

Abstract

Chemically crosslinked elastomers are a class of polymeric materials with properties that render them useful as adhesives, sealants, and in other engineering applications. Poly(γ-methyl-ε-caprolactone) (PγMCL) is a hydrolytically degradable and compostable aliphatic polyester that can be biosourced and exhibits competitive mechanical properties to traditional elastomers when chemically crosslinked. A typical limitation of chemically crosslinked elastomers is that they cannot be reprocessed; however, the incorporation of dynamic covalent bonds can allow for bonds to reversibly break and reform under an external stimulus, usually heat. In this work, we study the dynamic behavior and mechanical properties of PγMCL elastomers synthesized from aliphatic dianhydride crosslinkers. The crosslinked elastomers in this work were synthesized using the commercially available crosslinkers, 1,2,4,5-cyclohexanetetracarboxylic dianhydride, and 1,2,3,4-cyclobutanetetracarboxylic dianhydride and three-arm hydroxy-telechelic PγMCL star polymers. Stress relaxation experiments on the crosslinked networks showed an Arrhenius dependence of viscosity with temperature with an activation energy of 118 ± 8 kJ/mol, which agrees well with the activation energy of transesterification exchange chemistry obtained from small molecule model studies. Dynamic mechanical thermal analysis and rheological experiments confirmed the dynamic nature of the networks and provided insight into the mechanism of exchange (i.e., associative or dissociative). Tensile testing showed that these materials can exhibit high strains at break and low Young’s moduli, characteristic of soft and strong elastomers. By controlling the exchange chemistry and understanding the effect of macromolecular structure on mechanical properties, we prepared the high-performance elastomers that can be potentially reprocessed at moderately elevated temperatures.
Keywords: covalent adaptable network, polyester, internal catalysis, catalyst-free crosslinking, sustainable polymer
Introduction
Chemically crosslinked polymer networks can typically exhibit a variety of useful properties, including toughness, solvent resistance, and thermal stability, which make them versatile materials for the applications that include elastomers, adhesives, and foams. However, permanent covalent bonds in these crosslinked polymer networks prevent the implementation of traditional mechanical recycling strategies. This leads to an accumulation of waste upon end-of-use, which is disadvantageous from a sustainability perspective.1 In an effort to address this problem, replacing the permanent chemical crosslinks with dynamic bonds that can be activated with external stimuli, such as heat or light, to rearrange network bonds has been widely pursued.2−6 These covalent adaptable networks (CANs) provide a pathway for accessing tunable materials that can be recycled by reprocessing, thus increasing usage lifetime, an important element of sustainability in this class of materials.
CANs are generally divided into two categories based on their dynamic crosslink exchange mechanism: associative and dissociative.7 For associative mechanisms, bond breaking and reformation occur simultaneously, in which a pendant or end reactive group undergoes a substitution reaction with another chain.7,8 As a result, the number of crosslinks in an associative mechanism remains constant throughout the course of the exchange reactions.9 Materials that operate by associative mechanisms typically exhibit an Arrhenius-like (i.e., exponential) decrease in viscosity as a function of temperature and have been termed vitrimers.7,10,11 Conversely, in CANs that operate by dissociative mechanisms, bond breakage precedes bond formation, and, as a result, the crosslinking density decreases as bonds are broken at elevated temperature. Due to this loss of crosslink density, dissociative CANs typically demonstrate a more rapid decrease in viscosity with increasing temperature when compared to associative networks.9 Dissociative CANs also exhibit a gel to sol transition upon bond breaking that can lead to undesirable properties such as the lack of solvent resistance, creep, and a loss of material integrity.12 The consideration of the glass transition temperature (Tg), exchange reaction activation energy (Ea), identity of reactive groups, and polymer degradation temperatures is therefore important for selecting appropriate exchange chemistries.13 Some of the first vitrimers investigated were crosslinked epoxy-acid and epoxy-anhydride networks that underwent transesterification at elevated temperatures.14 These materials exhibit viscosities with an Arrhenius dependence on temperature and in the presence of 10 mol % zinc acetyl acetonate [Zn(acac)2] showed an activation energy of 88 kJ/mol.14 This transesterification reaction occurs between an existing ester bond and a free alcohol functional group to generate a new ester bond through a well-established associative mechanism. However, recent work has demonstrated that transesterification in CANs can be achieved through a dissociative mechanism when relying on neighboring carboxylic acid or sulfonic acid groups.15,16
For the practical reasons, the exchange reactions need to occur on reasonable timescales that allow macroscopic flow at desired reprocessing temperatures. To achieve this, a highly active catalyst is typically employed to lower the activation energy for exchange. Lewis and Brønsted acids, as well as metal-based catalysts such as zinc and tin derivatives, have been used in CANs.4,17−19 However, relying on external catalysts to facilitate the exchange reactions can lead to the limitations in material properties due to, for example, catalyst leeching over time.20 To move away from the use of external catalysts in CANs, new efforts have focused on catalyst-free systems that rely on an excess of exchanging functional groups, such as vinylogous urethane exchange,3,21−23 or on internal catalysts in the as-synthesized networks.24−26 Internal catalysts enable rapid exchange between reactive functional groups, but necessitate close proximity to reactive functional groups to be effective. Also called neighboring group participation, this method increases the reaction rate of exchange by lowering the activation energy as a result of this proximity effect.27,28 The neighboring group participation of carboxylic acids in transesterification of phthalate monoesters has been reported to follow a dissociative pathway and relies on carboxylic acid activating groups.15 This transesterification exchange can be applied to aliphatic polyester elastomers as a way to prepare renewable, reprocessable, and degradable CANs.
