

ABSTRACT
Proteolysis-targeting chimeras (PROTACs) based on the ubiquitin-proteasome system have made great progress in the field of drug discovery. There is mounting evidence that the accumulation of aggregation-prone proteins or malfunctioning organelles is associated with the occurrence of various age-related neurodegenerative disorders and cancers. However, PROTACs are inefficient for the degradation of such large targets due to the narrow entrance channel of the proteasome. Macroautophagy (hereafter referred to as autophagy) is known as a self-degradative process involved in the degradation of bulk cytoplasmic components or specific cargoes that are sequestered into autophagosomes. In the present study, we report the development of a generalizable strategy for the targeted degradation of large targets. Our results suggested that tethering large target models to phagophore-associated ATG16L1 or LC3 induced targeted autophagic degradation of the large target models. Furthermore, we successfully applied this autophagy-targeting degradation strategy to the targeted degradation of HTT65Q aggregates and mitochondria. Specifically, chimeras consisting of polyQ-binding peptide 1 (QBP) and ATG16L1-binding peptide (ABP) or LC3-interacting region (LIR) induced targeted autophagic degradation of pathogenic HTT65Q aggregates; and the chimeras consisting of mitochondria-targeting sequence (MTS) and ABP or LIR promoted targeted autophagic degradation of dysfunctional mitochondria, hence ameliorating mitochondrial dysfunction in a Parkinson disease cell model and protecting cells from apoptosis induced by the mitochondrial stress agent FCCP. Therefore, this study provides a new strategy for the selective proteolysis of large targets and enrich the toolkit for autophagy-targeting degradation.
Abbreviations: ABP: ATG16L1-binding peptide; ATG16L1: autophagy related 16 like 1; ATTEC: autophagy-tethering compound; AUTAC: autophagy-targeting chimera; AUTOTAC: autophagy-targeting chimera; Baf A1: bafilomycin A1; BCL2: BCL2 apoptosis regulator; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CASP3: caspase 3; CPP: cell-penetrating peptide; CQ: chloroquine phosphate; DAPI: 4’,6-diamidino-2-phenylindole; DCM: dichloromethane; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide; EBSS: Earle′s balanced salt solution; FCCP: carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone; FITC: fluorescein-5-isothiocyanate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; HEK293: human embryonic kidney 293; HEK293T: human embryonic kidney 293T; HPLC: high-performance liquid chromatography; HRP: horseradish peroxidase; HTT: huntingtin; LIR: LC3-interacting region; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MFF: mitochondrial fission factor; MTS: mitochondria-targeting sequence; NBR1: NBR1 autophagy cargo receptor; NLRX1: NLR family member X1; OPTN: optineurin; P2A: self-cleaving 2A peptide; PB1: Phox and Bem1p; PBS: phosphate-buffered saline; PE: phosphatidylethanolamine; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; PROTACs: proteolysis-targeting chimeras; QBP: polyQ-binding peptide 1; SBP: streptavidin-binding peptide; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis; SPATA33: spermatogenesis associated 33; TIMM23: translocase of inner mitochondrial membrane 23; TMEM59: transmembrane protein 59; TOMM20: translocase of outer mitochondrial membrane 20; UBA: ubiquitin-associated; WT: wild type.
KEYWORDS: ATG16L1, autophagy-targeting degradation, HTT65Q aggregates, LC3, mitochondria, proteolysis-targeting chimeras
Introduction
Targeted protein degradation has emerged as one of the most useful strategies to elucidate biological systems as well as to develop therapeutics for undruggable disease targets [1–3]. Proteolysis-targeting chimeras (PROTACs) utilize the ubiquitin-proteasome system to induce targeted degradation of soluble proteins but are inefficient for degradation of large targets, including protein aggregates and organelles [4–6]. The accumulation of aggregation-prone proteins or malfunctioning organelles has been shown to be associated with the development of age-related diseases, including neurodegenerative disorders and cancers [7–9]. Therefore, targeted degradation of large targets is expected to be a promising clinical strategy for treating such diseases but remains a major challenge [10,11].
Macroautophagy (hereafter referred to as autophagy) is a self-degradative process within eukaryotic cells and can be either nonselective or selective [12–14]. Nonselective autophagy drives turnover of bulk cytoplasm under starvation conditions, whereas selective autophagy degrades specific cargoes, such as protein aggregates and malfunctioning organelles [15–18]. Selective autophagy is mediated via cargo receptors that recognize and sequester specific cargoes into the autophagosome, which subsequently fuses with a lysosome, resulting in degradation of the cargoes by lysosomal acid hydrolases [19–21]. Owing to its properties of specific clearance of large substrates, the autophagy-lysosome pathway can be utilized as an ideal system for targeted degradation of large targets [22]. Indeed, several autophagy-targeting degradation strategies, including autophagy-tethering compound (ATTEC) and autophagy-targeting chimera (AUTAC/AUTOTAC), have been developed to eliminate specific large targets. Mechanistically, ATTEC works as a molecular glue or a bifunctional molecule that links large targets to the autophagy component MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) [23,24]; AUTAC triggers K63-linked polyubiquitination of the cargoes by a cGMP-based degradation tag, which is further recognized by autophagy receptors [25]; and AUTOTAC bridges the cargoes and SQSTM1/p62 by interacting with the ZZ domain of SQSTM1, leading to the oligomerization and activation of SQSTM1 [26]. These new strategies push targeted protein degradation to reach a new climax for targeted degradation of large targets [4,5,22]. Even so, to accelerate and expand the development of autophagy-based degradation inducers in various pathological contexts, there is an urgent need to find more exploitable autophagy components for autophagy-targeting degradation strategies.
ATG16L1 and LC3 are core autophagy-related proteins, and decorate on both the outer and inner membranes of the cup-shaped phagophore [27,28]. LC3 is converted to the membrane-bound phosphatidylethanolamine (PE)-conjugated form LC3-II (LC3–PE) [29,30]. ATG16L1 binds directly to membranes through separate N-terminal and C-terminal membrane-binding regions [31,32]. In most cases, cargo receptors, including SQSTM1 [33], CALCOCO2/NDP52 [34], NBR1 [35] and OPTN [36], recognize and bring cargoes to phagophores through binding with LC3 for autophagic degradation (Figure 1A). In addition, the cargo receptor SPATA33 delivers mitochondria into phagophores by binding with ATG16L1 [37]. Therefore, ATG16L1 and LC3 presenting on the inner membrane of the phagophore are supposed to serve as autophagy components for autophagy targeting; tethering ATG16L1 and LC3 could enable efficient autophagy targeting and subsequent degradation of large targets. Indeed, LC3 has been utilized as an autophagy targeting receptor in some cases [23,24].
Figure 1.
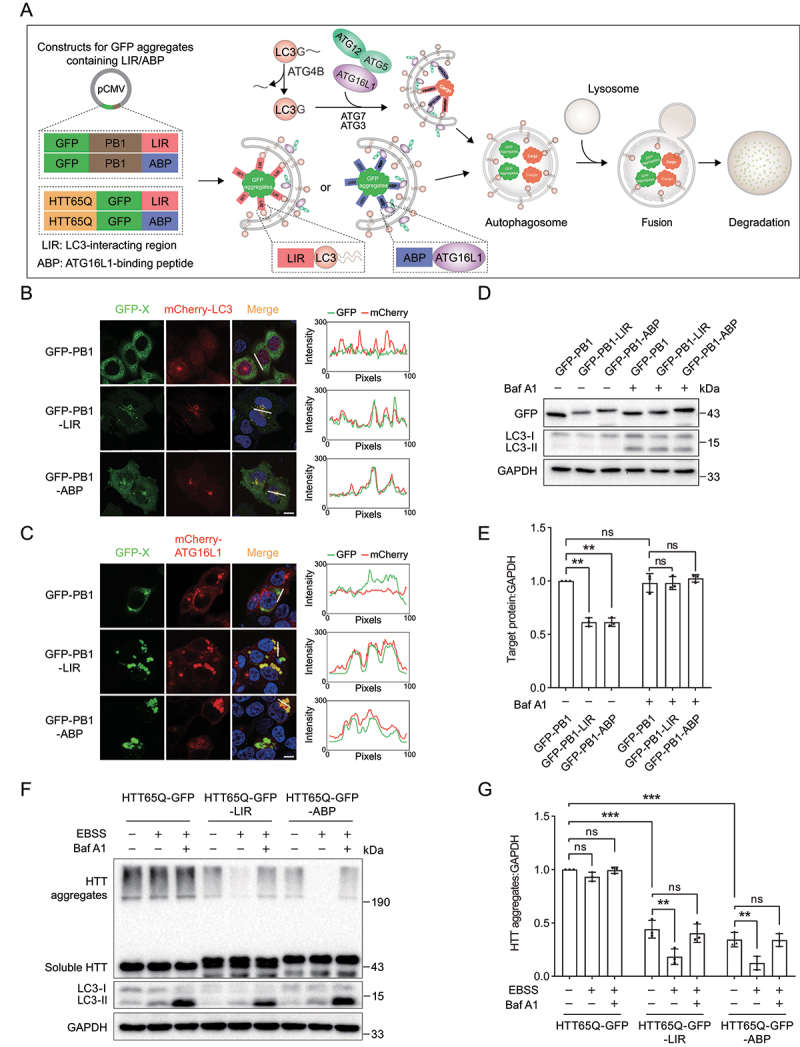
Tethering PB1 or HTT65Q aggregates to LC3 or ATG16L1 induces selective autophagy. (A) Scheme of autophagy-targeting degradation using LIR and ABP. Large targets exposing multivalent autophagy-targeting ligands (LIR or ABP) are recognized by the autophagy machineries LC3 and ATG16L1. (B) Representative images of mCherry-LC3 with GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP in HeLa cells. Cells were transiently cotransfected with mCherry-LC3 and GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP. The colocalization of mCherry-LC3 with GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP was determined by calculating fluorescence intensity of the areas marked with white lines. (C) Representative images of mCherry-ATG16L1 with GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP in HEK293T cells. The colocalization of mCherry-ATG16L1 with GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP was determined by calculating fluorescence intensity of the areas marked with white lines. (D) Immunoblot analysis of GFP-PB1, GFP-PB-LIR and GFP-PB1-ABP expressed in HEK293 cells. Cells were transiently transfected with GFP-PB1, GFP-PB-LIR or GFP-PB1-ABP for 24 h and then treated with Baf A1 (1 μM) for 12 h. (E) Quantification of the levels of target proteins as in (D). (F) Immunoblot analysis of HTT65Q-GFP, HTT65Q-GFP-LIR and HTT65Q-GFP-ABP under starvation condition or Baf A1 treatment. HTT65Q-GFP, HTT65Q-GFP-LIR and HTT65Q-GFP-ABP were transiently transfected into HeLa cells for 24 h, followed by incubation in EBSS for 2 h. The autophagy inhibition group was treated with EBSS containing 1 µM Baf A1. (G) Quantification of the levels of target proteins as in (F). Data in (E) and (G) are presented as the mean ± SEM of three independent experiments. “ns”, no significant difference; *P < 0.05, **P < 0.01, Student’s t test. Scale bar: 10 μm.
