HF, Hütter JC, Hu B, Thakore PI, Tabaka M, Fulco CP, et al. (2021). Cycling cancer persister cells arise from lineages with distinct programs. Nature 596, 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Delgado RN, Allen DE, Keefe MG, Mancia Leon WR, Ziffra RS, Crouch EE, Alvarez-Buylla A, and Nowakowski TJ (2022). Individual human cortical progenitors can produce excitatory and inhibitory neurons. Nature 601, 397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, Daza RM, McFaline-Figueroa JL, Packer JS, Christiansen L, et al. (2018). Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T, et al. (2020). Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 183, 1103–1116.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen S, Lake BB, and Zhang K (2019). High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol 37, 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, Lucero J, Behrens MM, Hu M, and Ren B (2019). An ultra high-throughput method for single-cell joint analysis of open chromatin and transcriptome. Nat. Struct. Mol. Biol 26, 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sierra A, Encinas JM, Deudero JJP, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, and Maletic-Savatic M (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sottocornola R, and Lo Celso C (2012). Dormancy in the stem cell niche. Stem Cell Res. Ther 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Theodoris CV, Xiao L, Chopra A, Chaffin MD, Al Sayed ZR, Hill MC, Mantineo H, Brydon EM, Zeng Z, Liu XS, and Ellinor PT (2023). Transfer learning enables predictions in network biology. Nature 618, 616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, Zeisel A, Borm LE, Stott SRW, Toledo EM, Villaescusa JC, et al. (2016). Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Liu DD, He JQ, Sinha R, Eastman AE, Toland AM, Morri M, Neff NF, Vogel H, Uchida N, and Weissman IL (2023). Purification and characterization of human neural stem and progenitor cells. Cell 186, 1179–1194.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Paap RH, Oosterbroek S, Wagemans CMRJ, von Oerthel L, Schellevis RD, Vastenhouw-van der Linden AJA, Groot Koerkamp MJA, Hoekman MFM, and Smidt MP (2016). FoxO6 affects Plxna4-mediated neuronal migration during mouse cortical development. Proc. Natl. Acad. Sci. USA 113, E7087–E7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Prosser HM, Bradley A, and Caldwell MA (2007). Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. Eur. J. Neurosci 26, 3339–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AMJ, Resch JM, McCarroll SA, et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nat. Neurosci 20, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, and Rinn JL (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol 32, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, Motamedi A, Shiau AK, Zhou X, Xie F, et al. (2021). Comprehensive analysis of single cell ATAC-seq data with SnapATAC. Nat. Commun 12, 1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schep AN, Wu B, Buenrostro JD, and Greenleaf WJ (2017). chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- 138.Speir ML, Bhaduri A, Markov NS, Moreno P, Nowakowski TJ, Papatheodorou I, Pollen AA, Raney BJ, Seninge L, Kent WJ, and Haeussler M (2021). UCSC Cell Browser: visualize your single-cell data. Bioinformatics 37, 4578–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmitt FA, Nelson PT, Abner E, Scheff S, Jicha GA, Smith C, Cooper G, Mendiondo M, Danner DD, Van Eldik LJ, et al. (2012). University of Kentucky Sanders-Brown healthy brain aging volunteers: donor characteristics, procedures and neuropathology. Curr. Alzheimer Res. 9, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nelson PT, Wang W-X, Janse SA, and Thompson KL (2018). MicroRNA expression patterns in human anterior cingulate and motor cortex: A study of dementia with Lewy bodies cases and controls. Brain Res. 1678, 374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Broad Institute (2019). Picard toolkit (Broad Institute, GitHub repository; ). [Google Scholar]
- 143.Cao J, O’Day DR, Pliner HA, Kingsley PD, Deng M, Daza RM, Zager MA, Aldinger KA, Blecher-Gonen R, Zhang F, et al. (2020). A human cell atlas of fetal gene expression. Science 370, eaba7721. 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rodriguez A, and Laio A (2014). Machine learning. Clustering by fast search and find of density peaks. Science 344, 1492–1496. [DOI] [PubMed] [Google Scholar]
- 147.Schep A (2017). motifmatchr: Fast Motif Matching in R. https://github.com/GreenleafLab/motifmatchr/.
- 148.Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.McIntyre RL, Molenaars M, Schomakers BV, Gao AW, Kamble R, Jongejan A, van Weeghel M, van Kuilenburg ABP, Possemato R, Houtkooper RH, and Janssens GE (2023). Anti-retroviral treatment with zidovudine alters pyrimidine metabolism, reduces translation, and extends healthy longevity via ATF-4. Cell Rep. 42, 111928. [DOI] [PubMed] [Google Scholar]
- 150.Solius GM, Maltsev DI, Belousov VV, and Podgorny OV (2021). Recent advances in nucleotide analogue-based techniques for tracking dividing stem cells: An overview. J. Biol. Chem 297, 101345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq and ATAC-seq data have been deposited at Gene Expression Omnibus. Accession numbers are listed in the key resources table. Interactive website for data exploration is available at UCSC cell browser138: https://adult-mouse-brain-newborn.cells.ucsc.edu.
