

Abstract

Late-stage functionalization (LSF) constitutes a powerful strategy for the assembly or diversification of novel molecular entities with improved physicochemical or biological activities. LSF can thus greatly accelerate the development of medicinally relevant compounds, crop protecting agents, and functional materials. Electrochemical molecular synthesis has emerged as an environmentally friendly platform for the transformation of organic compounds. Over the past decade, electrochemical late-stage functionalization (eLSF) has gained major momentum, which is summarized herein up to February 2023.
1. Introduction
The direct and site-selective late-stage diversification of structurally complex molecules is of great potential for drug discovery, materials science, crop protection, and other areas.1−8 This approach avoids a complete de novo synthesis of a target molecule, enables the rapid creation of large compound libraries, and hence offers the promise of a fast exploration of structure–activity relationships (SARs). Thereby, an improvement of pharmacokinetics properties as well as physicochemical drug characteristics, such as potency, stability, solubility, and selectivity, is frequently viable.9 The most synthetically useful late-stage functionalization (LSF) strategy is often the direct installation of fluorophores or small, noninvasive groups—inter alia methyl, hydroxyl, chloro, fluoro, or trifluoromethyl—with a selectivity control at a specific site of an existing biologically relevant molecule. The introduction of a small group can dramatically affect the bioactivity profiles of a structurally complex pharmaceutical molecule. For instance, Pfizer found that the installation of a methyl group to a morpholine-containing compound of mineralocorticoid receptor (MR) agonist, gave rise to a 45-fold potency increase.10 In the past decade, a large number of LSF approaches have been developed, including metal-catalyzed transformations,11−21 visible-light-induced photocatalysis22−34 and enzyme catalysis,35−43 among others (Scheme 1a).44−48
Scheme 1. Opportunities for Electrochemical Late-Stage Functionalization Strategies in Drug Discovery.
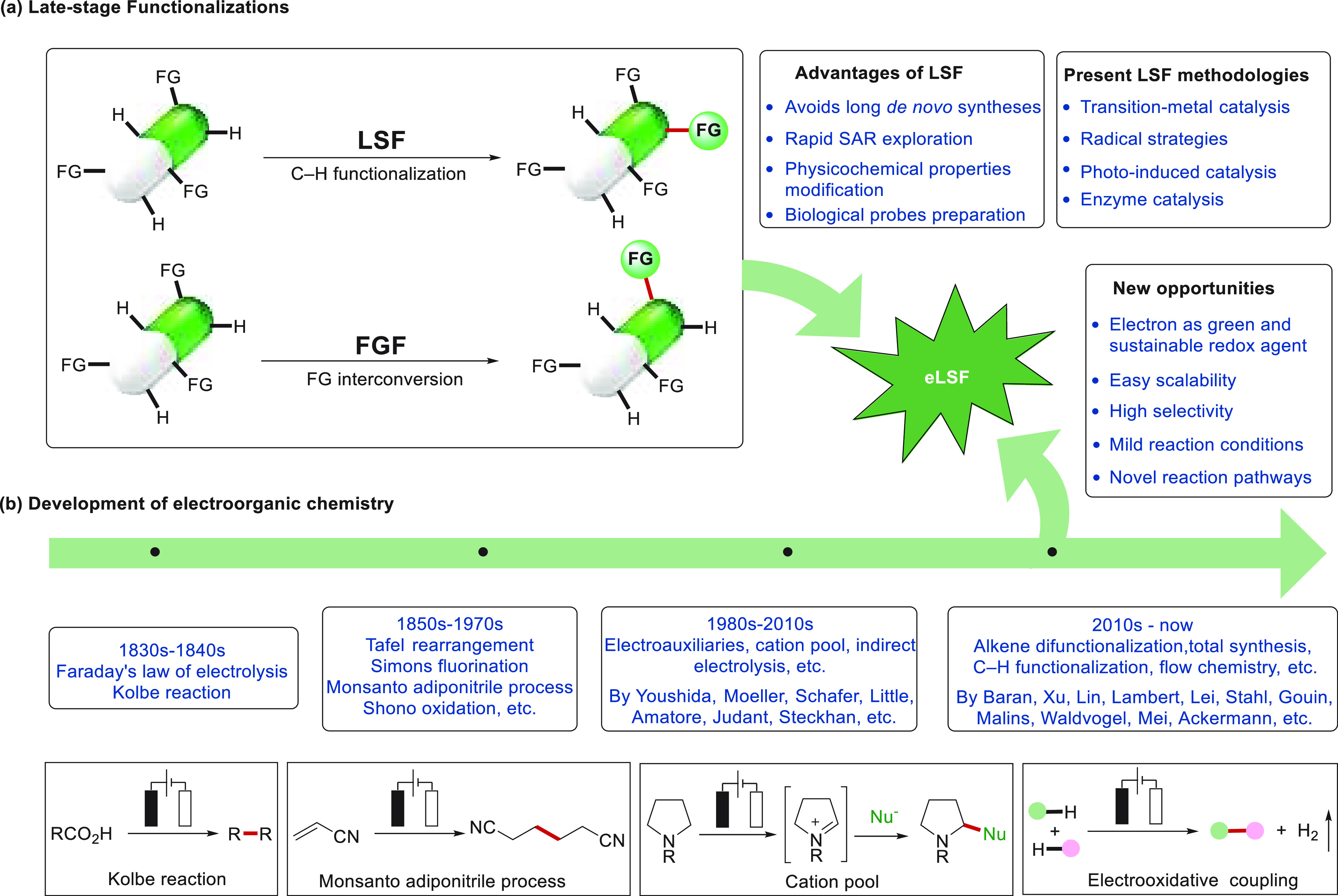
Electrochemical synthesis is a robust tool for sustainable molecular syntheses since it generally features mild reaction conditions, high selectivities, and facile scalability by flow techniques (Scheme 1b). Electrosynthesis has a history of nearly 200 years that can be traced back to Faraday’s conversion of acetic acid in the 1830s49 and Kolbe’s electrochemical decarboxylative dimerization,50 as well as industrially conducted processes, including the Simons fluorination process,51 the Monsanto adiponitrile process,52 and the Shono oxidation.53 Yoshida introduced the concepts of electroauxiliaries and cation pool to increase the electrosynthesis viability in the late 20th century.54−56 Meanwhile, Steckhan elegantly formalized the principles of indirect electrolysis, which thereafter brought forth numerous mediator-driven processes.57,58 Subsequent key achievements on the direct electrolysis were made by Schäfer,59 Lund,60 Little,61−63 Moeller,64 Jutand,65 and Amatore66 around the 21st century. On the basis of these pioneering contributions, electro-organic synthesis reemerged in the past decade, with major contributions by Baran,67 Xu,68 Lei,69 Ackermann,70 Lambert,71 Lin,72 Gouin,73 Waldvogel,74 Stahl,75 Malins,76 and Mei,77 among others.78−85 Thus, major advances have been achieved in various fields, including C–H activation, reductive cross-electrophile coupling, alkene difunctionalization, nitrogen-centered radical mediated chemistry, total synthesis, and the LSF of natural products and medicinally relevant molecules (Scheme 1b).
Over the years, a variety of articles have been published that summarized the impressive advances made in the field of electro-organic synthesis86−106 and late-stage functionalization,1−8,46,107−113 respectively. In contrast, comprehensive reviews of electrochemical late-stage functionalization has remained elusive.76 Thus, we herein aim at providing an overview on the advances in the area of electrochemical late-stage functionalization (eLSF), with a topical focus on biorelevant compounds. Notably, we define eLSF reactions as the direct, site-selective, and chemoselective functionalization of C–H bonds or endogenous functional groups on biologically relevant molecules, natural products, pharmaceuticals, or structurally complex molecules consisting of these moieties to provide their analogues. Such alterations may have the capacity to modulate properties, in a beneficial manner of binding affinity, drug metabolism, or pharmacokinetic properties, generally without loss of or even with an enhancement of the drug’s biological activity.
2. eLSF of C–H Bonds
Over the past decade, the merger of electrocatalysis with C–H activation has revolutionized the art of molecular synthesis. Particularly, a number of these electrochemical C–H functionalization techniques have been successfully applied for late-stage diversification of natural products and pharmaceuticals, and these key developments afforded tremendous opportunities in drug discovery programs.
2.1. eLSF of C(sp2)–H Bonds
2.1.1. Late-Stage C(sp2)–H Carbonization
Among numerous strategies for late-stage functionalization, the methylation reaction plays a unique role in the modulation of bioactive molecules.6 The incorporation of a simple methyl group can dramatically improve their potency by enhancing lipophilicity, metabolic stability, and binding interactions, among others, which are collectively referred as the “magic methyl effect”. A literature survey of >2000 cases revealed that 8% of methyl installations led to a > 10-fold potency boost, and >100-fold activity increases in 0.4% of cases.9 Consequently, and despite indisputable progress, new synthetic methylation strategies, particularly direct C–H methylation, are highly sought after.
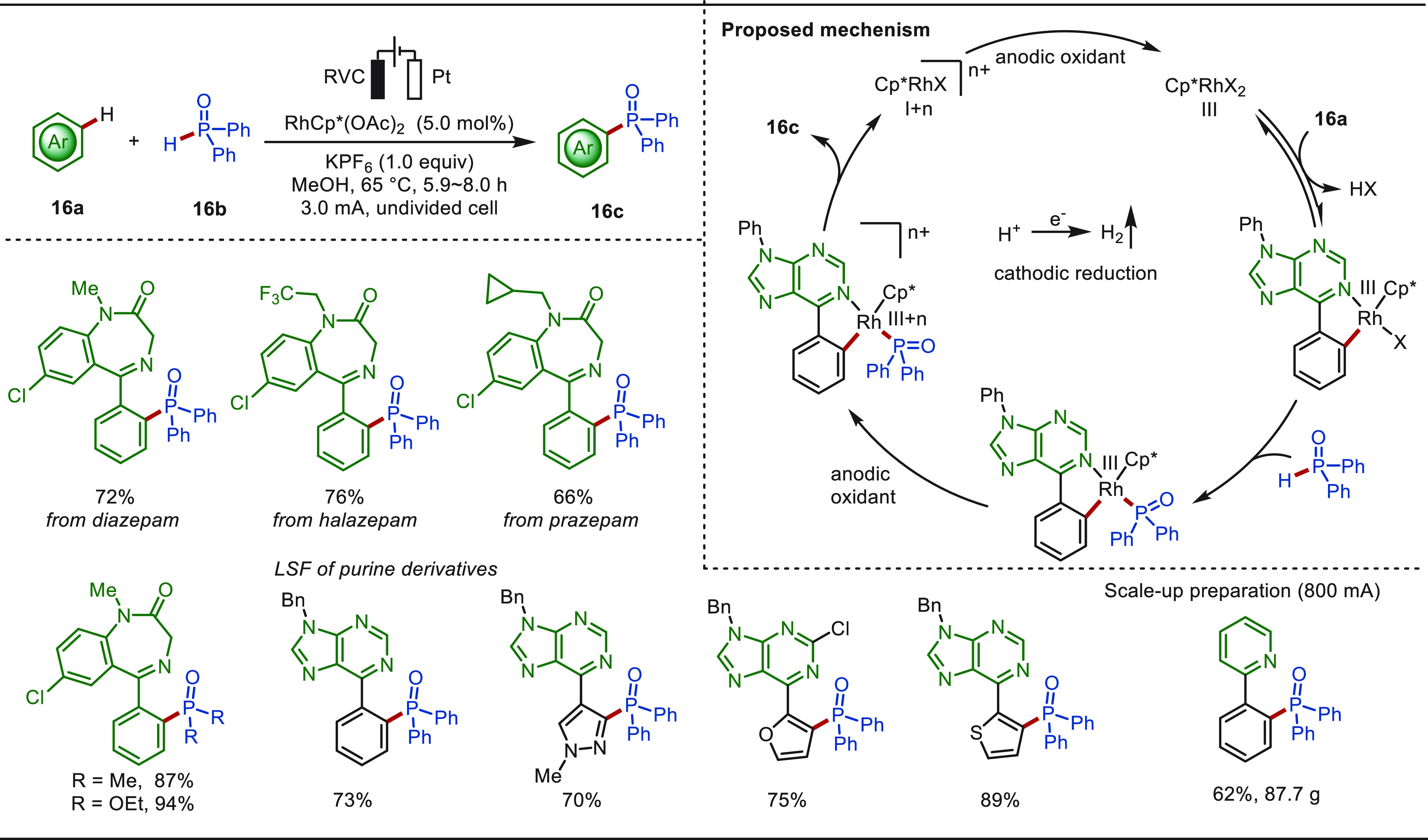
In 2017, Mei and co-workers first disclosed the electrochemical C(sp2)–H methylation via anodic oxidation with MeBF3K as the methyl source under the catalysis of Pd(OAc)2, offering an alternative methylation strategy to conventional method that requires strong chemical oxidants.114,115 In 2022, the Ackermann group reported on an electrochemical ortho C(sp2)–H methylation of N-heteroarenes with the aid of RhCp*(OAc)2 as a catalyst and MeBF3K as the methyl source in a mixture of nBuOH and H2O (Scheme 2a).116 This approach proceeded in a user-friendly undivided cell setup and has been successfully applied to various biologically molecules, including purines, diazepam, and amino acids with high levels of site- and monoselectivity. Shortly thereafter, Guo and co-workers described a similar transformation in MeOH. Besides MeBF3K, MeB(OH)2 and MeBPin were further used as coupling partners for this rhodaelectro-catalyzed C–H methylation (Scheme 2b).117 Hence, the eLSF of a variety of bioactive architectures including purines, estrone, nucleosides, and nucleotides were achieved under mild reaction conditions. Current was the only oxidant for this catalysis. Mechanistic studies indicated that an anodic-oxidation-induced reductive elimination occurred within a rhodium(III/IV/II) regime. Meanwhile, H2 was released as the byproduct at the cathode (Scheme 2).
Scheme 2. Electrochemical Rhodium-Catalyzed Late-Stage C(sp2)–H Methylation.
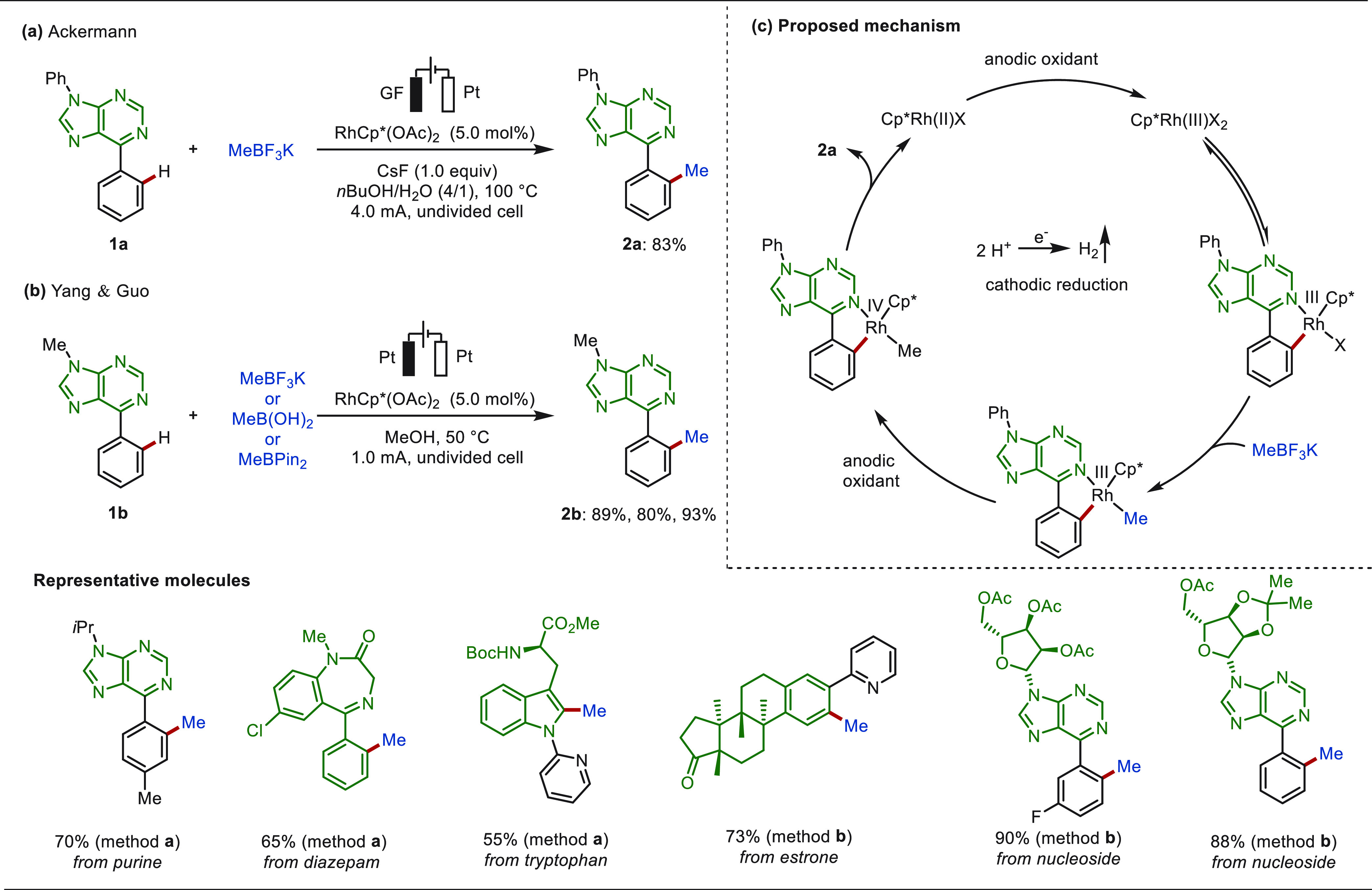
In addition, the monoselective C–H ethylation of purines and diazepam was achieved by the Ackermann group with VinBF3K by paired electrolysis, in which the reduction of in situ generated vinylated products takes place at the cathode to afford the ethylated products (Scheme 3a).116 Under Guo’s reaction conditions, different alkylation agents was examined for the LSF of purine derivatives (Scheme 3b).117 While potassium ethyltrifluoroborate and potassium benzylic trifluoroborates were identified as suitable substrates, giving the desired alkylation products, other alkyltrifluoroborates (nbutyl, trifluoromethyl, and cyclohexyl) were unsuccessful.
Scheme 3. Electrochemical Rhodium-Catalyzed Late-Stage C(sp2)–H Alkylation.

Approximately 20% of commercial drugs contain at least one fluorine atom.118 The introduction of fluorine-containing groups leads to a significant boost in the potency of bioactive compounds.119,120 Particularly, trifluoromethylated compounds are in high demand in pharmaceutical industries and medicinal chemistry, since they can display unique lipophilicity and bioactivity.121,122 Consequently, direct C–H trifluoromethylation occupies an important position in terms of LSF.
In 2014, Baran and co-workers reported an electrochemical C(sp2)–H trifluoromethylation of heterocyclics with Zn(CF3SO2)2 under constant current electrolysis (Scheme 4).123 This approach featured mild reaction conditions with high site-selectivity and offers a wide application for late-stage trifluoromethylation of molecular architectures, including metronidazole, pentoxifylline, caffeine, and ketorolac methyl ester. Notably, this strategy resulted in significantly improved yields compared to the traditional method using tert-butyl hydroperoxide (TBHP) as the radical initiator and oxidant. Mechanistic studies indicated a controlled electron transfer at the anode, giving a sulfinate radical, which was rapidly converted to fluoroalkyl radical by cleavage and releasing SO2. Difluoromethylation of complex molecules was also achieved with Zn(CF2HSO2)2 at an elevated temperature of 60 °C, albeit in lower yield because of the poor reactivity of the CF2H radical with heterocycles.123
Scheme 4. Electrochemical Controlled C(sp2)–H Late-Stage Trifluoromethylation and Difluoromethylation.

The transition-metal-catalyzed C–H alkynylation is a practical strategy in synthetic chemistry to install the versatile alkyne as a (transient) functional group.124−126 In 2020, Shi and Xie reported an electrochemical iridium-catalyzed directed C(sp2)–H alkynylation with terminal alkyne in an undivided cell (Scheme 5).127 Here, anodic oxidation was enabled by an iridium(III) intermediate to promote reductive elimination, affording the desired coupling products in excellent to good yields without the use of exogenous chemical oxidants. This transformation was amenable to various N-based directing groups, such as pyridyl, pyrazolyl, and isoquinolyl, enabling a high atom economy with H2 as the byproduct. The success of installing an alkyne on complex bioactive molecules, including derivatives of purine, diazepam, estrone, and coumarin, highlighted the potential application of this approach in late-stage functionalization of pharmaceuticals.
Scheme 5. Electrochemical Iridium-Catalyzed Late-Stage C(sp2)–H Alkynylation.

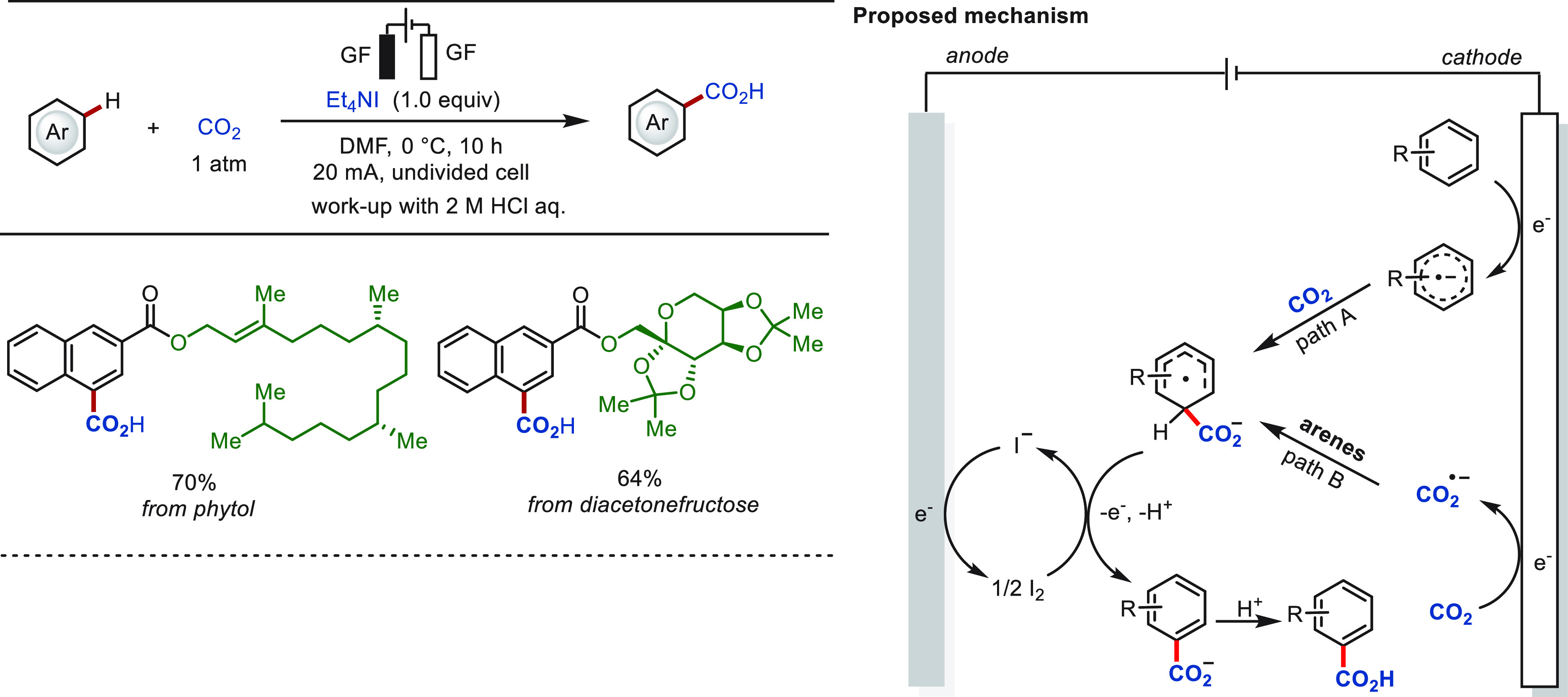
CO2 is an abundant C-1 source that has been widely used in electrochemical transformations to construct diverse carboxylic acid compounds or their derivatives.128−144 In 2022, the Qiu group reported a direct aromatic C(sp2)–H carboxylation approach with CO2 to access synthetically useful aryl carboxylic acids (Scheme 6).145 This transformation proceeded in an undivided cell, displaying high site selectivity and chemoselectivity and obviating the use of a transition-metal catalyst. An array of challenging arenes, including electron-deficient naphthalenes, as well as heteroarenes such as pyridines and substituted quinolines, proved to be suitable substrates. The late-stage carboxylation of bioactive molecules derived from phytol and diacetonefructose was achieved efficiently. For a substrate with a less negative reduction potential, the process commences with the arene reduction at the cathode to form the corresponding radical anion. By contrast, for a substrate with a more negative reduction potential than that of CO2, the transformation starts with the CO2 reduction to a CO2 radical anion.
Scheme 6. Electrochemical Late-Stage C(sp2)–H Carboxylation with CO2.
2.1.2. Late-Stage C(sp2)–H Oxygenation
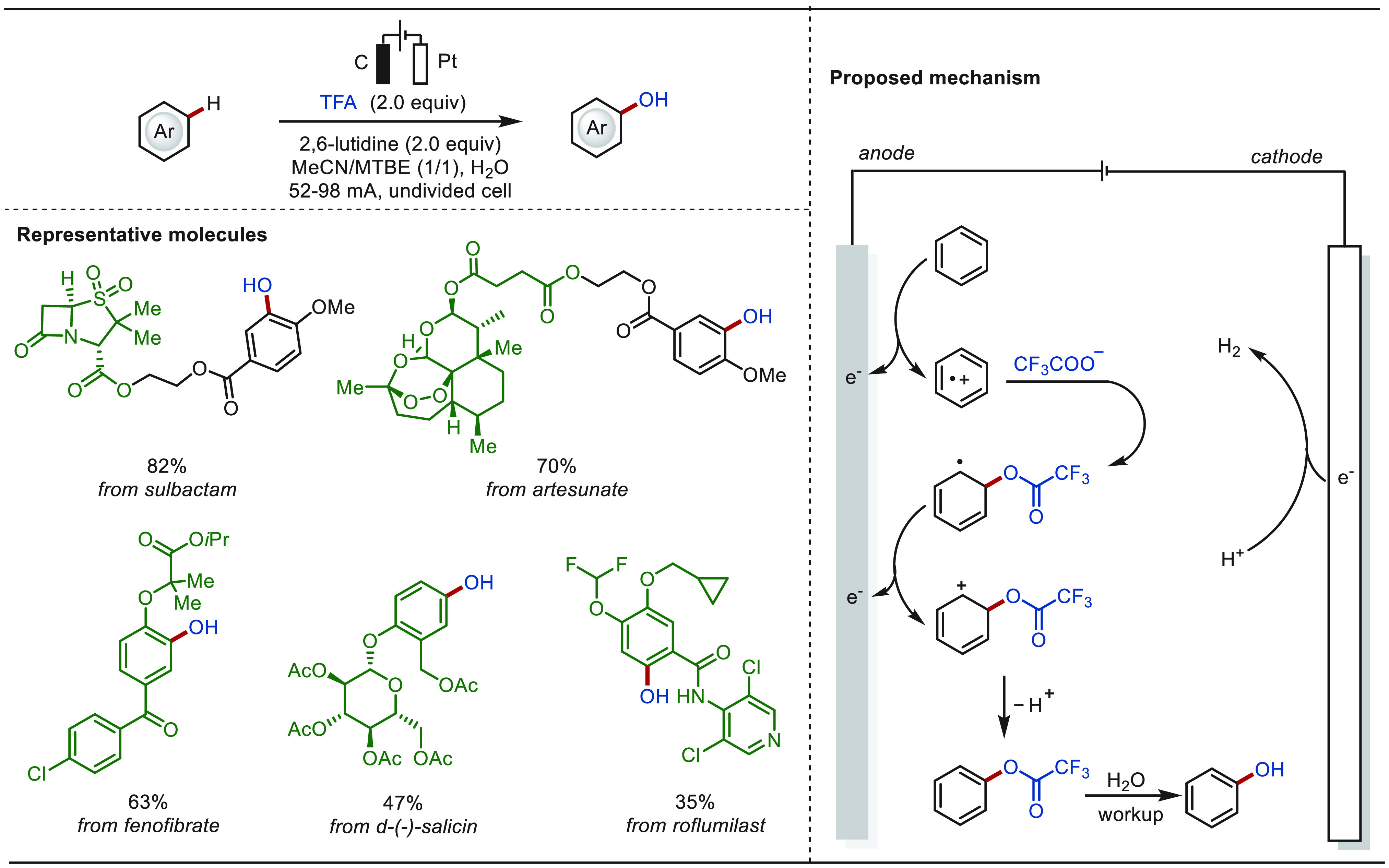
The direct hydroxylation of arene C(sp2)–H bonds is a highly sought-after transformation in the field of LSF, since the introduction of a small −OH group can dramatically improve inter alia the water solubility of the drug molecules.38 However, the controlled electrochemical hydroxylation of the aromatic C–H bond is challenging to achieve given the high propensity of the phenol to undergo overoxidation. To attenuate the overoxidation issue, trifluoroacetic acid (TFA) was taken into consideration as the oxygen donor to first generate aryl trifluoroacetate intermediates, followed by hydrolysis to release the hydroxylated products.146,147 Although this approach seems promising, the viable methods are limited to a few examples of structurally simple and electron-deficient/neutral arenes. Electron-rich arenes still typically suffer from overoxidation and self-coupling side reactions, and hence cannot efficiently converted to phenols.148
Continuous-flow electrochemical microreactors have the potential to increase the reaction efficiency and reducing overoxidation.149−155 In this context, the Xu group elegantly realized electrochemical C(sp2)–H hydroxylation of diverse arenes with high efficiency and selectivity in a continuous flow electrochemical microreactor (Scheme 7).156 The approach proceeded under mild conditions without chemical oxidants or transition-metal catalysts, featuring a broad scope of arenes with diverse electronic properties. The overoxidation reaction was greatly inhibited. The eLSF of a number of natural products and drug derivatives was achieved in an efficient and selective manner.
Scheme 7. Electrochemical Late-Stage C(sp2)–H Hydroxylation.
Besides the direct electrolysis method, the merger of electrocatalysis with organometallic C–H activation provides another sustainable strategy for the oxygenation of the aromatic C(sp2)–H bond.157−162 The Ackermann group has previously reported rhodaelectro- and ruthenaelectro-catalyzed hydroxylations of diverse arenes with TFA.159,161 Very recently, the same group achieved the ruthenaelectro-catalyzed late-stage C(sp2)–H acyloxylation of tyrosine-containing peptides with various aromatic acids (Scheme 8).162 Notably, attempted transformations with chemical oxidants, including AgOAc, K2S2O8, and PhI(OAc)2, proved to be ineffective — a strong testament to the robust nature of the electrochemical approach. A variety of di-, tri-, and tetrapeptides were efficiently acyloxylated without epimerization of the otherwise sensitive peptides. Remarkably, this electro-oxidative regime bypassed Shono-type manifolds even when employing proline-containing peptides. Mechanistic studies indicated that p-cymene dissociated during the catalytic cycle and the catalyst underwent a ruthenium II/IV regime likely involving a bis-cyclometalated complex intermediate.
Scheme 8. Electrochemical Late-Stage C–H Acyloxylation of Tyrosine-Containing Peptides.
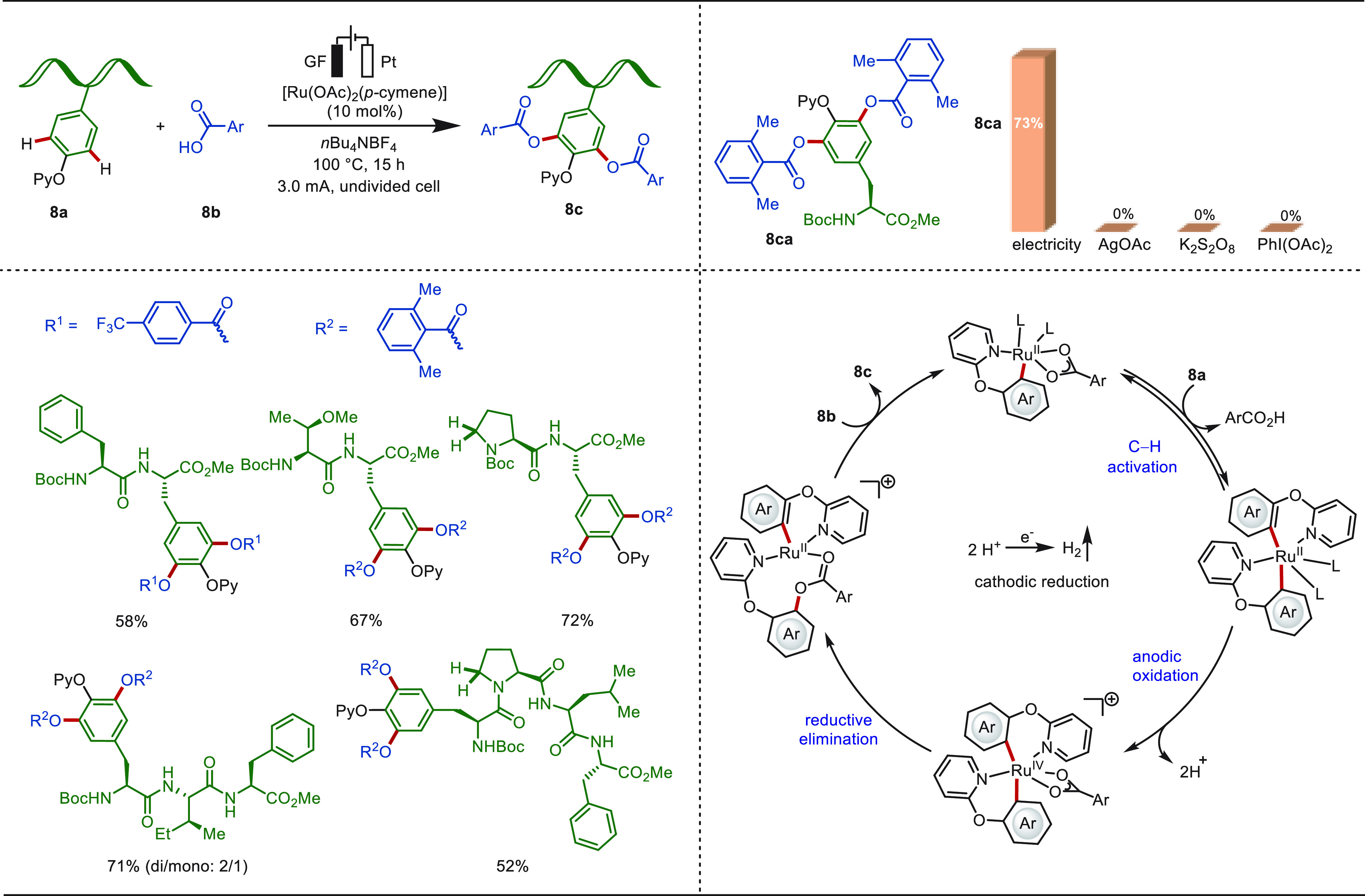
Reproduced with permission from ref (162). Copyright 2022, Royal Society of Chemistry.
In 2019, Ackermann reported a rare example of electro-oxidative nickel-catalyzed C(sp2)–H alkoxylation reaction with secondary alcohols (Scheme 9).158 This metallaelectrocatalysis exhibited high chemo- and positional-selectivity, and the plausible mechanism of this transformation involves a nickel(IV)-intermediate. Notably, various naturally occurring alcohols, such as menthol, cholesterol, and β-estradiol, were accommodated as coupling partners, delivering the corresponding aromatic ethers in excellent yields.
Scheme 9. Electro-oxidative C–H Alkoxylation of Arenes with Secondary Alcohols.

2.1.3. Late-Stage C(sp2)–H Amination
The electro-oxidative C–H/N–H cross-coupling is a straightforward and powerful tool to install nitrogen functionalities into aromatic compounds. In 2019, the Lei group reported an intermolecular cross-coupling between sulfonimides and aromatic arenes (Scheme 10).163 The transformation proceeded through a nitrogen-centered radical addition pathway under transition-metal-free and exogenous oxidant-free conditions. A variety of arenes, alkenes, heteroarenes, and pharmaceuticals, such as flavone, caffeine, and fenofibrate, were amenable scaffolds. Aryl sulfonamides or aniline derivatives could thus be obtained after the deprotection process. Mechanistic studies indicated that the nitrogen-centered radicals were generated via a proton-coupled electron transfer (PCET) process jointly mediated by nBu4NOAc and an anodic oxidation process.163,164 Concurrently, the Ackermann group achieved an electrochemical oxidation induced C(sp2)–H nitrogenation for a variety of heteroarenes, including pyrroles, indoles, benzothiophene, and benzofuran.165 In addition, metallaelectro-catalyzed C(sp2)–H aminations have been realized with diverse transition-metal catalysts (Ni, Co, Cu, etc.).166−169
Scheme 10. Electrochemical Late-Stage Aminations.

Introducing a functional group into a peptide or a protein under mild, user-friendly conditions is of great significance in the field of chemical biology, medical chemistry, and pharmacology.170−173 In this context, the post modification of the phenolic tyrosine side chain has become the most commonly used strategy due to its relatively high reactivity and low abundance in the proteome (2.9%). Thus, early in the 1990s, Walton and Heptinstall had reported on the modification of hen egg-white lysozyme proteins and horse heart myoglobin via electro-oxidative nitration in a mildly acidic buffer (Scheme 11).174−178
Scheme 11. Electrochemical Late-Stage Nitrogenation.

The development of efficient methods for the conjugation of native proteins is relevant for chemical biology and biotherapies, among others. In 2010, Barbas disclosed an ene-like reaction between the tyrosine residues and substituted phenyl-3H-1,2,4-triazole-3,5(4H)-diones (PTADs).179 Bulk chemical oxidant and cosolvents or scavengers (e.g., Tris) needed to be employed, which limited its application scope. In 2018, Gouin and co-workers discovered the electrochemical Y-click reaction at a mild oxidative potential (+0.36 V, constant voltage electrolysis) in an aqueous buffer (Scheme 12).73 At the low potential conditions, the in situ generated reactive species is immediately conjugated with tyrosine, hence minimizing undesired side reactions and leading to a higher selectivity. The utility of this protocol was highlighted by the functionalization of a remarkably broad range of substrates. Both the small peptide hormone oxytocin and epratuzumab, a 152 kDa monoclonal antibody, were selectively modified by this method. In addition, Huan and Li recently employed this strategy to cross-link peptides and proteins at tyrosine residues without the use of photoirradiation or a metal catalyst.180
Scheme 12. Electrochemical Peptide and Protein Modification at Tyrosine.
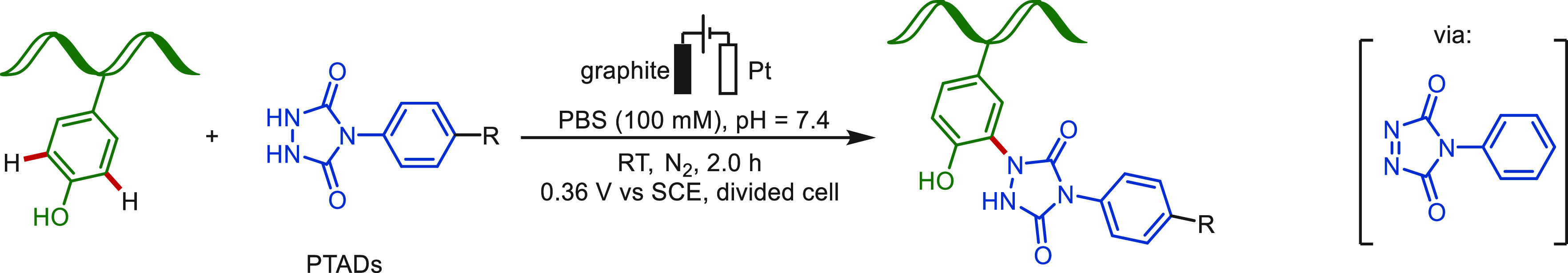
In 2020, Nakamura and co-workers likewise devised a modified version of the e-Y-click reaction for selective bioconjugation at tyrosine residues (Scheme 13).181 In their studies, N-methyl luminol and 1-methyl-4-phenylurazole derivatives were used as active small-molecules, which easily converted to the corresponding nitrogen radical species via the SET process under electro-oxidative conditions. A protected model octapeptide angiotensin II was successfully modified at the native tyrosine residue in a biological fashion. This approach employed purely aqueous buffer (Tris, 50 mM), neutral pH (7.4), and mild electrochemical conditions (400–700 mV), representing a truly biocompatible electrochemical modification strategies.
Scheme 13. Selective Peptide Modification at Tyrosine Using N-Methyl Luminol and 1-Methyl-4-Phenylurazole.
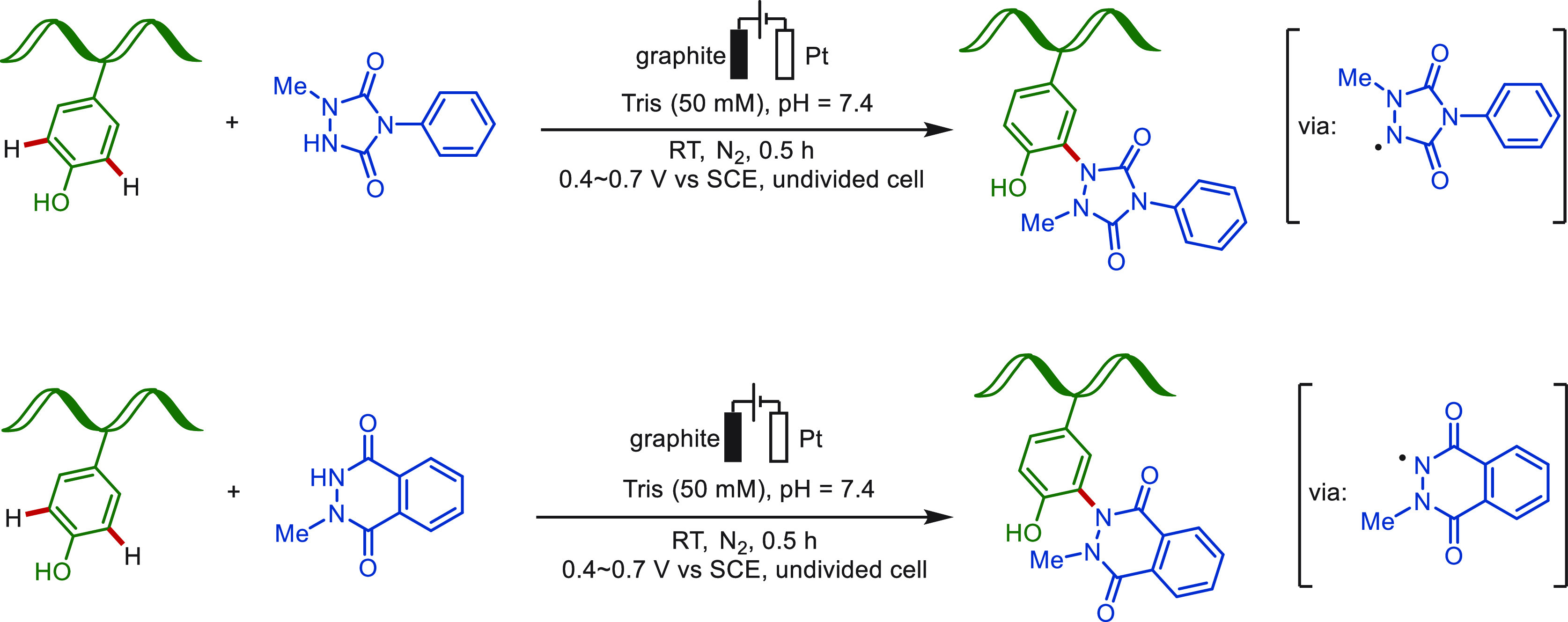
In contrast, the Lei group described an electrochemical method to execute the bioconjugation of a tyrosine side chain with phenothiazine derivatives in a simple and rapid manner (Scheme 14).182 This approach provided direct and efficient access to LSF of oligopeptides and proteins, featuring high chemo- and site-selectivity, without the use of transition-metal or chemical oxidants. Valuable bioactive compounds, such as angiotensin Y, tyrosine protein kinases, and MOG 35-55, selectively underwent the electro-oxidative bioconjugation process. It was also demonstrated that the phenothiazine-labeled peptide could be utilized as a fluorophore.
Scheme 14. Electrochemical Late-Stage C–H Nitrogenation of Tyrosine-Containing Peptides.
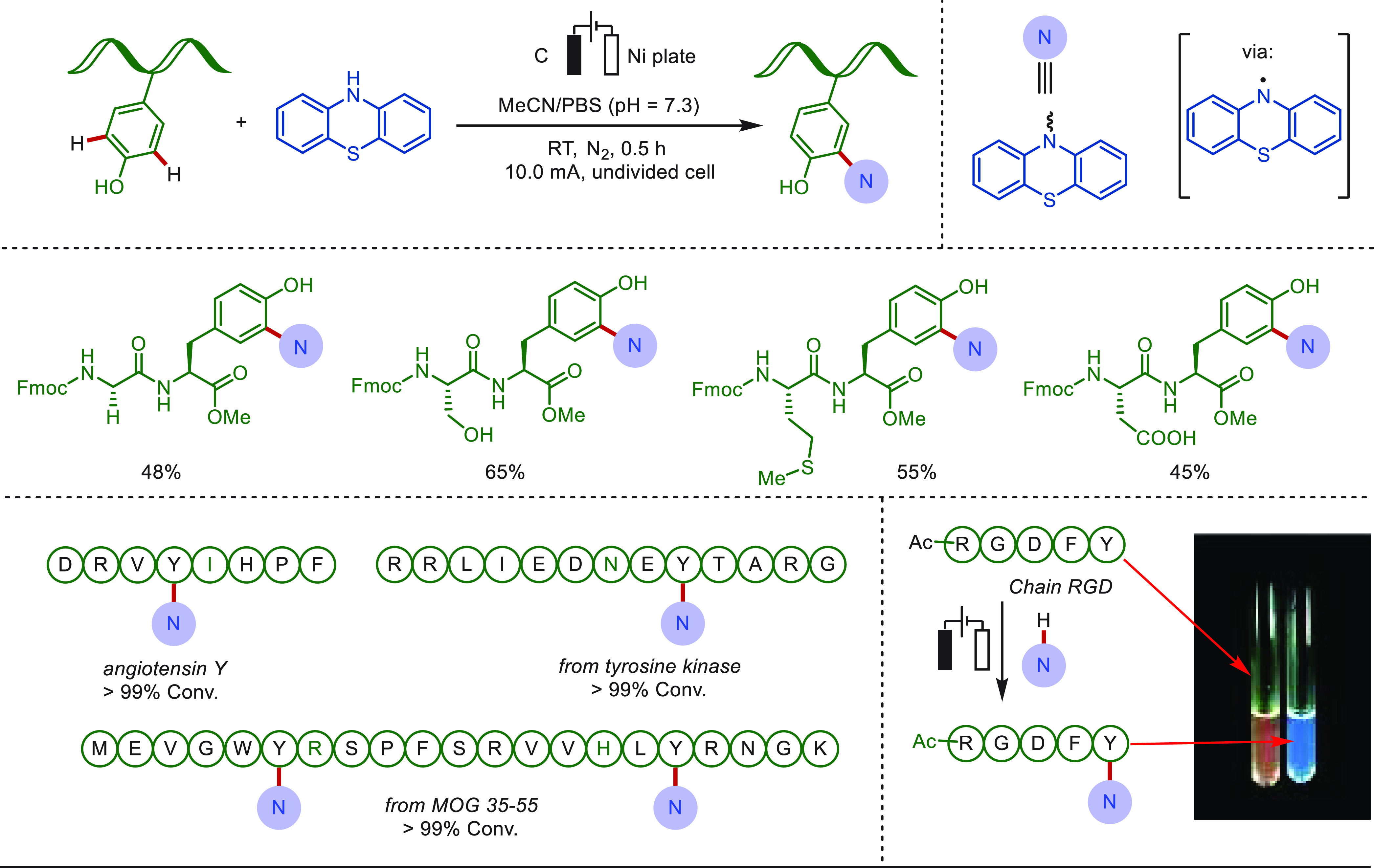
Reproduced with permission from ref (182). Copyright 2019, Royal Society of Chemistry.
2.1.4. Late-Stage C(sp2)–H Phosphorylation
The development of efficient late-stage phosphorylation methods is of considerable importance, given that organophosphorus compounds have wide utilities in medicinal chemistry.183 The past few years have seen significant development of electrochemical phosphorylation methodologies.184−191 In 2019, Budnikova and co-workers realized the metallaelectro-catalyzed coupling reactions of caffeine with dialkylphosphites (Scheme 15).185 Interestingly, diverse transition metals, including Pd(OAc)2, AgOAc, and Bipy3Ni(BF4)2 proved to be efficient for these transformations, affording the phosphorylated caffeine in good yields of 62–80%.
Scheme 15. Electrocatalytic Coupling Reactions of Caffeine with Dialkylphosphites.