In this study, a generalizable strategy for the targeted degradation of large targets was reported. Our results suggested that chimera consisting of a ligand for a large target and ATG16L1 or LC3-binding peptide, tethers a large target to phagophore-associated ATG16L1 or LC3, thereby inducing selective autophagic clearance of the large target. In addition, our results suggested that this autophagy-targeting degradation strategy shows high selectivity for large targets. Furthermore, we successfully applied this strategy to the targeted degradation of pathogenic polyglutamine (polyQ)-expanded HTT (huntingtin) protein aggregates as well as dysfunctional mitochondria. Therefore, this study provides a new autophagy-targeting degradation strategy for large targets.
Results
Tethering PB1 or polyQ aggregates to ATG16L1 or LC3 induces targeted selective autophagy
The most important feature of the selective autophagy pathway is the involvement of autophagy receptors [19]. Most autophagy receptors possess two domains: the LC3-interacting region (LIR) and cargo-binding domain [38,39]. Autophagy receptors serve as a bridge to link autophagic substrates via its cargo-binding domain, often the ubiquitin-binding domain, to the autophagic membrane-associated LC3 via its LIR [40]. Instead, the autophagy receptor SPATA33 directly binds to the autophagy machinery ATG16L1 [37]. Autophagy receptors assemble with autophagic substrates, resulting in the formation of large cargo complexes exposing multiple LIRs or ATG16L1-binding peptides (ABPs) [19,41]. Therefore, we proposed that cargo exposing multivalent autophagy-targeting ligands could be sufficient to trigger selective autophagy (Figure 1A). To verify this hypothesis, we selected the Phox1 and Bem1p (PB1) domain responsible for SQSTM1 oligomerization or polyQ-expanded HTT protein as a large target model and two peptide sequences, including the ABP derived from TMEM59 [42] and the LIR derived from SQSTM1 [33,43,44], as autophagy-targeting ligands (Table 1).
Table 1.
Peptide sequences used in this study.
| Peptide name | Amino acid sequence (N to C) |
Binding affinity | Reference |
|---|---|---|---|
| LIR (derived from SQSTM1, SQSTM1332–346) | SGGDDDWTHLSSKEV | 3.2 µM to LC3B | [43] |
| mLIR | SGGDDDAAAASSKEV | ||
| ABP (derived from TMEM59, TMEM59263–281) | TAVEQYVPSEKLSIYGDLE | n.d. | [42] |
| mABP | TAVEQAVPSEKLSIAGDAE | ||
| SBP | MDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREP | 2.5–4.9 nM to streptavidin | [45] |
| QBP | SNWKWWPGIFD | 5.7 μM to Q62 | [46] |
| MTS1 (derived from TOMM20, TOMM201–34) | MVGRNSAIAAGVCGALFIGYCIYFDRKRRSDPNF | [47] | |
| MTS2 (derived from MFF, MFF323–342) | VMYSITVAFWLLNSWLWFRR | [48] | |
| MTS3 (derived from BCL2, BCL2218–239) | KTLLSLALVGACITLGAYLGHK | [49] | |
| MTS4 (derived from NLRX1, NLRX11–86) | MRWGHHLPRASWGSGFRRALQRPDDRIPFLIHWSWPLQGERPFGP PRAFIRHHGSSVDSAPPPGRHGRLFPSASATEAIQRHRRNL |
[50] |
Abbreviations: LIR, LC3-interacting region; ABP, ATG16L1-binding peptide; SBP, streptavidin-binding peptide; QBP, polyQ-binding peptide 1; MTS, mitochondria-targeting sequence. Bold letters represent positions where mutations were introduced. n.d., not determined.
We first examined whether the selected ligands carry PB1 aggregates into autophagosomes. A plasmid encoding GFP-fused PB1 was constructed. As expected, we observed discrete GFP puncta in HeLa and HEK293T cells expressing GFP-PB1 (Figure 1B,C) [51]. Furthermore, we constructed GFP-PB1-LIR and GFP-PB1-ABP by fusing GFP-PB1 with the selected autophagy-targeting ligands LIR or ABP (Figure 1A). We then observed that GFP-PB1-LIR and GFP-PB1-ABP formed numerous GFP punctate structures, which showed strong colocalization with LC3 and ATG16L1 in WT and SQSTM1 knockout (SQSTM1-KO) cells (Figure 1B,C Figure S1A). In addition, we observed that LIR and ABP mutants in which key residues were mutated to disrupt the interaction of the ligand LIR or ABP with their respective receptor abolished the colocalization between the GFP punctate structure and LC3-positive membrane (Table 1, Figure S1B). Therefore, these results suggested that LIR and ABP are effective and selective ligands for autophagy targeting, thereby carrying PB1 aggregates to autophagosomes.
To further exclude the possibility that GFP-PB1 are recruited by autophagy, we evaluated colocalization of endogenous SQSTM1 and mCherry-LC3 with GFP-PB1 aggregates. To this purpose, mCherry-LC3 was cotransfected with GFP-PB1, GFP-PB1-LIR or GFP-PB1-ABP in HeLa cells, followed by immunofluorescent staining of endogenous SQSTM1. We observed weak colocalization of GFP-PB1 with endogenous SQSTM1, however, both of GFP-PB1 and endogenous SQSTM1 had no colocalization with mCherry-LC3 (Figure S1C). Moreover, GFP-PB1-LIR and GFP-PB1-ABP induced the formation of LC3 puncta, and showed strong colocalization with SQSTM1 and ATG16L1.
We then tested whether such PB1 aggregates containing autophagy-targeting ligands are degraded by the autophagy pathway. HEK293 cells were transfected with plasmids encoding GFP-PB1-LIR, GFP-PB1-ABP or GFP-PB1. We observed that the protein levels of GFP-PB1-LIR and GFP-PB1-ABP decreased significantly compared with the control GFP-PB1, which was recovered by the lysosome inhibitor bafilomycin A1 (Baf A1), whereas Baf A1 treatment had no effect on the protein level of GFP-PB1 (Figure 1D,E). These results strongly support our notion that tethering large target model PB1 aggregates to LC3 or ATG16L1 induces targeted autophagic degradation of PB1 aggregates, and PB1 domain itself does not recruit the autophagy machinery and thus cannot be degraded by autophagy.
Furthermore, to reconfirm this autophagy-targeting degradation strategy, we used polyQ-expanded HTT protein as a large target model. A distinct size > 190 kDa represents aggregated HTT65Q [52,53]. We observed that the protein levels of the aggregated HTT65Q-GFP-LIR and HTT65Q-GFP-ABP decreased significantly compared with the control HTT65Q-GFP. Furthermore, the aggregated HTT65Q-GFP-LIR and HTT65Q-GFP-ABP showed markedly reduced protein levels upon induction of autophagy with amino acid starvation (EBSS), which was recovered by Baf A1 treatment (Figure 1F,G).
Therefore, these results consistently suggested that tethering large target model PB1 or HTT65Q aggregates to LC3 or ATG16L1 induces targeted autophagic degradation of the protein aggregates.
Autophagy-targeting degradation strategy shows high selectivity for large targets
Our autophagy-targeting degradation strategy was initially designed to eliminate large targets. We therefore examined whether this strategy selectively degrades large targets. We constructed GFP-LIR and GFP-ABP plasmids encoding soluble proteins, and further performed microscopy and immunoblot analysis. We observed that fusing with LIR or ABP had no influence on the cellular distribution of GFP (Figure 2A). Furthermore, the protein levels of GFP-LIR and GFP-ABP were not reduced upon autophagy induction, suggesting that fusing with the LIR or ABP sequence cannot degrade soluble protein (Figure 2B,C). In addition, we also found that Baf A1 treatment did not change the level of GFP-3×LIR (Figure S1D), suggesting that tethering triple LC3 binding peptides does not induce the degradation of GFP proteins.
Figure 2.
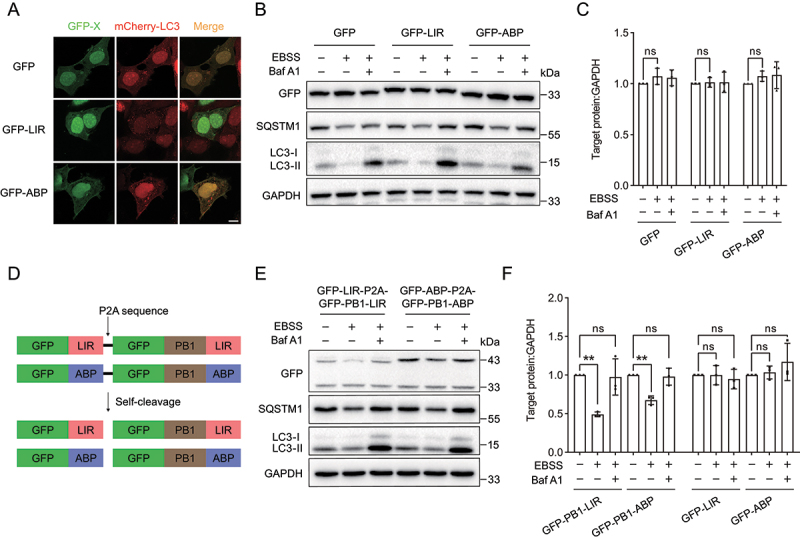
The autophagy-targeting degradation strategy shows high selectivity for large protein aggregates. (A) Representative images of mCherry-LC3 with GFP, GFP-LIR or GFP-ABP in HEK293 cells. mCherry-LC3 was transiently cotransfected with GFP, GFP-LIR or GFP-ABP in HEK293 cells. (B) Immunoblot analysis of GFP, GFP-LIR and GFP-ABP under starvation condition. GFP, GFP-LIR and GFP-ABP were transiently transfected into HeLa cells for 24 h, followed by incubation in EBSS for 12 h. The autophagy inhibition group was treated with EBSS containing 1 µM Baf A1. (C) Quantification of the levels of target proteins as in (B). (D) Scheme of GFP-LIR-P2A-GFP-PB1-LIR and GFP-ABP-P2A-GFP-PB1-ABP constructs. The self-cleavage sequence P2A enables the same amounts of soluble and PB1-generated protein aggregates. (E) Immunoblot analysis of GFP-LIR-P2A-GFP-PB1-LIR and GFP-ABP-P2A-GFP-PB1-ABP under starvation condition. The constructs were transiently transfected into HeLa cells for 24 h, followed by incubation in EBSS for 12 h. The autophagy inhibition group was treated with EBSS containing 1 µM Baf A1. (F) Quantification of the levels of target proteins as in (E). Data in (C) and (F) are presented as the mean ± SEM of three independent experiments. “ns”, no significant difference; **p < 0.01, ***p < 0.001, Student’s t test. Scale bar: 10 μm.
To exclude the possibility that the degradation results of PB1 aggregates were due to differences in transfection efficiency, we introduced a self-cleaved P2A peptide derived from porcine teschovirus-1 into between the large polymerizable model and soluble protein [54] (Figure 2D). The resulting chimeric proteins GFP-LIR-P2A-GFP-PB1-LIR and GFP-ABP-P2A-GFP-PB1-ABP enabled the same amount of soluble protein and PB1-generated polymerizable protein in cells. We observed that the aggregated forms of GFP-PB1-LIR and GFP-PB1-ABP were degraded under starvation condition, which was reversed by Baf A1 treatment (Figure 2E,F). However, the protein levels of soluble GFP-LIR and GFP-ABP were not changed upon starvation treatment.
