

Abstract
Members of the candidate phylum Dadabacteria, recently reassigned to the phylum Candidatus Desulfobacterota, are cosmopolitan in the marine environment found both free-living and associated with hosts that are mainly marine sponges. Yet, these microorganisms are poorly characterized, with no cultured representatives and an ambiguous phylogenetic position in the tree of life. Here, we performed genome-centric metagenomics to elucidate their phylogenomic placement and predict the metabolism of the sponge-associated members of this lineage. Rank-based phylogenomics revealed several new species and a novel family (Candidatus Spongomicrobiaceae) within a sponge-specific order, named here Candidatus Nemesobacterales. Metabolic reconstruction suggests that Ca. Nemesobacterales are aerobic heterotrophs, capable of synthesizing most amino acids, vitamins and cofactors and degrading complex carbohydrates. We also report functional divergence between sponge- and seawater-associated metagenome-assembled genomes. Niche-specific adaptations to the sponge holobiont were evident from significantly enriched genes involved in defense mechanisms against foreign DNA and environmental stressors, host-symbiont interactions and secondary metabolite production. Fluorescence in situ hybridization gave a first glimpse of the morphology and lifestyle of a member of Ca. Desulfobacterota. Candidatus Nemesobacterales spp. were found both inside sponge cells centred around sponge nuclei and in the mesohyl of the sponge Geodia barretti. This study sheds light on the enigmatic group Ca. Nemesobacterales and their functional characteristics that reflect a symbiotic lifestyle.
Subject terms: Marine microbiology, Microbial ecology, Metagenomics, Symbiosis, Bacterial genomics
Introduction
The candidate phylum Dadabacteria, formerly known as SBR1093 was initially detected in 16S rRNA gene cloning experiments from activated sludge in sequential batch reactors [1]. In 2014, the first draft genome was reconstructed from industrial-activated sludge samples using metagenome binning methods [2]. Hug et al. [3] later established SBR1093 as a novel phylum under the name “Dadabacteria” [3]. To date, this lineage remains uncultured and understudied displaying a yet-unresolved phylogenetic placement in the tree of life. Waite et al. [4] recently proposed the assignment of members of the Candidatus Dadabacteria to the candidate phylum Desulfobacterota [4]. However, phylogenetic analyses based on concatenated ribosomal proteins did not recover ”Dadabacteria” as a monophyletic group with members of the Ca. Desulfobacterota phylum [3, 4], highlighting the lack of confidence in assigning a taxonomic rank to this lineage. Currently, both the NCBI (NCBI Taxonomy Browser, November 2022) and SILVA databases (r138.1) contain sequences classified as Ca. Dadabacteria. Due to the explosion of novel taxa stemming from metagenome-assembled genomes (MAGs), taxonomic assignment has shifted towards whole-genome-based approaches to improve the classification of microorganisms recalcitrant to laboratory culture [5, 6]. In congruence with databases such as the Genome Taxonomy Database (GTDB), the candidate phylum Dadabacteria was united with the phylum Ca. Desulfobacterota (GTDB release 95) and remains without a designated nomenclature type referred to as “Desulfobacterota__D” (GTDB releases 202 and 207) [7]. For the purpose of the comparative genomics study described here, we decided to follow the classification as proposed by GTDB [7]. According to the latest version of GTDB (r207), the candidate phylum Dadabacteria is placed within Ca. Desulfobacterota as a class-level lineage (UBA1144) with three order- (UBA1144, UBA2774 and RKRQ01) and six family-level lineages (UBA1144, UBA2774, CSP1-2, N074bin48, RKRQ01 and GCA-014075295).
Members of Ca. Dadabacteria are, currently, all uncultured bacteria, and their MAGs have been recovered from various habitats (free-living and host-associated), such as hydrothermal vents, terrestrial environments, human-made ecosystems, seawater and marine sponges [8]. Most marine sponges harbour dense and phylogenetically diverse microbial consortia, which span all domains of life and have tight associations with their hosts [9]. The sponge host plus their associated microbiota has been coined with the term ”sponge holobiont” [10, 11]. Several genome-centric studies have shed light on the functional role of the sponge microbiome highlighting significant attributes of their metabolism, including elemental cycling, nutrient supply and host defense [12–20]. However, most of these studies have focused primarily on characterizing highly abundant sponge-associated lineages or the microbe-host interactions of specific sponge species. Therefore, less abundant, yet persistent, members of sponge microbiomes and their ecological functions have been largely overlooked. Members of Ca. Dadabacteria are a perfect example of that. Microbial community profiling of 81 sponge species using 16S rRNA gene amplicon sequencing data showed that Ca. Dadabacteria sequences were detected in 78 of those species, albeit at a low relative abundance (<1%) [9]. Despite their ubiquity, no studies have specifically investigated their role in the sponge microbiome.
To address this gap, we resolved the phylogeny of this lineage following a rank-normalized taxonomy and expanded their genomic representation with newly generated MAGs, recovered from different sponge species. To better understand the ecological importance of this group, the abundance of the MAGs was estimated and a meta-analysis was conducted using 16S rRNA gene sequences to assess their global environmental distribution. Metabolic reconstruction was used to elucidate the functional repertoire and symbiotic features of the sponge-derived MAGs. Comparative genomic analysis was further performed among members of this lineage isolated from sponges and seawater to examine potential mechanisms underlying habitat adaptation. Finally, we employed fluorescence in situ hybridization (FISH) to visualize Ca. Nemesobacterales inhabiting sponges.
Materials and methods
Sample collection and sequencing
Collection, processing and metagenome sequencing of samples of Aplysina aerophoba (Aply), Geodia atlantica (GA), Geodia barretti (GB), Petrosia ficiformis (PF) and other sponge genera (Caminus, Desmacella and Fascaplysinopsis) (DOM) presented here were performed in previous studies from our group [21–24]. Sponge specimens of Aply were collected by SCUBA-diving in Cala Montgó, Spain in June 2014. Samples of GB and GA were collected from Korsfjord, Norway in September and October 2017 by dredging onboard R/V Hans Brattström and PF samples were acquired by SCUBA-diving from a semi-submerged cave in Sfakia, Greece. All the aforementioned samples were treated with liquid nitrogen and stored at −80 °C. Collection of additional specimens of GB was performed by trawling from Davis Strait, Canada in September and October from 2011 to 2015 and stored at −20 °C until further processing. The sampling cruises were conducted by Fisheries and Oceans Canada onboard R/V Pâmiut of the Greenland Institute of Natural Resources. DOM samples were collected from Dominica by a submersible and stored at −20 °C in RNAlater (Thermo Fisher Scientific, Gerhard Menzel B.V. & Co. KG, Germany). One additional DOM sponge sample of the genus Scleritoderma (DOM43) was collected for the present study as described above. Metadata is available in Table S1.
Preprocessing and total DNA extraction for metagenome sequencing of the GA, GB and PF samples were performed in [23] and the Aply samples in [21]. In brief, all sponge samples were pulverized in liquid nitrogen using mortar and pestle. For the bead-beating step, 200 mg of sponge tissue were disrupted by 5 stainless steel beads of 2 mm diameter and 2 beads of 5 mm diameter. Shaking followed (2 × 20 s at 4000 rpm) using a Precellys 24 tissue homogenizer (Bertin GmbH, Germany) according to the protocol of [25]. DNA was then extracted from the tissue lysate using the AllPrep DNA/RNA/Protein Mini Kit (Qiagen, Germany). DNA samples of GA, GB and PF were sent for sequencing at Novogene (Hong Kong) with the HiSeq PE150 system (Illumina). For the Aply samples, sequencing was performed using the HiSeq PE100 system (Illumina). The DOM sponge samples were processed in [24], except for the Scleritoderma sample (DOM43) which was processed here as described in [24]. The tissue was first thawed and rinsed with artificial seawater. It was then added in a PowerBead Tube and shaking followed (3 × 60 s at 6000 × g) using a Precellys 24 Homogenizer (Bertin GmbH, Frankfurt, Germany). DNA extraction was performed with the DNeasy PowerSoil Pro Kit (Qiagen GmbH, Hilden, Germany). Sequencing was conducted by Novogene Europe (Cambridge, UK) using the Novaseq 6000 system (Illumina).
Metagenome assembly, binning and classification
Quality filtering, adaptor removal of raw reads and metagenome assembly of the GA, GB and PF sponge samples were performed in [23], the DOM samples in [24] and DOM43 in the present study. First, quality control of raw reads was performed with FASTQC 0.11.4 [26]. The BBTools suite 37.64 [27] was employed for adaptor removal and quality filtering. Minimum length was set at 50 for all sponge metagenomes, except for the Aply samples that was set at 30 bp. Sequencing artifacts and phiX contamination were removed using BBDuk [27]. Filtered reads were assembled with SPAdes 3.12 [28] with the –meta and –only-assembler modes. For the assembly and binning of the Aply metagenomes, only the filtered HiSeq reads generated in [23] were used.