In the same year, Xu and co-workers reported an rhodaelectro-catalyzed late-stage aryl C(sp2)–H phosphorylation reaction with various phosphine oxides (Scheme 16).186 The electrochemical approach was characterized by a broad scope and high functional group tolerance without using exogenous chemical oxidants. The method proved to be compatible with the eLSF of a variety of bioactive molecules such as diazepam and purine derivatives. Notably, this electrochemical reaction was also easily scaled up, remarkably yielding a phosphonate product in 87.7 g. Mechanistic interrogation suggested that the C–P bond was formed via an oxidation-induced reductive elimination process.
Scheme 16. Electrochemical Rhodium-Catalyzed C(sp2)–H Phosphorylation.
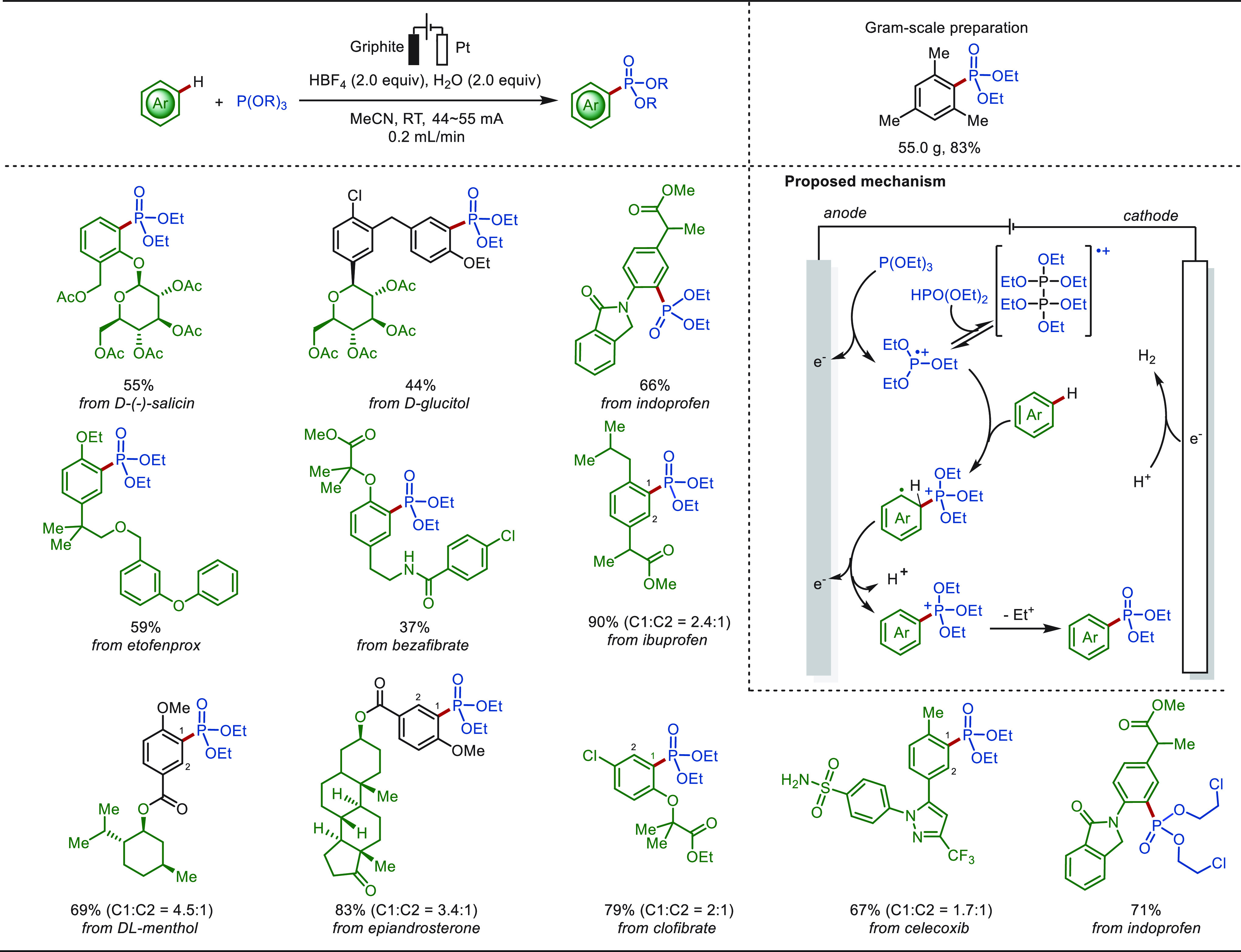
In 2021, the Xu group furthermore disclosed an electrochemical aromatic C(sp2)–H phosphorylation reaction using triethyl phosphite P(OR)3 in a continuous flow cell, obviating the use of exogenous chemical oxidants and transition-metal catalysts (Scheme 17).187 This continuous flow electrosynthesis was found to be compatible with both electron-rich and electron-deficient arenes. The practical utility of this electrochemical phosphorylation was further illustrated by the continuous production of one phosphonate product in 55.0 g. The C–P bond was formed through the reaction of arenes with in situ anodically generated P-radical cations. The selective late-stage functionalization of a series of bioactive compounds and natural products was accomplished through continuous flow electrosynthesis.
Scheme 17. Electrochemical Late-Stage C(sp2)–H Phosphorylation in Continuous Flow.
2.1.5. Late-Stage C(sp2)–H Halogenation
The incorporation of a halide atom into drug molecules may have a profound effect on enhancing their biological properties.192 In addition, halogenated arenes, particularly aryl bromides, can be quickly converted to radio-labeled compounds, showing great utilities in metabolism studies.193 Moreover, halogenated arenes and heterocycles are versatile intermediates in diverse organic transformations.194 Therefore, the development of efficient and atom-economical halogenation methodologies under mild conditions has been a long-term goal in molecular synthesis. Traditionally, strong corrosive, oxidizing reagents (X2, NXS) or halides (X−) combined with external strong chemical oxidants were needed for halogenation of arenes.195 In contrast, electrochemistry offers a powerful alternative for various halogenation reactions with simple halides salts or an aqueous HX solution as the halogenating source.196−203
Recently, Ackermann and co-workers disclosed electrochemical ruthenium-catalyzed distal C(sp2)–H bromination with an aqueous HBr solution as the brominating agent (Scheme 18).199 The regioselective meta-C–H bromination was conducted in an undivided cell by the catalysis of RuCl3·XH2O, under external ligand- and electrolyte-free conditions, featuring an ample substrate scope. Particularly, phenylpyrazole was readily brominated at the meta-position on the benzenoid moiety rather than at the commonly functionalized electron-rich pyrazole ring. Thus, and in sharp contrast, the bromination of pyrazolylarene under reported ruthenium/NBS conditions204 or electrochemical metal-free200 conditions proved to occur on the electron-rich pyrazole rings via a simple SEAr process. Purine derivatives were identified as suitable substrates for the ruthena-electrocatalyzed meta-C–H bromination. Mechanistic studies revealed that the bromide ion Br– was oxidized to molecular Br2, which equilibrated with the tribromide anion Br3– by combining and/or releasing a bromide ion. Then the bromination process occurred between Br2 and the in situ generated cycloruthenated complex. The desired meta-brominated product was released after ligand-to-ligand hydrogen transfer (LLHT) and a ligand exchange process.
Scheme 18. Ruthenaelectro-Catalyzed meta-C–H Bromination with HBr.

In 2017, Rivera and Liu disclosed an eLSF bromination method using NaBr in a mixture of water with acetonitrile/methanol (Scheme 19).202 The bromination reactions were conducted in a separate microflow electrochemical cell under mild conditions. Electrochemical bromination of drug molecules, including Cytidine, Sch 48793, Tenofovir, and MK-4618, gave rise to the corresponding aryl bromides. The brominated analogues of Tenofovir and MK-4618 were further converted to the corresponding tritium labeled products.
Scheme 19. Electrochemical Late-Stage Bromination of Drug Molecules with NaBr.

Jiao and co-workers elegantly achieved the electrochemical aromatic chlorination with common solvent DCE as the chloro source, producing vinyl chloride as a useful byproduct (Scheme 20).203 In this work, the electrochemical dehydrochlorination of DCE occurred by controlling the current intensity, producing vinyl chloride and HCl. This method opened a new avenue for the preparation of (hetero)aryl chlorides and vinyl chloride in an environmentally benign manner. The mild nature and practicality of the method was further demonstrated by its easily scaled-up and efficient eLSF chlorination of a number of bioactive molecules, such as (S)-naproxen methyl ester, leflunomide, and acetaminophen. Using a similar strategy, McNeil and co-workers recently realized the chlorination of arenes with waste poly(vinyl chloride) as a chloro source.83
Scheme 20. Paired Electrocatalysis for the Preparation of Aryl Chlorides Using DCE.

In addition to the electrochemical protein late-stage nitrogenation (vide supra), Heptinstall and co-workers have developed the selective electrochemical iodination of horse heart myoglobin with KI (Scheme 21).205 Since rapid anodic oxidation of an iodide anion led to persistent formation of the undesirable triiodide, the authors used an innovative “redox pulse” method (2.5 s at 0.4 V vs SCE and 5 s at 0.0 V vs SCE, 240 cycles) to enable mono- and double iodination of myoglobin with high levels of selectivity. Notably, this exquisitely controlled protein iodination strategy could proceed at both high and very low iodide concentrations, offering improved selectivity compared to those of reported chemical and enzymatic methods.
Scheme 21. Electrochemical Late-Stage C(sp2)–H Iodination of Tyrosine-Containing Protein.

2.1.6. Late-Stage Annulation Reactions via C(sp2)–H Activations
In the past two decades, annulations via C–H bonds activation have revolutionized the art of preparing cyclic compounds.206−208 Particularly, the electrochemical annulations have gained significant recent momentum without the use of sacrificial chemical oxidants, such as Cu(OAc)2 and AgOAc, avoiding the generation of undesired byproducts and increasing the atom economy.209−220 Diverse five-, six-, and seven-membered rings have been efficiently assembled through formal [3 + 2], [4 + 1], [4 + 2], or [5 + 2] cycloadditions. However, only a small part of these tools were exploited for chemo-selective eLSF.
In 2021, the Ackermann group reported on the rhodaelectro-catalyzed annulations of 2-hydroxybenzaldehydes with alkynes by electrochemical formyl C–H activation (Scheme 22).221 The strategy was applicable to the functionalization of tyrosine derivatives and hence enabled access to site-selective electrolabeling of tyrosine-derived fluorescent amino acids and peptides. A broad variety of dipeptides, even including oxidation-sensitive methionine and serine containing peptides as well as polypeptide, were efficiently converted to the desired products. Mechanistic studies provided strong support for an oxidation-induced reductive elimination within a rhodium(III/IV/II) manifold. Notably, a mediated photoelectrochemical oxidation of the modified amino acids allowed for access to π-extended peptide labels, which exhibited intense fluorescence and have great potential as fluorogenic probes.
Scheme 22. Rhodaelectro-Catalyzed Peptide Late-Stage Labeling via Formyl C–H Activation.
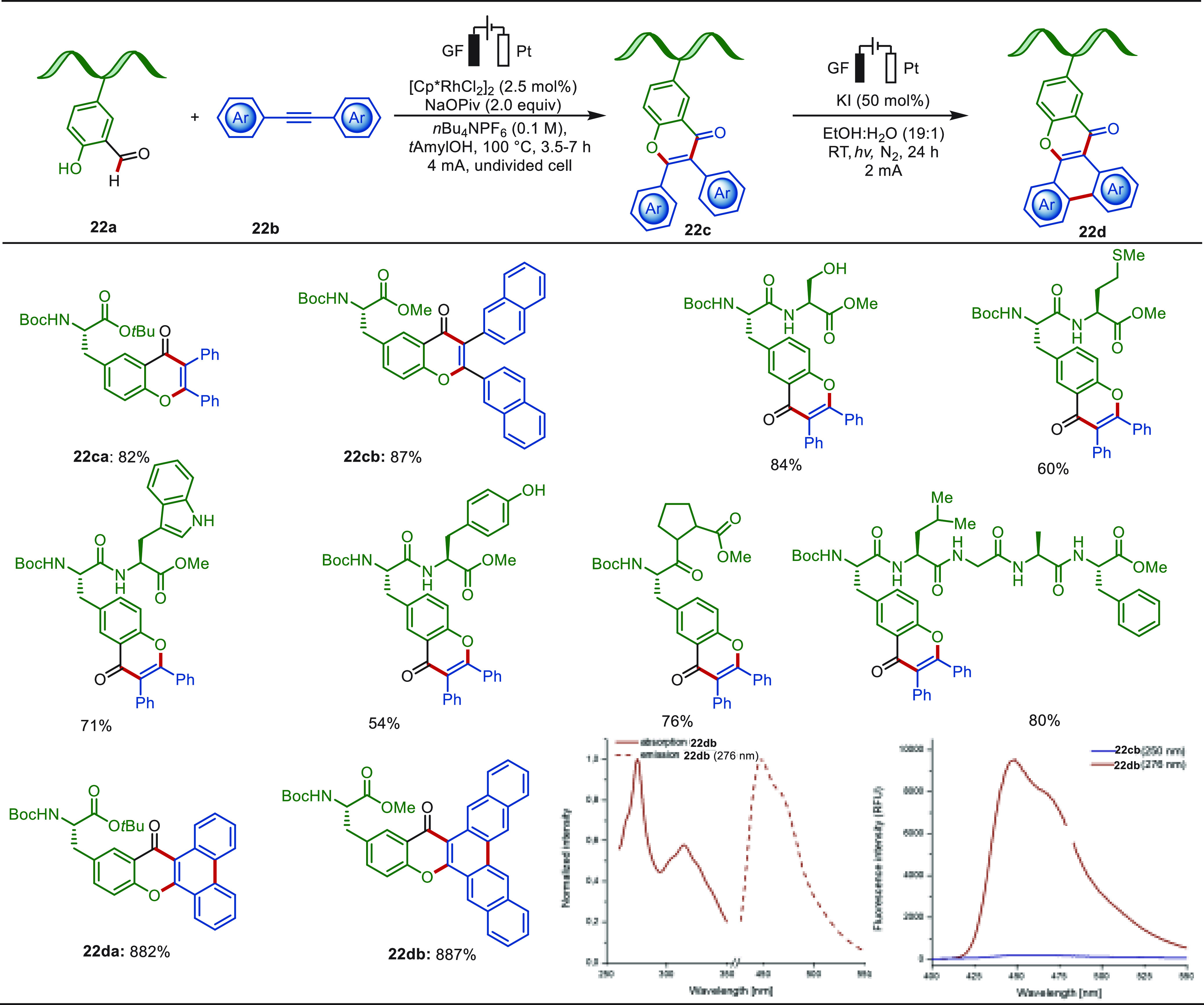
Reproduced with permission from ref (221). Copyright 2021, Springer Nature.
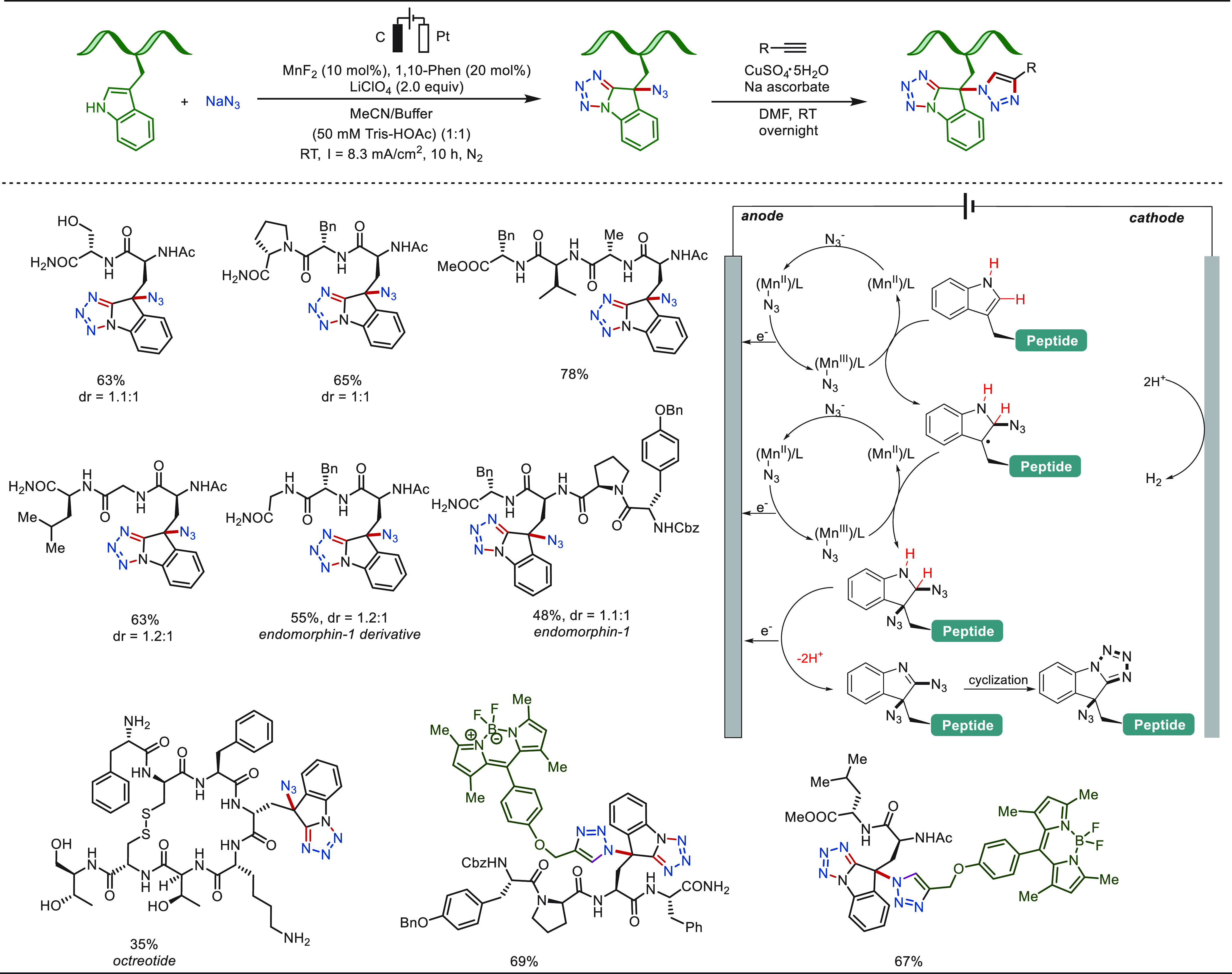
Very recently, Weng and co-workers developed an electrochemical LSF of tryptophan-containing peptides with NaN3 to afford azide-substituted tetrazolo[1,5-α]indolecontaining peptides (Scheme 23).222 This reaction used an earth abundant Mn catalyst under mild buffered conditions. This strategy was applicable for a wide range of peptides with good functional-group tolerance and high site-selectivity. In addition, the thus-obtained Trp-containing peptides with an azide group could be further derivatized to various triazole products by a copper-catalyzed “click” reaction. The reaction was proposed to proceed through manganese-mediated diazidation of the indole unit, followed by the dehydrogenation and heterocyclization to deliver the LSF products.
Scheme 23. Tandem Electrochemical Oxidative Azidation/Heterocyclization of Tryptophan-Containing Peptides.
The synthesis of macrocycles—abundant motifs in biologically relevant molecules and pharmaceuticals—continues to represent a popular arena for synthesis chemists.223−227 However, the construction of large ring systems is challenging, considering geometrical and thermodynamic constraints. Recently, electrochemical transformations have emerged as useful techniques in this regard. In 2015, Harran uncovered an electro-oxidative late-stage macrocyclization strategy for a scalable synthesis of antimitotic agent DZ-2384 (Scheme 24).228 The synthetic strategy used the dipeptide tert-Leu-5-F-Trp-OH as the precursor for the synthesis. After strategic modification over this dipeptide, advanced intermediate 24a was synthesized, which upon constant potential electrolysis realized an oxidative cyclization on the indole core to construct the DZ-2384 analogue in moderate yields. The oxidative cyclization involved SET oxidation of 24a, which then underwent a nucleophilic attack with the phenolic −OH functionality present in the molecule. Next a 5-exo-trig cyclization with the arene counterpart followed by aromatization generated the DZ-2384.
Scheme 24. Electrochemical Oxidative Macrocyclization for the Synthesis of DZ-2384.

2.2. eLSF of C(sp3)–H Bonds
2.2.1. Late-Stage Benzylic C(sp3)–H Functionalization
Benzylic C(sp3)–H bonds are ubiquitous in natural products and drug molecules. About 25% of the 200 best-selling drugs contain benzylic C–H bonds. The relatively low bond dissociation energy of benzylic C–H bonds enables high site-selectivity among various types of C–H bonds in structurally complex molecules.229 Therefore, benzylic C(sp3)–H functionalization has wide application foreground in the LSF field and has received a great deal of attention. In this context, significant progress was achieved for electrochemical benzylic C(sp3)–H functionalization.230 The reaction mechanism was proposed as following for most cases: Anodic oxidation of the hydrocarbon substrate gives an arene-centered radical cation, which undergoes rapid proton transfer and a second electron transfer oxidation to form a benzylic cation. Then, the intermediate was trapped by a nucleophilic reagent to afford the final product (Scheme 25). Meanwhile, hydrogen gas is generated at the cathode. Notably, the selection of a suitable solvent generally plays a key role in such transformation to modulate the oxidation potentials of the starting substrate and the product to avoid overoxidation.
Scheme 25. Proposed Mechanism for Electrochemical Benzylic C(sp3)–H Functionalization.

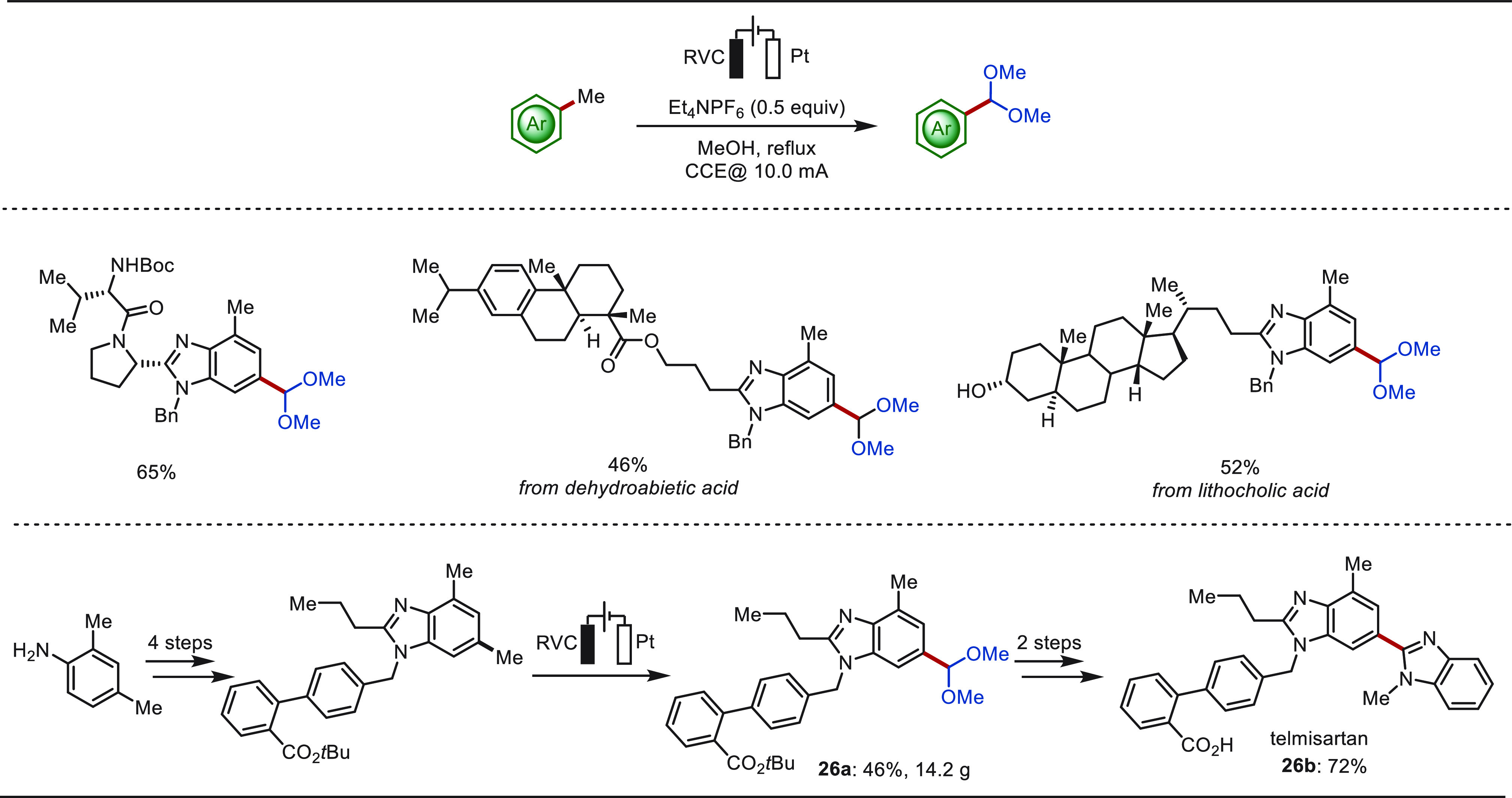
The formyl group is a synthetically versatile functional group that can be converted to a variety of functionalities. The oxygenation of methylarenes to benzaldehyde derivatives is of significant practical interest for LSF as the benzyl methyl motifs are widely present in drug molecules. However, control of chemo-selective oxidation with highly functionalized methylarenes remains a significant challenge due to the product overoxidation and selectivity issues for substrates featuring multiple oxidizable C–H bonds.231,232 Recently, the Xu group disclosed an electrochemical method that can site-selectively oxidize methyl benzoheterocycles to aromatic acetals in an undivided cell setup, without the utility of transition-metal catalysts and exogenous chemical oxidants (Scheme 26).233 The acetals could be easily hydrolyzed to the corresponding aldehydes in one-pot or in a separate step. This electro-oxidation approach was amenable to various functionalized benzoheterocycles and medicinally relevant molecules. The utility of this electro-oxidation reaction was further demonstrated by the efficient construction of the antihypertensive drug telmisartan 26b, in which the key dimethyl acetal intermediate 26a was obtained on a 14.2 g scale by site-selective electro-oxidation reaction.
Scheme 26. Electrochemical Late-Stage Oxidation of Various Methylarenes.
The oxidation of methylarenes is generally ineffective for electron-neutral and electron-deficient arenes since their higher redox potentials lead to poor selectivity or competitive solvent oxidation. A NHPI (N-hydroxypthalimide) mediated electrosynthetic method was developed by Stahl and co-workers to overcome these limitations (Scheme 27).234 In their studies, proton-coupled electrochemical oxidation of NHPI generated the PINO (phthalimide-N-oxyl) radical, which serves as a hydrogen-atom-transfer (HAT) mediator and as a metastable persistent radical to trap the in situ generated benzylic radicals. This PINOylation reaction operated at ∼0.5–1.5 V lower electrode potentials compared with the direct electrolysis methods, and hence enables the mediated electrolysis approach to tolerate a broad scope of methylarenes with diverse electronic properties and ancillary functional groups. The synthetic utility of this method was clearly reflected by facial conversion of the thus-obtained products into benzylic alcohols or aldehydes under photochemical conditions, both of which are compatible with LSF of the nonsteroidal anti-inflammatory pharmaceutical celecoxib.
Scheme 27. NHPI Mediated Late-Stage Benzylic Oxidation of Methylarenes.

In 2021, Xu and co-workers reported on a site-selective electrochemical benzylic C–H amination via the hydrogen evolution reaction (HER) without the need of exogenous oxidants or transition-metal catalysts (Scheme 28a).235 The practical utility of this electrochemical C–H amination reaction was illustrated by a gram-scale preparation with Celebrex as the aminating source, giving the corresponding C(sp3)–N coupling product in 78% yield. Meanwhile, the Ackermann group disclosed an effective method for electrochemical C–H aminations of 1,3-diarylpropenes via direct oxidative C(sp3)–H functionalizations with various substituted amides including the chiral auxiliary (−)-10,2-camphorsultam as well as the sulfonamide drugs Celebrex and Topiramate (Scheme 28b).236
Scheme 28. eLSF by Benzylic C–H Amination.

The Wang group also achieved a similar benzylic C–H amination reaction with diverse pyrazoles in a mixture of DCE and MeCN as solvents (Scheme 29).237 DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) was employed as a redox mediator to improve the electrolysis efficiency. The compatibility of this electrochemical strategy was demonstrated by the late-stage aminations of bioactive molecule substrates deriving from buluofen, abietic acid, epiandrosterone, and perillyl alcohol.
Scheme 29. DDQ-Mediated Late-Stage Benzylic C–H Azolation.

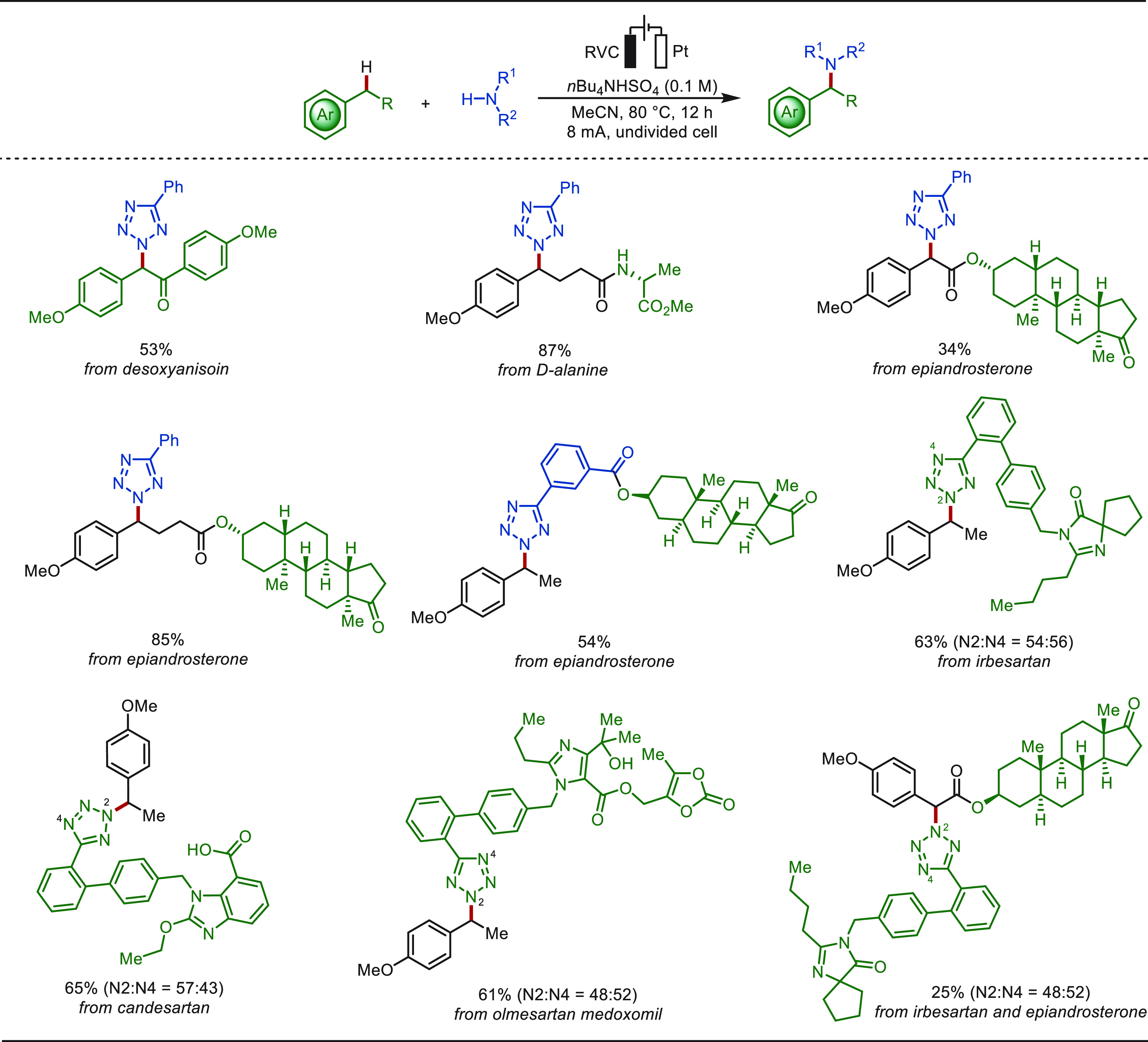
Later, Ruan and co-workers showed that azoles were suitable amination reagents for the electrochemical C–H/N–H cross-coupling reactions, with nBu4NHSO4 as the electrolyte and MeCN as the solvent in an undivided cell (Scheme 30).238 The azolation occurred efficiently and selectively at primary, secondary, and even challenging tertiary benzylic positions. This approach was directly exploited to install azole or benzyl motifs on a variety of structurally complex drug molecules.
Scheme 30. Electrochemical Late-Stage Benzylic C–H Azolation.
Direct C(sp3)–H isothiocyanations represent a straightforward strategy for the introduction of the versatile isothiocyanate functional group.239 Recently, Guo and Wen disclosed an electrochemical late stage benzylic C(sp3)–H isothiocyanation with TMSNCS (Scheme 31).240 A broad range of drug and bioactive molecules smoothly underwent the isothiocyanation under mild conditions with high chemo- and regio-selectivity. The chemoselectivity was attributed to the ready isomerization of in situ generated thiocyanates to isothiocyanates under the electrolysis conditions. In addition, with the electrochemical isothiocyanation strategy, two drug molecules—appetite suppressant 31a and herpesvirus inhibitor 31b—were prepared in a one-pot, two-step procedure from readily available alkylated arenes. In contrast, previous syntheses of these two compounds required four- and three-step processes, respectively.
Scheme 31. Electrochemical Late-Stage Benzylic C–H Isothiocyanation.

The straightforward and efficient introduction of fluorine is of great important in medicinal chemistry because of the unique properties of the C–F bond.241−245 In recent years, electrochemical fluorinations of C(sp3)–H bonds with nucleophilic fluoride sources have gained more attention.246−249 Recently, the Ackermann group developed a selective electrochemical C(sp3)–H fluorination with readily available NEt3·3HF, in lieu of alternative expensive electrophilic fluorine reagents (Scheme 32).249 External oxidants and transition-metal catalysts, as well as directing groups, were not required. The method displayed broad functional group tolerance, setting the stage for the late-stage fluorination of bioactive drugs. The practical utility was substantiated by fluorination of ibuprofen on a large scale of 2.5 g. Notably, adamantane was fluorinated at the tertiary position under otherwise identical electrolysis conditions, implying considerable potential for alkane modification. In addition, the synthetic utility of the C(sp3)–H fluorination could be further illustrated by a subsequent one-pot arylation of the generated benzylic fluorides.
Scheme 32. Electrochemical Late-Stage Benzylic C–H Fluorination.

Ketones are versatile functional groups and omnipresent in natural products and biologically active compounds.250 Recently, Liu and co-workers developed a sustainable protocol for direct benzylic C–H bond oxidation of alkylarenes to provide the corresponding ketone compounds with tert-butyl hydroperoxide as the radical- and oxygen-source (Scheme 33).251 The tert-butyl peroxyl radical was first generated by mild anodic oxidation. Then, the hydrogen atom transfer (HAT) occurred to form a benzylic radical, which reacts with tBuOOH, affording the corresponding ketone. This approach was successfully applied to the LSF of bioactive molecules, including celestolide, ibuprofen methyl ester, and papaverine, in synthetically useful yields without affecting other functional groups.
Scheme 33. Electrochemical Late-Stage Benzylic C–H Bonds Oxidation to Form Ketones.

2.2.2. Late-Stage Allylic C(sp3)–H Functionalization
Over the past decade, late-stage allylic C(sp3)–H functionalization has attracted substantial interest among organic chemists, benefiting from the fact that the carbon–carbon double bond is widely present in natural products and drug molecules. The allylic C(sp3)–H bond features relatively low bond dissociation energy, which enables high site selectivity in structurally complex molecules. In this context, electrochemical late-stage allylic C(sp3)–H functionalization has witnessed considerable recent progress.252−255
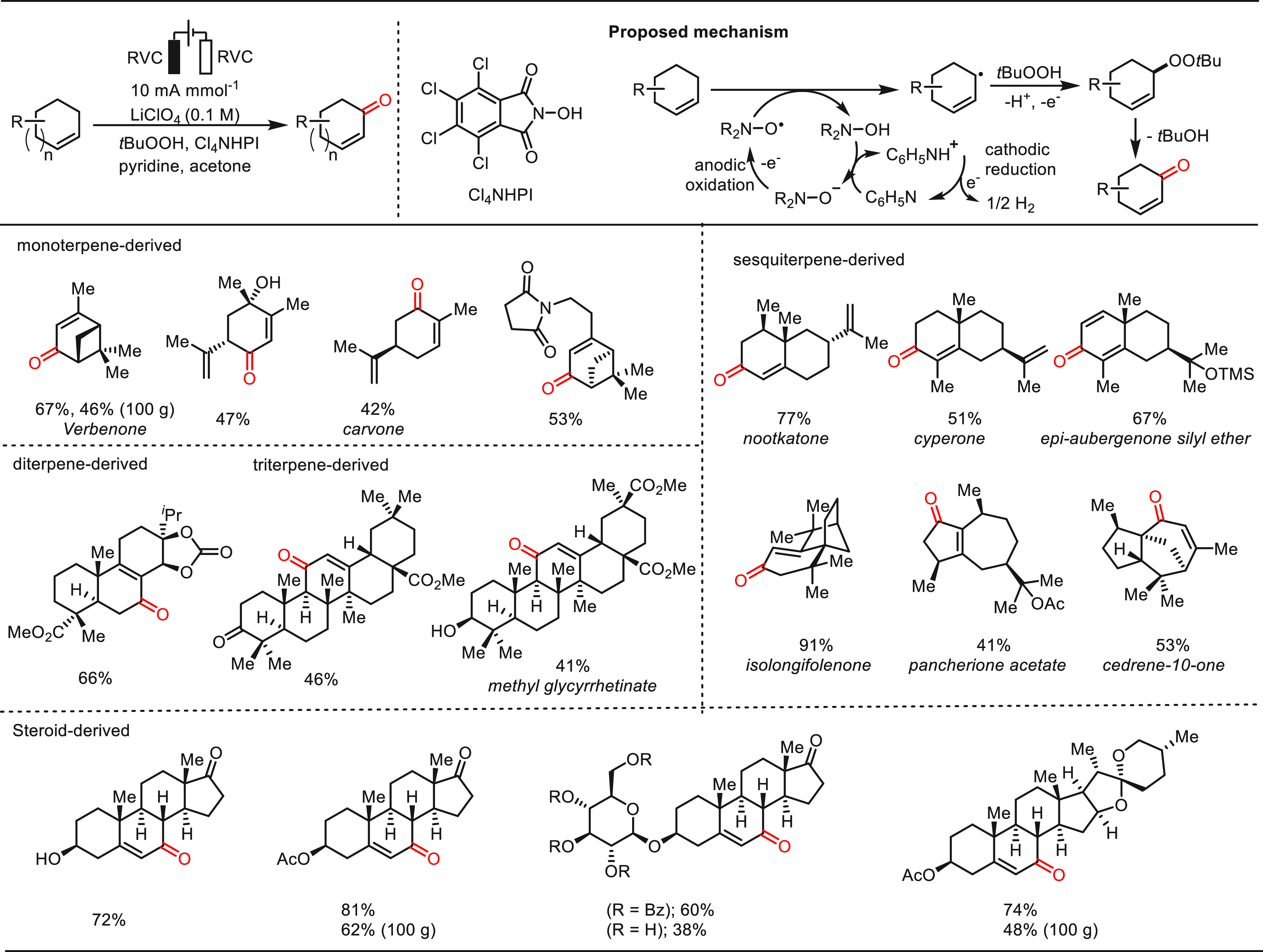
In 2016, Baran and co-workers described an elegant electrochemical allylic C(sp3)–H oxidation strategy using 20 mol % Cl4NHPI as a redox mediator, pyridine (2.0 equiv) as the base, tBuOOH (1.5 equiv) as a co-oxidant, and LiClO4 as the electrolyte (0.1 M) in acetone under constant-current conditions in an undivided cell (Scheme 34).255 This powerful electrochemical approach was characterized by a broad substrate scope, high chemoselectivity, and operational simplicity. A variety of representative terpenes were oxidized under the electrolysis conditions, affording corresponding versatile monoterpenes, sesquiterpenes, diterpenes, triterpenes, and steroids, which have outstanding utilities in food, fragrance, and pharmaceuticals industries. Notably, the user-friendly and robust nature of this electrochemical allylic C–H oxidation was demonstrated by 100 g preparation of several products with good efficiency.
Scheme 34. Electrochemical Late-Stage Allylic C(sp3)–H Oxidation.
Allylic amines are valuable building blocks in molecular synthesis and they are likewise prevalent in diverse biologically active molecules.256−258 In 2021, Wickens and co-workers developed a most user-friendly electrochemical strategy to prepare aliphatic allylic amines by the oxidative coupling of unactivated alkenes with secondary aliphatic amines (Scheme 35).259 This reaction proceeded via the electrochemical formation of a dicationic alkene-bis(thianthrene) adduct between thianthrene (TT) and the alkene substrate. Treatment of these adducts with aliphatic amines and base efficiently provides the corresponding linear, tertiary allylic amine products in high Z selectivity. Complex biologically active molecules are amenable to this transformation as both amine and alkene partners. Mechanistic studies revealed the vinylthianthrenium salts as the key reactive intermediates.
Scheme 35. Electrochemical Late-Stage Allylic C(sp3)–H Amination.

2.2.3. Late-Stage α-C(sp3)–H Functionalization of Carbonyls
The diversification of the C(sp3)–H bond adjacent to a carbonyl group is among the most basic transformations of utmost utility in molecular chemistry. Representative examples include the Claisen condensation, aldol reactions, or the Mannich reaction. In this context, α-C(sp3)–H functionalization of carbonyl compounds has been extensively explored.260−268 Particularly, significant recent momentum has been gained in eLSF and preparation of pharmaceutical derivatives.
In 2020, Li and Song reported a practical electro-oxidative dehydrogenative cross-coupling of ketones with xanthenes (Scheme 36).261 This transformation was performed under mild conditions, featuring a high atom economy and excellent functional-group tolerance. Drug molecules including dihydroprogesterone, progesterone, and canrenone proved to be compatible with the electrochemical C(sp3)–H/C(sp3)–H cross-coupling reactions, giving the corresponding products in excellent yields. Mechanistic studies indicated that a stabilized carbocation was first generated via anodic oxidation of xanthene. Then, the intermediate reacted with the nucleophilic enol to afford the cross-coupling product.
Scheme 36. Electrochemical Dehydrogenative Cross-Coupling of Ketones with Xanthenes (66).

Carbonyl desaturation to enone is a fundamental organic oxidation that was widely employed in organic synthesis.269 Established approaches to achieve this transformation generally rely on transition metals (Cu or Pd) or stoichiometric oxidative reagents.270−273 In 2021, Baran and co-workers disclosed an operationally simple electrochemical method to access such structures from enol phosphates or silanes, which can be readily formed from carbonyls (Scheme 37).262 This electrochemically driven desaturation (EDD) was characterized by a broad substrate scope including a variety of ketones and lactams. Notably, the late-stage site-selective desaturation of structurally complex molecules, which is difficult to achieve, afforded the desired enones in synthetically useful yields. In addition, the practical utility of the EDD was further illustrated by the desaturation of 4 g of cyclopentadecanone-derived silyl enol ether 37a to afford cyclopentadecenone 37b, which is easily converted to the valuable (R)-muscone 37c. Increasing the current from 10 to 300 mA and using alternating polarity enabled the EDD reaction to smoothly afforded compound 37b in a 66% isolated yield. By further increasing the current to 3.6 A, 100 g of 37a was successfully converted into 37b in a 61% yield in a flow apparatus that contained six reaction cells. Mechanistic studies suggested a radical-based manifold, involving two consecutive single-electron oxidations of enol silane to form oxonium, which released the desired enone after hydrolysis.
Scheme 37. Electrochemically Driven Late-Stage Desaturation of Carbonyl Compounds.