Taken together, these results suggested that our proposed autophagy-targeting degradation strategy employing the polymerizable PB1 domain induces selective targeted autophagic degradation of GFP-LIR and GFP-ABP.
Tethering PB1 aggregates to ATG16L1 or LC3 using streptavidin system
To bring our autophagy-targeting degradation strategy closer to being an autophagy-based degrader, we set up a streptavidin system in which GFP-PB1 was fused with streptavidin to form GFP-PB1-streptavidin as a large target. Streptavidin is known to bind to proteins containing a streptavidin-binding peptide (SBP) with high affinity [45,55] (Table 1). The chimeras SBP-LIR and SBP-ABP were expected to be autophagy-based degraders that specifically degrade GFP-PB1-streptavidin aggregates (Figure 3A). We therefore investigated whether the chimeras SBP-LIR and SBP-ABP induce degradation of GFP-PB1-streptavidin. We first established HEK293 cell lines stably expressing GFP-PB1-streptavidin or GFP-streptavidin and transiently transfected cells with SBP-LIR or SBP-ABP constructs. We observed that expression of SBP-LIR or SBP-ABP decreased the protein level of GFP-PB1-streptavidin but not GFP-streptavidin (Figure 3B,C), suggesting that autophagy-based degraders SBP-LIR and SBP-ABP induce specific degradation of aggregated GFP-PB1-streptavidin.
Figure 3.
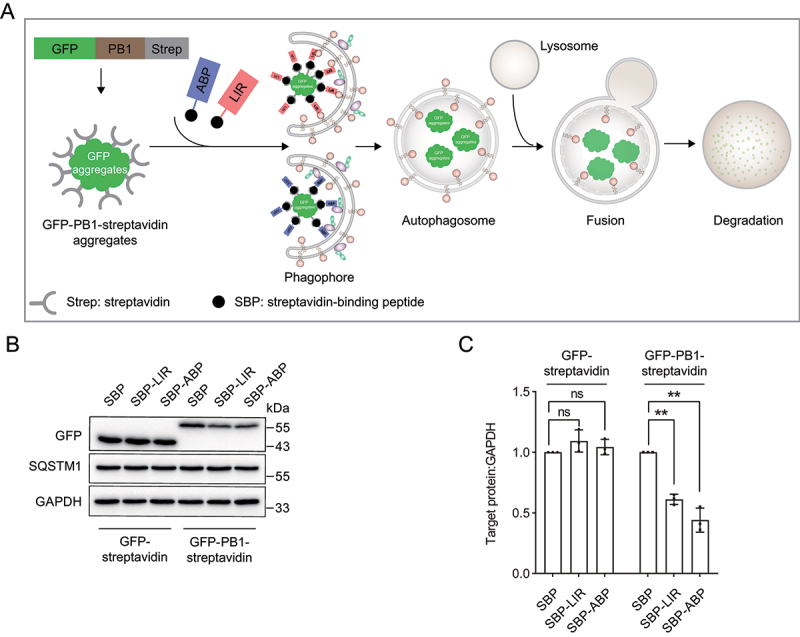
Tethering PB1 aggregates to ATG16L1 or LC3 induces targeted autophagic degradation of PB1-streptavidin aggregates using streptavidin system. (A) The concept of streptavidin system. The streptavidin system consisting of the ligand SBP for streptavidin and the autophagy-targeting ligand LIR or ABP are expected to induce targeted degradation of PB1-streptavidin aggregates. (B) Immunoblot analysis of GFP-PB1-streptavidin and GFP-streptavidin upon streptavidin system expression. HEK293 cells stably expressing GFP-PB1-streptavidin and GFP-streptavidin were transiently transfected with streptavidin system SBP-LIR and SBP-ABP constructs. (C) Quantification of the protein levels of GFP-PB1-streptavidin and GFP-streptavidin as in (B). Data in (B) are presented as the mean ± SEM of three independent experiments. “ns”, no significant difference; **p < 0.01, Student’s t test.
In cells, selective autophagic degradation of protein aggregates and malfunctioning organelles is mediated by cargo receptors that recognize and sequester specific cargoes into autophagosomes [19,20]. Mimicking cargo receptors, tethering LIR or ABP onto large targets enables the exposure of multiple LIRs or ABPs on the surface of large targets, which are further recognized by the autophagy machineries LC3 or ATG16L1 for autophagic degradation. Therefore, we proved a new autophagy-targeting degradation strategy that anchors large targets to the autophagy component LC3 or ATG16L1 for targeted autophagic degradation.
Tethering ATG16L1 or LC3 induces targeted degradation of pathogenic HTT aggregates
Aberrant protein aggregates are pathological hallmarks of many neurodegenerative diseases, which represent major health problems for the world’s aging population [10]. To expand the application of our autophagy-targeting degradation strategy in the targeted degradation of neurodegenerative disease-related protein aggregates, we tried to use the strategy to degrade HTT aggregates, the main cause of Huntington disease. We observed that HTT65Q-GFP (an N-terminally truncated HTT containing 65 tandem glutamine residues fused with GFP) formed discrete large GFP punctate structures, in line with a previous study [56]. We found that fusing LIR or ABP to HTT65Q-GFP decreased the numbers of GFP punctate structures (Figure 4A,B). Furthermore, immunoblot analysis showed that the aggregated protein levels of HTT65Q-GFP-LIR and HTT65Q-GFP-ABP were lower than that of HTT65Q-GFP (Figure 4C,D). These results suggested that fusing LIR or ABP to HTT65Q-GFP indeed induces the degradation of HTT65Q-GFP aggregates (Figure 1F,G).
Figure 4.
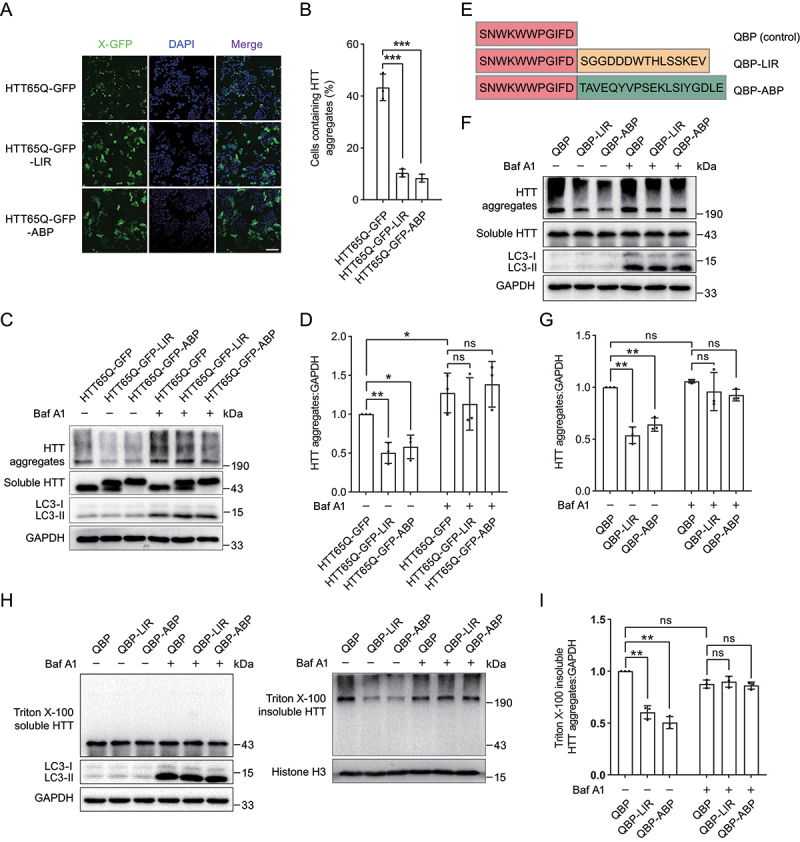
Tethering ATG16L1 or LC3 induces targeted autophagic degradation of pathogenic HTT aggregates. (A) Representative images of HTT65Q-GFP-LIR and HTT65Q-GFP-ABP puncta in HEK293 cells. (B) Quantification of the number of cells containing HTT65Q-GFP puncta as in (B). (C) Immunoblot analysis of HTT65Q-GFP-LIR and HTT65Q-GFP-ABP in HEK293 cells. (D) Quantification of the protein levels of HTT65Q-GFP-LIR and HTT65Q-GFP-ABP aggregates as in (C). (E) Scheme of QBP-LIR and QBP-ABP constructs. (F) Immunoblot analysis of HTT65Q-GFP in HEK293 cells upon expression of QBP-LIR and QBP-ABP. HEK293 cells stably expressing HTT65Q-GFP were transiently transfected with plasmids encoding QBP-LIR and QBP-ABP for 48 h. The autophagy inhibition group was treated with 1 µM Baf A1 for 12 h. (G) Quantification of the protein levels of HTT65Q-GFP aggregates as in (F). (H) Immunoblot analysis of Triton X-100 soluble (left panel) and insoluble fractions (right panel) of different HTT65Q proteins. (I) Quantification of the protein levels of Triton X-100 insoluble HTT65Q-GFP aggregates as in right panel of (H). Data in (B), (D), (G) and (I) are presented as the mean ± SEM of three independent experiments. **p < 0.01, Student’s t test. Scale bar: 100 μm.
PolyQ-binding peptide 1 (QBP) binds to polyQ stretch with strong binding affinity [46,56]. We further constructed the chimeras, including QBP-LIR and QBP-ABP (Table 1, Figure 4E). In HEK293 cells stably expressing HTT65Q-GFP, both the expression of QBP-LIR and QBP-ABP decreased the protein level of HTT65Q-GFP aggregates (Figure 4F,G). Moreover, Baf A1 treatment abolished the degradation effect of QBP-LIR and QBP-ABP on HTT65Q-GFP aggregates (Figure 4F,G), suggesting that QBP-LIR and QBP-ABP induce targeted degradation of HTT65Q-GFP aggregates dependent on the autophagy-lysosome pathway.
To further dissect degradation efficiency of aggregated HTT65Q, insoluble HTT65Q aggregates and soluble HTT65Q were isolated by extraction with Triton X-100. We found that QBP-LIR and QBP-ABP induced the degradation of Triton X-100 insoluble HTT aggregates, which was reversed by Baf A1 treatment (Figure 4H,I).
Taken together, these results suggested that our proposed autophagy-targeting degradation strategy can be applied in the targeted degradation of cellular pathogenic HTT aggregates.