For all sponges, contigs were binned using the metaWRAP 1.2 [29] binning module at 50% minimum completeness and 10% maximum contamination in accordance with the Minimum Information about a Metagenome-Assembled Genome (MIMAG) standards [30]. Metagenome binning of the PF samples was performed in [23]. Binning of GA and GB contigs was performed in the framework of this study with metaBAT2 2.9.1 [31] and MaxBin2 2.2.4 [32]. The binning of the remaining sponge metagenomes (Aply and DOM) was done with metaBAT2 2.9.1 [31], MaxBin2 2.2.4 [32] and CONCOCT 0.4.0 [33]. The bin refinement module of metaWRAP was later used which compared the original bins to each other and based on a scoring function (S = completion-5*contamintation) [34] consolidated the better bins into an improved bin set. Within this module, any duplicate contigs were removed and bins more than 70% complete and less than 5% contaminated composed the refined set. The refined bin set was further improved by metagenomic read mapping and subjected to individual reassembly with SPAdes 3.10.1 via the metaWRAP reassemble bins module [28]. After comparing the resulting bins (original and reassembled), the bin with the highest score was selected and added to the final bin set.
Taxonomic classification of all MAGs generated for this study was done with the Genome Taxonomy Database-Tool Kit 1.5.0 (GTDB-Tk) [35] classify workflow using default settings (with reference to GTDB R06-RS202). Only MAGs classified as ”Desulfobacterota__D” were used for downstream analysis. To support the genome-based classification, 16S rRNA gene sequences were retrieved from the sponge-associated MAGs with RNAmmer 1.2 [33]. SINA web aligner 1.2.11 [34] was used to align the MAG-derived 16S rRNA gene sequences which were later imported into ARB 6.0.2 [36] containing the SILVA 138 SSU Ref Nr99 database [37]. The alignment was manually refined in ARB and added by parsimony into the SILVA guide tree, allowing the classification of the sequences of interest and the identification of related sequences that were selected for subsequent phylogenetic analysis.
MAG collection and phylogenetic analysis
To ensure the robust representation of the studied lineage, all genomes publicly available at NCBI (February 2021) under the name Candidatus Dadabacteria were included in the initial dataset together with MAGs generated for this study and classified as ”Desulfobacterota__D” (Tables S2 and S3). An additional quality control step was performed with CheckM 1.1.2 [34] and thus, the final dataset consisted of MAGs with >80% estimated completeness and <5% contamination.
Phylogenetic inference was performed based on a set of 120 single-copy marker protein sequences identified in the MAGs and aligned using the GTDB-Tk 1.5.0 [35]. The resulting concatenated alignment was trimmed with trimAl 1.4.1 (-gappyout) [37]. A maximum-likelihood tree was generated from the trimmed alignment (5 030 positions) with IQ-TREE 2.0.6 [38] using extended model selection (-m MFP) [39]. The best-fit substitution model was LG + F + R6 and the final tree was created with 1000 standard nonparametric bootstrap replicates. To investigate the phylogenetic placement of the novel genomes, a 16S rRNA gene-based approach was also employed. MAG-derived and closely related 16S rRNA gene sequences identified as described above were aligned using MAFFT 7.453 with default settings (FFT-NS-2 strategy) [40]. The alignment was trimmed with trimAl 1.4.1 [37] using the ”gappyout” option yielding a final alignment of 1 528 positions. IQ-TREE 2.0.6 [38] was used to infer a maximum-likelihood tree using the ModelFinder Plus option (-m MFP). The chosen model was GTR + F + R3 and the tree was bootstrapped 1000x. Both trees were annotated and visualized in iTOL 6.3 [41] and Inkscape 0.92.3.
In order to assign taxonomic ranks to the newly generated MAGs, we employed a rank normalization approach based on relative evolutionary divergence (RED) [7, 42]. First, a phylogenetic tree was inferred from a concatenated alignment of 120 bacterial single-copy marker genes identified in all GTDB genomes spanning the bacterial domain and the ones generated in this study. This was performed with GTDB-Tk 1.5.0 [35] classify workflow and the tree was built with FastTree 2.1.11 [43] under the WAG + GAMMA model with 100 bootstrap replicates. The inferred tree and the taxonomic information obtained from the classify command were used as input for PhyloRank 0.1.10 (https://github.com/dparks1134/PhyloRank) in order to calculate the RED of the taxa of interest. Type material was selected according to the quality criteria proposed by [44]. Presence of 23S, 5S and tRNAs was detected using Barrnap 0.9 (https://github.com/tseemann/barrnap).
Global distribution
The distribution of representatives of the studied lineage across different environments was estimated as described in [45]. Briefly, the longest 16S rRNA gene sequences acquired from MAGs analyzed in this study were employed for a homology-based search against the 16S rRNA Public Assembled Metagenomes database (01-01-2021) of the Integrated Microbial Genomes and Microbiomes (IMG/M 5.4) system [46], using the IMG BLAST Tool (February 2021). BLAST hits with sequence identity greater than 75% (phylum threshold) [47] and the respective metadata (longitude, latitude, habitat type) were downloaded from IMG for further analysis.
Statistical analysis and data visualization
Data were analyzed and visualized with R 4.0.3 [48] in RStudio 1.4.1106 [49] using the R packages vegan 2.5.7 [50] and ggplot2 3.3.3 [51]. To assess the functional variation between sponge and seawater MAGs, Bray-Curtis dissimilarity matrices were generated based on the frequency of KOs, Pfams and TIGRFAMs with the ”vegdist” function of the vegan R package. Differences in the functional profiles between the two groups were evaluated by Permutational Multivariate Analysis of Variance (PERMANOVA) and Non-metric Multidimensional Scaling (NMDS) using the ”adonis2” and the ”metaMDS” function of the vegan R package, respectively. Global distribution map, ordination plots and heatmaps were generated with ggplot2 3.3.3 [51].
Metabolic reconstruction and comparative genomics
The MAGs were first dereplicated at 95% identity with dRep 2.6.2 [52] and then their relative abundance in the sponge metagenomes was estimated with CoverM 0.6.1 (https://github.com/wwood/CoverM). Dereplicated MAGs were annotated with EnrichM 0.5.0 (https://github.com/geronimp/enrichM) (annotate function) using the following databases: Kyoto Encyclopaedia of Genes and Genomes (KEGG) Orthologies (KOs) for metabolic reconstruction, Pfam and TIGRFAM for detecting proteins of interest. KEGG module completeness was estimated with EnrichM’s classify function, and only modules of completeness greater than 70% were taken for further consideration. Pfams enriched within and between MAGs from different environments (sponges vs. seawater) were identified with EnrichM’s enrichment function. Only significantly enriched Pfams (p < 0.05, after Benjamini–Hochberg correction) were considered for later comparison. Annotation of carbohydrate-active enzymes (CAZymes) was done by HMMER 3.0 [53] against the dbCAN2 database (r9.0) [54]. Secondary metabolite biosynthetic gene clusters were identified in the MAGs with the online tool ”Antibiotics and secondary metabolite analysis shell” (antiSMASH 6.0) [55] applying ”relaxed” detection strictness.
Oligonucleotide probe design
16S rRNA gene sequences recovered from the Geodia MAGs and classified as Ca. Nemesobacterales were aligned in SILVA Incremental Aligner [56] and manually curated in ARB 6.0.6 according to rRNA secondary structure [36]. To determine a monophyletic probe target group, a phylogenetic tree including all sequences in SILVA SSU Ref NR 99 138 [57] classified as Ca. Dadabacteria and 16S rRNA sequences from recovered MAGs was reconstructed with the RAxML 7 [58] maximum-likelihood method (GTR-GAMMA rate distribution model, rapid bootstrap algorithm, 100 repetitions) using a 50% positional conservation filter for all sequences. Oligonucleotide probe design was performed in the probe design tool of ARB 6.0.6 using the SILVA database 138. Optimum formamide concentration for the probe was determined in silico using the formamide curve generator tool implemented in mathFISH [59].
FISH and microscopy
G. barretti sponges were dissected in cubes of ~1 cm3 and fixed in 4% formaldehyde in Dulbecco’s Phosphate Buffer Saline (DPBS) overnight at 4 °C. Washing steps with DPBS, glycine (50 mM) in DPBS and MilliQ H2O followed. All buffers were pre-treated with diethyl pyrocarbonate (DEPC) (0.1%) for RNase inhibition. Sponge samples were dehydrated in a 30% and 50% ethanol series and stored in 50% ethanol at −20 °C. Processing of G. barretti sponge tissue for cryosectioning was done following the methods of [60] . Sponge cubes of G. barretti were washed briefly with sterile MilliQ H2O and embedded in cryomedium (KP-CryoCompound, Immunologic, VWR International B.V., Duiven, The Netherlands) overnight at 4 °C for infiltration. They were then embedded in fresh cryomedium in base moulds to solidify overnight at −80 °C. Longitudinal sections of 5–8 μm were made using a cryostat (Leica CM 3050 S, Leica Biosystems GmbH, Nussloch, Germany) with both chamber and object temperature set at −35 °C. Tissue sections were mounted on polylysine coated glass slides (Thermo Fisher Scientific) and stored at −20 °C.