Aliphatic polyesters are an attractive alternative to hydrocarbon-based polymers due to their ability to be derived from renewable feedstocks, recycled, and composted.29−31 Polyesters can be readily prepared through the ring-opening transesterification polymerization (ROTEP) of cyclic ester (i.e., lactone) monomers. ROTEP allows for a high degree of control over molar mass, narrow molar mass distributions, and the facile preparation of various molecular architectures, which can in turn significantly impact the physical and mechanical properties of resulting materials.29 The ester linkages along the polymer backbone also allow for ready hydrolysis and polymer degradation. A variety of organic catalysts including 1,8-diazobicyclo[5.4.0]undec-7-ene (DBU),32 diphenyl phosphate (DPP),33 and dimethyl phosphate34 have been used as greener replacements for tin-based catalysts, such as stannous octoate (Sn(Oct)2),35,36 in the synthesis of these polyesters. We have reported the synthesis of high-performance thermoplastic elastomers (TPEs)37,38 and chemically crosslinked elastomers from γ-methyl-ε-caprolactone (γMCL),39 a seven membered lactone that can be derived from p-cresol (that can in turn be sourced from biorenewable lignin feedstocks).40 Our group has also shown the efficient polymerization and control of molar mass of poly(γMCL) (PγMCL) with the use of such organocatalysts.34 Synthesized elastomers exhibited impressive mechanical properties with high ultimate tensile strengths and elongation at break, both attributed to the contributions from chemical crosslinks and trapped entanglements. However, reported syntheses of these crosslinked polyester elastomers relied on catalysts such as Sn(oct)2 and multistep syntheses for the formation of crosslinkers investigated. Analogous crosslinked polyesters made from PγMCL have also been shown to hydrolyze enzymatically and degrade to high extents under industrial composting conditions.41
In this work, we seek to understand the behavior of transesterification in polyester CANs of PγMCL crosslinked with readily available aliphatic dianhydrides. Taking advantage of the hydroxy end-functionalized PγMCL, crosslinking with aliphatic dianhydrides will allow for a rapid exchange due to the presence of neighboring carboxylic acid groups. A focus of this work is to understand the effect of these neighboring carboxylic acids in lowering the activation energy barrier for exchange through kinetics studies on model systems. We also utilize the facile synthesis of PγMCL using DPP as an organocatalyst and subsequent catalyst-free crosslinking.34 We discuss the differences in stress relaxation behavior and material properties on polyester networks synthesized from two commercially available dianhydrides and connect these results to the conclusions about the mechanism of exchange.
Experimental Summary
Synthetic details of model ester compounds, polymers, and elastomers along with kinetics of model studies are covered in the Supporting Information. Specifics regarding methodology for the characterization of polymers and crosslinked elastomers by NMR spectroscopy, Fourier transform infrared spectroscopy (FTIR), thermogravimetric analysis (TGA) dynamic mechanical thermal analysis (DMTA), stress relaxation, and tensile testing experiments along with characterization parameters and instrument details can also be found in the Supporting Information.
Results and Discussion
Model Reaction Studies
The kinetics of esterification between anhydrides and alcohols have been well studied.42,43 At elevated temperatures, cyclic anhydrides can react with hydroxyl moieties without added catalysts.44 The esterification of a cyclic anhydride by an alcohol results in the formation of an ester and a carboxylic acid in close proximity, which can influence subsequent transesterification of the newly formed ester through self-catalysis. However, the second esterification of this pendant carboxylic acid with concomitant dehydration to form the corresponding diester typically requires the addition of a strong acid catalyst. Such a strong acid protonates and activates the carboxylic acid for nucleophilic attack from alcohols, allowing for the formation of the diester.43 Therefore, in the absence of a strong acid, only the first esterification typically takes place even in the presence of excess alcohol. Given this, it is likely that the carboxylic acid formed during crosslinking of telechelic PγMCL with cyclic dianhydrides acts as a neighboring group catalyst during the transesterification of these networks. To gain insight to this, we investigated a model system using a cyclic aliphatic anhydride and long-chain alcohol to model crosslinking with a hydroxyl end-functionalized PγMCL chain as well as potential transesterification with free alcohols.
Esterification kinetics were conducted on model systems using 1,2-cyclohexanedicarboxylic anhydride (CHMA) and dodecanol. Excess dodecanol was used to promote a pseudo-first-order process (Figure 1a). The melting temperatures of dodecanol and CHMA are 34 and 24 °C, respectively, allowing for the kinetics to be readily conducted in bulk, which is relevant and convenient for modeling practical polyester elastomer synthesis. Aliquots taken at periodic intervals were quenched by dilution in CDCl3 below room temperature, and the reaction kinetics were tracked using 1H NMR spectroscopy (Figure S4). The formation of the monoester was observed at temperatures of 40 °C and above; at 120 °C, the formation of the ester was complete after 8 min. The slopes of the pseudo-first-order kinetics plot in Figure 1b were used to determine the apparent first-order rate constants, k, at each temperature (sample calculation in Supporting Information). These rate constants were then used in an Arrhenius analysis to give an activation energy for this ring-opening under neat conditions of 41 ± 2 kJ/mol (Figure 1c). This activation energy is similar to previously reported activation energies for the uncatalyzed esterification of similar cyclic anhydrides including phthalic anhydride with 2-(2-methoxyethoxy)ethanol which gave an activation energy of 28 ± 2 kJ/mol.45 The rapid formation of the monoester indicates that crosslinking should occur rapidly between dianhydrides and hydroxyl end-functionalized PγMCL star polymers at moderate temperatures. The second esterification is more demanding and often characterized by much higher activation energies (e.g., >180 kJ/mol) to obtain the diester even with excess alcohol and the presence of strong acid catalysts, as is the case in traditional Fischer esterification.46
Figure 1.

(a) Scheme describing the model reaction used for esterification, conducted in bulk (i.e., neat, no solvent). (b) Pseudo-first-order ring-opening reaction kinetics between excess dodecanol and CHMA where [M] is the concentration of anhydride. (c) Arrhenius analysis of dodecanol and CHMA ring-opening reaction kinetics from 40 to 120 °C.
While the esterification kinetics aid in understanding the rate of crosslinking, the kinetics that describe the rate of bond exchange in the networks are those of transesterification between the ester formed from anhydride ring-opening and a free hydroxyl group. In these polyester systems, it is also likely that interchain esters participate in transesterification as also seen in the synthesis of copolyesters.47 To explore transesterification, a similar method to the esterification model kinetics was employed. We first prepared and isolated the dodecyl ester cyclohexyl carboxylic acid product from the above reaction for the transesterification kinetics. An excess of benzyl alcohol was used to model this as a pseudo-first-order reaction (Figure 2a). Benzyl alcohol was chosen due to the distinct resonances in the 1H NMR spectrum for the protons α to the alcohol in benzyl alcohol and the protons α to the alcohol in dodecanol (Figure S5). The formation of the benzyl monoester was observed at temperatures above 120 °C. A first-order kinetics plot of the transesterification reaction in Figure 2b shows the slopes at each associated temperature, corresponding to the rate constants. Arrhenius analysis gave an activation energy of 83 ± 6 kJ/mol (Figure 2c). Additionally, this transesterification reaction is likely internally catalyzed, with an activation energy close to the previously reported transesterification in model systems using phthalic monoesters (Ea = 95 ± 5 kJ/mol).15
Figure 2.

