

Abstract
Almost all mitochondrial proteins are synthesized in the cytosol and subsequently targeted to mitochondria. The accumulation of nonimported precursor proteins occurring upon mitochondrial dysfunction can challenge cellular protein homeostasis. Here we show that blocking protein translocation into mitochondria results in the accumulation of mitochondrial membrane proteins at the endoplasmic reticulum, thereby triggering the unfolded protein response (UPRER). Moreover, we find that mitochondrial membrane proteins are also routed to the ER under physiological conditions. The level of ER-resident mitochondrial precursors is enhanced by import defects as well as metabolic stimuli that increase the expression of mitochondrial proteins. Under such conditions, the UPRER is crucial to maintain protein homeostasis and cellular fitness. We propose the ER serves as a physiological buffer zone for those mitochondrial precursors that cannot be immediately imported into mitochondria while engaging the UPRER to adjust the ER proteostasis capacity to the extent of precursor accumulation.
INTRODUCTION
The ability of cells to maintain protein homeostasis (proteostasis) is crucial for organismal health. Imbalances in protein synthesis, targeting, folding, and degradation are associated with numerous diseases and are also hallmarks of aging (Gidalevitz et al., 2006; Hipp et al., 2014; Klaips et al., 2018; Moehle et al., 2019; Aviner and Frydman, 2020). Cells constantly monitor their proteome to quickly sense proteotoxic perturbations and launch stress-reactive programs to restore homeostasis. Of particular importance are the compartment-specific stress responses to misfolded proteins of the cytosol and nucleus (heat-shock response) as well as of the endoplasmic reticulum (ER; unfolded protein response of the ER, UPRER) and mitochondria (UPRmt). Via the activation of dedicated transcription factors, these pathways elevate the levels of chaperones, proteases and other quality control factors in the compartment where protein misfolding is sensed (Münch, 2018; Karagöz et al., 2019; Naresh and Haynes, 2019; Pincus, 2020).
When misfolded proteins accumulate in the ER, the ER membrane kinase Ire1 dimerizes, autophosphorylates, and then splices the mRNA of XBP1 (HAC1 in yeast). This enables its efficient translation (Cox and Walter, 1996), giving rise to a potent transcription factor that induces the UPRER. Besides increasing the expression of ER chaperones and other biogenesis factors, the UPRER can considerably expand the ER volume of a cell. In yeast, the Ire1-Hac1 pathway is the only dedicated regulator of the UPRER, while mammalian cells have two additional branches of the UPRER that control transcription, translation, and eventually apoptosis via PERK and ATF6 (Walter and Ron, 2011).
Cellular organelles have clearly distinct organizations and functions, yet they are no independent entities; instead, they form tight physical contacts (Kornmann et al., 2009; Valm et al., 2017; Bohnert, 2020) and functionally cooperate in the synthesis of proteins, lipids, and metabolites (Hansen, Aviram, et al., 2018; Carreras-Sureda et al., 2019). Hence, they mutually influence and rely on the homeostasis of one another. In many protein-folding diseases, defects in proteostasis are observed in multiple organelles at the same time, even though the primary perturbation occurs in most cases in only one compartment (Hetz and Mollereau, 2014; Bäuerlein et al., 2017). As a consequence, the different stress response programs need to act in concert (Liu and Chang, 2008; Pincus et al., 2014; Lebeau et al., 2018; Schmidt et al., 2019; Boos et al., 2020). For instance, perturbations of mitochondrial proteostasis often compromise mitochondrial protein import so that nonimported precursor proteins accumulate in the cytosol (Nargund et al., 2012; Harbauer et al., 2014; Rolland et al., 2019; Boos et al., 2020). Thus, mitochondrial dysfunction not only activates mitochondrial quality control pathways, but also the expression of cytosolic chaperones and the ubiquitin-proteasome system, which mitigate the deleterious effects of mistargeted precursors (Wrobel et al., 2015; Kim et al., 2016; Boos et al., 2019). In addition, the synthesis of many mitochondrial proteins is muted by transcriptional repression as well as global translation attenuation to further reduce the burden on cytosolic proteostasis (Nargund et al., 2015; Wang and Chen, 2015; Boos et al., 2019).
While numerous pathways of cross-compartment communication under proteotoxic stress have been identified, our understanding of the connections between organellar stress-response programs is still incomplete. Here we show that defective mitochondrial protein import not only activates mitochondrial and cytosolic stress responses, but also triggers the UPRER . This is at least in part attributable to the targeting of mitochondrial membrane proteins to the ER. The UPRER is functionally relevant both under conditions of compromised protein import, and conditions that induce mitochondrial biogenesis such as metabolic adaptations. Thus, the UPRER supports mitochondrial biogenesis by buffering the adverse consequences of elevated levels of nonimported mitochondrial precursor proteins.
RESULTS
The UPR ER is triggered by long-lasting mitoprotein-induced stress
Cellular adaptations to imbalances in mitochondrial proteostasis have been studied using mutants of protein import components (Wrobel et al., 2015), chaperones (Kim et al., 2016), folding-incompetent mitochondrial proteins (Wang and Chen, 2015; Fiorese et al., 2016), or defects in the respiratory chain (Labbadia et al., 2017; Guo et al., 2020). Many of these perturbations converge on the impairment of mitochondrial protein import. Model systems in which protein import can be acutely blocked have proven particularly useful to decipher the mechanistic details of responses to such mitoprotein-induced stress. A way of achieving this is the overexpression of mitochondrial precursor proteins that are intrinsically prone to premature folding and stalling inside the narrow mitochondrial translocases (Weidberg and Amon, 2018). For instance, the well-characterized “clogger” protein b2-DHFR can be used for this purpose (Boos et al., 2019; Mårtensson et al., 2019). This fusion protein consists of the N-terminal 167 amino acids of cytochrome b2 (including its mitochondrial targeting signal) and the rapidly and tightly folding dihydrofolate reductase (DHFR) (Figure 1A; Eilers and Schatz, 1986; Rassow et al., 1989; Wienhues et al., 1991). Expression of the clogger results in accumulation of nonimported precursor proteins (Figure 1B) and impairs cell growth (Figure 1C). In baker’s yeast, the expression of b2-DHFR can be tightly controlled by using a GAL promoter that can be switched on by the addition of galactose to the lactate-based growth media. This allows for a tight temporal resolution and the discrimination between short-term and long-term responses to an acute and specific impairment of mitochondrial protein import.
FIGURE 1:

Mitoprotein-induced stress triggers the UPRER. (A) Fusion of DHFR to the C-terminus of the cytochrome b2 presequence generates a mitochondrial “clogger” that jams the protein import machinery. (B) The mitochondrial clogger b2-DHFR or cytosolic DHFR were expressed for 4.5 h. The precursor form of the mitochondrial proteins Mdj1 and Rip1 were detected by Western blotting. (C) Expression of b2-DHFR leads to attenuated growth. Mean values and standard deviations (n = 3) are shown, p values are calculated on differences in maximum growth rate (t test). (D) The mitoprotein-induced stress response encompasses an early transcriptional induction of chaperones and the proteasome and a down-regulation of cytosolic ribosomes and OXPHOS components. (E) Protein levels in clogger-expressing vs. control cells after 18 h of induction were measured by quantitative mass spectrometry (Boos et al., 2019). Highlighted are proteins which are reported UPRER target genes (Schmidt et al., 2019). Data from n = 3 independent biological replicates are shown. (F and G) The cellular transcriptome and translatome after 4.5 h of clogger induction were measured by RNA-Seq (n = 4) (Boos et al., 2019) and ribosome profiling (n = 3), respectively. Shown are log2 fold changes of b2-DHFR versus cytosolic DHFR. HAC1 transcripts are slightly reduced, but its translation is up-regulated. (H) Ribosome footprints along the HAC1 gene from cells expressing b2-DHFR or cytosolic DHFR for 4.5 h are shown. (I) Levels of spliced HAC1 mRNA in cells expressing b2-DHFR or cytosolic DHFR were measured by RT-qPCR over time (n = 3). The line connects mean values, error bars denote standard deviations and Benjamini-Hochberg-adjusted p values for pairwise comparisons at each timepoint are shown below the graph.
We previously characterized the immediate reactions of the cellular transcriptome and proteome to mitoprotein-induced stress (Boos et al., 2019). An induction of many chaperones and the proteasome and a repression of oxidative phosphorylation (OXPHOS) components and ribosomes all took place within 1.5 h of clogger expression, some of them even markedly earlier (Figure 1D). However, many cellular adaptations change when acute stress persists and becomes long-lasting (Morimoto, 1991; Tsaytler et al., 2011; Samluk et al., 2019). We therefore asked whether cells undergo additional adaptations when exposed to more long-term mitoprotein-induced stress. To this end, we reanalyzed our previously collected data to examine changes in the cellular proteome after up to 18 h of clogger expression (Boos et al., 2019). We queried for changes in the proteome that were evident at time points no earlier than 4.5 h, which corresponds to approximately one cell doubling in respiratory medium. Interestingly, this criterion identified several enzymes with antioxidant activity as well as a group of proteins that are associated with the UPRER (Figure 1E; Supplementary Figures 1 and 2A; Schmidt et al., 2019). Some individual targets of the UPRER, such as Ero1 and Kar2, were induced at earlier time points, presumably due to their responsiveness to the transcription factors Hsf1 and/or Rpn4 that form the first line of defense against mitoprotein-induced stress. Moreover, a small number of UPRER targets were decreased over time. These proteins (Hem15, Mdl1, Coq6, and Mgr1) almost exclusively localize to mitochondria and their levels are likely affected by the import block or the clogger-induced down-regulation of mitochondrial components. However, most UPRER targets showed a consistent up-regulation that was observed 9 h after clogger induction, and even more so after 18 h (Figure 1E; Supplementary Figure 2A).
In yeast, the UPRER is activated by splicing of an intron from the HAC1 mRNA in the cytosol through the ER-resident kinase Ire1. Only the spliced isoform of the mRNA (called HAC1i) can be translated and gives rise to a transcription factor (Cox and Walter, 1996). To test whether HAC1 was indeed spliced and translated under mitoprotein-induced stress, we analyzed clogger-expressing cells by ribosome profiling. Here, ribosome footprints from cells expressing b2-DHFR or cytosolic DHFR were sequenced 4.5 h after induction, and the changes in the translatome were compared with the changes in the transcriptome (Figure 1F). For the large majority of all genes, transcriptional and translational changes correlated tightly. For HAC1 however, we observed a slight reduction of mRNA levels, while we found four times more ribosome footprints on HAC1 mRNA in clogger-expressing than in control cells (Figure 1G). In fact, HAC1 was one of the most prominent outliers in this comparison, ranking as the gene with the second-highest gain in translational efficiency when mitochondrial import was blocked (Supplementary Figure 2B). The increase in ribosome occupancy was restricted to the exon region of the mRNA, while the intron region of HAC1 was free of ribosome densities in both conditions (Figure 1H).
