

Abstract
Non-homologous end-joining (NHEJ) and homologous recombination (HR) are the primary pathways for repairing DNA double-strand breaks (DSBs) during interphase, while microhomology-mediated end-joining (MMEJ) has been regarded as a backup mechanism. Through CRISPR/Cas9-based synthetic lethal screens, we identify subunits of the 9–1-1 complex (RAD9A-HUS1-RAD1) and its interacting partner, RHINO, as crucial MMEJ factors. We uncover an unexpected function for RHINO in restricting MMEJ to mitosis. RHINO accumulates in M phase, undergoes PLK1 phosphorylation, and interacts with polymerase theta (Polθ), enabling its recruitment to DSBs for subsequent repair. Additionally, we provide evidence that MMEJ activity in mitosis repairs persistent DSBs originating in S phase. Our findings offer insights into the synthetic lethal relationship between POLQ and BRCA1/2 and the synergistic effect of Polθ and PARP inhibitors.
One-Sentence Summary:
RHINO, an interacting partner of the 9-1-1 complex, stimulates error-prone MMEJ repair during mitosis to resolve DNA breaks.
Microhomology-mediated end-joining (MMEJ) is an intrinsically mutagenic repair pathway. Nevertheless, it mitigates the harmful effects of double-strand breaks (DSBs) by preventing the accumulation of large-scale DNA rearrangements. Repair by MMEJ is necessary for the survival of cells with compromised HR and NHEJ (1–3). Targeting this pathway has emerged as a promising therapeutic approach for cancer patients with defective HR, including ones carrying mutations in BRCA1 and BRCA2 (4–6). MMEJ is characterized by the presence of 2–6 base pairs of microhomology, as well as insertions and deletions (indels) that scar the repair sites (7). These indels are introduced by DNA polymerase theta (Polθ), encoded by the POLQ gene. Polθ is a low-fidelity polymerase with a helicase-like activity that plays a central role in MMEJ (8, 9).
The mutational signature associated with MMEJ has been found across different species, and the pathway is conserved from bacteria to humans (10). However, its mechanistic basis remains poorly defined. In mammalian cells, studies have demonstrated that following DSB formation, short-range DNA end resection by MRE11 and CtIP exposes flanking microhomologies that promote the annealing of opposite ends of the break (11–13). When internal homologies are base-paired, the resulting single-stranded DNA (ssDNA) flaps are cleaved by APEX2 and FEN1 (14–16). Annealed intermediates are extended by Polθ (17–19) and sealed by XRCC1/LIG3 to complete the end-joining (20). Polθ also acts on transient “snap-back” substrates formed when the overhang of resected DSBs folds back and anneals to itself. Ultimately, Polθ-mediated insertions contribute to the mutagenicity of MMEJ (19). While upregulated in many cancer types, Polθ is generally low in abundance and must be actively recruited to DSB sites (21). Yet, the mechanism by which the low-fidelity polymerase is recruited to break sites and the upstream factors that drive MMEJ remain unknown.
MMEJ was initially identified as an inefficient DNA end-joining activity in Ku-deficient Saccharomyces cerevisiae (22) and has been primarily regarded as a backup pathway that acts when preferred modes of DSB repair are absent (1–3). However, recent reports suggest that under certain conditions, MMEJ prevails. For instance, MMEJ is the primary repair mechanism for CRISPR/Cas9-induced DSBs in early zebrafish embryos (23). In human and mouse cells, MMEJ acts with NHEJ to promote the random integration of foreign DNA into the genome and repair CRISPR/Cas9-induced breaks at particular loci (24–26). Furthermore, recent evidence suggests that MMEJ plays a role in DSBs during mitosis, where HR and NHEJ are attenuated (27–30).
Due to the synthetic lethal relationship between MMEJ and HR, Polθ inhibitors are currently under investigation in phase I/II in the clinic as monotherapy and in combination with PARP inhibitors (PARPi). Preclinical studies demonstrated that Polθ inhibitors target BRCA-deficient tumors, are synergistic with PARPi, and eliminate a subset of PARPi-resistant tumors (4, 6, 31). Elucidating the underlying mechanism of MMEJ and its temporal and spatial regulation is critical to understanding when and how cells choose to use mutagenic MMEJ and potentially explain the synthetic lethal interaction between MMEJ and HR.
Results:
CRISPR/Cas9 synthetic lethal screen uncovers the full spectrum of MMEJ factors
To identify the full spectrum of MMEJ factors, we conducted a genome-wide CRISPR-based synthetic lethal screen in cells lacking HR and NHEJ (BRCA2−/−LIG4−/−TP53−/− cells or TKO cells). We hypothesized that since cells lacking these canonical DSB repair pathways are highly dependent on MMEJ for survival, this approach would reveal the full spectrum of potential MMEJ factors (Fig. 1A–C and Fig. S1A–D). The synthetic lethal screen identified a set of genes that were preferentially depleted in TKO cells, including hits previously reported to be essential for the survival of BRCA2 null cells, such as FEN1, RNaseH2A/B/C (32), CIP2A (33), and ALC1 (34), and known MMEJ factors, including POLQ, HMCES (35), and APEX2 (14, 16, 36) (Fig. S1E–G). Notably, we retrieved members of the 9-1-1 complex (RAD9A-HUS1-RAD1) and its interacting partner RHINO (encoded by RHNO1) as essential in TKO cells (Fig. 1D–E). To confirm the synthetic lethality, we individually targeted subunits of the complex using independent sgRNAs. We found that while depletion of 9-1-1 and RHINO had little impact on control cells, their loss compromised the survival of cells lacking HR and NHEJ (Fig. 1F, Fig. S1H–J).
Fig. 1. A genome-wide CRISPR-Cas9 screen uncovers an essential function for 9-1-1 and RHNO1 in cells lacking BRCA2 and LIG4.