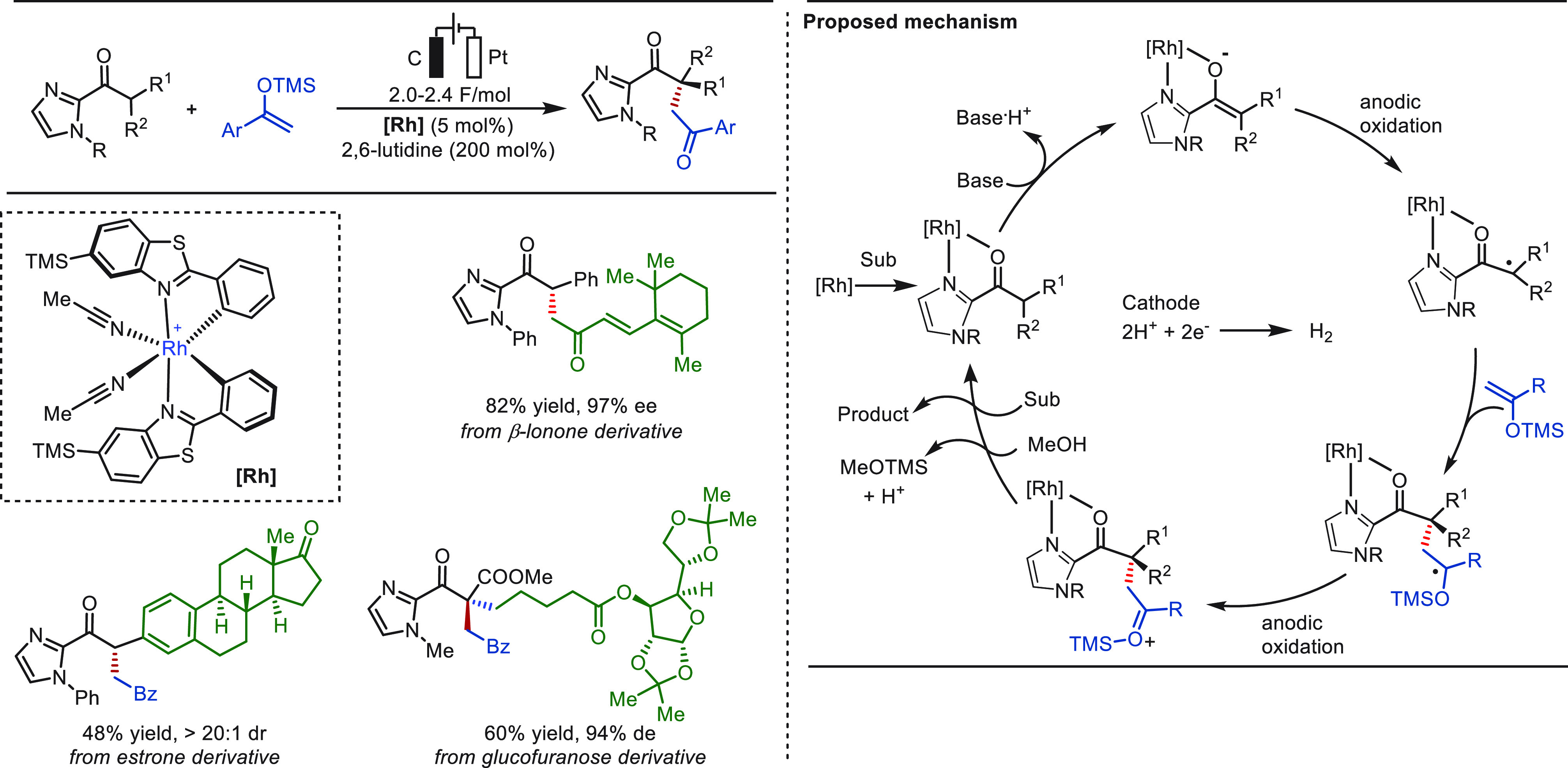
By the merger of organic electrosynthesis with asymmetric catalysis, the Meggers group introduced, in 2019, a versatile electricity-driven chiral Lewis acid catalyzed asymmetric coupling of 2-acyl imidazoles with silyl enol ethers to generate synthetically useful 1,4-dicarbonyls, which include products bearing all-carbon quaternary stereocenters (Scheme 38).263 The chiral-at-metal rhodium catalyst played a dual role in both the electrochemical step and to guarantee the asymmetric induction, enabling mild reaction conditions, a broad substrate scope, and high chemo- and enantioselectivities (up to >99% ee). The robustness of this approach is further demonstrated by the effective generation of complex products derived from β-ionone estrone and glucofuranose. Notably, the cleavage of the imidazolyl group could be achieved without a significant loss of optical purity.
Scheme 38. eLSF by α-C(sp3)–H Functionalization of Carbonyls.
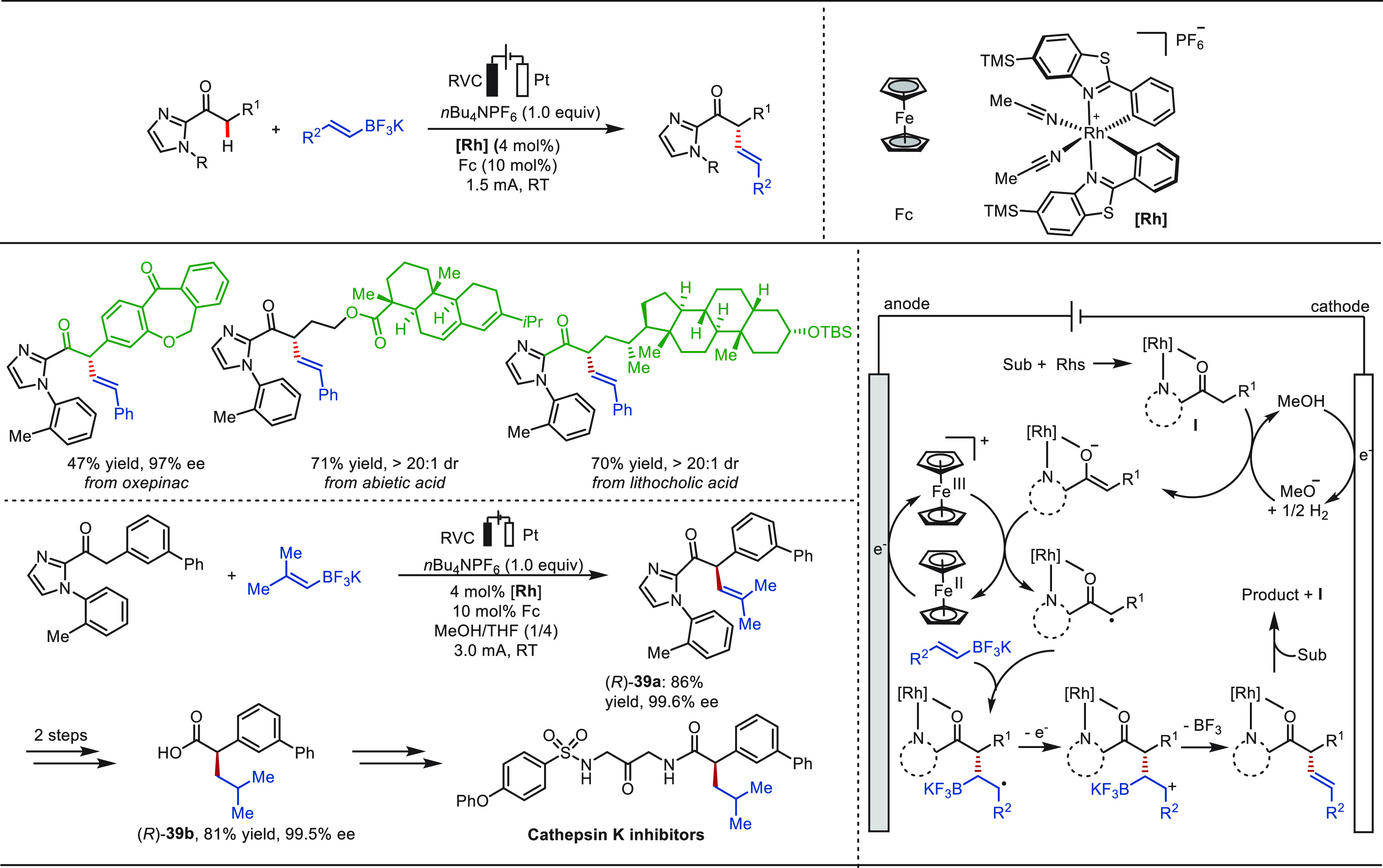
Recently, Meggers and co-workers reported another conceptually related approach to achieve enantioselective α-C(sp3)–H alkenylation of ketones with potassium alkenyl trifluoroborates (Scheme 39).264 The electrochemical asymmetric oxidative coupling reaction features a broad substrate scope, high yields (up to 94%), and exceptional enantioselectivities (≥99% ee). Catalytic amounts of ferrocene were used as the redox mediator, which enables the key chiral rhodium-involving single-electron transfer reaction to homogeneously occur in the solution rather than at the electrode surface, hence providing mild electrochemical conditions. The eLSF alkenylation of complex molecules derived from oxepinac, abietic acid, and lithocholic acid were accomplished in excellent yields via the electricity-driven asymmetric synthesis method. Moreover, this approach was applied to the straightforward assembly of intermediates (R)-39a of the cathepsin K inhibitor in 86% yield with an ee value up to 99.6%.
Scheme 39. Electrochemical Late-Stage α-C(sp3)–H Alkenylation of Carbonyls.
2.2.4. Late-Stage α-C(sp3)–H Functionalization of Amines
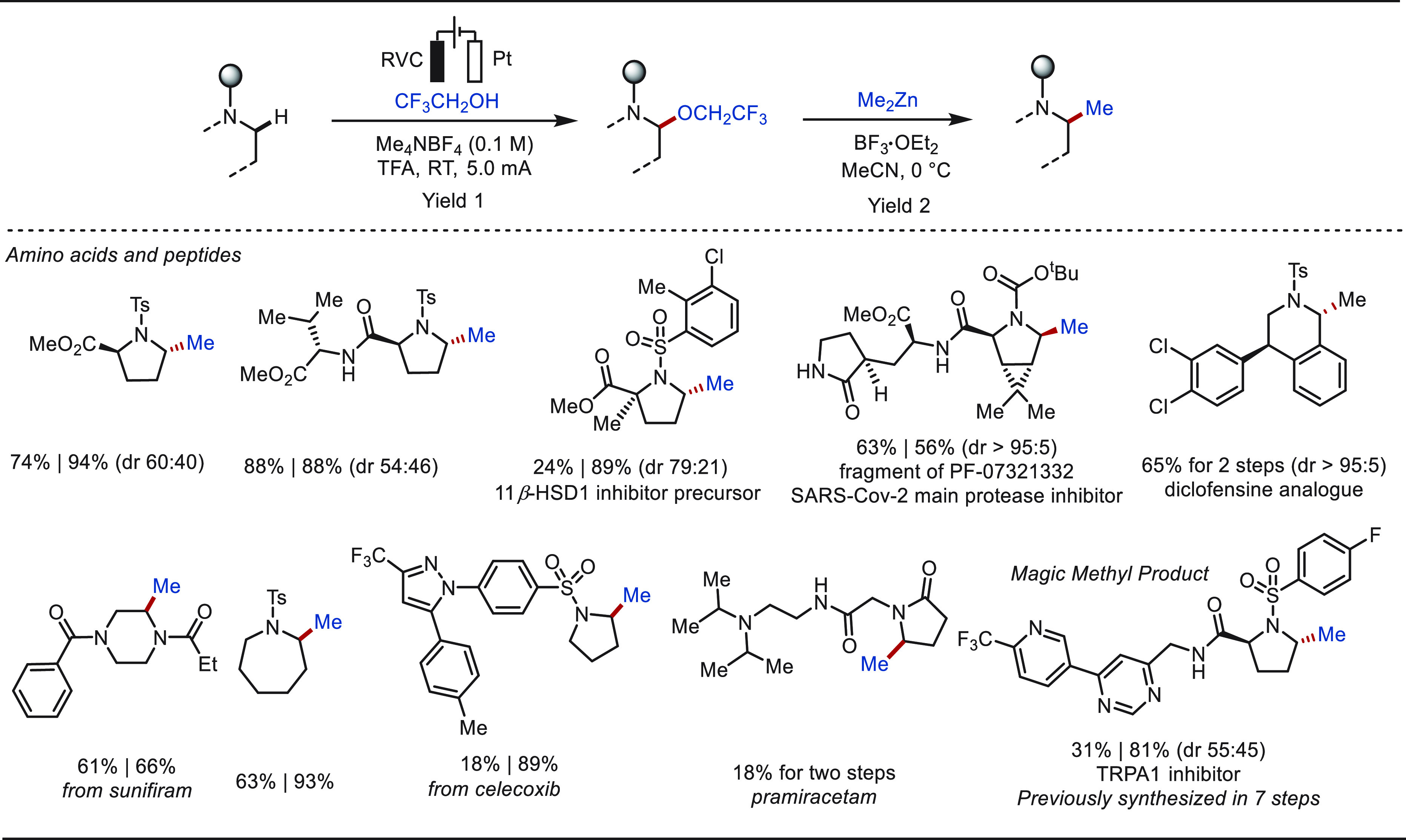
The electrochemical functionalization of C(sp3)–H bonds adjacent to nitrogen atoms, such as the well-esablished Shono oxidation, has been wildly applied in organic synthesis.274,275 However, until recently, it was primarily employed for the functionalization of structurally simple compounds. Based on the classic Shono oxidation reaction, Lin and Terrett recently reported a modular and practical strategy for eLSF α-methylation of structurally complex amines derivatives (Scheme 40).276 The electro-oxidation generated N,O-acetal readily reacted with organozinc reagents, enabling the facile installation of a methyl moiety as well as various other important groups. This improved electrochemical protocol features operational simplicity and high functional group compatibility. The site-selective late-stage methylation of a variety of bioactive targets, has been efficiently achieved. Notably, a drug molecule of TRPA1 inhibit, which has been explored for the “magic methyl” effect, presenting a >10-fold boost in potency, was previously synthesized using de novo routes in a total of 7 steps.277 In sharp contrast, this compound could be readily prepared from its parent inhibitor by this electron driven approach.
Scheme 40. Electrochemical Late-Stage α-Methylation of Amines.
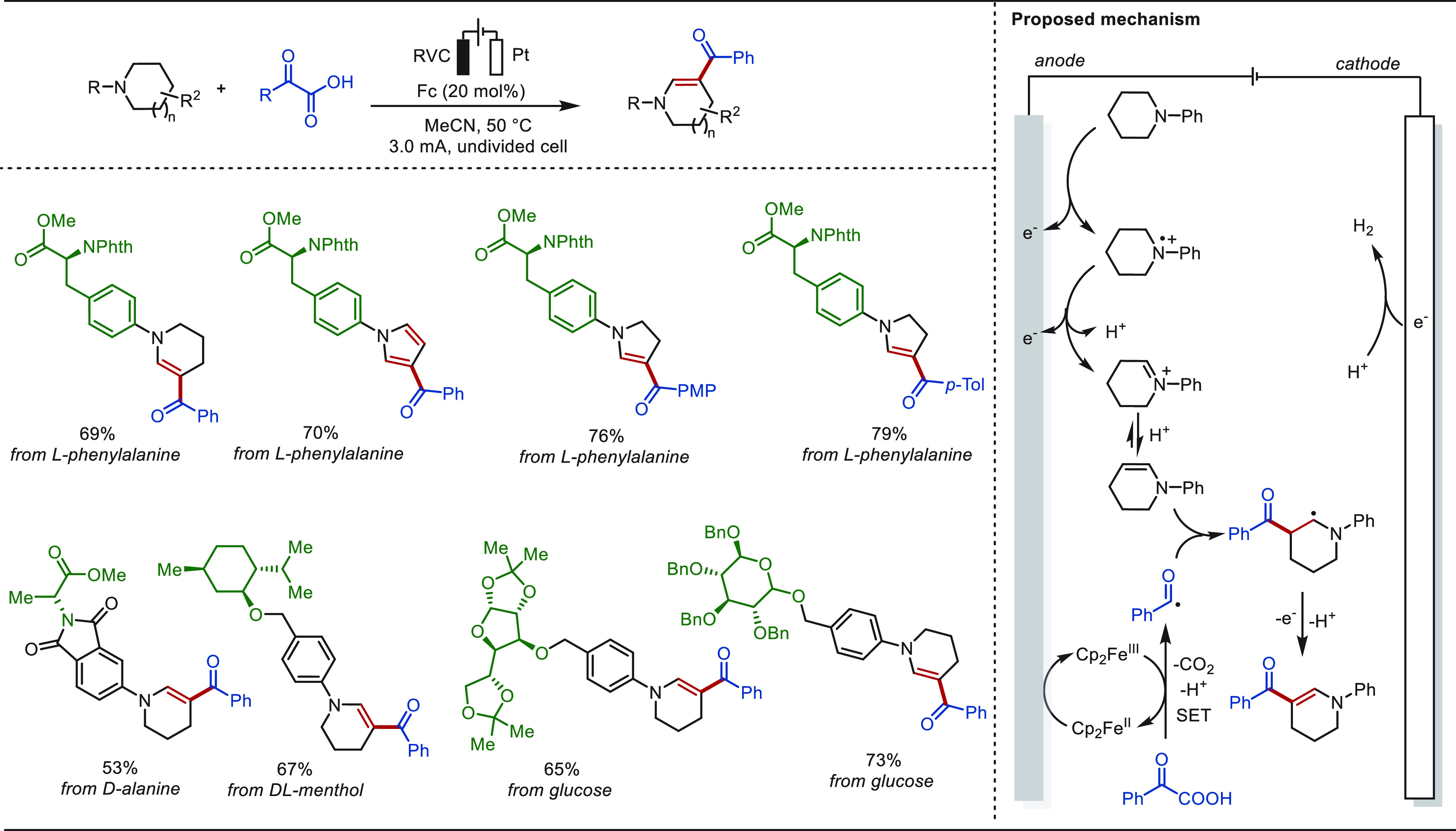
Electrochemical dehydrogenation based on the α-C(sp3)–H activation of amines represent an important organic transformation to ubiquitous unsaturated compounds.278−283 In 2018, the Lei group disclosed a TEMPO-mediated dehydrogenation of N-heterocycles in an undivided cell to access a variety of five- and six-membered nitrogen-heteroarenes without the usage of sacrificial hydrogen acceptors.282 Recently, Qiu and co-workers reported a straightforward and robust approach of electrochemically driven desaturative β-C(sp3)–H functionalization of cyclic amines (Scheme 41).284 Various β-substituted desaturated cyclic amines were obtained under constant current electrolysis in MeCN at 50 °C. This transformation was achieved via multiple single-electron oxidation processes with catalytic amounts of ferrocene as a redox mediator. The unique utility of this approach was clearly demonstrated by the eLSF of natural products and derivatives (Scheme 41). Diverse pyrrolidine- or piperidine-containing molecules deriving from l-phenylalanine, d-alanine, d,l-menthol, glucofuranose, and glucopyranose afforded the corresponding desaturated acylation products in excellent yields. Notably, the reaction of l-phenylalanine bearing a pyrrolidine motif with phenylacetic acid formed a pyrrole product through further electro-oxidation.
Scheme 41. Electrochemically Driven Desaturative β-C(sp3)–H Functionalization of Amines.
2.2.5. Late-Stage C(sp3)–H Functionalization of Sulfides
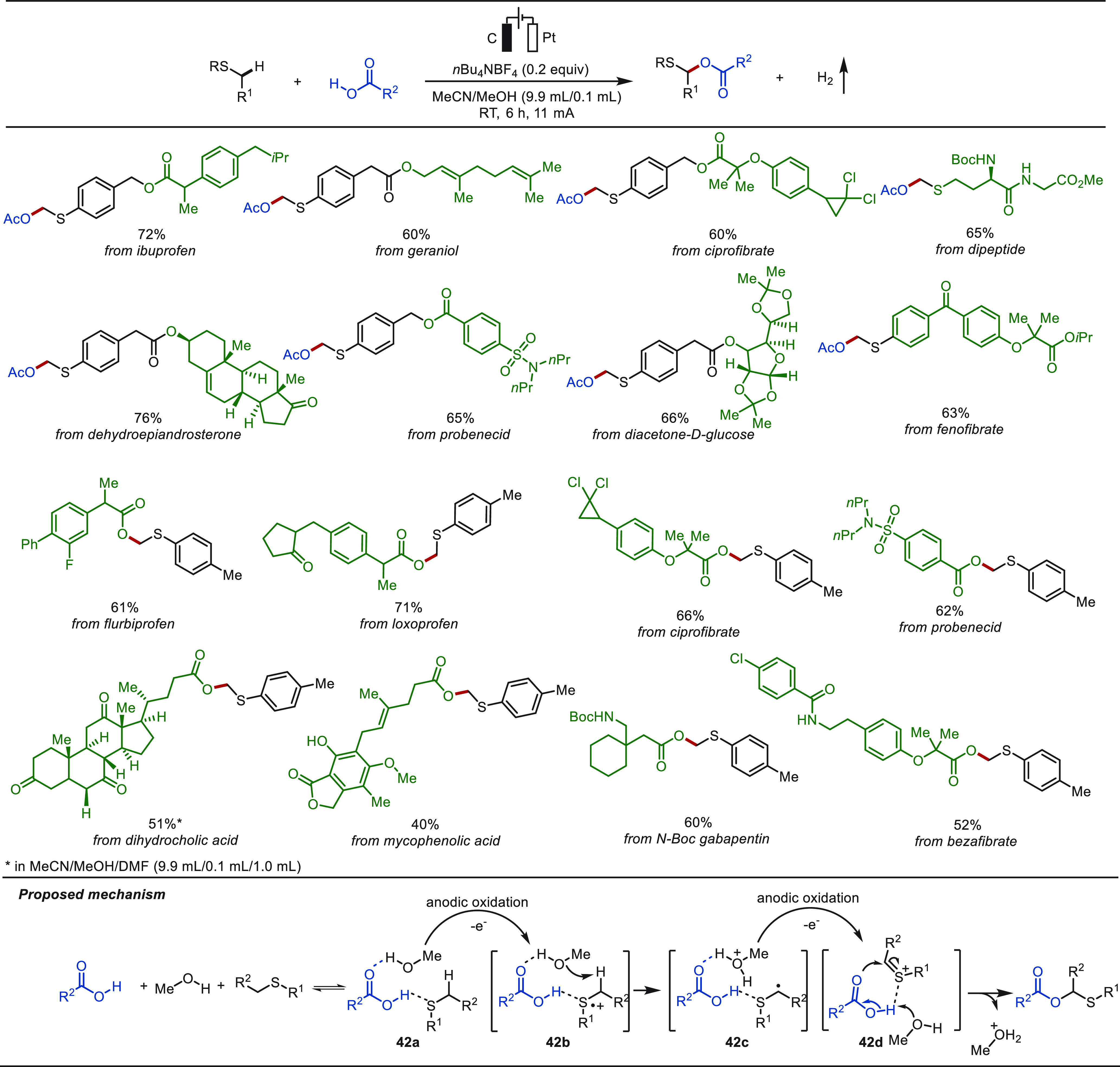
The precise and selective activation of oxidation-sensitive sulfur-containing compounds is a significant challenge due to its inherent activity and complicated valence states.285 In 2021, Lei and co-workers reported on an electrochemical protocol for the construction of α-acyloxy sulfides, which represent key structural motifs in agrochemicals and pharmaceuticals (Scheme 42).286 This electro-oxidized C(sp3)–H/O–H cross-coupling protocol was found to be environmentally friendly, highly selective, and scalable while featuring an exceptionally broad substrate scope. The robustness and utility of this protocol was demonstrated by the efficient eLSF of a wealth of bioactive molecules, including amino acids, peptides, and pharmaceuticals. Mechanistic studies suggested a synergistic effect of the self-assembly induced C(sp3)–H/O–H coupling pathway. Sulfide, AcOH, and MeOH assemble into an adduct, and hydrogen bonding between AcOH and the sulfur atom can facilitate a SET oxidation of sulfide. MeOH selectively captures the proton to form the state 42c with high regioselectivity. Then, a thionium ion is generated via the loss of a proton and an electron, and the desired product is finally delivered after the nucleophilic attack of AcOH to the thionium ion.
Scheme 42. Electrochemical Late-Stage α-C(sp3)–H Acyloxylation of Sulfides.
2.2.6. Late-Stage Unactivated C(sp3)–H Functionalization
Unactivated C(sp3)–H bonds generally feature a high redox potential of more than 3.0 V vs SCE.287 Thus, the direct electrolysis of the C(sp3)–H bond represents a formidable challenge, since oxidation of other functionalities or solvents is likely to occur prior to the desired C–H oxidation of simple alkanes. Despite the difficulties, scientists have made remarkable progress in electrochemical late-stage functionalization of unactivated C(sp3)–H bond by the use of redox mediators or transition-metal catalysts.288,289
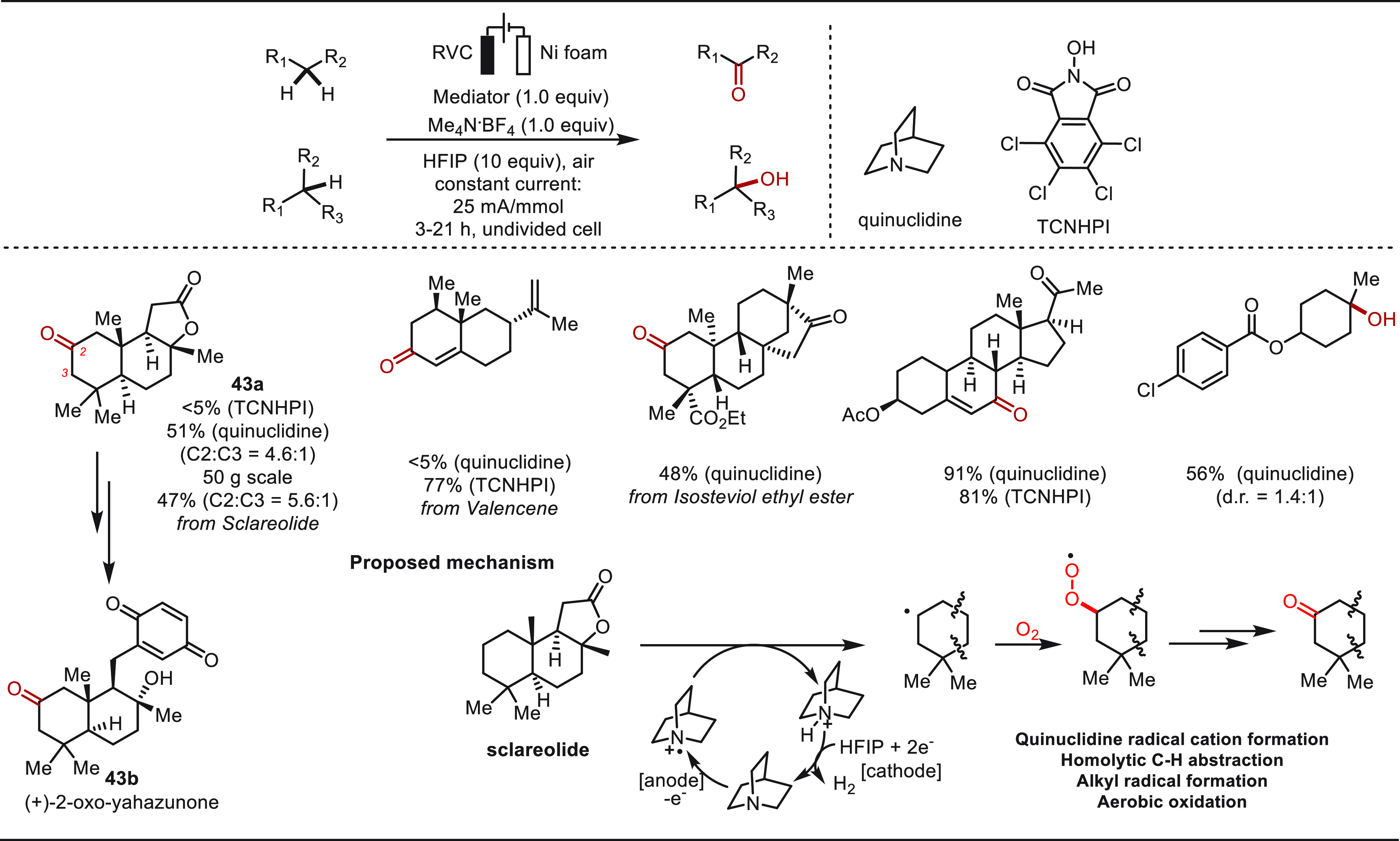
In 2017, the Baran group presented a practical electrochemical oxidation of otherwise unactivated C–H bonds in MeCN with Me4NBF4 as the electrolyte and HFIP as the additive (Scheme 43).288 Identification of a suitable redox mediator was the key to success for high yields and chemoselectivities. While using quinuclidine as a mediator allowed the selective late-stage oxidation of Sclareolide in a 51% yield at ca. 1.8 V vs SCE Ag/AgCl, the use of TCNHPI as mediator is superior on those bearing multiple olefin motifs, such as valencene. The quinuclidine-based redox mediator system further proved compatible for the eLSF of isosteviol ethyl ester and oxidation of a terpene to a relative steroid. In addition, a tertiary C–H bond is efficiently oxidized to the corresponding alcohol under the quinuclidine-mediated electrolysis. The utility of this protocol was illustrated with a 50 g scale late-stage oxidation of sclareolide to 43a, which is a key intermediate for the synthesis of (+)-2-oxo-yahazunone. Mechanistically, the in situ anodic oxidation generated quinuclidine radical cation served as a HAT reagent and O2 was involved in the later aerobic oxidation step.
Scheme 43. Electrochemical Late-Stage Oxidation of Unactivated C(sp3)–H Bonds.
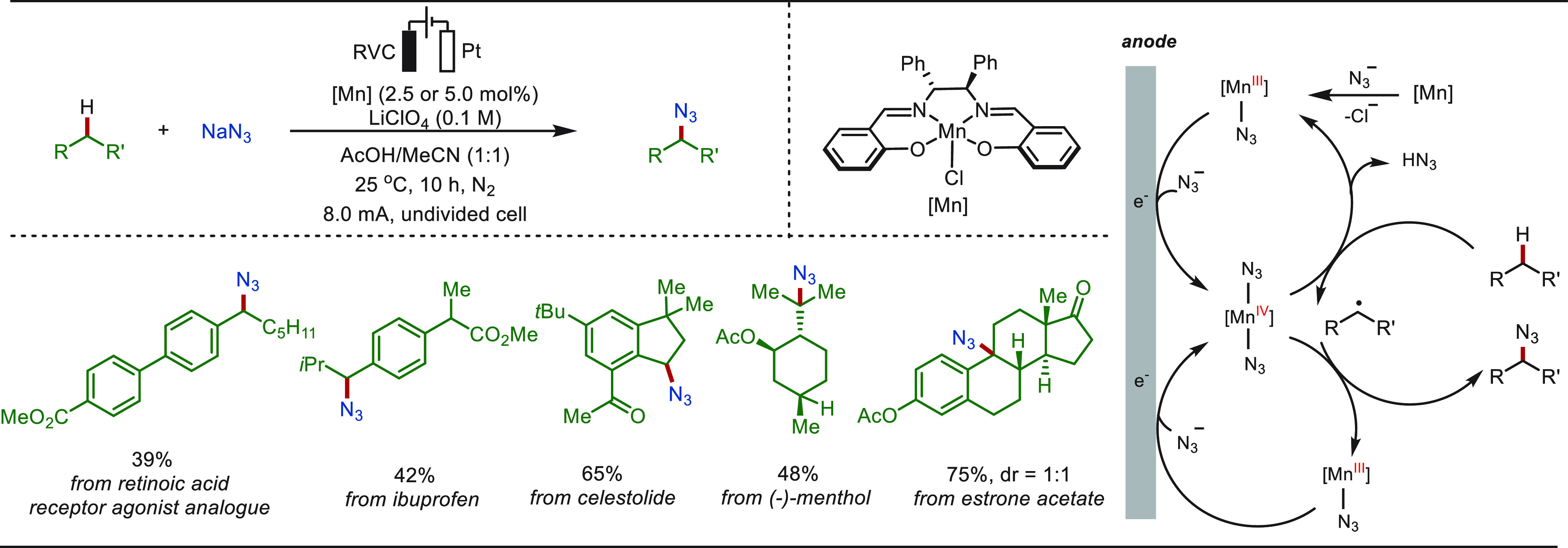
Organic azides are key intermediates for numerous transformations in medicinal chemistry, peptide chemistry, or molecular biology.290−292 Traditionally, stoichiometric amounts of strong indiscriminate chemical oxidants, such as NFSI and hypervalent iodine reagents, are required to install the azido group into C(sp3)–H bonds. By contrast, in 2021, the Ackermann group disclosed a manganaelectro-catalyzed C–H azidation of otherwise unactivated C(sp3)–H bonds with most user-friendly NaN3 as the nitrogen-source and traceless electrons as the sole redox-reagent (Scheme 44).289 The robustness and practicability of the resource-economic method was highlighted by the eLSF azidation of a variety of bioactive molecules. Detailed mechanistic studies supported a unique manganese(III/IV) regime, avoiding overoxidation to the carbocation and thus suppressing undesired side-reactions to oxygenated products.
Scheme 44. Electrochemical Late-Stage C(sp3)–H Azidation.
The merger of electrochemistry with organometallic catalysis has shown significant advances in C(sp3)–H activation. For example, Mei and Sanford, respectively, have achieved unactivated C(sp3)–H bond oxygenation by palladium catalysis under electrochemical conditions.293,294 This strategy is expected to be used in the late-stage modification of bioactive molecules in the future.
2.3. eLSF of C(sp)–H Bonds
Oxidative carbonylation of alkynes represents an important transformation in molecular synthesis that generally uses O2 as the oxidant. However, the explosibility of gas mixtures of CO/O2 (12.5–74.0%) deters scalable application of this process. In 2019, the Lei group disclosed an electro-oxidative palladium-catalyzed carbonylation of alkynes to 2-ynamides under copper- and O2-free conditions (Scheme 45). This transformation occurred under potentiostatic conditions, and the role of the current was to oxidize Pd(0) to Pd(II), which was also the rate-determining step of this process. The eLSF of propyzamide with CO (1 atm) and NH4NO3 furnished corresponding propiolamide in a 63% yield. Primary and secondary amines proved to be amenable for this electrochemical aminocarbonylation reaction. Drug molecules, including desloratadine, fluoxetine, and desbenzyl donepezil, smoothly underwent eLSF, affording desired 2-ynamides in satisfactory yields.
Scheme 45. Electrochemical Palladium-Catalyzed Aminocarbonylation of Terminal Alkynes.

In 2021, by employing arylhydrazines instead of amines, Lei and co-workers further accomplished the electrochemical palladium-catalyzed oxidative carbonylation of alkynes to synthesize ynones, which is an alternative supplement of the carbonylative Sonogashira–Hagihara reaction (Scheme 46).295 The LSF of bioactive molecules deriving from propyzamide, estrone, naproxen, ibuprofen, and levulinic acid afforded the corresponding ynones in excellent yields. Similarly, the process occurs via a proposed palladium(0)/palladium(II) regime, and the use of current as oxidant avoids the explosion hazard of CO.
Scheme 46. Electrochemical Palladium-Catalyzed Oxidative Sonogashira–Hagihara Carbonylation of Arylhydrazines and Alkynes.

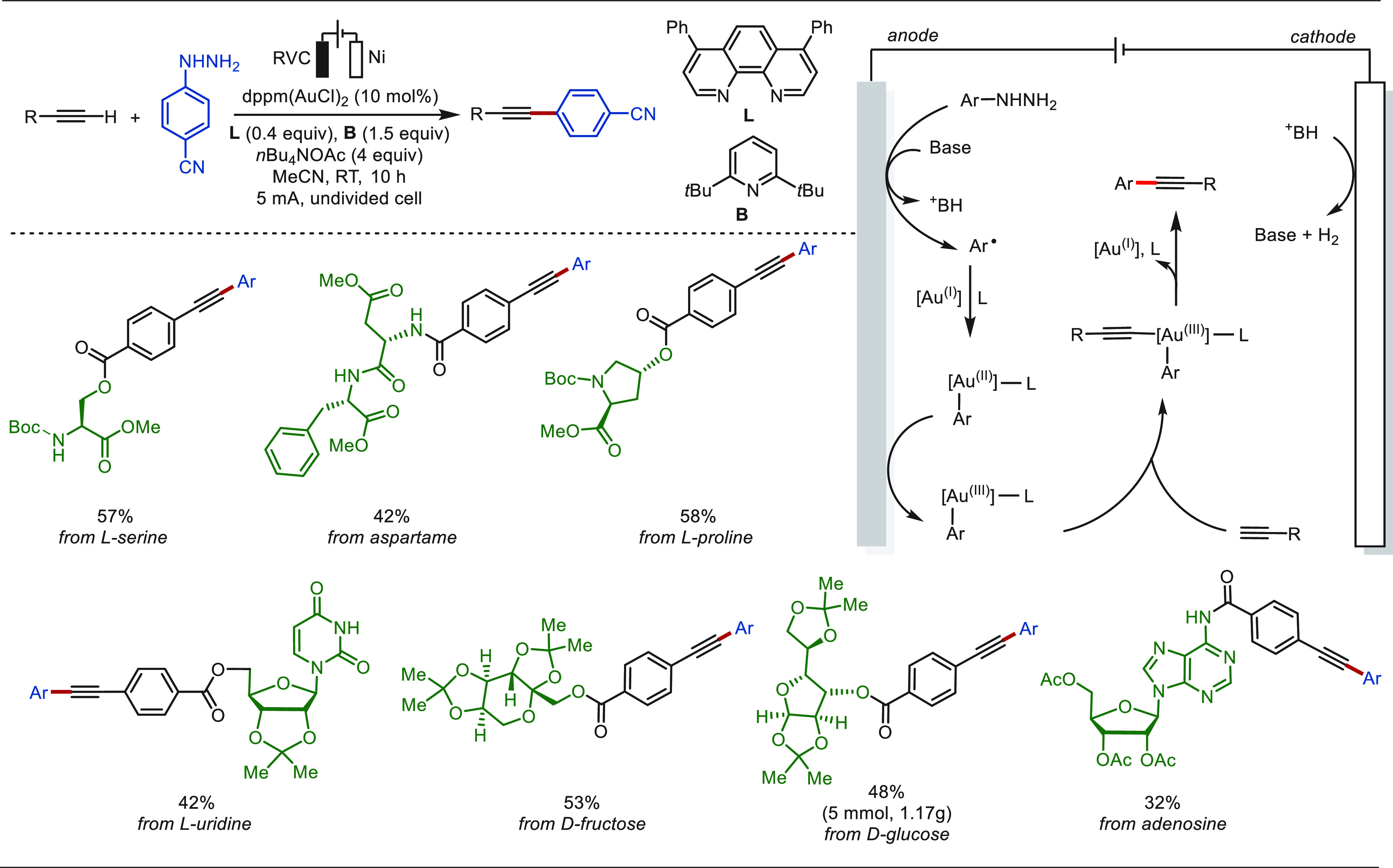
Recently, Xie and co-workers disclosed a electrochemical gold-catalyzed C(sp)–C(sp2) coupling reaction between structurally complex alkynes and arylhydrazines (Scheme 47).296 This approach exhibited broad functional group tolerance without the use of chemical oxidants. The robustness of this approach was further illustrated by the efficient late-stage modification of a variety of alkynes tethered to biomolecules. Mechanistic studies suggested the anodic oxidation of aromatic hydrazine to generate an aryl radical, which recombined with gold(I) and underwent further anodic oxidation to form the Ar–Au(III) species for subsequent σ-activation of alkynes.
Scheme 47. Electrochemical Gold-Catalyzed Oxidative C(sp)–C(sp2) Coupling.
3. eLSF of Functional Groups
Electrocatalytic interconversion of common organic functionalities bears unique potential for the advancement of organic synthesis. Interestingly, owing to their robust and mild conditions, these approaches are often adopted for the late-stage derivatization of complex organic molecules. In the following section, we will discuss the progress in the area of electrochemical late-stage functional group modification strategies.
3.1. eLSF of Alkenes and Alkynes
Olefins are prevalent structural motifs in various biologically relevant molecules and natural products.297,298 These moieties are quite reactive and are often garnered for incorporating new functional groups in the molecule.299−306 Synthetic manipulation of these substructures is thus used as a versatile strategy for late-stage functionalization reactions.
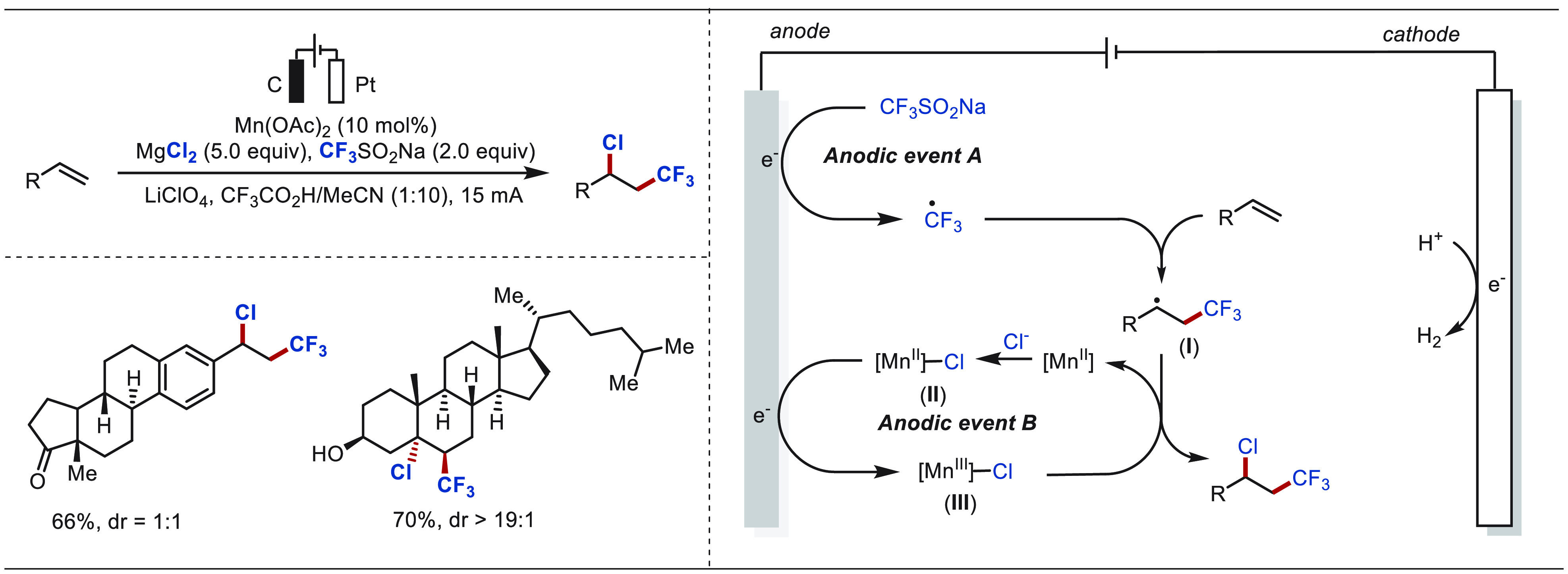
The Lin group has done great contributions in the field of metallaelectro-catalyzed functionalization of alkenes.301,302,304−309 In 2018, Lin described an electro-oxidative heterodifunctionalization of olefins enabled by anodic oxidation of CF3SO2Na (Scheme 48).307 The interception of the anodically generated trifluoromethyl radical with a terminal olefin formed a secondary alkyl radical intermediate, which was trapped with a chloride radical to form the heterodifunctionalized product. The use of catalytic Mn(OAc)2 assisted the electrochemical process through the formation of an alleged Mn(III)-Cl radical chlorinating agent, which helped the chloride radical recombination step. This anodically coupled electrocatalytic process was exploited for the late-stage functionalization of several natural product analogues.
Scheme 48. Electro-oxidative Chlorotrifluoromethylation of Olefins.
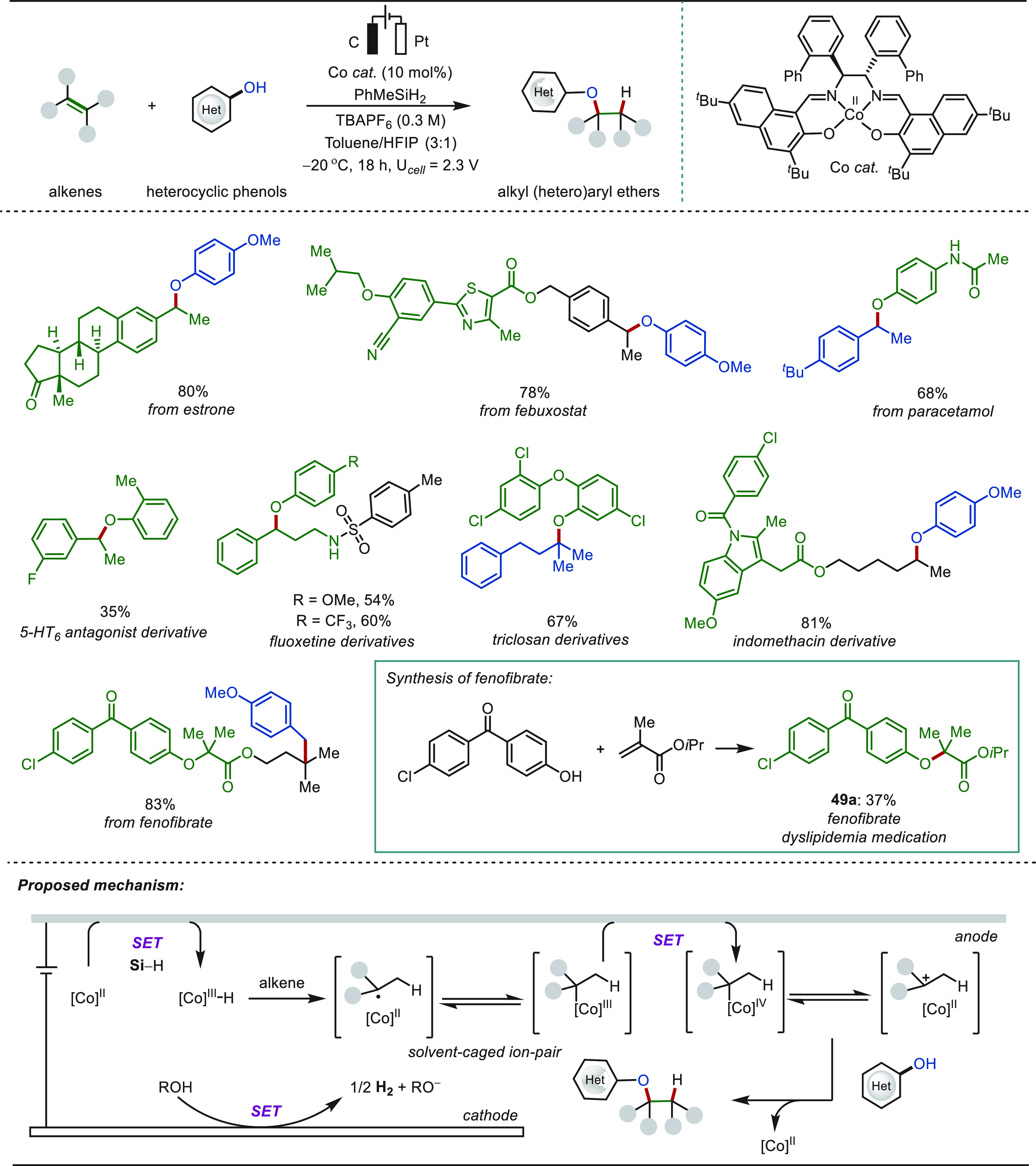
Later, a cobalt-salen-catalyzed hydroetherification strategy was demonstrated by Kim and Shin combining MHAT and anodic oxidation (Scheme 49).310 Generally, in MHAT strategies, weak nucleophiles exhibit poor reactivity owing to the formation of a “solvent-caged radical pair”, which deflates the nucleophilic entrapment process. Anodic oxidation of the caged intermediate detoured the detrimental bimetallic disproportionation pathway and enabled the nucleophilic displacement process. The electrocatalysis involved a plausible cobalt(II/III/IV) pathway for product formation. This versatile strategy was employed for the late-stage hydroetherification of estrone, febuxostat, paracetamol, fluoxetine, triclosan, indomethacin derivatives including many other important organic molecules. The versatility of the hydroetherification strategy was also highlighted through the synthesis of fenofibrate 49a, an oral medication for dyslipidemia.
Scheme 49. Electrochemical Cobalt-Catalyzed Late-Stage Hydroetherification of Olefins.
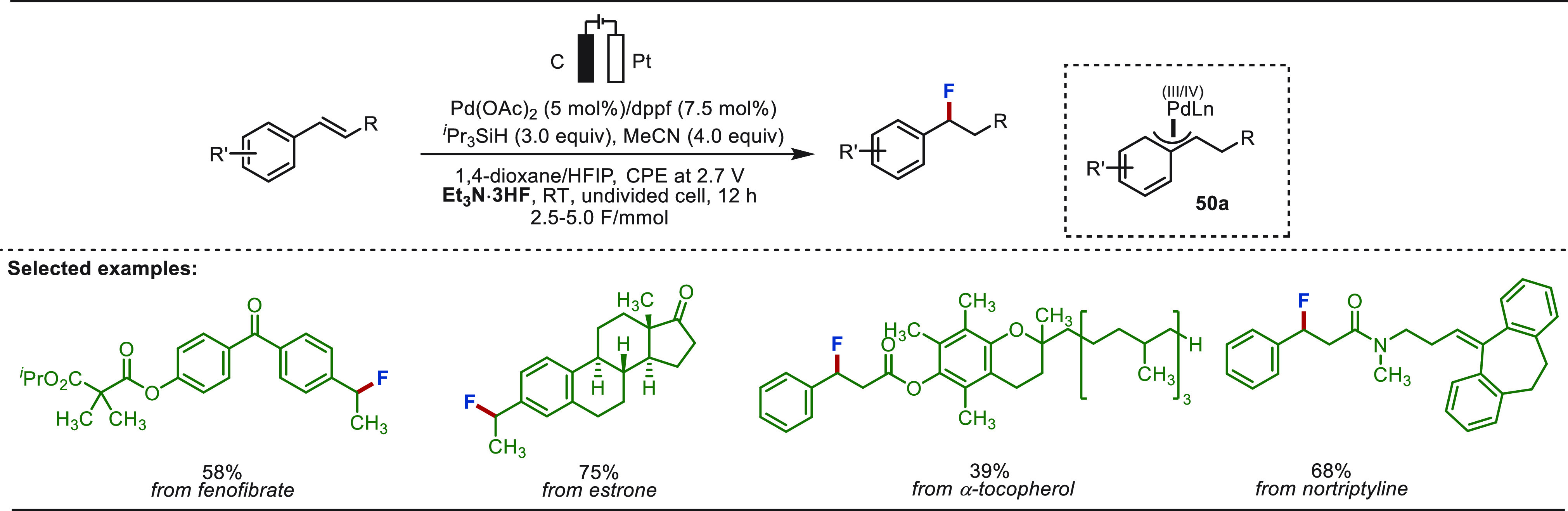
They further illustrated an electro-oxidative palladium-catalyzed approach very recently to realize benzylic fluorinations in a straightforward manner using Et3N·3HF as a nucleophilic fluorinating agent (Scheme 50).311 Similar to the prior findings, this strategy operated through a metal hydride intermediate, which after migratory insertion with the olefin formed a high-valent η3-benzylpalladium intermediate. This intermediate under electro-oxidative conditions guided a nucleophilic displacement reaction with the nucleophilic fluorinating agent. This hydrofluorination strategy employed the dppf ligand and silane as the hydride source. The formation of intermediate 50a was confirmed by cyclic voltammetry studies. This approach was employed for the selective benzylic fluorination of biologically relevant nortriptyline, fenofibrate, estrone, and α-tocopherol derivatives.
Scheme 50. Electrochemical Palladium-Catalyzed Late-Stage Hydrofluorination of Olefins.
In 2018, Ackermann reported the versatile electro-oxidative olefination/annulation approach under rhodium and iridium catalysis, respectively (Scheme 51).312,313 These chemo- and site-selective strategies harvested electricity as the renewable terminal oxidant, converting easily accessible aromatic carboxylic acids to synthetically meaningful phthalides. Various acrylate analogues embracing naturally occurring complex terpenoids and amino acids were compatible to the reaction conditions generating respective phthalides in high yields. While the rhoda-electrocatalyzed approach relied on direct anodic oxidation to regenerate the rhodium(III)-catalyst, catalytic amounts of benzoquinone redox-mediator were necessary to promote an iridium-catalyzed transformation.
Scheme 51. Electro-oxidative Late-Stage Annulation of Biologically Relevant Olefins.

The controlled isomerization of readily available terminal alkenes or reduction of alkynes is an effective and practical strategy to access internal olefins.314−319 Recently, Baran and co-workers established an electroreductive Co-catalyzed regioselective olefin isomerization strategy harnessing transition-metal hydride intermediates (Scheme 52).320 The cathodic reduction of high-valent Co(III)-species formed low-valent Co(I)-species, which can effectively reduce protons to form a Co(III)–H intermediate. This cobalt hydride intermediate when reacted with terminal olefins and alkynes, it selectively transformed them into corresponding internal olefins and Z-olefins, respectively. This simple and straightforward method proved to be applicable for the modification of a variety of substrates including the late-stage derivatization of structurally complex organic architectures.
Scheme 52. Electroreductive Cobalt-Catalyzed Late-Stage Functionalization of Olefins and Alkynes.

The electrochemical functionalization of alkenes under transition-metal-free conditions has also been extensively studied. In 2019, Fang and Hu reported a scalable difunctionalization of olefins harnessing anodic oxidation, in which the reaction presumably proceeded through a nucleophilic addition of dimethylformamide to the benzylic carbocation, formed after anodic oxidation of a benzylic radical (Scheme 53).321 This approach allowed for the bromination, chlorination, and trifluoromethylation-formyloxyation of naturally occurring steroids using bench-stable NaBr, NaCl, and NaSO2CF3 as corresponding radical sources.
Scheme 53. Late-Stage Difunctionalization of Steroid Derivatives.

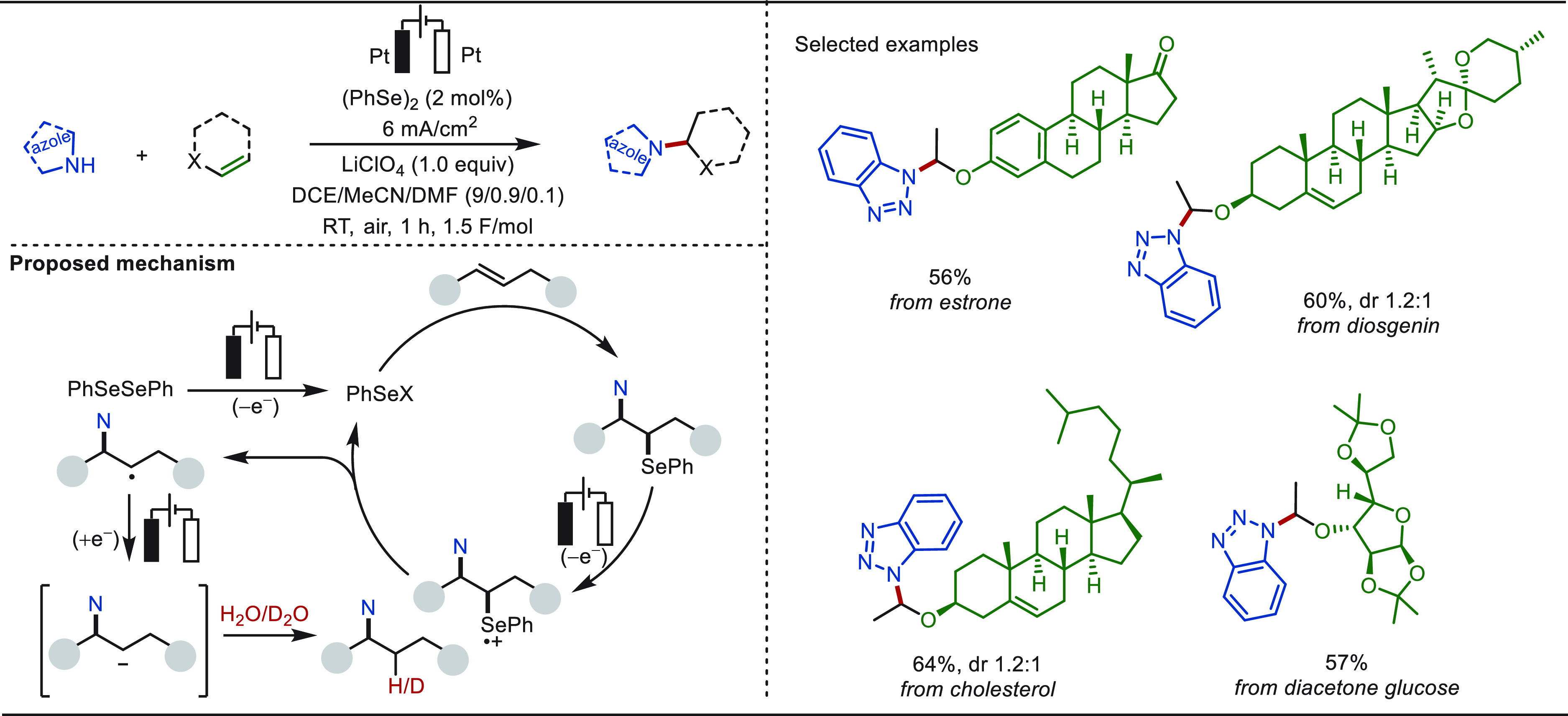
Recently, Xu and Zeng demonstrated a versatile electroseleno-catalytic hydroazolylation of olefins in the absence of external oxidants (Scheme 54).322,323 Electrochemical conditions acetivated the diselenide catalyst to PhSe+ or PhSe·, which triggered an electrophilic activation of the olefin followed by a nucleophilic addition with the azole substrate. The difunctionalized product then realized an anodic oxidation induced deselenylation generating the hydroaminated product. Deuterium labeling studies revealed the significance of the cathode in this transformation, which assisted in the formation of a carbanion. The role of the cathode was further concluded by executing the reaction in a divided cell, in which the substrate at the cathodic chamber was consumed and no product formation was observed in the anodic chamber. This electroseleno-catalyzed approach enabled the diversification of estrone, cholesterol, diosgenin, and diacetone glucose analogues with good yields.
Scheme 54. Electrochemical Selenium-Catalyzed Late-Stage Hydroazolation of Olefins.
In 2021, Han reported a (4 + 2)-annulation strategy for the construction of benzo[c]-[1,2]oxazines in good yields (Scheme 55).324 Anodic oxidation of hydroxamic acid produced an amidoxyl radical intermediate. This intermediate reacted with the olefin substrate, constructing the oxazine core structures in decent yields. This mild, external oxidant-free approach was used to execute late-stage functionalization of tryptophol, tryptamine, tryptophan and its analogous peptides, and various steroid derivatives in high efficacy.
Scheme 55. Electrochemical Late-Stage [4 + 2] Annulation of Olefins with Hydroxamic Acid.

Anodic oxidation-based electrochemical functionalization of olefins was also transcended for a late-stage labeling of biologically relevant olefins (Scheme 56).325 An oxidizable phenol derivative was used as the cross-linker, which upon anodic oxidation formed a phenoxionium cation. This phenoxionium intermediate underwent a facile [3 + 2] addition with the olefin to form a fluorescent active dihydrobenzofuran moiety. The electrochemical labeling approach was able to cross-link citronellol, citroneic acid, and amino acid derivatives with high efficacy.
Scheme 56. Late-Stage Electrochemical Labelling of Biologically Relevant Olefins.

Late-stage olefin functionalizations are not limited to electro-oxidative approaches. Indeed, electroreductive functionalizations of olefins are gaining significant momentum.326 In this context, Cheng reported selective electroreductive deuteration of α,β-unstaurated carbonyl compounds using D2O as the deuterium source (Scheme 57). This electroreductive approach used graphite-felt as the cathode as well as the anode and operated in the absence of an external catalyst, and thus obviated the need of stoichiometric metallic reductants unlike prior examples. An oxygen evolution was observed at the anode, confirmed using isotopically labeled water (H218O), which regulated the need of an additional reductant along with maintaining the pH of the medium during the process. This simple and versatile deuteration method exhibited tremendous potential enabling the late-stage deuteration of a large variety of biologically relevant molecules and pharmaceuticals.
Scheme 57. Electroreductive Late-Stage Hydrogenation of Olefins with D2O.

In 2021, Pan disclosed a straightforward electroreductive defluorinative functionalization of trifluoromethylated styrenes (Scheme 58).327 Notably, straightforward synthetic routes to C–C bonds harvesting sp3-hybridized carbon-centered radicals have always been considered as versatile approaches in organic synthesis. The authors used easily accessible Katritzky salt as a useful source for the generation of C(sp3)-centered radicals. This reductive deaminative approach required a sacrificial zinc anode and obviated the need for external electrolytes. Single electron reduction of Katrizky salts generated alkyl radicals, which were intercepted by the olefin forming benzylic radical. This benzylic radical under electroreductive conditions underwent SET reduction, which facilitated the defluorination of the CF3 unit, generating 1,1-difluoro substituted olefins. Interestingly the method was also operative under flow-electrochemical conditions. The synthetic utility of the method was reflected by the late-stage modification of alogliptin, isopexac, estrone, indomethacin, and fenbufen analogues.
Scheme 58. Electroreductive Late-Stage Defluorinative Alkylation of Trifluoromethylated Styrenes.

Recently, Cheng has developed an electroreductive cyclopropanation of olefins, where deuterated chloroform was used as the C1-synthon (Scheme 59).328 This electrochemical method employed a sacrificial Zn-anode for the SET reduction of CDCl3 forming a CDCl2 radical. The transient CDCl2 radical then reacted with the olefin and forged an alkyl radical, which upon eletroreduction constructed the deuterated cyclopropane derivative plausibly through the formation of a carbanion intermediate and subsequent substitution of a chloride group from the CDCl2 unit. Alternatively, the carbanion intermediate was also trapped using suitable proton/deuterium sources and CDCl3 for one-carbon elongation of terminal olefins. The current method was susceptible to afford various deuterated cyclopropane analogues with high labeling of deuterium. This cyclopropanation strategy enabled the late-stage functionalization of estrone, bexarotene, and fenofibrate analogues.
Scheme 59. Electroreductive Late-Stage Functionalization of Olefins with Deuterochloroform.

Alkynes are present in a variety of natural products and biologically relevant molecules.329 Thus, late-stage alkynylation reactions and the diversification of alkynes have also remained an attractive target in the synthetic regime. Wang delineated the conversion of 4-acyl-1,4-dihydropyridines (DHPs) into ynones through an anodic oxidation-based approach (Scheme 60).330 This reaction proceeded through the electro-oxidation of DHPs, which then produced acyl radicals. The acyl radical intermediate was then intercepted with a hypervalent iodine derived alkynyl group transfer reagent and bestowed the ynones in good to excellent yields. Notably, boron-doped diamond (BDD) was used as the electrode for this electrochemical process. Various pharmaceuticals and biologically relevant molecules were diversified by following this approach.
Scheme 60. Electrochemical Synthesis of Ynones.