We next sought to determine the timing of the UPRER activation more precisely. To this end, we set up an RT-qPCR assay which quantifies the spliced isoform of HAC1i by using a primer-probe combination which specifically recognizes the exon–exon junction of HAC1i (Supplementary Figure 2, C and D). We induced b2-DHFR by addition of 0.5% galactose to cultures that were previously grown in lactate medium and followed HAC1 splicing over time. The earliest time point at which we could detect a considerable difference between clogger-expressing and control cells was 3 h (Figure 1I). As a certain delay between the onset of HAC1 splicing and downstream changes in protein levels of UPRER targets is expected, this is consistent with our earlier observation that UPRER induction is a rather late event in mitoprotein-induced stress signaling.
We conclude that under long-term impairment of mitochondrial protein import, cells induce the UPRER via the canonical Ire1-Hac1 pathway.
UPR ER induction is required for cellular fitness under mitoprotein-induced stress
We asked whether UPRER induction is functionally relevant under sustained mitoprotein-induced stress, given that its magnitude is rather mild when compared with harsh ER insults such as treatment with tunicamycin (cf. Supplementary Figure 2D). To this end, we compared the fitness of UPRER-deficient cells with that of wild type cells when mitochondrial import was impaired. Indeed, when either HAC1 or IRE1 were deleted, cells exhibited synthetic growth defects upon clogger expression, both in liquid medium and on plates (Figure 2, A and B). Growth defects were not due to cell death, but instead reflected stress-induced slowdown or arrest of cell division (Supplementary Figure 3, A and B).
FIGURE 2:

The UPRER is required for cellular fitness under mitoprotein-induced stress conditions. (A) Wild-type and ∆ire1 cells were grown under non-inducing (left) or inducing (right) conditions, expressing either b2-DHFR or cytosolic DHFR. ∆ire1 cells are more susceptible to mitoprotein-induced stress. p values denote differences in maximum growth rate, assessed by ANOVA and post hoc testing of pairwise differences (corrected for multiple testing with Benjamini-Hochberg). (B) Tenfold serial dilutions of wild-type, ∆ire1 and ∆hac1 cultures were dropped on lactate plates with or without 0.5% galactose. The UPRER-deficient mutants show synthetic growth defects with expression of b2-DHFR. (C) HAC1 and IRE1 were deleted in temperature-sensitive mia40-4 mutants. Cells were grown in glucose medium and serial dilutions were spotted on glucose plates and incubated at the semipermissive temperature of 30°C. Loss of the UPRER results in synthetic growth defects. (D) Early cytonuclear adaptations to mitoprotein-induced stress are accompanied by the induction of the UPRER as a second line of defense.
We examined the relevance of UPRER signaling when mitochondrial import is impaired by an approach orthogonal to clogging the translocases. To this end, we deleted HAC1 or IRE1 in a strain that carries a temperature-sensitive mutation in the essential import component Mia40. Mia40 is responsible for the import and oxidative folding of cysteine-containing mitochondrial intermembrane space proteins (Chacinska et al., 2004; Mesecke et al., 2005). The import defects in the mia40-4 mutant were shown to trigger cytosolic adaptations (UPR activated by mistargeting of proteins, UPRam), similar to those elicited by the clogger (Wrobel et al., 2015). Indeed, mia40-4 cells grew worse at semipermissive growth conditions when IRE1 or HAC1 were deleted, demonstrating that UPRER signaling is relevant when protein import into the IMS is perturbed (Figure 2C). In conclusion, defects in mitochondrial protein import trigger the UPRER, which is required for cellular fitness under such conditions (Figure 2D).
Mitochondrial membrane proteins accumulate at the ER when mitochondrial protein import is impaired
What could be the cause for UPRER activation in situations when mitochondrial import is impaired? Given the induction of redox-active enzymes in parallel to UPRER genes, we asked whether mitochondrial import stress elevates the levels of reactive oxygen species, which could perturb the redox homeostasis of the ER. Therefore, we measured cytosolic levels of H2O2 upon clogger induction in wild type (WT) and ∆hac1 cells using the ultra-sensitive genetically encoded fluorescent peroxide probe roGFP2-Tsa2∆CR (Supplementary Figure 4A) (Morgan et al., 2016). However, oxidation of the probe tended to be lower upon expression of the clogger, both in the cytosol and in mitochondria (Supplementary Figure 4, B and C). Intriguingly, clogger-expressing ∆hac1 cells also exhibited significantly decreased probe oxidation in response to exogenous H2O2 addition compared with clogger-expressing WT cells (Supplementary Figure 4D). Hence, clogger-expressing cells clearly do not experience elevated oxidative stress at this point, and notably, the UPRER is also not required for protection against reactive oxygen species.
Blocking mitochondrial import should elevate levels of mitochondrial precursor proteins in the cytosol. Therefore, we reasoned that a portion of these nonimported precursors, perhaps comprising membrane proteins, may be targeted to the ER, where they would accumulate and engage folding and protein quality control systems, thus triggering UPRER activation.
To test this hypothesis, we labeled the mitochondrial inner membrane protein Oxa1 with ymNeonGreen, and coexpressed it with Sec63-ymScarletI as an ER marker, followed by analysis of their subcellular distribution by fluorescence microscopy. When we expressed cytosolic DHFR, the green Oxa1 signal and the red Sec63 signal partitioned into separate structures with no considerable colocalization. In contrast, when b2-DHFR was expressed for 4.5 h, we found that a fraction of Oxa1-ymNeonGreen colocalized with Sec63-ymScarletI in the typical ring-shaped structures of the perinuclear and peripheral ER (Supplementary Figure 5A). This ER colocalization was observed in around 30% of the clogger-expressing cells, but only in around 1% of control cells (Figure 3A). As expected for mitochondrial stress, we also observed a change in mitochondrial morphology upon b2-DHFR expression, an early event that preceded the ER localization of Oxa1 (Supplementary Figure 5B).
FIGURE 3:

Non-imported mitochondrial membrane proteins localize to the ER. (A) The mitochondrial inner membrane protein Oxa1 was genomically tagged with ymNeonGreen, the ER marker Sec63 was tagged with ymScarletI. Confocal fluorescence microscopy was performed after 4.5 h of expression of either b2-DHFR or cytosolic DHFR (cf. Supplementary Figure 5A). Quantification of the number of cells in which ER localization of Oxa1-ymNeonGreen was observed. (B) Schematic depiction of the split-GFP strategy to measure ER localization of mitochondrial proteins. (C) The GFP11 fragment was fused to Oxa1 and the GFP1-10 fragment was fused to Oxa1, Mia40, Ssa1 or Sec63. b2-DHFR or cytosolic DHFR were induced for 4.5 h and fluorescence was measured in a plate reader. Mean values and standard deviations are shown for n = 7 (Oxa1-GFP1-10, Sec63-GFP1-10) or n = 4 (Mia40-GFP1-10, Ssa1-GFP1–10) independent biological replicates. (D) Fluorescence microscopy of cells expressing Oxa1-GFP11 and Sec63-GFP1-10 and either b2-DHFR or cytosolic DHFR after 4.5 h of induction. Scale bar, 5 µm. (E) The GFP11 fragment was fused to Om45 and the GFP1-10 fragment to Sec63. Clogger expression for 4.5 h evoked an increase in fluorescence (mean values and standard deviations are shown, n = 3). (F) Cells expressing Oxa1-GFP11 and Sec63-GFP1-10 were cultured in lactate medium before either b2-DHFR or cytosolic DHFR were induced by addition of 0.5% galactose. Fluorescence was monitored in a Clariostar plate reader every 10 min for n = 6 biological replicates. Constitutively expressed ymScarletI was used to normalize for growth and overall translation rates. After around 3 h of induction, elevated split-GFP signals in clogger-expressing cells indicated accumulation of Oxa1 at the ER. Significance was assessed with a two-way mixed ANOVA followed by post hoc testing of pairwise comparisons at each timepoint (Benjamini-Hochberg-adjusted p values are shown as color code). (G) Model for the connection between mitochondrial import block and UPRER induction. Clogging the mitochondrial translocases leads to accumulation of precursor proteins in the cytosol as well as at the ER surface, which triggers the ER stress response.
Fusions with fluorescent proteins can interfere with the function, localization, folding, and stability of proteins (Waldo et al., 1999; Wigley et al., 2001; Weill et al., 2019b). In particular, a large, stably folding C-terminal moiety might generate a mitochondrial clogger, as exemplified by b2-DHFR itself, and interfere with import and localization. We therefore sought to verify our results by a method that avoids the fusion of large protein domains to mitochondrial precursors and also minimizes the need for manual categorization of microscopic images with high mitochondrial background signal. To this end, we adapted a split-GFP method specifically designed to assess protein localization in vivo (Smoyer et al., 2016). Superfolder GFP is split into two parts, GFP1–10 and GFP11, which only emit fluorescence when colocalized to the same compartment (Figure 3B). The GFP fragments do not alter the folding behavior of the fusion proteins and their affinity is high enough to promote self-association without the need for a direct protein–protein interaction of the fusion partners (Cabantous et al., 2005; Cabantous and Waldo, 2006; Pedelacq et al., 2006). The GFP11 tag consists of only 17 amino acid residues and is therefore unlikely to affect translocation across the mitochondrial membranes.
To first verify that the split-GFP assay captures the subcellular localization of mitochondrial proteins, we fused the GFP11 fragment to the C-terminus of Oxa1 (inner membrane, C-terminus at matrix side), Mia40, Dld1 (inner membrane, C-terminus at IMS side) and Om45 (outer membrane, C-terminus at IMS side) and the GFP1–10 fragment to Oxa1 (IM, matrix side), Mia40 (IM, IMS side), Sec63 (ER membrane, cytosolic side), and Ssa1 (cytosol) (Supplementary Figure 6A) (Rojo et al., 1998; Szyrach et al., 2003; Terziyska et al., 2005; Song et al., 2014; Wenz et al., 2014; Wu et al., 2019; Kater et al., 2020). In the absence of any stress, by far the strongest fluorescence signal was detected for the combinations that recapitulate the known localization and topology for all proteins tested (Oxa1-GFP11/Oxa1-GFP1-10, Mia40-GFP11/Mia40-GFP1-10, Dld1-GFP11/Mia40-GFP1-10, and Om45-GFP11/Mia40-GFP1-10), while all other combinations resulted in much lower fluorescent signals (Figure 3C; Supplementary Figure 6, B–D, control condition in blue). This showed that the approach can measure protein localization with suborganellar resolution.