The chimeric peptide CPP-QBP-LIR induces targeted degradation of HTT aggregates
To apply our autophagy-targeting degradation strategy for the targeted degradation of HTT aggregates as a chemical degrader, we designed a chimeric peptide consisting of QBP, LIR and polyarginine cell-penetrating peptide (CPP) (Figure 5A). Then we synthesized CPP-QBP-LIR peptide as well as the FITC-linked peptide employing an Fmoc solid-phase peptide synthesis strategy (Figure S2 and S3). By fluorescence analysis, we assessed the cell penetrating ability of this chimeric peptide using the FITC-linked CPP-QBP-LIR peptide, and observed that green fluorescence of the FITC-linked CPP-QBP-LIR peptide was distributed in whole cells, suggesting that the peptide can efficiently penetrate cell membranes (Figure 5B). Moreover, we treated HEK293 cells stably expressing HTT65Q-GFP with the chimeric peptide CPP-QBP-LIR and further checked the protein level of HTT65Q-GFP. We observed that the chimeric peptide CPP-QBP-LIR induced targeted degradation of HTT65Q-GFP aggregates in a concentration- and time-dependent manner, which was restored by treatment with Baf A1 and CQ but not MG132 (Figure 5C–F). These results suggested that the chimeric peptide can be applied to the targeted autophagic degradation of large protein aggregates, providing new perspectives for autophagy-targeting degradation strategies.
Figure 5.
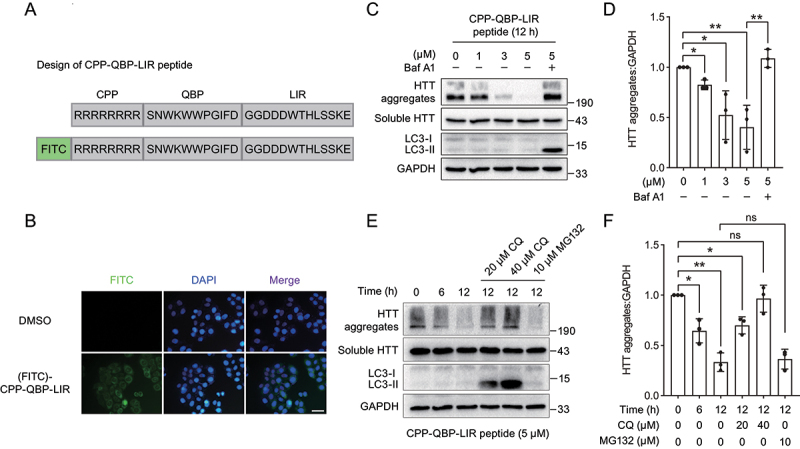
The chimeric peptide induces targeted degradation of HTT aggregates. (A) Scheme of the chimeric peptide for targeted degradation of HTT65Q aggregates. The chimeric peptide consists of CPP, QBP and LIR. (B) Representative images of HeLa cells treated with the FITC-linked chimeric peptide. Cells were treated with 5 µM the FITC-linked chimeric peptide for 6 h, followed by fluorescence analysis. (C) Immunoblot analysis of HTT65Q-GFP in HEK293 cells upon treatment with different concentrations of the chimeric peptide. HEK293 cells stably expressing HTT65Q-GFP were treated with different concentrations of the chimeric peptide (0, 1, 3 and 5 µM) for 12 h. The autophagy inhibition group was treated with 5 µM peptide accompanied by 1 µM Baf A1. (D) Quantification of the protein level of HTT65Q-GFP aggregates as in (C). (E) Immunoblot analysis of HTT65Q-GFP in HEK293 cells upon treatment with the chimeric peptide at different time points. HEK293 cells stably expressing HTT65Q-GFP were treated with the chimeric peptide (5 µM) at the indicated time points (0, 6 and 12 h). The autophagy inhibition group was treated with 5 µM peptide accompanied by 10 µM or 20 µM CQ for 12 h while the proteasome inhibition group was treated with 5 µM peptide accompanied by 10 µM MG132 for 12 h. (F) Quantification of the protein level of HTT65Q-GFP aggregates as in (E). Data in (D) and (F) are presented as the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test. Scale bar: 40 μm.
Tethering ATG16L1 or LC3 induces targeted degradation of mitochondria
Mitochondrial dysfunction is closely related to the occurrence of various neurodegenerative diseases [8,11,57]. We therefore investigated whether tethering ATG16L1 or LC3 to mitochondria is sufficient to induce targeted degradation of dysfunctional mitochondria. To screen a suitable mitochondria-targeting sequence (MTS) responsible for mitochondria targeting, we generated a series of chimeras consisting of LIR and different MTSs derived from NLRX1 (NLR family member X1) [50], outer mitochondrial membrane proteins including TOMM20 (translocase of outer mitochondrial membrane 20) [47] and MFF (mitochondrial fission factor) [48], and BCL2 (BCL2 apoptosis regulator) [49] (Table 1). We found that the chimera containing the transmembrane helix of TOMM20 as MTS1 most efficiently decreased levels of mitochondrial outer membrane protein TOMM20 and inner membrane protein TIMM23 upon FCCP treatment, suggesting that the chimera enables the degradation of damaged mitochondria (Figure S4A and S4B). The mitochondria targeting ability of the MTS1 derived from TOMM20 was also confirmed by immunofluorescence analysis, and MTS1 had no effect on the morphology of mitochondria (Figure S4C).
We then investigated the effect of MTS1-LIR and MTS1-ABP on mitochondria. We observed that GFP-tagged MTS1-LIR and MTS1-ABP were colocalized with autophagosomes and lysosomes (Figure 6A,B), suggesting that the expression of MTS1-LIR and MTS1-ABP facilitates the engulfment of mitochondria into autophagosomes and lysosomes through binding with ATG16L1 or LC3. Consistently, the expression of both MTS1-LIR and MTS1-ABP caused a significant decrease in the levels of mitochondrial proteins such as TIMM23 and TOMM20, in the presence, but not absence, of the mitochondrial stress agent FCCP (Figure 6C,D, Figure S4D and S4E), implying that the smaller size of fragmented mitochondria accelerated their engulfment by isolation membranes for autophagic degradation [25]. In addition, MTS1-LIR- or MTS1-ABP-induced mitochondria degradation effects were also detected under starvation condition in HeLa cells expressing no PRKN protein (Figure 6E,F).
Figure 6.
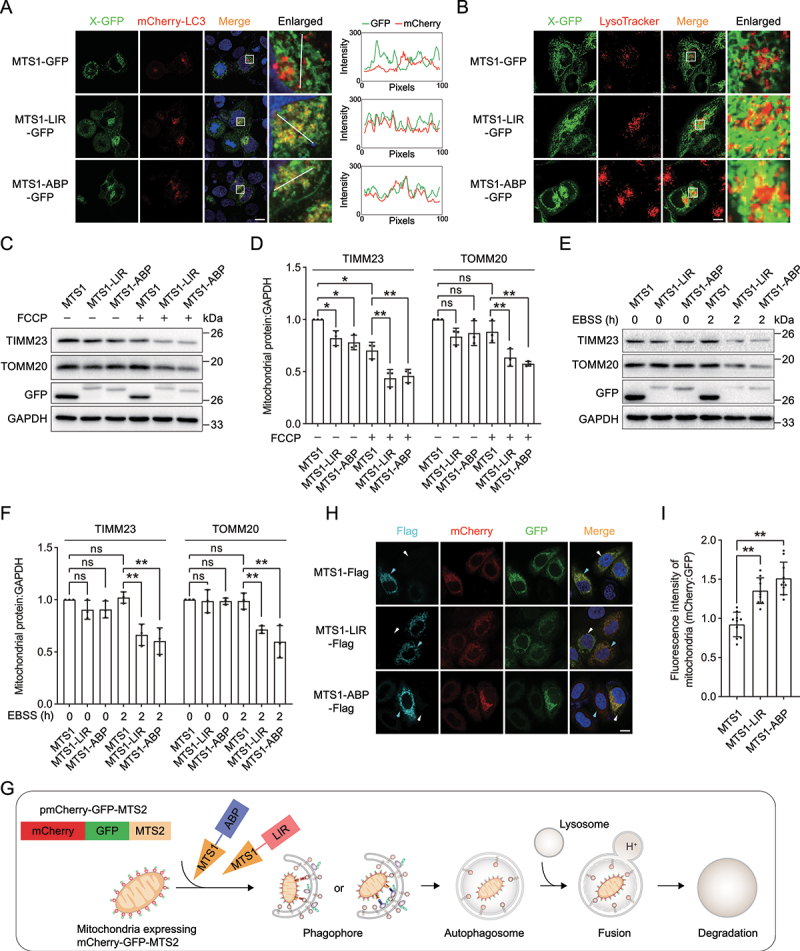
Tethering ATG16L1 or LC3 induces targeted degradation of mitochondria. (A) Representative images of MTS1-LIR-GFP and MTS1-ABP-GFP with mCherry-LC3 in HeLa cells. HeLa cells stably expressing mCherry-LC3 were transiently transfected with MTS1-GFP, MTS1-LIR-GFP or MTS1-ABP-GFP. The colocalization of mCherry-LC3 with MTS1-LIR-GFP or MTS1-ABP-GFP was determined by calculating fluorescence intensity of the areas marked with white lines. (B) Representative images of MTS1-LIR-GFP and MTS1-ABP-GFP with lysosomes in HeLa cells. MTS1-GFP, MTS1-LIR-GFP and MTS1-ABP-GFP were transiently transfected into HeLa cells, followed by lysosome staining with LysoTracker. (C) Immunoblot analysis of the mitochondrial proteins TIMM23 and TOMM20 upon the expression of MTS1-LIR and MTS1-ABP in HeLa cells in response to FCCP-induced mitochondrial dysfunction. MTS1-LIR and MTS1-ABP were transiently transfected into HeLa cells for 36 h, followed by treatment with 20 µM FCCP for 12 h. (D) Quantification of the levels of the mitochondrial proteins TIMM23 and TOMM20 as in (C). (E) Immunoblot analysis of the mitochondrial proteins TIMM23 and TOMM20 upon the expression of MTS1-LIR and MTS1-ABP in HeLa cells in response to starvation condition. (F) Quantification of the levels of the mitochondrial proteins TIMM23 and TOMM20 as in (E). (G) Scheme of mitophagy biosensor mCherry-GFP-MTS2. (H) Representative images of mCherry-GFP-MTS2 upon MTS1-LIR- and MTS1-ABP-induced mitophagy. Cells expressing mCherry-GFP-MTS2 were transiently transfected with MTS1-Flag, MTS1-LIR-Flag or MTS1-ABP-Flag for 24 h, followed by treatment with 20 µM FCCP for 6 h. The cyan arrows indicate cells that express MTS1, MTS1-LIR or MTS1-ABP, whereas the white arrows indicate cells that have no expression of these constructs. (I) Quantification of the relative fluorescence intensity as in (H) (n = 50). The fluorescence intensity of cells with the expression of MTS, MTS1-LIR or MTS1-ABP (indicated by the cyan arrows in (H)) was measured by ImageJ. Data in (d) and (F) are presented as the mean ± SEM of three independent experiments. *p < 0.05, **p < 0.01, Student’s t test. Scale bar: 10 μm.
To directly visualize the mitochondria degradation induced by MTS1-LIR and MTS1-ABP, we constructed a stable cell line expressing a mitophagy biosensor mCherry-GFP-MTS2 (Figure 6G). The mCherry-GFP-MTS2 is first anchored onto the mitochondria surface, enabling the mCherry and GFP signals of mitochondria. The mitophagy biosensor mCherry-GFP-MTS2 displays red and green fluorescence during normal conditions. In response to the MTS1-LIR- and MTS1-ABP-induced mitophagy, only the mCherry signal is stably detected as the mitochondria are delivered to the lysosome where the GFP signals are quenched due to the acidic condition of the lysosomes (Figure 6H). The relative intensity of the mCherry and GFP signals reveal the mitochondria degradation induced by MTS1-LIR and MTS1-ABP (Figure 6I).