3.2. eLSF of Organic Halides
For years, organic halides have served as convenient synthetic handles for a diverse range of functionalizations using transition-metal catalysis.331−333 Manipulations over these moieties result in controlled and selective functionalization processes. Notably, electrochemical conditions have also appeared as a unique transformative tool to harvest these functionalities for late-stage functionalization reactions, and several such strategies have been reported in recent years.
Amines and their analogues are of considerable synthetic relevance in terms of their medicinal properties.334 Notably, a large variety of pharmaceuticals or biologically relevant molecules are analogues of amines. Thus, the sustainable construction of C–N bonds have gained tremendous attention in synthetic chemists’ repertoire.256,335−338 Despite the presence of numerous synthetic strategies to gain access to these fundamental units, general and economic approaches for late-stage amination reactions have unfortunately remained elusive. In this context, Baran reported a versatile nickel-catalyzed amination of aryl halides under electrochemical conditions (Scheme 61).339 This electrocatalytic protocol harvested both cathodic reduction and anodic oxidation during the catalytic cycle to enable the transformation. The proposed catalytic cycle is initiated by the cathodic reduction of nickel(II)-precatalysts to form the nickel(I) active catalyst. The oxidative insertion of the aryl bromides onto the nickel(I) catalyst followed by comproportionation or cathodic reduction of nickel(III)-species generated nickel(II)-species. The nickel(II) intermediate after ligand exchange with the amine and reductive elimination yielded the desired aminated product. The amination reaction was successfully realized with diverse amino acids, peptides, and sugar derivatives.
Scheme 61. Nickel-Catalyzed Electrochemical Amination of Aryl Halides.

In 2021, Rueping reported an electrochemical amination of aryl bromides with weak N-centered nucleophiles (Scheme 62).340 This nickel-catalyzed cross-coupling strategy was also amenable to accommodate more challenging aryl tosylates as electrophiles, harnessing anilines, sulfonamides, sulfoximines, carbamates, and imines as nucleophiles. Interestingly, the protocol was proven to be applicable for the late-stage modification of fenofibrate, galactopyranose, and cholestanol derivatives.
Scheme 62. Nickel-Catalyzed Electrochemical Amination with Weak N-Centered Nucleophiles.

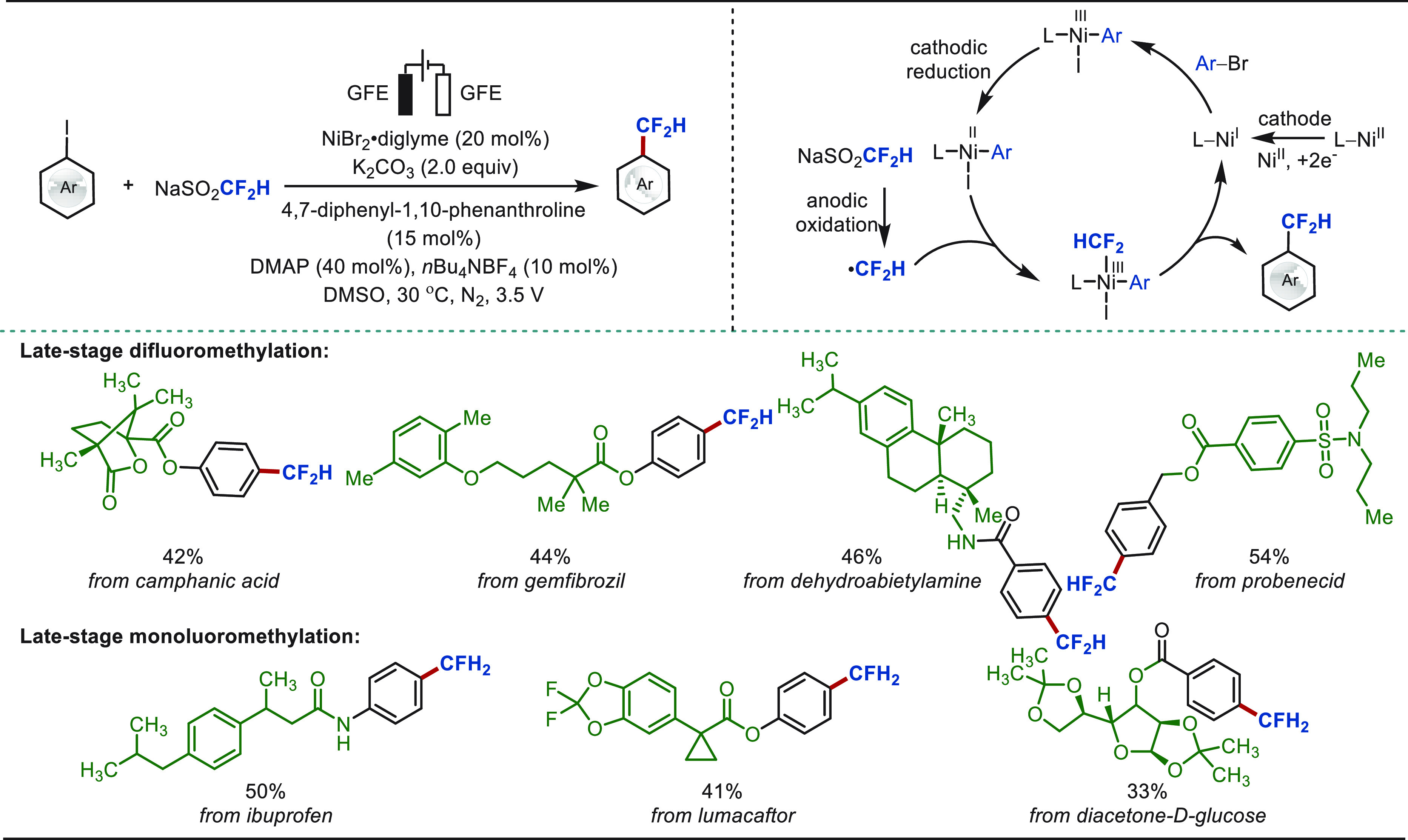
Later, Wang and Zhang developed a general nickel-catalyzed fluoroalkylation strategy with aryl iodides (Scheme 63).341 This strategy was operative via paired electrolysis, where the sulfinate salt was oxidized at the anode, forming a fluoroalkyl radical, while the cathodic reduction allowed the low-valent nickel catalysis. This method displayed good functional group compatibility and high substrate diversity. The synthetically meaningful strategy for the incorporation of difluoromethyl and monofluoromethyl groups to arenes enabled the functionalization of a broad range of natural product analogues and biologically relevant molecules.
Scheme 63. Ni-Catalyzed Late-Stage Fluoroalkylation of Aryl Iodides.
Electrochemical nickel-catalyzed functionalization strategies were not limited to organic halides. Indeed, the corresponding pseudohalides also served well as useful substrates for such transformations.342−344 In 2021, Wang and co-workers reported one such example, in which electrochemical nickel-catalysis was employed for deoxygenative thiolation of ketones (Scheme 64).345 In both of the cases the deoxygenation was facilitated through an initial activation of alcohols and ketones converting them to alkyl mesylate and vinyl triflate analogues, respectively. These reactive pseudohalide functionalities efficiently participated in the nickel-catalyzed thiolation reactions, generating the corresponding thioethers in high yields. Both approaches showed excellent functional group compatibility and enabled the late-stage diversification of a glucide derivative and various steroid analogues.
Scheme 64. Electrochemical Nickel-Catalyzed Deoxygenative Thiolation Reactions.
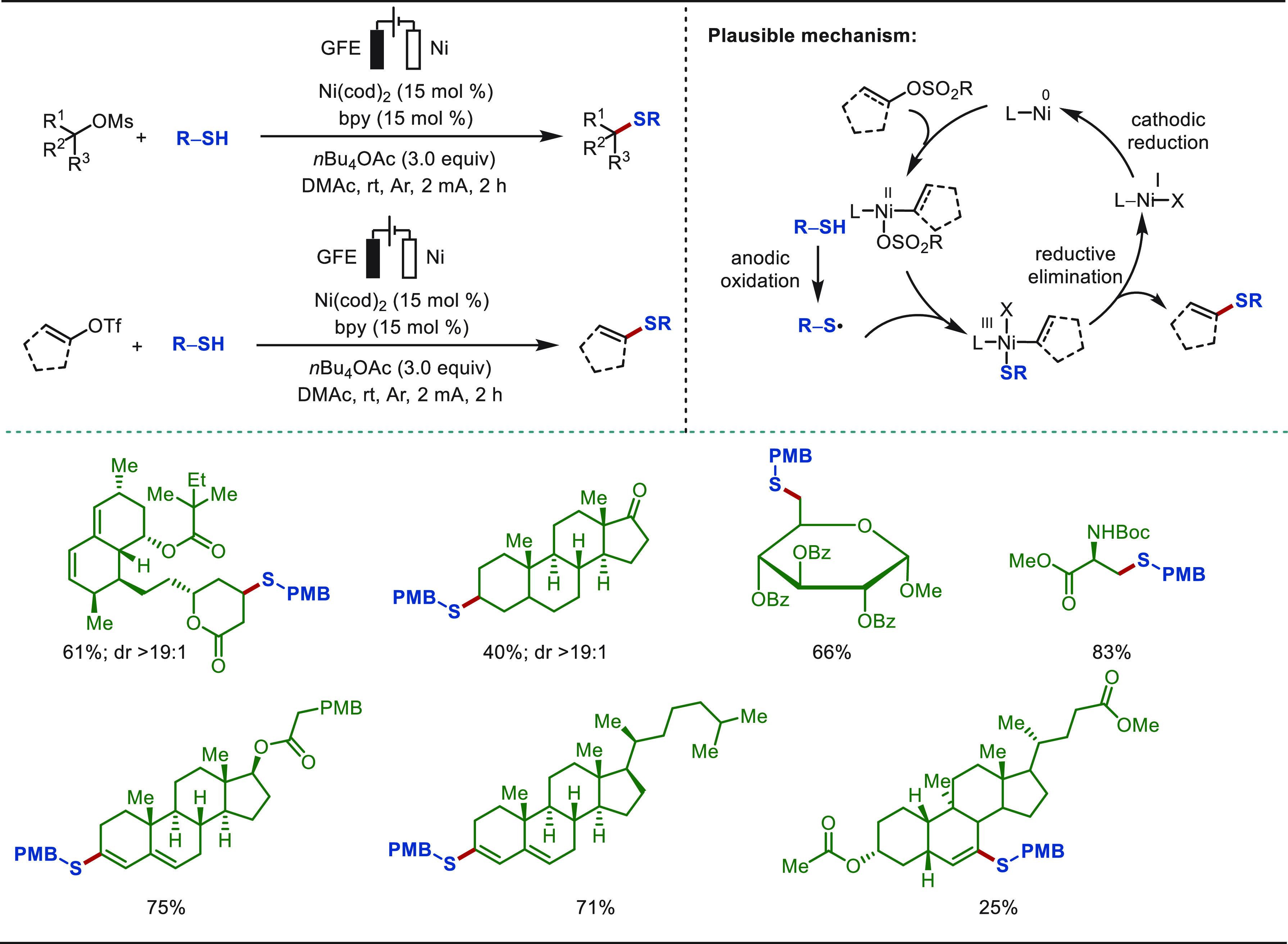
In 2021, Ye and Li reported an electrochemical nickel-catalyzed aminomethylation reaction of aryl bromides (Scheme 65).346 The reaction was proposed to proceed through a cathodic reduction of the nickel(II)-precatalyst, which upon oxidative addition to the aryl bromide gives intermediate 65e. Intermediate 65e after another cathodic reduction was intercepted by the aminomethyl radical, generated through anodic oxidation of the corresponding tertiary amine, producing intermediate 65g. Reductive elimination of 65g gave the desired aminomethylated product. This strategy was particularly powerful for the late-stage derivatization of benzobromarone, phenylalanine, clofibrate, and fenofibrate analogues.
Scheme 65. Nickel-Catalyzed Late-Stage Aminomethylation of Aryl Bromides.
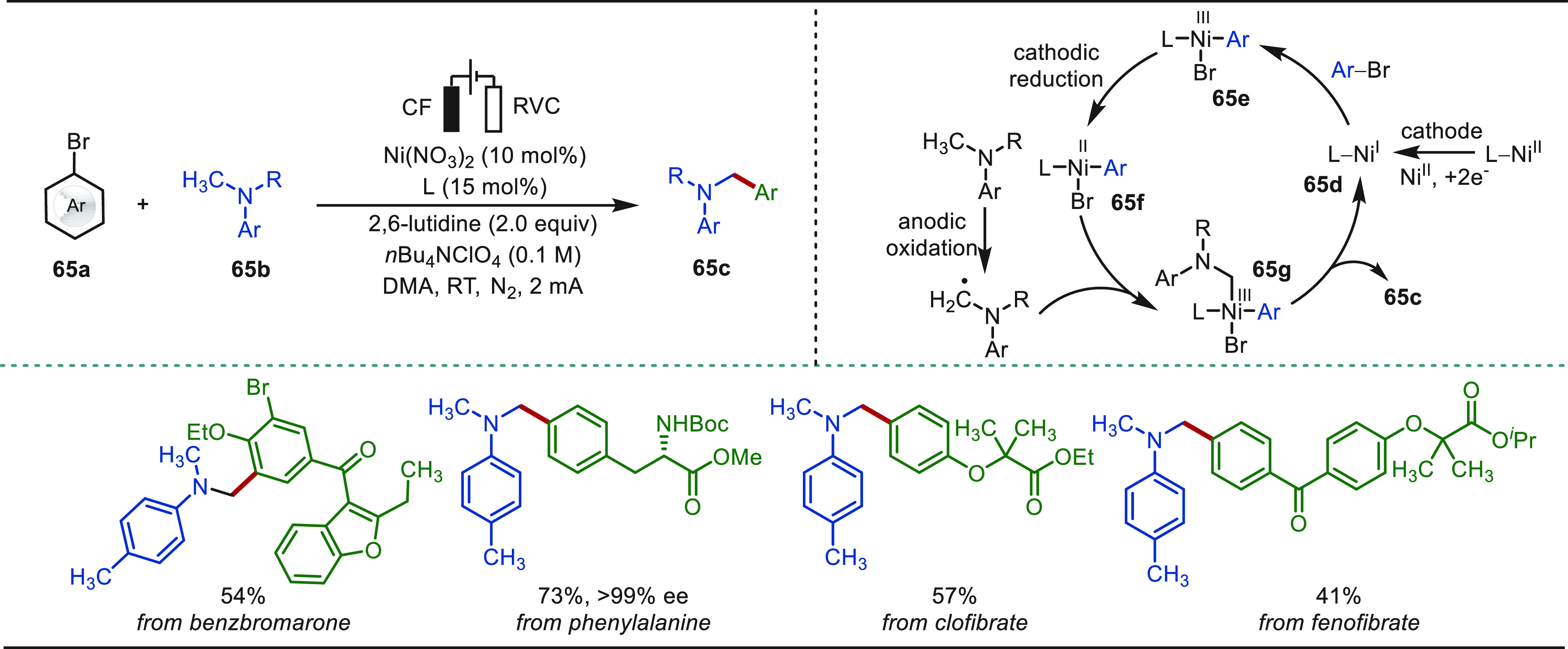
In 2021, a practical borylation was developed by Qi and Lu under electroreductive conditions (Scheme 66).347 The reduction of organic halides was induced with the aid of a sacrificial magnesium-anode, generating an alkyl radical, which was then intercepted with the diborane to construct the borylated product. The protocol operated under a high current (∼150 mA) with alkyl chlorides, bromides, and iodides. The efficiency of the approach was demonstrated through efficient late-stage borylation of natural products and drug analogues. Notably, the borylating agent served both as a boron source and a mediator controlling the reactivity of the process. The DMA stabilized B2cat2 mediated the single-electron reduction of the alkyl halide generating the alkyl radical, which then realized a barrierless radical–radical cross-coupling with B2cat2. Detailed DFT studies rendered the possibility of path b unlikely to be operative, with an activation energy barrier of 9.1 kcal/mol.
Scheme 66. Electrochemical Late-Stage Borylation of Alkyl Halides.
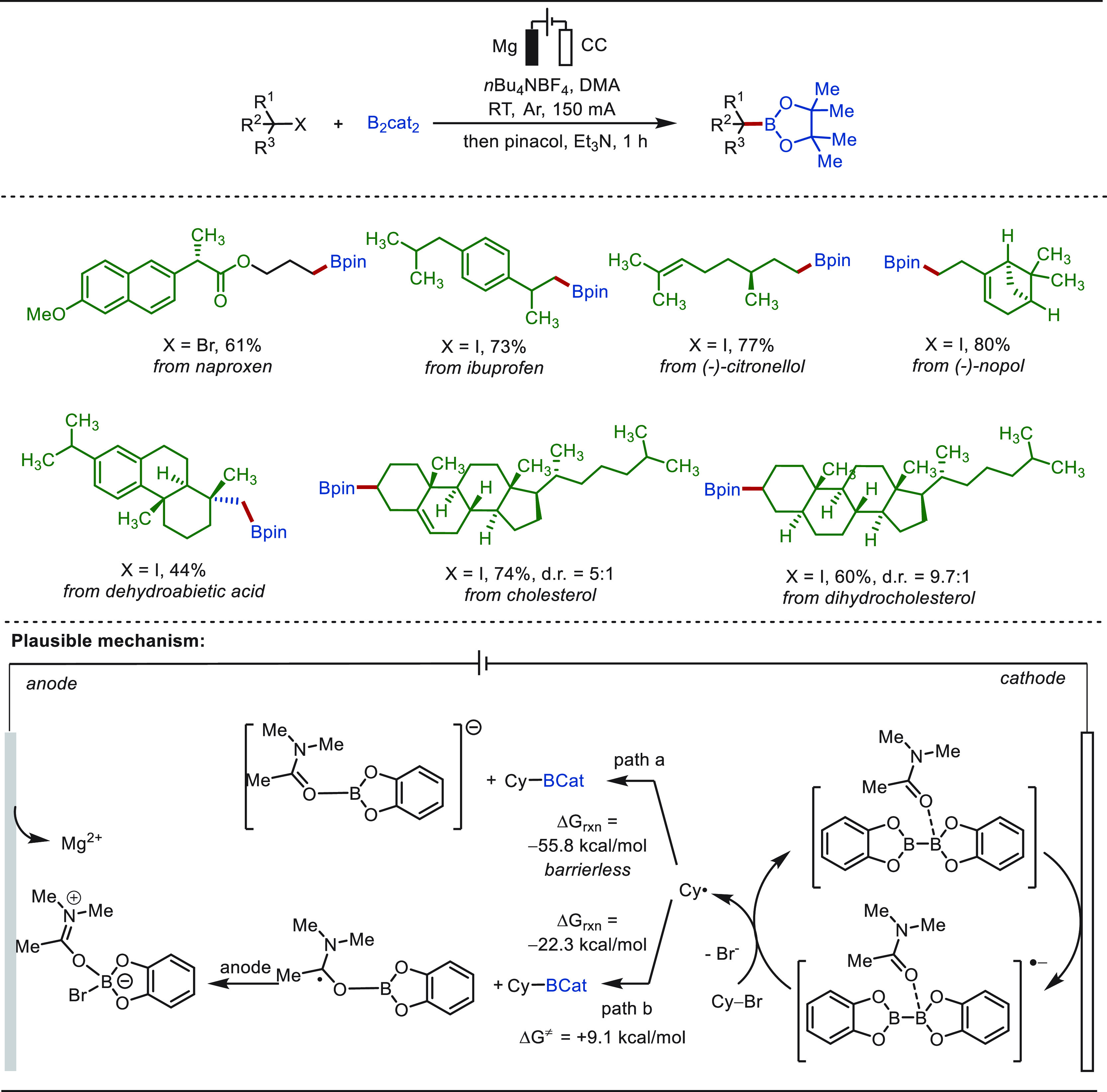
The presence of C(sp3)-rich organic molecules may improve efficacy for the drug-candidates in clinical trials.348 Thus, constant effort has been devoted to devising methods for selective formation of C(sp3)–C(sp3) bonds. In 2022, Lin and co-workers developed an electroreductive strategy for the construction of C(sp3)–C(sp3) bonds harvesting easily accessible alkyl halides as the alkyl source (Scheme 67).349 A selective cathodic reduction of the more substituted alkyl halide to the corresponding carbanion governed a preferential substitution of comparatively less substituted alkyl halide, forging the C(sp3)–C(sp3) bond with high precision. Altering the transition-metal-catalyzed approach with the direct electrolysis of alkyl halides avoided the typical β-hydride elimination pathway, offering a modular selective C(sp3)–C(sp3) bond formation. A sacrificial Mg-anode effectuated this transformation and the mild reaction conditions enabled the late-stage modification of complex organic molecules and drug derivatives. Further, a sequential photochemical chlorination of benzylic C–H bond followed by electroreductive methylation of methyl dehydroabietate exhibited the broad synthetic utility of this electrochemically driven cross-electrophile coupling (e-XEC). Interestingly, this sequential electrochemical alkylation process was also successful for the late-stage deuteromethylation of ibuprofen and retionic acid receptor agonist.
Scheme 67. Electrochemically Driven Late-Stage C(sp3)–C(sp3) Bond Formation.
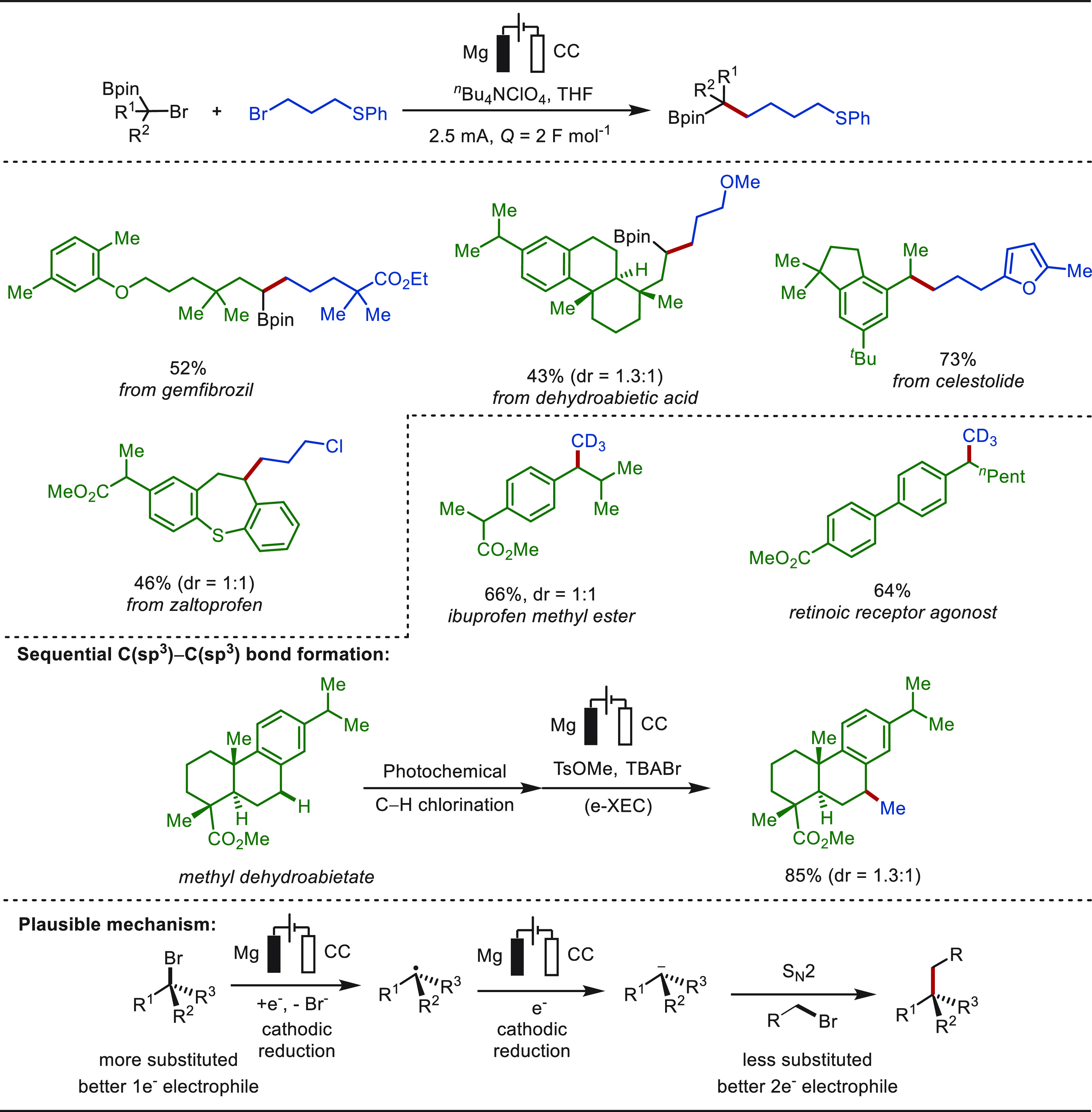
Reductive deuteration of organic halides331,350−352 constitutes a promising method to prepare deuterated molecules, which are widely used in pharmaceutical research.353−355 In 2022, Qiu described an interesting example of late-stage deuteration of organic halides, where high deuterium incorporation (up to 99%) in the product was achieved using simple D2O as the deuterium source (Scheme 68).356 The plausible reaction mechanism involved a 2-fold cathodic reduction of the organic halide forming a carbanion intermediate, which is quenched with the D2O present in the medium. This protocol was adopted for the deuterium labeling of various pharmaceuticals and their intermediates along with other complex substrates. The strategy also functioned well under 500 mA current with high selectivity, which intimated the applicability in industrial application. Under related conditions, the electrochemical reductive deuteration of aryl halides and benzylic chlorides was achieved by Lei357 and Lin,358 respectively.
Scheme 68. Electrochemical Late-Stage Deuteration of Alkyl Halides.
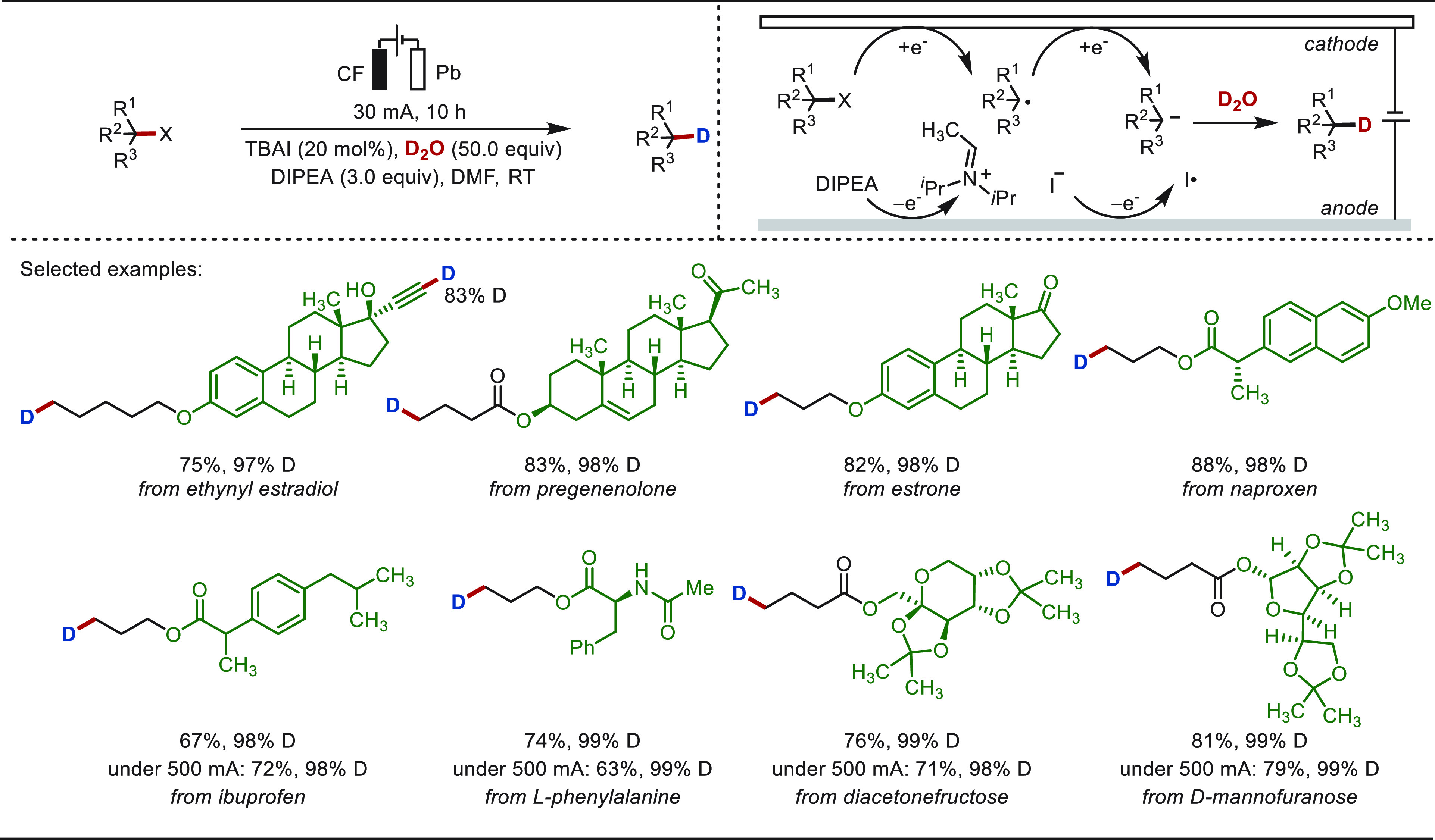
Recently, a metal-free electroreductive carboxylation of aryl halides was devised by Qiu (Scheme 69).143,359 This electroreducitve protocol functioned by utilizing naphthalene as a catalytic mediator without any sacrificial anode material. The naphthalene mediator under the catalytic conditions formed a strong reductant naphthalene anion radical, which reduced the aryl halide generating aryl radical. Consequently, the aryl radical was quenched with CO2 delivering the carboxylic acid. This simple dehalogenative strategy was effective for the late-stage carboxylation of several natural products, drugs, and bioactive compounds.
Scheme 69. Electrochemical Carboxylation of Aryl Halides.

3.3. eLSF of Organic Carboxylic Acid and Derivatives
Carboxylic acids are one of the most versatile feedstocks in modern organic synthesis.360−362 This functionality is also highly abundant in natural products, bioactive molecules, and pharmaceuticals.363 Thus, electrochemical decarboxylative strategies, since its inception from popular Kolbe electrolysis, have flourished significantly for the late-stage functionalization of valuable organic molecules.50,364
In 2019, Baran reported an electrochemical decarboxylative synthesis of hindered aliphatic dialkylethers harnessing electrogenerated carbocation intermediates (Scheme 70).365 Sterically hindered dialkylethers, though highly coveted motifs owing to their medicinal importance, are challenging to access through conventional synthetic approaches.366 However, the trapping of the carbocation intermediate, generated from the direct electrochemical oxidation of easily accessible carboxylic acids, with alcohols furnished these valuable organic molecules in a straightforward manner. This method operated under the most user-friendly and mild conditions, tolerating diverse common functional groups in the substrates. This electro-oxidative reaction was successful in accomplishing late-stage functionalization of a large variety of complex and biologically relevant organic molecules. Further, water was also an effective nucleophile under these conditions to deliver late-stage decarboxylative hydroxylation of complex organic molecules.
Scheme 70. Decarboxylative Synthesis of Sterically Hindered Ethers.

In this context, the Wang group reported a protodecarboxylation as well as a decarboxylative Giese reaction of aliphatic carboxylic acids, which were efficient for the functionalization of various amino acids and natural products (Scheme 71).367 The decarboxylation proceeded through the single-electron reduction of the redox-active ester (RAE), which upon decarboxylation generated the alkyl radical. The alkyl radical was then trapped with the activated olefin to produce the desired product. In the absence of the olefin, decarboxylated products were obtained.
Scheme 71. Electrochemical Decarboxylative Functionalization of Bioactive Carboxylic Acids.
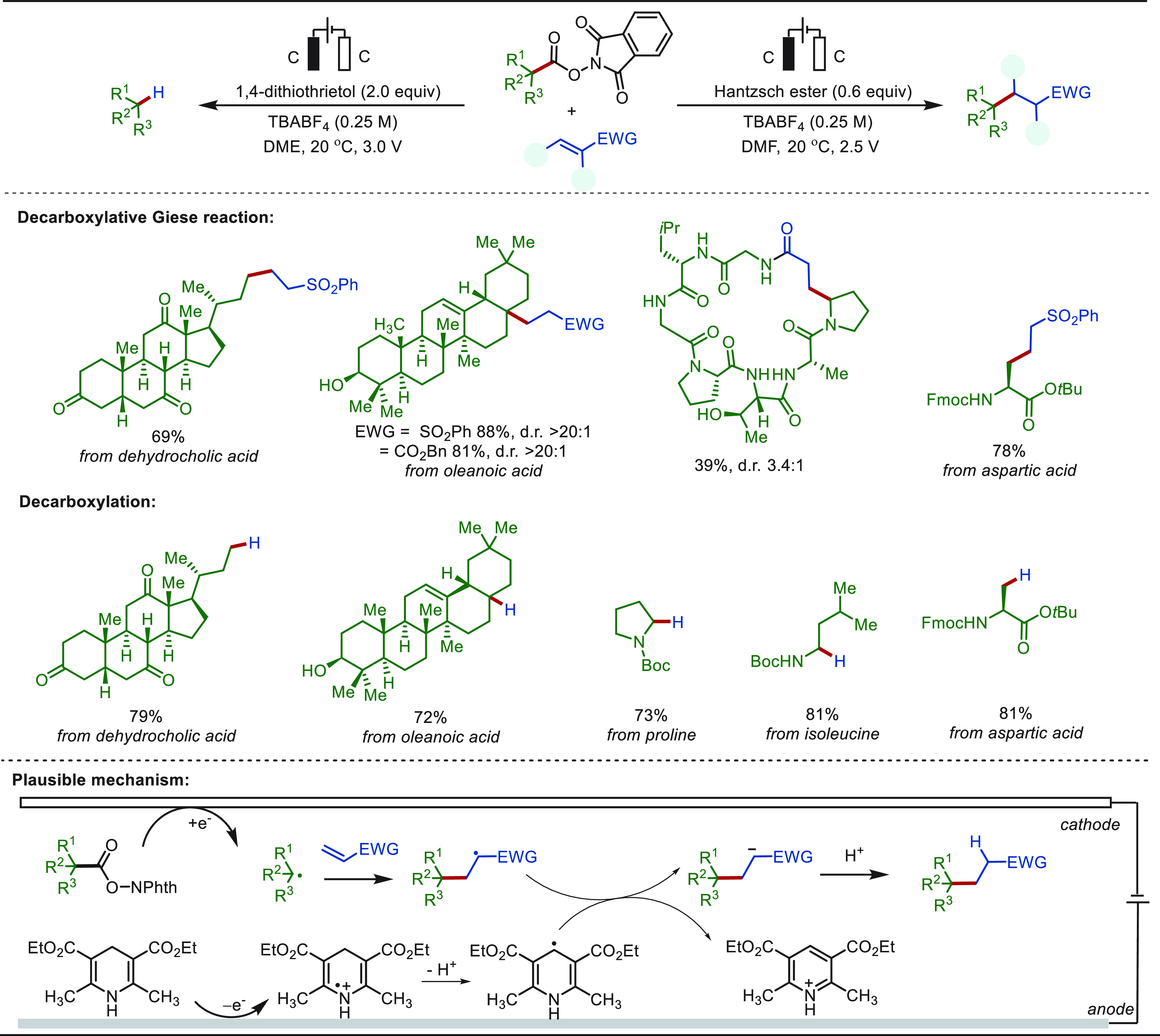
In 2021, Baran reported an electroreductive Nozaki–Hiyama–Kishi (NHK) reaction, a popular strategy often applied in natural product synthesis, generating complex allylic alcohols from easily accessible vinyl halides and aldehydes (Scheme 72).368 This approach used catalytic amounts of chromium salts, avoiding stoichiometric metallic reductants. While similar transformations had previously been achieved by Grigg, Tanaka, and Périchon/Durandetti, these approaches mainly suffered in terms of the broader synthetic applicability and complex reaction settings.369−372
Scheme 72. Late-Stage Modification through Electrochemical Nozaki–Hiyama–Kishi Reaction.
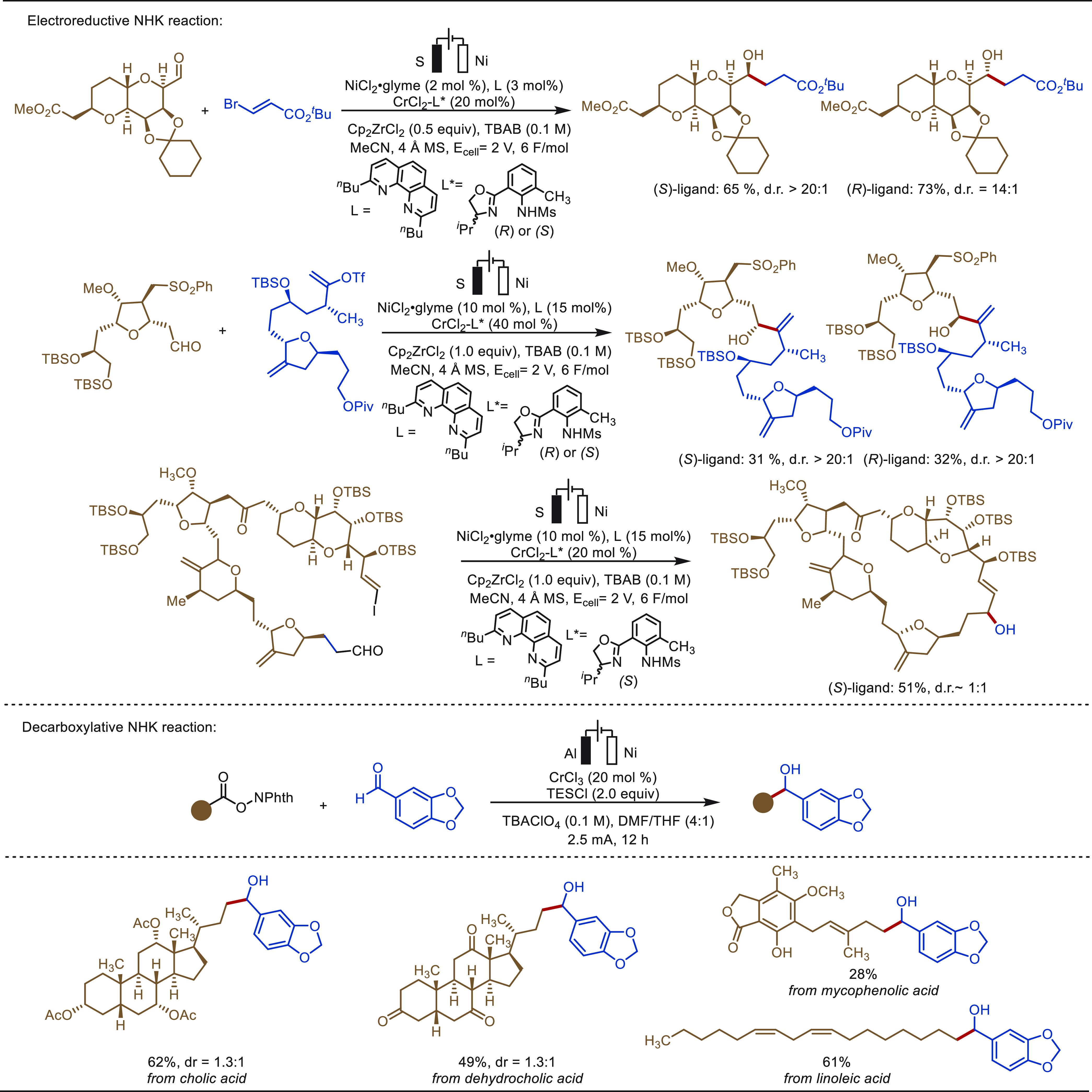
The combination of nickel- and chromium-salts under constant potential enabled this transformation, which was applied for the synthesis of complex chiral allylic alcohols including the synthesis of a Halaven intermediate. This electroreductive approach was also effective to harness noncannonical redox-active esters as useful substrates for the generation of secondary alcohols in a straightforward manner. Mechanistic investigations revealed that the reaction rate of the e-NHK reaction was faster compared to classical NHK reactions (Scheme 73). Spectro-electrochemical studies justified the presence of chromium(III)-species in the catalytic process, while the presence of Ni(II)-species significantly influenced the electron transfer process to chromium(III). The e-NHK reaction commenced with the cathodic reduction of chromium(III)-salt to chromium(II), which reduced the nickel(II) cocatalyst to nickel(0). This nickel(0) species underwent a facile oxidative addition with the vinyl halide to form respective intermediate, which upon transmetalation with chromium(III) followed by 1,2-addition with the aldehyde released the allyl alcohol analogue. The chromium(III)-catalyst was regenerated with another transmetalation with Cp2ZrCl2. The decarboxylative variant of this transformation also followed a similar mechanistic pathway, in which the Cr(II)-species facilitated the single-electron reduction of the redox-active ester to form an alkyl-Cr(III) species. This intermediate bestowed the product after 1,2-addition and silylation of the alkoxy-Cr(III) intermediate.
Scheme 73. Plausible Mechanism for Electrochemical NHK Reaction.

In 2021, Chiba described a green biphasic peptide synthesis protocol using electro-oxidative conditions (Scheme 74).373 Stoichiometric amounts of PPh3 were used as the additive, which under electrochemical oxidation formed a triphenylphosphine radical cation. This intermediate activated the terminal carboxylic acid of the amino acid, and then a nucleophilic displacement reaction at the carbonyl center with another amino acid constructed a peptide bond along with a reusable Ph3PO byproduct. This process proved viable to access a library of small peptides and successfully implemented for the synthesis of active pharmaceutical ingredient (API), leuprorelin without using traditional expensive peptide synthesis reagents.
Scheme 74. Electrochemical Synthesis of Peptides.
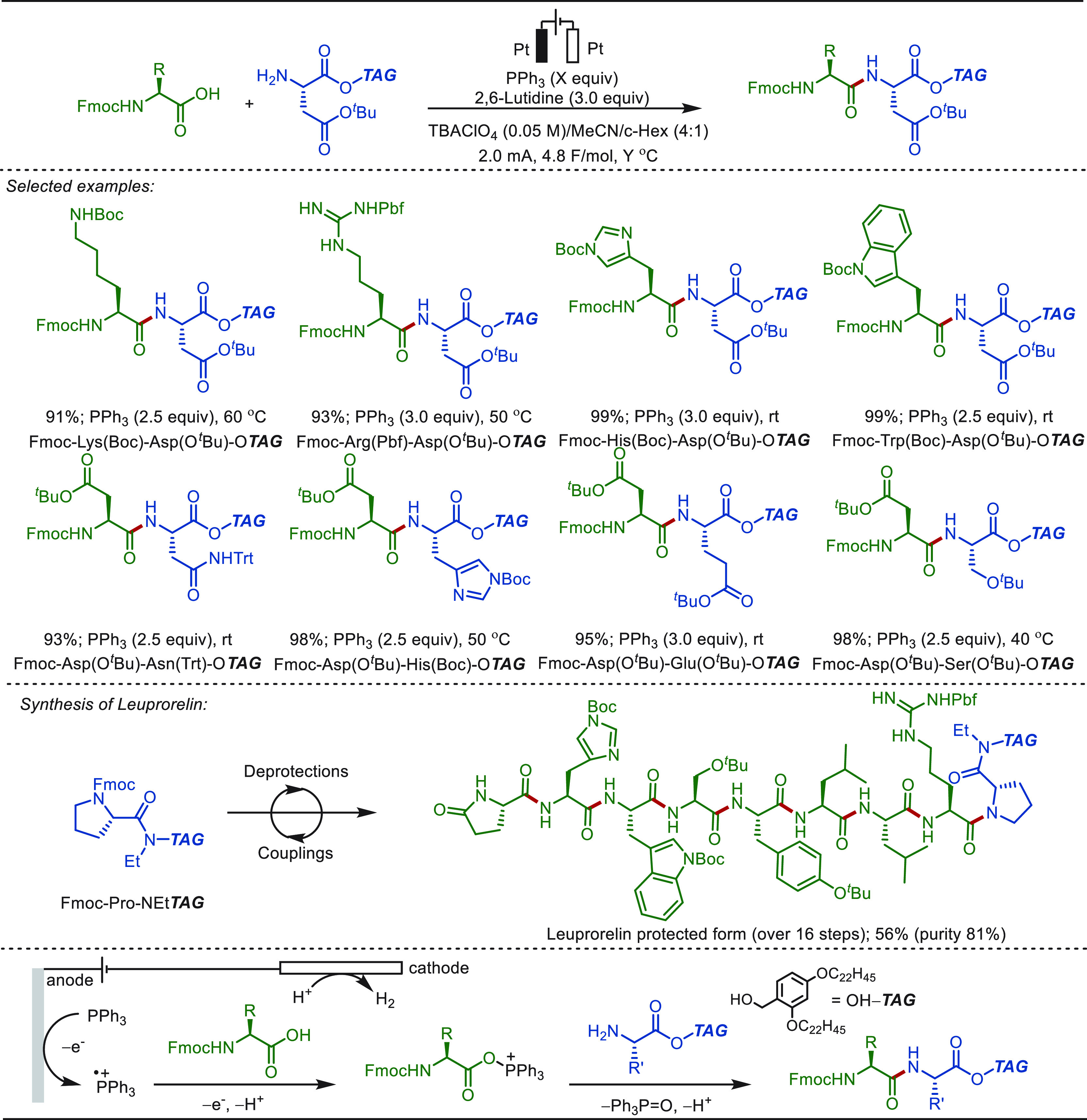
In 2020, Malins developed an electrochemical decarboxylation-nucleophilic addition approach (Scheme 75).374 First the oxidative decarboxylation at the C-terminal led to the formation of N,O-acetal intermediates, which after the treatment of nucleophiles under acidic conditions forged the functionalized product. The synthetic utility of this method was mirrored by the divergent synthesis of various bioactive peptides, including biseokeaniamide analogues 75a.
Scheme 75. Electrochemical Decarboxylative Functionalization of Peptides.
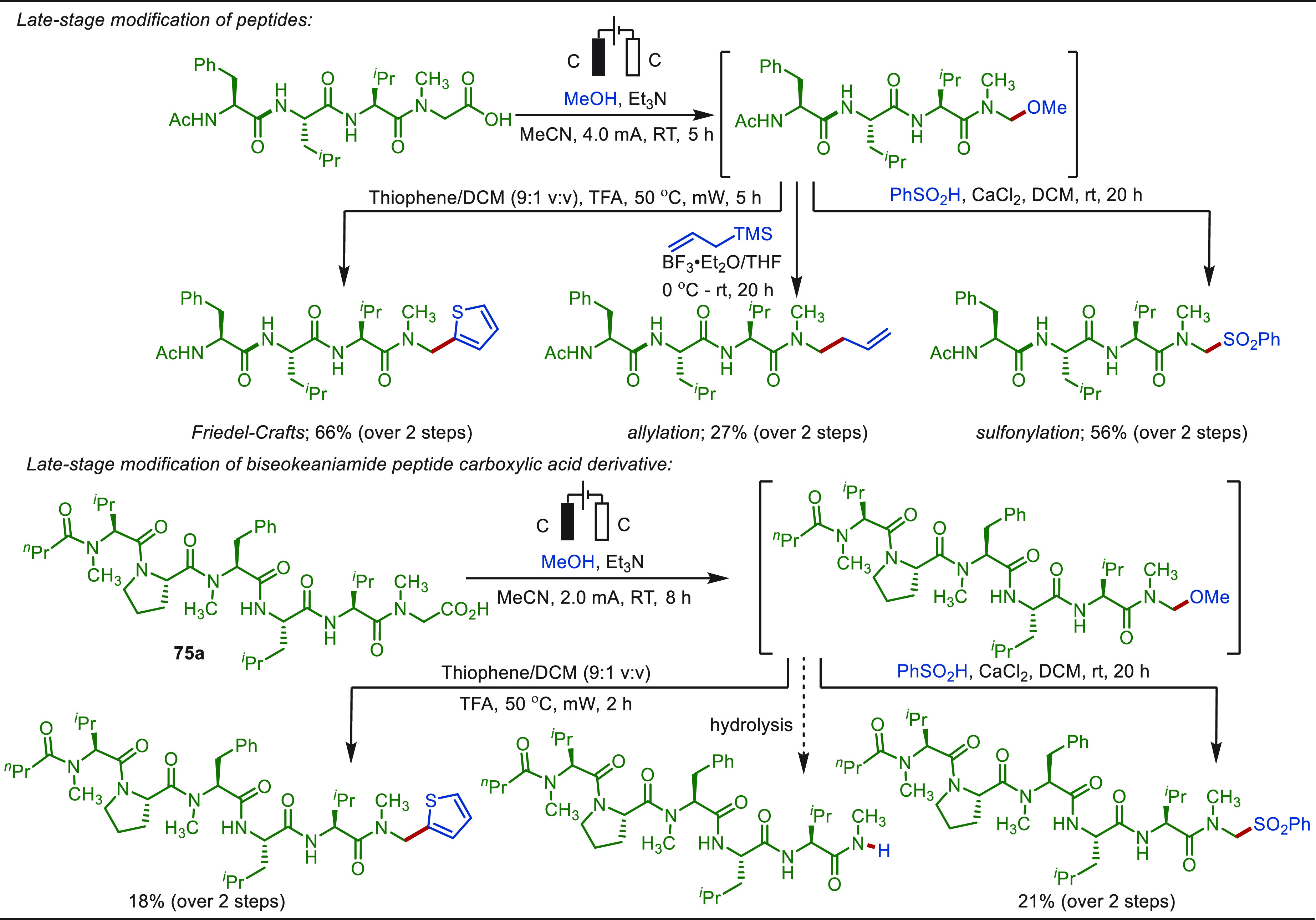
This oxidative decarboxylative approach was further extended by Malins for the synthesis of designer C-terminal peptides (Scheme 76).375 Decarboxylation followed by a reduction of the N,O-acetal under acidic conditions led to the formation of the desired peptides in decent yields. The innate reactivity of the C-terminal carboxylate analogue was exploited under robust conditions, where a large variety of proteinogenic functionalities were tolerated and the designer peptides were obtained without epimerization. Utilizing this electrochemical strategy, natural product acidiphilamide A as well as an anti-HIV peptide and the cancer therapeutic leuprolide were synthesized.
Scheme 76. Electrochemical Decarboxylative Modification of C-Terminal Peptides.
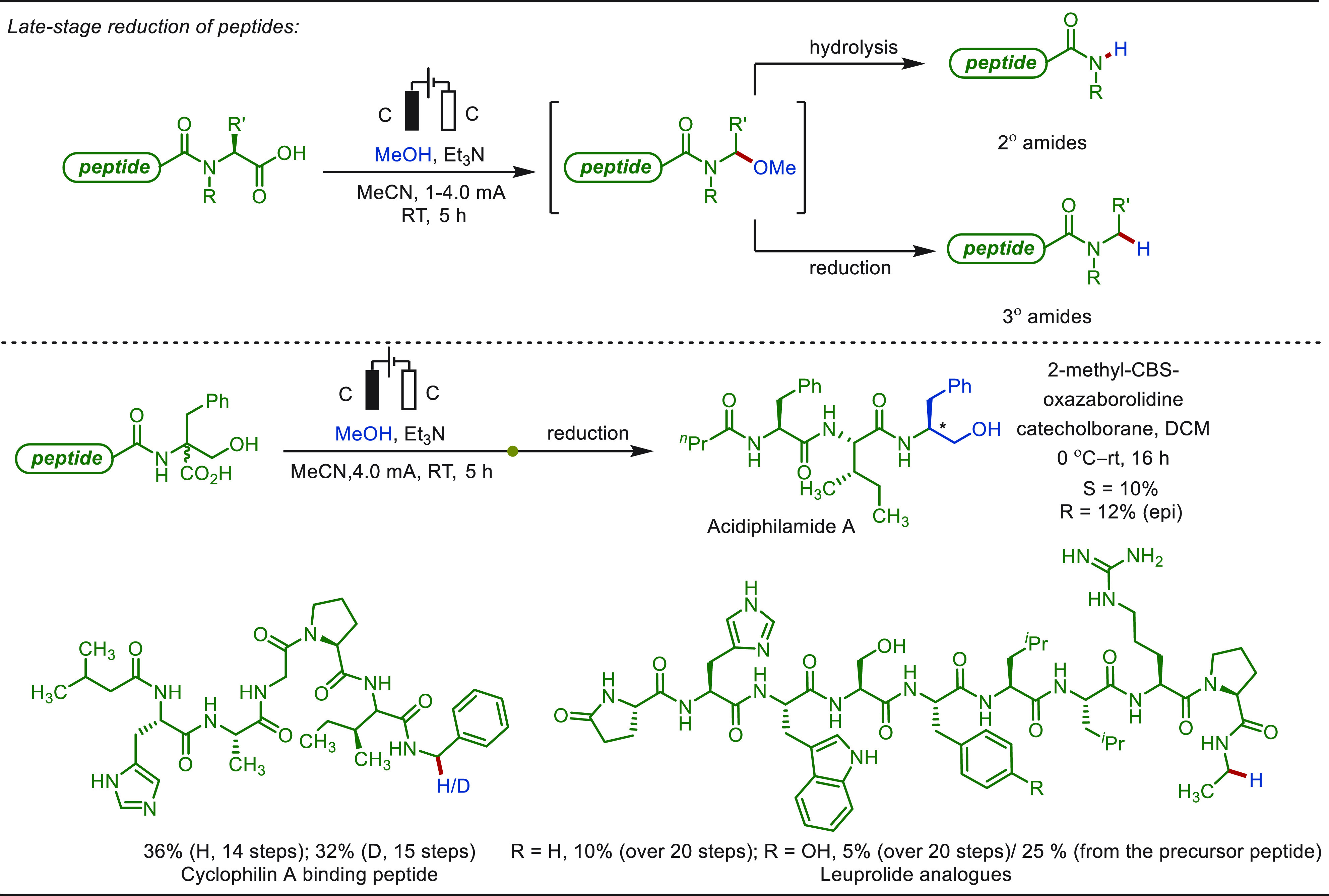
Recently, Malins has further extended the decarboxylative functionalization strategy combining with an acid promoted aromatization for late-stage modification of C-terminal hydroxyproline containing peptides to access a library of C-terminal N-acylpyrrole derivatives (Scheme 77).376 Identical to prior examples, this strategy was also operationally simple, compatible with various common protecting groups in the peptide-chain, and useful to incorporate bioisosteres and peptide labels. Respective aldehyde analogues were amenable through the reduction of the C-terminal N-acylpyrrole containing peptide derivatives.
Scheme 77. Electrochemical Decarboxylative Aromatization of C-Terminal Hydroxyproline Containing Peptides.
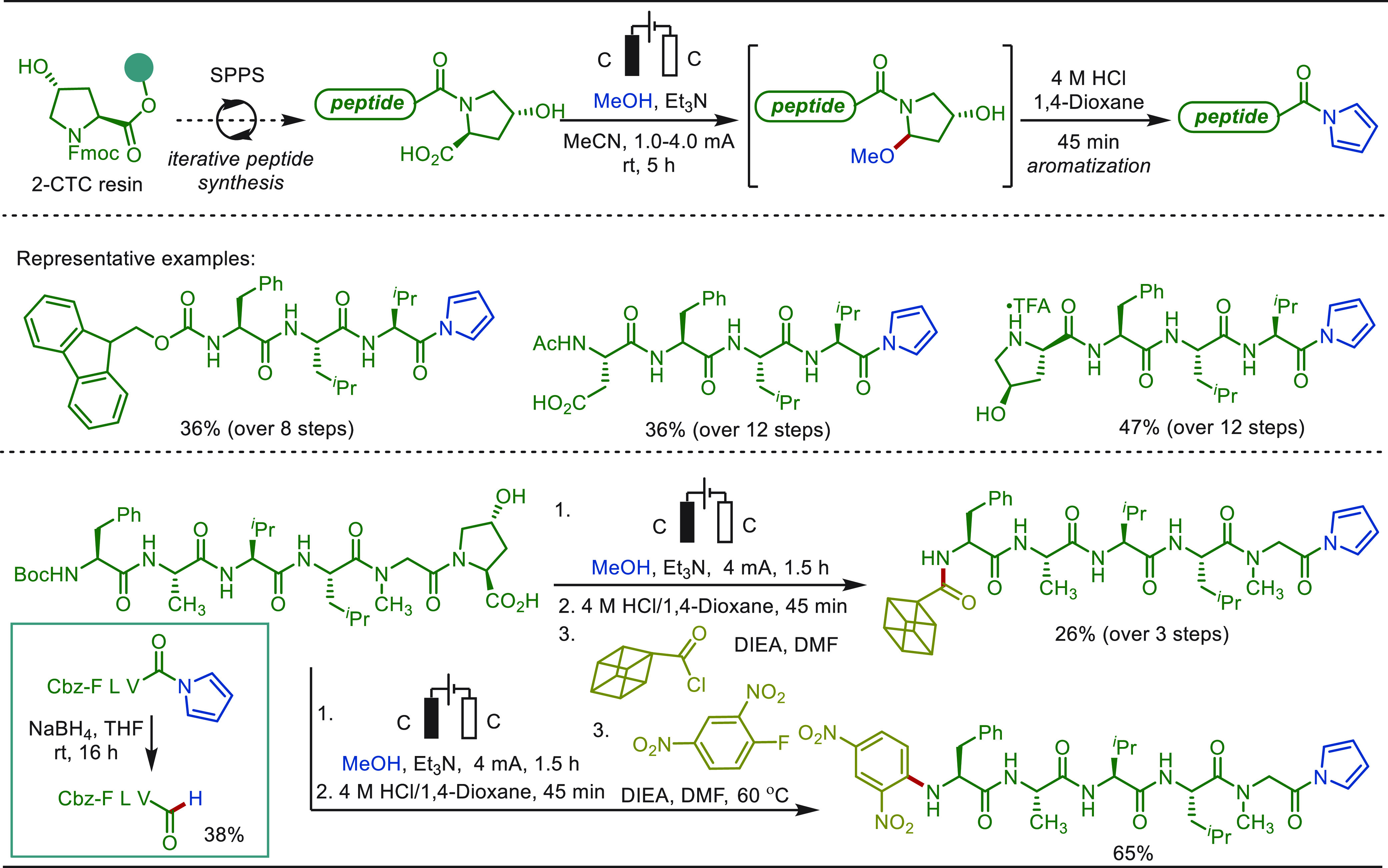
Baran further contributed in this area depicting a nickel-catalyzed decarboxylative C(sp3)–C(sp3) bond formation reaction (Scheme 78).377 The synthetic method used redox-active N-hydroxyphthalimide protected carboxylic acid ester derivatives as the alkyl source, where two distinct alkyl groups were stitched together by the nickel-catalyst. This electroreductive process effectively combined primary, secondary, and even tertiary alkyl radicals for selective C(sp3)–C(sp3) bond formation. This simple single-step process tolerated diverse common functional groups exploiting widely available carboxylic acid derivatives as useful synthons. Notably, several complex natural products and biologically relevant molecules were easily manipulated utilizing this nickel-catalyzed strategy.
Scheme 78. Electroreductive Nickel-Catalyzed Decarboxylative C(sp3)–C(sp3) Bond Formation.
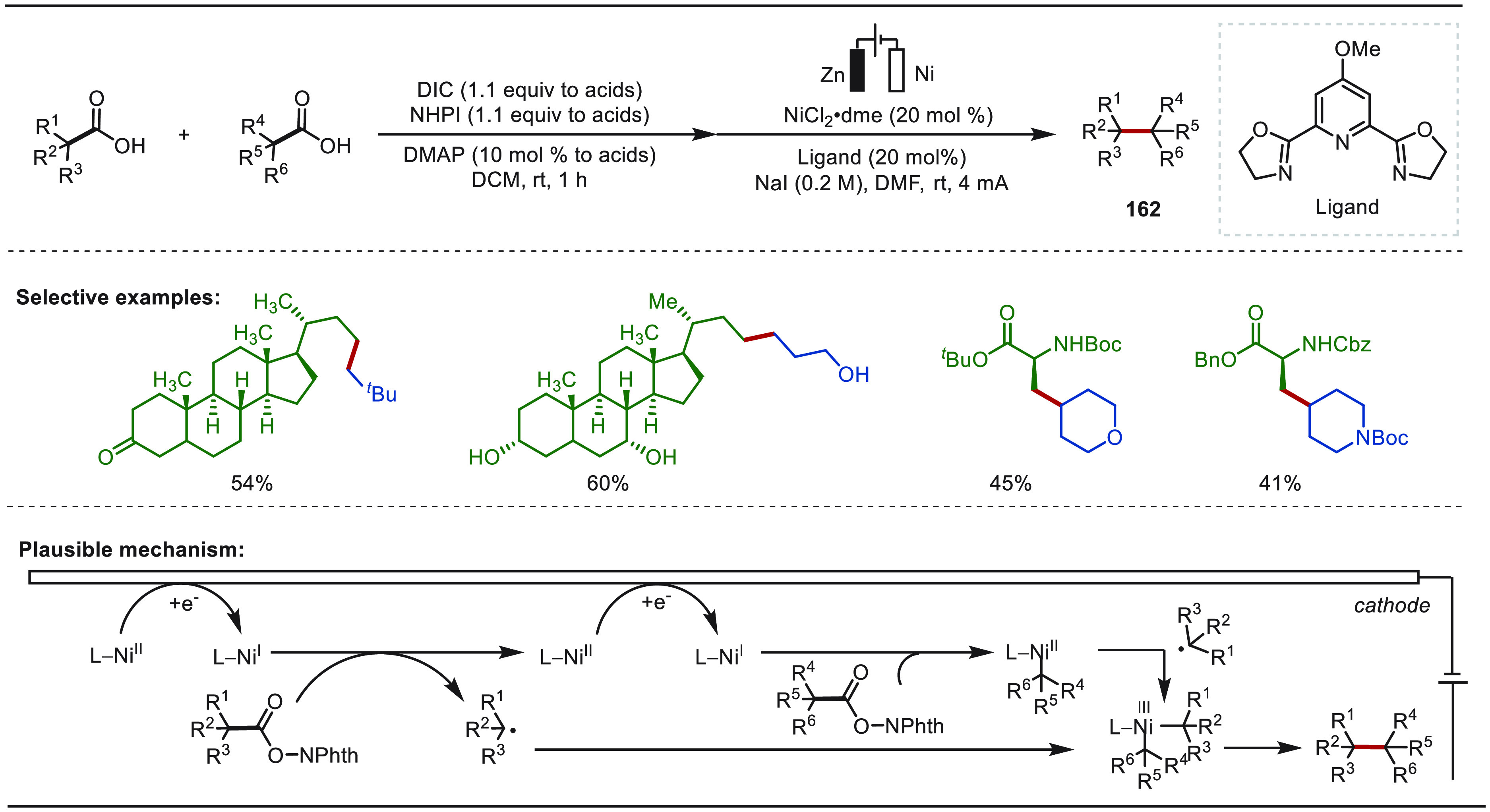
3.4. eLSF of Alcohols and Derivatives
Alcohols, and analogues thereof, have always remained in the general focus of synthetic chemists, as they are prevalent motifs in a multitude of natural products, pharmaceuticals, and biologically relevant molecules.378 Thus, the synthesis and derivatization of this prevalent functionality attract significant attention. However, direct functionalization over these synthetic handles is cumbersome due to the high C–O bond strength, which can be accomplished through the activation of the C–O bonds by other means. In this domain, electrochemical approaches have served well, providing some practical transformative alternatives, which are also extended for late-stage functionalization reactions.379 An early example by Ohmori and co-workers highlighted that anodic oxidation of triphenylphosphine could execute an efficient C–O bond activation through alkoxy phosphonium salt formation.380−382 Oxidation of PPh3 generated a triphenylphosphine radical cation, which was intercepted by the alcohol to form the alkoxy phosphonium salt intermediate 79a, nucleophilic substitution of which bestowed functionalized product 79b (Scheme 79). The method was pertinent in combination with a large variety of nucleophiles. This versatile process was applied for the late-stage deoxygenative functionalization of glycosides with fluoride and chloride nucleophiles.383 Recently, Wang and Tian harnessed this electro-oxidative approach for a deoxygenative C–N bond formation reaction, which thereby allowed for the glycosylation of azoles in straightforward manner under mild conditions (Scheme 80).384
Scheme 79. Late-Stage Dehydroxylative Halogenation and Alkoxylation.