We next expressed either b2-DHFR or cytosolic DHFR for 4.5 h in strains carrying the split-GFP reporters. b2-DHFR expression evoked a marked increase in signal for Oxa1-GFP11 with the Sec63-GFP1-10 and the Ssa1-GFP1-10 reporters, while with cytosolic DHFR, only very little signal was detected (Figure 3C). This points towards relocation of a fraction of newly synthesized Oxa1-GFP11 to the ER surface or, potentially, the cytosol. We used fluorescence microscopy to confirm that the fluorescence we measured in a plate reader setup indeed originated from ER-localized GFP complementation (Figure 3D; Supplementary Figure 6E). For the mitochondrial outer membrane protein Om45-GFP11, we found a similar redistribution to the ER under import stress (Figure 3E; Supplementary Figure 6B), while neither Mia40-GFP11 nor Dld1-GFP11 showed detectable ER localization (Supplementary Figure 6, C and D). Oxa1 also localized to the ER when we blocked import by dissipating the membrane potential with CCCP (Supplementary Figure 6F). In ∆hrd1, ∆doa10, or ∆yos9 mutants that are defective in ER-associated degradation, accumulation at the ER was not stronger than in wild-type cells (Supplementary Figure 6F), suggesting that precursor proteins might not be subject to rapid degradation at the ER under these conditions.
We next asked about the timing of precursor localization to the ER after mitochondrial import is inhibited. To assess this question, we grew cells expressing Oxa1-GFP11 and Sec63-GFP1-10 in a plate reader, induced b2-DHFR or cytosolic DHFR by addition of galactose, and monitored split-GFP fluorescence over time in living cells. Constitutively expressed ymScarletI was used to normalize for differences in cell growth and translation rates. Clogger-expressing cells showed elevated split-GFP signals from around 3 h after induction (Figure 3F). Notably, the induction of the UPRER and the detection of Oxa1 at the ER were apparent around the same timepoint (cf. Figure 1I).
In conclusion, when mitochondrial import is blocked, some mitochondrial preproteins accumulate at the ER membrane which likely evokes the UPRER (Figure 3G).
The UPR ER maintains cellular fitness during adaptation of mitochondrial biogenesis
Thus far, we have shown that the UPRER is important as a stress-reactive system that comes into play when mitochondrial import is defective. However, we were of course next interested to examine if it is of more general relevance for mitochondrial biogenesis. Accurate protein sorting is a challenging task for cells, and the ER might constantly encounter a baseline load of mitochondrial precursor proteins. To check whether there is evidence for mitochondrial proteins routed to the ER in the absence of stress, we reanalyzed several high-resolution datasets on protein targeting. Proximity labeling of ribosomes close to the ER or the mitochondrial outer membrane and subsequent ribosome profiling determined the “local translatome” at the ER and mitochondrial surface in yeast (Jan et al., 2014; Williams et al., 2014; Figure 4A). Interestingly, while most mitochondrial proteins were enriched in the vicinity of mitochondria, a subset of mitochondrial proteins was found to be translated close to the ER, notably including Oxa1 (Figure 4B). Also in human cells, mRNAs of some mitochondrial proteins were found at the ER surface (Supplementary Figure 7A; Fazal et al., 2019). Finally, we reanalyzed datasets from studies that determined which nascent chains interact with the signal recognition particle (SRP) in yeast by pulldown of SRP and subsequent sequencing of the bound transcripts (del Alamo et al., 2011; Chartron et al., 2016; Figure 4C). SRP is a major targeting factor for secretory proteins that carry a signal sequence or transmembrane domains (Walter and Blobel, 1981; Ng et al., 1996; Wild et al., 2004; Costa et al., 2018). While secretory proteins were clearly the most enriched among the SRP substrates, a subset of mitochondrial encoding ribosome-nascent chains were also bound by SRP to a lesser extent, but significantly above what was found for cytosolic proteins (Figure 4D). Both in yeast and in human cells, ER-localized mitochondrial transcripts include, but are not limited to proteins with known dual localization to mitochondria and ER. Apparently, some mitochondrial precursors tend to associate with the ER even in the absence of stress, possibly mediated by “low priority” SRP-binding to at least some of these precursors.
FIGURE 4:

Some mitochondrial precursor proteins are physiologically translated at the ER. (A) Study design from (Jan et al., 2014) on the localized translation near the mitochondrial and the ER surface. (B) For all mitochondrial proteins in the dataset (Jan et al., 2014), the log2 enrichment of ribosome-nascent chain complexes at the ER membrane (Sec63-BirA) and the mitochondrial outer membrane (Om45-BirA) over the total ribosomes are shown. While most translated mRNAs localize to the mitochondrial membrane, some transcripts are enriched near the ER surface (expansion shows genes with more than twofold enrichment at the ER). (C) Study design from Chartron et al. (2016) on the SRP-bound translatome. SRP was immune-purified from cell lysates and the coisolated ribosome-nascent chains complexes were analyzed by ribosome profiling. (D) Data from (Chartron et al., 2016). The distribution of the log2 fold enrichment SRP-bound ribosome-nascent chain complexes over total ribosomes is shown for cytonuclear and mitochondrial proteins and proteins that carry a signal sequence or transmembrane domain for ER targeting (SS/TMD). Some mitochondrial proteins, including Oxa1, are bound by SRP. Distributions for mitochondrial and cytonuclear proteins are significantly different as judged by a two-sample Kolmogorov-Smirnov test. (E) The ER localization of Oxa1 was determined with the split-GFP assay in WT and ∆djp1 cells that were grown in glucose medium. Oxa1 is trapped at the ER in ∆djp1. Mean values and standard deviations are shown (n = 3), significance was assessed with a paired Student’s t test (p value is shown).
We did not observe considerable fluorescence in our split-GFP assays without applying import stress. However, under steady state conditions, precursors might only very transiently localize to the ER because they can be efficiently rerouted to mitochondria with the help of the ER-resident J protein Djp1 in a process called ER-SURF (Hansen, Aviram, et al., 2018). Loss of this pathway does not impair mitochondrial import per se, but traps mitochondrial orphans at the ER. To test this, we employed our split-GFP assay in the ‘ER-trapping’ ∆djp1 mutant and found accumulation of Oxa1 at the ER even under optimal growth conditions (Figure 4E). Furthermore, we asked whether in wild-type cells, there is ER-localized Oxa1 at levels below the detection limit of the split-GFP assay. We reasoned that Oxa1 might get glycosylated if it is (partially) imported into the ER, given that it has three predicted N-glycosylation motifs in its sequence, two of which are predicted to be luminal if Oxa1 was inserted into the ER membrane (Supplementary Figure 7B). We expressed Oxa1 with a hemagglutinin (HA) tag (to exclude cross-reactions of our polyclonal Oxa1 antibody) in wild-type cells in the absence and presence of the clogger, lysed the cells and digested one half of the lysates with the endoglycosylase EndoH, which specifically cleaves N-linked mannose-rich oligosaccharides. Using a monoclonal HA antibody, we detected a higher running band that was sensitive to EndoH treatment (Supplementary Figure 7C), showing that a fraction of Oxa1 was indeed N-glycosylated. This fraction was already present without the clogger and accumulated to higher levels when mitochondrial import was blocked (Supplementary Figure 7, C and D). Hence, there is indeed a constitutive flux of mitochondrial precursors to the ER in the absence of stress.
We next asked whether the UPRER might be required to buffer fluctuations in the levels of ER-localized mitochondrial precursors under physiological conditions. Mitochondrial biogenesis is strongly dependent on the carbon source in the growth media: The levels and, hence, the synthesis of many mitochondrial proteins is repressed in the presence of glucose, but is considerably increased on raffinose, galactose, glycerol, or lactate (Paulo et al., 2015; Paulo et al., 2016; Morgenstern, Stiller, Lübbert, Peikert, et al., 2017). In fact, we observed that the extent of steady state HAC1 splicing was low when cells were grown on glucose, but elevated on all other carbon sources (Figure 5A). To assess the functional relevance of the UPRER under different states of mitochondrial metabolism, we grew wild-type, ∆hac1 and ∆ire1 cells to exponential phase in liquid medium containing glucose, galactose, or lactate as sole carbon source. Then we washed the cells, resuspended them in glucose, galactose and lactate medium in all possible combinations and monitored their growth (Figure 5B; Supplementary Figure 8A). While there was no difference between wild-type and UPRER-deficient strains when they remained in the media they were cultured in before, ∆hac1 and ∆ire1 mutants had problems to adapt when carbon sources were switched to a medium with higher levels of HAC1 splicing. Likewise, UPRER-deficient strains grew well during exponential phase in glucose, but exhibited a phenotype at high optical densities, shortly before the cultures entered the stationary phase. At this point, yeast cells respond to the depletion of glucose and switch to respiratory metabolism, a growth phase called diauxic shift in which mitochondrial biogenesis is strongly induced (Murphy et al., 2015; Di Bartolomeo et al., 2020). Hence, the UPRER is important when such a remodeling takes place. To assess whether this could be attributed to increased ER-localization of mitochondrial proteins under these conditions, we monitored the split GFP fluorescence of Oxa1-GFP11 and Sec63-GFP1-10 after cells were shifted from glucose to lactate media. Indeed, we observed an increase in fluorescence during the transition from fermentative to respiratory growth, and a slight but significant drop in fluorescence during the reverse transition (Figure 5C). This indicates that changes in metabolism alter the flux of mitochondrial proteins to the ER, likely due to the strong increase in synthesis of mitochondrial proteins during the transition to respiratory growth.
FIGURE 5:

The UPRER maintains cellular fitness during changes in mitochondrial biogenesis. (A) HAC1 splicing in wild-type cells grown to log phase in media with the indicated carbon sources was measured via RT-qPCR. HAC1i levels were normalized to total HAC1 levels. Mean values and standard deviations from n = 5 independent biological replicates are shown. Significance was assessed with Welch’s ANOVA (p = 0.0034) followed by post hoc tests of pairwise comparisons (p values corrected for multiple testing according to Benjamini-Hochberg are shown). (B) Wild-type, ∆ire1 and ∆hac1 cells were grown to log phase in glucose, galactose, and lactate media, washed and switched to glucose, galactose, and lactate media in all combinations. Growth was monitored by OD600 measurement in a plate reader. Both UPRER-deficient mutants showed impaired growth when the carbon source was switched to one that promotes higher levels of HAC1 splicing in wild-type cells. p values from an ANOVA on maximum growth rates are shown. (C) Cells expressing Oxa1-GFP11 and Sec63-GFP1-10 were cultured in glucose or lactate medium and then switched to either glucose or lactate medium. Fluorescence was monitored in a Clariostar plate reader every 10 min for n = 6 biological replicates. Constitutively expressed ymScarletI was used to normalize for growth and overall translation rates. When switching from fermentative to respiratory growth, the signal increased over time. Significance was assessed with a two-way mixed ANOVA followed by post hoc testing of pairwise comparisons at each timepoint (Benjamini-Hochberg-adjusted p values are shown as color code). (D) Wild-type, ∆ire1, and ∆hac1 cells were plated on glycerol and 10 µl of a 3-M solution of the UPRER-inducing agent dithiotreitol (DTT) were applied on a filter dish in the middle of the plate. Note the ring-like growth of the wild type around the filter dish (arrowhead), indicating better growth at intermediate DTT concentrations. (E) Wild-type cells and cells that express HAC1i from a GAL promoter driven by the β-estradiol-inducible artificial transcription factor Gal4-ER-Msn2 were grown to log phase in glucose medium. They were washed, resuspended in either glucose or lactate medium supplemented with the indicated concentration of β-estradiol. Ectopic expression of low levels of HAC1i result in better growth in lactate, but not in glucose medium. See Supplementary Figure 8, B and C for statistics. (F) Schematic model for the role of the UPRER in mitochondrial protein biogenesis. A fraction of mitochondrial precursor proteins constantly localizes to the ER. Global changes in expression of mitochondrial genes increase the influx of precursors to the ER. Defects in protein import also elevate the levels of ER-resident mitochondrial precursors. In both cases, activation of the UPRER adjusts the proteostasis capacity of the ER.