Computational pipeline scripts for processing data are available at this github repository: https://github.com/ZiyuLu041/TrackerSci_analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat anti-Iba1 | Novus Biologicals | Cat#NB100–1028; RRID:AB_521594 |
| Mouse anti-Ki67 | Invitrogen | Cat#14-5699-82; RRID:AB_2016711 |
| Biological samples | ||
| Whole mouse brains | This study | N/A |
| Aged human brain tissues | University of Kentucky AD Center Tissue Bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| 5-Ethynyl-2′-deoxyuridine | Lumiprobe | Cat#20540 |
| SUPERase•In™ RNase Inhibitor | Thermo Fisher Scientific | Cat#AM2696 |
| BSA | NEB | Cat#B90000S |
| TRIS 1.0M Sterile Solution, pH 7.5 | VWR | Cat#97062–936 |
| 16% methanol-free formaldehyde | Thermo Fisher Scientific | Cat#28906 |
| SuperScript™ IV Reverse Transcriptase | Invitrogen | Cat#18090200 |
| mRNA Second Strand Synthesis buffer and enzyme | NEB | Cat#E6111L |
| NEBNext High-Fidelity 2X PCR Master Mix | NEB | Cat#M0541L |
| EDTA-free Protease Inhibitor Cocktail | Sigma | Cat#11873580001 |
| 3′-Azido-3′-deoxythymidine | Thermo Fisher Scientific | Cat#J65127–06 |
| Critical commercial assays | ||
| Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat#C10634 |
| Nuclei Isolation Kit: Nuclei EZ Prep Millipore Sigma | Millipore Sigma | Cat#NUC101–1KT |
| Deposited data | ||
| Raw and analyzed data | This study | GEO: GSE212251 |
| Single-nucleus RNA-seq data of whole mouse brain across three age groups and two Alzheimer’s disease associated mutants | Sziraki et al.15 | GEO: GSE212606 |
| Single-cell RNA-seq data of the adolescent mouse nervous system | Zeisel et al.26 | http://mousebrain.org/ |
| Spatial transcriptomic Slide-seq data of adult mouse brain | Langlieb et al.27 | https://www.braincelldata.org/ |
| Single-cell RNA-seq data of adult mouse brain subventricular zone | Zywitza et al.39 | GEO: GSE111527 |
| Gene scores and effect sizes from in vitro genome-wide CRISPR screen in primary neural stem cells | Ruetz et al.54 | https://www.biorxiv.org/content/biorxiv/early/2021/11/23/2021.11.23.469762/DC1/embed/media-1.xlsx?download=true |
| Single-nucleus RNA-seq data of human brain tissues from patients with Alzheimer’s disease and healthy controls | Gerrits et al.90 | GEO: GSE148822 |
| Single-cell RNA-seq data from pituitary glands of 7-week-old male C57BL/6 mice | Cheung et al.65 | GEO: GSE120410 |
| Experimental models: Cell lines | ||
| HEK293T | Gift from J. Shendure, University of Washington | N/A |
| NIH/3T3 | Gift from J. Shendure, University of Washington | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: 5XFAD | The Jackson Laboratory | RRID:MMRRC_034840-JAX |
| Software and algorithms | ||
| R | R Core | https://www.r-project.org/ |
| Python | Python Software Foundation | https://www.python.org/ |
| bcl2fastq | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| Trim Galore | Babraham Institute | https://github.com/FelixKrueger/TrimGalore |
| STAR | Dobin et al.127 | https://github.com/alexdobin/STAR |
| Samtools | Li et al.128 | http://www.htslib.org/download/ |
| Picard MarkDuplicates | Broad Institute | https://broadinstitute.github.io/picard/ |
| Scanpy | Wolf et al.129 | https://github.com/scverse/scanpy |
| Scrublet | Wolock et al.130 | https://github.com/swolock/scrublet |
| Seurat | Stuart et al.131 | https://satijalab.org/seurat/ |
| Monocle | Trapnell et al.132 | http://cole-trapnell-lab.github.io/monocle-release/ |
| SnapTools | Fang et al.133 | https://github.com/r3fang/SnapTools |
| SnapATAC | Fang et al.133 | https://github.com/r3fang/SnapTools |
| MACS2 | Zhang et al.134 | https://github.com/macs3-project/MACS |
| bedtools | Quinlan and Hall135 | https://bedtools.readthedocs.io/en/latest/ |
| chromVar | Schep et al.136 | https://github.com/GreenleafLab/chromVAR |
| scVelo | Bergen et al.37 | https://github.com/theislab/scvelo |
| ComplexHeatmap | Gu et al.137 | https://github.com/jokergoo/ComplexHeatmap |
| miloR | Dann et al.50 | https://github.com/MarioniLab/miloR |