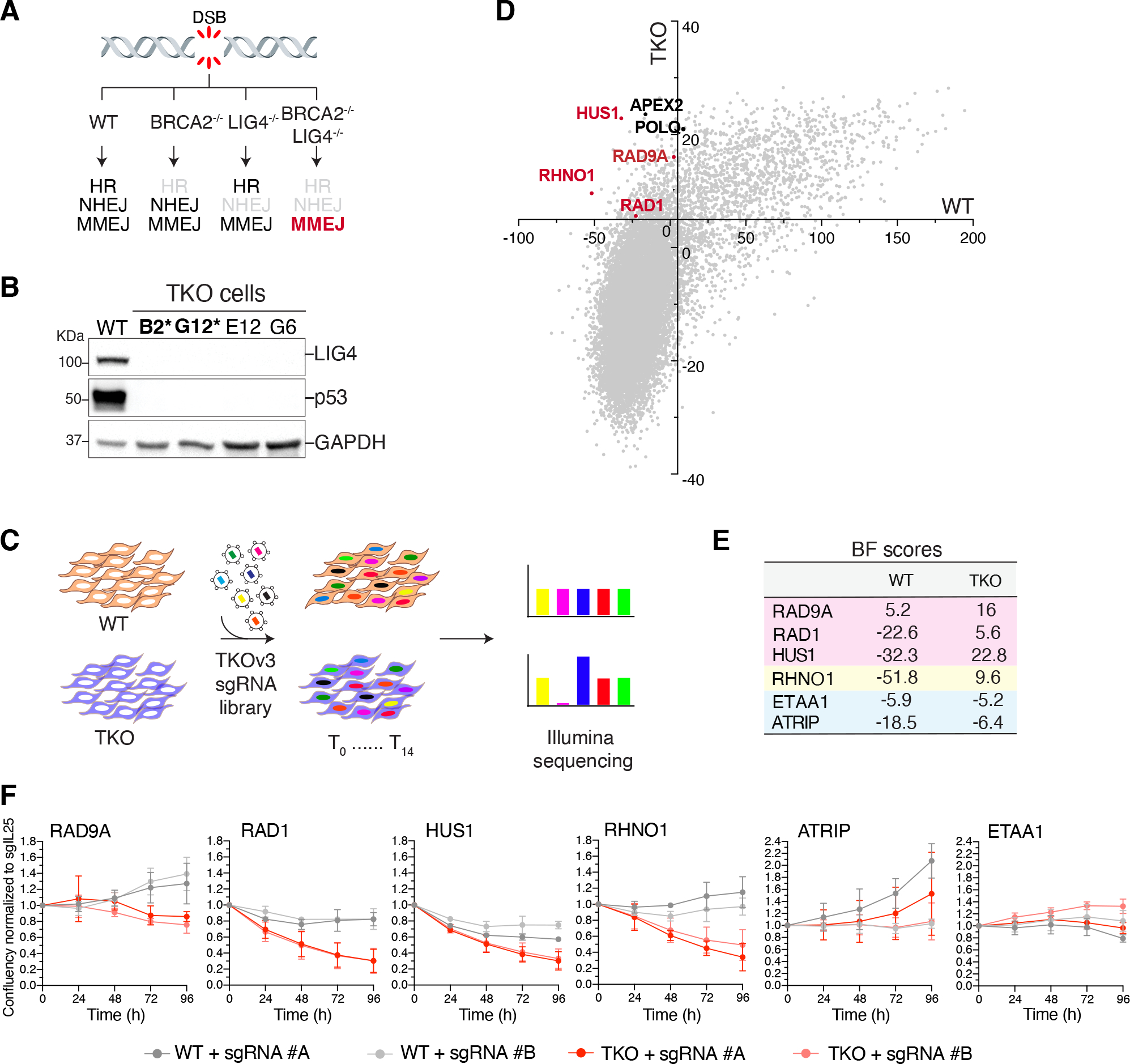
(A) Schematic of the three major DSB repair pathways in mammalian cells. (B) Western blot analysis of LIG4 and p53 in clonally derived BRCA2−/− LIG4−/− TP53−/− DLD1 cells (TKO). The asterisk indicates clones used in the screen. (C) Schematic of the dropout CRISPR/Cas9 screen to identify synthetic lethal interactions. (D) Genome-wide CRISPR-Cas9 screen result in TKO cells. Genes with a Bayes factor (BF) score >5 (intersection and x- and y-axes) were considered essential. (E) BF scores for the indicated genes. (F) Growth curve of TKO and TP53−/− cells treated with the indicated sgRNAs. Data are mean ± s.d. of three independent experiments normalized to time point zero (one day after seeding) and a control sgRNA (sgIL25).
RAD9-HUS1-RAD1 (9-1-1) and RHINO are critical MMEJ factors
To directly test whether the 9-1-1 complex and RHINO are required for MMEJ, we investigated the repair of dysfunctional telomeres. The six-subunit shelterin complex protects telomeres from being recognized as DSBs. When shelterin is absent, telomeres become deprotected, and the DNA damage response is activated at chromosome ends (37). In cells where telomeres are unprotected and the NHEJ factors Ku70/80 are absent, MMEJ is the primary repair pathway leading to chromosome end-to-end fusions (37) (Fig. 2A). We targeted subunits of the 9-1-1 complex and RHINO in TRF1/2Δ/ΔKu80−/− cells (Fig. S2A–B) and noted a significant reduction in the frequency of MMEJ-dependent telomere fusions (Fig. 2B–C). Depletion of the 9-1-1 subunits and RHINO in NHEJ-proficient settings had no impact on telomere fusions, suggesting that the activity of 9-1-1 and RHINO is specific to MMEJ (Fig. 2D). Despite the reduced MMEJ activity, the absence of 9-1-1 and RHINO did not prevent the accumulation of 53BP1 at shelterin-free telomeres (Fig. S2C–D). This implies that the canonical role of the 9-1-1 complex in activating DNA damage signaling through ATR kinase (38) does not completely account for its function in MMEJ. In an orthogonal approach, we measured MMEJ activities at an I-SceI-induced break using the traffic light reporter (TLR) system (39) (Fig. S2E). Findings based on the fluorescent DSB reporter corroborated the results derived from the telomere fusion assay and are consistent with Repair-seq (40), which demonstrated a correlation between POLQ and the 9-1-1 complex as well as the Rad17-RFC clamp loader (Fig. 2B–D, Fig. S2E–I and Fig. S1G).
Fig. 2. A non-canonical function for 9-1-1/RHINO in MMEJ.
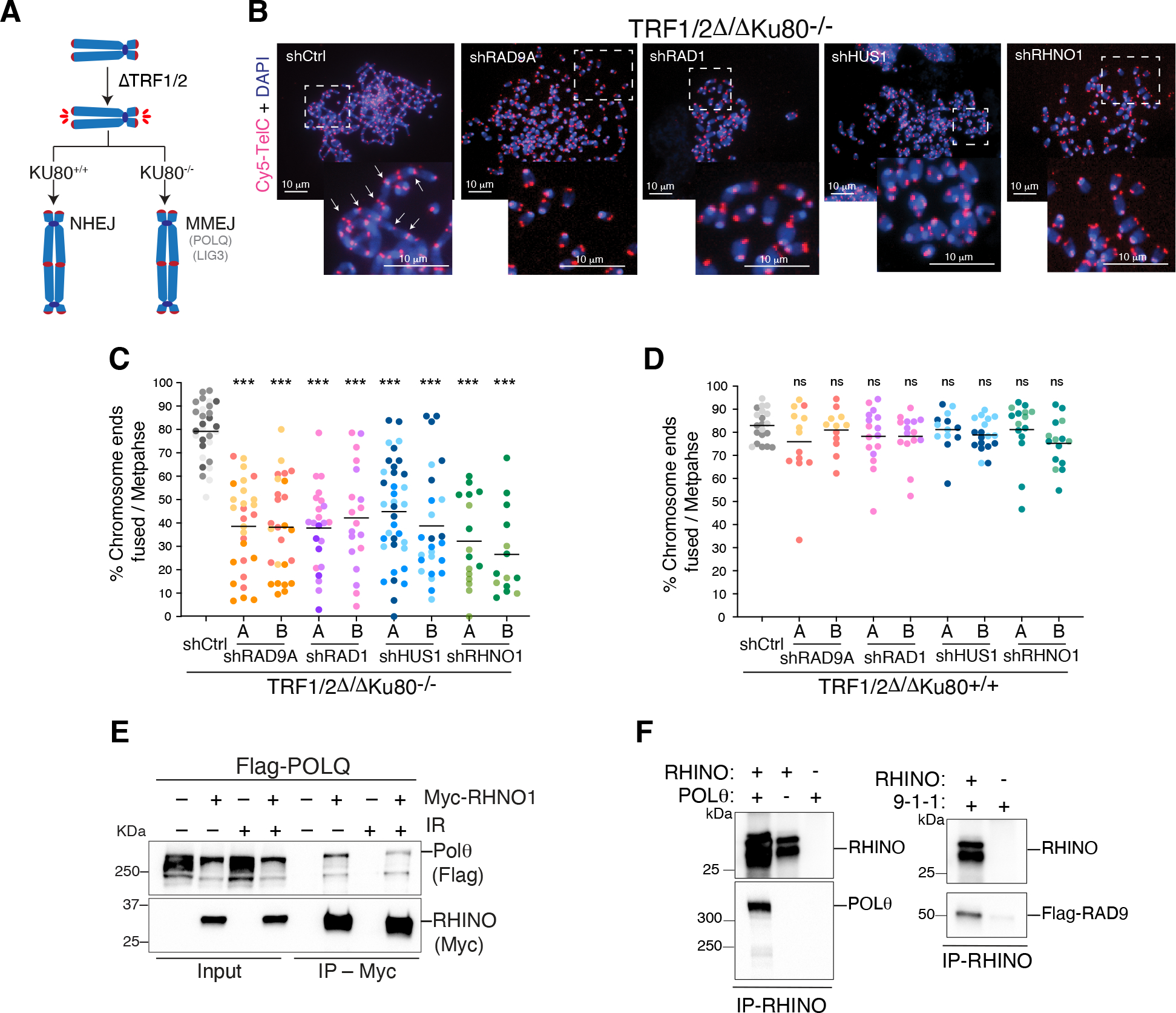
(A) Schematic of the shelterin-free assay (37) to monitor MMEJ frequency at deprotected telomeres. (B) Representative images of metaphase spreads from TRF1/2Δ/ΔKu80−/− cells depleted for the subunits of the 9-1-1 complex and RHNO1 with two independent shRNAs. Telomeres are marked by FISH using a Cy5-[CCCTAA]3 PNA probe (red), and chromosomes are counterstained with DAPI (blue). White arrows indicate examples of telomeric fusions in the control sample. (C) Quantification of telomeric fusions mediated by MMEJ as shown in panel B. (D) Quantification of telomere fusions by NHEJ in TRF1/2Δ/ΔKu80+/+. Data in (C, D) are the mean of at least two independent experiments. One-way ANOVA (***p<0.001, **p<0.01). (E) Co-IP experiment depicting Polθ and RHINO interaction in whole-cell extracts from HEK293T cells co-transfected with plasmids expressing FLAG-Polθ and RHINO-MYC. Co-IPs were performed in cells treated with ionizing radiation (+IR, 20 Gy) and control cells. (F) Co-IP experiments showing Polθ/RHINO and 9-1-1/RHINO interaction with purified proteins. Polθ was purified from HEK293T cells, RHINO from E. coli, and 9-1-1 from S. cerevisiae.
A non-canonical function for 9-1-1 and RHINO in MMEJ
9-1-1 is a heterotrimeric complex loaded onto 5’ ends of resected DNA and single-stranded DNA gaps in response to replication stress (38). Its interaction with RHINO induces DNA damage signaling by ATR (41, 42). ATR is also activated by ETAA1 and its interacting partner ATRIP (43, 44). Notably, in the synthetic lethal screen, ATRIP and ETAA1 did not emerge as potential hits (Fig. 1E), and CRISPR/Cas9-mediated deletion of these genes did not result in growth defects in TKO cells (Fig. 1F, Fig. S1K–N). Moreover, while the loss of ATRIP had a minor impact on telomere fusions, ETAA1 depletion did not impair MMEJ (Fig. S3A–E). In contrast, depletion of 9-1-1, RHINO, and Polθ significantly reduced telomere fusion events (Fig. 2B–C, Fig. S3A–E). These findings suggest that the established role of 9-1-1/RHINO in ATR signaling does not entirely explain their function in MMEJ.
The 9-1-1 subunits assemble into a ring-shaped complex structurally resembling proliferating cell nuclear antigen (PCNA), which recruits DNA polymerases to the replisome (45). We speculated that 9-1-1/RHINO might facilitate MMEJ by interacting with Polθ and recruiting it to break sites. We performed Co-IP experiments in HEK293T cells co-expressing 9-1-1 proteins, RHINO, and Polθ. Although we could not detect an interaction between Polθ and any of the subunits of the 9-1-1 complex (Fig. S3F), we observed an interaction between Polθ and RHINO, independent of DNA damage (Fig. 2E). In addition, using purified proteins, we found that RHINO bound full-length Polθ in vitro (Fig. 2F and Fig. S3G).
RHINO is predominantly expressed in mitosis, and its PLK1 phosphorylation stimulates the interaction with Polθ
To gain better insight into the function of RHINO, we sought to determine its genetic interactors. We carried out synthetic lethal screens in three clonally derived RHNO1−/− cell lines and the parental RHNO1+/+ cells (Fig. 3A, Fig. S4A–G). Pathway analysis of top genes essential in RHNO1−/− cells revealed enrichment in several pathways related to mitosis (Fig. 3B), including cyclin-dependent kinase CDK1, members of the ESCRT complex, spindle checkpoint proteins, and the kinetochore factor ZWILCH (Fig. 3A, Fig. S4F–G, and Fig. S5A–B). Independently, we analyzed the genetic dependencies in DepMap (46) and found that RHNO1 correlated with CIP2A, MDC1, and TOPBP1, which form a complex that tethers mitotic DSBs together (33, 47, 48). DepMap analysis also uncovered a correlation between the essentiality scores of POLQ, CIP2A, and RHNO1 (Fig. 3C).
Fig. 3. RHINO is predominantly expressed in mitosis.