SQSTM1 is a main selective autophagy receptor for mitochondria degradation. To exclude the possibility that endogenous SQSTM1 could involve in the MTS1-LIR and MTS1-ABP-induced mitochondria degradation, we evaluated the degradation effect of MTS1-LIR and MTS1-ABP on mitochondria in SQSTM1-KO cells (Figure S4F). We observed that MTS1-LIR and MTS1-ABP still induces a decrease of mitochondrial proteins TIMM23 and TOMM20 upon FCCP treatment in SQSTM1-KO HeLa cells (Figure S4G and S4H), suggesting that mitochondria degradation induced by MTS1-LIR and MTS1-ABP is independent of the autophagy receptor SQSTM1.
Therefore, these results indicated that MTS1-LIR and MTS1-ABP induce targeted clearance of dysfunctional mitochondria.
Targeted degradation of mitochondria ameliorates mitochondria dysfunction in a cell model
The canonical PINK1 (PTEN induced kinase 1)-PRKN (parkin RBR E3 ubiquitin protein ligase) pathway plays an important role in mitochondrial quality control by sensing and removing damaged mitochondria [58,59]. Mutations in PINK1 and PRKN are associated with Parkinson disease in different model systems [60–62]. Our results have demonstrated that MTS1-LIR and MTS1-ABP deliver cargoes into autophagosomes by directly interacting with ATG16L1 or LC3 without the assistance of autophagy receptors and the PINK1-PRKN-dependent ubiquitination process. PINK1-deficient cells are generally used as a Parkinson disease cell model [63,64]. We therefore examined whether MTS1-LIR and MTS1-ABP could ameliorate mitochondrial dysfunction in PINK1-KO HEK293 cells (Figure 7A). In response to FCCP-induced mitochondrial dysfunction, we observed that the expression of MTS1-LIR and MTS1-ABP decreased the protein level of TIMM23 and TOMM20 in PINK1-KO cells (Figure 7B,C). To further gain evidence of the independence from the ubiquitin system, we performed the experiment with the E1 inhibitor TAK-243 (MLN7243) treatment. We found that MTS1-LIR and MTS1-ABP induce targeted clearance of damaged mitochondria, which was not reversed by TAK-243 treatment (Figure 7D,E). Combination with results from PINK1-KO cells, we conclude that MTS1-LIR and MTS1-ABP directly anchor damaged mitochondria to autophagosomes via the autophagy-targeting peptide LIR or ABP, which is independent of PINK1-PRKN pathway and ubiquitin signaling.
Figure 7.
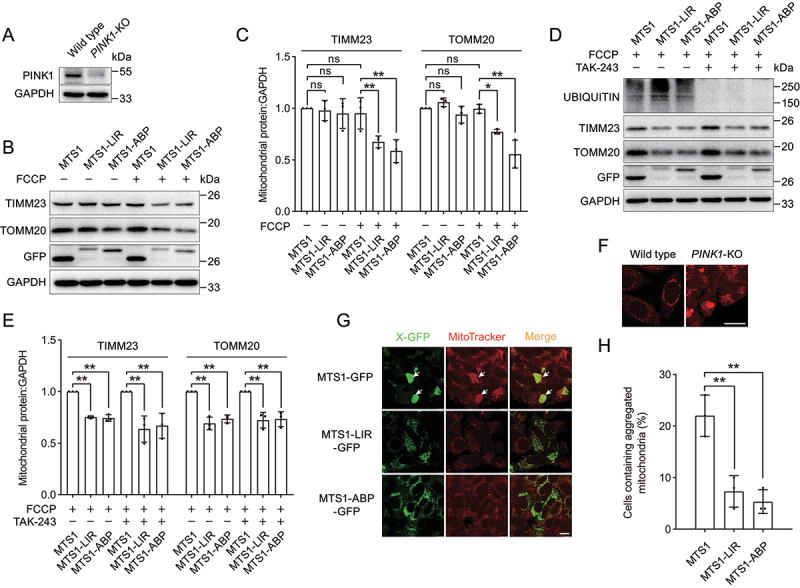
Targeted degradation of mitochondria ameliorates mitochondria dysfunction in a Parkinson disease cell model. (A) Immunoblot analysis of PINK1 in HEK293 WT or PINK1-KO cells. (B) Immunoblot analysis of the mitochondrial proteins TIMM23 and TOMM20 upon the expression of MTS1-LIR and MTS1-ABP in PINK1-KO HEK293 cells. MTS1-LIR and MTS1-ABP were transiently transfected into PINK1-KO HEK293 cells for 36 h, followed by treatment with 20 µM FCCP for 12 h. (C) Quantification of the levels of the mitochondrial proteins TIMM23 and TOMM20 as in (B). (D) Immunoblot analysis of the mitochondrial proteins TIMM23 and TOMM20 upon the expression of MTS1-LIR and MTS1-ABP in HeLa cells in response to FCCP-induced mitochondrial dysfunction in the presence of E1 inhibitor TAK-243. The MTS1-LIR and MTS1-ABP were transiently transfected into HeLa cells for 36 h. Following by pre-treatment with 1 µM TAK-243 for 4 h, cells were treated with 20 µM FCCP for 12 h. (E) Quantification of the levels of the mitochondrial proteins TIMM23 and TOMM20 as in (D). (F) Representative images of aggregated mitochondria in PINK1-KO HEK293 cells. Mitochondria were stained with MitoTracker. The aggregated mitochondria in PINK1-KO HEK293 cells were indicated by white arrows. Scale bar: 25 μm. (G) Representative images of mitochondria upon expression of MTS1-LIR and MTS1-ABP in PINK1-KO HEK293 cells. PINK1-KO HEK293 cells were transiently transfected with MTS1-LIR and MTS1-ABP for 72 h, followed by staining of mitochondria with MitoTracker. The aggregated mitochondria in PINK1-KO HEK293 cells were indicated by white arrows. Scale bar: 10 μm. (H) Quantification of the numbers of cells containing aggregated mitochondria as in (G). Data in (C) and (E) are presented as the mean ± SEM of three independent experiments. “ns”, no significant difference, **p < 0.01, Student’s t test.
Moreover, the deletion of PINK1 caused the accumulation and fragmentation of mitochondria (Figure 7F), which is in line with previous studies [65]. Interestingly, we observed that the expression of MTS1-LIR and MTS1-ABP reduced the numbers of aggregated mitochondria and abolished the aggregation of mitochondria in PINK1-KO cells (Figure 7G,H). Thus, MTS1-LIR and MTS1-ABP induce targeted degradation of mitochondria, thereby ameliorating mitochondrial dysfunction caused by PINK1 deletion.
Targeted degradation of mitochondria protects cells from apoptosis
Upon exposure to apoptotic stimuli, mitochondria are damaged, and release CYCS/cytochrome c to the cytoplasm for caspase cascade activation, leading to cell apoptosis [66,67]. We then assessed the physiological effects of MTS1-LIR and MTS1-ABP-mediated mitochondrial clearance in response to mitochondrial damage. We observed that FCCP treatment induced the activation of caspase 3 (CASP3) (Figure 8A,B), which is consistent with previous studies [68,69]. Interestingly, the expression of MTS1-LIR and MTS1-ABP suppressed the activation of CASP3 under FCCP treatment (Figure 8C,D). Taken together, these results demonstrated that tethering mitochondria to ATG16L1 or LC3 promotes targeted clearance of damaged mitochondria by autophagic degradation, thus protecting cells from apoptosis (Figure 8E).
Figure 8.

Targeted degradation of mitochondria protects cells from apoptosis. (A) Workflow for MTS1-LIR- and MTS1-ABP-mediated autophagic clearance of mitochondria in response to mitochondrial damage. (B) Immunoblot analysis of CASP3 upon FCCP treatment at different time points. (C) Immunoblot analysis of CASP3 upon the expression of MTS1-LIR and MTS1-ABP in response to FCCP-induced cell apoptosis. HeLa cells were transiently transfected with MTS1-LIR and MTS1-ABP for 36 h, followed by treatment with 40 µM FCCP for 12 h. After removal of FCCP compounds, cells were incubated in fresh medium for 4 h. (D) Quantification of the protein levels of activated CASP3 as in (C). (E) Scheme of targeted degradation of mitochondria induced by MTS1-LIR and MTS1-ABP.
Discussion
Here, we report the development of a generalizable chimera consisting of three parts, including a ligand for a large target, autophagy-targeting ligand LIR or ABP and a linker between the two peptides. The chimera links large targets to the autophagic membrane via LIR or ABP binding with core autophagy machineries LC3 or ATG16L1, respectively, thereby leading to large target recruitment into autophagosomes for degradation. We also demonstrated that this autophagy-targeting degradation strategy induces targeted autophagic degradation of protein aggregates but not soluble proteins, suggesting that the strategy shows high selectivity for large targets. In addition, we proved the concept of the autophagy-targeting degradation strategy using PB1 aggregates as a large target model in a streptavidin system. Then we successfully applied autophagy-targeting degradation strategy to the targeted degradation of HTT65Q aggregates and mitochondria. Specifically, the chimeric peptide works as an autophagy-based degrader to induce targeted autophagic degradation of HTT65Q aggregates. More importantly, our results showed that ATG16L1 can serve as an accommodable receptor for autophagy targeting and that binding with ATG16L1 enables efficient targeted degradation of large targets, which is line with a previous study that tethering of the region of ATG16L1 responsible for RB1CC1/FIP200 binding to mitochondria is enough to induce mitophagy [70]. Overall, this study demonstrated a new autophagy-targeting degradation strategy, which has been shown to efficiently degrade HTT65Q aggregates and mitochondria, thereby providing a generalizable strategy for targeted degradation of large targets.
In cells, selective autophagy degrades specific cargoes, such as protein aggregates and malfunctioning organelles, which is mediated via cargo receptors that recognize and sequester specific cargo into the autophagosome [16,18]. SQSTM1 is the best characterized cargo receptor that binds to ubiquitinated proteins via its ubiquitin-associated (UBA) domain and interacts with LC3 on phagophores via its LC3-interacting region (LIR), ultimately leading to ubiquitinated protein degradation by fusing with lysosomes [33,71,72]. In general, selective autophagy is initiated when a cellular signal, such as ubiquitin, tags protein aggregates or organelles for degradation [15,21,40]. Our autophagy-targeting degradation strategy directly anchor large targets to autophagosomes via the autophagy-targeting peptide LIR or ABP, which is independent of ubiquitin signaling and endogenous autophagy receptor SQSTM1. Supporting this notion, we observed that MTS1-LIR and MTS1-ABP induced targeted degradation of mitochondria in PINK1-KO or SQSTM1-KO cells as well as in the presence of E1 inhibitor. The PINK1-PRKN pathway is responsible for the ubiquitination of mitochondrial proteins in mitophagy. Therefore, MTS1-LIR and MTS1-ABP directly sequester largo targets to autophagosomes, thereby inducing targeted degradation of large targets, which is independent of PINK1-PRKN pathway and autophagy receptor.