Scheme 80. Electrochemical Late-Stage Dehydroxylative Azolation.

The epoxide functionality is a highly reactive synthetic handle for the diversification of organic molecules.385 While nucleophilic displacement reactions are one of the most common approaches in modifying epoxides, recently electroreductive approaches are also gaining significant momentum for the late-stage functionalization of epoxides of biologically relevant molecules. In 2022, Lu and Qi described an electrochemical transition-metal free reduction of epoxide to generate primary, secondary, and tertiary alcohols (Scheme 81).386 This approach delivered both regioisomeric ring-opening products, where the thermodynamic stability of the benzylic radical was decisive for aryl epoxides and the alkyl epoxides realized ring-opening following a kinetic manifold. The electroreductive method was able to deliver the corresponding alcohols of natural products α-pinene, betulin, and pregnenolone. A plausible mechanism of this reaction involved the single-electron reduction of the epoxide in the presence of a Lewis acid followed by protonation.
Scheme 81. Electrochemical Transition-Metal Free Reduction of Epoxides.

Recently, efficient electroreductive protocols to access β-hydroxycaboxylic acids from readily available aryl epoxides were reported by Qiu387 and Zhang,388 independently (Scheme 82). The strategy used a sacrificial magnesium anode to promote the cathodic reduction of epoxide, forming a benzylic radical, which upon another cathodic reduction generated a carbanion intermediate. The carbanion intermediate was quenched by CO2 constructing the desired product. The formation of carbanion intermediate was confirmed by a deuterium labeling study. This method was able to functionalize aryl epoxides derived from drug molecules and amino acids giving access to corresponding β-hydroxycaboxylic acids in good to excellent yields.
Scheme 82. Electrochemical Transition-Metal Free Carboxylation of Epoxides.

3.5. Late-Stage Reduction of Arenes and Ketones
The Birch reduction is an important method for the dearomatization of arenes into sp3-rich organic molecules.389,390 However, the direct use of hazardous alkali metals is necessary for typical Birch reduction. In 2019, Baran reported an exquisite example of electrochemical Birch reduction harvesting Li-ion battery materials and additives (Scheme 83).391 The e-Birch reduction method operated by consuming a combination of sacrificial anode (Mg or Al), inexpensive proton source dimethylurea, and tris(pyrrolidino)phosphoramide additive for overcharge protection. This method was operationally simple, avoided the direct use of alkali metals maintaining a similar reactivity trend, and exhibited high functional group compatibility along with the application toward the late-stage manipulation of pharmaceutically relevant molecules.
Scheme 83. Electrochemical Late-Stage Birch Reduction.

The vast majority of electrocatalyzed reactions are enabled by direct current, where the polarity of the electrodes remains constant over time and the flow of electrons in the reaction media is unidirectional. While alternating current (AC) was used to realize decarboxylation, nitro reduction, and electrolysis of propylene in the early 20th century,392−395 it was rarely explored in mainstream organic synthesis. In 2021, Baran studied the use of AC for a controlled reduction of phthalimides (Scheme 84).396 The use of alternating current offered precise control on the transformation, and various sensitive functional groups remained untouched, only selectively reducing the phthalimide motif. Overall, this transformation displayed a broad scope and was employed for the synthesis of PROTAC-relevant molecules.
Scheme 84. Selective Reduction of Phthalimides under Alternating Current.

3.6. Other eLSF of Functional Groups
The direct modification of amino acids and peptides is cumbersome owing to a similar range of oxidation potentials in most of the peptide linkages, leading to site-selectivity issues. The incorporation of a silyl group to the α-position of the amine functionality provides an alternative to execute late-stage modification of peptides. In 2002, Moeller devised an elegant electro-oxidative modification of silylated amino acids, where monocyclic or bicyclic peptidomimetics were easily constructed through an anodic oxidation-based approach (Scheme 85).397,398 The electrochemical oxidation of the silylated amino acid led to the formation of acyliminium ion intermediates. The presence of the silyl electroauxiliary reduced the general oxidation potential for the synthesis of acyliminium intermediates from the respective variants without having the electroauxiliary, which amplified the selectivity for the ring construction. These acyliminium intermediates then underwent intramolecular nucleophilic attack with nucleophilic functionalities present in the molecule, forging mono- or bicyclic peptidomimetics.
Scheme 85. eLSF of Silylated Amino Acids.

Later, Moeller reported on a two-step method involving anodic oxidation as a key step to convert a sugar derivative into C-glycosides consisting of a masked aldehyde functionality (Scheme 86).399 The reaction sequence involved a Wittig reaction forming enol ethers, which under electro-oxidative conditions realized an intramolecular nucleophilic attack with the free hydroxy functionality present in the molecule, constructing five- and six-membered C-glycosides in moderate to good yields.
Scheme 86. Electrochemical Synthesis of C-Glycosides.

In 2012, Chiba described an electro-oxidative soluble-support assisted synthesis of disulfide bonds in peptides (Scheme 87).400 The method involved bromide ion assisted electron transfer, where the oxidized bromide ion led to disulfide bond formation. Alternatively, the strategy also worked in the absence of bromide ion, where direct oxidation of the substrate was relevant. After the transformation was completed, dilution with acetonitrile, followed by simple filtration, was sufficient to recover the product.
Scheme 87. Electrochemical Construction of Disulfide Linkages in Peptides.

Another electro-oxidative C–C bond forming approach was unveiled by Chiba group for the modification of C- and N-terminal proline containing peptides (Scheme 88).401 The electrochemical incorporation of the 2,4,6-trimethoxyphenyl (TMP) moiety in the C-5 position of proline led to the selective generation of N-acyl iminium intermediates through electro-oxidation of the TMP moiety, which was then trapped with allylTMS to fabricate the allylated products in good yields.
Scheme 88. Selective C-5 Functionalization of Proline and Analogues.

Recently, Lei and Huang demonstrated AC promoted C–O/O–H cross-metathesis reactions (Scheme 89).402 This AC-based approach allowed for easy transformation of 4-alkoxyanilines into high-value products through electro-oxidation. Single-electron oxidation of these electron-rich arenes generated quinonoid intermediates, which realized a facile nucleophilic attack with the alcohol present in the medium, replacing the methoxy functionality with a new alkoxy functionality. The reaction was operationally simple and chemo- and regioselective. This cross-metathesis reaction was successfully employed for the late-stage diversification of pharmaceuticals and their derivatives.
Scheme 89. Selective C–O/O–H Metathesis under Alternating Current.
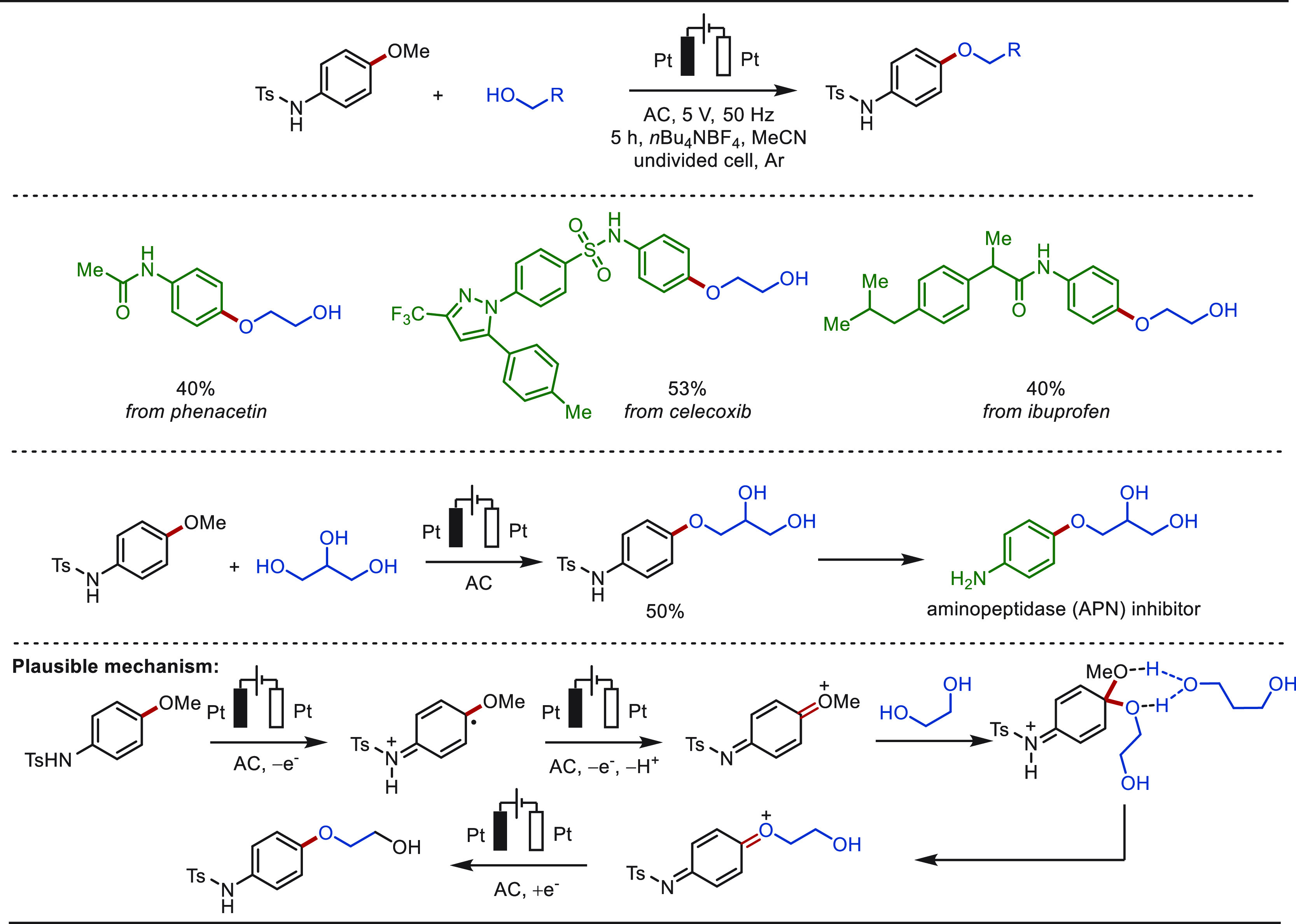
Very recently, Malins and Connal described an electroauxiliary-assisted late-stage diversification strategy of glutamine residues of peptides (Scheme 90).403 Peptides consisting of N,S-acetals in the glutamine residues under electro-oxidative conditions realized a facile cleavage of the thio-functionality to form an iminium intermediate, which in the presence of an alcohol produced respective N,O-acetals in decent yields. The oxidation potential of electron-rich N,S-acetals is significantly low, which allowed the strategy to be mild and to tolerate various common oxidation-sensitive functional groups in the peptide chain. This electroauxiliary-based approach served well for the late-stage functionalization of various bioactive peptides.
Scheme 90. Electroauxiliary-Assisted Late-Stage Functionalization of Peptides.
4. Photoelectrochemical LSF of Drug-like Molecules
The photoelectrochemistry is an efficient and sustainable tool for organic synthesis.98,404−411 The merger of electrochemistry with photocatalysis411−417 combines their advantages and enhances the utility, which has grown rapidly in the past few years and inaugurated a new frontier in synthetic chemistry. Photoelectrochemical catalysis, which requires no exogenous chemical oxidant and generally exhibits a broad functional group compatibility and high selectivity, is ideally suited for the LSF of structurally complex molecules.418−435
4.1. Photoelectrochemical LSF of C(sp2)–H Bonds
In 2020, Ackermann developed a mild photoelectrochemical C(sp2)–H trifluoromethylation of arenes with CF3SO2Na (Scheme 91).436 This approach featured a broad substrate scope and high functional groups tolerance. The pe-LSF of natural products, including pentoxifylline, doxofylline, theobromine, methyl estrone, and tryptophan, occurred efficiently. Notably, this photoelectrochemical transformation was also achieved in a flow setup with operationally simple online NMR-monitoring. Mechanistic studies indicated that irradiation of photocatalyst Mes-Acr+ led to its highly oxidizing excited state Mes-Acr+*. Then, SET between CF3SO2Na and Mes-Acr+* gave the acridinyl radical Mes-Acr· and a controlled sulfinate radical, which was rapidly converted to a fluoroalkyl radical by cleavage and releasing SO2. The stable radical Mes-Acr· was oxidized at the anode to regenerate Mes-Acr+, while the alkyl radical was trapped by the heteroarene to give a radical cation, which lost a proton and an electron to give the final product.
Scheme 91. Photoelectrochemical C(sp2)–H Trifluoromethylation via Minisci-Type Reactions.
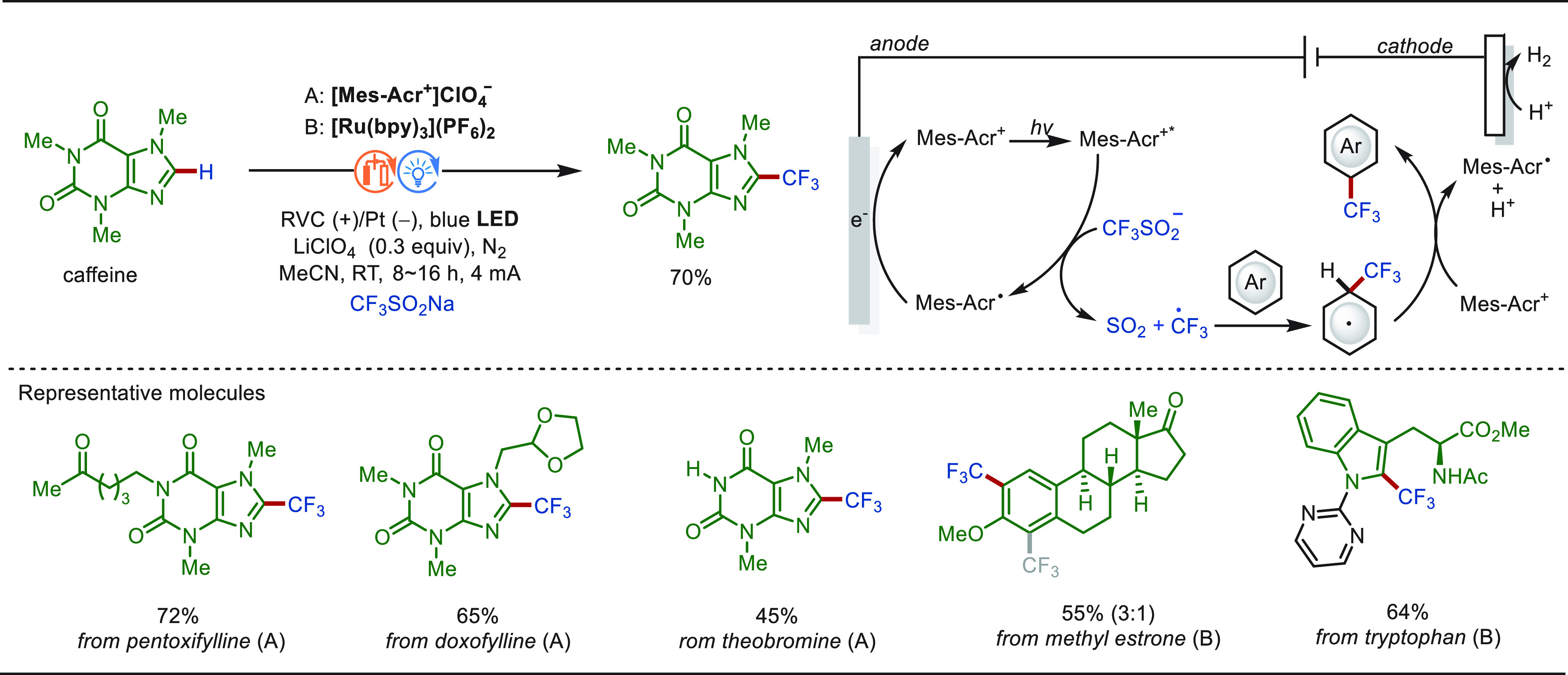
Minisci-type reactions, which furnish net C–H alkylation in heteroarenes by addition of carbon-centered radicals to electron-deficient heterocycles, have attracted broad interest as they provide rapid and direct access to functionalized heterocycles without the need for de novo synthesis.437,438 In 2019, Xu and co-workers uncovered a photoelectrochemical approach for the late-stage C(sp2)–H alkylation of bioactive heteroarenes with stable and easily available organotrifluoroborates (Scheme 92).439 This approach featured the generation of alkyl radicals from organotrifluoroborates without an exogenous chemical oxidant, and various heteroarenes were functionalized with excellent site selectivity and chemoselectivity. Notably, the challenging α-alkoxyl and tertiary radicals also reacted successfully under mild conditions.440 The catalytic cycle is initiated by the irradiation and oxidation of Mes-Acr+, and thus they are prone to undergo single-electron transfer with organotrifluoroborates.
Scheme 92. Photoelectrochemical C–H Alkylation via Minisci Reactions.

In 2020, Xu and co-workers described a photoelectrocatalytic decarboxylative C(sp2)–H alkylation of heteroarenes under the catalysis of CeCl3·H2O and 4CzlPN in a mixture of HFIP/TFE (Scheme 93).441 Carboxylic acids and oxamic acids were used as the alkyl and carbamoyl sources, respectively. These reactions proceed through photoinduced ligand-to-metal charge transfer (LMCT),442−444 which upon decarboxylation generate the alkyl or carbamoyl radicals. The radicals are then trapped with the protonated lepidine and undergo the HER to produce the desired product. Advantageously, this efficient method was scalable to decagram amounts and applicable for the direct C–H alkylation and carbamoylation of drug molecules, such as fasudil, quinoxyfen, quinine, and voriconazole.
Scheme 93. Photoelectrochemical Decarboxylative C–H Alkylation.
Subsequently, the same group reported an elegant dehydrogenative alkylation of heteroarenes with aliphatic C(sp3)–H bonds under photoelecrtochemical conditions (Scheme 94).445 This strategy obviated the use of transition-metal catalysts or chemical oxidants and achieved efficient coupling of a variety of C(sp3)–H donors with a wide range of heteroaromatic drug molecules including quinoxyfen, roflumilast, and fasudil. Mechanistic studies indicated that the C(sp3)–H donor was transformed to a nucleophilic carbon radical through hydrogen-atom transfer (HAT) with chlorine radical, which was generated by light irradiation of anodically produced Cl2.446 The key carbon radical then underwent radical substitution to the protonated heteroarene to afford the alkylated products.
Scheme 94. Photoelectrochemical C–H Alkylation with Alkanes.
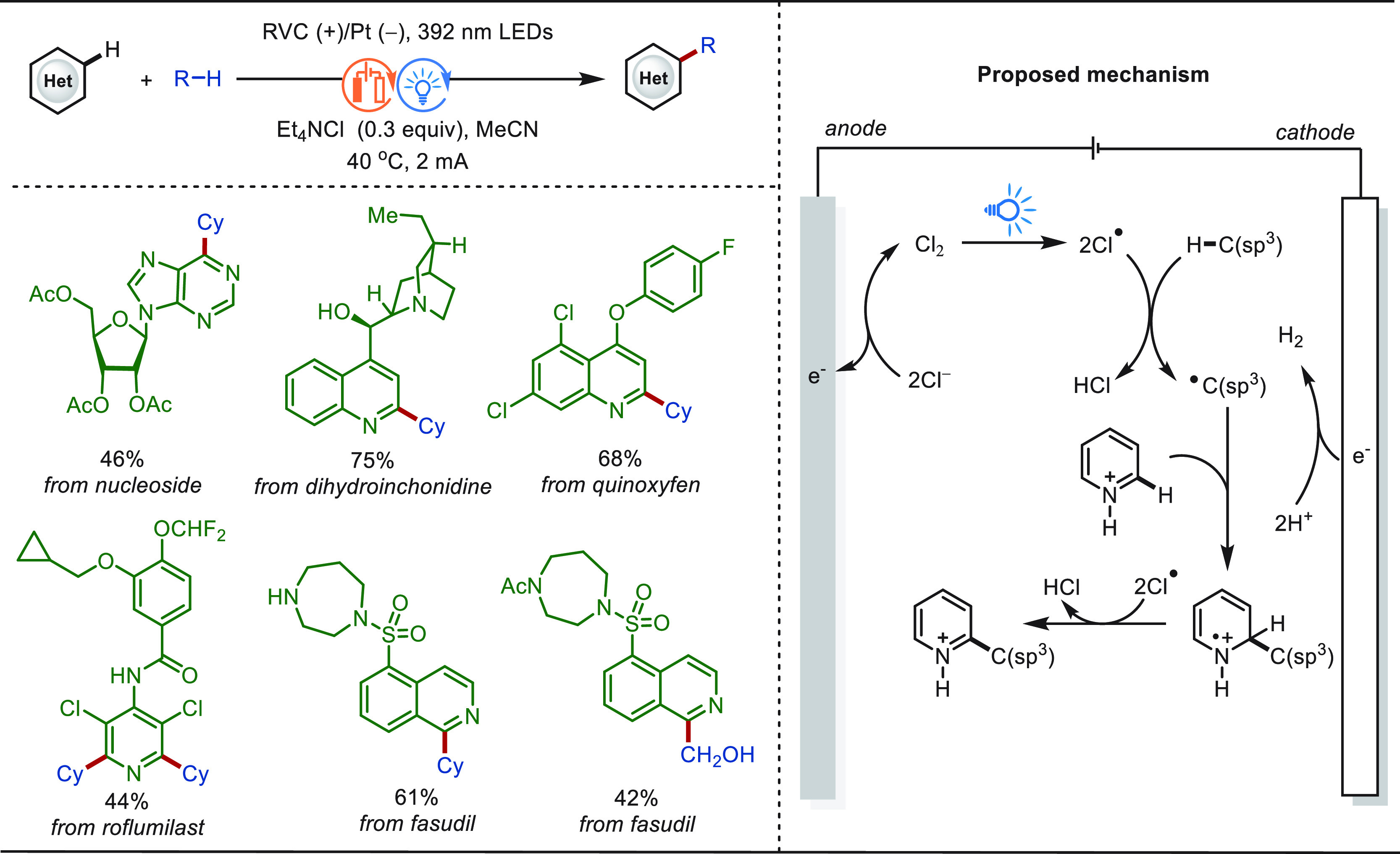
Bench-stable Katritzky salts have been used as an alkylation reagent in various transformations. In this context, Chen and co-workers recently developed a photoelectrocatalytic deaminative alkylation approach (Scheme 95).447 Mechanistic studies indicated that the homogeneously dispersed photocatalyst was essential for the efficient generation of alkyl radicals. Notably, the practicability and robustness of this method were highlighted by the late-stage functionalization of a great number of bioactive molecules.
Scheme 95. Photoelectrochemical C–H Alkylation with Bench-Stable Katritzky Salts.
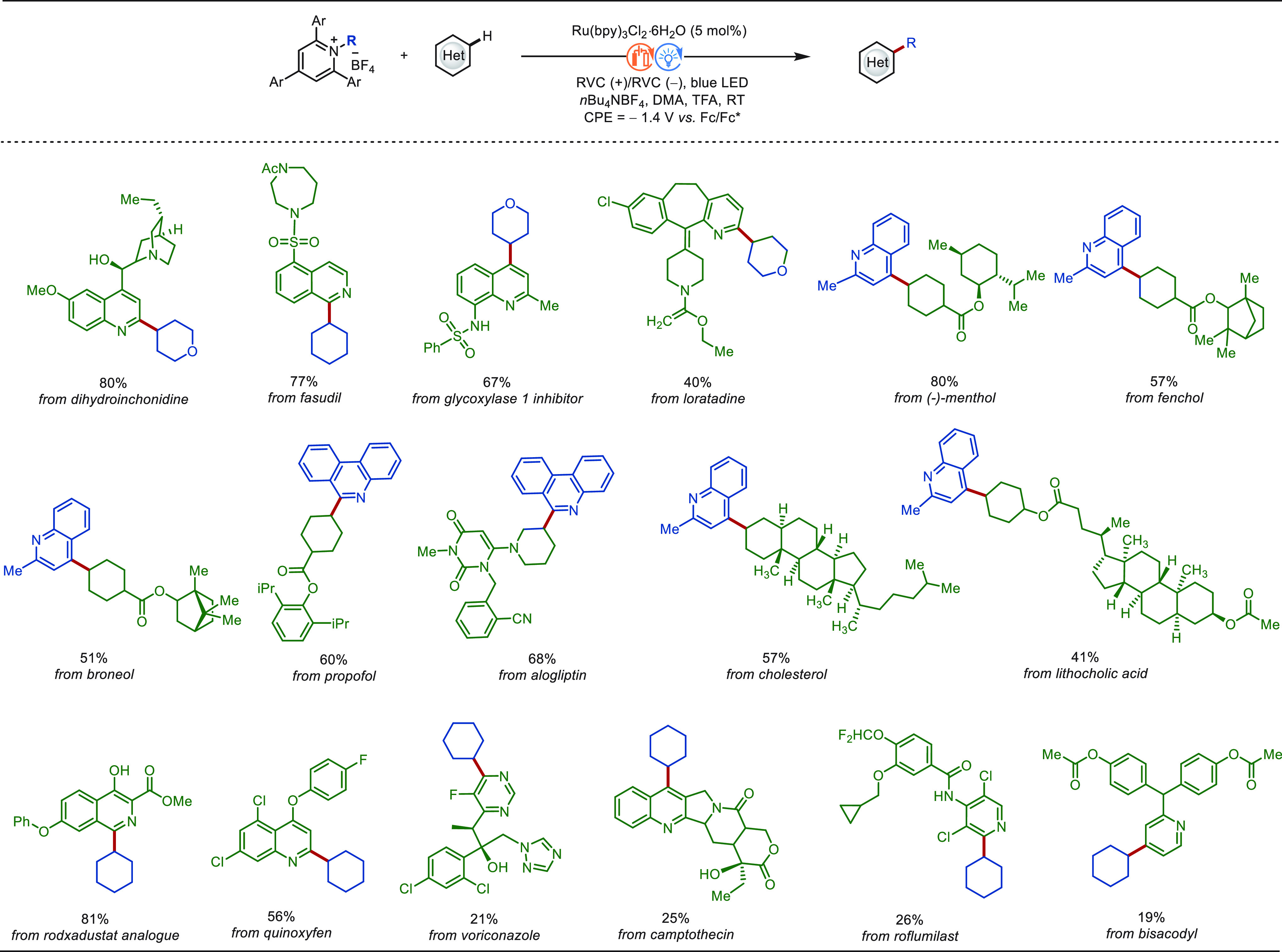
Recently, Wang and Hou developed a photoelectrocatalytic C–H silylation of heteroarenes by dehydrogenative cross-coupling with H2 evolution, which employed 9,10-phenanthrenequinone (PQ) as a photoelectrocatalyst (Scheme 96).448 This strategy avoided the use of an external oxidant or HAT reagent, and a variety of heteroaromatic molecules can be compatible with excellent site-selectivity and desirable yields. Mechanistic studies indicated that the dual function of 9,10-phenanthrenequinone (PQ) enabled the process of the photoelectrocatalytic cycle and hydrogen atom transfer from tBuMe2SiH to afford tBuMe2Si· and PQH·, which later reacted with heteroarenes to afford the ultimate product. The photoelectrocatalytic late-stage C–H silylated tactics could be well wielded in the formation of heteroaromatic bioactive molecules, including fasudil, cinchonidine, famciclovir, desloratadine, fenazaquin, and purine.
Scheme 96. Photoelectrochemical C–H Silylation of Heteroarenes.
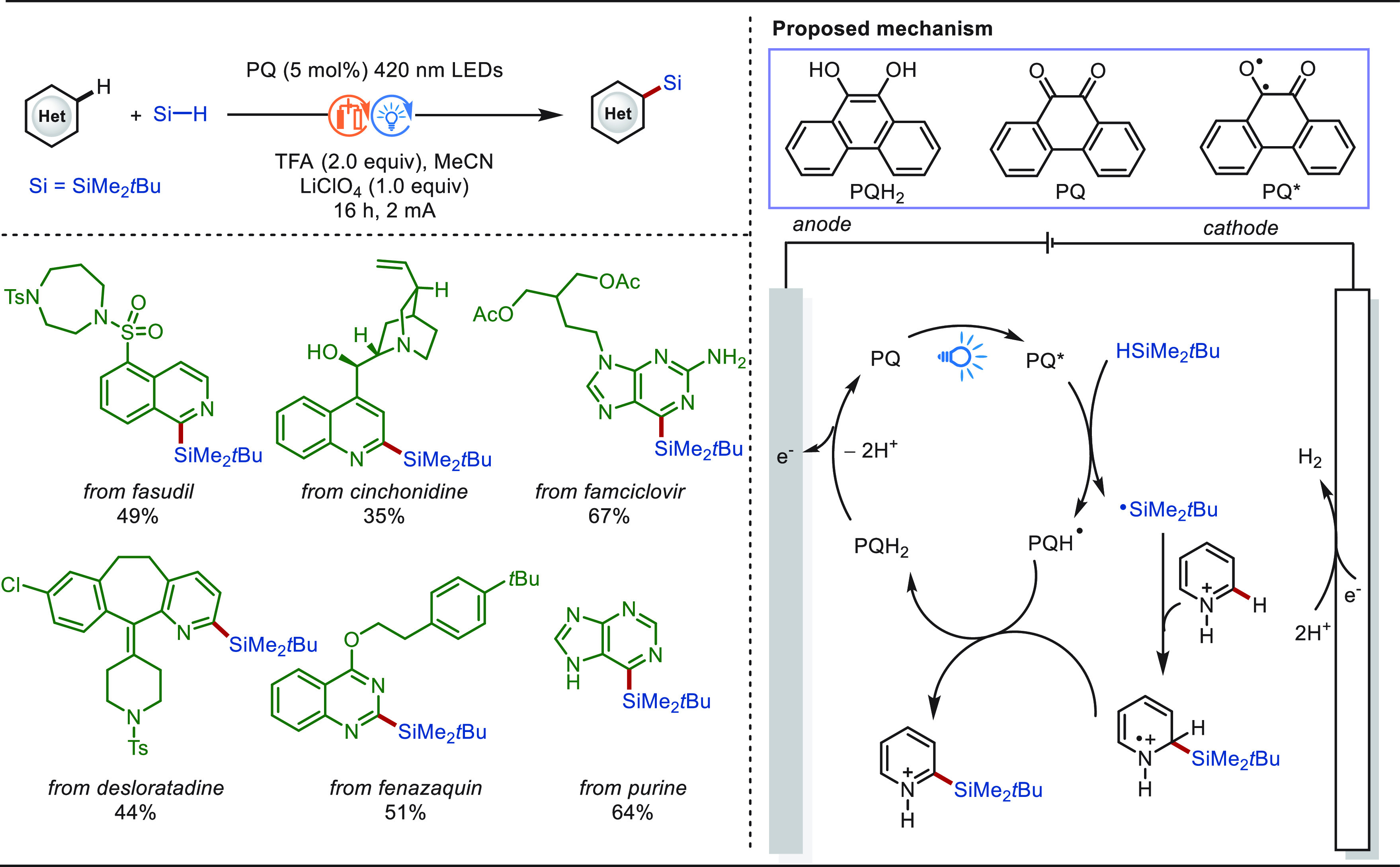
In 2021, Lambert reported a photoelectrocatalytic hydroxylation of arenes by the catalysis of 2,3-dichloro-5,6-dicyanoquinone (DDQ) with visible-light irradiation (Scheme 97).449 Mechanistic studies indicated that the process would be implemented by recycling of DDQ, wherein photoexcited DDQ oxidized arenes through single-electron transfer (SET) to furnish a radical cation that underwent nucleophilic capture. DDQ would be regenerated by anodic oxidation, with H2 released at the cathode.
Scheme 97. Photoelectrochemical Hydroxylation of Aryl C–H bonds.

4.2. Photoelectrochemical LSF of C(sp3)–H Bonds
In 2021, Lambert demonstrated a versatile photoelectrocatalytic diamination of vicinal C–H bonds under the catalysis of trisaminocyclopropenium (TAC) ion in the absence of external oxidants (Scheme 98).450 Photoelectrochemical conditions activate the TAC catalyst to a stable radical dication by anodic oxidation, while the cathodic reaction reduces protons to H2. Irradiation of the TAC radical dication with a light generates a strongly oxidizing photoexcited intermediate, which was an extremely potent oxidant and underwent an electron transfer with olefin followed by a Ritter-type functionalization of C–H bonds with the acetonitrile, a common solvent, as the nitrogen source. Notably, depending on the nature of the electrolyte, both 3,4-dihydroimidazole and 2-oxazoline products were obtained by using simply visible light and a mild electrochemical reaction condition. This photoelectrocatalyzed approach enabled the difunctionalization of a number of antitumor and antiviral active medicinal compound.
Scheme 98. Photoelectrochemical Diamination of Vicinal C–H Bond.
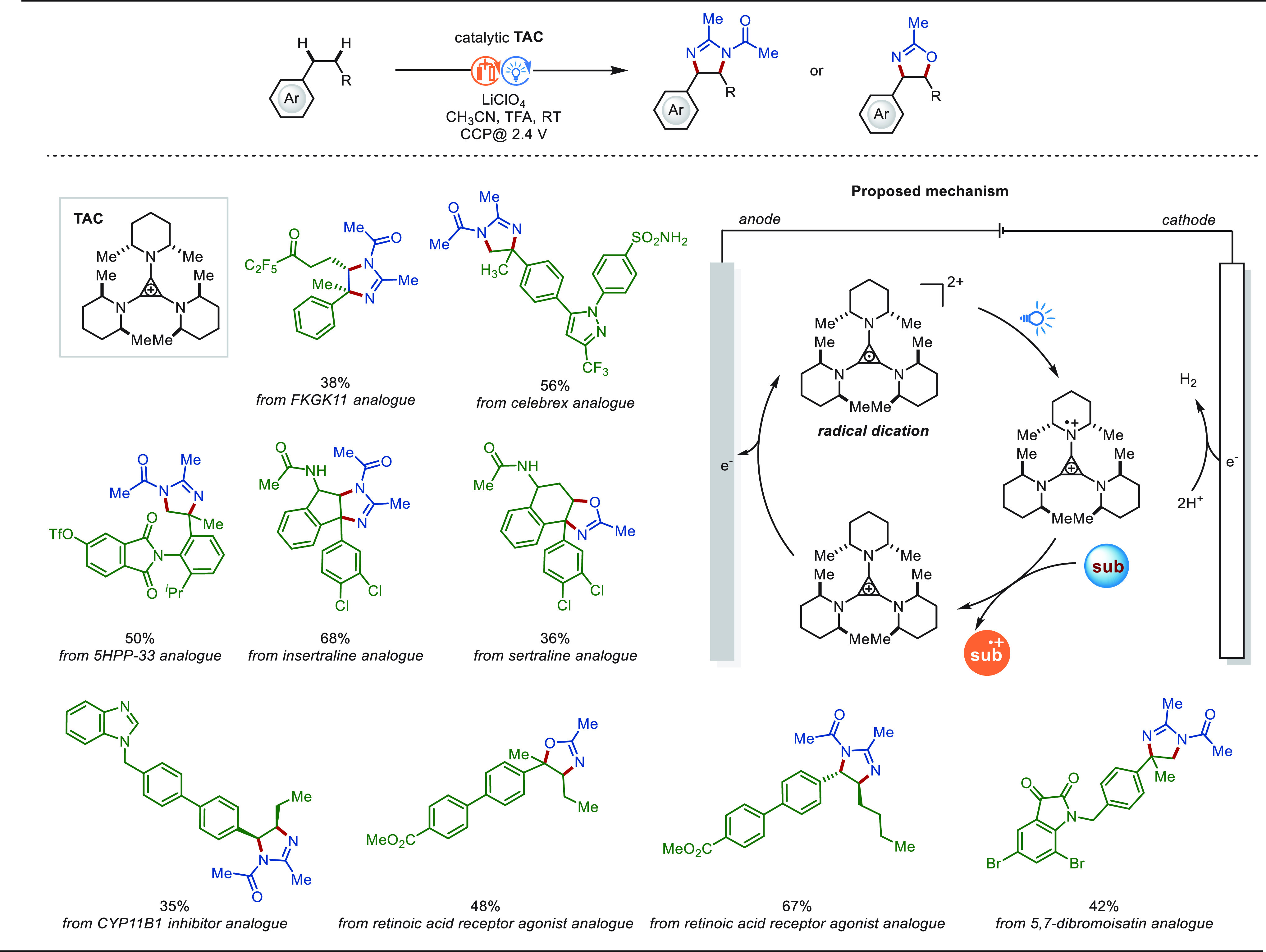
On a different note, Lambert reported an elegant C–H bond amination by the catalysis of trisaminocyclopropenium (TAC) ion in an electrochemical divided cell under white-light compact fluorescent light, where the reaction proceeded through anodic oxidation generating the stable radical dication (Scheme 99).451 Irradiation of radical dication leads to the photoexcited intermediate TAC*, which engages in single-electron oxidation of the arene substrate to generate the key radical cation. Subsequently, the reaction proceeds through the classic Ritter steps with an acetonitrile solvent to form the aminated product. It should be stressed that the introduction of an acetamide moiety could mitigate the kinetic rate of single-electron oxidation by TAC, which would avoid the risk of overoxidation of the C–H amination chemistry. Photoelectrocatalytic processes are more efficient and suitable for the late-stage functionalization of complex natural products than the direct electrochemical one.
Scheme 99. Photoelectrochemical Amination of Benzylic C(sp3)–H Bond.