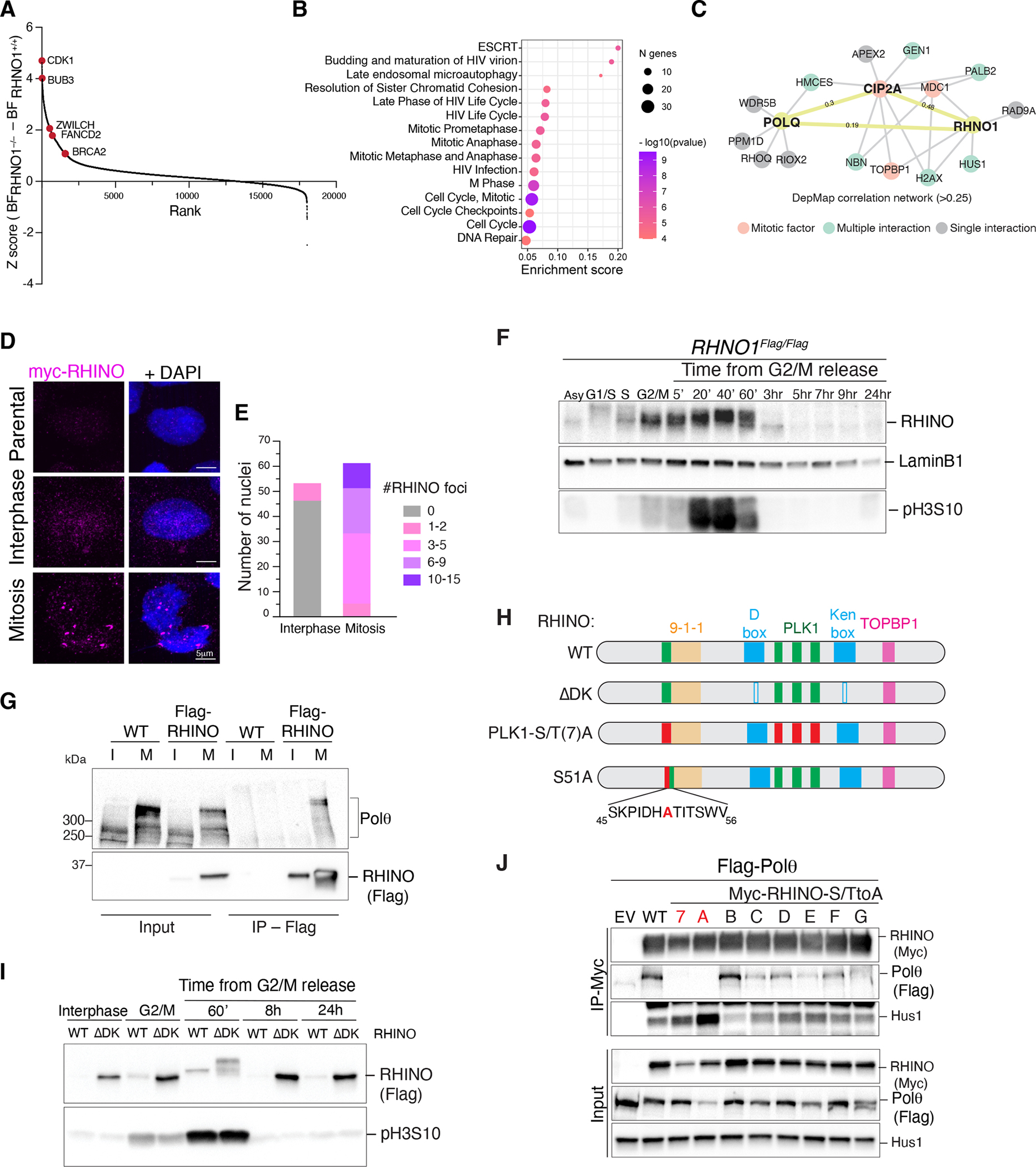
(A) Results from the CRISPR/Cas9 dropout screen in RHNO1−/− and isogenic RHNO1+/+ cells. Ranked z-scores of the difference in Bayes factor (BF) scores. (B) Reactome pathway overrepresentation analysis of synthetic lethal genes with RHNO1−/−. The fold enrichment of each pathway is plotted on the x-axis. The number of associated genes with each pathway is indicated by the size of the circle, while the color shade indicates the p-value. (C) Network analysis for POLQ and RHNO1 based on Pearson’s correlation of dependency scores derived from DepMap. (D) Representative immunofluorescence images of RHINO in interphase and mitotic cells. (E) Quantification of RHINO foci from panel D. (F) Western blot analysis of endogenous RHINO at different stages of the cell cycle. Extracts from RHNO1FLAG/FLAG cells at the indicated time points. pH3S10 antibody is used as a mitotic marker, and Lamin B1 as a loading control. (G) Control cells and ones expressing RHINO-MYC-FLAG were synchronized in mitosis and subjected to anti-FLAG immunoprecipitation followed by western blot for endogenous Polθ. I = interphase; M = mitosis. (H) Schematic of RHINO protein highlighting the binding domains for 9-1-1 and TOPBP1, D- and Ken-boxes (ΔDK), and the PLK1 phosphorylation sites (PLK1(S/T)7A and S51A). RHINO PLK1(S/T)A harbors alanine mutations in all 7 predicted PLK1 sites. RHINO S51A harbors a single mutation of serine 51 (conserved among primates and rodents) to alanine. (I) Western blot analysis of RHINO and RHINOΔDK during the cell cycle. (J) Co-IP experiments in HEK293T cells co-transfected with plasmids expressing FLAG-Polθ and RHINO-MYC mutants. RHINO mutants with a single S/T mutation to alanine are A through G.
The results from the synthetic lethal screen and DepMap analysis underscored a previously unrecognized role for RHINO in repairing DNA damage in mitosis. This observation was substantiated by the accumulation of large RHINO foci in cells arrested in M phase (Fig. 3D–E and Fig. S5C–D). Furthermore, western blot analysis of samples collected at different cell cycle stages revealed that RHINO accumulated in mitosis and was apidly degraded upon mitotic exit (Fig. 3F, Fig. S6A–D). In contrast, subunits of the 9-1-1 complex were expressed throughout all cell cycle stages (Fig. S6C–E). As anticipated by the strict expression of RHINO, Polθ interacted with Flag-RHINO, specifically in mitosis (Fig. 3G). Coincident with its stabilization, RHINO was phosphorylated in mitosis (Fig. S6F).
RHINO contains two recognition sequences for the anaphase-promoting complex (APC/C), namely the Ken-box and D-box domains (49) (Fig. 3H). Expression of RHINO-ΔDK that carries deletions in both degrons resulted in RHINO stabilization beyond mitosis (Fig. 3I). Furthermore, the overexpression of the APC/C adaptor protein Cdh1, but not Cdc20, led to RHINO depletion, suggesting that its degradation is a late event in mitosis (Fig. S6G) (49). Although RHINO-ΔDK remained stable during interphase, it only interacted with Polθ in mitosis (Fig. S6H). Based on this observation, we explored whether the interaction between RHINO and Polθ could be stimulated by phosphorylation. We found that inhibiting the major mitotic kinase, CDK1, and to a lesser extent, PLK1, hindered RHINO phosphorylation (Fig. S6I–K). RHINO contains seven Ser/Thr-Pro motifs susceptible to CDK1 targeting and seven Ser/Thr residues embedded within the consensus motif of PLK1. Using phospho-tag gels, we determined that RHINO phosphorylation was altered in the context of RHINO-PLK1S/T(7)A and abrogated in RHINO-CDK1S/T(7)A allele (Fig. S6J–K). These findings are consistent with CDK1 phosphorylation being a priming event for subsequent PLK1 phosphorylation of RHINO. Such sequential modification is commonly observed in PLK1 targets, including BUB1, BUBR1, and CLASP2 (50–52). Notably, Co-IP analysis showed that RHINO-PLK1S/T(7)A failed to bind Polθ (Fig. 3J, Fig. S6K). Furthermore, by individually mutating seven PLK1 sites, we identified a single phosphorylation residue on RHINO (S51) that, when mutated to alanine, exhibited a reduced interaction with Polθ (Fig. 3J, S6L).