(a) Scheme describing the model reaction used for transesterification (b) Pseudo-first-order transesterification reaction kinetics between excess dodecyl monoester and benzyl alcohol where [M] is the concentration of dodecyl monoester. (c) Arrhenius plot of dodecyl monoester and benzyl alcohol exchange kinetics.
The mechanism and kinetics of exchange can be correlated with the viscoelastic behavior of CANs.48 We explored additional model reactions to understand whether dissociative or associative pathways were favored.49 In a dissociative pathway, the rate-limiting step would be the reformation of the cyclic anhydride through a cleavage of the ester bond with concomitant release of alcohol, followed by the rapid reopening of the anhydride with another alcohol. An associative mechanism requires the addition of a free alcohol to the ester near the carboxylic acid to form the usual tetrahedral intermediate in the rate-determining step, followed by the reformation of the carbonyl bond and expulsion of the alcohol group from the original ester. A dissociative pathway has been reported in model systems using phthalate monoesters, but has not been explored with aliphatic monoesters.15 While we anticipate the transesterification mechanism to be similar between aromatic and aliphatic anhydrides, we aimed to confirm the supposition that activation energies found from our model system were consistent with those we found from stress relaxation on our crosslinked materials. In an associative mechanism, increasing the concentration of free alcohol functional groups should increase the overall rate of transesterification. While for a dissociative mechanism, the rate would largely be unaffected if the rate-limiting step was the initial reformation of the anhydride, providing that the alcohol was not involved in anhydride reformation.
By 1H NMR spectroscopy, we studied the effect on initial reaction rate when mono-1-dodecyl cyclohexyl carboxylic acid is reacted with 0.5, 1, or 2 equivalents of monoester at 140 °C. The data are shown in Figure S6, and the exact neat concentrations are provided in Table S1. The initial rate of ester exchange at approximately equimolar conditions is about 6.0 × 10–4 M min–1. At 2 equiv. of benzyl alcohol to 1 equiv. of monoester, the concentration of the monoester was lower by about 52% under these neat conditions. The rate was lower at 3.7 × 10–4 M min–1, but not significantly different than that under equimolar conditions. Conversely, at 2 equiv. of monoester to 1 equiv. of alcohol, the rate increased to 9.9 × 10–4 M min–1. Combined, these data are consistent with a rate-limiting step being cyclic anhydride formation early stages of the reaction. This is further supported with pseudo-first-order kinetics for both the monoester consumption and benzyl alcohol consumption under the conditions that dodecyl monoester was held in a 5:1 molar ratio to benzyl alcohol. Figure S7 shows the apparent rates of the reaction with excess dodecyl monoester to be greater than the reaction with excess benzyl alcohol.
To investigate the effect of the carboxylic acid on the rate of transesterification, a reference model reaction was studied wherein the carboxylic acid at the β position was replaced with a hydrogen as seen in Figure 3a. Figure 3b,c show the difference in reaction rate when the group is replaced from a carboxylic acid to a hydrogen. Over an order of magnitude increase in the rate constant of reaction from 5.9 × 10–4 to 6.7 × 10–3 min–1 was seen for the carboxylic acid derivative, indicating that the presence of the internal acid facilitates the exchange reaction as expected.
Figure 3.

(a) Scheme describing the model reaction used for transesterification replacing carboxylic acid with hydrogen (b) kinetics of transesterification reaction. (c) Pseudo-first-order transesterification reaction kinetics between excess dodecyl monoester with carboxylic acid or hydrogen at β position and benzyl alcohol where [M] is the concentration of dodecyl monoester.
Synthesis of Crosslinked Elastomers
The results from the model reactions helped inform the design of dynamic polyester elastomers with the rapid transesterification facilitated by the neighboring carboxylic acids. Low molar mass PγMCL prepolymer was synthesized through ROTEP using trimethylol propane, a trifunctional initiator and DPP as an organocatalyst according to previous work.34 The resultant star polymer with a Mn of 9.7 kDa with hydroxyl functional groups at each chain end was subsequently reacted with a dianhydride crosslinker in an equimolar ratio of alcohol to anhydride functional groups (Scheme 1). Two commercially available aliphatic dianhydrides were chosen as crosslinking reagents due to their solubility and our understanding of kinetics from model systems. The differences between 1,2,4,5-cyclohexanetetracarboxylic dianhydride (CHDA) and 1,2,3,4-cyclobutanetetracarboxylic dianhydride (CBDA) were explored to understand the differences in material properties and mechanism of exchange.
Scheme 1. Synthesis of Crosslinked Polyester Networks.

Two dianhydrides were investigated during the catalyst-free crosslinking step.
Solution casting the polymer and crosslinker from a mixture of THF (2 mL) and dimethylformamide (1 mL) was required to solubilize the crosslinker. The films were dried under a steady flow of nitrogen for 16 h before they were reacted at 120 °C for 24 h under nitrogen atmosphere. The resulting films were clear and colorless, void of any obvious macroscopic defects (Figure S9). Consumption of the anhydride was determined by FTIR spectroscopy for both CHDA and CBDA (Figures S10 and S11). The disappearance of the anhydride carbonyl stretch at 1780 cm–1 indicates near complete consumption of the anhydride during crosslinking and thus a high degree of alcohol consumption under these stoichiometric conditions. The degree of crosslinking was evaluated by swelling the films in THF for 24 h and extracting the soluble fraction. The theoretical extent of conversion required for the gelation of a stoichiometrically-balanced A3 + B2 system is 0.73.50 The resultant gel fractions were greater than 0.85 showing high degrees of crosslinking and are listed in Table S3. The glass transition (Tg) of the crosslinked elastomers was −56 °C, close to the prepolymer Tg of −60 °C (Tables S2 and S3).
When the crosslinking temperature of the network crosslinked with CHDA was increased to 180 °C and the film was left for 2 h, we observed the anhydride stretch in the FTIR (Figures 4 and S10–S12). This supports a dissociative cleavage of crosslink junctions leading to a reappearance of cyclic anhydride intermediates at these higher temperatures. However, upon cooling, the gel fractions of the films increased after this high temperature treatment, consistent with more extensive crosslinking, suggesting that dissociation may not be the only reaction taking place. It is possible that at 180 °C, dehydration and formation of esters from esterification between neighboring carboxylic acid groups generated from the ring-opened anhydrides is also ocurring. This would result in the formation of tri- or tetrafunctional crosslink junctions with no pendant carboxylic acid groups, thus limiting further exchange behavior (Scheme 2). We postulate at elevated temperatures there are contributions from both dissociation of crosslinks and formation of anhydride junctions from dehydration of carboxylic acids. Figures S13 and S14 show a TGA isotherm of an analogous PγMCL elastomer held at 180 °C with material mass loss likely coming from dehydration in the network. The FTIR spectrum of this experiment before and after exposure is shown in Figure S15.