Subsequently, Lambert and Ye reported photoelectrocatalytic selective oxygenation of either two or three contiguous C(sp3)–H bonds, which exploited a trisaminocyclopropenium (TAC) ion intermediate as a potent oxidative catalyst with excellent selectivity to restrain overoxidative decomposition (Scheme 100).452 Mechanistic investigations reveal that the process involves the sequential oxidation of C(sp3)–H bonds through a relay mechanism. In this intricate process, electro-oxidative and photoexcited triplet aryl cation (TAC) species facilitate the oxidation of C(sp3)–H bonds through a single-electron transfer (SET) process to form radical cations capable of nucleophilic trapping, followed by the giving rise to monooxygen products. In the presence of acidic conditions, the monooxygenated intermediate could undergo a gradual and reversible elimination process, leading to the formation of an olefin. As a result, it can further react to form the dioxygenated adduct or even undergo multiple oxidation events, leading to the formation of di- or trioxygenated products. Notably, the choice of acid (TFA or HOTf) allows for the selective synthesis of two or three contiguous C–O bonds. E1-type elimination was believed to be a critical step and is able to achieve a third C–H oxygenation by using the stronger HOTf acid. The utility of this method was demonstrated by the late-stage modification of bioactive molecules.
Scheme 100. Photoelectrocatalytic Multiple C–H Bonds Oxygenations.
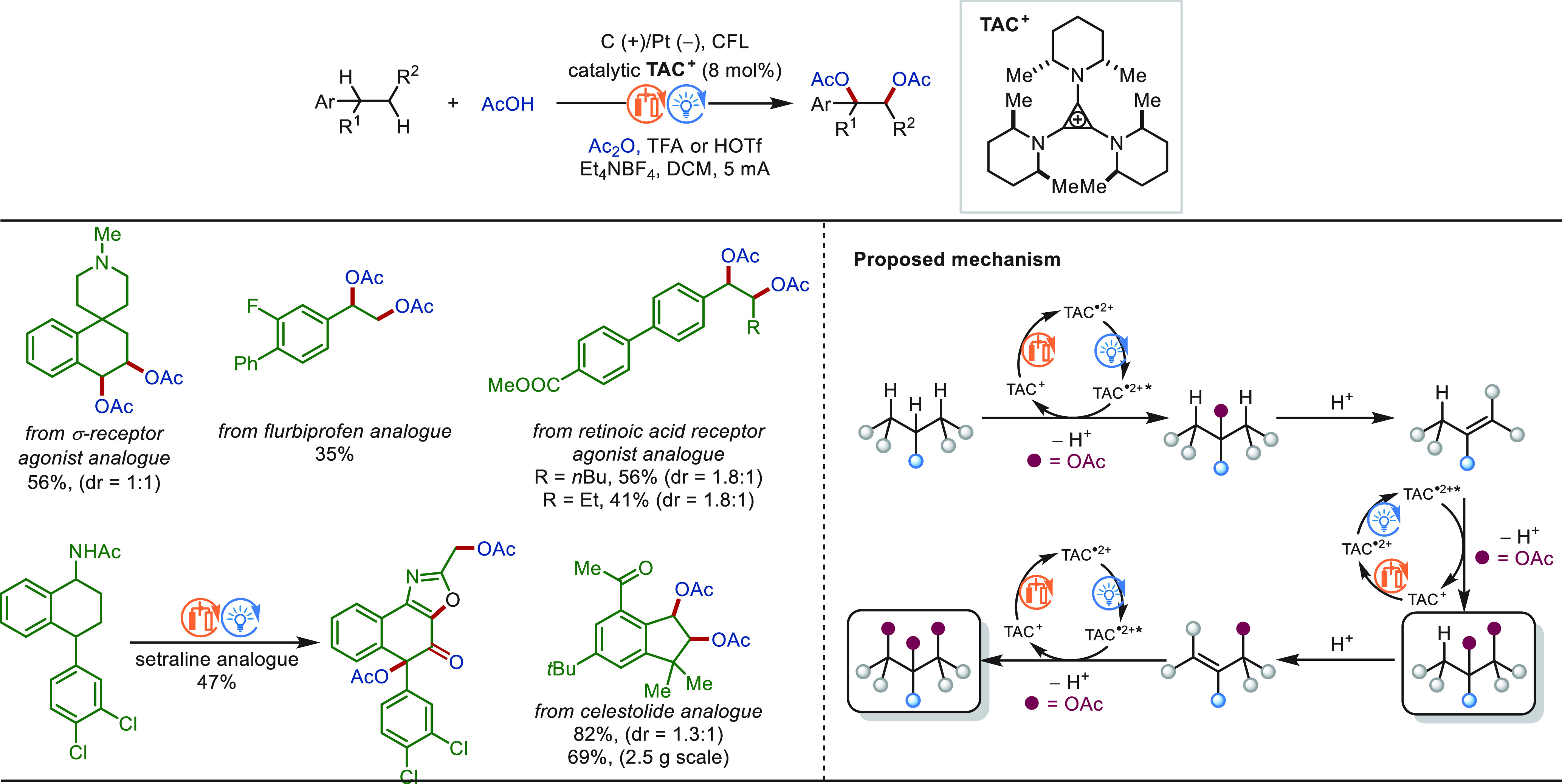
The azide group is a versatile moiety that can be easily reduced to free primary amines, which are featured in pharmaceutical discovery and late-stage functionalization. In 2020, Lei and co-workers have disclosed a manganese-catalyzed azidation of C(sp3)–H bond by merging of visible-light catalysis and electrochemical oxidation (Scheme 101).453 The ketone photocatalysts (DDQ, 9-fluorenone, or 4,4′-dimethoxybenzophenone) were used to generate a C(sp3)-centered radical intermediate via a HAT pathway. In this process, coordination of NaN3 to the manganese(II) followed by anodic oxidation of manganese(II)/L–N3 intermediate generated the manganese(III)/L–N3 intermediate, which furnished a suitable azide radical to generate the azide product. Meanwhile, molecular hydrogen was evaluated at the cathode. Under the photoelectrocatalytic conditions the late-stage azidation of various valuable drug-like molecules could be achieved.
Scheme 101. Photoelectrochemical Manganese-Catalyzed C(sp3)–H Azidation.
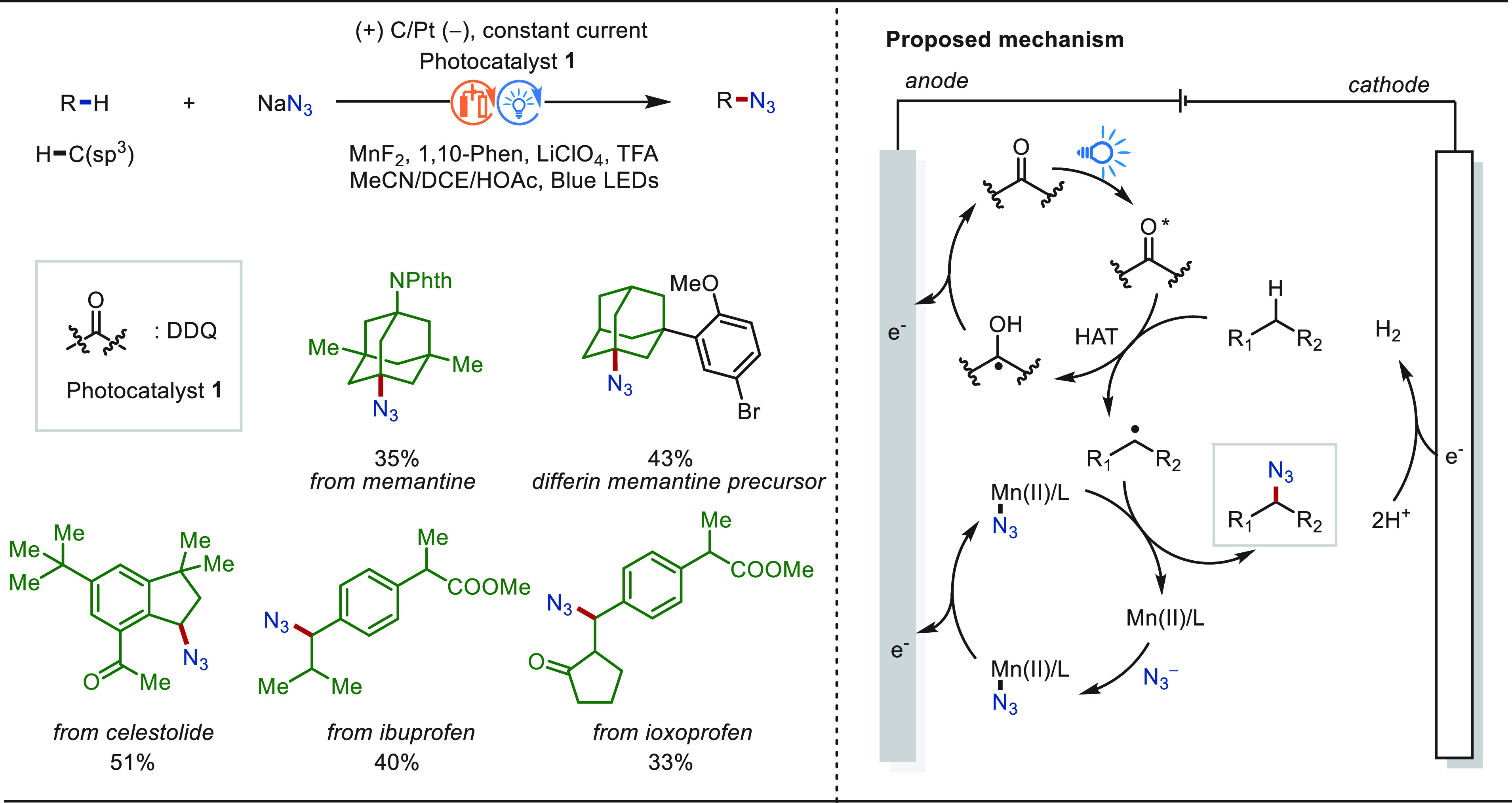
Asymmetric C(sp3)–H functionalization has proven to substantially shorten the process of late-stage modification of complex molecules.4,454,455 Recently, Xu and Liu independently disclosed the first photoelectrocatalytic asymmetric functionalization of C–H bonds (Scheme 102).456,457 The asymmetric photoelectrocatalytic cyanation reaction proceeded in the presence of a copper catalyst in combination with a photocatalyst, featuring a broad substrate scope without the use of any chemical oxidant. The late-stage cyanation of a range of complex structures was achieved in excellent yields with high site selectivity and enantioselectivity. Mechanistic studies suggested the following mode of action: photocatalyst AQDS upon irradiation formed the electronically excited photocatalyst ADQS*, which underwent a single-electron oxidation generating a benzylic radical. The benzylic radical then combined with the chiral copper complex (L*)copper(II)(CN)2 and then after reductive elimination released the desired cyanation product.458,459 The reduced (L*)copper(I)(CN) and intermediate (AQDS–H)· underwent anodic oxidation to regenerate (L1*)copper(II)(CN)2 and AQDS, along with H2 evolution at the cathode. This merger of photoelectrocatalysis and asymmetric copper-catalyzed radical cyanation460 enabled modular control to the radical relay catalysis of various C–H functionalizations.461,462
Scheme 102. Photoelectrochemical Asymmetric Cyanation of Benzylic C–H Bonds.
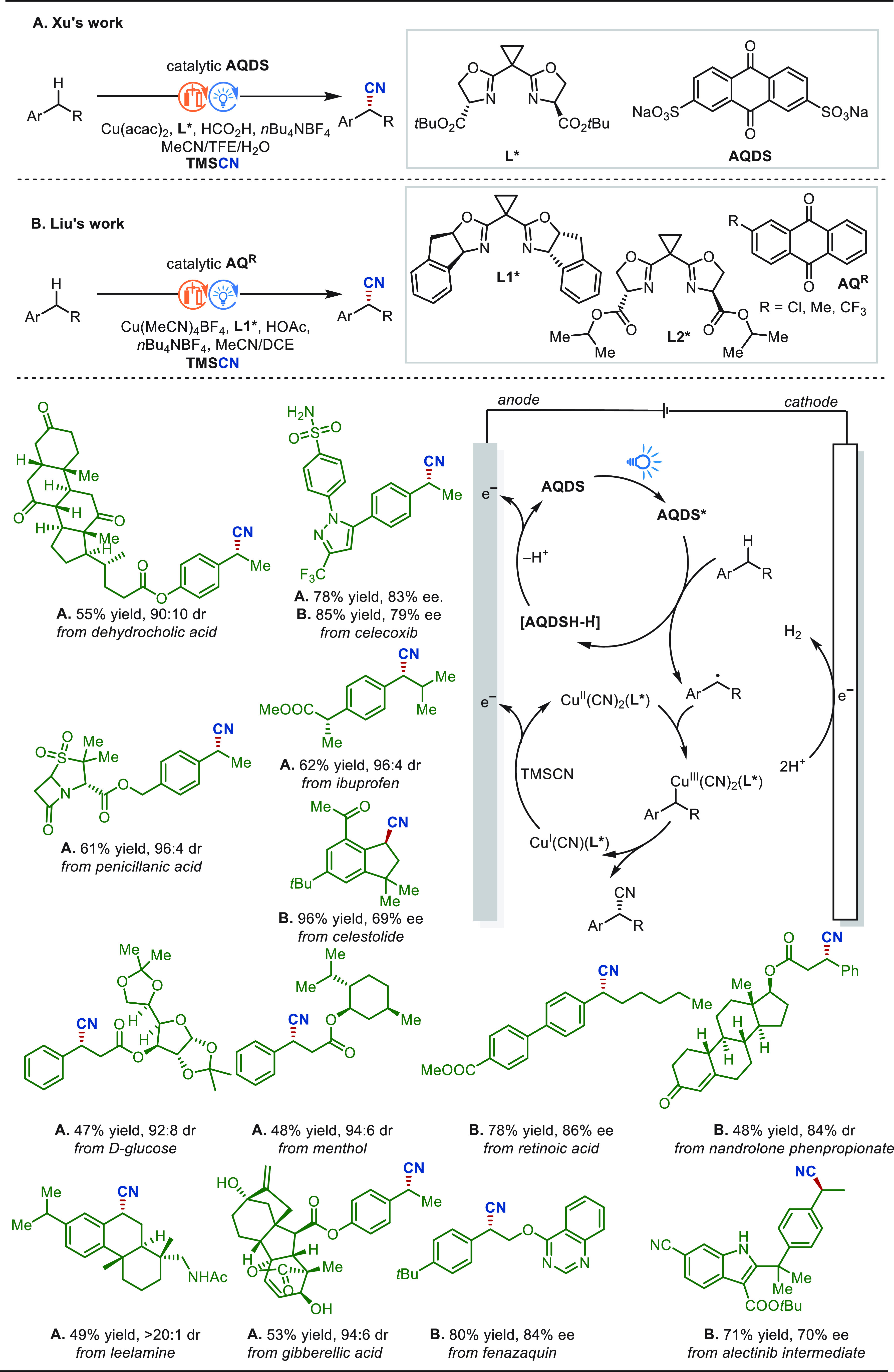
4.3. Miscellaneous Photoelectrochemical LSF
In 2019, Lambert exhibited an elegant approach to acetoxyhydroxylation of aryl olefins with the photoelectrocatalytic trisaminocyclopropenium (TAC) ion intermediate with a controlled electrochemical potential under visible light irradiation (Scheme 103).463 Specifically, this method is triggered by single-electron transfer of the olefin substrate by strongly oxidizing intermediate TAC·2+* to generate an olefin radical cation, which is subsequently trapped with AcOH and further oxidized to an oxocarbenium intermediate, followed by hydrolysis to release the acetoxyhydroxylative product. This approach was applied to the derivatization of complex structures and a range of functional modification.
Scheme 103. Photoelectrochemical Acetoxyhydroxylation of Aryl Olefins.

In 2020, Lambert disclosed an photoelectrocatalytic nucleophilic aromatic substitution reactions of unactivated aryl fluorides under exceedingly mild conditions (Scheme 104).464 Mechanistic investigations revealed that the photoexcitation of DDQ produced an excited state species, which is sufficient to facilitate the single-electron oxidation of fluoroarene. The resulting radical cation processed nucleophilic attack by pyrazole and then generated an aryl radical, which underwent cathodic reduction and the fluoride group left to furnish the corresponding product. The applicability of this approach was demonstrated by the late-stage diversification and syntheses of the drug molecules.
Scheme 104. Photoelectrocatalytic Nucleophilic Aromatic Substitution Reaction.
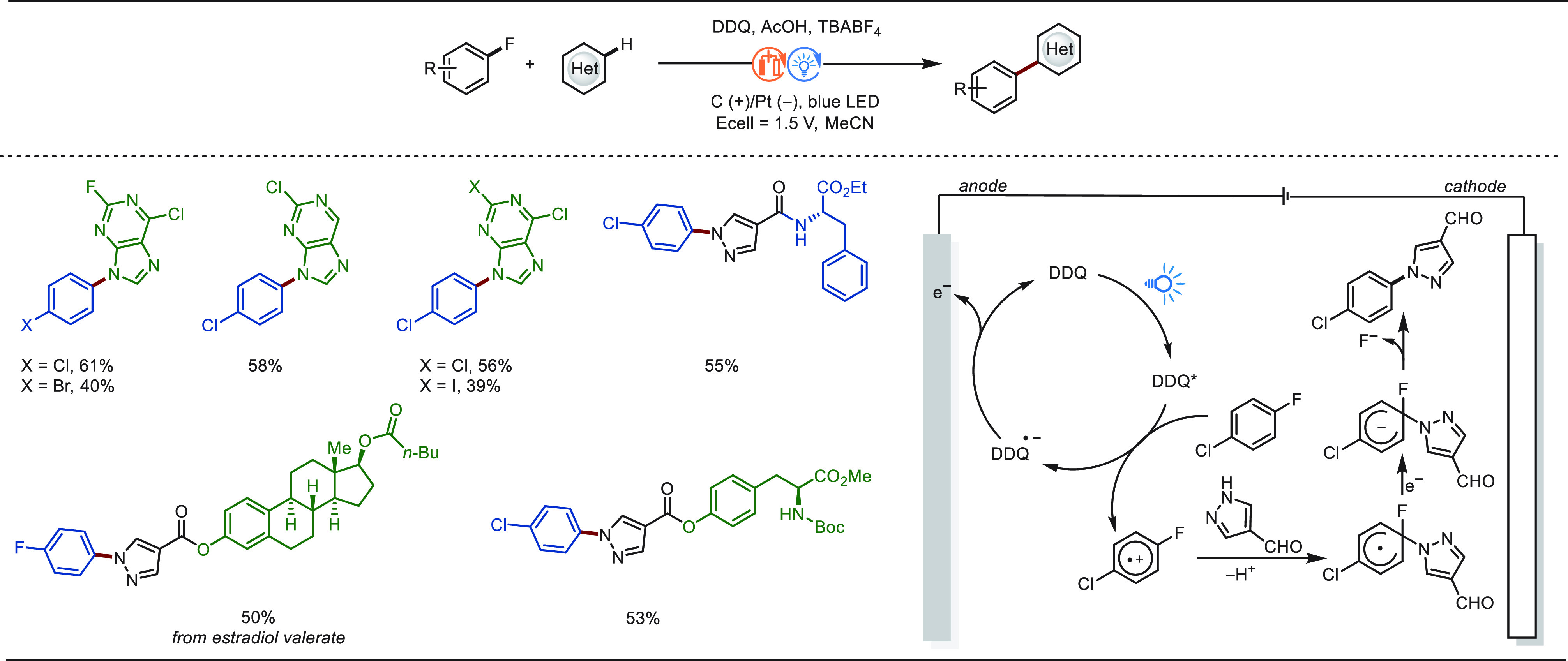
In 2021, Barham and co-workers reported an efficient photoelectrocatalytic method in the reductions of phosphinates derived from α-chloroketones toward olefination and deoxygenation by using 2,6-diisopro-pylphenyl-containing naphthalenemonoimide (NpMI) as a catalyst (Scheme 105).465 A plausible mechanism of the photoelectrocatalysis cycle involves cathodic reduction and photoexcitation to afford a strongly potent reductant. Subsequently, the SET reduction of phosphinates species to its radical anion followed by C(sp3)–O bond cleavage generates a benzyl radical.466 The benzyl radical undergoing reduction to the corresponding carbanion intermediate would further enable either an olefination or a deoxygenation. Surprisingly the photoelectrocatalytic method tolerated aryl chlorides/bromides with similar or more accessible reductive potentials and operated under mild conditions, which favored the late-stage functionalization of complex drug molecules.
Scheme 105. Photoelectrochemical C(sp3)–O Cleavages of Phosphinated Alcohols to Carbanions.

Based on the success of the proof-of-concept asymmetric photoelectrocatalytic approach, Xu subsequently devised a photoelectrochemical enantioselective decarboxylative cyanation, which converted racemic carboxylic acids directly to enantioenriched nitriles (Scheme 106A).467 This method employed a cerium(III) salt and a chiral copper(II)(L*) as the relay catalysis, which proceeded through the cerium salt for photoelectrocatalytic decarboxylation and a chiral copper complex for enantioselectivity control. Key to the success was the ideal incorporation of photoelectrocatalytic tactics with asymmetric copper catalytic cycle, and both catalysts were regenerated by anodic oxidation. The photoelectrochemical asymmetric approach converted straightforwardly commercial carboxylic-acid-based drug molecules to their corresponding enantioenriched nitriles, including flurbiprofen, ketoprofen, loxoprofen, naproxen, zaltoprofen, and pranoprofen. Coincidentally, the same photoelectrochemical strategy, in which cerium(III) salt and copper(II)(BOX*) were employed as cocatalysts to promote the catalytic decarboxylation and asymmetric cyanation was reported by Zhang (Scheme 106B).468
Scheme 106. Photoelectrochemical Asymmetric Decarboxylative Cyanation.