Would a stronger UPRER help cells to adapt to respiratory growth conditions? Using a halo assay, we exposed cells growing on respiratory media to a gradient of the reducing agent dithiothreitol (DTT), which is a known trigger of the UPRER. Intriguingly, wild-type cells that were exposed to moderate amounts of DTT grew better than those exposed to lower doses, visible as a ring of larger colonies around the halo. The critical DTT concentration that boosted growth for wild-type cells was toxic for UPR-deficient strains, showing that it is indeed in a range that triggers the UPRER (Figure 5D). However, the beneficial effect of DTT on respiratory growth might – at least in part – also result from UPRER-independent effects. We therefore sought to induce the UPRER directly without any stress treatment by expressing the spliced isoform of HAC1 from a β-estradiol-inducible GAL promoter (Pincus et al., 2014). Cells were precultured in glucose medium and, upon shift to either glucose or lactate medium, exposed to various β-estradiol concentrations, that is, to different levels of HAC1i expression. Indeed, cells grew better in lactate when GAL-HAC1i was induced with up to 50 nM β-estradiol, while they were not affected when grown in glucose (Figure 5E; Supplementary Figure 8, B and C). Higher concentrations of β-estradiol delayed growth, consistent with earlier reports that overshooting UPRER activation can be toxic (Chawla et al., 2011; Rubio et al., 2011).
We also tested whether overexpression of HAC1i can benefit cells during clogger-induced stress but did not observe any growth advantage at any concentration tested (Supplementary Figure 8, D–F), likely because the UPRER is already induced and its protective effect cannot be further boosted by additional HAC1i.
Obviously, a functional UPRER is not only important when mitochondrial protein import is impaired, but also maintains cellular fitness under physiological conditions with elevated mitochondrial biogenesis. We propose that a fraction of mitochondrial precursor proteins is always localizing to the ER, either transiently as part of the ER-SURF pathway or terminally mistargeted. When the influx of precursors is altered due to changes in gene expression or by mitochondrial dysfunction, the UPRER acts as a ‘rheostat’ and adjusts the protein folding and quality control components of the ER accordingly (Figure 5F).
DISCUSSION
Precursor proteins that accumulate outside mitochondria impose a burden on cellular proteostasis. Many precursors remain in the cytosol (Wang and Chen, 2015) or end up in the nucleus (Shakya et al., 2021), where chaperones and the proteasome mitigate the adverse effects of mistargeted proteins and eventually degrade them (Wrobel et al., 2015; Kowalski et al., 2018; Boos et al., 2019; Nowicka et al., 2021). Membrane proteins are particularly prone to misfolding and aggregation in an aqueous environment. Hence, their prolonged presence in the cytosol can be very hazardous for cells (Liu et al., 2019; Backes et al., 2021). In this study, we found that cells can adsorb precursors of mitochondrial membrane proteins to the surface of the ER and employ the UPRER to buffer their elevated levels at the ER. Apparently, mitochondrial proteins associate with the ER even under physiological conditions. However, the accumulation of mitochondrial precursors, especially certain membrane proteins, at the ER is exacerbated by import defects as well as by metabolic stimuli that increase the expression of abundant mitochondrial enzymes. Our observations identify the UPRER as an important cellular response to promote cellular fitness under such conditions, especially during the phase of adaptation.
There are numerous reasons why engaging the ER as a venue for buffering mitochondrial membrane proteins can be beneficial: 1) The large ER membrane provides a favorable environment for proteins with hydrophobic transmembrane domains that would otherwise misfold in the aqueous cytosol. 2) The ER has a remarkable capacity to prevent protein aggregation, even exceeding that of the cytosol for some classes of proteins (Rousseau et al., 2004; Vincenz-Donnelly et al., 2018). 3) Besides having chaperones that promote protein folding, the ER harbors an elaborate machinery for ER-associated protein degradation (ERAD). While our data don’t suggest that ERAD degrades ER-localized Oxa1, ER components have been found to participate in the degradation of cytosolic and, more recently, mitochondrial proteins (Metzger et al., 2008; Laborenz et al., 2021). 4) ER and mitochondria share many components in their protein biogenesis and quality control systems, e.g. the Hsp40 cochaperone Ydj1 or Cdc48/VCP/p97 and many of its cofactors (Caplan et al., 1992; Heo et al., 2010; Tran et al., 2011; Mårtensson et al., 2019). In addition, some organelle-specific factors of ER and mitochondria physically interact and functionally cooperate with each other (Opaliński et al., 2018). 5) Protein transfer between mitochondrial and ER membranes is possible via dedicated machineries that can extract mislocalized proteins from the membrane and set them back en route to the respective other organelle (Hansen, Aviram, et al., 2018; Dederer et al., 2019; Matsumoto et al., 2019; McKenna et al., 2020; Xiao et al., 2021). 6) The close proximity of mitochondria and ER at membrane contact sites might facilitate the exchange of proteins between the two organelles. Interestingly, ER-mitochondria contact sites are enriched with ER chaperones and other UPRER effectors (Hayashi et al., 2009) and their loss activates the UPRER (Munoz et al., 2013). In addition, contact sites are crucial for the initiation of autophagy (Hamasaki et al., 2013; Böckler and Westermann, 2014).
Based on the above considerations, it is possible that routing of mitochondrial precursors to the ER could be more than a mere “mistake” in protein targeting, but rather an actively regulated quality control pathway. In line with this idea, our reanalysis of recent ribosome profiling data (Jan et al., 2014; Chartron et al., 2016) show that SRP recognizes and binds nascent chains of some mitochondrial proteins, suggesting that a portion of the mitochondrial proteome is synthesized at the ER surface. In addition, the GET pathway (guided entry of tail-anchored proteins) was recently identified to be involved in ER targeting of mitochondrial tail-anchored proteins and some carrier proteins (Vitali et al., 2018; Xiao et al., 2021).
Our findings add to a significant body of observations linking the stress responses and homeostasis mechanisms of mitochondria and ER (Rainbolt et al., 2014). Several processes connect mitochondrial and ER homeostasis in the context of stress: the flux of lipids between mitochondrial and ER membranes (Eiyama et al., 2021); the generation of ATP as well as reactive oxygen species by the respiratory chain (Tran et al., 2019; Yong et al., 2019); the transport of calcium (Carreras-Sureda et al., 2019; Costa et al., 2019); or the availability of building blocks for glycosylation of secretory proteins, provided by mitochondrial carbohydrate metabolism (Balsa et al., 2019). We propose that mitochondria and ER are also linked in the management of mitochondrial protein biogenesis.
Our observations pose several questions: Which of the many components and pathways that are reinforced by the UPRER are the most important for the management of ER-localized mitochondrial proteins? Do elevated levels of mitochondrial precursor proteins perturb ER homeostasis directly through localization to the ER, or also indirectly via sequestration of chaperones used by clients from both organelles? What happens to mitochondrial precursors at the ER – storage and sequestration, transfer to mitochondria, or degradation at the ER surface? Which pathways direct mitochondrial proteins to the ER, and is this specifically regulated or a general consequence of an overwhelmed import machinery?
We suggest that cells engage the ER and its proteostasis capacity – augmented by the UPRER when necessary – as a buffer for proteins that can’t be immediately imported into mitochondria. From there, they can be either degraded, permanently sequestered, or kept on hold for a second attempt of mitochondrial import. Therefore, we should consider rethinking the classical concept of ‘mislocalization’ as a problem that cells need to avoid. Rather, spatial sequestration (transient or terminal) of proteins to compartments other than those they are primarily targeted to might be a productive step in protein biogenesis. It will be exciting to explore this concept and the components that are involved in the future.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Yeast strains and plasmids
All yeast strains used in this study are listed in Supplementary Table S1 and were based on the wild-type strain W303, YPH499, or BY4742 (Sikorski and Hieter, 1989; Thomas and Rothstein, 1989). The mia40-4 strain was a gift from Agnieszka Chacinska (Chacinska et al., 2004). Yeast strains were grown on YP medium (1% yeast extract, 2% peptone) or synthetic medium (0.17% yeast nitrogen base and 0.5% [NH4]2SO4) containing 2% glucose, 2% galactose, 2% raffinose, 2% glycerol, or 2% lactate and supplemented with appropriate amounts of amino acids and nucleobases for selection.
pFA6a-ymNeongreen-CaURA3 and pFA6a-ymScarletI-CaURA3 were kindly provided by Bas Teusink (Addgene plasmids # 125703 and # 118457) (Botman et al., 2019). Genomic tagging with ymNeonGreen was performed by amplifying the ym-NeonGreen-CaURA3 cassette with overhangs homologous to the OXA1 locus and transforming yeast cells using the lithium acetate/ss carrier DNA/PEG method (Janke et al., 2004). Genomic deletion of IRE1 and HAC1 in the mia40-4 background was performed by amplifying a kanMX4 cassette from a pFA6a plasmid with overhangs homologous to the sequences up- and downstream of the genomic open reading frames of the target genes (Janke et al., 2004). Yeast cells were transformed with the PCR product and grown on plates containing 150 µg/ml G418 for selection. Deletions were confirmed by colony PCR on the targeted genomic loci.