Figure 4.

(a) Scheme of likely bond exchange occurring at 120 and 180 °C (b) IR spectrum of films crosslinked with CHDA before and after heating in oven under flow of nitrogen air at 120 °C for 24 h and subsequently exposed 180 °C. The peak at 1780 cm–1 corresponds to the stretch of the anhydride while the peak at 1726 cm–1 corresponds to the ester stretch of the polymer backbone.
Scheme 2. Formation of Anhydride from Carboxylic Acid Dehydration at Crosslink Junction Leading to a Trifunctional Crosslink Junction.

The results for similar experiments with the CBDA networks can be found in Figure S16, in which some evidence of anhydride is seen at elevated temperatures. When the networks were directly crosslinked at 180 °C, the gel fractions and glass transition temperatures increased while stress relaxation times decreased, indicating a higher degree of crosslinking (Table S3 and Figure S17).
Mechanical Properties of Crosslinked Elastomers
The temperature–modulus relationships in the PγMCL elastomers were investigated by DMTA where the storage and loss moduli of both networks were monitored from −70 to 200 °C under uniaxial extension at a heating rate of 5 °C min–1 (Figure 5). The storage modulus was greater than the loss modulus at all temperatures above the glass transition, consistent with crosslinked networks. The networks with both crosslinkers exhibit glass transitions around −57 °C and the rubbery plateau modulus then remained constant until dramatic material softening or the end of the experiment, and both networks maintained mechanical integrity during testing. Specifically, the CBDA network maintains its plateau modulus over the entire temperature range investigated (T > 200 °C) and even a slight increase in modulus is observed at higher temperatures. However, in the CHDA networks, the modulus begins to rapidly drop at approximately 150 °C. This precipitous drop in the modulus reflects a softening behavior and demonstrates that the viscosity of the system is decreasing likely through a loss of crosslink density. The more rapid decrease in modulus observed in the CHDA system at high temperature system further supports the claim that these networks are exchanging through a dissociative mechanism, as has been observed in Diels–Alder polymer networks.51 We conclude this because the energy barrier to reform the cyclohexyl anhydride is lower than the cyclobutyl anhydride due to ring strain.52 Additionally, the position of the anhydrides as cis or trans relative to the ring can contribute to the difficulty or ease to reform the anhydride. While the CBDA crosslinker is cis, the CHDA crosslinker used is a mixture of cis and trans isomers. The dissociative pathway is also supported by the recent work from DuPrez and coworkers in pyromellitic dianhydride polyester networks.15
Figure 5.

DMTA plot of a three-arm 9.7 kDa PγMCL prepolymer crosslinked with CHDA (blue) and CBDA (black). Samples were heated at a rate of 5 °C min–1 from −70 to 150 °C. ω = 6.28 rad/s.
The theoretical molar mass between crosslinks (Mx) for the crosslinked elastomers was 6.5 kg/mol. Contributions from both entanglements and crosslinks contribute to the rubbery plateau modulus (EN′), and this value can be used to estimate the effective molar mass between crosslinks Mx,eff using eq 1 where E′ is the storage modulus under tension, T is the absolute temperature, taken at the minimum of the tan δ data, and ρ is the density of the PγMCL films, which was estimated to be 1.066. From this, the calculated values of Mx,eff were 7.1 and 7.9 kg/mol for the CHDA and CBDA networks, respectively. These values are closer to the predicted Mx than the theoretical entanglement molar mass (Me) of 2.9 kg/mol for PγMCL, indicating that at low strains, more contribution comes from crosslinks than entanglements, likely due to the molar mass of the prepolymer is not high enough to lead to significant contributions from entanglements.
| 1 |
One of the ways to probe exchange mechanisms in CANs is through stress relaxation experiments. These stress relaxation experiments measure the stress response of a viscoelastic material placed under a step (fixed) strain. In crosslinked elastomers, contributions from both entanglements and crosslinks contribute to the modulus, and thus, there may be several relaxation pathways present during stress relaxation experiments. However, well above the Tg in crosslinked CANs, viscous flow is limited by the rate of bond exchange in the networks.53 The Arrhenius dependence of viscosity on temperature in CANs has been observed in both networks undergoing associative and dissociative bond exchange mechanisms.7 The activation energy of this bond exchange can be related to the terminal relaxation time found from stress relaxation experiments, as shown in eq 2 where τ0 is the pre-exponential factor, or the terminal relaxation time without thermal constraints,7,54EaS is the activation energy from stress relaxation, R is the gas constant, and T is the temperature at which the experiment was conducted. The characteristic relaxation time (τ*) is the time at which the modulus (E′) has decreased to 1/e of its initial value. Plotting the natural log of these relaxation times against temperature and calculation of the slope allows for the estimation of the activation energy for stress relaxation.
| 2 |
Stress relaxation experiments were conducted on two crosslinked systems comprised of a 9.6 kg/mol prepolymer crosslinked with an equimolar ratio of either CHDA or CBDA to alcohol functional groups. Figure 6a,c show the stress relaxation curves of the two systems investigated at temperatures ranging from 100 to 180 °C. All data, including samples that did not fully relax past 1/e, were fit to a stretched exponential decay function to obtain a value of τ* (eq S1, fitting parameters listed in Table S4). An Arrhenius plot of the characteristic relaxation times allowed for the calculation of activation energies (EaS) of 118 ± 8 and 83 ± 17 kJ/mol for the CHDA and CBDA networks, respectively (Figure 6b,d). This falls within the range of 30–160 kJ/mol as has been observed in most transesterification vitrimers.27 The stress relaxation plots in Figure 6 and the non-normalized stress relaxation plots seen in Figures S19 and S20 show more rapid relaxation in the CHDA system compared to the CBDA systems at analogous temperatures. The differences between these relaxation times at various temperatures indicate that there may be more than one relaxation pathway contributing to a higher activation energy for the CHDA over CBDA system. It takes over 3000 s in the CBDA network for the modulus to relax to 1/e at 180 °C while it only takes 70 s in the CHDA network at the same temperature. Additionally, the shape of the curves in the CBDA networks differs at higher temperatures than those observed at lower probed temperatures. This could suggest that different relaxation processes are occurring at different temperatures, making correlation between temperature and viscosity in this network more difficult. Deviations from temperature–viscosity linearity have been observed in transesterification vitrimers with two distinct slopes in the Arrhenius plot, indicating two mechanisms of relaxation: bond exchange through chemically limited transesterification at low temperatures and diffusion limited dynamic rearrangement of network strands at high temperatures.55 In PDMS vitrimers, three regimes arise from temperature–viscosity relationships. At temperatures Tg + 200 K, bond exchange dynamics control viscosity, and at temperatures < Tg + 200 K, polymer segmental dynamics control viscosity.56
Figure 6.