5. Conclusion and Perspective
Over the past decade, electrocatalysis has undergone a remarkable renaissance and has been identified as an increasingly robust tool in molecular sciences. Consequentially, significant recent momentum has been gained by applications of molecular electrochemistry to late-stage functionalization (LSF). Thus, eLSF was accomplished with a variety of natural products, drugs, peptides, proteins, as well as other bioactive molecules bearing complex structures, typically featuring exceedingly mild conditions with a high level of selectivity control. These eLSFs are characterized by remarkable advantages, namely, the use of electrons and protons as sustainable redox agents, obviating the need of chemical oxidants/reductants; new mechanistic routes enabling reactions to proceed under ambient temperature rather than superheated conditions; direct formation of target molecules via electrolysis, greatly shortening the synthetic steps; exhibiting increased selectivity or different selectivity compared to traditional methods; accomplishing specific transformations that could not be achieved before; and so on. Notably, the application of electrochemical continuous-flow techniques, which feature improved mass transfer, low residence times, and high surface-to-volume ratios, has greatly accelerated the development of the LSF field by increasing reaction efficiency, reducing overoxidation, and simplifying scale-up. In addition, the merger of electrochemistry with photochemistry, so-called photoelectrochemistry, shows huge application potential and has yet to make several dazzling achievements.
Despite the remarkable recent progress, the electro-organic chemistry as well as the eLSF arena are arguably still in their infancies. Here we address the present limitations of eLSF, which might also represent some valuable research directions in the future.
-
i)
The established eLSF methodologies are still rather limited considering the huge demand in drug-discovery programs. For instance, the reported late-stage methylation only occurs in the presence of several restricted nitrogen-oriented directing groups. The development of general strategies for the electrochemical late-stage methylation of complex molecules bears a significant synthetic space. Similarly, strategies describing straightforward and selective inclusion of functionalities such as trifluoromethyl, halogens, methoxy, amino, hydroxy, nitro, methylamino, ethoxy, carbonyl, etc., which comprise high relevance in medicinal chemistry and determine the structure–activity relationship, are limited. Thus, there is a need for further exploration toward increasing the scope of substrate classes and their widespread application in eLSF reactions. The incorporation of a fluorine atom onto an aromatic ring of a drug-like molecule, which is undoubtedly of great significance, has not yet been reported. Furthermore, the development of eLSF strategies for the functionalization with bioisosteres is underexplored and requires immediate consideration.
-
ii)
The labeling or modification of peptides, proteins as well as sugars plays a key role in modern drug discovery. To date, only a limited number of electrochemical peptides/proteins LSF approaches have been disclosed, and these methods mainly focus on the modification of tyrosine or tryptophan side motifs or require prefunctionalization with oxidation-sensitive electroauxiliaries for selectivity control. Developing electrochemical strategies beyond this prefunctionalization would obviously alleviate the synthetic prospects of late-stage peptide and protein modification reactions. Meanwhile, practical eLSF of sugar derivatives remains largely elusive, which needs special attention to resolve.
-
iii)
While a large proportion of drug molecules bear chiral structures, the enantioselective eLSF, or even the electrochemical asymmetric synthesis, has thus far been met with limited success. Hence, more efforts should be devoted toward the development of full selectivity control in asymmetric electrocatalysis.
-
iv)
Electrode materials show great influence on electrochemical LSF reactions. While traditional electrode materials such as graphite, carbon, and metal electrodes have been extensively used in electrochemical transformations, there is a growing interest in exploring new materials with tailored properties. For instance, the development of novel electrode materials with improved catalytic activity and stability, such as modified electrodes, nanomaterials, catalyst-supported electrodes, and metal–organic frameworks (MOFs) may expand the scope of electrochemical LSF in drug discovery.
-
v)
Electrolyte selection is crucial in electrochemical LSF reactions as it influences the reaction kinetics and efficiency. Evaluating the impact of different electrolytes, including ionic liquids or redox mediators, on reaction outcomes and exploring tailored electrolytes, including the optimization of their composition and redox potential, could broaden the applicability of electrochemistry in drug discovery.
-
vi)
Constant current electrolysis (CCE) is the most commonly used electrochemical method. It allows for controlled reaction rates and provides simpler experimental setups, but suffers from low selectivity and competitive reactions in some cases. By contrast, controlled potential electrolysis (CPE) allows precise control over the reaction potential, enabling selective transformations, and hence deserves more attention in the eLSF arena. In addition, the recent surge of alternating current (AC) shows its meticulous control of specific reaction pathways and exact chemoselectivity. A profound comprehension of the merits and constraints of each method can aid in the development of more efficient electrochemical LSF strategies.
In summary, organic electrosynthesis has emerged as an increasingly viable and powerful platform with ideal levels of the resource economy for late-stage functionalization. We expect major advances of eLSF in drug discovery, materials science, crop protection, as well as other areas in the near future.
Acknowledgments
The authors gratefully acknowledge support from DZHK and the DFG (Gottfried-Wilhelm-Leibniz award to L.A.), the ERC Advanced (Grant no. 101021358), the Alexander-von-Humboldt Foundation (fellowship to Y.W.), and the CSC (fellowships to Y.X.).
Biographies
Yulei Wang received his M.Sc. degree from Xiamen University and Ph.D. from Shanghai Institute of Organic Chemistry under the supervision of Prof. Zheng Huang. Since 2019, he has been conducting postdoctoral research with Prof. Lutz Ackermann at Georg-August University Göttingen. His research interests mainly include organometallic catalysis, C–H activation, and electrochemistry.
Suman Dana was born in 1992 in Bankura, West Bengal. He obtained his Ph.D. from Indian Institute of Technology Madras, India, in 2021 under the supervision of Prof. Mahiuddin Baidya. He joined Georg-August University Göttingen in September, 2021 in the group of Prof. Lutz Ackermann as a postdoctoral fellow. His research focuses on the development of electro-oxidative asymmetric C–H activation reactions.
Hao Long received his Ph.D. from Xiamen University under the supervision of Professor Hai-chao Xu. He has engaged in postdoctoral research in the group of Prof. Lutz Ackermann at Georg-August University Göttingen since November 2022. His research interest mainly focuses on photoelectrocatalytic C–H bond functionalization reactions.
Yang Xu received his M.Sc. from Zhengzhou University. He joined Georg-August University Göttingen in February 2022 in the group of Prof. Lutz Ackermann as a Ph.D. candidate. His research focuses on the development of electro-oxidative asymmetric C–H activation reactions.
Yanjun Li was born in 1992 in Hubei, China. He received his Ph.D. in Organic Chemistry from Xiamen University in 2020 under the supervision of Prof. Lei Gong. He then joined the group of Prof. Lei Gong as a research assistant. Since 2021, he has been conducting postdoctoral research with Prof. Lutz Ackermann at Georg-August University Göttingen. His research interest mainly focuses on asymmetric C–H activation.
Nikolaos Kaplanaris was born in 1992 in Athens, Greece. He received his B.Sc. degree in Chemistry from the National and Kapodistrian University of Athens in 2014. He obtained his M.Sc. degree in Organic Chemistry from the same university in 2016 following studies in the area of organocatalysis and photochemistry under the supervision of Prof. C. G. Kokotos. In 2016, he joined the research group of Prof. Lutz Ackermann at the Georg-August-Universität Göttingen as an Onassis fellow, and obtained his Ph.D. degree in 2021, working on late-stage peptide diversification and remote functionalization. After a short postdoctoral stay at the same group, working on electrochemical modifications of biomolecules, he joined the group of Prof. T. Noël at the University of Amsterdam where he is developing photochemical methodologies in flow.
Lutz Ackermann studied Chemistry at the Christian-Albrechts University Kiel and received his Ph.D. in 2001 for research under the supervision of Prof. Dr. Alois Fürstner from Max-Planck-Institut für Kohlenforschung in Mülheim/Ruhr. He was a postdoctoral co-worker in the laboratory of Prof. Dr. Robert G. Bergman at the University of California at Berkeley before initiating his independent research career in 2003 at the Ludwig Maximilians-University München supported by the Emmy Noether programme of the DFG. In 2007, he was appointed as a Full (W3) Professor at the Georg-August University Göttingen. His recent awards include an ERC Advanced Grant (2021) and the Gottfried Wilhelm Leibniz-Preis (2017) as well as visiting professorships at the Università degli Studi di Milano, the University of Wisconsin at Madison, and the Università di Pavia. The development and application of novel concepts for sustainable catalysis constitute his major research interests, with a current topical focus on C–H activation.
Author Contributions
† Y.W., S.D., and H.L. contributed equally.
The authors declare no competing financial interest.
Special Issue
Published as part of the Chemical Reviewsvirtual special issue “Remote and Late Stage Functionalization”.
References
- Guillemard L.; Kaplaneris N.; Ackermann L.; Johansson M. J. Late-Stage C–H Functionalization Offers New Opportunities in Drug Discovery. Nat. Rev. Chem. 2021, 5, 522–545. 10.1038/s41570-021-00300-6. [DOI] [PubMed] [Google Scholar]
- Lasso J. D.; Castillo-Pazos D. J.; Li C.-J. Green Chemistry Meets Medicinal Chemistry: A Perspective on Modern Metal-Free Late-Stage Functionalization Reactions. Chem. Soc. Rev. 2021, 50, 10955–10982. 10.1039/D1CS00380A. [DOI] [PubMed] [Google Scholar]
- Dominguez-Huerta A.; Dai X.-J.; Zhou F.; Querard P.; Qiu Z.; Ung S.; Liu W.; Li J.; Li C.-J. Exploration of New Reaction Tools for Late-Stage Functionalization of Complex Chemicals. Can. J. Chem. 2019, 97, 67–85. 10.1139/cjc-2018-0357. [DOI] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. W. The Medicinal Chemist’s Toolbox for Late Stage Functionalization of Drug-Like Molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- Moir M.; Danon J. J.; Reekie T. A.; Kassiou M. An Overview of Late-Stage Functionalization in Today’s Drug Discovery. Expert Opin. Drug Discovery 2019, 14, 1137–1149. 10.1080/17460441.2019.1653850. [DOI] [PubMed] [Google Scholar]
- Aynetdinova D.; Callens M. C.; Hicks H. B.; Poh C. Y. X.; Shennan B. D. A.; Boyd A. M.; Lim Z. H.; Leitch J. A.; Dixon D. J. Installing the “Magic Methyl” – C–H Methylation in Synthesis. Chem. Soc. Rev. 2021, 50, 5517–5563. 10.1039/D0CS00973C. [DOI] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Ravelli D. Photocatalytic Hydrogen Atom Transfer: The Philosopher’s Stone for Late-Stage Functionalization?. Green Chem. 2020, 22, 3376–3396. 10.1039/D0GC01035A. [DOI] [Google Scholar]
- Boström J.; Brown D. G.; Young R. J.; Keserü G. M. Expanding the Medicinal Chemistry Synthetic Toolbox. Nat. Rev. Drug Discovery 2018, 17, 709–727. 10.1038/nrd.2018.116. [DOI] [PubMed] [Google Scholar]
- Leung C. S.; Leung S. S. F.; Tirado-Rives J.; Jorgensen W. L. Methyl Effects on Protein–Ligand Binding. J. Med. Chem. 2012, 55, 4489–4500. 10.1021/jm3003697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski D. W.; Futatsugi K.; Casimiro-Garcia A.; Wei L.; Sammons M. F.; Herr M.; Jiao W.; Lavergne S. Y.; Coffey S. B.; Wright S. W.; et al. Identification of Morpholino-2H-Pyrido[3,2-b][1,4]Oxazin-3(4H)-Ones as Nonsteroidal Mineralocorticoid Antagonists. J. Med. Chem. 2018, 61, 1086–1097. 10.1021/acs.jmedchem.7b01515. [DOI] [PubMed] [Google Scholar]
- Friis S. D.; Johansson M. J.; Ackermann L. Cobalt-Catalysed C–H Methylation for Late-Stage Drug Diversification. Nat. Chem. 2020, 12, 511–519. 10.1038/s41557-020-0475-7. [DOI] [PubMed] [Google Scholar]
- Dai H.-X.; Stepan A. F.; Plummer M. S.; Zhang Y.-H.; Yu J.-Q. Divergent C–H Functionalizations Directed by Sulfonamide Pharmacophores: Late-Stage Diversification as a Tool for Drug Discovery. J. Am. Chem. Soc. 2011, 133, 7222–7228. 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- Simonetti M.; Cannas D. M.; Just-Baringo X.; Vitorica-Yrezabal I. J.; Larrosa I. Cyclometallated Ruthenium Catalyst Enables Late-Stage Directed Arylation of Pharmaceuticals. Nat. Chem. 2018, 10, 724–731. 10.1038/s41557-018-0062-3. [DOI] [PubMed] [Google Scholar]
- Kaplaneris N.; Rogge T.; Yin R.; Wang H.; Sirvinskaite G.; Ackermann L. Late-Stage Diversification through Manganese-Catalyzed C–H Activation: Access to Acyclic, Hybrid, and Stapled Peptides. Angew. Chem., Int. Ed. 2019, 58, 3476–3480. 10.1002/anie.201812705. [DOI] [PubMed] [Google Scholar]
- Wang J.; Li R.; Dong Z.; Liu P.; Dong G. Complementary Site-Selectivity in Arene Functionalization Enabled by Overcoming the Ortho Constraint in Palladium/Norbornene Catalysis. Nat. Chem. 2018, 10, 866–872. 10.1038/s41557-018-0074-z. [DOI] [PubMed] [Google Scholar]
- Rodrigalvarez J.; Nappi M.; Azuma H.; Flodén N. J.; Burns M. E.; Gaunt M. J. Catalytic C(sp3)–H Bond Activation in Tertiary Alkylamines. Nat. Chem. 2020, 12, 76–81. 10.1038/s41557-019-0393-8. [DOI] [PubMed] [Google Scholar]
- He Z.-T.; Li H.; Haydl A. M.; Whiteker G. T.; Hartwig J. F. Trimethylphosphate as a Methylating Agent for Cross Coupling: A Slow-Release Mechanism for the Methylation of Arylboronic Esters. J. Am. Chem. Soc. 2018, 140, 17197–17202. 10.1021/jacs.8b10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pony Yu R.; Hesk D.; Rivera N.; Pelczer I.; Chirik P. J. Iron-Catalysed Tritiation of Pharmaceuticals. Nature 2016, 529, 195–199. 10.1038/nature16464. [DOI] [PubMed] [Google Scholar]
- Shi H.; Lu Y.; Weng J.; Bay K. L.; Chen X.; Tanaka K.; Verma P.; Houk K. N.; Yu J.-Q. Differentiation and Functionalization of Remote C–H Bonds in Adjacent Positions. Nat. Chem. 2020, 12, 399–404. 10.1038/s41557-020-0424-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Lorion M. M.; Martinazzoli O.; Ackermann L. Bodipy Peptide Labeling by Late-Stage C(sp3)–H Activation. Angew. Chem., Int. Ed. 2018, 57, 10554–10558. 10.1002/anie.201804654. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wu J.; Kuniyil R.; Kopp A.; Lima R. N.; Ackermann L. Peptide Late-Stage Diversifications by Rhodium-Catalyzed Tryptophan C7 Amidation. Chem. 2020, 6, 3428–3439. 10.1016/j.chempr.2020.10.026. [DOI] [Google Scholar]
- Le C.; Liang Y.; Evans R. W.; Li X.; MacMillan D. W. C. Selective sp3 C–H Alkylation via Polarity-Match-Based Cross-Coupling. Nature 2017, 547, 79–83. 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls T. P.; Leonori D.; Bissember A. C. Applications of Visible Light Photoredox Catalysis to the Synthesis of Natural Products and Related Compounds. Nat. Prod. Rep. 2016, 33, 1248–1254. 10.1039/C6NP00070C. [DOI] [PubMed] [Google Scholar]
- Douglas J. J.; Sevrin M. J.; Stephenson C. R. J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev. 2016, 20, 1134–1147. 10.1021/acs.oprd.6b00125. [DOI] [Google Scholar]
- Ruffoni A.; Juliá F.; Svejstrup T. D.; McMillan A. J.; Douglas J. J.; Leonori D. Practical and Regioselective Amination of Arenes Using Alkyl Amines. Nat. Chem. 2019, 11, 426–433. 10.1038/s41557-019-0254-5. [DOI] [PubMed] [Google Scholar]
- Loh Y. Y.; Nagao K.; Hoover A. J.; Hesk D.; Rivera N. R.; Colletti S. L.; Davies I. W.; MacMillan D. W. C. Photoredox-Catalyzed Deuteration and Tritiation of Pharmaceutical Compounds. Science 2017, 358, 1182–1187. 10.1126/science.aap9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Chen J.; Sang R.; Ham W.-S.; Plutschack M. B.; Berger F.; Chabbra S.; Schnegg A.; Genicot C.; Ritter T. Photoredox Catalysis with Aryl Sulfonium Salts Enables Site-Selective Late-Stage Fluorination. Nat. Chem. 2020, 12, 56–62. 10.1038/s41557-019-0353-3. [DOI] [PubMed] [Google Scholar]
- Chen W.; Huang Z.; Tay N. E. S.; Giglio B.; Wang M.; Wang H.; Wu Z.; Nicewicz D. A.; Li Z. Direct Arene C–H Fluorination with 18F– via Organic Photoredox Catalysis. Science 2019, 364, 1170–1174. 10.1126/science.aav7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Lee J. W.; Morales-Rivera C. A.; Liu P.; Ngai M.-Y. Redox-Active Reagents for Photocatalytic Generation of the OCF3 Radical and (Hetero)Aryl C–H Trifluoromethoxylation. Angew. Chem., Int. Ed. 2018, 57, 13795–13799. 10.1002/anie.201808495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Guo X.; Shi Y.; He C.; Duan C. Dye-Incorporated Coordination Polymers for Direct Photocatalytic Trifluoromethylation of Aromatics at Metabolically Susceptible Positions. Nat. Commun. 2018, 9, 4024–4032. 10.1038/s41467-018-05919-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.; MacMillan D. W. C. Alcohols as Alkylating Agents in Heteroarene C–H Functionalization. Nature 2015, 525, 87–90. 10.1038/nature14885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiRocco D. A.; Dykstra K.; Krska S.; Vachal P.; Conway D. V.; Tudge M. Late-Stage Functionalization of Biologically Active Heterocycles through Photoredox Catalysis. Angew. Chem., Int. Ed. 2014, 53, 4802–4806. 10.1002/anie.201402023. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Xiang X.-X.; Chen J.; Yang C.; Pan Y.-L.; Cheng J.-P.; Meng Q.; Li X. Direct C–H Difluoromethylation of Heterocycles via Organic Photoredox Catalysis. Nat. Commun. 2020, 11, 638–647. 10.1038/s41467-020-14494-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.-X.; Morales-Rivera C. A.; Wang Y.; Gao F.; He G.; Liu P.; Chen G. Photoredox-Mediated Minisci C–H Alkylation of N-Heteroarenes Using Boronic Acids and Hypervalent Iodine. Chem. Sci. 2016, 7, 6407–6412. 10.1039/C6SC02653B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawayama A. M.; Chen M. M. Y.; Kulanthaivel P.; Kuo M.-S.; Hemmerle H.; Arnold F. H. A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chem. Eur. J. 2009, 15, 11723–11729. 10.1002/chem.200900643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. C.; Coelho P. S.; Arnold F. H. Enzymatic Functionalization of Carbon–Hydrogen Bonds. Chem. Soc. Rev. 2011, 40, 2003–2021. 10.1039/C0CS00067A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryszkowska A.; Devine P. N. Biocatalysis in Drug Discovery and Development. Curr. Opin. Chem. Biol. 2020, 55, 151–160. 10.1016/j.cbpa.2020.01.012. [DOI] [PubMed] [Google Scholar]
- Chakrabarty S.; Wang Y.; Perkins J. C.; Narayan A. R. H. Scalable Biocatalytic C–H Oxyfunctionalization Reactions. Chem. Soc. Rev. 2020, 49, 8137–8155. 10.1039/D0CS00440E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolev J. N.; O’Dwyer K. M.; Jordan C. T.; Fasan R. Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C—H Functionalization. ACS Chem. Bio. 2014, 9, 164–173. 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Shafer B. M.; Demars M. D. II; Stern H. A.; Fasan R. Controlled Oxidation of Remote sp3 C–H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J. Am. Chem. Soc. 2012, 134, 18695–18704. 10.1021/ja3073462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loskot S. A.; Romney D. K.; Arnold F. H.; Stoltz B. M. Enantioselective Total Synthesis of Nigelladine a Via Late-Stage C–H Oxidation Enabled by an Engineered P450 Enzyme. J. Am. Chem. Soc. 2017, 139, 10196–10199. 10.1021/jacs.7b05196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell A. N.; DeMars M. D. II; Slocum S. T.; Yu F.; Anand K.; Chemler J. A.; Korakavi N.; Priessnitz J. K.; Park S. R.; Koch A. A.; et al. Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C–H Functionalization. J. Am. Chem. Soc. 2017, 139, 7913–7920. 10.1021/jacs.7b02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Ligibel M.; Sager E.; Voss M.; Hunziker J.; Schroer K.; Snajdrova R.; Buller R. Evolved Aliphatic Halogenases Enable Regiocomplementary C–H Functionalization of a Pharmaceutically Relevant Compound. Angew. Chem., Int. Ed. 2019, 58, 18535–18539. 10.1002/anie.201907245. [DOI] [PubMed] [Google Scholar]
- Wu G.; Zhao T.; Kang D.; Zhang J.; Song Y.; Namasivayam V.; Kongsted J.; Pannecouque C.; De Clercq E.; Poongavanam V.; Liu X.; Zhan P. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. 10.1021/acs.jmedchem.9b00359. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Ritter T. A Perspective on Late-Stage Aromatic C–H Bond Functionalization. J. Am. Chem. Soc. 2022, 144, 2399–2414. 10.1021/jacs.1c10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noisier A. F. M.; Johansson M. J.; Knerr L.; Hayes M. A.; Drury W. J. III; Valeur E.; Malins L. R.; Gopalakrishnan R. Late-Stage Functionalization of Histidine in Unprotected Peptides. Angew. Chem., Int. Ed. 2019, 58, 19096–19102. 10.1002/anie.201910888. [DOI] [PubMed] [Google Scholar]
- Berger F.; Plutschack M. B.; Riegger J.; Yu W.; Speicher S.; Ho M.; Frank N.; Ritter T. Site-Selective and Versatile Aromatic C–H Functionalization by Thianthrenation. Nature 2019, 567, 223–228. 10.1038/s41586-019-0982-0. [DOI] [PubMed] [Google Scholar]
- Fier P. S.; Hartwig J. F. Selective C-H Fluorination of Pyridines and Diazines Inspired by a Classic Amination Reaction. Science 2013, 342, 956–960. 10.1126/science.1243759. [DOI] [PubMed] [Google Scholar]
- Faraday M. Siebente Reihe Von Experimental-Untersuchungen über Elektricität. Ann. Phys. 1834, 109, 481–520. 10.1002/andp.18341093102. [DOI] [Google Scholar]
- Kolbe H. Beobachtungen über Die Oxydirende Wirkung Des Sauerstoffs, Wenn Derselbe Mit Hülfe Einer Elektrischen Säule Entwickelt Wird. J. Prakt. Chem. 1847, 41, 137–139. 10.1002/prac.18470410118. [DOI] [Google Scholar]
- Simons J. H. Production of Fluorocarbons: I. The Generalized Procedure and Its Use with Nitrogen Compounds. J. Electrochem. Soc. 1949, 95, 47–67. 10.1149/1.2776733. [DOI] [Google Scholar]
- Baizer M. M. Electrolytic Reductive Coupling: I. Acrylonitrile. J. Electrochem. Soc. 1964, 111, 215–222. 10.1149/1.2426086. [DOI] [Google Scholar]
- Shono T.; Hamaguchi H.; Matsumura Y. Electroorganic Chemistry. XX. Anodic Oxidation of Carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. 10.1021/ja00848a020. [DOI] [Google Scholar]
- Yoshida J.-i.; Murata T.; Isoe S. Electrochemical Oxidation of Organosilicon Compounds I. Oxidative Cleavage of Carbon-Silicon Bond in Allylsilanes and Benzylsilanes. Tetrahedron Lett. 1986, 27, 3373–3376. 10.1016/S0040-4039(00)84799-1. [DOI] [Google Scholar]
- Yoshida J.-i.; Shimizu A.; Hayashi R. Electrogenerated Cationic Reactive Intermediates: The Pool Method and Further Advances. Chem. Rev. 2018, 118, 4702–4730. 10.1021/acs.chemrev.7b00475. [DOI] [PubMed] [Google Scholar]
- Yoshida J.-i.; Suga S.; Suzuki S.; Kinomura N.; Yamamoto A.; Fujiwara K. Direct Oxidative Carbon–Carbon Bond Formation Using the “Cation Pool” Method. 1. Generation of Iminium Cation Pools and Their Reaction with Carbon Nucleophiles. J. Am. Chem. Soc. 1999, 121, 9546–9549. 10.1021/ja9920112. [DOI] [Google Scholar]
- Steckhan E.Organic Syntheses with Electrochemically Regenerable Redox Systems. In Electrochemistry I; Steckhan E., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1987; pp 1–69.
- Steckhan E. Indirect Electroorganic Syntheses—A Modern Chapter of Organic Electrochemistry. Angew. Chem. Inter. Ed. Engl. 1986, 25, 683–701. 10.1002/anie.198606831. [DOI] [Google Scholar]
- Schäfer H.-J.Recent Contributions of Kolbe Electrolysis to Organic Synthesis. In Electrochemistry IV; Steckhan E., Ed.; Springer, 2005; pp 91–151.
- Lund H. A Century of Organic Electrochemistry. J. Electrochem. Soc. 2002, 149, S21–S33. 10.1149/1.1462037. [DOI] [Google Scholar]
- Francke R.; Little R. D. Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev. 2014, 43, 2492–2521. 10.1039/c3cs60464k. [DOI] [PubMed] [Google Scholar]
- Little R. D.; Schwaebe M. K.. Reductive Cyclizations at the Cathode. In Electrochemistry VI Electroorganic Synthesis: Bond Formation at Anode and Cathode; Steckhan E., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 1997; pp 1–48.
- Little R. D.; Fox D. P.; Van Hijfte L.; Dannecker R.; Sowell G.; Wolin R. L.; Moens L.; Baizer M. M. Electroreductive Cyclization. Ketones and Aldehydes Tethered To.Alpha.,.Beta.-Unsaturated Esters (Nitriles). Fundamental Investigations. J. Org. Chem. 1988, 53, 2287–2294. 10.1021/jo00245a029. [DOI] [Google Scholar]
- Moeller K. D. Synthetic Applications of Anodic Electrochemistry. Tetrahedron 2000, 56, 9527–9554. 10.1016/S0040-4020(00)00840-1. [DOI] [Google Scholar]
- Jutand A. Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev. 2008, 108, 2300–2347. 10.1021/cr068072h. [DOI] [PubMed] [Google Scholar]
- Amatore C.; Cammoun C.; Jutand A. Pd(OAc)2/p-Benzoquinone-Catalyzed Anaerobic Electrooxidative Homocoupling of Arylboronic Acids, Arylboronates and Aryltrifluoroborates in DMF and/or Water. Eur. J. Org. Chem. 2008, 2008, 4567–4570. 10.1002/ejoc.200800631. [DOI] [Google Scholar]
- Rosen B. R.; Werner E. W.; O’Brien A. G.; Baran P. S. Total Synthesis of Dixiamycin B by Electrochemical Oxidation. J. Am. Chem. Soc. 2014, 136, 5571–5574. 10.1021/ja5013323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong P.; Xu H.-C. Chemistry with Electrochemically Generated N-Centered Radicals. Acc. Chem. Res. 2019, 52, 3339–3350. 10.1021/acs.accounts.9b00472. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Lei A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 2019, 52, 3309–3324. 10.1021/acs.accounts.9b00512. [DOI] [PubMed] [Google Scholar]
- Ackermann L. Metalla-Electrocatalyzed C–H Activation by Earth-Abundant 3d Metals and Beyond. Acc. Chem. Res. 2020, 53, 84–104. 10.1021/acs.accounts.9b00510. [DOI] [PubMed] [Google Scholar]
- Huang H.; Steiniger K. A.; Lambert T. H. Electrophotocatalysis: Combining Light and Electricity to Catalyze Reactions. J. Am. Chem. Soc. 2022, 144, 12567–12583. 10.1021/jacs.2c01914. [DOI] [PubMed] [Google Scholar]
- Siu J. C.; Fu N.; Lin S. Catalyzing Electrosynthesis: A Homogeneous Electrocatalytic Approach to Reaction Discovery. Acc. Chem. Res. 2020, 53, 547–560. 10.1021/acs.accounts.9b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Dorta D.; Thobie-Gautier C.; Croyal M.; Bouzelha M.; Mével M.; Deniaud D.; Boujtita M.; Gouin S. G. Electrochemically Promoted Tyrosine-Click-Chemistry for Protein Labeling. J. Am. Chem. Soc. 2018, 140, 17120–17126. 10.1021/jacs.8b09372. [DOI] [PubMed] [Google Scholar]
- Barjau J.; Schnakenburg G.; Waldvogel S. R. Diversity-Oriented Synthesis of Polycyclic Scaffolds by Modification of an Anodic Product Derived from 2,4-Dimethylphenol. Angew. Chem., Int. Ed. 2011, 50, 1415–1419. 10.1002/anie.201006637. [DOI] [PubMed] [Google Scholar]
- Wang F.; Stahl S. S. Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron–Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. 10.1021/acs.accounts.9b00544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay A. S.; Payne R. J.; Malins L. R. Electrochemistry for the Chemoselective Modification of Peptides and Proteins. J. Am. Chem. Soc. 2022, 144, 23–41. 10.1021/jacs.1c11185. [DOI] [PubMed] [Google Scholar]
- Jiao K.-J.; Xing Y.-K.; Yang Q.-L.; Qiu H.; Mei T.-S. Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis. Acc. Chem. Res. 2020, 53, 300–310. 10.1021/acs.accounts.9b00603. [DOI] [PubMed] [Google Scholar]
- Ke J.; Liu W.; Zhu X.; Tan X.; He C. Electrochemical Radical Silyl-Oxygenation of Activated Alkenes. Angew. Chem., Int. Ed. 2021, 60, 8744–8749. 10.1002/anie.202016620. [DOI] [PubMed] [Google Scholar]
- Mei Y.; Yan Z.; Liu L. L. Facile Synthesis of the Dicyanophosphide Anion via Electrochemical Activation of White Phosphorus: An Avenue to Organophosphorus Compounds. J. Am. Chem. Soc. 2022, 144, 1517–1522. 10.1021/jacs.1c11087. [DOI] [PubMed] [Google Scholar]
- Chen M.; Zhao D.; Xu J.; Li C.; Lu C.; Yan H. Electrooxidative B–H Functionalization of Nido-Carboranes. Angew. Chem., Int. Ed. 2021, 60, 7838–7844. 10.1002/anie.202015299. [DOI] [PubMed] [Google Scholar]
- Fried A. D.; Wilson B. J.; Galan N. J.; Brantley J. N. Electroediting of Soft Polymer Backbones. J. Am. Chem. Soc. 2022, 144, 8885–8891. 10.1021/jacs.2c02098. [DOI] [PubMed] [Google Scholar]
- Siddiqi Z.; Sarlah D. Electrochemical Dearomatization of Commodity Polymers. J. Am. Chem. Soc. 2021, 143, 21264–21269. 10.1021/jacs.1c11546. [DOI] [PubMed] [Google Scholar]
- Fagnani D. E.; Kim D.; Camarero S. I.; Alfaro J. F.; McNeil A. J. Using Waste Poly(Vinyl Chloride) to Synthesize Chloroarenes by Plasticizer-Mediated Electro(de)chlorination. Nat. Chem. 2023, 15, 222–229. 10.1038/s41557-022-01078-w. [DOI] [PubMed] [Google Scholar]
- Peterson B. M.; Lin S.; Fors B. P. Electrochemically Controlled Cationic Polymerization of Vinyl Ethers. J. Am. Chem. Soc. 2018, 140, 2076–2079. 10.1021/jacs.8b00173. [DOI] [PubMed] [Google Scholar]
- Dong X.; Roeckl J. L.; Waldvogel S. R.; Morandi B. Merging Shuttle Reactions and Paired Electrolysis for Reversible Vicinal Dihalogenations. Science 2021, 371, 507–514. 10.1126/science.abf2974. [DOI] [PubMed] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Ang N. W. J.; Meyer T. H.; Qiu Y.; Ackermann L. Organic Electrochemistry: Molecular Syntheses with Potential. ACS Cent. Sci. 2021, 7, 415–431. 10.1021/acscentsci.0c01532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Y.; Zhu C.; Stangier M.; Struwe J.; Ackermann L. Rhodaelectro-Catalyzed C–H and C–C Activation. CCS Chem. 2021, 3, 1529–1552. 10.31635/ccschem.020.202000365. [DOI] [Google Scholar]
- Meyer T. H.; Choi I.; Tian C.; Ackermann L. Powering the Future: How Can Electrochemistry Make a Difference in Organic Synthesis?. Chem. 2020, 6, 2484–2496. 10.1016/j.chempr.2020.08.025. [DOI] [Google Scholar]
- Ma C.; Fang P.; Mei T.-S. Recent Advances in C–H Functionalization Using Electrochemical Transition Metal Catalysis. ACS Catal. 2018, 8, 7179–7189. 10.1021/acscatal.8b01697. [DOI] [Google Scholar]
- Tang S.; Zeng L.; Lei A. Oxidative R1–H/R2–H Cross-Coupling with Hydrogen Evolution. J. Am. Chem. Soc. 2018, 140, 13128–13135. 10.1021/jacs.8b07327. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Lei A.; Mei T.-S.; Xu H.-C.; Xu K.; Zeng C. Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis. CCS Chem. 2022, 4, 1120–1152. 10.31635/ccschem.021.202101451. [DOI] [Google Scholar]
- Wang Z.; Ma C.; Fang P.; Xu H.; Mei T. Advances in Organic Electrochemical Synthesis. Acta Chim. Sinica 2022, 80, 1115–1134. 10.6023/A22060260. [DOI] [Google Scholar]
- Jiao K.-J.; Wang Z.-H.; Ma C.; Liu H.-L.; Cheng B.; Mei T.-S. The Applications of Electrochemical Synthesis in Asymmetric Catalysis. Chem. Catal. 2022, 2, 3019–3047. 10.1016/j.checat.2022.09.039. [DOI] [Google Scholar]
- Ackermann L.; You S.-L.; Oestreich M.; Meng S.; MacFarlane D.; Yin Y. Seeds for the Future: Growing Chemistry Trends. Trends Chem. 2020, 2, 275–277. 10.1016/j.trechm.2020.02.006. [DOI] [Google Scholar]
- Waldvogel S. R.; Lips S.; Selt M.; Riehl B.; Kampf C. J. Electrochemical Arylation Reaction. Chem. Rev. 2018, 118, 6706–6765. 10.1021/acs.chemrev.8b00233. [DOI] [PubMed] [Google Scholar]
- Gieshoff T.; Kehl A.; Schollmeyer D.; Moeller K. D.; Waldvogel S. R. Insights into the Mechanism of Anodic N–N Bond Formation by Dehydrogenative Coupling. J. Am. Chem. Soc. 2017, 139, 12317–12324. 10.1021/jacs.7b07488. [DOI] [PubMed] [Google Scholar]
- Tay N. E. S.; Lehnherr D.; Rovis T. Photons or Electrons? A Critical Comparison of Electrochemistry and Photoredox Catalysis for Organic Synthesis. Chem. Rev. 2022, 122, 2487–2649. 10.1021/acs.chemrev.1c00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P.; Zha Z.; Wang Z. Recent Advances in C–H Functionalization with Electrochemistry and Various Iodine-Containing Reagents. ChemElectroChem. 2020, 7, 2527–2544. 10.1002/celc.202000252. [DOI] [Google Scholar]
- Meng W.; Xu K.; Guo B.; Zeng C. Recent Advances in Minisci Reactions under Electrochemical Conditions. Chin. J. Org. Chem. 2021, 41, 2621–2635. 10.6023/cjoc202102001. [DOI] [Google Scholar]
- Novaes L. F. T.; Liu J.; Shen Y.; Lu L.; Meinhardt J. M.; Lin S. Electrocatalysis as an Enabling Technology for Organic Synthesis. Chem. Soc. Rev. 2021, 50, 7941–8002. 10.1039/D1CS00223F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing Q.; Moeller K. D. From Molecules to Molecular Surfaces. Exploiting the Interplay between Organic Synthesis and Electrochemistry. Acc. Chem. Res. 2020, 53, 135–143. 10.1021/acs.accounts.9b00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S.; Liu Y.; Lei A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution: A Green and Sustainable Way for Bond Formation. Chem. 2018, 4, 27–45. 10.1016/j.chempr.2017.10.001. [DOI] [Google Scholar]
- Wirtanen T.; Prenzel T.; Tessonnier J.-P.; Waldvogel S. R. Cathodic Corrosion of Metal Electrodes–How to Prevent It in Electroorganic Synthesis. Chem. Rev. 2021, 121, 10241–10270. 10.1021/acs.chemrev.1c00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajada M. A.; Sanjosé-Orduna J.; Di Liberto G.; Tosoni S.; Pacchioni G.; Noël T.; Vilé G. Interfacing Single-Atom Catalysis with Continuous-Flow Organic Electrosynthesis. Chem. Soc. Rev. 2022, 51, 3898–3925. 10.1039/D2CS00100D. [DOI] [PubMed] [Google Scholar]
- Schotten C.; Nicholls T. P.; Bourne R. A.; Kapur N.; Nguyen B. N.; Willans C. E. Making Electrochemistry Easily Accessible to the Synthetic Chemist. Green Chem. 2020, 22, 3358–3375. 10.1039/D0GC01247E. [DOI] [Google Scholar]
- White M. C.; Zhao J. Aliphatic C–H Oxidations for Late-Stage Functionalization. J. Am. Chem. Soc. 2018, 140, 13988–14009. 10.1021/jacs.8b05195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannalire R.; Pelliccia S.; Sancineto L.; Novellino E.; Tron G. C.; Giustiniano M. Visible Light Photocatalysis in the Late-Stage Functionalization of Pharmaceutically Relevant Compounds. Chem. Soc. Rev. 2021, 50, 766–897. 10.1039/D0CS00493F. [DOI] [PubMed] [Google Scholar]
- Jana R.; Begam H. M.; Dinda E. The Emergence of the C–H Functionalization Strategy in Medicinal Chemistry and Drug Discovery. Chem. Commun. 2021, 57, 10842–10866. 10.1039/D1CC04083A. [DOI] [PubMed] [Google Scholar]
- Budhwan R.; Yadav S.; Murarka S. Late Stage Functionalization of Heterocycles Using Hypervalent Iodine(III) Reagents. Org. Biomol. Chem. 2019, 17, 6326–6341. 10.1039/C9OB00694J. [DOI] [PubMed] [Google Scholar]
- Kelly C. B.; Padilla-Salinas R. Late Stage C–H Functionalization via Chalcogen and Pnictogen Salts. Chem. Sci. 2020, 11, 10047–10060. 10.1039/D0SC03833D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash G.; Paul N.; Oliver G. A.; Werz D. B.; Maiti D. C–H Deuteration of Organic Compounds and Potential Drug Candidates. Chem. Soc. Rev. 2022, 51, 3123–3163. 10.1039/D0CS01496F. [DOI] [PubMed] [Google Scholar]
- Bellotti P.; Huang H.-M.; Faber T.; Glorius F. Photocatalytic Late-Stage C–H Functionalization. Chem. Rev. 2023, 123, 4237–4352. 10.1021/acs.chemrev.2c00478. [DOI] [PubMed] [Google Scholar]
- Ma C.; Zhao C.-Q.; Li Y.-Q.; Zhang L.-P.; Xu X.-T.; Zhang K.; Mei T.-S. Palladium-Catalyzed C–H Activation/C–C Cross-Coupling Reactions via Electrochemistry. Chem. Commun. 2017, 53, 12189–12192. 10.1039/C7CC07429H. [DOI] [PubMed] [Google Scholar]
- Yang Q.-L.; Li C.-Z.; Zhang L.-W.; Li Y.-Y.; Tong X.; Wu X.-Y.; Mei T.-S. Palladium-Catalyzed Electrochemical C–H Alkylation of Arenes. Organometallics 2019, 38, 1208–1212. 10.1021/acs.organomet.8b00550. [DOI] [Google Scholar]
- Kuciński K.; Simon H.; Ackermann L. Rhoda-Electrocatalyzed C–H Methylation and Paired Electrocatalyzed C–H Ethylation and Propylation. Chem. Eur. J. 2022, 28, e202103837 10.1002/chem.202103837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.-L.; Liu Y.; Liang L.; Li Z.-H.; Qu G.-R.; Guo H.-M. Facilitating Rh-Catalyzed C–H Alkylation of (Hetero)Arenes and 6-Arylpurine Nucleosides (Nucleotides) with Electrochemistry. J. Org. Chem. 2022, 87, 6161–6178. 10.1021/acs.joc.2c00391. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Sumii Y.; Shibata N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. 10.1021/acsomega.0c00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hird M. Fluorinated Liquid Crystals – Properties and Applications. Chem. Soc. Rev. 2007, 36, 2070–2095. 10.1039/b610738a. [DOI] [PubMed] [Google Scholar]
- Purser S.; Moore P. R.; Swallow S.; Gouverneur V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. 10.1039/B610213C. [DOI] [PubMed] [Google Scholar]
- Gouverneur V.; Seppelt K. Introduction: Fluorine Chemistry. Chem. Rev. 2015, 115, 563–565. 10.1021/cr500686k. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- O’Brien A. G.; Maruyama A.; Inokuma Y.; Fujita M.; Baran P. S.; Blackmond D. G. Radical C–H Functionalization of Heteroarenes under Electrochemical Control. Angew. Chem., Int. Ed. 2014, 53, 11868–11871. 10.1002/anie.201407948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda B. Joy and Challenges of Alkynylation of Arenes and Heteroarenes through Double C–H Functionalizations. Asian J. Org. Chem. 2020, 9, 492–507. 10.1002/ajoc.201900733. [DOI] [Google Scholar]
- Dudnik A. S.; Gevorgyan V. Formal Inverse Sonogashira Reaction: Direct Alkynylation of Arenes and Heterocycles with Alkynyl Halides. Angew. Chem., Int. Ed. 2010, 49, 2096–2098. 10.1002/anie.200906755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspers L. D.; Nachtsheim B. J. Directing-Group-Mediated C–H-Alkynylations. Chem. Asian J. 2018, 13, 1231–1247. 10.1002/asia.201800102. [DOI] [PubMed] [Google Scholar]
- Ye X.; Wang C.; Zhang S.; Wei J.; Shan C.; Wojtas L.; Xie Y.; Shi X. Facilitating Ir-Catalyzed C–H Alkynylation with Electrochemistry: Anodic Oxidation-Induced Reductive Elimination. ACS Catal. 2020, 10, 11693–11699. 10.1021/acscatal.0c03207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Feng T.; Wang Y.; Qiu Y. Recent Advances in Electrocarboxylation with CO2. Chem. Asian J. 2022, 17, e202200543 10.1002/asia.202200543. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Park G. D.; Balamurugan M.; Seo J.; Min B. K.; Nam K. T. Electrochemical Β-Selective Hydrocarboxylation of Styrene Using CO2 and Water. Adv. Sci. 2020, 7, 1900137–1900144. 10.1002/advs.201900137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkayal A.; Tabas V.; Montanaro S.; Wright I. A.; Malkov A. V.; Buckley B. R. Harnessing Applied Potential: Selective Β-Hydrocarboxylation of Substituted Olefins. J. Am. Chem. Soc. 2020, 142, 1780–1785. 10.1021/jacs.9b13305. [DOI] [PubMed] [Google Scholar]
- Sheta A. M.; Mashaly M. A.; Said S. B.; Elmorsy S. S.; Malkov A. V.; Buckley B. R. Selective α,δ-Hydrocarboxylation of Conjugated Dienes Utilizing CO2 and Electrosynthesis. Chem. Sci. 2020, 11, 9109–9114. 10.1039/D0SC03148H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheta A. M.; Alkayal A.; Mashaly M. A.; Said S. B.; Elmorsy S. S.; Malkov A. V.; Buckley B. R. Selective Electrosynthetic Hydrocarboxylation of α,β-Unsaturated Esters with Carbon Dioxide. Angew. Chem., Int. Ed. 2021, 60, 21832–21837. 10.1002/anie.202105490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Yuan G.; Jiang H. Electrocarboxylation of Alkynes with Carbon Dioxide in the Presence of Metal Salt Catalysts. Chin. J. Chem. 2010, 28, 1685–1689. 10.1002/cjoc.201090285. [DOI] [Google Scholar]
- Katayama A.; Senboku H. Sequential Vinyl Radical Cyclization/Fixation of Carbon Dioxide through Electrochemical Reduction of Vinyl Bromide in the Presence of an Electron-Transfer Mediator. ChemElectroChem. 2016, 3, 2052–2057. 10.1002/celc.201600508. [DOI] [Google Scholar]
- Zhang K.; Wang H.; Zhao S.-F.; Niu D.-F.; Lu J.-X. Asymmetric Electrochemical Carboxylation of Prochiral Acetophenone: An Efficient Route to Optically Active Atrolactic Acid Via Selective Fixation of Carbon Dioxide. J. Electroanal. Chem. 2009, 630, 35–41. 10.1016/j.jelechem.2009.02.013. [DOI] [Google Scholar]
- You Y.; Kanna W.; Takano H.; Hayashi H.; Maeda S.; Mita T. Electrochemical Dearomative Dicarboxylation of Heterocycles with Highly Negative Reduction Potentials. J. Am. Chem. Soc. 2022, 144, 3685–3695. 10.1021/jacs.1c13032. [DOI] [PubMed] [Google Scholar]
- Ang N. W. J.; Oliveira J. C. A.; Ackermann L. Electroreductive Cobalt-Catalyzed Carboxylation: Cross-Electrophile Electrocoupling with Atmospheric CO2. Angew. Chem., Int. Ed. 2020, 59, 12842–12847. 10.1002/anie.202003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.-T.; Zhu M.; Schiffer Z. J.; Williams K.; Song X.; Liu X.; Manthiram K. Direct Electrochemical Carboxylation of Benzylic C–N Bonds with Carbon Dioxide. ACS Catal. 2019, 9, 4699–4705. 10.1021/acscatal.9b00818. [DOI] [Google Scholar]
- Zhang Y.-F.; Mellah M. Samarium(II)-Electrocatalyzed Chemoselective Reductive Alkoxylation of Phthalimides. Org. Chem. Front. 2022, 9, 1308–1314. 10.1039/D1QO01760H. [DOI] [Google Scholar]
- Jiao K.-J.; Li Z.-M.; Xu X.-T.; Zhang L.-P.; Li Y.-Q.; Zhang K.; Mei T.-S. Palladium-Catalyzed Reductive Electrocarboxylation of Allyl Esters with Carbon Dioxide. Org. Chem. Front. 2018, 5, 2244–2248. 10.1039/C8QO00507A. [DOI] [Google Scholar]
- Sun G.-Q.; Zhang W.; Liao L.-L.; Li L.; Nie Z.-H.; Wu J.-G.; Zhang Z.; Yu D.-G. Nickel-Catalyzed Electrochemical Carboxylation of Unactivated Aryl and Alkyl Halides with CO2. Nat. Commun. 2021, 12, 7086–7095. 10.1038/s41467-021-27437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börjesson M.; Moragas T.; Gallego D.; Martin R. Metal-Catalyzed Carboxylation of Organic (Pseudo)Halides with CO2. ACS Catal. 2016, 6, 6739–6749. 10.1021/acscatal.6b02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Lin Z.; Maksso I.; Struwe J.; Ackermann L. Electrochemical Cross-Electrophile-Coupling for Transition-Metal-Free Allylic Carboxylation with Ambient CO2. ChemElectroChem. 2022, 9, e202200989 10.1002/celc.202200989. [DOI] [Google Scholar]
- Sun G.-Q.; Yu P.; Zhang W.; Zhang W.; Wang Y.; Liao L.-L.; Zhang Z.; Li L.; Lu Z.; Yu D.-G.; Lin S. Electrochemical Reactor Dictates Site Selectivity in N-Heteroarene Carboxylations. Nature 2023, 615, 67–72. 10.1038/s41586-022-05667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Liu Y.; Wang S.; Tang S.; Ma D.; Zhu Z.; Guo C.; Qiu Y. Site-Selective Electrochemical C–H Carboxylation of Arenes with CO2. Angew. Chem., Int. Ed. 2023, 62, e202214710 10.1002/anie.202214710. [DOI] [PubMed] [Google Scholar]
- So Y.-H.; Becker J. Y.; Miller L. L. Hydroxylation and Amidation of Aromatic Carbonyl Compounds. J. Chem. Soc., Chem. Commun. 1975, 262–263. 10.1039/c39750000262. [DOI] [Google Scholar]
- Fujimoto K.; Tokuda Y.; Maekawa H.; Matsubara Y.; Mizuno T.; Nishiguchi I. Selective and One-Pot Formation of Phenols by Anodic Oxidation. Tetrahedron 1996, 52, 3889–3896. 10.1016/S0040-4020(96)00056-7. [DOI] [Google Scholar]
- Kreh R. P.; Tadros M. E.; Hand H. M.; Cockerham M. P.; Smith E. K. Indirect Hydroxylation of Aromatic Rings Using Electrochemical Methods. J. Appl. Electrochem. 1986, 16, 440–446. 10.1007/BF01008855. [DOI] [Google Scholar]
- Pletcher D.; Green R. A.; Brown R. C. D. Flow Electrolysis Cells for the Synthetic Organic Chemistry Laboratory. Chem. Rev. 2018, 118, 4573–4591. 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]
- Atobe M.; Tateno H.; Matsumura Y. Applications of Flow Microreactors in Electrosynthetic Processes. Chem. Rev. 2018, 118, 4541–4572. 10.1021/acs.chemrev.7b00353. [DOI] [PubMed] [Google Scholar]
- Tanbouza N.; Ollevier T.; Lam K. Bridging Lab and Industry with Flow Electrochemistry. iScience 2020, 23, 101720. 10.1016/j.isci.2020.101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noël T.; Cao Y.; Laudadio G. The Fundamentals Behind the Use of Flow Reactors in Electrochemistry. Acc. Chem. Res. 2019, 52, 2858–2869. 10.1021/acs.accounts.9b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsherbini M.; Wirth T. Electroorganic Synthesis under Flow Conditions. Acc. Chem. Res. 2019, 52, 3287–3296. 10.1021/acs.accounts.9b00497. [DOI] [PubMed] [Google Scholar]
- Laudadio G.; Barmpoutsis E.; Schotten C.; Struik L.; Govaerts S.; Browne D. L.; Noël T. Sulfonamide Synthesis through Electrochemical Oxidative Coupling of Amines and Thiols. J. Am. Chem. Soc. 2019, 141, 5664–5668. 10.1021/jacs.9b02266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y.; Lu Z.; Rughoobur G.; Patil P.; Gershenfeld N.; Akinwande A. I.; Buchwald S. L.; Jensen K. F. Microfluidic Electrochemistry for Single-Electron Transfer Redox-Neutral Reactions. Science 2020, 368, 1352–1357. 10.1126/science.aba3823. [DOI] [PubMed] [Google Scholar]
- Long H.; Chen T.-S.; Song J.; Zhu S.; Xu H.-C. Electrochemical Aromatic C–H Hydroxylation in Continuous Flow. Nat. Commun. 2022, 13, 3945–3951. 10.1038/s41467-022-31634-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauermann N.; Meyer T. H.; Tian C.; Ackermann L. Electrochemical Cobalt-Catalyzed C–H Oxygenation at Room Temperature. J. Am. Chem. Soc. 2017, 139, 18452–18455. 10.1021/jacs.7b11025. [DOI] [PubMed] [Google Scholar]
- Zhang S.-K.; Struwe J.; Hu L.; Ackermann L. Nickela-Electrocatalyzed C–H Alkoxylation with Secondary Alcohols: Oxidation-Induced Reductive Elimination at Nickel(III). Angew. Chem., Int. Ed. 2020, 59, 3178–3183. 10.1002/anie.201913930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massignan L.; Tan X.; Meyer T. H.; Kuniyil R.; Messinis A. M.; Ackermann L. C–H Oxygenation Reactions Enabled by Dual Catalysis with Electrogenerated Hypervalent Iodine Species and Ruthenium Complexes. Angew. Chem., Int. Ed. 2020, 59, 3184–3189. 10.1002/anie.201914226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.-Q.; Yang Q.-L.; Fang P.; Mei T.-S.; Zhang D. Palladium-Catalyzed C(sp2)–H Acetoxylation via Electrochemical Oxidation. Org. Lett. 2017, 19, 2905–2908. 10.1021/acs.orglett.7b01138. [DOI] [PubMed] [Google Scholar]
- Tan X.; Massignan L.; Hou X.; Frey J.; Oliveira J. C. A.; Hussain M. N.; Ackermann L. Rhoda-Electrocatalyzed Bimetallic C–H Oxygenation by Weak O-Coordination. Angew. Chem., Int. Ed. 2021, 60, 13264–13270. 10.1002/anie.202017359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.; Kaplaneris N.; Yuan B.; Frey J.; Ohyama T.; Messinis A. M.; Ackermann L. Ruthenaelectro-Catalyzed C–H Acyloxylation for Late-Stage Tyrosine and Oligopeptide Diversification. Chem. Sci. 2022, 13, 3461–3467. 10.1039/D1SC07267F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Zhang G.; Nie L.; Kong T.; Lei A. Electrochemical Oxidation Induced Intermolecular Aromatic C-H Imidation. Nat. Commun. 2019, 10, 5467–5476. 10.1038/s41467-019-13524-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Zhang G.; Bu F.; Nie L.; Lei A. Electrochemical-Oxidation-Induced Site-Selective Intramolecular C(sp3)–H Amination. ACS Catal. 2018, 8, 9370–9375. 10.1021/acscatal.8b02847. [DOI] [Google Scholar]
- Zhang Y.; Lin Z.; Ackermann L. Electrochemical C–H Amidation of Heteroarenes with N-Alkyl Sulfonamides in Aqueous Medium. Chem. Eur. J. 2021, 27, 242–246. 10.1002/chem.202004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.-K.; Samanta R. C.; Sauermann N.; Ackermann L. Nickel-Catalyzed Electrooxidative C–H Amination: Support for Nickel(iv). Chem. Eur. J. 2018, 24, 19166–19170. 10.1002/chem.201805441. [DOI] [PubMed] [Google Scholar]
- Sauermann N.; Mei R.; Ackermann L. Electrochemical C–H Amination by Cobalt Catalysis in a Renewable Solvent. Angew. Chem., Int. Ed. 2018, 57, 5090–5094. 10.1002/anie.201802206. [DOI] [PubMed] [Google Scholar]
- Gao X.; Wang P.; Zeng L.; Tang S.; Lei A. Cobalt(II)-Catalyzed Electrooxidative C–H Amination of Arenes with Alkylamines. J. Am. Chem. Soc. 2018, 140, 4195–4199. 10.1021/jacs.7b13049. [DOI] [PubMed] [Google Scholar]
- Yang Q.-L.; Wang X.-Y.; Lu J.-Y.; Zhang L.-P.; Fang P.; Mei T.-S. Copper-Catalyzed Electrochemical C–H Amination of Arenes with Secondary Amines. J. Am. Chem. Soc. 2018, 140, 11487–11494. 10.1021/jacs.8b07380. [DOI] [PubMed] [Google Scholar]
- Boutureira O.; Bernardes G. J. L. Advances in Chemical Protein Modification. Chem. Rev. 2015, 115, 2174–2195. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- Koniev O.; Wagner A. Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation. Chem. Soc. Rev. 2015, 44, 5495–5551. 10.1039/C5CS00048C. [DOI] [PubMed] [Google Scholar]
- Hu Q.-Y.; Berti F.; Adamo R. Towards the Next Generation of Biomedicines by Site-Selective Conjugation. Chem. Soc. Rev. 2016, 45, 1691–1719. 10.1039/C4CS00388H. [DOI] [PubMed] [Google Scholar]
- Cohen D. T.; Zhang C.; Fadzen C. M.; Mijalis A. J.; Hie L.; Johnson K. D.; Shriver Z.; Plante O.; Miller S. J.; Buchwald S. L.; Pentelute B. L. A Chemoselective Strategy for Late-Stage Functionalization of Complex Small Molecules with Polypeptides and Proteins. Nat. Chem. 2019, 11, 78–85. 10.1038/s41557-018-0154-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton D.; Campbell C.; Richards P.; Heptinstall J. Electrooxidative Nitration of Phenols at Copper Electrodes. Electrochim. Acta 1997, 42, 3499–3507. 10.1016/S0013-4686(97)00036-4. [DOI] [Google Scholar]
- Walton D. J.; Richards P. G.; Heptinstall J.; Coles B. Electrosynthetic Modification of Proteins: Electrooxidations at Methionine and Tryptophan in Hen Egg-White Lysozyme. Electrochim. Acta 1997, 42, 2285–2294. 10.1016/S0013-4686(96)00419-7. [DOI] [Google Scholar]
- Richards P. G.; Coles B.; Heptinstall J.; Walton D. J. Electrochemical Modification of Lysozyme: Anodic Reaction of Tyrosine Residues. Enzyme Microb. Technol. 1994, 16, 795–801. 10.1016/0141-0229(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Kendall G.; Cooper H. J.; Heptinstall J.; Derrick P. J.; Walton D. J.; Peterson I. R. Specific Electrochemical Nitration of Horse Heart Myoglobin. Arch. Biochem. Biophys. 2001, 392, 169–179. 10.1006/abbi.2001.2451. [DOI] [PubMed] [Google Scholar]
- Matters D.; Cooper H. J.; McDonnell L.; Iniesta J.; Heptinstall J.; Derrick P.; Walton D.; Peterson I. Mass Spectrometry in Demonstrating the Site-Specific Nitration of Hen Egg White Lysozyme by an Improved Electrochemical Method. Anal. Biochem. 2006, 356, 171–181. 10.1016/j.ab.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Ban H.; Gavrilyuk J.; Barbas C. F. III Tyrosine Bioconjugation through Aqueous Ene-Type Reactions: A Click-Like Reaction for Tyrosine. J. Am. Chem. Soc. 2010, 132, 1523–1525. 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- Cui L.; Ma Y.; Li M.; Wei Z.; Huan Y.; Li H.; Fei Q.; Zheng L. Tyrosine-Reactive Cross-Linker for Probing Protein Three-Dimensional Structures. Anal. Chem. 2021, 93, 4434–4440. 10.1021/acs.analchem.0c04337. [DOI] [PubMed] [Google Scholar]
- Sato S.; Matsumura M.; Kadonosono T.; Abe S.; Ueno T.; Ueda H.; Nakamura H. Site-Selective Protein Chemical Modification of Exposed Tyrosine Residues Using Tyrosine Click Reaction. Bioconjugate Chem. 2020, 31, 1417–1424. 10.1021/acs.bioconjchem.0c00120. [DOI] [PubMed] [Google Scholar]
- Song C.; Liu K.; Wang Z.; Ding B.; Wang S.; Weng Y.; Chiang C.-W.; Lei A. Electrochemical Oxidation Induced Selective Tyrosine Bioconjugation for the Modification of Biomolecules. Chem. Sci. 2019, 10, 7982–7987. 10.1039/C9SC02218J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner P.; Hehn J. P.; Gnamm C. Phosphine Oxides from a Medicinal Chemist’s Perspective: Physicochemical and in Vitro Parameters Relevant for Drug Discovery. J. Med. Chem. 2020, 63, 7081–7107. 10.1021/acs.jmedchem.0c00407. [DOI] [PubMed] [Google Scholar]
- Budnikova Y. H.; Dudkina Y. B. Progress of Electrochemical C(sp2)-H Phosphonation. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 415–419. 10.1080/10426507.2018.1540480. [DOI] [Google Scholar]
- Yurko E. O.; Gryaznova T. V.; Budnikova Y. H. Electrochemical C-H Phosphonation of Caffeine. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 343–344. 10.1080/10426507.2018.1541897. [DOI] [Google Scholar]
- Wu Z.-J.; Su F.; Lin W.; Song J.; Wen T.-B.; Zhang H.-J.; Xu H.-C. Scalable Rhodium(III)-Catalyzed Aryl C–H Phosphorylation Enabled by Anodic Oxidation Induced Reductive Elimination. Angew. Chem., Int. Ed. 2019, 58, 16770–16774. 10.1002/anie.201909951. [DOI] [PubMed] [Google Scholar]
- Long H.; Huang C.; Zheng Y.-T.; Li Z.-Y.; Jie L.-H.; Song J.; Zhu S.; Xu H.-C. Electrochemical C–H Phosphorylation of Arenes in Continuous Flow Suitable for Late-Stage Functionalization. Nat. Commun. 2021, 12, 6629–6635. 10.1038/s41467-021-26960-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Qiao J.; Cao Y.; Tang J.; Wang M.; Ke G.; Lu Y.; Liu X.; Lei A. Exogenous-Oxidant-Free Electrochemical Oxidative C–H Phosphonylation with Hydrogen Evolution. Chem. Commun. 2019, 55, 4230–4233. 10.1039/C9CC00975B. [DOI] [PubMed] [Google Scholar]
- Wang S.; Xue Q.; Guan Z.; Ye Y.; Lei A. Mn-Catalyzed Electrooxidative Undirected C–H/P–H Cross-Coupling between Aromatics and Diphenyl Phosphine Oxides. ACS Catal. 2021, 11, 4295–4300. 10.1021/acscatal.1c00549. [DOI] [Google Scholar]
- Zhang S.-K.; Del Vecchio A.; Kuniyil R.; Messinis A. M.; Lin Z.; Ackermann L. Electrocatalytic C–H Phosphorylation through Nickel(III/IV/II) Catalysis. Chem. 2021, 7, 1379–1392. 10.1016/j.chempr.2021.04.009. [DOI] [Google Scholar]
- Strekalova S. O.; Grinenko V. V.; Gryaznova T. V.; Kononov A. I.; Dolengovski E. L.; Budnikova Y. H. Electrochemical Phosphorylation of Arenes Catalyzed by Cobalt under Oxidative and Reductive Conditions. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 506–509. 10.1080/10426507.2018.1540488. [DOI] [Google Scholar]
- Latham J.; Brandenburger E.; Shepherd S. A.; Menon B. R. K.; Micklefield J. Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev. 2018, 118, 232–269. 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]
- Isin E. M.; Elmore C. S.; Nilsson G. N.; Thompson R. A.; Weidolf L. Use of Radiolabeled Compounds in Drug Metabolism and Pharmacokinetic Studies. Chem. Res. Toxicol. 2012, 25, 532–542. 10.1021/tx2005212. [DOI] [PubMed] [Google Scholar]
- Hassan J.; Sévignon M.; Gozzi C.; Schulz E.; Lemaire M. Aryl–Aryl Bond Formation One Century after the Discovery of the Ullmann Reaction. Chem. Rev. 2002, 102, 1359–1470. 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- Petrone D. A.; Ye J.; Lautens M. Modern Transition-Metal-Catalyzed Carbon–Halogen Bond Formation. Chem. Rev. 2016, 116, 8003–8104. 10.1021/acs.chemrev.6b00089. [DOI] [PubMed] [Google Scholar]
- Kakiuchi F.; Kochi T.; Mutsutani H.; Kobayashi N.; Urano S.; Sato M.; Nishiyama S.; Tanabe T. Palladium-Catalyzed Aromatic C–H Halogenation with Hydrogen Halides by Means of Electrochemical Oxidation. J. Am. Chem. Soc. 2009, 131, 11310–11311. 10.1021/ja9049228. [DOI] [PubMed] [Google Scholar]
- Aiso H.; Kochi T.; Mutsutani H.; Tanabe T.; Nishiyama S.; Kakiuchi F. Catalytic Electrochemical C–H Iodination and One-Pot Arylation by On/Off Switching of Electric Current. J. Org. Chem. 2012, 77, 7718–7724. 10.1021/jo3012286. [DOI] [PubMed] [Google Scholar]
- Konishi M.; Tsuchida K.; Sano K.; Kochi T.; Kakiuchi F. Palladium-Catalyzed Ortho-Selective C–H Chlorination of Benzamide Derivatives under Anodic Oxidation Conditions. J. Org. Chem. 2017, 82, 8716–8724. 10.1021/acs.joc.7b01137. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Simon H.; Chen X.; Lin Z.; Chen S.; Ackermann L. Distal Ruthenaelectro-Catalyzed Meta-C–H Bromination with Aqueous Hbr. Angew. Chem., Int. Ed. 2022, 61, e202201595 10.1002/anie.202201595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Yao A.; Zheng Y.; Gao M.; Zhou Z.; Qiao J.; Hu J.; Ye B.; Zhao J.; Wen H.; Lei A. Electrochemical Oxidative Clean Halogenation Using HX/NaX with Hydrogen Evolution. iScience 2019, 12, 293–303. 10.1016/j.isci.2019.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.-L.; Wang X.-Y.; Wang T.-L.; Yang X.; Liu D.; Tong X.; Wu X.-Y.; Mei T.-S. Palladium-Catalyzed Electrochemical C–H Bromination Using NH4Br as the Brominating Reagent. Org. Lett. 2019, 21, 2645–2649. 10.1021/acs.orglett.9b00629. [DOI] [PubMed] [Google Scholar]
- Tan Z.; Liu Y.; Helmy R.; Rivera N. R.; Hesk D.; Tyagarajan S.; Yang L.; Su J. Electrochemical Bromination of Late Stage Intermediates and Drug Molecules. Tetrahedron Lett. 2017, 58, 3014–3018. 10.1016/j.tetlet.2017.06.019. [DOI] [Google Scholar]
- Liang Y.; Lin F.; Adeli Y.; Jin R.; Jiao N. Efficient Electrocatalysis for the Preparation of (Hetero)Aryl Chlorides and Vinyl Chloride with 1,2-Dichloroethane. Angew. Chem., Int. Ed. 2019, 58, 4566–4570. 10.1002/anie.201814570. [DOI] [PubMed] [Google Scholar]
- Yu Q.; Hu L. a.; Wang Y.; Zheng S.; Huang J. Directed Meta-Selective Bromination of Arenes with Ruthenium Catalysts. Angew. Chem., Int. Ed. 2015, 54, 15284–15288. 10.1002/anie.201507100. [DOI] [PubMed] [Google Scholar]
- Iniesta J.; Cooper H. J.; Marshall A. G.; Heptinstall J.; Walton D. J.; Peterson I. R. Specific Electrochemical Iodination of Horse Heart Myoglobin at Tyrosine 103 as Determined by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Arch. Biochem. Biophys. 2008, 474, 1–7. 10.1016/j.abb.2008.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.; Kim Y.; Chang S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- Gandeepan P.; Müller T.; Zell D.; Cera G.; Warratz S.; Ackermann L. 3d Transition Metals for C–H Activation. Chem. Rev. 2019, 119, 2192–2452. 10.1021/acs.chemrev.8b00507. [DOI] [PubMed] [Google Scholar]
- Rej S.; Ano Y.; Chatani N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. 10.1021/acs.chemrev.9b00495. [DOI] [PubMed] [Google Scholar]
- Xu F.; Li Y.-J.; Huang C.; Xu H.-C. Ruthenium-Catalyzed Electrochemical Dehydrogenative Alkyne Annulation. ACS Catal. 2018, 8, 3820–3824. 10.1021/acscatal.8b00373. [DOI] [Google Scholar]