MMEJ is a dominant DSB repair pathway in mitosis
Unresolved damage during S and G2 phases of the cell cycle can be carried over to mitosis. However, it is well established that NHEJ and HR are repressed during M phase (53, 54). Recently, a tethering complex comprising MDC1-CIP2A-TOPBP1 was reported to hold broken mitotic DNA ends together until cells progress into the next G1 (33, 47, 48). Multiple studies implicated Polθ activity in repairing DNA damage in mitosis (27–30). An investigation using Xenopus egg extract showed that entry into mitosis before the completion of DNA replication leads to complex rearrangements driven by Polθ (29). In addition, Polθ was linked to the formation of Sister Chromatid Exchanges (SCE) as under-replicated DNA is transferred into mitosis (30). Last, it was shown that MMEJ activity is delayed until mitosis in cells lacking BRCA2, where Rad52 blocked Polθ activity in G2 (27). We conducted individual and combined depletion of RHINO and CIP2A in BRCA2−/− cells and observed an additive effect on growth, suggesting that RHINO-mediated MMEJ is independent of mitotic tethering of DNA breaks (Fig. S7A–B). Consistent with the involvement of Polθ in mitotic repair, POLQ−/− cells were more sensitive to DNA damage when treated with ionizing radiation in mitosis relative to interphase (Fig. S7C–D).
Given the critical function of RHINO in MMEJ, its enrichment in mitosis, and its interaction with Polθ, we hypothesized that RHINO could promote mitotic MMEJ by facilitating Polθ recruitment to condensed chromosomes. To assay MMEJ activity in mitosis, we synchronized POLQ−/− and POLQ+/+ cells at the G2/M boundary using a CDK1 inhibitor. We then released cells into M phase in the presence of nocodazole, which prevented mitotic exit. We irradiated cells 30 minutes after release from CDK1 inhibition and monitored the dissolution of phosphorylated γ-H2AX (Fig. 4A). Wild-type cells accumulated maximal γ-H2AX one-hour post-irradiation that significantly decreased after five hours (Fig. 4B–C, Fig. S7E). As a control, we showed a similar resolution of γ-H2AX foci LIG4−/− cells relative to wild-type cells, confirming the lack of NHEJ activity in mitosis (Fig. S7F). In contrast, POLQ−/− cells and ones carrying inactivating mutations in the polymerase domain of Polθ (POLQΔPol/ΔPol) failed to resolve γ-H2AX foci (Fig. 4B–C and Fig. S7F). Furthermore, treatment of cells with Polθ inhibitor (Polθi–RP6685) (31) during mitosis, but not interphase, led to the persistence of γ-H2AX foci post-irradiation (Fig. S7G–H). Similarly, the absence of RHINO prevented the resolution of γ-H2AX after radiation treatment (Fig. S7I). Last, we corroborated the essentiality of MMEJ activity in mitotic DSB repair by employing the inducible restriction enzyme, AsiSI (55), and observed a defect in γ-H2AX resolution in cells treated with Polθ inhibitor (Fig. S7J–L).
Fig. 4. MMEJ is the predominant DSB repair pathway during mitosis.
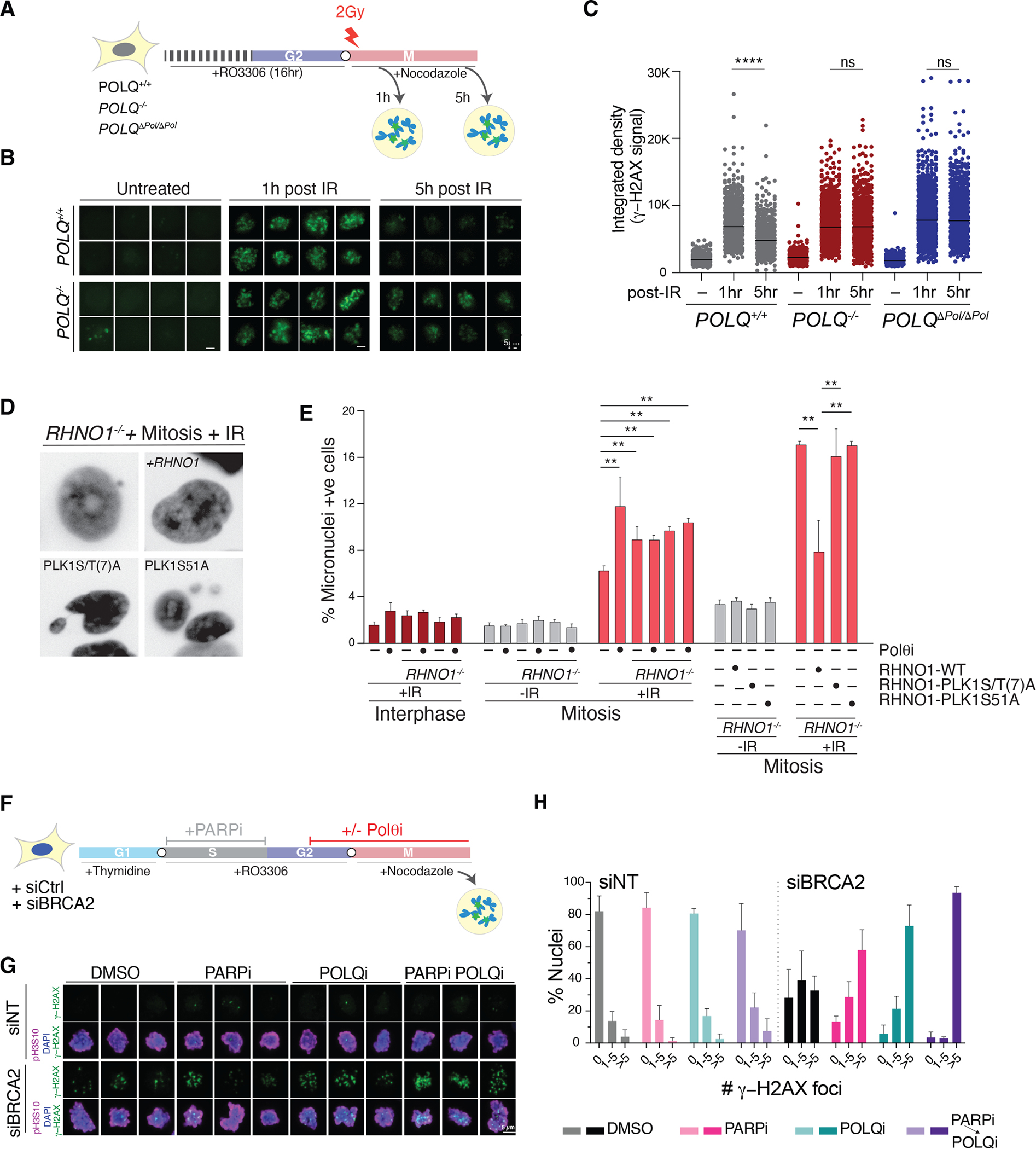
(A) Schematic of the experimental design to detect MMEJ in mitosis for panel B-C. (B) Representative images of γ-H2AX in cells treated as described in panel A. (C) Quantification of γ-H2AX intensity in mitotic cells with the indicated genotype. (n>450 cells; paired t-test.). (D) Representative images of micronuclei in cells with the indicated treatment and genotype. (E) Quantification of micronuclei formation after irradiation during interphase and mitosis as in panel D. Bars represent the mean of three independent experiments (n> 250 cells; paired t-test). (F) Schematic of the experimental pipeline for panels G-H. (G) Representative immunofluorescence images of mitotic cells treated as described in panel F and stained with anti-γ-H2AX. pH3S10 is used to mark mitotic chromosomes. DNA is stained with DAPI. (H) Quantification of γ-H2AX foci in mitotic cells with the indicated treatment. Bars represent the mean of three independent experiments (n> 50 cells).