After sample fixation, FISH was performed as described previously [61] with minor modifications. Briefly, the slides were dehydrated in decreasing ethanol series of 95, 80 and 70% (v/v) for 10 min each. They were then washed in 200 mM HCl for 10 min and 20 mM Tris-HCl for 10 min. For permeabilization, the sections were incubated in 1 µg/ml proteinase K (Sigma–Aldrich) solution for 5 min at 37 °C and washed into 20 mM Tris-HCl for 10 min. Hybridization was done using a 4-times-Atto594-labelled DADA393 probe (5′-TCA TCC CTC ACG CGA CAT CGC-3′ T) (10 pmol/µl, Biomers) together with unlabelled helper and competitor probes (10 pmol/µl, Biomers) at 46 °C for 3 h. After washing at 48 °C for 10 min, the slides were incubated in 1× phosphate-buffered saline (PBS) for 15 min and shortly dipped into MilliQ water. Sponge tissues were then stained with 4′,6-diamidino-2-phenylindole (DAPI, 1 µg/ml) (Thermo Fisher) and mounted in Citifluor (Electron Microscopy Sciences): Vecta Shield (Vector Laboratories) (4:1 v:v) medium. Four-times-Atto488-labelled EUB338 and NON338 probes were used as positive and negative controls, respectively.
Imaging was performed in a Nikon Eclipse Ti2-E inverted microscope equipped with Nikon DS-Qi2 camera and SOLA white light engine. DAPI and FISH signals were illuminated using different fluorescence filter cubes with corresponding excitation and emission spectra. Images were taken with two objectives: a plan-Apochromat 100 ×1.45 oil immersion and a plan-Fluor 10 × 0.3. Maximum intensity projection of multiple images acquired in z-stack were constructed in NIS-Element AR software 5.21.03.
Results
MAG collection and phylogenomic placement
In total, 29 MAGs obtained from selected sponge metagenomes (Table S1) were classified by the GTDB-Tk as ”Desulfobacterota__D”. Accordingly, insertion of the 16S rRNA gene sequences obtained from the respective MAGs by parsimony into the SILVA guide tree (r138) placed them all within Ca. Dadabacteria [57, 62]. Twenty-eight MAGs passed the quality control with average estimated completeness at 92.2 ± 4.5% and contamination at 1.2 ± 0.6% and were used for subsequent analysis (Table S3). Their predicted genome sizes varied from 1.2 to 1.9 Mbp (average 1.5 ± 0.2 Mbp) with an average GC content of 49.9 ± 2.5%. An additional 92 MAGs were obtained from NCBI and other studies (Table S2). After quality screening, we excluded 47 genomes due to either low completeness (<80%) and/or high contamination (>5%). The final dataset included 73 high-quality MAGs according to the MIMAG standards [30] (average completeness 91.0 ± 4.9% and contamination 1.5 ± 1.0%) representing the ”Desulfobacterota__D” (Table S3).
To reconstruct their phylogeny, we first employed a phylogenomic approach using publicly available genomes under the name “Candidatus Dadabacteria” or ”Desulfobacterota__D” in addition to MAGs generated for this study (Table S3). Inference of a maximum-likelihood tree on the basis of a concatenated alignment of 120 single-copy bacterial marker proteins supported the GTDB taxonomy of the class UBA1144 into three order-level lineages (UBA1144, UBA2774 and RKRQ01) with high bootstrap values (>98%). MAGs mainly derived from seawater (only one from a hydrothermal vent) were assigned to the GTDB order UBA1144, while MAGs from diverse environmental sources, except for seawater and marine sponges comprised the GTDB order UBA2774 (Fig. 1). All 49 sponge-associated MAGs (including 28 generated for this study) were placed within the GTDB order RKRQ01 and further clustered into three well supported (bootstrap values > 81%) subclades I, II and III (Fig. 1), revealing an order-level lineage exclusively comprised of sponge-derived sequences. The 16S rRNA gene tree reflected the phylogenetic placement of the genome tree, clustering the 16S rRNA gene sequences retrieved from MAGs into three clades within the GTDB order RKRQ01 represented only by MAGs recovered from marine sponges (Fig. S1).
Fig. 1. Phylogenomic tree of Ca. Dadabacteria (GTDB-Desulfobacterota__D) based on the concatenated alignment of 120 bacterial single-copy marker protein sequences (5 037 positions).

Outer coloured arcs indicate the environmental source of the MAGs. Different colours on the clades represent assigned taxonomic orders according to GTDB taxonomy. Dark red numbers show subclades I (Ca. Spongomicrobiaceae), II (Ca. Mycalebacteriaceae) and III (Ca. Nemesobacteraceae). Names in bold represent MAGs generated for this study. Bootstrap values over 80% are shown by grey circles. The collapsed clade indicates the outgroup and includes MAGs representing members of the Thermodesulfobacteria class of the Ca. Desulfobacterota phylum. The scale bar represents 0.1 substitutions per site. Abbreviations of the sponge-derived MAGs are as follows: Aply Aplysina aerophoba, DOM sponges collected from Dominica, GA Geodia atlantica, GB Geodia barretti, PF Petrosia ficiformis.
Taxonomic rank assignment and proposal of type material
Based on the inferred phylogeny and RED values, the RKRQ01 was best described as an order-level group (median RED = 0.636, median RED for orders = 0.634) falling within the ±0.1 RED interval for all taxa belonging at that rank [63] (Table S4). A putative family (subclade I) was identified that fell within the GTDB order RKRQ01 represented by a single MAG (DOM43_bin18). After bin dereplication at 95% average nucleotide identity (ANI), three novel MAGs were added to GTDB family GCA-014075295, which is comprised of a single genus (subclade II) and were resolved into two novel species represented by the MAGs SCOP_bin1 and CLI1_bin1 (Fig. 1). In addition, 34 MAGs were placed into GTDB family RKRQ01 (subclade III) representing 14 novel species-level taxa of the GTDB genus RKRQ01 (Table S5). Following the recommendations for defining species and higher ranks of yet-uncultured bacteria using MAGs as type material [5, 6, 44, 64], we propose the following names: Ca. Spongomicrobium dominicola sp. nov. (DOM43_bin18), Ca. Mycalebacterium zealandia sp. nov. (MH-Pat-all_metabat2_32), and Ca. Nemesobacter australis sp. nov. (SB0662_bin19) as type species representing subclades I, II and III, respectively. Furthermore, we propose the families Ca. Spongomicrobiaceae fam. nov. (subclade I), Ca. Mycalebacteriaceae fam. nov. (subclade II), Ca. Nemesobacteraceae fam. nov. (subclade III) and the order Ca. Nemesobacterales ord. nov. (Fig. 1). More details on the proposed taxonomic outline and etymological description can be found in the Supplementary Information (Table S5).
MAG abundance and global distribution
Read mapping of 26 sponge metagenomes generated here against the Ca. Nemesobacterales genomes showed that the dereplicated set (19 MAGs) represented a small fraction of the metagenomes, accounting for a relative abundance of 0.22% (±0.07%) on average and ranging from 0.002 to 1.29% of the metagenomic reads (including host DNA) (Table S6). The environmental distribution of Ca. Nemesobacterales was investigated by screening all available metagenomes from the IMG/M database for the presence of homologous 16S rRNA genes. This search showed 283 unique occurrences according to the sampling location of the metagenomic samples. Members of this lineage were distributed across a wide geographical range and were observed in 29 different types of environments, including aquatic, terrestrial, hydrothermal, host-associated and engineered systems (Fig. 2). The majority of the homologous sequences belonged to metagenomes from marine sponges (23.2%), followed by seawater (13.7%) and marine sediments (5.7%).
Fig. 2. Schematic map representing the global distribution of Ca. Nemesobacterales.

The dataset used corresponds to metagenome-derived 16S rRNA gene sequences homologous to Ca. Nemesobacterales. Different shapes represent the geographic location of the respective metagenomic samples. Members of this lineage are found in diverse environments worldwide, highlighted by different shape colours.