(a) Stress relaxation curves of 9 K PγMCL network crosslinked with CHDA. Samples were held at 5% strain for 3 h or until complete relaxation. (b) Arrhenius plot of PγMCL-CHDA network. (c) Stress relaxation curves of 9 K PγMCL network crosslinked with CBDA. (d) Arrhenius plot of PγMCL-CBDA network. Dashed lines indicate the fits of data to eq S1.
The prefactor 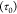 obtained in both these systems
is quite
different. In the CHDA network, τ0 is lower than
the CBDA network while in the CHDA network has a higher EaS. This prefactor can be understood
as an extrapolation of the Arrhenius plot to infinite temperature
and is what the characteristic relaxation time would be if there are
no contributions from network rearrangements. A wider range of temperatures
would need to be examined to obtain a complete understanding of this
effect.
obtained in both these systems
is quite
different. In the CHDA network, τ0 is lower than
the CBDA network while in the CHDA network has a higher EaS. This prefactor can be understood
as an extrapolation of the Arrhenius plot to infinite temperature
and is what the characteristic relaxation time would be if there are
no contributions from network rearrangements. A wider range of temperatures
would need to be examined to obtain a complete understanding of this
effect.
The more rapid decrease in modulus for the CHDA elastomer over the CBDA elastomer is also consistent with a dissociative mechanism for bond exchange. In a dissociative mechanism, the rate-limiting step is the reformation of the cyclic anhydride. There is an increased energy barrier to reform the cyclic anhydride in the cyclobutyl system because we anticipate that the ring strain would be greater than in the cyclohexyl dianhydride. An associative mechanism would only be rate limited by the formation of a tetrahedral intermediate in the unstrained structures. However, above 140 °C, there could also be competing effects between network dissociation and dehydration, leading to the formation of tri and tetra esters at crosslink junctions, which would limit bond exchange and result in longer relaxation times (Figures S21 and S22).
Rheological Analysis
The reversibility of network dissociation was studied through cyclic DMTA ramps under shear. Prior to the experiment, dynamic strain sweeps were conducted to determine the appropriate strain in the linear viscoelastic regime for each network (Figure S23). The temperature sweep experiments aimed to probe the change in modulus of the crosslinked elastomer over a range of temperatures (T = 100–180 °C) above the temperature at which bond exchange is substantively activated (Figure 7). At 120 °C, the modulus of the CHDA network is initially around 87 kPa while that of the CBDA network is approximately 100 kPa. These differences could be attributed to a greater percentage of crosslinks being dissociated upon the first heating cycle in the CHDA system. During the first heating cycle, the modulus of the CHDA system decreases over an order of magnitude. This is attributed to the dissociation of crosslinks leading to a less dense network, and thus, a drop in modulus is anticipated. Upon the cooling cycles, the modulus of the material begins to increase again, indicating the reformation of network bonds. However, the modulus does not return to its initial value after the first heating cycle, indicating that not all the crosslinks have been reformed. More bonds may be reformed if the sample was cooled back to room temperature. The second and third heating cycles follow similar trends where the modulus decreases upon heating but increases upon cooling. More bonds may be reformed if the sample was cooled back to room temperature (Figure S24).
Figure 7.

Reversibility of network dissociation through oscillatory shear experiments for 3 cycles of heating and cooling from 120 to 180 °C. CHDA (left) and CBDA (right). Samples were heated and cooled at a rate of 5 °C/min and held at 1% strain and ω = 6.28 rad/s.
These results differ in the CBDA system, particularly that over each of the heating and cooling cycles besides the first, the modulus increases. The first heating cycle results in a decrease in modulus but not as much as the CHDA system. Interestingly, the subsequent heating and cooling cycles see the increases in the modulus. This could be attributed to the dehydration in the network releasing water and forming ester junctions between carboxylic acids. The formation of more crosslinks would increase the modulus of the material. This was confirmed with gel fractions on the recovered material from the rheological experiment, which increased from 0.86 to 0.94 after rheological testing, which was not seen in the CHDA sample.
Tensile Properties
The elastic performance of the synthesized networks was investigated by linear and cyclical tensile tests. The Young’s modulus was determined by the slope of the stress–strain curve in the low strain limit. The two elastomers demonstrated low Young’s moduli, as seen by the inset in Figure 8. Though the theoretical molecular weight between crosslinks (Mx) of both networks is the same, there is a difference in the Young’s modulus between the CHDA and CBDA network. The lower Young’s modulus in the CBDA network could be attributed to the network defects and loops which are difficult to quantify.57 It has been shown that the effect of aromatic or aliphatic crosslinker structure in epoxy-resins has a strong effect on the elastic modulus and there may be a contribution here due to the choice of the crosslinker.58 The strain at break was greater for the CBDA network while the stress at break was greater than the CHDA network. Overall, these elastomers exhibited higher stresses and strains at break when compared to analogous elastomers crosslinked with a similar PγMCL prepolymer molar mass and bis-β-lactone crosslinker.39
Figure 8.

Representative stress–strain curves comparing crosslinked elastomers from PγMCL with a theoretical Mx of 6.5 kg/mol with different crosslinkers. Samples were extended at a 50 mm min–1 rate until the sample broke. The inset shows the difference in Young’s moduli from 0 to 10% strain.
Cyclical loading and unloading experiments on the dianhydride polyester networks were conducted to understand energy dissipation and exhibited low energy loss over 20 cycles (Figure 9). The largest energy loss was found for the first cycle. The hysteresis energy loss of the CBDA network was slightly greater than that of the CHDA network.
Figure 9.

Cyclical uniaxial extension tests for a 10 kg/mol PγMCL three-arm star prepolymer crosslinked with CHDA (left) and CBDA (right). Samples were extended to 50% strain at a rate of 50 mm min–1.
Conclusion
We have demonstrated the efficient polymerization of γMCL using an organocatalyst to prepare star-shaped polyesters with hydroxyl functional end groups that can readily react with commercially available dianhydrides without the need of an exogeneous catalyst. Internal carboxylic acids generated during crosslinking facilitate in catalyzing transesterification in these systems leading to interesting rheological properties. Kinetics on model systems demonstrate that rapid exchange kinetics occur with excess alcohol moieties. We suggest a dissociative mechanism to be the leading mechanism for the CHDA network, due to the decrease in modulus at elevated temperatures which is not observed by DMTA in the CBDA network. Activation energies from model reactions agree well with those found from stress relaxation experiments. Both systems exhibit a strong temperature response with viscosity. The tensile properties of the networks also varied slightly when changing the structure of the dianhydride crosslinker. High stresses and strains at break were reported for both systems, with CHDA having a higher modulus.