Unrepaired mitotic DSBs are especially toxic, as they can result in lagging chromosome fragments that accumulate in micronuclei and trigger chromothripsis (56). We tested whether MMEJ activity prevents micronuclei formation following irradiation of cells in mitosis. Polθ inhibition and RHINO deletion significantly increased micronuclei formation (Fig. 4D–E). Blocking Polθ in RHNO1−/− cells had no additive effect on the accumulation of micronuclei, suggesting that the two factors are epistatic (Fig. 4E). Furthermore, we expressed RHINO-WT, RHINO-PLK1-S/T(7)A, and RHINO-S51A alleles in RHNO1−/− cells and found that both mutants failed to rescue micronuclei formation following mitotic irradiation (Fig. 4D–E). Similarly, RHINO−/− cells and ones complemented with the RHINO-S51A allele failed to resolve mitotic γ-H2AX accumulation (Fig. S7I). In conclusion, our findings implicate the RHINO–Polθ interaction in stimulating MMEJ during mitosis.
Mitotic MMEJ resolves DNA breaks originating in S phase in BRCA2 mutant cells
We next tested whether the activity of RHINO and Polθ in mitosis was necessary to repair DNA lesions that arise in S and G2 phases but persist into M phase (Fig. 4F). We utilized siRNA against BRCA2 (Fig. S8A) to block repair by HR, and induced DNA damage in S phase by incubating cells with PARPi (Olaparib) (57). As cells progressed beyond S phase, we withdrew PARPi from the culture medium. Towards the end of G2, we added Polθi, which was continuously present as cells entered mitosis. To prevent mitotic exit, cells were treated with nocodazole before fixing them for subsequent analysis for γ-H2AX (Fig. 4F–H and Fig. S8B). We observed a baseline increase in γ-H2AX foci in mitotic cells treated with siBRCA2 compared to control siRNA. As expected, the levels of γ-H2AX were more elevated in cells treated with either Olaparib in S phase or Polθ inhibitor in G2 and M phase. Cells treated with PARP inhibitor in S phase and Polθ inhibitor in G2/M displayed a synergistic increase in γ-H2AX accumulation (Fig. 4F–H). Furthermore, this synergy was observed when the polymerase was inhibited in mitosis but not during S phase (Fig. S8C–E). To further substantiate the role of mitotic MMEJ in repairing unresolved S phase damage, we treated RHNO1−/− cells with siRNA against BRCA2 and observed a similar synergistic effect with PARPi treatment (Fig. S8F).
RHINO recruits Polθ to DSB sites in mitosis
Having established a role for MMEJ in repairing breaks in mitosis, we next investigated if RHINO recruits Polθ to damage sites to facilitate mitotic repair. We utilized a two-step CRISPR/Cas9 targeting strategy to establish cells where POLQ was endogenously tagged with Halo at the N-terminus (Fig. S9A–C). We treated POLQHalo/Halo cells with a Halo-tag ligand (JFX650) and traced Polθ single-particles using live-cell imaging in interphase and mitosis (Fig. S9D and Supplemental movies 1–2). To test if RHINO acts upstream of Polθ, we arrested POLQHalo/Halo cells treated with siRHNO1 at the G2/M boundary by CDK1 inhibition and released them into mitosis. Treatment of mitotic cells with Zeocin resulted in large and static Polθ foci. Polθ foci were significantly reduced upon RHNO1 depletion but not affected by MDC1 loss and ATR inhibition (Fig. 5A–B, S9E–I, S10A–E). Last, we investigated the static Polθ foci in the context of unresolved S phase damage that persisted into mitosis. We treated BRCA2-depleted POLQHalo/Halo cells with PARPi in S phase and showed that Polθ dynamics and co-localization with RPA were largely unchanged when monitored in S phase (Fig. S11A). However, this same treatment led to the accumulation of static Polθ foci in mitosis (Fig. 5A–C, Fig. S9E and S11B–C). These findings provide further evidence for the role of RHINO in recruiting Polθ to break sites during mitosis.
Fig. 5. RHINO recruits Polθ to damaged sites in mitosis.
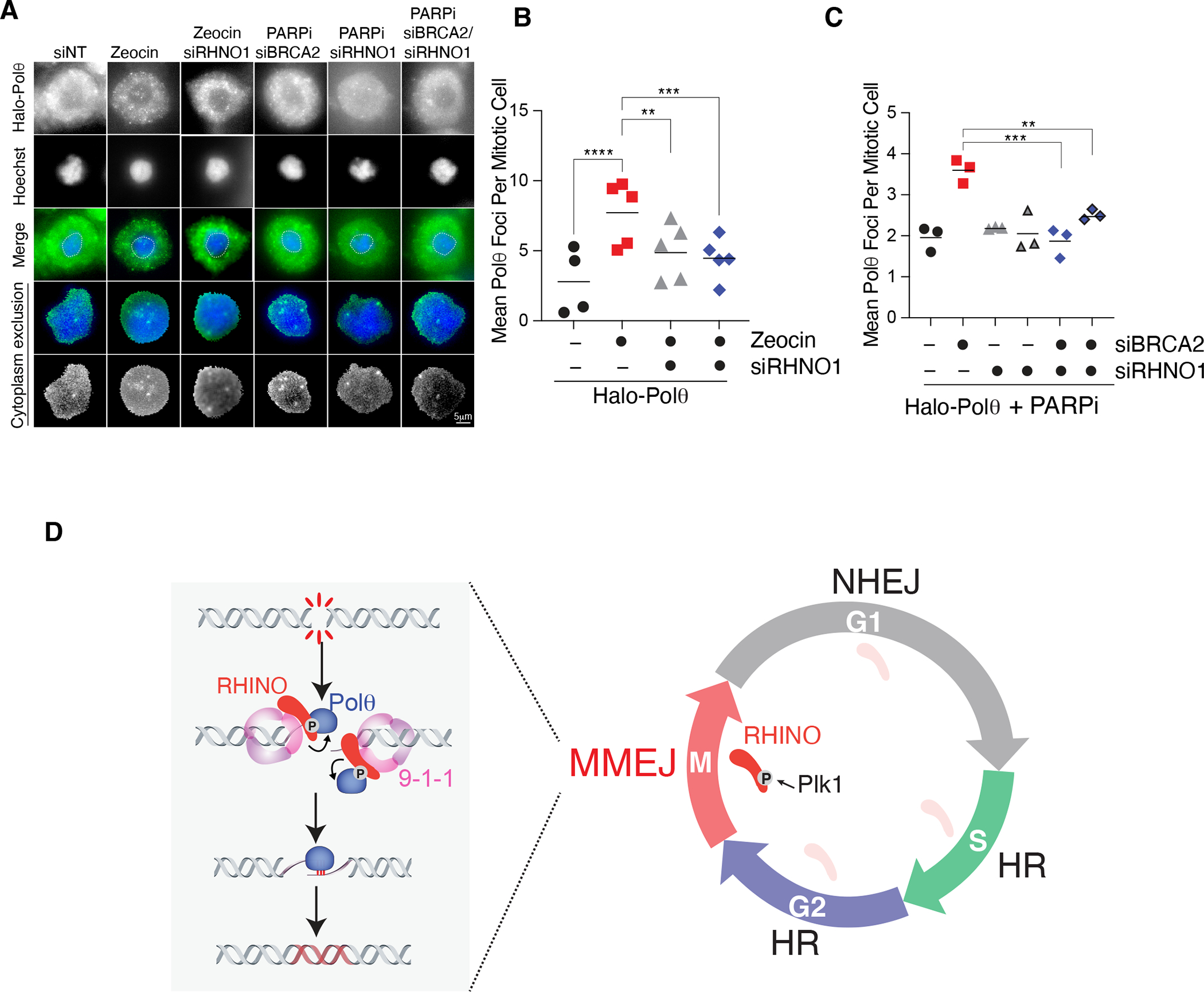
(A) Representative images of Halo-Polθ foci in mitosis. Cells with the indicated siRNA treatment were synchronized according to the scheme in Fig. 4A. Cells in mitosis were treated with Zeocin for 1 hour. To monitor mitotic Polθ foci in cells with persistent damage from S phase, cells expressing siRNA against BRCA2 and RHNO1 were treated with Olaparib according to the schematic in Fig. 4F. (B) Quantification of mitotic Halo-Polθ foci in live-cells treated with Zeocin and depleted of RHNO1. Bars represent the mean of three independent experiments. n>40 nuclei (one-way ANOVA (***p<0.001, **p<0.01)). (C) Quantification of mitotic Halo-Polθ foci in live-cell imaging experiments in nocodazole-arrested cells treated with PARPi during S phase. Bars represent the mean of three independent experiments (n>40 nuclei; one-way ANOVA; ***p<0.001, **p<0.01). (D) NHEJ dominates in G1, and HR is preferred in S and G2. The confinement of MMEJ to mitosis occurs due to the accumulation of RHINO during M phase, PLK1- phosphorylation, and the recruitment of Polθ to DNA breaks