Niche adaptation and functional traits
Comparative genomic analysis was conducted to delineate the functional repertoire of Ca. Nemesobacterales using both in-house and publicly available MAGs. Core pathways for carbon metabolism such as glycolysis, tricarboxylic acid cycle (TCA), pentose phosphate pathway (non-oxidative phase) and phosphoribosyl diphosphate (PRPP) biosynthesis pathway were mostly complete, whereas no autotrophic carbon fixation pathways were detected (Table S7). Most of the aerobic respiration complexes were found (NADH:quinone oxidoreductase, cytochrome c oxidase and F-type ATPase) and hence, oxygen is likely to be the terminal electron acceptor. No genes for anaerobic respiration (e.g., respiration with nitrate, nitric oxide, sulfate) were found in the genomes. The majority of MAGs included in our study contained nearly all the essential genes for the biosynthesis of 13 amino acids, 6 cofactors and 4 vitamins, such as proline, lysine, biotin, riboflavin, coenzyme A and heme (Fig. 3, Fig. S2 and Table S7).
Fig. 3. Schematic overview of the inferred metabolism of Ca. Nemesobacterales.

Pathways affiliated with nitrogen and carbohydrate metabolism are highlighted in different colours. Dashed arrows indicate pathways and transporters for which not all responsible genes were annotated. Abbreviations: AA amino acid, CoA coenzyme A, FA fatty acid, GS-GOGAT glutamine synthase (GS)-glutamine oxoglutarate aminotransferase (GOGAT), PEP phosphoenolpyruvate, PPP pentose phosphate pathway. (Created with https://biorender.com/).
Their putative capacity to degrade carbohydrates was assessed by screening the MAGs for carbohydrate-active enzymes (CAZymes) belonging to the following families: glycoside hydrolases (GHs), carbohydrate esterases (CEs), polysaccharide lyases (PLs), carbohydrate-binding modules (CBMs) and auxiliary activities (AAs). On average, Ca. Nemesobacterales possessed 15.8 ± 2.9 CAZymes/Mbp. Overall, 40 different CAZY families were identified. The most frequently observed CAZymes (average CAZymes/Mbp > 1.0) were related to enzymes cleaving either chitin or peptidoglycans (GH23 and CBM50) and glycosyltransferases involved in the biosynthesis of different molecules, such as cellulose and chitin (GT2 and GT4) (Table S8).
To obtain a better overview of the functions of the host-associated bacteria, a set of 19 highly complete MAGs derived from sponges (Ca. Nemesobacterales) was compared to 16 seawater-derived MAGs (order UBA1144). Sponge and seawater MAGs shared 72.7% of the annotated KEGG modules related to carbohydrate metabolism (i.e., glycolysis, citrate cycle, pentose phosphate pathway) (Fig. S3). Accordingly, MAGs derived from both habitats seemed to have the potential of synthesizing a similar range of amino acids (93.3% shared KEGG modules), cofactors and vitamins (83.3% shared KEGG modules) (Fig. S3). Distinct for some sponge-derived MAGs (16% of total) was the ability to produce cobalamin (vitamin B12) that was absent from the seawater MAGs. Despite their highly similar carbohydrate, amino acid, cofactors and vitamins metabolism, only 42.9% of KEGG modules related to environmental information processing (i.e., membrane transport, signal transduction) were shared between sponge and seawater MAGs (Fig. S3). Annotation of ATP-binding cassette (ABC) transporters showed that only sponge MAGs encoded genes for the transport of L-amino acids, lipoproteins and oligopeptides (Fig. S2). They shared the same phosphate starvation and nitrogen regulation system, possibly used under nutrient-limiting conditions. For the case of iron, the host-associated MAGs carried different transport systems, as well as the potential of using the glyoxylate shunt, recently proposed as an acclimation strategy to iron limitation [65] (Fig. S2). CAZyme analysis showed that MAGs from the two different habitats harboured almost the same average number of CAZymes/Mbp (16.2 ± 2.3). Similarly, the most frequent CAZY families (average CAZymes/Mbp > 1) in the seawater MAGs were the same as the ones in the sponge MAGs (Fig. 4A). However, the CAZymes repertoire was less diverse for the seawater MAGs. Almost 60% of the total identified CAZY families (28 out of 47) were detected only in MAGs from sponges whereas only 6 CAZY families were exclusively found in seawater MAGs (Table S8).
Fig. 4. Functional comparison of Ca. Dadabacteria (GTDB-Desulfobacterota__D) MAGs associated with marine sponges and seawater.

A Heatmap illustrating the abundance of carbohydrate-active enzymes (CAZymes) families in sponge- and seawater-associated MAGs. Abbreviations: PLs polysaccharide lyases, GTs glycosyltransferases, GHs glycoside hydrolases, CEs carbohydrate esterases, CBMs carbohydrate-binding modules, AAs auxiliary activities. B Non-metric multidimensional scaling plot (NMDS) based on Bray–Curtis dissimilarity scores calculated from annotated Pfams. C Mean number of Pfams related to symbiosis factors and defense mechanisms identified in MAGs from each habitat. Functional variability is displayed between sponge and seawater MAGs (different shapes) and different taxonomic groups (different colours).
Functional profiles based on annotated Pfams, KEGG orthologs (KOs) and TIGRFAMs were used in order to assess correlation between taxonomy (different clades) and niche (sponge vs. seawater) with the predicted traits (Fig. S4). MAGs belonging to the different order-level lineages varied significantly at the functional level based on a relative abundance analysis of all three annotations (PERMANOVA, p < 0.001; Fig. 4B and S4). Similarly, highly divergent functional profiles (PERMANOVA, p < 0.001; Fig. 4B and S4) were observed between bacterial genomes from sponges and seawater. Enrichment analysis showed that sponge-derived MAGs were enriched in Pfams associated with defense mechanisms and symbiosis-related factors (Fig. 4C and Table S9).
To further investigate the distribution of these functions in the MAGs, screening was done for 230 Pfams encoding restriction-modification systems (RMs), clustered regularly interspaced short palindromic repeat systems (CRISPRs), secretion systems (SSs), toxin-antitoxin systems (TAs), transposases, eukaryotic-like proteins (ELPs) and others (Table S9). In total, 166 Pfams of the aforementioned types were identified either in sponge or seawater MAGs from which 94% were significantly enriched in the Ca. Nemesobacterales. Only 26 Pfams were shared between MAGs from both habitats, while 137 Pfams were specific to the sponge symbionts (Table S9). Most Pfams enriched in Ca. Nemesobacterales were related to RMs (45 Pfams), ELPs (42 Pfams), TAs (32 Pfams) and SSs (12 Pfams). More specifically, Pfams associated with methyltransferases, helicases, the putative ABI (abortive infection) toxin, type III restriction enzymes, Fic/DOC (filamentation induced by cAMP/death on curing) proteins and tetratricopeptide repeats were among the most abundant ones in Ca. Nemesobacterales (average abundance > 5.0). Certain Pfams, such as type I restriction enzyme, leucine rich and tetratricopeptide repeats were overrepresented, even more than tenfold in sponges compared to seawater (Table S9).
The potential for production of secondary metabolites was also investigated by screening the investigated MAGs for secondary metabolite biosynthetic gene clusters (BGCs). In general, four different BGC types were identified in the genomes from both habitats coding for type I polyketide synthases (T1PKS), type III polyketide synthases (T3PKS) and for enzymes involved in the production of beta-lactones and terpenes (Table S10). Genome mining showed that both BGC abundance and diversity were higher for the sponge-associated MAGs, which harboured on average almost twice as many BGCs (1.11 ± 0.19 BGCs/MAG) compared to the free-living MAGs (0.63 ± 0.15 BGCs/MAG). The distribution of these BGC types was uneven as terpene BGCs were encoded exclusively by seawater MAGs while the others were mainly found in the sponge-derived MAGs (Fig. S5). BGCs coding for beta-lactones and T1PKS showed the highest abundance in Ca. Nemesobacterales MAGs (Table S10).
Visualization of Ca. Nemesobacterales
We investigated the localization and morphology of symbiotic bacteria in the sponge G. barretti using fluorescence in situ hybridization (FISH). Labelling with the general bacterial probe EUB338 revealed a widespread occurrence and even distribution of bacteria throughout the mesohyl of G. barretti (Fig. S6). Novel oligonucleotide probes were designed (Fig. S7) to specifically label Ca. Nemesobacterales spp. in the sponge tissue, which allowed their first-time visualization in environmental samples. Ca. Nemesobacterales spp. cells featured a rod-shaped with a length of 1.04 ± 0.21 μm, and were located in the sponge mesohyl (Fig. 5A). The cells often surrounded sponge nuclei resembling bacteriocyte formation (Fig. 5B, C), indicating a close association with host cells.
Fig. 5. Imaging of Ca. Nemesobacterales spp. in G. barretti.