The networks investigated are promising renewable elastomers, with a biodegradation potential under appropriate conditions. Additionally, the rapid transesterification at elevated temperatures makes these systems the viable candidates for recycling and reprocessing. This chemistry can be expanded to other crosslinked systems taking advantage of internal catalysis of carboxylic acid groups to prepare materials that are industrially competitive with incumbent materials. Varying the molar mass between crosslinks, crosslink functionality and crosslink structure are the mechanical properties of these systems that can be tuned to afford stronger materials. The properties observed in these elastomers expand the scope of catalyst free CANs. We have demonstrated herein a promising candidate for sustainably derived and synthesized elastomers taking advantage of commercially available dianhydride crosslinkers. Exploring other avenues of architectural modifications including varying the number of arms in the prepolymer and exploring the chemical composition of these networks with different polyesters or polyanhydrides and additional crosslinkers will provide a broader understanding of the impact of these systems.
Acknowledgments
We would like to thank Dr. David Giles for the advice on DMTA and stress relaxation experiments. Additionally, we would like to acknowledge Yoon-Jung Jang, Dr. Satu Häkkinen, and Dr. Stephanie Liffland for their helpful discussions. This work was supported by the National Science Foundation Center for Sustainable Polymers headquartered at the University of Minnesota, CHE-1901635.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acspolymersau.3c00004.
Materials, methods, experimental controls, and supplementary characterization data including 1H NMR spectra, 13C NMR spectra, IR spectra, SEC data, TGA data, and rheological stress relaxation data (PDF).
The authors declare no competing financial interest.
Notes
All primary data files are available at https://doi.org/10.13020/qewn-zh14.
Supplementary Material
References
- Post W.; Susa A.; Blaauw R.; Molenveld K.; Knoop R. J. I. A Review on the Potential and Limitations of Recyclable Thermosets for Structural Applications. Polym. Rev. 2019, 60, 359–388. 10.1080/15583724.2019.1673406. [DOI] [Google Scholar]
- Kloxin C. J.; Bowman C. N. Covalent Adaptable Networks: Smart, Reconfigurable and Responsive Network Systems. Chem. Soc. Rev. 2013, 42, 7161–7173. 10.1039/c3cs60046g. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; Cramer C. J.; Hillmyer M. A.; Dichtel W. R. Mechanically Activated, Catalyst-Free Polyhydroxyurethane Vitrimers. J. Am. Chem. Soc. 2015, 137, 14019–14022. 10.1021/jacs.5b08084. [DOI] [PubMed] [Google Scholar]
- Snyder R. L.; Fortman D. J.; De Hoe G. X.; Hillmyer M. A.; Dichtel W. R. Reprocessable Acid-Degradable Polycarbonate Vitrimers. Macromolecules 2018, 51, 389–397. 10.1021/acs.macromol.7b02299. [DOI] [Google Scholar]
- Brutman J. P.; Delgado P. A.; Hillmyer M. A. Polylactide Vitrimers. ACS Macro Lett. 2014, 3, 607–610. 10.1021/mz500269w. [DOI] [PubMed] [Google Scholar]
- Ishibashi J. S. A.; Kalow J. A. Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum’s Acid-Derived Cross-Links. ACS Macro Lett. 2018, 7, 482–486. 10.1021/acsmacrolett.8b00166. [DOI] [PubMed] [Google Scholar]
- Scheutz G. M.; Lessard J. J.; Sims M. B.; Sumerlin B. S. Adaptable Crosslinks in Polymeric Materials: Resolving the Intersection of Thermoplastics and Thermosets. J. Am. Chem. Soc. 2019, 141, 16181–16196. 10.1021/jacs.9b07922. [DOI] [PubMed] [Google Scholar]
- Chakma P.; Konkolewicz D. Dynamic Covalent Bonds in Polymeric Materials. Angew. Chem., Int. Ed. 2019, 58, 9682–9695. 10.1002/anie.201813525. [DOI] [PubMed] [Google Scholar]
- Elling B. R.; Dichtel W. R. Reprocessable Cross-Linked Polymer Networks: Are Associative Exchange Mechanisms Desirable?. ACS Cent. Sci. 2020, 6, 1488–1496. 10.1021/acscentsci.0c00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloxin C. J.; Scott T. F.; Adzima B. J.; Bowman C. N. Covalent Adaptable Networks (CANs): A Unique Paradigm in Cross-Linked Polymers. Macromolecules 2010, 43, 2643–2653. 10.1021/ma902596s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winne J. M.; Leibler L.; Du Prez F. E. Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective. Polym. Chem. 2019, 10, 6091–6108. 10.1039/c9py01260e. [DOI] [Google Scholar]
- Yang Y.; Urban M. W. Self-Healing Polymeric Materials. Chem. Soc. Rev. 2013, 42, 7446. 10.1039/c3cs60109a. [DOI] [PubMed] [Google Scholar]
- Wu J.-B.; Li S.-J.; Liu H.; Qian H.-J.; Lu Z.-Y. Dynamics and Reaction Kinetics of Coarse-Grained Bulk Vitrimers: A Molecular Dynamics Study. Phys. Chem. Chem. Phys. 2019, 21, 13258. 10.1039/c9cp01766f. [DOI] [PubMed] [Google Scholar]
- Montarnal D.; Capelot M.; Tournilhac F.; Leibler L. Silica-like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. 10.1126/science.1212648. [DOI] [PubMed] [Google Scholar]