Discussion
NHEJ predominates in G1 phase of the cell cycle, while HR is preferred for repairing DSBs in S and G2 (58–60). Here, we demonstrate that in mitosis, where both HR and NHEJ are repressed (53, 54), MMEJ is the sole DSB repair pathway. MMEJ activity in M phase is driven by the accumulation of RHINO to promote Polθ recruitment to damage sites (Fig. 5D). The decoupling of DNA repair pathways during different stages of the cell cycle has implications for maintaining genome stability. Mitotic MMEJ may have evolved as a failsafe mechanism that operates on highly condensed chromosomes and ensures that cells do not commit to cellular division with unrepaired lesions that trigger genome instability. Conversely, suppression of MMEJ in G1 and S/G2 could protect our genome from the intrinsic mutagenic potential of this pathway.
RHINO was previously identified as a 9-1-1 interacting protein required for activating ATR signaling (41), but now emerges as a critical factor promoting MMEJ during mitosis. We show that RHINO, but not 9-1-1 complex members, is greatly stabilized in mitosis and degraded by the APC/C complex upon mitotic exit (Fig. 3H–I). Based on our data, we propose that PLK1-dependent phosphorylation of RHINO facilitates its interaction with Polθ to stimulate MMEJ. A recent study has also identified PLK1 phosphorylation sites on Polθ that are critical for mitotic MMEJ (61). Furthermore, it has been established that the CDK1-PLK1 signaling axis attenuates NHEJ and HR, by phosphorylating and inhibiting 53BP1 and BRCA2, respectively (62–64). These studies highlight a multifaceted role for PLK1 in controlling repair pathway choice in mitosis.
RHINO is highly unstructured and interacts with RAD1 and TOPBP1 through distinct domains (41, 42). Unrepaired S phase damage may be marked by 9-1-1 through mitosis, where RHINO accumulates. Tethering RHINO to the 9-1-1 complex in M phase would subsequently lead to Polθ recruitment to break sites, thereby enabling MMEJ. RHINO-TOPBP1 interaction in mitosis could stabilize RHINO at break sites and form a complex that recruits Polθ. Further structural studies could provide a deeper insight into 9-1-1/RHINO–POLQ–TOPBP1 complex formation and probe the impact of PLK1 phosphorylation.
Our study does not rule out a role for Polθ in filling ssDNA gaps post-replication, nor do we exclude the possibility of RHINO-independent MMEJ activity during S phase (65–68). However, our findings provide evidence suggesting that robust MMEJ activity in mitosis accounts for the synthetic lethal interaction between Polθ and BRCA2. Uncoupling DNA repair activities during different cell cycle stages provides a rationale for the reported synergy of Polθ inhibitors with PARP inhibitors and potentially other antineoplastic therapies that induce DNA damage during S-phase (4, 6).
Supplementary Material
Acknowledgments:
We thank Dan Durocher, Robert Weiss, and Roger Greenberg for providing critical reagents for this study. We thank Dirk Remus for providing 9-1-1 proteins and assisting with in vitro assays. Stephen Morris and Marie-Claude Mathieu from Repare Therapeutics generously provided reagents. We are grateful to Eros Lazzerini-Denchi and members of the Sfeir lab for commenting on the manuscript. We thank Sarah Deng, Toby Lieber, Yi Fu, Monica Selvaraj, and Hina Shah for their experimental assistance.
Funding:
This work was supported by the following grants:
National Institutes of Health grant R01CA229161 (AS)
National Institutes of Health grant DP2CA195767-01 (AS)
Pershing Square-Sohn Cancer Foundation grant (AS)
V-foundation grant (AS)
New York Stem Cell Foundation training grant (AB)
National Institutes of Health grant F32GM139292 (JH)
National Institutes of Health grant DP2GM142307 (JS)
Footnotes
Competing interest: Agnel Sfeir is a co-founder, consultant, and shareholder of Repare Therapeutics. All other authors have no competing interests
Data and material availability:
All data are available in the main text and supplemental materials. All material will be shared upon request.