The sequences of GFP11 (pSJ1321, pRS315-NOP1pr-GFP11-mCherry-PUS1) and GFP1-10 (pSJ2039, pRS316-NOP1pr-GFP1-10-SCS2TM) were a gift from Sue Jaspersen (Addgene plasmids # 86413 and # 86418; Smoyer et al., 2016). Cloning of the split-GFP constructs into the pYX122, pYX142, and pNH605 plasmids used in this study was performed by Gibson Assembly with the HiFi DNA Assembly Master Mix (New England Biolabs, #E2621L) according to the manufacturer’s instructions. The GFP11 part was fused to the different proteins by integration of the sequence (5′ AGA GAT CAT ATG GTT TTG CAT GAA TAT GTT AAT GCT GCT GGT ATT ACT TAA 3′) into the corresponding primers. GFP1-10 was amplified from the plasmid (pSJ2039) with overhangs homologous to the end of the fusion partner and the plasmid pYX122.
The W303 yeast strain expressing the LexA-ER transcription factor and plasmid pFA6a-kanMX6-p8LexOCYC1 carrying the lexO promoter (Ottoz et al., 2014) were kindly provided by Elçin Ünal.
Isolation of RNA and RT-qPCR
RNA was extracted from yeast cells using either the acid phenol-chloroform method or an RNeasy Mini kit with on-column removal of DNA (Qiagen), both as previously described (Boos et al., 2019). In either case, 3 OD600 × ml of cells were collected by centrifugation (17,000 × g, 3 min, 2°C), washed with prechilled water, snap-frozen in liquid nitrogen and stored at −80°C.
For acid phenol-chloroform extraction, cell lysates were prepared in lysis buffer (50 mM Tris/HCl [pH 7.0], 130 mM NaCl, 5 mM EDTA, 5% [wt/vol] SDS) with a FastPrep-24 5 G homogenizer [MP Biomedicals] with 3 cycles of 20 s, speed 6.0 m s–1, 120 s breaks, lysis matrix Y). RNA was purified with repeated extraction with acid phenol–chloroform (5:1, pH 4.5, two times) and 24:1 chloroform–isoamylalcohol (24:1). Then, 0.3 M sodium acetate (pH 5.5) was added, RNA was precipitated with ethanol and solubilized in water. DNA was removed using a Turbo DNA Free kit (Ambion) following the manufacturer’s instructions. RNA purity and concentration were assessed using a DeNovix DS-11 FX+ Fluorometer.
Reverse transcription-quantitative PCR (RT-qPCR) was performed with a CFX96 Touch Real-Time PCR Detection System (Bio-Rad). 100-ng total RNA per 20-µl reaction were analyzed using the Luna Universal Probe One-Step RT-qPCR Kit (NEB, # E3006) in technical triplicates. cDNA was generated by reverse transcription for 10 min at 55°C. PCR amplification was then carried out under the following conditions: initial denaturation for 1 min at 95°C, followed by 45 cycles of 10 s at 95°C (denaturation) and 30 s at 60°C (extension). Primer-probe combinations for qPCR are listed in Supplementary Table 3. For the specific detection of the spliced isoform of HAC1, primers were chosen to flank the intron and the fluorescent probe spans the exon–exon junction (Supplementary Figure 2C). Primer efficiency was determined by measuring serial dilutions of pooled cDNA and only primer-probe combinations with an efficiency within 90 and 110% were used. Cq values were obtained with the Bio-Rad CFX Manager 3.1 with Cq Determination Mode set to “Single Threshold” and Baseline Setting set to “Baseline Subtracted Curve Fit.” Gene expression was normalized to the geometric mean of the expression values of the reference gene TFC1 (Livak and Schmittgen, 2001). Statistical significance was assessed with paired two-tailed Student’s t test.
Growth Assays
Growth curves were performed automated in a 96 well plate in technical triplicates using the ELx808 Absorbance Microplate Reader (BioTek). Precultures of 100 μl were inoculated at an OD600 of 0.1 in round bottom microtiter plates and sealed with an air-permeable membrane (Breathe-Easy; Sigma-Aldrich, St. Louis, MO). The growth curves started at OD600 0.1 and incubated at 30°C for 72 h under constant shaking. The OD600 was measured every 10 min. Individual data points that were obvious outliers from technical problems with the OD600 measurements (spike considerably above the immediate neighboring data points) were excluded from the analysis to reduce technical noise. These data points are highlighted in the source data in Supplementary Table 4. Their removal from the plot did not alter the biological conclusions in any case.
Statistical analysis was performed by fitting the standard form of a logistic model to the data using the growthcurver package within R. We calculated maximum growth rate r and carrying capacity k for all individual replicates and assessed significance of differences between samples by ANOVA, followed by post hoc testing of pairwise differences (where relevant). P values were adjusted for multiple hypothesis testing with the Benjamini-Hochberg procedure.
For the Halo assay, strains were grown in liquid YPD media to mid-log phase, washed and plated on YPG plates. A filter plate was placed onto the plate and soaked with 10 µl of a 3-M DTT solution. Plates were incubated at 30°C for 2 d.
Flow cytometry viability analysis
Yeast strains were cultivated in selective lactate media. Clogger expression was induced by adding 0.5% galactose. After 4.5 h, 1 OD600 × ml of cells was harvested by centrifugation (4,000 g, 1.5 min, RT) and washed with PBS. Cells were resuspended in 500-µl FxCycle PI/RNase staining solution (Life Technologies, #F10797) and incubated at room temperature in the dark for 30 min. Samples were measured on an Attune NxT flow cytometer (Life Technologies). Data analyses were performed using the FlowJo software, version 10.8.0. Samples were gated for single cells (SSC-A/SSC-H) and a histogram of Forward scatter (FSC-A) against propidium iodide (PI) intensity was plotted. Unstained cells and stained cells that were killed by incubation at 70°C for 30 min were used as controls to set the threshold for gating PI positive and PI negative cells. The percentage of PI negative cells over total (PI negative and PI positive) cells was taken as measure of viability.
Preparation of cell extracts for Western blotting
For whole cell lysates yeast strains were cultivated in selective lactate media. Clogger expression was induced by adding 0.5% galactose. After 4.5 h, 2 OD600 × ml of cells were harvested by centrifugation (5,000 g, 5 min, RT) and washed with water. The cells were resuspended in 40-µl/OD600 1× Laemmli buffer (125-mM Tris/HCl [pH 6.8], 5% SDS [wt/vol], 25% glycerol, 0.0005% bromophenol blue) and transferred to a screw-cap tube containing glass beads (0.5 mm). Cell were lysed using a FastPrep-24 5 G homogenizer (MP Biomedicals) with 3 cycles of 20 s, speed 6.0 m/s, 120 s breaks, lysis matrix Y. Cell extracts were boiled for 10 min at 96°C. Samples were stored at −20°C until usage. An equal amount of lysate corresponding to 0.4 OD600 × ml per sample was loaded on an SDS gel.
Immunoblotting
Proteins were separated by size using discontinuous SDS–PAGE. They were transferred to a nitrocellulose membrane by semidry Western blotting with blotting buffer (20-mM Tris, 150-mM glycine, 0.08% SDS [wt/vol], 20% methanol). To visualize the transferred proteins, the membrane was stained with Ponceau S solution (0.2% (wt/vol) Ponceau S, 3% (wt/vol) acetic acid) for 5 min. The membrane was cut in pieces to decorate against several antibodies at once and unspecific binding was blocked by incubation for 30 min in 5% milk in 1X TBS buffer (10-mM Tris/HCl [pH 7.5], 150 mM NaCl). The first antibodies were incubated over night at 4°C. The membrane was washed extensively with 1X TBS Buffer. Afterward, the membrane was incubated for 90 min at room temperature with the secondary antibody containing the horseradish peroxidase (anti-Rabbit). The membrane was again washed extensively before ECL1 (100-mM Tris/HCl [pH 8.5], 0.044% [wt/vol] luminol, 0.0066% p-coumaric acid) and ECL2 (100-mM Tris/HCl [pH 8.5], 0.03% H2O2) solutions were mixed 1:1 and poured onto the membrane. Thereby chemo luminescence is produced by horseradish peroxidase coupled to the secondary antibody, which was detected on Super RX Medical X-Ray Films (Fuji) using the Optimax Type TR-developer.
Antibodies
The antibodies for the use in immunoblotting of Saccharomyces cerevisiae (S. cerevisiae) cell extracts were raised in rabbits using purified recombinant proteins. The secondary antibody was ordered from Biorad (Goat anti-Rabbit IgG (H+L)-HRP Conjugate #172-1019). Antibodies were diluted in 5% (wt/vol) nonfat dry milk-TBS (Roth T145.2) with the following dilutions: anti-Sod1 1:1000, Anti-HA 1:500, anti-Rip1 1:750, anti-Mdj1 1:125, anti-Rabbit 1:10,000. anti-Rip1 and anti-Mdj1 sera were a gift from Thomas Becker.
roGFP2-Tsa2∆C R-based monitoring of intracellular H 2O 2 levels
WT or ∆hac1 cells, harboring a pYX223 plasmid encoding either cytosolic DHFR or b2-DHFR were cotransformed with a p416TEF plasmid encoding roGFP2-Tsa2∆CR (cytosolic) or Su9-roGFP2-Tsa2∆CR (mitochondrial), ultra-sensitive, fluorescent H2O2 sensors (Morgan et al., 2016).
For roGFP2-Tsa2∆CR measurements, cells were grown in lactate medium lacking histidine and uracil to ensure plasmid retention. Strains were grown until they reached logarithmic phase. The culture was split and expression of either cytosolic DHFR or the b2-DHFR construct was induced by the addition of 0.5% galactose with uninduced cultures serving as controls. All cultures were then incubated with shaking at 30°C for a further 4.5 h before harvesting by centrifugation at 5000 g for 6 min. Cells were resuspended in 100-mM MES Tris pH 6 to an OD600 = 7.5. 200-µl aliquots of cells were transferred to the wells of a flat-bottomed 96-well imaging plate (BD Falcon). The plate was centrifuged at 30 g for 5 min. For every strain measured control wells were utilized, which were treated with either 20-mM diamide (Sigma Aldrich) or 100-mM DTT (AppChem) to calibrate full probe oxidation or reduction respectively.
Fluorescence of roGFP2 was followed for 4 min before H2O2 was added at final concentration of 10, 50, or 100 µM. Responses to the treatment were monitored for one more hour in a BMG Labtech CLARIOstar fluorescence plate reader. Cells harboring an empty p416TEF plasmid were used for background subtraction. Reduction-oxidation-sensitive GFP2 (roGFP2) contains two cysteine residues adjacent to the GFP chromophore. Reversible formation of an intramolecular disulfide bond between these cysteine residues changes the protonation state of the chromophore. The anionic form dominates in reduced roGFP2 and has an excitation maximum at ∼490 nm. In contrast, the neutral chromophore, with an excitation maximum at ∼400 nm dominates in oxidized roGFP2. Excitation at either wavelength results in fluorescence emission at ∼510 nm. roGFP2 oxidation (OxDroGFP2) during the measurements was determined according to the equation below, based on the emission at 510 nm of the sample as well as the fully oxidized and fully reduced controls after excitation at both 405 and 490 nm.