- Delahaye M.; Winne J. M.; Du Prez F. E. Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry. J. Am. Chem. Soc. 2019, 141, 15277–15287. 10.1021/jacs.9b07269. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Majumdar S.; van Benthem R. A. T. M.; Sijbesma R. P.; Heuts J. P. A. Intramolecularly Catalyzed Dynamic Polyester Networks Using Neighboring Carboxylic and Sulfonic Acid Groups. ACS Macro Lett. 2020, 9, 272–277. 10.1021/acsmacrolett.9b01023. [DOI] [PubMed] [Google Scholar]
- Self J. L.; Dolinski N. D.; Zayas M. S.; Read De Alaniz J.; Bates C. M. Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks. ACS Macro Lett. 2018, 7, 817–821. 10.1021/acsmacrolett.8b00370. [DOI] [PubMed] [Google Scholar]
- Capelot M.; Montarnal D.; Tournilhac F.; Leibler L. Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. 10.1021/ja302894k. [DOI] [PubMed] [Google Scholar]
- Demongeot A.; Groote R.; Goossens H.; Hoeks T.; Tournilhac F.; Leibler L. Cross-Linking of Poly(Butylene Terephthalate) by Reactive Extrusion Using Zn(II) Epoxy-Vitrimer Chemistry. Macromolecules 2017, 50, 6117–6127. 10.1021/acs.macromol.7b01141. [DOI] [Google Scholar]
- Argyle M. D.; Bartholomew C. H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. 10.3390/catal5010145. [DOI] [Google Scholar]
- Lessard J. J.; Garcia L. F.; Easterling C. P.; Sims M. B.; Bentz K. C.; Arencibia S.; Savin D. A.; Sumerlin B. S. Catalyst-Free Vitrimers from Vinyl Polymers. Macromolecules 2019, 52, 2105–2111. 10.1021/acs.macromol.8b02477. [DOI] [Google Scholar]
- Pan X.; Sengupta P.; Webster D. C. High Biobased Content Epoxy-Anhydride Thermosets from Epoxidized Sucrose Esters of Fatty Acids. Biomacromolecules 2011, 12, 2416–2428. 10.1021/bm200549c. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu T.; Zhang S.; Shao L.; Fei M.; Yu H.; Zhang J. Catalyst-Free Vitrimer Elastomers Based on a Dimer Acid: Robust Mechanical Performance, Adaptability and Hydrothermal Recyclability. Green Chem. 2020, 22, 870–881. 10.1039/c9gc04080c. [DOI] [Google Scholar]
- Van Lijsebetten F.; Spiesschaert Y.; Winne J. M.; Du Prez F. E. Reprocessing of Covalent Adaptable Polyamide Networks through Internal Catalysis and Ring-Size Effects. J. Am. Chem. Soc. 2021, 143, 15834–15844. 10.1021/jacs.1c07360. [DOI] [PubMed] [Google Scholar]
- Berne D.; Cuminet F.; Lemouzy S.; Joly-Duhamel C.; Poli R.; Caillol S.; Leclerc E.; Ladmiral V. Catalyst-Free Epoxy Vitrimers Based on Transesterification Internally Activated by an α-CF3Group. Macromolecules 2022, 55, 1669–1679. 10.1021/acs.macromol.1c02538. [DOI] [Google Scholar]
- Hayashi M.; Inaba T. Achievement of a Highly Rapid Bond Exchange for Self-Catalyzed Polyester Vitrimers by Incorporating Tertiary Amino Groups on the Network Strands. ACS Appl. Polym. Mater. 2021, 3, 4424–4429. 10.1021/acsapm.1c00724. [DOI] [Google Scholar]
- Cuminet F.; Caillol S.; Dantras É.; Leclerc E. ´ R.; Ladmiral V. Neighboring Group Participation and Internal Catalysis Effects on Exchangeable Covalent Bonds: Application to the Thriving Field of Vitrimer Chemistry. Macromolecules 2021, 54, 3927–3961. 10.1021/acs.macromol.0c02706. [DOI] [Google Scholar]
- Li Q.; Ma S.; Li P.; Wang B.; Yu Z.; Feng H.; Liu Y.; Zhu J. Fast Reprocessing of Acetal Covalent Adaptable Networks with High Performance Enabled by Neighboring Group Participation. Macromolecules 2021, 54, 8423–8434. 10.1021/acs.macromol.1c01046. [DOI] [Google Scholar]
- Hillmyer M. A.; Tolman W. B. Aliphatic Polyester Block Polymers: Renewable, Degradable, and Sustainable. Acc. Chem. Res. 2014, 47, 2390–2396. 10.1021/ar500121d. [DOI] [PubMed] [Google Scholar]
- Albertsson A.-C.; Varma I. K.. Aliphatic Polyesters: Synthesis, Properties and Applications. In Degradable Aliphatic Polyesters; Springer: Berlin, 2002; pp 1–44. [Google Scholar]
- Lecomte P.; Jérôme C.; Zachmann H. G.; Rieger B.; Künkel A.; Coates W. G.; Reichardt R.; Dinjus E.; Zevaco A. T.. Recent Developments in Ring-Opening Polymerization of Lactones. In Synthetic Biodegradable Polymers; Springer, 2012; pp 173–217. [Google Scholar]
- Kemo V. M.; Schmidt C.; Zhang Y.; Beuermann S. Low Temperature Ring-Opening Polymerization of Diglycolide Using Organocatalysts with PEG as Macroinitiator. Macromol. Chem. Phys. 2016, 217, 842–849. 10.1002/macp.201500491. [DOI] [Google Scholar]
- Schneiderman D. K.High Performance Materials from Renewable Aliphatic Polyesters; Ph.D. Dissertation, University of Minnesota: Twin Cities, 2016. [Google Scholar]
- Batiste D. C.; Meyersohn M. S.; Watts A.; Hillmyer M. A. Efficient Polymerization of Methyl- ε -Caprolactone Mixtures To Access Sustainable Aliphatic Polyesters. Macromolecules 2020, 53, 1795–1808. 10.1021/acs.macromol.0c00050. [DOI] [Google Scholar]
- Leenslag J. W.; Pennings A. J. Synthesis of High-Molecular-Weight Poly(L-Lactide) Initiated with Tin 2-Ethylhexanoate. Makromol. Chem. 1987, 188, 1809–1814. 10.1002/macp.1987.021880804. [DOI] [Google Scholar]
- Kowalski A.; Duda A.; Penczek S. Mechanism of Cyclic Ester Polymerization Initiated with Tin(II) Octoate. 2. Macromolecules Fitted with Tin(II) Alkoxide Species Observed Directly in MALDI–TOF Spectra. Macromolecules 2000, 33, 689–695. 10.1021/ma9906940. [DOI] [Google Scholar]
- Watts A.; Kurokawa N.; Hillmyer M. A. Strong, Resilient, and Sustainable Aliphatic Polyester Thermoplastic Elastomers. Biomacromolecules 2017, 18, 1845–1854. 10.1021/acs.biomac.7b00283. [DOI] [PubMed] [Google Scholar]