Due to that fact that defects in organelle autophagy are lined to various neurodegenerative diseases and aging [73,74], therefore strategies for targeted degradation of organelles have been attracted attention. There are several reports using tethering of different autophagy proteins to organelles to induce their degradation. In yeast, tethering Atg11, a scaffold protein that interacts with various other autophagy proteins to peroxisomes, triggers degradation of peroxisomes by selective autophagy [75]. In mammals, linear Ub chains are fused to the N-terminal transmembrane segment of TOMM20, the resulting chimeric protein bypasses the PINK1-PRKN pathway and induces mitophagy [76]. In another example, PINK1 is targeted to mitochondria using rapamycin-dependent FRB-FKBP system, resulting in phosphorylation of mitochondrial ubiquitin and then recruit autophagy receptors to induce mitophagy [77]. Our findings are in line with the former, suggesting that tethering ATG16L1 or LC3 induces targeted degradation of protein aggregates and mitochondria, bypassing selective autophagy receptors. These approaches for targeted degradation of organelles need to be further optimized as small bifunctional molecules, provide new strategies for treating organelle-related diseases.
PROTACs have also been used to degrade amyloid proteins with no specificity to the functional monomeric form and pathological toxic aggregates, and the clearance of the functional monomeric form could cause unexpected results [78–80]. Recent studies have proven that monomeric Tau and α-synuclein exert important physiological functions [81,82]. Thus, it is imperative to develop a new type of targeted degradation chimeric strategy for toxic aggregates rather than functional monomeric forms. Recently, ATTEC molecular glues that interact with both LC3 and polyQ-HTT have been identified using small-molecule-microarray-based screening [23]. ATTEC compounds target polyQ-HTT to autophagosomes, reducing polyQ-HTT levels and thus rescuing disease-relevant phenotypes. ATTEC and our autophagy-targeting degradation strategy overcome limitation of PROTACs and induce targeted degradation of protein aggregates but not soluble proteins, thereby providing a promising strategy for the degradation of amyloid protein aggregates. In addition, ATTEC employs LC3 as an autophagy targeting protein, which could increase the risk of autophagy inhibition at the high concentration of ATTEC due to the importance of LC3 protein in autophagy pathway. Our strategy employing ATG16L1 could over this limitation and provide a compensatory approach for autophagy-targeting degradation.
During mitophagy, PRKN is recruited to the outer membrane of mitochondria by PINK1, which enables the ubiquitination of mitochondrial outer membrane proteins [83,84]. The ubiquitinated mitochondria are further recognized by autophagy receptors [85,86]. The mutations in PINK1 and PRKN have been shown to be associated with Parkinson disease [87,88]. In the PINK1-KO cell line, we observed the accumulation of aggregated mitochondrial fragments, which was ameliorated by the expression of MTS1-LIR and MTS1-ABP. Furthermore, our results showed that tethering mitochondria to ATG16L1 or LC3 promotes targeted clearance of damaged mitochondria by autophagic degradation, thus protecting cells from apoptosis induced by FCCP. Therefore, MTS1-LIR- or MTS1-ABP-induced mitochondria degradation ameliorates mitochondrial dysfunction caused by PINK1 deletion and protects cells from apoptosis.
Collectively, our proposed autophagy-targeting degradation strategy will provide new insights into the field of targeted degradation based on autophagy and lay a foundation for the development of targeted degradation of large targets.
Materials and Methods
Cell culture, antibodies and reagents
The human-derived HEK293 (ATCC, CRL-1573), HEK293T (ATCC, CRL-11268) and HeLa cells (ATCC, CCL-2) used in this study were cultured in Dulbecco’s Modified Eagle Medium (Basal Media, P110L7) containing 10% fetal bovine serum (Biological Industries, 04–001-1ACS), 1% penicillin-streptomycin solution (Biosharp, BL505A; 10 mg/mL streptomycin and 10,000 units/mL penicillin) in an incubator at 37°C with 5% CO2. For starvation treatment, cells were cultured in EBSS (Sigma Aldrich, E2888) for the indicated times.
The primary antibodies used in this study were as follows: rabbit anti-TOMM20 (382451), rabbit anti-GFP (300943) and mouse anti-GAPDH (250133) from Zen Bioscience; mouse anti-TIMM23 (sc -514463) and mouse anti-ubiquitin (sc-8017) from Santa Cruz Biotechnology; rabbit anti-LC3B (2775S) from Cell Signaling Technology (CST); rabbit anti-SQSTM1/p62 (PM045) from Medical Biological Laboratories (MBL); mouse anti-Flag (F1804) from Sigma-Aldrich; and rabbit anti-CASP3 (19677–1-AP) from Proteintech Group. HRP-labeled goat anti-mouse IgG (H+L) (A0216) and HRP-labeled goat anti-rabbit IgG (H+L) (A0208) secondary antibodies were purchased from Beyotime Biotechnology. Alexa Fluor 647-labeled goat anti-mouse IgG (H+L) antibody (A0473) and Alexa Fluor 647-labeled goat anti-rabbit IgG (H+L) antibody (A0468) were purchased from Beyotime Biotechnology.
The reagents used in this study were as follows: Lipofectamine 3000 transfection reagent (L3000015) from Invitrogen; Protease Inhibitor Cocktail, mini-Tablet (EDTA-Free) (HY-K0011), polybrene (HY-112735), puromycin (HY-B1743A), FCCP (HY-100410), TAK-243 (MLN7243) (HY-100487), chloroquine phosphate (CQ) (HY-17589) and bafilomycin A1 (Baf A1) (HY-100558) from MedChemExpress; MG132 (S2619) from Selleck; MitoTracker (C1035) and LysoTracker (C1046) from Beyotime Biotechnology; trypsin solution (BL512A), DAPI (BL105A) and 4% paraformaldehyde (BL539A) from Biosharp. Chemicals, building blocks and solvents for peptide synthesis and purification were obtained from Aldrich, GL Biochem and Aladdin and used without further purification.
Peptide synthesis and purification
Peptides were synthesized following an Fmoc solid-phase peptide synthesis strategy using Rink amide 4-methylbenzhydrylamine (MBHA) resin. Briefly, the resin (initial loading 0.57 mmol/g; Guoping Pharmaceutical, GP011153–2) was swelled in DCM for 10 min. The initial Fmoc group was cleaved by shaking the resin in a solution of piperidine:DMF (1:4 v:v) twice for 10 min each. Next, the subsequent N-terminal peptide chain elongation was achieved by a standard Fmoc strategy employing commercially available amino acid building blocks. Peptides were released using a cocktail cleavage solution (95% TFA, 2.5% TIS, 2.5% water). The crude peptides were purified by semipreparative HPLC. The peptide identities were confirmed by analytical HPLC and MALDI-TOF mass spectrometry. HPLC was performed on a HITACHI Primaide HPLC system using a reversed-phase C18 analytical column (HITACHI LaChrom C18, 8915055; 2.5 μm, flow 1.0 mL/min, from 10% B to 100% B over 20 min) and semipreparative column (ZONRAN Bondysil C18, 820201111; 5 μm, flow 3.0 mL/min, from 10% B to 100% B over 20 min) with detection at 220 nm. Buffer A – water +0.1% TFA, Buffer B – acetonitrile +0.1% TFA.
Plasmids construction and transfection
To verify the autophagy targeting of the LC3-interacting region (LIR) and ATG16L1-binding peptide (ABP), the cDNA sequences of the Phox1 and Bem1p (PB1) domain, LIR and ABP were cloned into the pEGFP-C1 vector (Clontech, HG-VYC0084) as required. For the generation of various stable cell lines, the cDNA sequences encoding PB1, streptavidin or the N-terminal truncated HTT were cloned into the lentiviral overexpression pLJM1-GFP vector (Addgene, 19319; deposited by David Sabatini) to generate plasmids pLJM1-GFP-streptavidin, pLJM1-GFP-PB1-streptavidin and pLJM1-HTT65Q-GFP, respectively. The LIR or ABP cDNA sequence was further cloned into pLJM1-HTT65Q-GFP to generate plasmids pLJM1-HTT65Q-GFP-LIR and pLJM1-HTT65Q-GFP-ABP. The cDNA sequence of GFP in the pLJM1-GFP vector was replaced by mCherry-LC3 to construct the pLJM1-mCherry-LC3 plasmid. The cDNA sequence of MTS2 and mCherry were cloned into pLJM1-GFP to generate the pLJM1-mCherry-GFP-MTS2 plasmid. The cDNA sequences of the streptavidin-binding peptide (SBP), polyQ-binding peptide 1 (QBP), LIR or ABP were cloned into the pHA-C1 vector (Clontech, 631604). For screening of mitochondria-targeting sequence (MTS), the cDNA sequences of LIR and different MTSs were cloned into the pHA-C1 vector. The cDNA sequences of MTS1 from TOMM20, LIR or ABP were cloned into the pEGFP-C1 vector and pCMV-Flag (Addgene, 182659; deposited by Ivana Nikić-Spiegel). Plasmids were transfected into cells using the Lipofectamine 3000 Transfection Reagent according to the manufacturer’s instructions. All primers used for plasmids construction in this study are shown in Table S1. LIR, ABP and QBP were inserted into vector using annealing oligonucleotides, as shown in Table S2.
Construction of stable mammalian cell lines
We generated HEK293 cells stably expressing GFP-PB1-streptavidin, GFP-streptavidin, HTT65Q-GFP or mCherry-LC3 and HeLa cells expressing mCherry-GFP-MTS2. The above constructed lentiviral plasmid pLJM1 containing the open reading frames of the above recombinant proteins and the packaging plasmids pMD2.G (Addgene, 12259; deposited by Didier Trono) and psPAX2 (Addgene, 12260; deposited by Didier Trono) were cotransfected into HEK293T cells with Lipofectamine 3000 transfection reagent in a 6 cm cell dish for 48 h. The lentivirus was collected and further used to infect cells in the presence of 8 μg/mL polybrene for 24 h. The infected cells were selected with 1 μg/mL puromycin.
Immunoblot assay
Cultured cells were washed twice with ice-cold phosphate-buffered saline (PBS; Biosharp, BL601A) and resuspended in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate [Sangon Biotech, NB0669], 1% Triton X-100 [Biofroxx, 1139ML100], 0.1% SDS) containing Protease Inhibitor Cocktail to prepare whole cell lysates. The protein concentration of the cell lysates was measured using a Bicinchoninic Acid Protein Assay Kit (Beyotime Biotechnology, P0009). For immunoblot, 20 µg of protein was loaded for SDS-PAGE, and the separated proteins were then transferred onto a 0.45 µm PVDF membrane (Millipore, IPVH00010). The resulting membrane was blocked with TBST (20 mM Tris-HCl, 150 mM NaCl, 0.1% [v:v] Tween 20 [Biofroxx, 1247ML500]) containing 5% nonfat dry milk for 1 h and incubated with the corresponding primary antibody overnight at 4°C. The dilutions of the primary antibodies used in this study were as follows: rabbit anti-TOMM20, 1:1000; rabbit anti-GFP, 1:10000; rabbit anti-CASP3, 1:1000; rabbit anti-SQSTM1/p62, 1:3000; rabbit anti-LC3B, 1:1000; mouse anti-TIMM23, 1:1000; and mouse anti-GAPDH, 1:5000. After three washes with TBST for 10 min each, the membrane was incubated with HRP-labeled goat anti-mouse IgG (H+L) or HRP-labeled goat anti-rabbit IgG (H+L) secondary antibody (dilution 1:5000). Immunoreactive signals were visualized with ECL Western Blot Detection Reagent (Beyotime Biotechnology, P0018FS) by a Western Blot Imager (GelDoc XR, Bio-Rad).