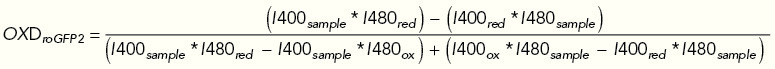 |
Significance was assessed with a two-way mixed ANOVA, followed by a post hoc test (pairwise tests at all timepoints corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure).
Split-GFP Assay
Cells were transformed with one of the plasmids pYX142-Oxa1-GFP11, pYX142-Om45-GFP11, pYX142-Dld1-GFP11, or pYX142-Mia40-GFP11 in combination with either pYX122-Sec63-GFP1-10, pYX122-Oxa1-GFP1-10, pYX122-Mia40-GFP1-10, or pYX122-Ssa1-GFP1-10. All combinations also contained either the plasmid pYX233-b2-DHFR or the control plasmid pYX233-cyt DHFR. Cells were grown in selective medium containing 2% lactate to mid log phase. Mitoprotein-induced stress was induced by addition of 0.5% galactose for 4.5 h. Three OD600 × ml were harvested, resuspended in 100-µl medium containing 2% lactate, transferred into a black 96 well plate and centrifuged (5 min at 30 g). The fluorescence was measured with the excitation/emission wavelengths 485±15/530±20 nm in a fluorescence microplate reader (Clariostar, BMG labtech). Significance of the results was assessed with a paired Student’s t test (corrected for multiple testing using the Benjamini-Hochberg procedure where necessary).
For the time-course measurement of split-GFP fluorescence in a growing culture, the split-GFP cassette with Oxa1-GFP11 and Sec63-GFP1-10 was genomically integrated into the LEU2 locus of yeast cells. In addition, a constitutively expressed TEF1p-ymScarletI was integrated into the HIS3 locus. The cells were transformed with a pYX233 plasmid for either cytosolic DHFR or b2-DHFR expression and grown to mid-log phase in synthetic lactate medium. The cells were then diluted to an OD600 of 0.4 in 100-µl lactate medium with (inducing) or without (noninducing) 0.5% galactose in a microtiter plate sealed with an air-permeable membrane (Breathe-Easy; Sigma-Aldrich, St. Louis, MO) in n = 6 replicates. A WT strain not expressing no fluorescent protein was used for correction of the background fluorescence of cells or media. The plate was incubated at 30°C under recurrent shaking in a ClarioStar spectrofluorometer (BMG Labtech) and fluorescence was measured every 10 min with the following excitation/emission wavelengths: 485 ± 15/530 ± 20 nm for split-GFP and 580 ± 15/631 ± 36 nm for ymScarletI. Background fluorescence was subtracted and the split-GFP signal was divided by the ymScarletI signal to control for growth and overall translation. The average fluorescence intensity at timepoint 0 was set to 1. Significance was assessed with a two-way mixed ANOVA, followed by a post hoc test (pairwise tests at all timepoints corrected for multiple hypothesis testing using the Benjamini-Hochberg procedure).
Fluorescence microcopy
To analyze the localization of the fluorescence signal in the different split-GFP combinations, mid-log phase cultures were shifted to media containing 0.5% galactose to induce the expression of the b2-DHFR clogger or cytosolic DHFR as control. After centrifugation of 1 OD600 × ml of cells (1 min at 16,000 g at RT), the cells were resuspended in 50-µl sterile water. The cell suspension was transferred to a microscope slide for fluorescence imaging using the HCX PL APO 63× oil immersion objective of a Leica TCS SP5II confocal laser scanning microscope or, for the time course experiment shown in Supplementary Figure 5B, the HC PL APO 100x/1.44 OIL UV objective of a Leica DMi8 microscope. GFP was excited at 488 nm (Leica SP5II) or 475 nm (Leica DMi8) and emission was detected by a photomultiplier through a 530/30-nm band pass filter. Microscopy images were processed using Leica software LAS X (v3.3) and Fiji (v2.1.0).
Glycosylation assay
Glycosylation sites in the Oxa1 sequence were predicted using the NetNGlyc 1.0 server with standard settings (Gupta and Brunak, 2002). Topology prediction of Oxa1 was performed with nine prediction algorithms (TMHMM, Topcons, Octopus, Phobius, PolyPhobius, SCAMPI, Spoctopus, HMMtop, and memsat svm) using the TopologYeast interface (http://www.weizmann.ac.il/molgen/TopologYeast/) (Weill et al., 2019a). Luminal localization of a motif was called when all prediction algorithms agreed on the topology at this position.
Cell lysates were prepared in 1xLaemmli buffer and mixed with 10× G5 reaction buffer and with or without 1000 units of Endo H from New England Biolabs (P0702S) and incubated for 1 h at 37°C. The enzyme was inactivated by boiling for 3 min at 96°C, protein were separated by SDS–PAGE and analyzed by Western blotting. Biological triplicates were used for the quantification of the glycosylated protein amount.
Ribosome profiling
Library preparation.
Yeast cultures were grown to mid-log phase in minimal medium containing 2% lactate. Expression of b2-DHFR or cytosolic DHFR was induced by addition of 0.5% galactose for 4.5 h. Cells were harvested by vacuum filtration (pore size 0.45 µm). In one out of three independent replicates, 100-µg/ml cycloheximide (CHX) was added to the yeast culture 2 min before harvesting and lysis to inhibit translation elongation, while in the other replicate, cells were not in contact with CHX before cell lysis. Cells were flash-frozen in liquid nitrogen and lysed in a mixer mill (Retsch, MM 301) in lysis buffer (20-mM Tris/HCl [pH 7.4], 140-mM KCl, 1.5-mM MgCl2, 0.5-mM DTT, 100-µg/ml CHX, 1% [vol/vol] Triton X-100) in 50 ml stainless steel grinding chambers under cryogenic conditions for 1 min at 20 Hz. Lysates were thawed in a water bath at room temperature, immediately followed by centrifugation at 15,000 g at 4°C for 10 min. RNA concentration was quantified with a NanoDrop fluorometer (absorbance at 260 nm) and RNA digestion was performed with RNase I (Ambion, #AM2294, 2.5-µl/mg RNA) for 45 min at room temperature. Digestion was stopped by the addition of SUPERase·In RNase inhibitor (Ambion, #AM2696, 2-µl/100-µl digestion). Ribosomes were isolate by centrifugation through a 25% (wt/vol) sucrose cushion in a TLA 100.2 rotor (Beckman) at 72,000 rpm for 20 min at 4°C. RNA was extracted from the ribosomal pellet using the hot SDS-Phenol-Chloroform method and 24–35 nt ribosome footprints were size selected on a 15% (wt/vol) polyacrylamide TBE-urea gel. Ribosomal RNA was removed with the RiboZero Gold kit (Illumina). Sequencing libraries were then prepared as previously described (Ingolia et al., 2012). Libraries were quantified by qPCR (Kapa Biosystems) and sequenced using a HiSeq 4000 (Illumina).
Data analysis.
Sequencing reads were demultiplexed with Illumina CASAVA v1.8 and adaptor sequences were trimmed using Cutadapt v2.8. Reads that mapped to ribosomal RNAs were removed using Bowtie v.1.2.3 (Langmead et al., 2009) and remaining reads were aligned to the yeast reference genome obtained from the Saccharomyces genome database (genome release R64-2-1).
For each read, reads were summed at each nucleotide by customized python scripts. Metagene analysis was performed separately on each fragment length to remove lengths that did not exhibit the 3-nucleotide periodicity that is characteristic for ribosome footprints. Each of the remaining reads was assigned to the first A-site nucleotide. To this end, a nucleotide offset from the 5′ end of each fragment length was empirically determined, using the characteristic high ribosome density at the start codon. Nucleotide reads at each codon were then summed and used for all downstream analysis.
Gene-level differential expression analysis was performed using HTSeq (Anders et al., 2015) and the DESeq2 package (Love et al., 2014) within the Bioconductor v3.12 project in the statistical programming language R v.4.0.3 (Huber et al., 2015).
GO enrichment analysis
Gene ontology (GO) enrichment analysis on proteomics data from Boos et al. (2019) was performed in RStudio using clusterProfiler (Wu et al., 2021). Proteins that changed significantly with Benjamini-Hochberg-adjusted p value smaller 0.05 after 4.5 h or 9 h were selected. These were further filtered for log2 fold changes greater 0.5 or smaller –0.5 for increased or decreased proteins respectively. The significantly increased or decreased proteins were tested against all identified proteins in the mass spec experiment. The function enrichGO for molecular function (MF) was run with the following settings: keyType = UNIPROT, OrgDb = org.Sc.sgd.db, ont = “MF”, pAdjustMethod = “BH”, pvalueCutoff = 0.05, qvalueCutoff = 0.05, readable = FALSE. p values were adjusted using Benjamini-Hochberg (BH). To reduce redundancy in GO terms the simplify function was used with the following settings: cutoff = 0.7 and by = ‘p adjust’. The Wang method was used to measure the similarity of GO terms. The dotplot function was used to plot the top 10 overrepresented GO terms based.
Analysis of published datasets on mRNA localization
The dataset on translation close to the ER and mitochondrial surface in yeast was obtained from Jan et al. (2014) . In this study, the authors fused the biotin ligase BirA to Sec63 (ER) or Om45 (mitochondrial) and pulled down ribosomes that were biotinylated after a short pulse of biotin and translation inhibition with CHX. The genes were filtered for those that code for mitochondrial proteins according to (Morgenstern, Stiller, Lübbert, Peikert, et al., 2017) and log2 fold enrichments of ribosome footprints at the ER (7-min CHX) or mitochondrial membranes (2-min CHX) over total ribosome footprints were plotted.
The dataset on transcript localization in human cells was obtained from Fazal et al. (2019). Here, the authors used the biotin ligase APEX2 fused to proteins of different cellular localizations to directly biotinylate RNA. Mitochondrial genes were filtered according to MitoCarta 3.0 (Rath et al., 2021) and log2 fold enrichment of ER- or mitochondria-localized transcripts over total transcripts were plotted.
The dataset on the SRP-bound translatome in yeast was obtained from Chartron et al. (2016) . The authors compared ribosome-nascent chain complexes purified by pulldown of SRP to total ribosomes by ribosome profiling. Genes coding for secretory, cytosolic, and mitochondrial proteins were filtered according to the author’s categorization and the distribution of the log2 fold enrichment of SRP-bound polysomes over total ribosome footprints was plotted.
Data and material availability
The data produced in this study are presented in this published article and its supplementary material. Source data are provided with this paper in Supplementary Table 4. The ribosome profiling data on clogger-expressing yeast cells are deposited into GEO (Barrett et al., 2013) with accession number GSE172017. The flow cytometry data are deposited to FlowRepository with accession number FR-FCM-Z4KF.
