
Figure 3.
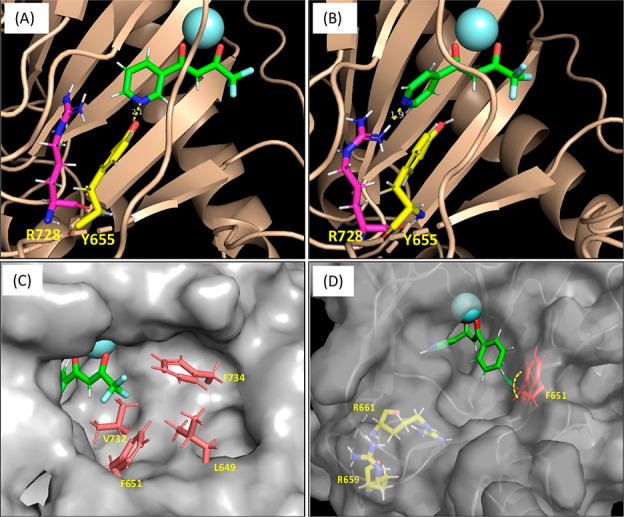
Binding interactions between HTS hit compounds 1 (A and C), 2 (B), and 5 (C) and the LH2 active site predicted by molecular docking studies and observed in MD and QM/MM simulations. (A) Calculated distance between the phenol hydrogen (Y655) and the pyridine nitrogen of 1 is 2.1 Å. (B) Calculated distance between the guanidine hydrogen (R728) and the pyridine nitrogen of 2 is 1.9 Å. (C) The entry of the active site of LH2 associated with 1 shows the hydrophobic environment in proximity to trifluoromethyl (−CF3) group of 1. (D) Docked model of 5 presenting the surface environment of the benzene ring of the compound.