Preparation of Triton X-100 soluble and insoluble fractions of HTT65Q
Cells were lysed in Triton X-100 soluble cell lysate buffer (PBS supplemented with 1% Triton X-100 and Protease Inhibitor Cocktail) for 30 min. Then cells were centrifuged at 20,000 × g for 30 min at 4°C, and the supernatants were collected as the Triton X-100 soluble fractions. After three washes with PBS, the cell pellets fractions were dissolved in RIPA buffer and sonicated three times on ice for 5 s each time, and were collected as the Triton X-100 insoluble fractions. To detect the HTT aggregates, proteins in both the separating and stacking gels were transferred onto the PVDF membrane.
Immunofluorescence assay and cell imaging
Cells were seeded in confocal dishes (Biosharp, BS-15-GJM) and transfected with the indicated plasmids. Then, transfected cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. For immunofluorescence analysis, the paraformaldehyde-fixed cells were permeabilized with 0.5% Triton X-100 for 20 min, and followed by blocking with 10% fetal bovine serum for 1 h at room temperature. Then cells were incubated with the indicated primary antibody overnight at 4°C. The dilutions of the primary antibodies used in this study were as follows: rabbit anti-SQSTM1/p62, 1:500; mouse anti-Flag, 1:1000; After three washes with 1×PBS containing 0.01% Tween 20, cells were incubated with the Alexa Fluor 647-labeled goat anti-mouse or goat anti-rabbit IgG (H+L) antibody (dilution 1:500). Cells were visualized using a Leica TCS SP8 DIVE confocal microscope (Leica) equipped with an Airyscan detector and a 63× oil immersion objective (1.4 numerical aperture; Leica). Images were acquired in Airyscan mode and analyzed with LAS X software (Leica). As indicated, mitochondria and lysosomes were stained with MitoTracker and LysoTracker, respectively, according to the manufacturer’s instructions.
Targeted degradation of protein aggregates and mitochondria
For verification of streptavidin system, HEK293 cells expressing GFP-PB1-streptavidin or GFP-streptavidin were transfected with SBP-LIR or SBP-ABP constructs for 48 h, and immunoblot was performed to detect the protein levels of targets. To degrade HTT aggregates, HEK293 cells expressing HTT65Q-GFP were transfected with QBP-LIR and QBP-ABP for 36 h, followed by treatment with or without Baf A1 for 12 h. Whole cell lysates were prepared to detect the protein levels of HTT65Q-GFP.
To degrade mitochondria, cells were transfected with MTS1-LIR and MTS1-ABP for 36 h, followed by treatment with 20 µM FCCP for 12 h. The levels of mitochondria were measured by immunoblot for mitochondrial proteins, including TIMM23 and TOMM20. To examine the mitochondria degradation mediated by MTS1-LIR and MTS1-ABP upon autophagy inhibition, 1 µM Baf A1 was added in MTS1-LIR- and MTS1-ABP-transfected cells. To evaluate the effects of ubiquitin signaling on the degradation of mitochondria induced by MTS1-LIR and MTS1-ABP, the transfected cells were pre-treated 1 µM TAK-243 (MLN7243) for 4 h, and followed by the treatment of with 20 µM FCCP for 12 h. For apoptosis detection affected by MTS1-LIR and MTS1-ABP, the transfected cells were incubated in fresh medium for 4 h after FCCP treatment, and whole cell lysates were prepared to detect the protein level of activated-CASP3 by immunoblot.
Targeted degradation of HTT aggregates by the chimeric peptide CPP-QBP-LIR
The chimeric peptide CPP-QBP-LIR was dissolved in DMSO to 10 mM as a stock solution. HEK293 cells expressing HTT65Q-GFP were preseeded in 6-well dishes for 12 h, and cells were treated with CPP-QBP-LIR peptide under the indicated conditions. For concentration-dependent degradation of HTT aggregates, the HEK293 stable cell line expressing HTT65Q-GFP was treated with the chimeric peptide CPP-QBP-LIR for 12 h at the indicated concentration. For time-dependent degradation of HTT aggregates, the HEK293 stable cell line expressing HTT65Q-GFP was treated with 5 µM CPP-QBP-LIR peptide for the indicated time.
Statistical analysis
Data are presented as the mean ± SEM of three independent experiments (Data S1). Statistical significance was analyzed using Student’s t test.
Supplementary Material
Acknowledgements
We thank Dr. Jieqiong Tan (Central South University, China) for PINK1-KO HEK293 cells and Dr. Qiming Sun (Zhejiang University, China) for SQSTM1-KO HeLa cells. We are also thankful for the technical support by the Analytical and Testing Center of Chongqing University.
Funding Statement
This study was supported by the National Natural Science Foundation of China (NSFC) (91854101, 32170185, 31801166 and 22011530161), the Natural Science Foundation of Chongqing, China (CSTB2022NSCQ-MSX0463), the Venture Innovation Support Program for Chongqing Overseas Returnees (cx2022066) and the Fundamental Research Funds for the Central Universities (2022CDJYGRH-002).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
All study data are included in the manuscript and its Supplementary Material files.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2234797
References
- [1].Wu T, Yoon H, Xiong Y, et al. Targeted protein degradation as a powerful research tool in basic biology and drug target discovery. Nat Struct Mol Biol. 2020. Jul;27(7):605–614. doi: 10.1038/s41594-020-0438-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Samarasinghe KTG, Crews CM.. Targeted protein degradation: a promise for undruggable proteins. Cell Chem Biol. 2021 Jul 15;28(7):934–951. doi: 10.1016/j.chembiol.2021.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chamberlain PP, Hamann LG. Development of targeted protein degradation therapeutics. Nat Chem Biol. 2019. Oct;15(10):937–944. doi: 10.1038/s41589-019-0362-y [DOI] [PubMed] [Google Scholar]
- [4].Ding Y, Fei Y, Lu B. Emerging new concepts of degrader technologies. Trends Pharmacol Sci. 2020. Jul;41(7):464–474. doi: 10.1016/j.tips.2020.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alabi SB, Crews CM. Major advances in targeted protein degradation: PROTACs, LYTACs, and MADTACs. J Biol Chem. 2021. Jan-Jun;296:100647. doi: 10.1016/j.jbc.2021.100647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Salami J, Crews CM. Waste disposal-An attractive strategy for cancer therapy. Science. 2017 Mar 17;355(6330):1163–1167. doi: 10.1126/science.aam7340 [DOI] [PubMed] [Google Scholar]
- [7].Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013 Sep 5;501(7465):45–51. doi: 10.1038/nature12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006 Oct 19;443(7113):787–795. doi: 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- [9].Hou YJ, Dan XL, Babbar M, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. 2019. Oct;15(10):565–581. doi: 10.1038/s41582-019-0244-7 [DOI] [PubMed] [Google Scholar]
- [10].Boland B, Yu WH, Corti O, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat Rev Drug Discov. 2018. Sep;17(9):660–688. doi: 10.1038/nrd.2018.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lou G, Palikaras K, Lautrup S, et al. Mitophagy and neuroprotection. Trends Mol Med. 2020. Jan;26(1):8–20. doi: 10.1016/j.molmed.2019.07.002 [DOI] [PubMed] [Google Scholar]
- [12].Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014. Jan;24(1):9–23. doi: 10.1038/cr.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mizushima N. A brief history of autophagy from cell biology to physiology and disease. Nat Cell Biol. 2018. May;20(5):521–527. doi: 10.1038/s41556-018-0092-5 [DOI] [PubMed] [Google Scholar]
- [14].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014. Jan;24(1):24–41. doi: 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019 Nov 15;366(6467):818–822. doi: 10.1126/science.aax3769 [DOI] [PubMed] [Google Scholar]
- [16].Lamark T, Johansen T. Mechanisms of selective autophagy. Annu Rev Cell Dev Biol. 2021 Oct 6;37(1):143–169. doi: 10.1146/annurev-cellbio-120219-035530 [DOI] [PubMed] [Google Scholar]
- [17].Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. doi: 10.1080/15548627.2017.1378838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014. Jun;16(6):495–501. doi: 10.1038/ncb2979 [DOI] [PubMed] [Google Scholar]
- [19].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018. Mar;20(3):233–242. doi: 10.1038/s41556-018-0037-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019 Jan 10;176(1–2):11–42. doi: 10.1016/j.cell.2018.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018. Jun;19(6):349–364. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [22].Takahashi D, Arimoto H. Selective autophagy as the basis of autophagy-based degraders. Cell Chem Biol. 2021 Jul 15;28(7):1061–1071. doi: 10.1016/j.chembiol.2021.05.006 [DOI] [PubMed] [Google Scholar]
- [23].Li ZY, Wang C, Wang ZY, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019 Nov 7;575:203–209. doi: 10.1038/s41586-019-1722-1 [DOI] [PubMed] [Google Scholar]
- [24].Fu Y, Chen N, Wang Z, et al. Degradation of lipid droplets by chimeric autophagy-tethering compounds. Cell Res. 2021. Sep;31(9):965–979. doi: 10.1038/s41422-021-00532-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Takahashi D, Moriyama J, Nakamura T, et al. Autacs: cargo-specific degraders using selective autophagy. Mol Cell. 2019 Dec 5;76:797–810. doi: 10.1016/j.molcel.2019.09.009 [DOI] [PubMed] [Google Scholar]
- [26].Ji CH, Kim HY, Lee MJ, et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat Commun. 2022 Feb 16;13(1):1–14. doi: 10.1038/s41467-022-28520-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mizushima N, Kuma A, Kobayashi Y, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003 May 1;116(Pt 9):1679–1688. doi: 10.1242/jcs.00381 [DOI] [PubMed] [Google Scholar]
- [28].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000 Nov 1;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Huang X, Yao J, Liu L, et al. Atg8-PE protein-based in vitro biochemical approaches to autophagy studies. Autophagy. 2022. Sep;18(9):2020–2035. doi: 10.1080/15548627.2022.2025572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ichimura Y, Kirisako T, Takao T, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000 Nov 23;408(6811):488–492. doi: 10.1038/35044114 [DOI] [PubMed] [Google Scholar]
- [31].Lystad AH, Carlsson SR, de la Ballina LR, et al. Distinct functions of ATG16L1 isoforms in membrane binding and LC3B lipidation in autophagy-related processes. Nat Cell Biol. 2019. Mar;21(3):372–383. doi: 10.1038/s41556-019-0274-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dudley LJ, Cabodevilla AG, Makar AN, et al. Intrinsic lipid binding activity of ATG16L1 supports efficient membrane anchoring and autophagy. Embo J. 2019 May 2;38(9):e100554. doi: 10.15252/embj.2018100554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pankiv S, Clausen TH, Lamark T, et al. P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007 Aug 17;282(33):24131–24145. doi: 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [34].Viret C, Rozieres A, Faure M. Novel insights into NDP52 autophagy receptor functioning. Trends Cell Biol. 2018. Apr;28(4):255–257. doi: 10.1016/j.tcb.2018.01.003 [DOI] [PubMed] [Google Scholar]