- Liffland S.; Hillmyer M. A. Enhanced Mechanical Properties of Aliphatic Polyester Thermoplastic Elastomers through Star Block Architectures. Macromolecules 2021, 54, 9327–9340. 10.1021/acs.macromol.1c01357. [DOI] [Google Scholar]
- De Hoe G. X. De; Zumstein M. T.; Tiegs B. J.; Brutman J. P.; Mcneill K.; Sander M.; Coates W.; Hillmyer M. A. Sustainable Polyester Elastomers from Lactones: Synthesis, Properties, and Enzymatic Hydrolyzability. J. Am. Chem. Soc. 2018, 140, 963–973. 10.1021/jacs.7b10173. [DOI] [PubMed] [Google Scholar]
- Lundberg D. J.; Lundberg D. J.; Hillmyer M. A.; Dauenhauer P. J. Techno-Economic Analysis of a Chemical Process To Manufacture Methyl-ε-Caprolactone from Cresols. ACS Sustainable Chem. Eng. 2018, 6, 15316–15324. 10.1021/acssuschemeng.8b03774. [DOI] [Google Scholar]
- Reisman L.; Siehr A.; Horn J.; Batiste D. C.; Kim H. J.; De Hoe G. X.; Ellison C. J.; Shen W.; White E. M.; Hillmyer M. A. Respirometry and Cell Viability Studies for Sustainable Polyesters and Their Hydrolysis Products. ACS Sustainable Chem. Eng. 2021, 9, 2736–2744. 10.1021/acssuschemeng.0c08026. [DOI] [Google Scholar]
- Kulawska M.; Sadłowski J. Z.; Skrzypek J. Kinetics of the Esterification of Maleic Anhydride with Octyl, Decyl or Dodecyl Alcohol over Dowex Catalyst. React. Kinet. Catal. Lett. 2005, 85, 51–56. 10.1007/s11144-005-0242-1. [DOI] [Google Scholar]
- Widell R.; Karlsson H. T. Autocatalytic Behaviour in Esterification between Anhydrides and Alcohols. Thermochim. Acta 2006, 447, 57–63. 10.1016/j.tca.2006.05.003. [DOI] [Google Scholar]
- Kienle R. H.; Hovey A. G. The Polyhydric Alcohol-Polybasic Acid Reaction. I. Glycerol-Phthalic Anhydride. J. Am. Chem. Soc. 1929, 51, 509–519. 10.1021/ja01377a021. [DOI] [Google Scholar]
- Hinde N. J.; Hall D. Kinetics and Mechanism of the Formation of Mono-and Di-Phthalate Esters Catalysed by Titanium and Tin Alkoxides. J. Chem. Soc., Perkin Trans. 1 1998, 2, 1249–1256. 10.1039/a708468d. [DOI] [Google Scholar]
- Skrzypek J.; Sadłowski J. Z.; Lachowska M.; Jaroszyński M. Kinetics of the Esterification of Phthalic Anhydride with 2-Ethylhexanol Part III. Tetrabutyl Titanate as a Catalyst. Chem. Eng. Process. 1996, 35, 283–286. 10.1016/0255-2701(95)04130-3. [DOI] [Google Scholar]
- Economy J.; Schneggenburger L. A.; Frich D. Interchain Transesterification Reactions in Copolyesters. Transreact. Condens. Polym. 1999, 1, 195–217. 10.1002/9783527613847.ch5. [DOI] [Google Scholar]
- Spiesschaert Y.; Taplan C.; Stricker L.; Guerre M.; Winne J. M.; Du Prez F. E. Influence of the Polymer Matrix on the Viscoelastic Behaviour of Vitrimers. Polym. Chem. 2020, 11, 5377. 10.1039/d0py00114g. [DOI] [Google Scholar]
- Mazo P.; Estenoz D.; Sponton M.; Rios L. Kinetics of the Transesterification of Castor Oil with Maleicanhydride Using Conventional and Microwave Heating. J. Am. Oil Chem. Soc. 2012, 89, 1355–1361. 10.1007/s11746-012-2020-3. [DOI] [Google Scholar]
- Stockmayer W. H. Theory of Molecular Size Distribution and Gel Formation in Branched-Chain Polymers. J. Chem. Phys. 1943, 11, 45–55. 10.1063/1.1723803. [DOI] [Google Scholar]
- Sheridan R. J.; Bowman C. N. A Simple Relationship Relating Linear Viscoelastic Properties and Chemical Structure in a Model Diels-Alder Polymer Network. Macromolecules 2012, 45, 7634–7641. 10.1021/ma301329u. [DOI] [Google Scholar]
- Eberson L.; Landström L. Studies on Cyclic Anhydrides. Acta Chem. Scand. 1972, 26, 239–249. 10.3891/acta.chem.scand.26-0239. [DOI] [Google Scholar]
- Podgórski M.; Fairbanks B. D.; Kirkpatrick B. E.; McBride M.; Martinez A.; Dobson A.; Bongiardina N. J.; Bowman C. N. Toward Stimuli-Responsive Dynamic Thermosets through Continuous Development and Improvements in Covalent Adaptable Networks (CANs). Adv. Mater. 2020, 32, 1906876. 10.1002/adma.201906876. [DOI] [PubMed] [Google Scholar]
- El-Zaatari B. M.; Ishibashi J. S. A.; Kalow J. A. Cross-Linker Control of Vitrimer Flow. Polym. Chem. 2020, 11, 5339–5345. 10.1039/d0py00233j. [DOI] [Google Scholar]
- Hubbard A. M.; Ren Y.; Konkolewicz D.; Sarvestani A.; Picu C. R.; Kedziora G. S.; Roy A.; Varshney V.; Nepal D. Vitrimer Transition Temperature Identification: Coupling Various Thermomechanical Methodologies. ACS Appl. Polym. Mater. 2021, 3, 1756–1766. 10.1021/acsapm.0c01290. [DOI] [Google Scholar]
- Porath L.; Huang J.; Ramlawi N.; Derkaloustian M.; Ewoldt R. H.; Evans C. M. Relaxation of Vitrimers with Kinetically Distinct Mixed Dynamic Bonds. Macromolecules 2022, 55, 4450–4458. 10.1021/acs.macromol.1c02613. [DOI] [Google Scholar]
- Zhong M.; Wang R.; Kawamoto K.; Olsen B. D.; Johnson J. A. Quantifying the Impact of Molecular Defects on Polymer Network Elasticity. Science 2016, 353, 1264–1268. 10.1126/science.aag0184. [DOI] [PubMed] [Google Scholar]
- Lv G.; Jensen E.; Shan N.; Evans C. M.; Cahill D. G. Effect of Aromatic/Aliphatic Structure and Cross-Linking Density on the Thermal Conductivity of Epoxy Resins. ACS Appl. Polym. Mater. 2021, 3, 1555–1562. 10.1021/acsapm.0c01395. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.