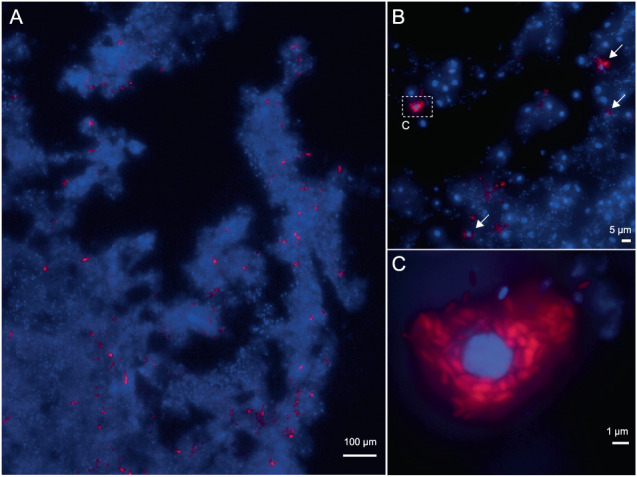
DNA is stained with DAPI (blue) and Ca. Nemesobacterales spp. cells are visualized with the 4-times-Alexa594 labelled oligonucleotide probe DADA392 (red). Maximum intensity projection of z-stack fluorescence images that were acquired in A ×150, B ×1000, and C ×1500 magnification are shown. B Some Ca. Nemesobacterales spp. cells cluster around a sponge nucleus (arrows). The signals shown in the dashed rectangle are magnified in (C). Representative images of 47 images taken in three individual experiments using two different sponge specimens (Gb5 and Gb10) are depicted.
Discussion
Members of the ”Desulfobacterota__D” (or ”Dadabacteria”) lineage are lacking cultured representatives and their phylogeny and metabolic traits have remained elusive despite the fact that they are widely found. Since the first published MAG in 2014 [2], several MAGs of this group have been recovered from a plethora of environmental samples, both free-living and host-associated [8], in particular from marine sponges [14, 18, 19]. However, to date no information exists on the precise phylogenetic placement and functional characteristics of the sponge-dwelling representatives. To this end, our study incorporated a large number of ”Desulfobacterota__D” MAGs, including published and newly obtained ones which were assigned to the three existing GTDB orders of (UBA1144, UBA2774 and RKRQ01). Previous phylogenetic analyses of the ”Dadabacteria” diversity revealed three main clades based on their environmental origin representing marine pelagic, hydrothermal and organic carbon-associated systems. The latter clade included only two sponge-derived MAGs [8]. Here, the addition of 49 MAGs affiliated with marine sponges altered the previously described architecture of the lineage revealing a sponge-specific clade (GTDB order RKRQ01). The remaining MAGs originated mainly from seawater (GTDB order UBA1144) and other marine and non-marine environments (GTDB order UBA2774). Our phylogenomic investigation identified one novel lineage within the sponge-specific order, which based on RED values corresponds to a here-proposed family, Ca. Spongomicrobiaceae. It should be mentioned that Ca. Spongomicrobiaceae is comprised of only one MAG (DOM43_bin18) obtained from a Scleritoderma species. In addition, 17 dereplicated MAGs were affiliated with the other two families, denoted here as Ca. Mycalebacteriaceae and Ca. Nemesobacteraceae. Thus, our findings considerably expand the ”Desulfobacterota__D” species tree and highlight the current underrepresentation of this lineage.
The relative abundance of Ca. Nemesobacterales in metagenomes of different sponge species based on read recruitment analysis, ranged from 0.002 to 1.3% with an average of 0.22%. These findings are in line with the relative abundance of ”SBR1093” OTUs (former name of the Ca. Dadabacteria phylum) in the global sponge microbiome, based on amplicon sequencing data [9]. It should be noted that in our case the total reads included host-derived reads and thus, the relative abundance of Ca. Nemesobacterales in the sponge microbiome is underestimated. Based on the above numbers and compared to genomic studies on other groups of sponge-associated bacteria [16, 17, 20, 66], Ca. Nemesobacterales are widespread but low in abundance (relative) members of the sponge microbiome. Specifically, Ca. Nemesobacterales were recovered here from 26 sponge metagenomes belonging to nine different sponge species collected from different geographical locations while in many cases, the same Ca. Nemesobacterales MAG was present in more than one host species (Table S6). This reflects a wide distribution in taxonomically diverse sponge species which might suggest a generalistic role within the sponge microbiome [9]. Besides marine sponges, metagenomes from various environments around the world contained homologous sequences of Ca. Nemesobacterales, including hydrothermal systems, sediment, seawater, soil and wetlands, further highlighting their broad occurrence at a global scale (Fig. 2). Moreover, its members were also detected in tunicates and marine worms, yet the majority of the homologous sequences were sponge-associated indicating their close association and importance of Ca. Nemesobacterales within marine sponges.
The sponge holobiont has been studied substantially and sponge-microbe interactions are considered a prime example of symbiosis in the animal realm [10, 11, 67–71]. Yet, many bacteria remain undescribed, and their contribution to host success has not been determined. A wide range of core functions which shape the sponge microbiome have been previously reported concerning nutrient and vitamin metabolism as well as defense features that protect microbial populations from viruses, pathogens and/or the host allowing sustained colonization of the host by the symbionts [10, 69]. Here, we provide insights into the functional potential of the sponge-dwelling Ca. Nemesobacterales using genome-centric metagenomics. The metabolism reconstruction showed that carbon acquisition is likely performed via heterotrophy as all major pathways for carbon metabolism were present, which is consistent with previous analyses of the marine pelagic clade [8]. Even though carbon fixation has been reported before [2], no evidence for autotrophy was found in Ca. Nemesobacterales. An almost complete electron transport chain for aerobic respiration reflects an aerobic, heterotrophic lifestyle (Fig. 3). In contrast to other members of this lineage [8], Ca. Nemesobacterales lack complete pathways for inorganic nitrogen and sulfur metabolism. Therefore, the presence of ABC transporters for L-amino acids, oligopeptides and lipoproteins might indicate a preference for organic nitrogen and sulfur compounds derived from the host or other members of the microbial community. Ca. Nemesobacterales also showed the potential to degrade and transform complex carbohydrates. For example, certain glycosylases (GH23) and other related proteins (CBM50) which act on bacterial peptidoglycans or animal chitin were widely present in Ca. Nemesobacterales. These enzymes have been previously reported as highly abundant in other sponge symbionts belonging to Chloroflexota, Bacteroidetota and Candidatus Poribacteria [66, 72–74] and are thought to play a role in cell adhesion, aggregation and allorecognition [72].
To investigate any adaptations related to host association, we compared the genomic features of the sponge-derived MAGs to MAGs isolated from seawater samples. Ca. Nemesobacterales shared equivalent pathways for carbohydrate and energy metabolism as well as amino acid and cofactor biosynthesis with their seawater counterparts. This supports recent findings on the possible nutrient provisioning of symbionts to the host without being key processes in the holobiont since sponges could also obtain these nutrients through feeding on seawater microbes [19]. In contrast, only sponge-associated MAGs had the ability of synthesizing cobalamin (vitamin B12) which highlights a potential interplay between microbes and host that might concern specific molecules that sponges are not able to synthesize, such as vitamins and certain amino acids [18, 66, 69, 72, 75, 76]. MAGs from both habitats showed a similar CAZyme repertoire in terms of abundance. However, Ca. Nemesobacterales were predicted to be capable of degrading a greater diversity of complex carbohydrates than their free-living counterparts, probably due to exposure to a larger variety of carbohydrates sourced from the host, other symbionts and/or the incoming seawater [69].
Further exploration of their functional profiles provided additional support that Ca. Nemesobacterales are metabolically divergent from seawater-derived members of the same class (Fig. 4B) [77, 78]. In fact, genomic signatures significantly enriched among members of Ca. Nemesobacterales were related to host-symbiont interactions and prokaryotic defense. Sponge symbionts have long been demonstrated to own a pronounced molecular toolbox of mechanisms in order to maintain a stable relationship with the host by evading phagocytosis, viral infections and coping with temporal variations in the sponge niche [14, 78]. It has been proposed that sponge-associated bacteria either evolved these mechanisms in the longstanding process of adaptation to the host environment or acquired them via horizontal gene transfer from the host or other symbionts [77–79]. Here, the majority of enriched Pfams were specific to Ca. Nemesobacterales and were affiliated with restriction-modification systems (RMs), toxin-antitoxin systems (TAs) and eukaryotic-like proteins (ELPs) often prevalent in sponge-associated bacteria [12, 14, 16–20, 66, 69, 77, 78, 80]. RMs are mechanisms that contribute to the bacterial defense by recognizing and targeting foreign DNA while TAs cause cell dormancy or apoptosis in response to environmental stressors and eventually protect the cell population [81, 82]. For example, a domain belonging to an ATPase associated with the ABI toxin (PF13304), which inhibits viral replication by programming cell death, was the most abundant TA in Ca. Nemesobacterales [75]. Furthermore, ELPs are proteins containing domains of eukaryotic origin in prokaryotes that have been previously proposed to modulate host behaviour by mediating protein-protein interactions and thus, facilitate the establishment of a successful symbiosis [79]. A large number of sponge-associated MAGs carry genes encoding ELPs [18–20] that likely aid them in escaping digestion by sponge cells [79, 83]. Similarly, in this study we show that Ca. Nemesobacterales inhabiting phylogenetically diverse sponge species had several types of ELPs in their genomes with TPRs showing the highest abundance [15, 69, 84]. TPR-containing proteins are thought to participate in various cellular processes including virulence of bacterial pathogens [85]. However, the mechanism of interaction between bacterial ELPs and host cells remains unknown. Evidence on co-expression of ELPs with transport systems suggest that they are delivered into the extracellular environment via transporters or secretion systems [15]. Recent genomic findings indicate that only a few lineages in the sponge microbiome (e.g., Gammaproteobacteria, Acidobacteriota, Gemmatimonadota) encode genes related to secretion systems [18, 19]. Ca. Nemesobacterales might be one of these taxa that interact with the sponge host via secretion systems since they were enriched in several Pfams affiliated with type II, III and IV secretion systems.