- [35].Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009 Feb 27;33(4):505–516. doi: 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- [36].Qiu Y, Wang J, Li H, et al. Emerging views of OPTN (optineurin) function in the autophagic process associated with disease. Autophagy. 2022. Jan;18(1):73–85. doi: 10.1080/15548627.2021.1908722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang Y, Xu X, Hu MX, et al. SPATA33 is an autophagy mediator for cargo selectivity in germline mitophagy. Cell Death Differ. 2021. Mar;28(3):1076–1090. doi: 10.1038/s41418-020-00638-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kirkin V, Rogov VV., Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019 Oct 17;76:268–285. doi: 10.1016/j.molcel.2019.09.005 [DOI] [PubMed] [Google Scholar]
- [39].Johansen T, Lamark T. Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol. 2020 Jan 3;432(1):80–103. doi: 10.1016/j.jmb.2019.07.016 [DOI] [PubMed] [Google Scholar]
- [40].Gubas A, Dikic I. A guide to the regulation of selective autophagy receptors. FEBS J. 2022. Jan;289(1):75–89. doi: 10.1111/febs.15824 [DOI] [PubMed] [Google Scholar]
- [41].Lu K, den Brave F, Jentsch S. Receptor oligomerization guides pathway choice between proteasomal and autophagic degradation. Nat Cell Biol. 2017. Jun;19(6):732–739. doi: 10.1038/ncb3531 [DOI] [PubMed] [Google Scholar]
- [42].Boada-Romero E, Letek M, Fleischer A, et al. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. Embo J. 2013 Feb 20;32(4):566–582. doi: 10.1038/emboj.2013.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Goode A, Butler K, Long J, et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy. 2016 Jul 2;12(7):1094–1104. doi: 10.1080/15548627.2016.1170257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wirth M, Zhang W, Razi M, et al. Molecular determinants regulating selective binding of autophagy adapters and receptors to ATG8 proteins. Nat Commun. 2019 May 3;10(1):2055. doi: 10.1038/s41467-019-10059-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Barrette-Ng IH, Wu SC, Tjia WM, et al. The structure of the SBP-Tag-streptavidin complex reveals a novel helical scaffold bridging binding pockets on separate subunits. Acta Crystallogr D Biol Crystallogr. 2013. May;69(Pt 5):879–887. doi: 10.1107/S0907444913002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Okamoto Y, Nagai Y, Fujikake N, et al. Surface plasmon resonance characterization of specific binding of polyglutamine aggregation inhibitors to the expanded polyglutamine stretch. Biochem Biophys Res Commun. 2009 Jan 16;378(3):634–639. doi: 10.1016/j.bbrc.2008.11.094 [DOI] [PubMed] [Google Scholar]
- [47].Kanaji S, Iwahashi J, Kida Y, et al. Characterization of the signal that directs TOM20 to the mitochondrial outer membrane. J Cell Bio. 2000 Oct 16;151(2):277–288. doi: 10.1083/jcb.151.2.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yamada T, Murata D, Adachi Y, et al. Mitochondrial stasis reveals p62-mediated ubiquitination in Parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018 Oct 2;28:588–604. doi: 10.1016/j.cmet.2018.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Strappazzon F, Vietri-Rudan M, Campello S, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. Embo J. 2011 Apr 6;30(7):1195–1208. doi: 10.1038/emboj.2011.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang Y, Yao Y, Qiu X, et al. Listeria hijacks host mitophagy through a novel mitophagy receptor to evade killing. Nat Immunol. 2019. Apr;20(4):433–446. doi: 10.1038/s41590-019-0324-2 [DOI] [PubMed] [Google Scholar]
- [51].Tobias IS, Newton AC. Protein scaffolds control localized protein kinase czeta activity. J Biol Chem. 2016 Jun 24;291:13809–13822. doi: 10.1074/jbc.M116.729483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000 Feb 15;97(4):1589–1594. doi: 10.1073/pnas.97.4.1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kim YE, Hosp F, Frottin F, et al. Soluble oligomers of polyQ-expanded huntingtin target a multiplicity of key cellular factors. Mol Cell. 2016 Sep 15;63(6):951–964. doi: 10.1016/j.molcel.2016.07.022 [DOI] [PubMed] [Google Scholar]
- [54].Szymczak AL, Workman CJ, Wang Y, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004. May;22(5):589–594. doi: 10.1038/nbt957 [DOI] [PubMed] [Google Scholar]
- [55].Boncompain G, Divoux S, Gareil N, et al. Synchronization of secretory protein traffic in populations of cells. Nat Methods. 2012 Mar 11;9(5):493–498. doi: 10.1038/nmeth.1928 [DOI] [PubMed] [Google Scholar]
- [56].Bauer PO, Goswami A, Wong HK, et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010. Mar;28(3):256–263. doi: 10.1038/nbt.1608 [DOI] [PubMed] [Google Scholar]
- [57].Kerr JS, Adriaanse BA, Greig NH, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017. Mar;40(3):151–166. doi: 10.1016/j.tins.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy. 2011. Mar;7(3):266–278. doi: 10.4161/auto.7.3.14348 [DOI] [PubMed] [Google Scholar]
- [59].Narendra D, Tanaka A, Suen DF, et al. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Bio. 2008 Dec 1;183(5):795–803. doi: 10.1083/jcb.200809125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Yang YF, Gehrke S, Imai Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused inactivation of Drosophila PINK1 is rescued by by Parkin. P Natl Acad Sci USA. 2006 Jul 11;103:10793–10798. doi: 10.1073/pnas.0602493103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pickrell AM, Youle RJ. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015 Jan 21;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chung SY, Kishinevsky S, Mazzulli JR, et al. Parkin and PINK1 patient Ipsc-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Rep. 2016 Oct 11;7:664–677. doi: 10.1016/j.stemcr.2016.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].van der Merwe C, van Dyk HC, Engelbrecht L, et al. Curcumin rescues a PINK1 knock down SH-SY5Y cellular model of Parkinson’s disease from mitochondrial dysfunction and cell death. Mol Neurobiol. 2017. May;54(4):2752–2762. doi: 10.1007/s12035-016-9843-0 [DOI] [PubMed] [Google Scholar]
- [64].Gandhi S, Wood-Kaczmar A, Yao Z, et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009 Mar 13;33(5):627–638. doi: 10.1016/j.molcel.2009.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lutz AK, Exner N, Fett ME, et al. Loss of Parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem. 2009 Aug 21;284(34):22938–22951. doi: 10.1074/jbc.M109.035774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Ow YP, Green DR, Hao Z, et al. Cytochrome c: functions beyond respiration. Nat Rev Mol Cell Biol. 2008. Jul;9:532–542. doi: 10.1038/nrm2434 [DOI] [PubMed] [Google Scholar]
- [67].Kagan VE, Tyurin VA, Jiang J, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005. Sep;1(4):223–232. doi: 10.1038/nchembio727 [DOI] [PubMed] [Google Scholar]
- [68].Livingston MJ, Wang J, Zhou J, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019. Dec;15(12):2142–2162. doi: 10.1080/15548627.2019.1615822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhou B, Zhang JY, Liu XS, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018. Dec;28(12):1171–1185. doi: 10.1038/s41422-018-0090-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vargas JNS, Wang C, Bunker E, et al. Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell. 2019 Apr 18;74(2):347–362. doi: 10.1016/j.molcel.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Long J, Gallagher TR, Cavey JR, et al. Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J Biol Chem. 2008 Feb 29;283:5427–5440. doi: 10.1074/jbc.M704973200 [DOI] [PubMed] [Google Scholar]
- [72].Sanchez-Martin P, Komatsu M. P62/SQSTM1 - steering the cell through health and disease. J Cell Sci. 2018 Nov 5;131(21):jcs222836. doi: 10.1242/jcs.222836 [DOI] [PubMed] [Google Scholar]
- [73].Lang M, Pramstaller PP, Pichler I. Crosstalk of organelles in Parkinson’s disease - MiT family transcription factors as central players in signaling pathways connecting mitochondria and lysosomes. Mol Neurodegener. 2022 Jul 16;17(1):50. doi: 10.1186/s13024-022-00555-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zhang S, Zhao J, Quan Z, et al. Mitochondria and other organelles in neural development and their potential as therapeutic targets in neurodegenerative diseases. Front Neurosci. 2022;16:853911. doi: 10.3389/fnins.2022.853911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Torggler R, Papinski D, Brach T, et al. Two independent pathways within selective autophagy converge to activate Atg1 kinase at the vacuole. Mol Cell. 2016 Oct 20;64(2):221–235. doi: 10.1016/j.molcel.2016.09.008 [DOI] [PubMed] [Google Scholar]
- [76].Yamano K, Kikuchi R, Kojima W, et al. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Bio. 2020 Sep 7;219(9):e201912144. doi: 10.1083/jcb.201912144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016 Apr 12;113(15):4039–4044. doi: 10.1073/pnas.1523926113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Chu TT, Gao N, Li QQ, et al. Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem Biol. 2016 Apr 21;23(4):453–461. doi: 10.1016/j.chembiol.2016.02.016 [DOI] [PubMed] [Google Scholar]
- [79].Qu J, Ren X, Xue F, et al. Specific knockdown of alpha-synuclein by peptide-directed proteasome degradation rescued its associated neurotoxicity. Cell Chem Biol. 2020 Jun 18;27:751–762. doi: 10.1016/j.chembiol.2020.03.010 [DOI] [PubMed] [Google Scholar]
- [80].Lu M, Liu T, Jiao Q, et al. Discovery of a Keap1-dependent peptide PROTAC to knockdown Tau by ubiquitination-proteasome degradation pathway. Eur J Med Chem. 2018 Feb 25;146:251–259. doi: 10.1016/j.ejmech.2018.01.063 [DOI] [PubMed] [Google Scholar]
- [81].Ludtmann MH, Angelova PR, Ninkina NN, et al. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J Neurosci. 2016 Oct 12;36:10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang Y, Mandelkow E. Tau in physiology and pathology. Nat Rev Neurosci. 2016. Jan;17:5–21. doi: 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- [83].Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010 Jan 5;107(1):378–383. doi: 10.1073/pnas.0911187107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lazarou M, Jin SM, Kane LA, et al. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell. 2012 Feb 14;22(2):320–333. doi: 10.1016/j.devcel.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Geisler S, Holmstrom KM, Skujat D, et al. Pink1/parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010. Feb;12(2):119–131. doi: 10.1038/ncb2012 [DOI] [PubMed] [Google Scholar]
- [86].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-Parkin mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell. 2015 Oct 1;60(1):7–20. doi: 10.1016/j.molcel.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Malpartida AB, Williamson M, Narendra DP, et al. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends Biochem Sci. 2021. Apr;46(4):329–343. doi: 10.1016/j.tibs.2020.11.007 [DOI] [PubMed] [Google Scholar]
- [88].Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson’s disease. Trends Mol Med. 2011. Mar;17:158–165. doi: 10.1016/j.molmed.2010.11.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All study data are included in the manuscript and its Supplementary Material files.