All yeast strains, plasmids and primers used in this study are listed in Supplementary Tables 1–3 and are available from the authors upon request. The plasmids pYX233 DHFR and pYX233 b2-DHFR for expression of the mitochondrial clogger are available via Addgene (plasmids #163761 and #163759).
Code availability
All code used for the analysis of data and generation of figures is available from the authors upon request.
Supplementary Material
Acknowledgments
Sequencing was performed at the University of California, San Francisco Center for Advanced Technology. We thank Sabine Knaus, Andrea Trinkaus, and Natalia M. Barbosa for assistance with the experiments, Torsten Möhlmann for help with the microscopy, and Prince S. Amponsah and Diana Ordonez for help with the flow cytometry. We thank Christiane Brune and Elçin Ünal for providing the ER-LexA expression system, and Jingxun Chen, Basile Jacquel, Gilles Charvin, Nir Cohen, T. Kelly Rainbolt, and Sebastian Schuck for discussions and comments on the manuscript. This project was funded by grants from the Landesforschungsinitiative Rheinland-Pfalz BioComp (to F.B. and J.M.H.), the Deutsche Forschungsgemeinschaft (DE2803/11-1 to J.M.H.), the US National Institutes of Health (NIH-GM56433 to J.F. and AG047126 to K.C.S.), the Glenn Foundation for Medical Research (Postdoctoral Fellowship to K.C.S.), the Company of Biologists (Travelling Fellowship JCSTF181148 to F.B.), the Damon Runyon Cancer Research Foundation (Postdoctoral Fellowship DRG-2461-22 to F.B.), the European Molecular Biology Organization (Long-Term Fellowship ALT 762-2019 to K.G.H. and Postdoctoral Fellowship ALTF-867-2021 to F.B.), the Helen Hay Whitney Foundation (F-1240 to K.G.H.) and the Joachim Herz Stiftung (to F.B. and C.G.).
Abbreviations used:
- ER
endoplasmic reticulum
- GO
gene ontology
- OXPHOS
oxidative phosphorylation
- RT-qPCR
reverse transcription-quantitative PCR
- sfGFP
superfolder green fluorescent protein
- UPR ER
unfolded protein response of the endoplasmic reticulum
- UPR mt
unfolded protein response of the mitochondria
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E23-05-0205) on June 28, 2023.
REFERENCES
Boldface names denote co–first authors.
- Anders S, Pyl PT, Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviner R, Frydman J (2020). Proteostasis in viral infection: unfolding the complex virus-chaperone interplay. Cold Spring Harb Perspect Biol 12, a034090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes S, Bykov YS, Flohr T, Räschle M, Zhou J, Lenhard S, Kramer L, Mühlhaus T, Bibi C, Jann C, et al. (2021). The chaperone-binding activity of the mitochondrial surface receptor Tom70 protects the cytosol against mitoprotein-induced stress. Cell Rep 35, 108936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa E, Soustek MS, Thomas A, Cogliati S, Garcia-Poyatos C, Martin-Garcia E, Jedrychowski M, Gygi SP, Enriquez JA, Puigserver P (2019). ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK-eIF2α axis. Mol Cell 74, 877–890 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. (2013). NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41, D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäuerlein FJB, Saha I, Mishra A, Kalemanov M, Martinez-Sanchez A, Klein R, Dudanova I, Hipp MS, Hartl FU, Baumeister W, Fernandez-Busnadiego R (2017). In situ architecture and cellular interactions of PolyQ inclusions. Cell 171, 179–187 e10. [DOI] [PubMed] [Google Scholar]
- Böckler S, Westermann B (2014). Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 28, 450–458. [DOI] [PubMed] [Google Scholar]
- Bohnert M (2020). Tether me, tether me not-dynamic organelle contact sites in metabolic rewiring. Dev Cell 54, 212–225. [DOI] [PubMed] [Google Scholar]
- Boos F, Krämer L, Groh C, Jung F, Haberkant P, Stein F, Wollweber F, Gackstatter A, Zöller E, van der Laan M, et al. (2019). Mitochondrial protein-induced stress triggers a global adaptive transcriptional programme. Nat Cell Biol 21, 442–451. [DOI] [PubMed] [Google Scholar]
- Boos F, Labbadia J, Herrmann JM (2020). How the mitoprotein-induced stress response safeguards the cytosol: a unified view. Trends Cell Biol 30, 241–254. [DOI] [PubMed] [Google Scholar]
- Botman D, de Groot DH, Schmidt P, Goedhart J, Teusink B (2019). In vivo characterisation of fluorescent proteins in budding yeast. Sci Rep 9, 2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS (2005). Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol 23, 102–107. [DOI] [PubMed] [Google Scholar]
- Cabantous S, Waldo GS (2006). In vivo and in vitro protein solubility assays using split GFP. Nat Methods 3, 845–854. [DOI] [PubMed] [Google Scholar]
- Caplan AJ, Cyr DM, Douglas MG (1992). YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell 71, 1143–1155. [DOI] [PubMed] [Google Scholar]
- Carreras-Sureda A, Jana F, Urra H, Durand S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos-Fernandez E, Sassano ML, et al. (2019). Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol 21, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A, Pfannschmidt S, Wiedemann N, Kozjak V, Sanjuan Szklarz LK, Schulze-Specking A, Truscott KN, Guiard B, Meisinger C, Pfanner N (2004). Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J 23, 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, Hunt KC, Frydman J (2016). Cotranslational signal-independent SRP preloading during membrane targeting. Nature 536, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Chakrabarti S, Ghosh G, Niwa M (2011). Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol 193, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa EA, Subramanian K, Nunnari J, Weissman JS (2018). Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science 359, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Peruzzo R, Bachmann M, Monta GD, Vicario M, Santinon G, Mattarei A, Moro E, Quintana-Cabrera R, Scorrano L, et al. (2019). Impaired mitochondrial atp production downregulates Wnt signaling via ER stress induction. Cell Rep 28, 1949–1960.e1946. [DOI] [PubMed] [Google Scholar]
- Cox JS, Walter P (1996). A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell 87, 391–404. [DOI] [PubMed] [Google Scholar]
- Dederer V, Khmelinskii A, Huhn AG, Okreglak V, Knop M, Lemberg MK (2019). Cooperation of mitochondrial and ER factors in quality control of tail-anchored proteins. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Alamo M, Hogan DJ, Pechmann S, Albanèse V, Brown PO, Frydman J (2011). Defining the specificity of cotranslationally acting chaperones by systematic analysis of mRNAs associated with ribosome-nascent chain complexes. PLoS Biol 9, e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bartolomeo F, Malina C, Campbell K, Mormino M, Fuchs J, Vorontsov E, Gustafsson CM, Nielsen J (2020). Absolute yeast mitochondrial proteome quantification reveals trade-off between biosynthesis and energy generation during diauxic shift. Proc Natl Acad Sci USA 117, 7524–7535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M, Schatz G (1986). Binding of a specific ligand inhibits import of a purified precursor protein into mitochondria. Nature 322, 228–232. [DOI] [PubMed] [Google Scholar]
- Eiyama A, Aaltonen MJ, Nolte H, Tatsuta T, Langer T (2021). Disturbed intramitochondrial phosphatidic acid transport impairs cellular stress signaling. J Biol Chem 296, 100335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, Chang HY, Ting AY (2019). Atlas of subcellular RNA localization revealed by APEX-Seq. Cell 178, 473–490 e426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorese CJ, Schulz AM, Lin YF, Rosin N, Pellegrino MW, Haynes CM (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr Biol 26, 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI (2006). Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474. [DOI] [PubMed] [Google Scholar]
- Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA, Kampmann M (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Brunak S (2002). Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput, 310–322. [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, et al. (2013). Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393. [DOI] [PubMed] [Google Scholar]
- Hansen KG, Aviram N, Laborenz J, Bibi C, Meyer M, Spang A, Schuldiner M, Herrmann JM (2018). An ER surface retrieval pathway safeguards the import of mitochondrial membrane proteins in yeast. Science 361, 1118–1122. [DOI] [PubMed] [Google Scholar]
- Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C (2014). The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19, 357–372. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009). MAM: more than just a housekeeper. Trends Cell Biol 19, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Livnat-Levanon N, Taylor EB, Jones KT, Dephoure N, Ring J, Xie J, Brodsky JL, Madeo F, Gygi SP, et al. (2010). A stress-responsive system for mitochondrial protein degradation. Mol Cell 40, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Mollereau B (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15, 233–249. [DOI] [PubMed] [Google Scholar]
- Hipp MS, Park SH, Hartl FU (2014). Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24, 506–514. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T, et al. (2015). Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods 12, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan CH, Williams CC, Weissman JS (2014). Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 346, 1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Karagöz GE, Acosta-Alvear D, Walter P (2019). The unfolded protein response: detecting and responding to fluctuations in the protein-folding capacity of the endoplasmic reticulum. Cold Spring Harb Perspect Biol 11, a033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kater L, Wagener N, Berninghausen O, Becker T, Neupert W, Beckmann R (2020). Structure of the Bcs1 AAA-ATPase suggests an airlock-like translocation mechanism for folded proteins. Nat Struct Mol Biol 27, 142–149. [DOI] [PubMed] [Google Scholar]
- Kim HE, Grant AR, Simic MS, Kohnz RA, Nomura DK, Durieux J, Riera CE, Sanchez M, Kapernick E, Wolff S, Dillin A (2016). Lipid biosynthesis coordinates a mitochondrial-to-cytosolic stress response. Cell 166, 1539–1552 e1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaips CL, Jayaraj GG, Hartl FU (2018). Pathways of cellular proteostasis in aging and disease. J Cell Biol 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009). An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 325, 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski L, Bragoszewski P, Khmelinskii A, Glow E, Knop M, Chacinska A (2018). Determinants of the cytosolic turnover of mitochondrial intermembrane space proteins. BMC Biol 16, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Brielmann RM, Neto MF, Lin YF, Haynes CM, Morimoto RI (2017). Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell Rep 21, 1481–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laborenz J, Bykov YS, Knoringer K, Raschle M, Filker S, Prescianotto-Baschong C, Spang A, Tatsuta T, Langer T, Storchova Z, et al. (2021). The ER protein Ema19 facilitates the degradation of non-imported mitochondrial precursor proteins. Mol Biol Cell 32, 664-674. mbcE20110748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau J, Saunders JM, Moraes VWR, Madhavan A, Madrazo N, Anthony MC, Wiseman RL (2018). The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep 22, 2827–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chang A (2008). Heat shock response relieves ER stress. EMBO J 27, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang X, Coyne LP, Yang Y, Qi Y, Middleton FA, Chen XJ (2019). Mitochondrial carrier protein overloading and misfolding induce aggresomes and proteostatic adaptations in the cytosol. Mol Biol Cell 30, 1272–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, Becker T (2019). Mitochondrial protein translocation-associated degradation. Nature 569, 679–683. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Nakatsukasa K, Kakuta C, Tamura Y, Esaki M, Endo T (2019). Msp1 Clears mistargeted proteins by facilitating their transfer from mitochondria to the ER. Mol Cell 76, 191–205 e10. [DOI] [PubMed] [Google Scholar]