Another line of prokaryotic defense is the biosynthesis of secondary metabolites. Sponge holobionts are well known chemical factories and their microbial symbionts are thought to contribute to the production of bioactive molecules in the face of predation, competition and microbial infections [10, 67, 86]. Several representatives of the sponge microbial community have exhibited in vitro bioactivity and possess a wide range of secondary metabolites [13, 66, 74, 87–91]. Ca. Nemesobacterales MAGs carried a greater diversity and abundance of BGCs compared to the seawater-derived MAGs of the same lineage confirming the prominent secondary metabolite biosynthesis potential of sponge-dwelling bacteria. Beta-lactone and T1PKS BGCs were the most predominant in the genomic repertoire of Ca. Nemesobacterales. Compounds resulting from these classes of BGCs have shown different properties such as antimicrobial and anticancer activity [92, 93]. Even though their exact ecological function remains elusive, it seems that Ca. Nemesobacterales are participating in the chemical defense or communication within the sponge holobiont.
Metabolic divergence was also evident from the analysis of the genomic features which showed that Ca. Nemesobacterales MAGs had relatively larger genomes (1.5 ± 0.2 Mbp) and higher GC content (50.4 ± 2.6%) compared to seawater members of the same phylum (1.0 ± 0.5 Mbp and 30.7 ± 0.8%, respectively) (Table S3). This is in accordance with a previous study that reported genome streamlining for free-living, seawater “Desulfobacterota__D” [8]. Genome reduction is a common phenomenon for marine bacteria and is typical with two types of lifestyles, free-living oligotrophy and association with a host [94]. In general, Ca. Nemesobacterales had small genomes of moderately low average GC content which might explain a dependency on the host. However, they were enriched with defence- and symbiosis-related mechanisms (e.g., ELPs, TA systems, BGCs) likely reflecting the difference in genome architecture compared to seawater “Desulfobacterota__D”.
Localization of the Ca. Nemesobacterales spp. in the tissue of G. barretti revealed that their cells were scattered in the sponge mesohyl and often clustered in vacuole-like structures surrounding sponge nuclei. These structures resemble bacteriocytes which are specialized host cells that accommodate intracellular bacteria that have previously been reported in other marine sponge species, such as P. ficiformis, Crambe crambe and Tethya stolonifera [20, 95, 96]. Bacteriocyte association in eukaryotes has been presumed to be an event of mutualistic infection between bacterial symbionts and eukaryotic hosts with a yet-unknown physiological role for either of the partners [97]. In sponges, bacteriocytes might enclose one or several bacterial morphotypes and it has been hypothesized as a mechanism for the host to control microbial populations or facilitate certain metabolic exchanges between the symbionts and the host [95]. In certain cases, symbionts require an enclosed environment to survive. Based on the observed localization patterns, members of Ca. Nemesobacterales seem to have the ability to reside both intracellularly and extracellularly. Accordingly, Usher et al. [98] reported cyanobacterial symbionts in Chondrilla australiensis being occasionally intracellular. Besides conventional bacteriocytes, microbes can also be found enclosed in atypical bacteriocytes which are not true endocytoplasmic vesicles but cell “pockets” formed by epithelial cells [95, 99, 100]. For Ca. Nemesobacterales, additional evidence is needed to prove the formation and nature of bacteriocytes, such as imaging using transmission electron microscopy. Nevertheless, a lifestyle presumably associated with bacteriocytes further supports the aforementioned metabolic predictions that Ca. Nemesobacterales exist in close symbiosis with marine sponges.
The current study provides unprecedented insights into the phylogeny and metabolism of Ca. Nemesobacterales, a sponge-specific order of the newly classified Desulfobacterota phylum. We propose the division of the order Ca. Nemesobacterales into three families, namely Ca. Nemesobacteraceae, Ca. Spongomicrobiaceae (newly described) and Ca. Mycalebacteriaceae. Despite their low abundance, Ca. Nemesobacterales display a wide distribution in different sponge species. Our analysis showed that they can also be found worldwide in diverse environments, albeit with the majority occurring in marine sponges. Metabolism reconstruction revealed that Ca. Nemesobacterales appear to be aerobic, heterotrophic microorganisms with the potential of degrading various complex carbohydrates. Our results highlight that Ca. Nemesobacterales are functionally divergent from seawater-associated members of the same lineage. This is mainly attributed to the overrepresentation of several genomic signatures related to nutritional provisioning, host-microbe interactions, phage defense and biosynthesis of bioactive molecules, which clearly reflects a host-associated lifestyle. We infer that Ca. Nemesobacterales are in a close relationship with sponges indicated by their presence inside cells of the sponge G. barretti in combination with their quite distinct gene repertoire and niche-specific adaptations mentioned above. Future investigations involving additional genomes, metatranscriptomic analysis and imaging data of other Ca. Nemesobacterales in sponges could potentially enhance our understanding of the ecological function of Ca. Nemesobacterales in the sponge holobiont.
Supplementary information
Acknowledgements
The authors would like to thank the late Hans Tore Rapp for his invaluable help in collecting G. barretti samples from Norway, Ellen Kenchington for the Canadian G. barretti samples, Vasilis Gerovasileiou for performing the sampling of P. ficiformis and Andriaan Schrier for supporting the sponge collection in Dominica. Henk Schipper is acknowledged for helping with the sponge tissue processing. The authors would like to thank Catarina Loureiro for performing the preprocessing of the reads, the metagenome assembly and binning of the A. aerophoba samples and for her feedback regarding the manuscript. We thank Paco Cárdenas and Karin Steffen for the identification of G. atlantica. We also thank Torsten Thomas for providing us with additional data for our analysis. Maria Chuvochina, Donovan Parks and Philip Hugenholtz are acknowledged for their advice on taxonomy and rank assignment. This research was financially supported by the European Commission through the SponGES project (Grant agreement ID: 679849) to DS and AsG, a Marie Skłodowska-Curie Individual Fellowship COSMos (Grant agreement ID: 897121) to MAS, and by grants from European Research Council (ERC consolidator grant 817834), the Dutch Research Council (NWO-VICI grant VI.C.192.016), and the Moore–Simons Project on the Origin of the Eukaryotic Cell (Simons Foundation 735925LPI) to TJGE.
Author contributions
AsG, CJI, HS and DS conceived the study. AsG, AnG and DS collected the sponge samples. AsG, AnG and MAS processed the samples for sequencing. AnG and MAS did the quality filtering of raw reads, metagenome assemblies, binning and taxonomic classification (Aply, DOM and PF samples). AsG performed the binning and taxonomic classification of the GA and GB samples. AsG performed the MAG collection, phylogenetic analysis, metabolic reconstruction, comparative genomic analysis, statistical analysis and visualization. BA performed the oligonucleotide probe design, FISH and microscopy. All authors contributed to the interpretation of the results. AsG wrote the first draft and all authors edited and approved the manuscript.
Data availability
Raw reads, metagenome assemblies and MAGs generated for this study can be found under the European Nucleotide Archive (ENA) project accession numbers PRJEB54590, PRJEB51534 and PRJEB51535. All accession numbers for the data included in this study are included in the supplementary information of this article.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Asimenia Gavriilidou, Email: asimenia.gavriilidou@wur.nl.
Detmer Sipkema, Email: detmer.sipkema@wur.nl.
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-023-01484-z.