- Shen H.; Liu T.; Cheng D.; Yi X.; Wang Z.; Liu L.; Song D.; Ling F.; Zhong W. Ruthenium-Catalyzed Electrochemical Synthesis of Indolines through Dehydrogenative [3 + 2] Annulation with H2 Evolution. J. Org. Chem. 2020, 85, 13735–13746. 10.1021/acs.joc.0c01879. [DOI] [PubMed] [Google Scholar]
- Zeng L.; Li H.; Tang S.; Gao X.; Deng Y.; Zhang G.; Pao C.-W.; Chen J.-L.; Lee J.-F.; Lei A. Cobalt-Catalyzed Electrochemical Oxidative C–H/N–H Carbonylation with Hydrogen Evolution. ACS Catal. 2018, 8, 5448–5453. 10.1021/acscatal.8b00683. [DOI] [Google Scholar]
- Tian C.; Dhawa U.; Scheremetjew A.; Ackermann L. Cupraelectro-Catalyzed Alkyne Annulation: Evidence for Distinct C–H Alkynylation and Decarboxylative C–H/C–C Manifolds. ACS Catal. 2019, 9, 7690–7696. 10.1021/acscatal.9b02348. [DOI] [Google Scholar]
- Huang P.; Wang P.; Wang S.; Tang S.; Lei A. Electrochemical Oxidative [4 + 2] Annulation of Tertiary Anilines and Alkenes for the Synthesis of Tetrahydroquinolines. Green Chem. 2018, 20, 4870–4874. 10.1039/C8GC02463D. [DOI] [Google Scholar]
- Tang S.; Wang D.; Liu Y.; Zeng L.; Lei A. Cobalt-Catalyzed Electrooxidative C-H/N-H [4 + 2] Annulation with Ethylene or Ethyne. Nat. Commun. 2018, 9, 798–804. 10.1038/s41467-018-03246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei R.; Sauermann N.; Oliveira J. C. A.; Ackermann L. Electroremovable Traceless Hydrazides for Cobalt-Catalyzed Electro-Oxidative C–H/N–H Activation with Internal Alkynes. J. Am. Chem. Soc. 2018, 140, 7913–7921. 10.1021/jacs.8b03521. [DOI] [PubMed] [Google Scholar]
- Meyer T. H.; Oliveira J. C. A.; Sau S. C.; Ang N. W. J.; Ackermann L. Electrooxidative Allene Annulations by Mild Cobalt-Catalyzed C–H Activation. ACS Catal. 2018, 8, 9140–9147. 10.1021/acscatal.8b03066. [DOI] [Google Scholar]
- Kong W.-J.; Finger L. H.; Messinis A. M.; Kuniyil R.; Oliveira J. C. A.; Ackermann L. Flow Rhodaelectro-Catalyzed Alkyne Annulations by Versatile C–H Activation: Mechanistic Support for Rhodium(III/IV). J. Am. Chem. Soc. 2019, 141, 17198–17206. 10.1021/jacs.9b07763. [DOI] [PubMed] [Google Scholar]
- Yang Q.-L.; Xing Y.-K.; Wang X.-Y.; Ma H.-X.; Weng X.-J.; Yang X.; Guo H.-M.; Mei T.-S. Electrochemistry-Enabled Ir-Catalyzed Vinylic C–H Functionalization. J. Am. Chem. Soc. 2019, 141, 18970–18976. 10.1021/jacs.9b11915. [DOI] [PubMed] [Google Scholar]
- Yang L.; Steinbock R.; Scheremetjew A.; Kuniyil R.; Finger L. H.; Messinis A. M.; Ackermann L. Azaruthena(ii)-Bicyclo[3.2.0]Heptadiene: Key Intermediate for Ruthenaelectro(II/III/I)-Catalyzed Alkyne Annulations. Angew. Chem., Int. Ed. 2020, 59, 11130–11135. 10.1002/anie.202000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Oliveira J. C. A.; Lin Z.; Ackermann L. Electrooxidative Rhodium-Catalyzed [5 + 2] Annulations via C–H/O–H Activations. Angew. Chem., Int. Ed. 2021, 60, 6419–6424. 10.1002/anie.202016895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangier M.; Messinis A. M.; Oliveira J. C. A.; Yu H.; Ackermann L. Rhodaelectro-Catalyzed Access to Chromones via Formyl C–H Activation Towards Peptide Electro-Labeling. Nat. Commun. 2021, 12, 4736–4743. 10.1038/s41467-021-25005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y.; Xu X.; Chen H.; Zhang Y.; Zhuo X. Tandem Electrochemical Oxidative Azidation/Heterocyclization of Tryptophan-Containing Peptides under Buffer Conditions. Angew. Chem., Int. Ed. 2022, 61, e202206308 10.1002/anie.202206308. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The Exploration of Macrocycles for Drug Discovery — an Underexploited Structural Class. Nat. Rev. Drug Discovery 2008, 7, 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Yu X.; Sun D. Macrocyclic Drugs and Synthetic Methodologies toward Macrocycles. Molecules 2013, 18, 6230–6268. 10.3390/molecules18066230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begnini F.; Poongavanam V.; Over B.; Castaldo M.; Geschwindner S.; Johansson P.; Tyagi M.; Tyrchan C.; Wissler L.; Sjö P.; et al. Mining Natural Products for Macrocycles to Drug Difficult Targets. J. Med. Chem. 2021, 64, 1054–1072. 10.1021/acs.jmedchem.0c01569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Mallinson J.; Collins I. Macrocycles in New Drug Discovery. Future Medicinal Chemistry 2012, 4, 1409–1438. 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- Ding H.; DeRoy P. L.; Perreault C.; Larivée A.; Siddiqui A.; Caldwell C. G.; Harran S.; Harran P. G. Electrolytic Macrocyclizations: Scalable Synthesis of a Diazonamide-Based Drug Development Candidate. Angew. Chem., Int. Ed. 2015, 54, 4818–4822. 10.1002/anie.201411663. [DOI] [PubMed] [Google Scholar]
- Blanksby S. J.; Ellison G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- Wang H.; Liang K.; Xiong W.; Samanta S.; Li W.; Lei A. Electrochemical Oxidation-Induced Etherification via C(sp3)—H/O—H Cross-Coupling. Sci. Adv. 2020, 6, eaaz0590 10.1126/sciadv.aaz0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino Y.; Hayashi Y.; Iwahama T.; Sakaguchi S.; Ishii Y. Catalytic Oxidation of Alkylbenzenes with Molecular Oxygen under Normal Pressure and Temperature by N-Hydroxyphthalimide Combined with Co(OAc)2. J. Org. Chem. 1997, 62, 6810–6813. 10.1021/jo9708147. [DOI] [Google Scholar]
- Ishii Y.; Sakaguchi S.; Iwahama T. Innovation of Hydrocarbon Oxidation with Molecular Oxygen and Related Reactions. Adv. Synth. Catal. 2001, 343, 393–427. . [DOI] [Google Scholar]
- Xiong P.; Zhao H.-B.; Fan X.-T.; Jie L.-H.; Long H.; Xu P.; Liu Z.-J.; Wu Z.-J.; Cheng J.; Xu H.-C. Site-Selective Electrooxidation of Methylarenes to Aromatic Acetals. Nat. Commun. 2020, 11, 2706–2713. 10.1038/s41467-020-16519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M. A.; Twilton J.; Zhu J.; Graaf M. D.; Harper K. C.; Tuca E.; DiLabio G. A.; Stahl S. S. Electrochemical Pinoylation of Methylarenes: Improving the Scope and Utility of Benzylic Oxidation through Mediated Electrolysis. J. Am. Chem. Soc. 2022, 144, 15295–15302. 10.1021/jacs.2c05974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z.-W.; Liu D.-J.; Xiong P.; Lai X.-L.; Song J.; Xu H.-C. Site-Selective Electrochemical Benzylic C–H Amination. Angew. Chem., Int. Ed. 2021, 60, 2943–2947. 10.1002/anie.202013478. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Lin Z.; Oliveira J. C. A.; Ackermann L. Electro-Oxidative Intermolecular Allylic C(sp3)–H Aminations. J. Org. Chem. 2021, 86, 15935–15945. 10.1021/acs.joc.1c00682. [DOI] [PubMed] [Google Scholar]
- Hou Z.-W.; Li L.; Wang L. Organocatalytic Electrochemical Amination of Benzylic C–H Bonds. Org. Chem. Front. 2021, 8, 4700–4705. 10.1039/D1QO00746G. [DOI] [Google Scholar]
- Ruan Z.; Huang Z.; Xu Z.; Zeng S.; Feng P.; Sun P.-H. Late-Stage Azolation of Benzylic C–H Bonds Enabled by Electrooxidation. Sci. China Chem. 2021, 64, 800–807. 10.1007/s11426-020-9938-9. [DOI] [Google Scholar]
- Mukerjee A. K.; Ashare R. Isothiocyanates in the Chemistry of Heterocycles. Chem. Rev. 1991, 91, 1–24. 10.1021/cr00001a001. [DOI] [Google Scholar]
- Zhang S.; Li Y.; Wang T.; Li M.; Wen L.; Guo W. Electrochemical Benzylic C(sp3)–H Isothiocyanation. Org. Lett. 2022, 24, 1742–1746. 10.1021/acs.orglett.2c00415. [DOI] [PubMed] [Google Scholar]
- O’Hagan D. Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev. 2008, 37, 308–319. 10.1039/B711844A. [DOI] [PubMed] [Google Scholar]
- Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Sánchez-Roselló M.; Aceña J. L.; del Pozo C.; Sorochinsky A. E.; Fustero S.; Soloshonok V. A.; Liu H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- Hagmann W. K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- Fujiwara T.; O’Hagan D. Successful Fluorine-Containing Herbicide Agrochemicals. J. Fluorine Chem. 2014, 167, 16–29. 10.1016/j.jfluchem.2014.06.014. [DOI] [Google Scholar]
- Tajima T.; Ishii H.; Fuchigami T. Anodic Benzylic Fluorination of Toluene, Ethylbenzene, and Cumene Derivatives. Electrochem. Commun. 2002, 4, 589–592. 10.1016/S1388-2481(02)00381-8. [DOI] [Google Scholar]
- Fuchigami T.; Inagi S. Selective Electrochemical Fluorination of Organic Molecules and Macromolecules in Ionic Liquids. Chem. Commun. 2011, 47, 10211–10223. 10.1039/c1cc12414e. [DOI] [PubMed] [Google Scholar]
- Fuchigami T.; Inagi S. Recent Advances in Electrochemical Systems for Selective Fluorination of Organic Compounds. Acc. Chem. Res. 2020, 53, 322–334. 10.1021/acs.accounts.9b00520. [DOI] [PubMed] [Google Scholar]
- Stangier M.; Scheremetjew A.; Ackermann L. Chemo- and Site-Selective Electro-Oxidative Alkane Fluorination by C(sp3)–H Cleavage. Chem. Eur. J. 2022, 28, e202201654 10.1002/chem.202201654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recupero F.; Punta C. Free Radical Functionalization of Organic Compounds Catalyzed by N-Hydroxyphthalimide. Chem. Rev. 2007, 107, 3800–3842. 10.1021/cr040170k. [DOI] [PubMed] [Google Scholar]
- Marko J. A.; Durgham A.; Bretz S. L.; Liu W. Electrochemical Benzylic Oxidation of C–H Bonds. Chem. Commun. 2019, 55, 937–940. 10.1039/C8CC08768G. [DOI] [PubMed] [Google Scholar]
- Masui M.; Hosomi K.; Tsuchida K.; Ozaki S. Electrochemical Oxidation of Olefins Using N-Hydroxyphthalimide as a Mediator. Chem. Pharm. Bull. 1985, 33, 4798–4802. 10.1248/cpb.33.4798. [DOI] [Google Scholar]
- Masui M.; Hara S.; Ueshima T.; Kawaguchi T.; Ozaki S. Anodic Oxidation of Compounds Having Benzylic or Allylic Carbon and α-Carbon to Hetero Atom Using N-Hydroxyphthalimide as a Mediator. Chem. Pharm. Bull. 1983, 31, 4209–4211. 10.1248/cpb.31.4209. [DOI] [Google Scholar]
- Shono T.; Ikeda A. Electroorganic Chemistry. X. Anodic Allylic Substitution. J. Am. Chem. Soc. 1972, 94, 7892–7898. 10.1021/ja00777a036. [DOI] [Google Scholar]
- Horn E. J.; Rosen B. R.; Chen Y.; Tang J.; Chen K.; Eastgate M. D.; Baran P. S. Scalable and Sustainable Electrochemical Allylic C–H Oxidation. Nature 2016, 533, 77–81. 10.1038/nature17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowbridge A.; Walton S. M.; Gaunt M. J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. 10.1021/acs.chemrev.9b00462. [DOI] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545, 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Wang D. J.; Targos K.; Wickens Z. K. Electrochemical Synthesis of Allylic Amines from Terminal Alkenes and Secondary Amines. J. Am. Chem. Soc. 2021, 143, 21503–21510. 10.1021/jacs.1c11763. [DOI] [PubMed] [Google Scholar]
- Hu K.; Qian P.; Su J.-H.; Li Z.; Wang J.; Zha Z.; Wang Z. Multifunctionalization of Unactivated Cyclic Ketones via an Electrochemical Process: Access to Cyclic Α-Enaminones. J. Org. Chem. 2019, 84, 1647–1653. 10.1021/acs.joc.8b02930. [DOI] [PubMed] [Google Scholar]
- Yang Y.-Z.; Wu Y.-C.; Song R.-J.; Li J.-H. Electrochemical Dehydrogenative Cross-Coupling of Xanthenes with Ketones. Chem. Commun. 2020, 56, 7585–7588. 10.1039/D0CC02580A. [DOI] [PubMed] [Google Scholar]
- Gnaim S.; Takahira Y.; Wilke H. R.; Yao Z.; Li J.; Delbrayelle D.; Echeverria P.-G.; Vantourout J. C.; Baran P. S. Electrochemically Driven Desaturation of Carbonyl Compounds. Nat. Chem. 2021, 13, 367–372. 10.1038/s41557-021-00640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Zhang Q.; Lin J.; Harms K.; Meggers E. Electricity-Driven Asymmetric Lewis Acid Catalysis. Nat. Catal. 2019, 2, 34–40. 10.1038/s41929-018-0198-y. [DOI] [Google Scholar]
- Xiong P.; Hemming M.; Ivlev S. I.; Meggers E. Electrochemical Enantioselective Nucleophilic α-C(sp3)–H Alkenylation of 2-Acyl Imidazoles. J. Am. Chem. Soc. 2022, 144, 6964–6971. 10.1021/jacs.2c01686. [DOI] [PubMed] [Google Scholar]
- Liang K.; Zhang Q.; Guo C. Nickel-Catalyzed Switchable Asymmetric Electrochemical Functionalization of Alkenes. Sci. Adv. 2022, 8, eadd7134. 10.1126/sciadv.add7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Liang K.; Guo C. Enantioselective Nickel-Catalyzed Electrochemical Radical Allylation. Angew. Chem., Int. Ed. 2022, 61, e202210632 10.1002/anie.202210632. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Liang K.; Guo C. Asymmetric Electrochemical Arylation in the Formal Synthesis of (+)-Amurensinine. CCS Chem. 2021, 3, 338–347. 10.31635/ccschem.021.202000720. [DOI] [Google Scholar]
- Zhang Q.; Chang X.; Peng L.; Guo C. Asymmetric Lewis Acid Catalyzed Electrochemical Alkylation. Angew. Chem., Int. Ed. 2019, 58, 6999–7003. 10.1002/anie.201901801. [DOI] [PubMed] [Google Scholar]
- Turlik A.; Chen Y.; Newhouse T. R. Dehydrogenation Adjacent to Carbonyls Using Palladium–Allyl Intermediates. Synlett 2016, 27, 331–336. 10.1055/s-0035-1561282. [DOI] [Google Scholar]
- Diao T.; Stahl S. S. Synthesis of Cyclic Enones via Direct Palladium-Catalyzed Aerobic Dehydrogenation of Ketones. J. Am. Chem. Soc. 2011, 133, 14566–14569. 10.1021/ja206575j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M.; Dong G. Copper-Catalyzed Desaturation of Lactones, Lactams, and Ketones under PH-Neutral Conditions. J. Am. Chem. Soc. 2019, 141, 14889–14897. 10.1021/jacs.9b07932. [DOI] [PubMed] [Google Scholar]
- Huang D.; Szewczyk S. M.; Zhang P.; Newhouse T. R. Allyl-Nickel Catalysis Enables Carbonyl Dehydrogenation and Oxidative Cycloalkenylation of Ketones. J. Am. Chem. Soc. 2019, 141, 5669–5674. 10.1021/jacs.9b02552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaou K. C.; Zhong Y. L.; Baran P. S. A New Method for the One-Step Synthesis of α,β-Unsaturated Carbonyl Systems from Saturated Alcohols and Carbonyl Compounds. J. Am. Chem. Soc. 2000, 122, 7596–7597. 10.1021/ja001825b. [DOI] [Google Scholar]
- Jones A. M.; Banks C. E. The Shono-Type Electroorganic Oxidation of Unfunctionalised Amides. Carbon–Carbon Bond Formation via Electrogenerated N-Acyliminium Ions. Beilstein J. Org. Chem. 2014, 10, 3056–3072. 10.3762/bjoc.10.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeshov M. A.; Musio B.; Murray P. R. D.; Browne D. L.; Ley S. V. Expedient Preparation of Nazlinine and a Small Library of Indole Alkaloids Using Flow Electrochemistry as an Enabling Technology. Org. Lett. 2014, 16, 4618–4621. 10.1021/ol502201d. [DOI] [PubMed] [Google Scholar]
- Novaes L. F. T.; Ho J. S. K.; Mao K.; Liu K.; Tanwar M.; Neurock M.; Villemure E.; Terrett J. A.; Lin S. Exploring Electrochemical C(sp3)–H Oxidation for the Late-Stage Methylation of Complex Molecules. J. Am. Chem. Soc. 2022, 144, 1187–1197. 10.1021/jacs.1c09412. [DOI] [PubMed] [Google Scholar]
- Chen H.; Volgraf M.; Do S.; Kolesnikov A.; Shore D. G.; Verma V. A.; Villemure E.; Wang L.; Chen Y.; Hu B.; et al. Discovery of a Potent (4R,5S)-4-Fluoro-5-Methylproline Sulfonamide Transient Receptor Potential Ankyrin 1 Antagonist and Its Methylene Phosphate Prodrug Guided by Molecular Modeling. J. Med. Chem. 2018, 61, 3641–3659. 10.1021/acs.jmedchem.8b00117. [DOI] [PubMed] [Google Scholar]
- Shono T.; Matsumura Y.; Nakagawa Y. Electroorganic Chemistry. XII. Anodic Oxidation of Enol Esters. J. Am. Chem. Soc. 1974, 96, 3532–3536. 10.1021/ja00818a028. [DOI] [Google Scholar]
- Shono T.; Okawa M.; Nishiguchi I. Electroorganic Chemistry. XXI. Selective Formation Of.Alpha.-Acetoxy Ketones and General Synthesis of 2,3-Disubstituted 2-Cyclopentenones through the Anodic Oxidation of Enol Acetates. J. Am. Chem. Soc. 1975, 97, 6144–6147. 10.1021/ja00854a030. [DOI] [Google Scholar]
- Reddy S. H. K.; Chiba K.; Sun Y.; Moeller K. D. Anodic Oxidations of Electron-Rich Olefins: Radical Cation Based Approaches to the Synthesis of Bridged Bicyclic Ring Skeletons. Tetrahedron 2001, 57, 5183–5197. 10.1016/S0040-4020(01)00358-1. [DOI] [Google Scholar]
- Perkins R. J.; Feng R.; Lu Q.; Moeller K. D. Anodic Cyclizations, Seven-Membered Rings, and the Choice of Radical Cation VS. Radical Pathways. Chin. J. Chem. . 2019, 37, 672–678. 10.1002/cjoc.201900132. [DOI] [Google Scholar]
- Wu Y.; Yi H.; Lei A. Electrochemical Acceptorless Dehydrogenation of N-Heterocycles Utilizing Tempo as Organo-Electrocatalyst. ACS Catal. 2018, 8, 1192–1196. 10.1021/acscatal.7b04137. [DOI] [Google Scholar]
- Stuck F.; Dietl M. C.; Meißner M.; Sebastian F.; Rudolph M.; Rominger F.; Krämer P.; Hashmi A. S. K. Modular Two-Step Access to Π-Extended Naphthyridine Systems—Potent Building Blocks for Organic Electronics. Angew. Chem., Int. Ed. 2022, 61, e202114277 10.1002/anie.202114277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T.; Wang S.; Liu Y.; Liu S.; Qiu Y. Electrochemical Desaturative β-Acylation of Cyclic N-Aryl Amines. Angew. Chem., Int. Ed. 2022, 61, e202115178 10.1002/anie.202115178. [DOI] [PubMed] [Google Scholar]
- Kaiser D.; Klose I.; Oost R.; Neuhaus J.; Maulide N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. 10.1021/acs.chemrev.9b00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; He M.; Li Y.; Zhang H.; Yang D.; Nagasaka M.; Lv Z.; Guan Z.; Cao Y.; Gong F.; Zhou Z.; Zhu J.; Samanta S.; Chowdhury A. D.; Lei A. Electrochemical Oxidation Enables Regioselective and Scalable α-C(sp3)-H Acyloxylation of Sulfides. J. Am. Chem. Soc. 2021, 143, 3628–3637. 10.1021/jacs.1c00288. [DOI] [PubMed] [Google Scholar]
- Eberson L.; Nyberg K. Synthetic Uses of Anodic Substitution Reactions. Tetrahedron 1976, 32, 2185–2206. 10.1016/0040-4020(76)85132-0. [DOI] [Google Scholar]
- Kawamata Y.; Yan M.; Liu Z.; Bao D.-H.; Chen J.; Starr J. T.; Baran P. S. Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc. 2017, 139, 7448–7451. 10.1021/jacs.7b03539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T. H.; Samanta R. C.; Del Vecchio A.; Ackermann L. Mangana(III/IV)Electro-Catalyzed C(sp3)–H Azidation. Chem. Sci. 2021, 12, 2890–2897. 10.1039/D0SC05924B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Billington R. A.; Canonico P. L.; Sorba G.; Genazzani A. A. Click Chemistry Reactions in Medicinal Chemistry: Applications of the 1,3-Dipolar Cycloaddition between Azides and Alkynes. Med. Res. Rev. 2008, 28, 278–308. 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]
- Smith D. B.; Kalayanov G.; Sund C.; Winqvist A.; Maltseva T.; Leveque V. J. P.; Rajyaguru S.; Pogam S. L.; Najera I.; Benkestock K.; et al. The Design, Synthesis, and Antiviral Activity of Monofluoro and Difluoro Analogues of 4′-Azidocytidine against Hepatitis C Virus Replication: The Discovery of 4′-Azido-2′-Deoxy-2′-Fluorocytidine and 4′-Azido-2′-Dideoxy-2′,2′-Difluorocytidine. J. Med. Chem. 2009, 52, 2971–2978. 10.1021/jm801595c. [DOI] [PubMed] [Google Scholar]
- Geurink P. P.; van der Linden W. A.; Mirabella A. C.; Gallastegui N.; de Bruin G.; Blom A. E. M.; Voges M. J.; Mock E. D.; Florea B. I.; van der Marel G. A.; Driessen C.; van der Stelt M.; Groll M.; Overkleeft H. S.; Kisselev A. F. Incorporation of Non-Natural Amino Acids Improves Cell Permeability and Potency of Specific Inhibitors of Proteasome Trypsin-Like Sites. J. Med. Chem. 2013, 56, 1262–1275. 10.1021/jm3016987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.-L.; Li Y.-Q.; Ma C.; Fang P.; Zhang X.-J.; Mei T.-S. Palladium-Catalyzed C(sp3)—H Oxygenation via Electrochemical Oxidation. J. Am. Chem. Soc. 2017, 139, 3293–3298. 10.1021/jacs.7b01232. [DOI] [PubMed] [Google Scholar]
- Shrestha A.; Lee M.; Dunn A. L.; Sanford M. S. Palladium-Catalyzed C–H Bond Acetoxylation via Electrochemical Oxidation. Org. Lett. 2018, 20, 204–207. 10.1021/acs.orglett.7b03559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Zeng L.; Li H.; Cao Y.; Hu J.; Xu M.; Shi R.; Yi H.; Lei A. Electrochemical Palladium-Catalyzed Oxidative Sonogashira Carbonylation of Arylhydrazines and Alkynes to Ynones. J. Am. Chem. Soc. 2021, 143, 12460–12466. 10.1021/jacs.1c06036. [DOI] [PubMed] [Google Scholar]
- Liang H.; Julaiti Y.; Zhao C.-G.; Xie J. Electrochemical Gold-Catalysed Biocompatible C(sp2)–C(sp) Coupling. Nature Synthesis 2023, 2, 338–347. 10.1038/s44160-022-00219-w. [DOI] [Google Scholar]
- Liu J.; Liu X.; Wu J.; Li C.-C. Total Synthesis of Natural Products Containing a Bridgehead Double Bond. Chem. 2020, 6, 579–615. 10.1016/j.chempr.2019.12.027. [DOI] [Google Scholar]
- Ertl P.; Schuhmann T. A Systematic Cheminformatics Analysis of Functional Groups Occurring in Natural Products. J. Nat. Prod. 2019, 82, 1258–1263. 10.1021/acs.jnatprod.8b01022. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Lin S. Electroreductive Carbofunctionalization of Alkenes with Alkyl Bromides via a Radical-Polar Crossover Mechanism. J. Am. Chem. Soc. 2020, 142, 20661–20670. 10.1021/jacs.0c08532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Siu J. C.; Lai Y.; Lin S. An Electroreductive Approach to Radical Silylation via the Activation of Strong Si–Cl Bond. J. Am. Chem. Soc. 2020, 142, 21272–21278. 10.1021/jacs.0c10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu N.; Song L.; Liu J.; Shen Y.; Siu J. C.; Lin S. New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc. 2019, 141, 14480–14485. 10.1021/jacs.9b03296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu N.; Sauer G. S.; Saha A.; Loo A.; Lin S. Metal-Catalyzed Electrochemical Diazidation of Alkenes. Science 2017, 357, 575–579. 10.1126/science.aan6206. [DOI] [PubMed] [Google Scholar]
- Fu N.; Sauer G. S.; Lin S. Electrocatalytic Radical Dichlorination of Alkenes with Nucleophilic Chlorine Sources. J. Am. Chem. Soc. 2017, 139, 15548–15553. 10.1021/jacs.7b09388. [DOI] [PubMed] [Google Scholar]
- Sauer G. S.; Lin S. An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes. ACS Catal. 2018, 8, 5175–5187. 10.1021/acscatal.8b01069. [DOI] [Google Scholar]
- Siu J. C.; Sauer G. S.; Saha A.; Macey R. L.; Fu N.; Chauviré T.; Lancaster K. M.; Lin S. Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO–N3 Charge-Transfer Complex. J. Am. Chem. Soc. 2018, 140, 12511–12520. 10.1021/jacs.8b06744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu J. C.; Parry J. B.; Lin S. Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc. 2019, 141, 2825–2831. 10.1021/jacs.8b13192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K.-Y.; Pombar G.; Fu N.; Sauer G. S.; Keresztes I.; Lin S. Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes. J. Am. Chem. Soc. 2018, 140, 2438–2441. 10.1021/jacs.7b13387. [DOI] [PubMed] [Google Scholar]
- Fu N.; Shen Y.; Allen A. R.; Song L.; Ozaki A.; Lin S. Mn-Catalyzed Electrochemical Chloroalkylation of Alkenes. ACS Catal. 2019, 9, 746–754. 10.1021/acscatal.8b03209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Fu N.; Lin S. Three-Component Chlorophosphinoylation of Alkenes via Anodically Coupled Electrolysis. Synlett 2019, 30, 1199–1203. 10.1055/s-0039-1689934. [DOI] [Google Scholar]
- Park S. H.; Jang J.; Shin K.; Kim H. Electrocatalytic Radical-Polar Crossover Hydroetherification of Alkenes with Phenols. ACS Catal. 2022, 12, 10572–10580. 10.1021/acscatal.2c02436. [DOI] [Google Scholar]
- Mandal A.; Jang J.; Yang B.; Kim H.; Shin K. Palladium-Catalyzed Electrooxidative Hydrofluorination of Aryl-Substituted Alkenes with a Nucleophilic Fluorine Source. Org. Lett. 2023, 25, 195–199. 10.1021/acs.orglett.2c04045. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Kong W.-J.; Struwe J.; Sauermann N.; Rogge T.; Scheremetjew A.; Ackermann L. Electrooxidative Rhodium-Catalyzed C–H/C–H Activation: Electricity as Oxidant for Cross-Dehydrogenative Alkenylation. Angew. Chem., Int. Ed. 2018, 57, 5828–5832. 10.1002/anie.201803342. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Stangier M.; Meyer T. H.; Oliveira J. C. A.; Ackermann L. Iridium-Catalyzed Electrooxidative C–H Activation by Chemoselective Redox-Catalyst Cooperation. Angew. Chem., Int. Ed. 2018, 57, 14179–14183. 10.1002/anie.201809611. [DOI] [PubMed] [Google Scholar]
- Kapat A.; Sperger T.; Guven S.; Schoenebeck F. E-Olefins through Intramolecular Radical Relocation. Science 2019, 363, 391–396. 10.1126/science.aav1610. [DOI] [PubMed] [Google Scholar]
- Fu S.; Chen N.-Y.; Liu X.; Shao Z.; Luo S.-P.; Liu Q. Ligand-Controlled Cobalt-Catalyzed Transfer Hydrogenation of Alkynes: Stereodivergent Synthesis of Z- and E-Alkenes. J. Am. Chem. Soc. 2016, 138, 8588–8594. 10.1021/jacs.6b04271. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Huang Z.; Liu G.; Huang Z. A New Paradigm in Pincer Iridium Chemistry: PCN Complexes for (De)Hydrogenation Catalysis and Beyond. Acc. Chem. Res. 2022, 55, 2148. 10.1021/acs.accounts.2c00311. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Qin C.; Jia X.; Leng X.; Huang Z. An Agostic Iridium Pincer Complex as a Highly Efficient and Selective Catalyst for Monoisomerization of 1-Alkenes to Trans-2-Alkenes. Angew. Chem., Int. Ed. 2017, 56, 1614–1618. 10.1002/anie.201611007. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Huang Z.; Huang Z. Catalyst as Colour Indicator for Endpoint Detection to Enable Selective Alkyne Trans-Hydrogenation with Ethanol. Nat. Catal. 2019, 2, 529–536. 10.1038/s41929-019-0299-2. [DOI] [Google Scholar]
- Huang Z.; Wang Y.; Leng X.; Huang Z. An Amine-Assisted Ionic Monohydride Mechanism Enables Selective Alkyne Cis-Semihydrogenation with Ethanol: From Elementary Steps to Catalysis. J. Am. Chem. Soc. 2021, 143, 4824–4836. 10.1021/jacs.1c01472. [DOI] [PubMed] [Google Scholar]
- Gnaim S.; Bauer A.; Zhang H.-J.; Chen L.; Gannett C.; Malapit C. A.; Hill D. E.; Vogt D.; Tang T.; Daley R. A.; et al. Cobalt-Electrocatalytic HAT for Functionalization of Unsaturated C–C Bonds. Nature 2022, 605, 687–695. 10.1038/s41586-022-04595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Ma H.-X.; Mei T.-S.; Fang P.; Hu Y. Electrochemical Radical Formyloxylation–Bromination, – Chlorination, and – Trifluoromethylation of Alkenes. Org. Lett. 2019, 21, 3167–3171. 10.1021/acs.orglett.9b00867. [DOI] [PubMed] [Google Scholar]
- Tan Z.; Xiang F.; Xu K.; Zeng C. Electrochemical Organoselenium-Catalyzed Intermolecular Hydroazolylation of Alkenes with Low Catalyst Loadings. Org. Lett. 2022, 24, 5345–5350. 10.1021/acs.orglett.2c01983. [DOI] [PubMed] [Google Scholar]
- Wilken M.; Ortgies S.; Breder A.; Siewert I. Mechanistic Studies on the Anodic Functionalization of Alkenes Catalyzed by Diselenides. ACS Catal. 2018, 8, 10901–10912. 10.1021/acscatal.8b01236. [DOI] [Google Scholar]
- Wei B.-Y.; Xie D.-T.; Lai S.-Q.; Jiang Y.; Fu H.; Wei D.; Han B. Electrochemically Tuned Oxidative [4 + 2] Annulation and Dioxygenation of Olefins with Hydroxamic Acids. Angew. Chem., Int. Ed. 2021, 60, 3182–3188. 10.1002/anie.202012209. [DOI] [PubMed] [Google Scholar]
- Kim S.; Hirose K.; Uematsu J.; Mikami Y.; Chiba K. Electrochemically Active Cross-Linking Reaction for Fluorescent Labeling of Aliphatic Alkenes. Chem. Eur. J. 2012, 18, 6284–6288. 10.1002/chem.201103630. [DOI] [PubMed] [Google Scholar]
- Liu X.; Liu R.; Qiu J.; Cheng X.; Li G. Chemical-Reductant-Free Electrochemical Deuteration Reaction Using Deuterium Oxide. Angew. Chem., Int. Ed. 2020, 59, 13962–13967. 10.1002/anie.202005765. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Tao X.; Mao Y.; Yuan X.; Qiu J.; Kong L.; Ni S.; Guo K.; Wang Y.; Pan Y. Electrochemical C–N Bond Activation for Deaminative Reductive Coupling of Katritzky Salts. Nat. Commun. 2021, 12, 6745–6751. 10.1038/s41467-021-27060-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. Electrochemical Reductive Functionalization of Alkenes with Deuterochloroform as a One-Carbon Deuteration Block. Org. Lett. 2022, 24, 8645–8650. 10.1021/acs.orglett.2c03443. [DOI] [PubMed] [Google Scholar]
- Chai Q.-Y.; Yang Z.; Lin H.-W.; Han B.-N. Alkynyl-Containing Peptides of Marine Origin: A Review. Marine Drugs 2016, 14, 216–233. 10.3390/md14110216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X.; Wang P. Ynonylation of Acyl Radicals by Electroinduced Homolysis of 4-Acyl-1,4-Dihydropyridines. Org. Lett. 2021, 23, 4960–4965. 10.1021/acs.orglett.1c01243. [DOI] [PubMed] [Google Scholar]
- Alonso F.; Beletskaya I. P.; Yus M. Metal-Mediated Reductive Hydrodehalogenation of Organic Halides. Chem. Rev. 2002, 102, 4009–4092. 10.1021/cr0102967. [DOI] [PubMed] [Google Scholar]
- Alberico D.; Scott M. E.; Lautens M. Aryl–Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174–238. 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- Ackermann L.Modern Arylation Methods; Wiley Online Library, 2009. [Google Scholar]
- Froidevaux V.; Negrell C.; Caillol S.; Pascault J.-P.; Boutevin B. Biobased Amines: From Synthesis to Polymers; Present and Future. Chem. Rev. 2016, 116, 14181–14224. 10.1021/acs.chemrev.6b00486. [DOI] [PubMed] [Google Scholar]
- Kim J. E.; Choi S.; Balamurugan M.; Jang J. H.; Nam K. T. Electrochemical C–N Bond Formation for Sustainable Amine Synthesis. Trends Chem. 2020, 2, 1004–1019. 10.1016/j.trechm.2020.09.003. [DOI] [Google Scholar]
- Hager A.; Vrielink N.; Hager D.; Lefranc J.; Trauner D. Synthetic Approaches Towards Alkaloids Bearing Α-Tertiary Amines. Nat. Prod. Rep. 2016, 33, 491–522. 10.1039/C5NP00096C. [DOI] [PubMed] [Google Scholar]
- Crozet D.; Urrutigoïty M.; Kalck P. Recent Advances in Amine Synthesis by Catalytic Hydroaminomethylation of Alkenes. ChemCatChem. 2011, 3, 1102–1118. 10.1002/cctc.201000411. [DOI] [Google Scholar]
- Nugent T. C.; El-Shazly M. Chiral Amine Synthesis – Recent Developments and Trends for Enamide Reduction, Reductive Amination, and Imine Reduction. Adv. Synth. Catal. 2010, 352, 753–819. 10.1002/adsc.200900719. [DOI] [Google Scholar]
- Kawamata Y.; Vantourout J. C.; Hickey D. P.; Bai P.; Chen L.; Hou Q.; Qiao W.; Barman K.; Edwards M. A.; Garrido-Castro A. F.; et al. Electrochemically Driven, Ni-Catalyzed Aryl Amination: Scope, Mechanism, and Applications. J. Am. Chem. Soc. 2019, 141, 6392–6402. 10.1021/jacs.9b01886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Kale A. P.; Yue H.; Rueping M. Redox-Neutral Cross-Coupling Amination with Weak N-Nucleophiles: Arylation of Anilines, Sulfonamides, Sulfoximines, Carbamates, and Imines via Nickelaelectrocatalysis. JACS Au 2021, 1, 1057–1065. 10.1021/jacsau.1c00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z.; Li H.; Huang M.; Zhang W.; Zhi S.; Wang Y.; Pan Y. Electrochemical-Promoted Nickel-Catalyzed Oxidative Fluoroalkylation of Aryl Iodides. Org. Lett. 2021, 23, 8252–8256. 10.1021/acs.orglett.1c02997. [DOI] [PubMed] [Google Scholar]
- Korch K. M.; Watson D. A. Cross-Coupling of Heteroatomic Electrophiles. Chem. Rev. 2019, 119, 8192–8228. 10.1021/acs.chemrev.8b00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves E. K.; Entz E. D.; Neufeldt S. R. Chemodivergence between Electrophiles in Cross-Coupling Reactions. Chem. Eur. J. 2021, 27, 6161–6177. 10.1002/chem.202004437. [DOI] [PubMed] [Google Scholar]
- Ackermann L.; Althammer A.; Born R. Catalytic Arylation Reactions by C–H Bond Activation with Aryl Tosylates. Angew. Chem., Int. Ed. 2006, 45, 2619–2622. 10.1002/anie.200504450. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Wang Y.; Wang Y.; Pan Y. Electrochemical Deoxygenative Thiolation of Preactivated Alcohols and Ketones. Org. Lett. 2021, 23, 7524–7528. 10.1021/acs.orglett.1c02738. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Hong J.; Yao X.; Liu C.; Zhang L.; Fu Y.; Sun M.; Cheng R.; Li Z.; Ye J. Aminomethylation of Aryl Bromides by Nickel-Catalyzed Electrochemical Redox Neutral Cross Coupling. Org. Lett. 2021, 23, 9387–9392. 10.1021/acs.orglett.1c03500. [DOI] [PubMed] [Google Scholar]
- Wang B.; Peng P.; Ma W.; Liu Z.; Huang C.; Cao Y.; Hu P.; Qi X.; Lu Q. Electrochemical Borylation of Alkyl Halides: Fast, Scalable Access to Alkyl Boronic Esters. J. Am. Chem. Soc. 2021, 143, 12985–12991. 10.1021/jacs.1c06473. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Lu L.; Zhang W.; Wang Y.; Ware S. D.; Mondragon J.; Rein J.; Strotman N.; Lehnherr D.; See K. A.; Lin S. Electrochemically Driven Cross-Electrophile Coupling of Alkyl Halides. Nature 2022, 604, 292–297. 10.1038/s41586-022-04540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Chen Z.; Su C.; Zhao X.; Gao Q.; Ning G.-H.; Zhu H.; Tang W.; Leng K.; Fu W.; Tian B.; Peng X.; Li J.; Xu Q.-H.; Zhou W.; Loh K. P. Controllable Deuteration of Halogenated Compounds by Photocatalytic D2O Splitting. Nat. Commun. 2018, 9, 80–88. 10.1038/s41467-017-02551-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z.-Z.; Zhao J.-H.; Gou X.-Y.; Chen X.-M.; Liang Y.-M. Visible-Light-Mediated Hydrodehalogenation and Br/D Exchange of Inactivated Aryl and Alkyl Halides with a Palladium Complex. Org. Chem. Front. 2019, 6, 1649–1654. 10.1039/C9QO00240E. [DOI] [Google Scholar]
- Soulard V.; Villa G.; Vollmar D. P.; Renaud P. Radical Deuteration with D2O: Catalysis and Mechanistic Insights. J. Am. Chem. Soc. 2018, 140, 155–158. 10.1021/jacs.7b12105. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Yesilcimen A.; Cao M.; Zhang Y.; Zhang B.; Chan J. Z.; Wasa M. Catalytic Deuterium Incorporation within Metabolically Stable β-Amino C–H Bonds of Drug Molecules. J. Am. Chem. Soc. 2019, 141, 14570–14575. 10.1021/jacs.9b08662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gant T. G. Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem. 2014, 57, 3595–3611. 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- Schmidt C. First Deuterated Drug Approved. Nat. Biotechnol. 2017, 35, 493–495. 10.1038/nbt0617-493. [DOI] [PubMed] [Google Scholar]
- Li P.; Guo C.; Wang S.; Ma D.; Feng T.; Wang Y.; Qiu Y. Facile and General Electrochemical Deuteration of Unactivated Alkyl Halides. Nat. Commun. 2022, 13, 3774. 10.1038/s41467-022-31435-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L.; Li H.; Zheng Y.; Bu F.; Lei A. Facile and Economical Electrochemical Dehalogenative Deuteration of (Hetero)Aryl Halides. CCS Chem. 2021, 3, 2669–2675. 10.31635/ccschem.020.202000512. [DOI] [Google Scholar]
- Wood D.; Lin S. Deuterodehalogenation under Net Reductive or Redox-Neutral Conditions Enabled by Paired Electrolysis. Angew. Chem., Int. Ed. 2023, 135, e202218858 10.1002/ange.202218858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhao Z.; Pan D.; Wang S.; Jia K.; Ma D.; Yang G.; Xue X.-S.; Qiu Y. Metal-Free Electrochemical Carboxylation of Organic Halides in the Presence of Catalytic Amounts of an Organomediator. Angew. Chem., Int. Ed. 2022, 61, e202210201 10.1002/anie.202210201. [DOI] [PubMed] [Google Scholar]
- Coyle J. D. Photochemistry of Carboxylic Acid Derivatives. Chem. Rev. 1978, 78, 97–123. 10.1021/cr60312a002. [DOI] [Google Scholar]
- Ballatore C.; Huryn D. M.; Smith A. B. III Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem. 2013, 8, 385–395. 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A.-L.; Alaimo P. J. Useful Products from Complex Starting Materials: Common Chemicals from Biomass Feedstocks. Chem. Eur. J. 2010, 16, 4970–4980. 10.1002/chem.200903028. [DOI] [PubMed] [Google Scholar]
- Gooßen L. J.; Rodríguez N.; Gooßen K. Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chem., Int. Ed. 2008, 47, 3100–3120. 10.1002/anie.200704782. [DOI] [PubMed] [Google Scholar]
- Leech M. C.; Lam K. Electrosynthesis Using Carboxylic Acid Derivatives: New Tricks for Old Reactions. Acc. Chem. Res. 2020, 53, 121–134. 10.1021/acs.accounts.9b00586. [DOI] [PubMed] [Google Scholar]
- Xiang J.; Shang M.; Kawamata Y.; Lundberg H.; Reisberg S. H.; Chen M.; Mykhailiuk P.; Beutner G.; Collins M. R.; Davies A.; et al. Hindered Dialkyl Ether Synthesis with Electrogenerated Carbocations. Nature 2019, 573, 398–402. 10.1038/s41586-019-1539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roughley S. D.; Jordan A. M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- Chen X.; Luo X.; Peng X.; Guo J.; Zai J.; Wang P. Catalyst-Free Decarboxylation of Carboxylic Acids and Deoxygenation of Alcohols by Electro-Induced Radical Formation. Chem. Eur. J. 2020, 26, 3226–3230. 10.1002/chem.201905224. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Hill D. E.; Hao W.; McNicholas B. J.; Vantourout J. C.; Hadt R. G.; Reisman S. E.; Blackmond D. G.; Baran P. S. Electrochemical Nozaki–Hiyama–Kishi Coupling: Scope, Applications, and Mechanism. J. Am. Chem. Soc. 2021, 143, 9478–9488. 10.1021/jacs.1c03007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durandetti M.; Nédélec J.-Y.; Périchon J. An Electrochemical Coupling of Organic Halide with Aldehydes, Catalytic in Chromium and Nickel Salts. The Nozaki–Hiyama–Kishi Reaction. Org. Lett. 2001, 3, 2073–2076. 10.1021/ol016033g. [DOI] [PubMed] [Google Scholar]
- Durandetti M.; Périchon J.; Nédélec J.-Y. Nickel- and Chromium-Catalysed Electrochemical Coupling of Aryl Halides with Arenecarboxaldehydes. Tetrahedron Lett. 1999, 40, 9009–9013. 10.1016/S0040-4039(99)01674-3. [DOI] [Google Scholar]
- Grigg R.; Putnikovic B.; Urch C. J. Electrochemically Driven Catalytic Pd(0)/Cr(II) Mediated Coupling of Organic Halides with Aldehydes. The Nozaki-Hiyama-Kishi Reaction. Tetrahedron Lett. 1997, 38, 6307–6308. 10.1016/S0040-4039(97)01415-9. [DOI] [Google Scholar]
- Kuroboshi M. Electrochemical Regeneration of Chromium(II). Alkenylation of Carbonyl Compounds. Synlett 1999, 1999, 69–70. 10.1055/s-1999-2555. [DOI] [Google Scholar]
- Nagahara S.; Okada Y.; Kitano Y.; Chiba K. Biphasic Electrochemical Peptide Synthesis. Chem. Sci. 2021, 12, 12911–12917. 10.1039/D1SC03023J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Malins L. R. Total Synthesis of Biseokeaniamides A–C and Late-Stage Electrochemically-Enabled Peptide Analogue Synthesis. Chem. Sci. 2020, 11, 10752–10758. 10.1039/D0SC03701J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Malins L. R. An Electrochemical Approach to Designer Peptide α-Amides Inspired by α-Amidating Monooxygenase Enzymes. J. Am. Chem. Soc. 2021, 143, 11811–11819. 10.1021/jacs.1c05718. [DOI] [PubMed] [Google Scholar]
- Lin Y.; Malins L. R. Synthesis of Peptide N-Acylpyrroles via Anodically Generated N,O-Acetals. Synthesis 2022, 54, 3558–3567. 10.1055/s-0041-1737411. [DOI] [Google Scholar]
- Zhang B.; Gao Y.; Hioki Y.; Oderinde M. S.; Qiao J. X.; Rodriguez K. X.; Zhang H.-J.; Kawamata Y.; Baran P. S. Ni-Electrocatalytic Csp3–Csp3 Doubly Decarboxylative Coupling. Nature 2022, 606, 313–318. 10.1038/s41586-022-04691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J.; Sager C. P.; Ernst B. Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group. J. Med. Chem. 2019, 62, 8915–8930. 10.1021/acs.jmedchem.9b00179. [DOI] [PubMed] [Google Scholar]
- Villo P.; Shatskiy A.; Kärkäs M. D.; Lundberg H. Electrosynthetic C–O Bond Activation in Alcohols and Alcohol Derivatives. Angew. Chem., Int. Ed. 2023, 62, e202211952 10.1002/anie.202211952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmori H.; Nakai S.; Sekiguchi M.; Masui M. Anodic Oxidation of Organophosphorus Compounds. III. Anodic Alkoxylation and Thioalkoxylation of Triphenylphosphine. Chem. Pharm. Bull. 1980, 28, 910–915. 10.1248/cpb.28.910. [DOI] [Google Scholar]
- Ohmori H.; Nakai S.; Miyasaka H.; Masui M. An Improved Electrochemical Preparation of Alkoxytriphenylphosphonium Salts. Chem. Pharm. Bull. 1982, 30, 4192–4194. 10.1248/cpb.30.4192. [DOI] [Google Scholar]
- Yoshida H.; Usami N.; Ohishi Y.; Watanabe K.; Yamamoto I.; Yoshimura H. Synthesis and Pharmacological Effects in Mice of Halogenated Cannabinol Derivatives. Chem. Pharm. Bull. 1995, 43, 335–337. 10.1248/cpb.43.335. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Matsumoto S.; Koide T.; Ohmori H. Dehydroxy Substitution Reactions of the Anomeric Hydroxy Groups in Some Protected Sugars Initiated by Anodic Oxidation of Triphenylphosphine. Chem. Pharm. Bull. 1998, 46, 939–943. 10.1248/cpb.46.939. [DOI] [Google Scholar]
- Xu Z.; Zheng Y.; Wang Z.; Shao X.; Tian L.; Wang Y. Triphenylphosphine-Assisted Dehydroxylative Csp3–N Bond Formation Via Electrochemical Oxidation. Chem. Commun. 2019, 55, 15089–15092. 10.1039/C9CC08622F. [DOI] [PubMed] [Google Scholar]
- Huang C.-Y.; Doyle A. G. The Chemistry of Transition Metals with Three-Membered Ring Heterocycles. Chem. Rev. 2014, 114, 8153–8198. 10.1021/cr500036t. [DOI] [PubMed] [Google Scholar]
- Huang C.; Ma W.; Zheng X.; Xu M.; Qi X.; Lu Q. Epoxide Electroreduction. J. Am. Chem. Soc. 2022, 144, 1389–1395. 10.1021/jacs.1c11791. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tang S.; Yang G.; Wang S.; Ma D.; Qiu Y. Electrocarboxylation of Aryl Epoxides with CO2 for the Facile and Selective Synthesis of Β-Hydroxy Acids. Angew. Chem., Int. Ed. 2022, 61, e202207746 10.1002/anie.202207746. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Ren B.-H.; Liu X.-F.; Wang L.-L.; Zhang M.; Ren W.-M.; Lu X.-B.; Zhang W.-Z. Direct and Selective Electrocarboxylation of Styrene Oxides with CO2 for Accessing Β-Hydroxy Acids. Angew. Chem., Int. Ed. 2022, 61, e202207660 10.1002/anie.202207660. [DOI] [PubMed] [Google Scholar]
- Birch A. J. Reduction by Dissolving Metals. Part III. J. Chem. So. 1946, 593–597. 10.1039/jr9460000593. [DOI] [Google Scholar]
- Birch A. J. Reduction by Dissolving Metals. Nature 1946, 158, 585–585. 10.1038/158585c0. [DOI] [Google Scholar]
- Peters B. K.; Rodriguez K. X.; Reisberg S. H.; Beil S. B.; Hickey D. P.; Kawamata Y.; Collins M.; Starr J.; Chen L.; Udyavara S.; et al. Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 2019, 363, 838–845. 10.1126/science.aav5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh J. C. Alternating Current Electrolysis. J. Am. Chem. Soc. 1914, 36, 2333–2346. 10.1021/ja02188a007. [DOI] [Google Scholar]
- Srivastava S.; Shukla S. Electrolysis of Sodium Salts of Aliphatic Acids with ac. Electrochim. Acta 1967, 12, 1049–1057. 10.1016/0013-4686(67)80101-4. [DOI] [Google Scholar]
- Fleischmann M.; Goodridge F. Anodic Oxidation under Pulse Conditions. Discuss. Faraday Soc. 1968, 45, 254–260. 10.1039/df9684500254. [DOI] [Google Scholar]
- Alkire R. C.; Tsai J. E. Electrosynthesis of Propylene Oxide with Alternating Current. J. electrochem. Soc. 1982, 129, 1157–1158. 10.1149/1.2124048. [DOI] [Google Scholar]
- Kawamata Y.; Hayashi K.; Carlson E.; Shaji S.; Waldmann D.; Simmons B. J.; Edwards J. T.; Zapf C. W.; Saito M.; Baran P. S. Chemoselective Electrosynthesis Using Rapid Alternating Polarity. J. Am. Chem. Soc. 2021, 143, 16580–16588. 10.1021/jacs.1c06572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Martin C.; Kesselring D.; Keller R.; Moeller K. D. Building Functionalized Peptidomimetics: Use of Electroauxiliaries for Introducing N-Acyliminium Ions into Peptides. J. Am. Chem. Soc. 2006, 128, 13761–13771. 10.1021/ja064737l. [DOI] [PubMed] [Google Scholar]
- Sun H.; Moeller K. D. Silyl-Substituted Amino Acids: New Routes to the Construction of Selectively Functionalized Peptidomimetics. Org. Lett. 2002, 4, 1547–1550. 10.1021/ol025776e. [DOI] [PubMed] [Google Scholar]
- Xu G.; Moeller K. D. Anodic Coupling Reactions and the Synthesis of C-Glycosides. Org. Lett. 2010, 12, 2590–2593. 10.1021/ol100800u. [DOI] [PubMed] [Google Scholar]
- Kitada S.; Takahashi M.; Yamaguchi Y.; Okada Y.; Chiba K. Soluble-Support-Assisted Electrochemical Reactions: Application to Anodic Disulfide Bond Formation. Org. Lett. 2012, 14, 5960–5963. 10.1021/ol302863r. [DOI] [PubMed] [Google Scholar]
- Shoji T.; Kim S.; Yamamoto K.; Kawai T.; Okada Y.; Chiba K. Anodic Substitution Reaction of Proline Derivatives Using the 2,4,6-Trimethoxyphenyl Leaving Group. Org. Lett. 2014, 16, 6404–6407. 10.1021/ol503198p. [DOI] [PubMed] [Google Scholar]
- Wang D.; Jiang T.; Wan H.; Chen Z.; Qi J.; Yang A.; Huang Z.; Yuan Y.; Lei A. Alternating Current Electrolysis Enabled Formal C–O/O–H Cross-Metathesis of 4-Alkoxy Anilines with Alcohols. Angew. Chem., Int. Ed. 2022, 61, e202201543 10.1002/anie.202201543. [DOI] [PubMed] [Google Scholar]
- Karipal Padinjare Veedu D.; Connal L. A.; Malins L. R. Tunable Electrochemical Peptide Modifications: Unlocking New Levels of Orthogonality for Side-Chain Functionalization. Angew. Chem., Int. Ed. 2023, 62, e202215470 10.1002/anie.202215470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Ravelli D. Merging Photocatalysis with Electrochemistry: The Dawn of a New Alliance in Organic Synthesis. Angew. Chem., Int. Ed. 2019, 58, 17508–17510. 10.1002/anie.201910348. [DOI] [PubMed] [Google Scholar]
- Wu S.; Kaur J.; Karl T. A.; Tian X.; Barham J. P. Synthetic Molecular Photoelectrochemistry: New Frontiers in Synthetic Applications, Mechanistic Insights and Scalability. Angew. Chem., Int. Ed. 2022, 61, e202107811 10.1002/anie.202107811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y.; Shao M. Photoelectrocatalysis for High-Value-Added Chemicals Production. Chin. J. Catal. 2022, 43, 595–610. 10.1016/S1872-2067(21)63923-2. [DOI] [Google Scholar]
- Li P.; Zhang T.; Mushtaq M. A.; Wu S.; Xiang X.; Yan D. Research Progress in Organic Synthesis by Means of Photoelectrocatalysis. Chem. Rec. 2021, 21, 841–857. 10.1002/tcr.202000186. [DOI] [PubMed] [Google Scholar]
- Yang G.; Wang Y.; Qiu Y. Advances in Organic Photoelectrochemical Synergistic Catalysis. Chin. J. Org. Chem. 2021, 41, 3935–3947. 10.6023/cjoc202105054. [DOI] [Google Scholar]
- Yu Y.; Guo P.; Zhong J.-S.; Yuan Y.; Ye K.-Y. Merging Photochemistry with Electrochemistry in Organic Synthesis. Org. Chem. Front. 2020, 7, 131–135. 10.1039/C9QO01193E. [DOI] [Google Scholar]
- Barham J. P.; König B. Synthetic Photoelectrochemistry. Angew. Chem., Int. Ed. 2020, 59, 11732–11747. 10.1002/anie.201913767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buglioni L.; Raymenants F.; Slattery A.; Zondag S. D. A.; Noël T. Technological Innovations in Photochemistry for Organic Synthesis: Flow Chemistry, High-Throughput Experimentation, Scale-up, and Photoelectrochemistry. Chem. Rev. 2022, 122, 2752–2906. 10.1021/acs.chemrev.1c00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanam J. M. R.; Stephenson C. R. J. Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102–113. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]
- Romero N. A.; Nicewicz D. A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]
- Corbin D. A.; Miyake G. M. Photoinduced Organocatalyzed Atom Transfer Radical Polymerization (O-Atrp): Precision Polymer Synthesis Using Organic Photoredox Catalysis. Chem. Rev. 2022, 122, 1830–1874. 10.1021/acs.chemrev.1c00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K. P. S.; Sarkar S.; Gevorgyan V. Visible Light-Induced Transition Metal Catalysis. Chem. Rev. 2022, 122, 1543–1625. 10.1021/acs.chemrev.1c00403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo L.; Ravelli D.; Fagnoni M. Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration. Chem. Rev. 2022, 122, 1875–1924. 10.1021/acs.chemrev.1c00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A. Y.; Perry I. B.; Bissonnette N. B.; Buksh B. F.; Edwards G. A.; Frye L. I.; Garry O. L.; Lavagnino M. N.; Li B. X.; Liang Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Liardet L.; Luo J.; Ren D.; Grätzel M.; Hu X. Photoelectrocatalytic Arene C–H Amination. Nat. Catal. 2019, 2, 366–373. 10.1038/s41929-019-0231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-J.; Deng W.-H.; Guo J.-D.; Ci R.-N.; Zhou C.; Chen B.; Li X.-B.; Guo X.-N.; Liao R.-Z.; Tung C.-H.; Wu L.-Z. Transition-Metal-Free, Site-Selective C–F Arylation of Polyfluoroarenes via Electrophotocatalysis. J. Am. Chem. Soc. 2022, 144, 17261–17268. 10.1021/jacs.2c08068. [DOI] [PubMed] [Google Scholar]
- Tan Z.; He X.; Xu K.; Zeng C. Electrophotocatalytic C–H Functionalization of N-Heteroarenes with Unactivated Alkanes under External Oxidant-Free Conditions. ChemSusChem 2022, 15, e202102360 10.1002/cssc.202102360. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li L.; Fu N. Electrophotochemical Decarboxylative Azidation of Aliphatic Carboxylic Acids. ACS Catal. 2022, 12, 10661–10667. 10.1021/acscatal.2c02934. [DOI] [Google Scholar]
- Ortiz E.; Chang Y.-H.; Shezaf J. Z.; Shen W.; Krische M. J. Stereo- and Site-Selective Conversion of Primary Alcohols to Allylic Alcohols via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by 2-Butyne. J. Am. Chem. Soc. 2022, 144, 8861–8869. 10.1021/jacs.2c03614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernowsky C. P.; Chmiel A. F.; Wickens Z. K. Electrochemical Activation of Diverse Conventional Photoredox Catalysts Induces Potent Photoreductant Activity. Angew. Chem., Int. Ed. 2021, 60, 21418–21425. 10.1002/anie.202107169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y.; Xu K.; Zeng C. Electrophotocatalytic Si–H Activation Governed by Polarity-Matching Effects. CCS Chem. 2022, 4, 1796–1805. 10.31635/ccschem.021.202101010. [DOI] [Google Scholar]
- Chen Y.-J.; Lei T.; Hu H.-L.; Wu H.-L.; Zhou S.; Li X.-B.; Chen B.; Tung C.-H.; Wu L.-Z. Tandem Photoelectrochemical and Photoredox Catalysis for Efficient and Selective Aryl Halides Functionalization by Solar Energy. Matter 2021, 4, 2354–2366. 10.1016/j.matt.2021.05.004. [DOI] [Google Scholar]
- Cowper N. G. W.; Chernowsky C. P.; Williams O. P.; Wickens Z. K. Potent Reductants via Electron-Primed Photoredox Catalysis: Unlocking Aryl Chlorides for Radical Coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. 10.1021/jacs.9b12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Kim H.; Lambert T. H.; Lin S. Reductive Electrophotocatalysis: Merging Electricity and Light to Achieve Extreme Reduction Potentials. J. Am. Chem. Soc. 2020, 142, 2087–2092. 10.1021/jacs.9b10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Carpenter K. L.; Lin S. Electrochemistry Broadens the Scope of Flavin Photocatalysis: Photoelectrocatalytic Oxidation of Unactivated Alcohols. Angew. Chem., Int. Ed. 2020, 59, 409–417. 10.1002/anie.201910300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo L.; Quadri L. L.; Merli D.; Ravelli D. Photoelectrochemical Cross-Dehydrogenative Coupling of Benzothiazoles with Strong Aliphatic C–H Bonds. Chem. Commun. 2021, 57, 4424–4427. 10.1039/D1CC01012C. [DOI] [PubMed] [Google Scholar]
- Huang H.; Strater Z. M.; Lambert T. H. Electrophotocatalytic C–H Functionalization of Ethers with High Regioselectivity. J. Am. Chem. Soc. 2020, 142, 1698–1703. 10.1021/jacs.9b11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Žurauskas J.; Domański M.; Hitzfeld P. S.; Butera V.; Scott D. J.; Rehbein J.; Kumar A.; Thyrhaug E.; Hauer J.; Barham J. P. Hole-Mediated Photoredox Catalysis: Tris(p-substituted)Biarylaminium Radical Cations as Tunable, Precomplexing and Potent Photooxidants. Org. Chem. Front. 2021, 8, 1132–1142. 10.1039/D0QO01609H. [DOI] [Google Scholar]
- Yan H.; Song J.; Zhu S.; Xu H.-C. Synthesis of Acridinium Photocatalysts via Site-Selective C–H Alkylation. CCS Chem. 2021, 3, 317–325. 10.31635/ccschem.021.202000743. [DOI] [Google Scholar]
- Huang H.; Strater Z. M.; Rauch M.; Shee J.; Sisto T. J.; Nuckolls C.; Lambert T. H. Electrophotocatalysis with a Trisaminocyclopropenium Radical Dication. Angew. Chem., Int. Ed. 2019, 58, 13318–13322. 10.1002/anie.201906381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Stahl S. S. Merging Photochemistry with Electrochemistry: Functional-Group Tolerant Electrochemical Amination of C(sp3)–H Bonds. Angew. Chem., Int. Ed. 2019, 58, 6385–6390. 10.1002/anie.201813960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno H.; Iguchi S.; Miseki Y.; Sayama K. Photo-Electrochemical C–H Bond Activation of Cyclohexane Using a WO3 Photoanode and Visible Light. Angew. Chem., Int. Ed. 2018, 57, 11238–11241. 10.1002/anie.201805079. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Scheremetjew A.; Finger L. H.; Ackermann L. Electrophotocatalytic Undirected C–H Trifluoromethylations of (Het)Arenes. Chem. Eur. J. 2020, 26, 3241–3246. 10.1002/chem.201905774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor R. S. J.; Phipps R. J. Recent Advances in Minisci-Type Reactions. Angew. Chem., Int. Ed. 2019, 58, 13666–13699. 10.1002/anie.201900977. [DOI] [PubMed] [Google Scholar]
- Duncton M. A. J. Minisci Reactions: Versatile CH–Functionalizations for Medicinal Chemists. MedChemComm 2011, 2, 1135–1161. 10.1039/c1md00134e. [DOI] [Google Scholar]
- Yan H.; Hou Z.-W.; Xu H.-C. Photoelectrochemical C–H Alkylation of Heteroarenes with Organotrifluoroborates. Angew. Chem., Int. Ed. 2019, 58, 4592–4595. 10.1002/anie.201814488. [DOI] [PubMed] [Google Scholar]
- Suzuki J.; Tanigawa M.; Inagi S.; Fuchigami T. Electrochemical Oxidation of Organotrifluoroborate Compounds. ChemElectroChem. 2016, 3, 2078–2083. 10.1002/celc.201600451. [DOI] [Google Scholar]
- Lai X.-L.; Shu X.-M.; Song J.; Xu H.-C. Electrophotocatalytic Decarboxylative C–H Functionalization of Heteroarenes. Angew. Chem., Int. Ed. 2020, 59, 10626–10632. 10.1002/anie.202002900. [DOI] [PubMed] [Google Scholar]
- Hu A.; Guo J.-J.; Pan H.; Zuo Z. Selective Functionalization of Methane, Ethane, and Higher Alkanes by Cerium Photocatalysis. Science 2018, 361, 668–672. 10.1126/science.aat9750. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Kochi J. K. Photochemical and Thermal Reduction of Cerium(IV) Carboxylates. Formation and Oxidation of Alkyl Radicals. J. Am. Chem. Soc. 1968, 90, 6688–6698. 10.1021/ja01026a022. [DOI] [Google Scholar]
- Shirase S.; Tamaki S.; Shinohara K.; Hirosawa K.; Tsurugi H.; Satoh T.; Mashima K. Cerium(IV) Carboxylate Photocatalyst for Catalytic Radical Formation from Carboxylic Acids: Decarboxylative Oxygenation of Aliphatic Carboxylic Acids and Lactonization of Aromatic Carboxylic Acids. J. Am. Chem. Soc. 2020, 142, 5668–5675. 10.1021/jacs.9b12918. [DOI] [PubMed] [Google Scholar]
- Xu P.; Chen P.-Y.; Xu H.-C. Scalable Photoelectrochemical Dehydrogenative Cross-Coupling of Heteroarenes with Aliphatic C–H Bonds. Angew. Chem., Int. Ed. 2020, 59, 14275–14280. 10.1002/anie.202005724. [DOI] [PubMed] [Google Scholar]
- Rohe S.; Morris A. O.; McCallum T.; Barriault L. Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem., Int. Ed. 2018, 57, 15664–15669. 10.1002/anie.201810187. [DOI] [PubMed] [Google Scholar]
- Wang K.; Liu X.; Yang S.; Tian Y.; Zhou M.; Zhou J.; Jia X.; Li B.; Liu S.; Chen J. In Situ Alkyl Radical Recycling-Driven Decoupled Electrophotochemical Deamination. Org. Lett. 2022, 24, 3471–3476. 10.1021/acs.orglett.2c01022. [DOI] [PubMed] [Google Scholar]
- Wan Q.; Hou Z.-W.; Zhao X.-R.; Xie X.; Wang L. Organoelectrophotocatalytic C–H Silylation of Heteroarenes. Org. Lett. 2023, 25, 1008–1013. 10.1021/acs.orglett.3c00144. [DOI] [PubMed] [Google Scholar]
- Huang H.; Lambert T. H. Electrophotocatalytic C–H Heterofunctionalization of Arenes. Angew. Chem., Int. Ed. 2021, 60, 11163–11167. 10.1002/anie.202100222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T.; Lambert T. H. Electrophotocatalytic Diamination of Vicinal C–H Bonds. Science 2021, 371, 620–626. 10.1126/science.abf2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T.; Lambert T. H. C–H Amination via Electrophotocatalytic Ritter-Type Reaction. J. Am. Chem. Soc. 2021, 143, 8597–8602. 10.1021/jacs.1c03718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T.; Li Y.-L.; Ye K.-Y.; Lambert T. H. Electrophotocatalytic Oxygenation of Multiple Adjacent C–H Bonds. Nature 2023, 614, 275–280. 10.1038/s41586-022-05608-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L.; Jiang C.; Liang Y.; Liu D.; Bu F.; Shi R.; Chen H.; Chowdhury A. D.; Lei A. Manganese-Catalyzed Oxidative Azidation of C(sp3)–H Bonds under Electrophotocatalytic Conditions. J. Am. Chem. Soc. 2020, 142, 17693–17702. 10.1021/jacs.0c08437. [DOI] [PubMed] [Google Scholar]
- Woźniak Ł.; Tan J.-F.; Nguyen Q.-H.; Madron du Vigné A.; Smal V.; Cao Y.-X.; Cramer N. Catalytic Enantioselective Functionalizations of C–H Bonds by Chiral Iridium Complexes. Chem. Rev. 2020, 120, 10516–10543. 10.1021/acs.chemrev.0c00559. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Beckwith R. E. J. Catalytic Enantioselective C–H Activation by Means of Metal–Carbenoid-Induced C–H Insertion. Chem. Rev. 2003, 103, 2861–2904. 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]
- Cai C.-Y.; Lai X.-L.; Wang Y.; Hu H.-H.; Song J.; Yang Y.; Wang C.; Xu H.-C. Photoelectrochemical Asymmetric Catalysis Enables Site- and Enantioselective Cyanation of Benzylic C–H Bonds. Nat. Catal. 2022, 5, 943–951. 10.1038/s41929-022-00855-7. [DOI] [Google Scholar]
- Fan W.; Zhao X.; Deng Y.; Chen P.; Wang F.; Liu G. Electrophotocatalytic Decoupled Radical Relay Enables Highly Efficient and Enantioselective Benzylic C–H Functionalization. J. Am. Chem. Soc. 2022, 144, 21674–21682. 10.1021/jacs.2c09366. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Wang F.; McCann S. D.; Wang D.; Chen P.; Stahl S. S.; Liu G. Enantioselective Cyanation of Benzylic C–H Bonds via Copper-Catalyzed Radical Relay. Science 2016, 353, 1014–1018. 10.1126/science.aaf7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Fu N.; Ernst B. G.; Lee W. H.; Frederick M. O.; DiStasio R. A.; Lin S. Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. Nat. Chem. 2020, 12, 747–754. 10.1038/s41557-020-0469-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Chen P.; Liu G. Copper-Catalysed Asymmetric Radical Cyanation. Nat. Synth. 2022, 1, 107–116. 10.1038/s44160-021-00016-x. [DOI] [Google Scholar]
- Zhang Z.; Chen P.; Liu G. Copper-Catalyzed Radical Relay in C(sp3)–H Functionalization. Chem. Soc. Rev. 2022, 51, 1640–1658. 10.1039/D1CS00727K. [DOI] [PubMed] [Google Scholar]
- Wang F.; Chen P.; Liu G. Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res. 2018, 51, 2036–2046. 10.1021/acs.accounts.8b00265. [DOI] [PubMed] [Google Scholar]
- Huang H.; Lambert T. H. Electrophotocatalytic Acetoxyhydroxylation of Aryl Olefins. J. Am. Chem. Soc. 2021, 143, 7247–7252. 10.1021/jacs.1c01967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.; Lambert T. H. Electrophotocatalytic Snar Reactions of Unactivated Aryl Fluorides at Ambient Temperature and without Base. Angew. Chem., Int. Ed. 2020, 59, 658–662. 10.1002/anie.201909983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X.; Karl T. A.; Reiter S.; Yakubov S.; de Vivie-Riedle R.; König B.; Barham J. P. Electro-Mediated Photoredox Catalysis for Selective C(sp3)–O Cleavages of Phosphinated Alcohols to Carbanions. Angew. Chem., Int. Ed. 2021, 60, 20817–20825. 10.1002/anie.202105895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieth A. J.; Gonzalez M. I.; Kudisch B.; Nava M.; Nocera D. G. How Radical Are “Radical” Photocatalysts? A Closed-Shell Meisenheimer Complex Is Identified as a Super-Reducing Photoreagent. J. Am. Chem. Soc. 2021, 143, 14352–14359. 10.1021/jacs.1c06844. [DOI] [PubMed] [Google Scholar]
- Lai X.-L.; Chen M.; Wang Y.; Song J.; Xu H.-C. Photoelectrochemical Asymmetric Catalysis Enables Direct and Enantioselective Decarboxylative Cyanation. J. Am. Chem. Soc. 2022, 144, 20201–20206. 10.1021/jacs.2c09050. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Yang J.; Zhang J. Cu-Catalyzed Enantioselective Decarboxylative Cyanation via the Synergistic Merger of Photocatalysis and Electrochemistry. Chem. Sci. 2023, 14, 705–710. 10.1039/D2SC05428K. [DOI] [PMC free article] [PubMed] [Google Scholar]