- McKenna MJ, Sim SI, Ordureau A, Wei L, Harper JW, Shao S, Park E (2020). The endoplasmic reticulum P5A-ATPase is a transmembrane helix dislocase. Science 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM (2005). A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 121, 1059–1069. [DOI] [PubMed] [Google Scholar]
- Metzger MB, Maurer MJ, Dancy BM, Michaelis S (2008). Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J Biol Chem 283, 32302–32316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehle EA, Shen K, Dillin A (2019). Mitochondrial proteostasis in the context of cellular and organismal health and aging. J Biol Chem 294, 5396–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B, Van Laer K, Owusu TN, Ezerina D, Pastor-Flores D, Amponsah PS, Tursch A, Dick TP (2016). Real-time monitoring of basal H2O2 levels with peroxiredoxin-based probes. Nat Chem Biol 12, 437–443. [DOI] [PubMed] [Google Scholar]
- Morgenstern M, Stiller SB, Lübbert P, Peikert CD, Dannenmaier S, Drepper F, Weill U, Hoss P, Feuerstein R, Gebert M, et al. (2017). Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep 19, 2836–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (1991). Heat shock: the role of transient inducible responses in cell damage, transformation, and differentiation. Cancer Cells 3, 295–301. [PubMed] [Google Scholar]
- Münch C (2018). The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol 16, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, Diaz-Ramos A, Hernandez-Alvarez MI, Sebastian D, Mauvezin C, et al. (2013). Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32, 2348–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JP, Stepanova E, Everley RA, Paulo JA, Gygi SP (2015). Comprehensive Temporal Protein Dynamics during the Diauxic Shift in Saccharomyces cerevisiae. Mol Cell Proteomics 14, 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naresh NU, Haynes CM (2019). Signaling and Regulation of the Mitochondrial Unfolded Protein Response. Cold Spring Harb Perspect Biol 11, a033944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P, Haynes CM (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DT, Brown JD, Walter P (1996). Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol 134, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka U, Chroscicki P, Stroobants K, Sladowska M, Turek M, Uszczynska-Ratajczak B, Kundra R, Goral T, Perni M, Dobson CM, et al. (2021). Cytosolic aggregation of mitochondrial proteins disrupts cellular homeostasis by stimulating the aggregation of other proteins. eLife 10, e65484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opaliński Ł, Song J, Priesnitz C, Wenz LS, Oeljeklaus S, Warscheid B, Pfanner N, Becker T (2018). Recruitment of cytosolic J-Proteins by TOM receptors promotes mitochondrial protein biogenesis. Cell Rep 25, 2036–2043 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottoz DS, Rudolf F, Stelling J (2014). Inducible, tightly regulated and growth condition-independent transcription factor in Saccharomyces cerevisiae. Nucleic Acids Res 42, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA, Gygi SP (2016). Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J Proteomics 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo JA, O’Connell JD, Gaun A, Gygi SP (2015). Proteome-wide quantitative multiplexed profiling of protein expression: carbon-source dependency in Saccharomyces cerevisiae. Mol Biol Cell 26, 4063–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol 24, 79–88. [DOI] [PubMed] [Google Scholar]
- Pincus D (2020). Regulation of Hsf1 and the heat shock response. Adv Exp Med Biol 1243, 41–50. [DOI] [PubMed] [Google Scholar]
- Pincus D, Aranda-Diaz A, Zuleta IA, Walter P, El-Samad H (2014). Delayed Ras/PKA signaling augments the unfolded protein response. Proc Natl Acad Sci USA 111, 14800–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, Wiseman RL (2014). Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab 25, 528–537. [DOI] [PubMed] [Google Scholar]
- Rassow J, Guiard B, Wienhues U, Herzog V, Hartl FU, Neupert W (1989). Translocation arrest by reversible folding of a precursor protein imported into mitochondria. a means to quantitate translocation contact sites. J Cell Biol 109, 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, et al. (2021). MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 49, D1541–D1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo EE, Guiard B, Neupert W, Stuart RA (1998). Sorting of D-lactate dehydrogenase to the inner membrane of mitochondria. Analysis of topogenic signal and energetic requirements. J Biol Chem 273, 8040–8047. [DOI] [PubMed] [Google Scholar]
- Rolland SG, Schneid S, Schwarz M, Rackles E, Fischer C, Haeussler S, Regmi SG, Yeroslaviz A, Habermann B, Mokranjac D, et al. (2019). Compromised mitochondrial protein import acts as a signal for UPR(mt). Cell Rep 28, 1659–1669 e5. [DOI] [PubMed] [Google Scholar]
- Rousseau E, Dehay B, Ben-Haiem L, Trottier Y, Morange M, Bertolotti A (2004). Targeting expression of expanded polyglutamine proteins to the endoplasmic reticulum or mitochondria prevents their aggregation. Proc Natl Acad Sci USA 101, 9648–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P (2011). Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol 193, 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samluk L, Urbanska M, Kisielewska K, Mohanraj K, Kim MJ, Machnicka K, Liszewska E, Jaworski J, Chacinska A (2019). Cytosolic translational responses differ under conditions of severe short-term and long-term mitochondrial stress. Mol Biol Cell 30, 1864–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RM, Schessner JP, Borner GH, Schuck S (2019). The proteasome biogenesis regulator Rpn4 cooperates with the unfolded protein response to promote ER stress resistance. Elife 8, e43244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakya VP, Barbeau WA, Xiao T, Knutson CS, Schuler MH, Hughes AL (2021). A nuclear-based quality control pathway for non-imported mitochondrial proteins. Elife 10, e61230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P (1989). A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoyer CJ, Katta SS, Gardner JM, Stoltz L, McCroskey S, Bradford WD, McClain M, Smith SE, Slaughter BD, Unruh JR, Jaspersen SL (2016). Analysis of membrane proteins localizing to the inner nuclear envelope in living cells. J Cell Biol 215, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Tamura Y, Yoshihisa T, Endo T (2014). A novel import route for an N-anchor mitochondrial outer membrane protein aided by the TIM23 complex. EMBO Rep 15, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM (2003). Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J 22, 6448–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terziyska N, Lutz T, Kozany C, Mokranjac D, Mesecke N, Neupert W, Herrmann JM, Hell K (2005). Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett 579, 179–184. [DOI] [PubMed] [Google Scholar]
- Thomas BJ, Rothstein R (1989). Elevated recombination rates in transcriptionally active DNA. Cell 56, 619–630. [DOI] [PubMed] [Google Scholar]
- Tran DM, Ishiwata-Kimata Y, Mai TC, Kubo M, Kimata Y (2019). The unfolded protein response alongside the diauxic shift of yeast cells and its involvement in mitochondria enlargement. Sci Rep 9, 12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran JR, Tomsic LR, Brodsky JL (2011). A Cdc48p-associated factor modulates endoplasmic reticulum-associated degradation, cell stress, and ubiquitinated protein homeostasis. J Biol Chem 286, 5744–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaytler P, Harding HP, Ron D, Bertolotti A (2011). Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 332, 91–94. [DOI] [PubMed] [Google Scholar]
- Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott-Schwartz J (2017). Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz-Donnelly L, Holthusen H, Korner R, Hansen EC, Presto J, Johansson J, Sawarkar R, Hartl FU, Hipp MS (2018). High capacity of the endoplasmic reticulum to prevent secretion and aggregation of amyloidogenic proteins. EMBO J 37, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali DG, Sinzel M, Bulthuis EP, Kolb A, Zabel S, Mehlhorn DG, Figueiredo Costa B, Farkas A, Clancy A, Schuldiner M, Grefen C, et al. (2018). The GET pathway can increase the risk of mitochondrial outer membrane proteins to be mistargeted to the ER. J Cell Sci 131, jcs211110. [DOI] [PubMed] [Google Scholar]
- Waldo GS, Standish BM, Berendzen J, Terwilliger TC (1999). Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol 17, 691–695. [DOI] [PubMed] [Google Scholar]
- Walter P, Blobel G (1981). Translocation of proteins across the endoplasmic reticulum III. Signal recognition protein (SRP) causes signal sequence-dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang X, Chen XJ (2015). A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Amon A (2018). MitoCPR-a surveillance pathway that protects mitochondria in response to protein import stress. Science 360, eaan4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill U, Cohen N, Fadel A, Ben-Dor S, Schuldiner M (2019). Protein topology prediction algorithms systematically investigated in the yeast Saccharomyces cerevisiae. Bioessays 41, e1800252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill U, Krieger G, Avihou Z, Milo R, Schuldiner M, Davidi D (2019). Assessment of GFP tag position on protein localization and growth fitness in yeast. J Mol Biol 431, 636–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz LS, Opaliński Ł, Schuler MH, Ellenrieder L, Ieva R, Bottinger L, Qiu J, van der Laan M, Wiedemann N, Guiard B, et al. (2014). The presequence pathway is involved in protein sorting to the mitochondrial outer membrane. EMBO Rep 15, 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienhues U, Becker K, Schleyer M, Guiard B, Tropschug M, Horwich AL, Pfanner N, Neupert W (1991). Protein folding causes an arrest of preprotein translocation into mitochondria in vivo. J Cell Biol 115, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley WC, Stidham RD, Smith NM, Hunt JF, Thomas PJ (2001). Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat Biotechnol 19, 131–136. [DOI] [PubMed] [Google Scholar]
- Wild K, Halic M, Sinning I, Beckmann R (2004). SRP meets the ribosome. Nat Struct Mol Biol 11, 1049–1053. [DOI] [PubMed] [Google Scholar]
- Williams CC, Jan CH, Weissman JS (2014). Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science 346, 748–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, Varabyova A, Lirski M, Chroscicki P, Mroczek S, et al. (2015). Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488. [DOI] [PubMed] [Google Scholar]
- Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation (Camb) 2, 100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Cabanos C, Rapoport TA (2019). Structure of the post-translational protein translocation machinery of the ER membrane. Nature 566, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Shakya VP, Hughes AL (2021). ER targeting of non-imported mitochondrial carrier proteins is dependent on the GET pathway. Life Sci Alliance 4, e202000918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong J, Bischof H, Burgstaller S, Siirin M, Murphy A, Malli R, Kaufman RJ (2019). Mitochondria supply ATP to the ER through a mechanism antagonized by cytosolic Ca(2). Elife 8, e49682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.