References and Notes:
- 1.Mateos-Gomez PA et al. , Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 518, 254–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccaldi R et al. , Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature 518, 258–262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wyatt DW et al. , Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol Cell 63, 662–673 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zatreanu D et al. , Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun 12, 3636 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schrempf A, Slyskova J, Loizou JI, Targeting the DNA Repair Enzyme Polymerase theta in Cancer Therapy. Trends Cancer 7, 98–111 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Zhou J et al. , A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat Cancer 2, 598–610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sfeir A, Symington LS, Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci 40, 701–714 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Black SJ et al. , Molecular basis of microhomology-mediated end-joining by purified full-length Poltheta. Nat Commun 10, 4423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mateos-Gomez PA et al. , The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat Struct Mol Biol 24, 1116–1123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsden DA, Carvajal-Garcia J, Gupta GP, Mechanism, cellular functions and cancer roles of polymerase-theta-mediated DNA end joining. Nat Rev Mol Cell Biol 23, 125–140 (2022). [DOI] [PubMed] [Google Scholar]
- 11.Truong LN et al. , Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad Sci U S A 110, 7720–7725 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie A, Kwok A, Scully R, Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol 16, 814–818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J, CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol 18, 75–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleury H et al. , The APE2 nuclease is essential for DNA double strand break repair by microhomology-mediated end-joining. BioRxiv, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma S et al. , Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis 6, e1697 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mengwasser KE et al. , Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol Cell 73, 885–899 e886 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan SH, Yu AM, McVey M, Dual roles for DNA polymerase theta in alternative end-joining repair of double-strand breaks in Drosophila. PLoS Genet 6, e1001005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arana ME, Seki M, Wood RD, Rogozin IB, Kunkel TA, Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res 36, 3847–3856 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kent T, Mateos-Gomez PA, Sfeir A, Pomerantz RT, Polymerase theta is a robust terminal transferase that oscillates between three different mechanisms during end-joining. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audebert M, Salles B, Calsou P, Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279, 55117–55126 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Seki M, Marini F, Wood RD, POLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res 31, 6117–6126 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boulton SJ, Jackson SP, Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15, 5093–5103 (1996). [PMC free article] [PubMed] [Google Scholar]
- 23.Thyme SB, Schier AF, Polq-Mediated End Joining Is Essential for Surviving DNA Double-Strand Breaks during Early Zebrafish Development. Cell Rep 15, 1611–1613 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Schimmel J, Kool H, van Schendel R, Tijsterman M, Mutational signatures of non-homologous and polymerase theta-mediated end-joining in embryonic stem cells. EMBO J 36, 3634–3649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saito S, Maeda R, Adachi N, Dual loss of human POLQ and LIG4 abolishes random integration. Nat Commun 8, 16112 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corneo B et al. , Rag mutations reveal robust alternative end joining. Nature 449, 483–486 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Llorens-Agost M et al. , POLtheta-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis. Nat Cell Biol 23, 1095–1104 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H et al. , PLK1 targets CtIP to promote microhomology-mediated end joining. Nucleic Acids Res 46, 10724–10739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng L et al. , Mitotic CDK Promotes Replisome Disassembly, Fork Breakage, and Complex DNA Rearrangements. Mol Cell 73, 915–929 e916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heijink AM et al. , Sister chromatid exchanges induced by perturbed replication can form independently of BRCA1, BRCA2 and RAD51. Nat Commun 13, 6722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bubenik M et al. , Identification of RP-6685, an Orally Bioavailable Compound that Inhibits the DNA Polymerase Activity of Poltheta. J Med Chem 65, 13198–13215 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmermann M et al. , CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature 559, 285–289 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adam S et al. , The CIP2A-TOPBP1 axis safeguards chromosome stability and is a synthetic lethal target for BRCA-mutated cancer. Nat Cancer 2, 1357–1371 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Verma P et al. , ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat Cell Biol 23, 160–171 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shukla V et al. , HMCES Functions in the Alternative End-Joining Pathway of the DNA DSB Repair during Class Switch Recombination in B Cells. Mol Cell 77, 1154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez-Quilon A et al. , Endogenous DNA 3’ Blocks Are Vulnerabilities for BRCA1 and BRCA2 Deficiency and Are Reversed by the APE2 Nuclease. Mol Cell 78, 1152–1165 e1158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sfeir A, de Lange T, Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melo JA, Cohen J, Toczyski DP, Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15, 2809–2821 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Certo MT et al. , Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods 8, 671–676 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hussmann JA et al. , Mapping the genetic landscape of DNA double-strand break repair. Cell 184, 5653–5669 e5625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cotta-Ramusino C et al. , A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332, 1313–1317 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindsey-Boltz LA, Kemp MG, Capp C, Sancar A, RHINO forms a stoichiometric complex with the 9-1-1 checkpoint clamp and mediates ATR-Chk1 signaling. Cell Cycle 14, 99–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bass TE et al. , ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mordes DA, Glick GG, Zhao R, Cortez D, TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22, 1478–1489 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eichinger CS, Jentsch S, 9-1-1: PCNA’s specialized cousin. Trends Biochem Sci 36, 563–568 (2011). [DOI] [PubMed] [Google Scholar]
- 46.B. Institute. (Broad Institute, 2021). [Google Scholar]
- 47.Leimbacher PA et al. , MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol Cell 74, 571–583 e578 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Marco Zompit M et al. , The CIP2A-TOPBP1 complex safeguards chromosomal stability during mitosis. Nat Commun 13, 4143 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan M, Morgan DO, Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol 8, 894–903 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Singh P et al. , BUB1 and CENP-U, Primed by CDK1, Are the Main PLK1 Kinetochore Receptors in Mitosis. Mol Cell 81, 67–87 e69 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Elia AE et al. , The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115, 83–95 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Maia AR et al. , Cdk1 and Plk1 mediate a CLASP2 phospho-switch that stabilizes kinetochore-microtubule attachments. J Cell Biol 199, 285–301 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blackford AN, Stucki M, How Cells Respond to DNA Breaks in Mitosis. Trends Biochem Sci 45, 321–331 (2020). [DOI] [PubMed] [Google Scholar]
- 54.Zirkle RE, Bloom W, Irradiation of parts of individual cells. Science 117, 487–493 (1953). [DOI] [PubMed] [Google Scholar]
- 55.Iacovoni JS et al. , High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29, 1446–1457 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang CZ et al. , Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murai J et al. , Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 72, 5588–5599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beucher A et al. , ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 28, 3413–3427 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karanam K, Kafri R, Loewer A, Lahav G, Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 47, 320–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakamura K et al. , H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat Cell Biol 21, 311–318 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gelot M. T. K. Camille, Miron Simona, Mylne1 Emilie, Ghouil Rania,, Popova F. D. Tatiana, Loew Damarys, Guirouilh-Barbat Josée, Elaine, S. Z.-J. a. R. C. 6 Del Nery, Polθ is phosphorylated by Polo-like kinase 1 1 (PLK1) to enable repair of DNA double strand breaks in mitosis. BioRxiv, (2023). [Google Scholar]
- 62.Esashi F et al. , CDK-dependent phosphorylation of BRCA2 as a regulatory mechanism for recombinational repair. Nature 434, 598–604 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Lee DH et al. , Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol Cell 54, 512–525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Orthwein A et al. , Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Roerink SF, van Schendel R, Tijsterman M, Polymerase theta-mediated end joining of replication-associated DNA breaks in C. elegans. Genome Res 24, 954–962 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belan O et al. , POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol Cell 82, 4664–4680 e4669 (2022). [DOI] [PubMed] [Google Scholar]
- 67.Schrempf A et al. , POLtheta processes ssDNA gaps and promotes replication fork progression in BRCA1-deficient cells. Cell Rep 41, 111716 (2022). [DOI] [PubMed] [Google Scholar]
- 68.Mann A et al. , POLtheta prevents MRE11-NBS1-CtIP-dependent fork breakage in the absence of BRCA2/RAD51 by filling lagging-strand gaps. Mol Cell 82, 4218–4231 e4218 (2022). [DOI] [PubMed] [Google Scholar]
- 69.Joshua R MM. Heyza, Aastha Bahl, David Broadbent, Jens C. Schmidt, Systematic analysis of the molecular and biophysical properties of key DNA damage response factors. BioRxiv, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grimm JB et al. , A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature Methods 12, 244-+ (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hart T et al. , Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 (Bethesda) 7, 2719–2727 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim E, Hart T, Improved analysis of CRISPR fitness screens and reduced off-target effects with the BAGEL2 gene essentiality classifier. Genome Med 13, 2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castaneda JC, Schrecker M, Remus D, Hite RK, Mechanisms of loading and release of the 9-1-1 checkpoint clamp. Nat Struct Mol Biol 29, 369–375 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hansen AS et al. , Robust model-based analysis of single-particle tracking experiments with Spot-On. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text and supplemental materials. All material will be shared upon request.