References
- 1.Bond PL, Hugenholtz P, Keller J, Blackall LL. Bacterial community structures of phosphate-removing and non-phosphate-removing activated sludges from sequencing batch reactors. Appl Environ Microbiol. 1995;61:1910–6. doi: 10.1128/aem.61.5.1910-1916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Z, Guo F, Liu L, Zhang T. Evidence of carbon fixation pathway in a bacterium from candidate phylum SBR1093 revealed with genomic analysis. PLoS ONE. 2014;9:e109571. doi: 10.1371/journal.pone.0109571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hug LA, Thomas BC, Sharon I, Brown CT, Sharma R, Hettich RL, et al. Critical biogeochemical functions in the subsurface are associated with bacteria from new phyla and little studied lineages. Environ Microbiol. 2016;18:159–73. doi: 10.1111/1462-2920.12930. [DOI] [PubMed] [Google Scholar]
- 4.Waite DW, Chuvochina M, Pelikan C, Parks DH, Yilmaz P, Wagner M, et al. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int J Syst Evol Microbiol. 2020;70:5972–6016. doi: 10.1099/ijsem.0.004213. [DOI] [PubMed] [Google Scholar]
- 5.Konstantinidis KT, Rossello-Mora R, Amann R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017;11:2399–406. doi: 10.1038/ismej.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hugenholtz P, Chuvochina M, Oren A, Parks DH, Soo RM. Prokaryotic taxonomy and nomenclature in the age of big sequence data. ISME J. 2021;15:1879–92. doi: 10.1038/s41396-021-00941-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38:1079–86. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 8.Graham ED, Tully BJ. Marine Dadabacteria exhibit genome streamlining and phototrophy-driven niche partitioning. ISME J. 2021;15:1248–56. doi: 10.1038/s41396-020-00834-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas T, Moitinho-Silva L, Lurgi M, Bjork JR, Easson C, Astudillo-Garcia C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pita L, Rix L, Slaby BM, Franke A, Hentschel U. The sponge holobiont in a changing ocean: from microbes to ecosystems. Microbiome. 2018;6:46. doi: 10.1186/s40168-018-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Webster NS, Taylor MW. Marine sponges and their microbial symbionts: love and other relationships. Environ Microbiol. 2012;14:335–46. doi: 10.1111/j.1462-2920.2011.02460.x. [DOI] [PubMed] [Google Scholar]
- 12.Podell S, Blanton JM, Neu A, Agarwal V, Biggs JS, Moore BS, et al. Pangenomic comparison of globally distributed Poribacteria associated with sponge hosts and marine particles. ISME J. 2019;13:468–81. doi: 10.1038/s41396-018-0292-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lackner G, Peters EE, Helfrich EJ, Piel J. Insights into the lifestyle of uncultured bacterial natural product factories associated with marine sponges. Proc Natl Acad Sci USA. 2017;114:E347–56. doi: 10.1073/pnas.1616234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slaby BM, Hackl T, Horn H, Bayer K, Hentschel U. Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 2017;11:2465–78. doi: 10.1038/ismej.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diez-Vives C, Moitinho-Silva L, Nielsen S, Reynolds D, Thomas T. Expression of eukaryotic-like protein in the microbiome of sponges. Mol Ecol. 2017;26:1432–51. doi: 10.1111/mec.14003. [DOI] [PubMed] [Google Scholar]
- 16.Astudillo-Garcia C, Slaby BM, Waite DW, Bayer K, Hentschel U, Taylor MW. Phylogeny and genomics of SAUL, an enigmatic bacterial lineage frequently associated with marine sponges. Environ Microbiol. 2018;20:561–76. doi: 10.1111/1462-2920.13965. [DOI] [PubMed] [Google Scholar]
- 17.Sizikov S, Burgsdorf I, Handley KM, Lahyani M, Haber M, Steindler L. Characterization of sponge-associated Verrucomicrobia: microcompartment-based sugar utilization and enhanced toxin-antitoxin modules as features of host-associated Opitutales. Environ Microbiol. 2020;22:4669–88. doi: 10.1111/1462-2920.15210. [DOI] [PubMed] [Google Scholar]
- 18.Engelberts JP, Robbins SJ, de Goeij JM, Aranda M, Bell SC, Webster NS. Characterization of a sponge microbiome using an integrative genome-centric approach. ISME J. 2020;14:1100–10. doi: 10.1038/s41396-020-0591-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robbins SJ, Song W, Engelberts JP, Glasl B, Slaby BM, Boyd J, et al. A genomic view of the microbiome of coral reef demosponges. ISME J. 2021;15:1641–54. doi: 10.1038/s41396-020-00876-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor JA, Palladino G, Wemheuer B, Steinert G, Sipkema D, Williams TJ, et al. Phylogeny resolved, metabolism revealed: functional radiation within a widespread and divergent clade of sponge symbionts. ISME J. 2021;15:503–19. doi: 10.1038/s41396-020-00791-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaib De Mares M, Jimenez DJ, Palladino G, Gutleben J, Lebrun LA, Muller EEL, et al. Expressed protein profile of a Tectomicrobium and other microbial symbionts in the marine sponge Aplysina aerophoba as evidenced by metaproteomics. Sci Rep. 2018;8:11795. doi: 10.1038/s41598-018-30134-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steffen K, Indraningrat AAG, Erngren I, Haglöf J, Becking LE, Smidt H, et al. Oceanographic setting influences the prokaryotic community and metabolome in deep-sea sponges. Sci Rep. 2022;12:3356. doi: 10.1038/s41598-022-07292-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loureiro C, Galani A, Gavriilidou A, Chaib de Mares M, van der Oost J, Medema MH, et al. Comparative metagenomic analysis of biosynthetic diversity across sponge microbiomes highlights metabolic novelty, conservation, and diversification. mSystems. 2022;7:e00357–22. doi: 10.1128/msystems.00357-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters EE, Cahn JKB, Lotti A, Gavriilidou A, Steffens UAE., Loureiro C, et al. Distribution and diversity of ‘Tectomicrobia’, a deep-branching uncultivated bacterial lineage harboring rich producers of bioactive metabolites. ISME Commun. 2023;3:50. [DOI] [PMC free article] [PubMed]
- 25.Roume H, Heintz-Buschart A, Muller EE, Wilmes P. Sequential isolation of metabolites, RNA, DNA, and proteins from the same unique sample. Methods Enzymol. 2013;531:219–36. doi: 10.1016/B978-0-12-407863-5.00011-3. [DOI] [PubMed] [Google Scholar]
- 26.Andrews S FASTQC: a quality control tool for high throughput sequence data. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 27.Bushnell B, Rood J, Singer E. BBMerge - Accurate paired shotgun read merging via overlap. PLoS ONE. 2017;12:e0185056. doi: 10.1371/journal.pone.0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:1–13. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2:26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 34.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6. doi: 10.1093/nar/gkab301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rinke C, Chuvochina M, Mussig AJ, Chaumeil PA, Davin AA, Waite DW, et al. A standardized archaeal taxonomy for the Genome Taxonomy Database. Nat Microbiol. 2021;6:946–59. doi: 10.1038/s41564-021-00918-8. [DOI] [PubMed] [Google Scholar]
- 43.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chuvochina M, Rinke C, Parks DH, Rappe MS, Tyson GW, Yilmaz P, et al. The importance of designating type material for uncultured taxa. Syst Appl Microbiol. 2019;42:15–21. doi: 10.1016/j.syapm.2018.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Zhou Z, Tran PQ, Kieft K, Anantharaman K. Genome diversification in globally distributed novel marine Proteobacteria is linked to environmental adaptation. ISME J. 2020;14:2060–77. doi: 10.1038/s41396-020-0669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen IA, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2019;47:D666–77. doi: 10.1093/nar/gky901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 48.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020.
- 49.RStudio Team. RStudio: integrated development of R. Boston, MA: RStudio, PBC; 2020.
- 50.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: community ecology package. R package version 2.5-2. 2018. https://CRAN.R-project.org/package=vegan.
- 51.Wickham H. ggplot2: Elegant graphics for data analysis. Verlag New York: Springer; 2016.
- 52.Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–8. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eddy SR. HMMER User’s guide: biological sequence analysis using profile hidden Markov models. 3.2.1 ed2018.
- 54.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:W95–101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blin K, Shaw S, Kloosterman AM, Charlop-Powers Z, van Wezel GP, Medema MH, et al. antiSMASH 6.0: improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021;49:W29–35. doi: 10.1093/nar/gkab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 59.Yilmaz LS, Parnerkar S, Noguera DR. mathFISH, a web tool that uses thermodynamics-based mathematical models for in silico evaluation of oligonucleotide probes for fluorescence in situ hybridization. Appl Environ Microbiol. 2011;77:1118–22. doi: 10.1128/AEM.01733-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Radax R, Rattei T, Lanzen A, Bayer C, Rapp HT, Urich T, et al. Metatranscriptomics of the marine sponge Geodia barretti: tackling phylogeny and function of its microbial community. Environ Microbiol. 2012;14:1308–24. doi: 10.1111/j.1462-2920.2012.02714.x. [DOI] [PubMed] [Google Scholar]
- 61.Schimak MP, Kleiner M, Wetzel S, Liebeke M, Dubilier N, Fuchs BM. MiL-FISH: multilabeled oligonucleotides for fluorescence in situ hybridization improve visualization of bacterial cells. Appl Environ Microbiol. 2016;82:62–70. doi: 10.1128/AEM.02776-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, et al. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014;42:D643–8. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 64.Whitman WB. Genome sequences as the type material for taxonomic descriptions of prokaryotes. Syst Appl Microbiol. 2015;38:217–22. doi: 10.1016/j.syapm.2015.02.003. [DOI] [PubMed] [Google Scholar]
- 65.Koedooder C, Guéneuguès A, Van Geersdaële R, Vergé V, Bouget F-Y, Labreuche Y, et al. The role of the glyoxylate shunt in the acclimation to iron limitation in marine heterotrophic bacteria. Front Mar Sci. 2018;5:435.
- 66.Bayer K, Jahn MT, Slaby BM, Moitinho-Silva L, Hentschel U. Marine sponges as Chloroflexi hot spots: Genomic insights and high-resolution visualization of an abundant and diverse symbiotic clade. mSystems. 2018;3:e00150–18. doi: 10.1128/mSystems.00150-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor MW, Hill RT, Piel J, Thacker RW, Hentschel U. Soaking it up: the complex lives of marine sponges and their microbial associates. ISME J. 2007;1:187–90. doi: 10.1038/ismej.2007.32. [DOI] [PubMed] [Google Scholar]
- 69.Thomas T, Rusch D, DeMaere MZ, Yung PY, Lewis M, Halpern A, et al. Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 2010;4:1557–67. doi: 10.1038/ismej.2010.74. [DOI] [PubMed] [Google Scholar]
- 70.Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Webster NS, et al. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci USA. 2012;109:E1878–87. doi: 10.1073/pnas.1203287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Webster NS, Thomas T. The sponge hologenome. MBio. 2016;7:e00135–16. doi: 10.1128/mBio.00135-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kamke J, Sczyrba A, Ivanova N, Schwientek P, Rinke C, Mavromatis K, et al. Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 2013;7:2287–300. doi: 10.1038/ismej.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bennke CM, Kruger K, Kappelmann L, Huang S, Gobet A, Schuler M, et al. Polysaccharide utilisation loci of Bacteroidetes from two contrasting open ocean sites in the North Atlantic. Environ Microbiol. 2016;18:4456–70.. doi: 10.1111/1462-2920.13429. [DOI] [PubMed] [Google Scholar]
- 74.Gavriilidou A, Gutleben J, Versluis D, Forgiarini F, van Passel MWJ, Ingham CJ, et al. Comparative genomic analysis of Flavobacteriaceae: insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genomics. 2020;21:569. doi: 10.1186/s12864-020-06971-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haber M, Burgsdorf I, Handley KM, Rubin-Blum M, Steindler L. Genomic insights into the lifestyles of Thaumarchaeota inside sponges. Front Microbiol. 2020;11:622824. doi: 10.3389/fmicb.2020.622824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Munroe S, Sandoval K, Martens DE, Sipkema D, Pomponi SA. Genetic algorithm as an optimization tool for the development of sponge cell culture media. Vitr Cell Dev Biol Anim. 2019;55:149–58. doi: 10.1007/s11626-018-00317-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fan L, Reynolds D, Liu M, Stark M, Kjelleberg S, Websterf NS, et al. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc Natl Acad Sci USA. 2012;109:1879–87. doi: 10.1073/pnas.1203287109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horn H, Slaby BM, Jahn MT, Bayer K, Moitinho-Silva L, Forster F, et al. An enrichment of CRISPR and other defense-related features in marine aponge-associated microbial metagenomes. Front Microbiol. 2016;7:1751. doi: 10.3389/fmicb.2016.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reynolds D, Thomas T. Evolution and function of eukaryotic-like proteins from sponge symbionts. Mol Ecol. 2016;25:5242–53. doi: 10.1111/mec.13812. [DOI] [PubMed] [Google Scholar]
- 80.Moreno-Pino M, Cristi A, Gillooly JF, Trefault N. Characterizing the microbiomes of Antarctic sponges: a functional metagenomic approach. Sci Rep. 2020;10:645. doi: 10.1038/s41598-020-57464-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Makarova KS, Wolf YI, Koonin EV. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013;41:4360–77. doi: 10.1093/nar/gkt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Harms A, Brodersen DE, Mitarai N, Gerdes K. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell. 2018;70:768–84. doi: 10.1016/j.molcel.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen MT, Liu M, Thomas T. Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol Ecol. 2014;23:1635–45. doi: 10.1111/mec.12384. [DOI] [PubMed] [Google Scholar]
- 84.Fiore CL, Labrie M, Jarett JK, Lesser MP. Transcriptional activity of the giant barrel sponge, Xestospongia muta Holobiont: molecular evidence for metabolic interchange. Front Microbiol. 2015;6:364. doi: 10.3389/fmicb.2015.00364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, et al. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun. 2013;81:629–35. doi: 10.1128/IAI.01035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Piel J. Metabolites from symbiotic bacteria. Nat Prod Rep. 2009;26:338–62. doi: 10.1039/b703499g. [DOI] [PubMed] [Google Scholar]
- 87.Indraningrat AA, Smidt H, Sipkema D. Bioprospecting sponge-associated microbes for antimicrobial compounds. Mar Drugs. 2016;14:87. doi: 10.3390/md14050087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brinkmann CM, Marker A, Ipek Kurtböke D. An overview on marine sponge-symbiotic bacteria as unexhausted sources for natural product discovery. Diversity. 2017;9:40.
- 89.Karimi E, Ramos M, Goncalves JMS, Xavier JR, Reis MP, Costa R. Comparative metagenomics reveals the distinctive adaptive features of the Spongia officinalis endosymbiotic consortium. Front Microbiol. 2017;8:2499. doi: 10.3389/fmicb.2017.02499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Konstantinou D, Mavrogonatou E, Zervou SK, Giannogonas P, Gkelis S. Bioprospecting sponge-associated marine Cyanobacteria to produce bioactive compounds. Toxins. 2020;12:73. doi: 10.3390/toxins12020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gavriilidou A, Mackenzie TA, Sanchez P, Tormo JR, Ingham C, Smidt H, et al. Bioactivity screening and gene-trait matching across marine sponge-associated bacteria. Mar Drugs. 2021;19:75. doi: 10.3390/md19020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Piel J, Hui D, Wen G, Butzke D, Platzer M, Fusetani N, et al. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc Natl Acad Sci USA. 2004;101:16222–7. doi: 10.1073/pnas.0405976101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robinson SL, Christenson JK, Wackett LP. Biosynthesis and chemical diversity of β-lactone natural products. Nat Prod Rep. 2019;20:458–75. doi: 10.1039/c8np00052b. [DOI] [PubMed] [Google Scholar]
- 94.Kirchberger PC, Schmidt ML, Ochman H. The ingenuity of bacterial genomes. Ann Rev Microbiol. 2020;74:815–34. doi: 10.1146/annurev-micro-020518-115822. [DOI] [PubMed] [Google Scholar]
- 95.Maldonado M. Intergenerational transmission of symbiotic bacteria in oviparous and viviparous demosponges, with emphasis on intracytoplasmically-compartmented bacterial types. J Mar Biol Assoc. 2007;87:1701–13. [Google Scholar]
- 96.Vacelet J, Donadey C. Electron microscope study of the association between some sponges and bacteria. J Exp Mar Biol Ecol. 1977;30:301–14. [Google Scholar]
- 97.Moran NA, Wernegreen JJ. Lifestyle evolution in symbiotic bacteria: insights from genomics. Trends Ecol Evol. 2000;15:321–6. doi: 10.1016/s0169-5347(00)01902-9. [DOI] [PubMed] [Google Scholar]
- 98.Usher KM, Kuo J, Fromont J, Sutton DC. Vertical transmission of cyanobacterial symbionts in the marine sponge Chondrilla australiensis (Demospongiae) Hydrobiologia. 2001;461:9–13. [Google Scholar]
- 99.Diez-Vives C, Koutsouveli V, Conejero M, Riesgo A. Global patterns in symbiont selection and transmission strategies in sponges. Front Ecol Evol. 2022;10:1015592.
- 100.Carrier TJ, Maldonado M, Schmittmann L, Pita L, Bosch TCG, Hentschel U. Symbiont transmission in marine sponges: reproduction, development, and metamorphosis. BMC Biol. 2022;20:100. doi: 10.1186/s12915-022-01291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw reads, metagenome assemblies and MAGs generated for this study can be found under the European Nucleotide Archive (ENA) project accession numbers PRJEB54590, PRJEB51534 and PRJEB51535. All accession numbers for the data included in this study are included in the supplementary information of this article.