

Summary
The role of alternative polyadenylation (APA) in tumor development is becoming increasingly evident, but the impact of APA events on the prognosis of LUAD patients is unclear. Therefore, in the present study, we aimed to analyze specific APA events in LUAD to identify novel prognostic biomarkers for LUAD. We first identified prognostic candidate genes for LUAD associated with APA events and validated them in both the East Asian and the USA cohorts, finding that five genes (DCUN1D5, PSMC4, TFAM, THRA, and TMEM100) were of prognostic significance in both populations. Based on this, an APA-based prognostic signature was constructed for the East Asian population. The predictive accuracy of the prognostic signature was further evaluated by the time-dependent ROC, with 1-, 2-, and 3-year AUCs of 0.86, 0.81, and 0.71, respectively. This study may provide new markers for individualized diagnosis and prognostic assessment of LUAD and potential targets for precision treatment.
Subject areas: Medical science, Biochemistry, Physiology
Graphical abstract
Highlights
-
•
We utilize PDUI values to quantify dynamic APA events
-
•
We construct APA-based LUAD prognostic signatures in East Asian populations
-
•
Our results highlight the prognostic value of APA events in LUAD
Medical science; Biochemistry; Physiology
Introduction
Non-small cell lung cancer (NSCLC) is the most common subtype of lung cancer and the leading cause of cancer-related deaths worldwide.1 In recent years, the number of lung adenocarcinoma (LUAD) cases has increased rapidly and has surpassed squamous cell carcinoma (LUSC) to become the most common clinicopathologic type of NSCLC.2,3 LUAD generally has mild early symptoms, late onset of clinical symptoms, and a high degree of malignancy. The side effects and drug resistance of conventional chemotherapy and other treatment modalities lead to an overall 5-year survival rate of less than 20% for patients with LUAD.1,4 However, stage I lung cancer patients who underwent surgical resection showed a high 5-year survival rate of 92%,5 which proves the importance of early diagnosis and timely treatment for the prognosis of LUAD patients. The prognosis of LUAD is associated with various factors, such as TNM stage, degree of tumor differentiation, and pathologic subtype.6 These factors are widely used to guide clinical decision-making. However, they are not yet sufficient to accurately assess the prognosis of patients with this disease due to the lack of understanding of the biological characteristics of LUAD, limiting the further improvement of treatment outcomes. Therefore, it is necessary to elucidate the underlying molecular mechanisms of LUAD in order to identify relevant prognostic genes for early detection, early diagnosis, and better prognosis.
Alternative polyadenylation (APA) is a widespread post-transcriptional regulatory mechanism for genes. Over 70% of human genes have multiple poly(A) sites and not all of them.7 The APA event for most genes occurs at the 3′UTR of the transcript, known as the untranslated region APA (UTR-APA), which produces transcripts encoding the same protein but containing different lengths of the 3′UTR. This is because genes usually have multiple polyadenylation sites. Different sites are selected for cleavage and addition of poly(A) tails, catalyzed by polyadenylate polymerase so that different transcripts have different lengths. The length of the 3′UTR usually alters the secondary structure of the RNA and contains additional regulatory elements such as microRNA (miRNA) binding sites and RNA-binding protein (RBP) recognition sites. As a result, UTR-APA may affect mRNA’s stability, localization and translation efficiency.8,9
In recent years, the role of APA in the occurrence of tumors, cancer development, and phenotypes is increasingly apparent. A series of studies have revealed that the shorter 3′UTRs are more conducive to releasing proto-oncogenes from the repressive effects of miRNAs, which in turn enhances mRNA stability and expression levels to promote cancer development.10,11 For example, widespread proximal poly(A) site utilization has been observed in cancer cells from various tissues, including the thymus, colon, liver, kidney and lung.12 However, compared with some tumor types, lung cancer has more shortening 3′UTR events,13 APA site-switching of 3′UTR is prevalent in NSCLC, such as CSTF2 may play a vital role in the regulation of 3′UTR length in cancer cells as an oncogene driving NSCLC carcinogenesis.14 Oncogene CSNK1D in lung cancer tissues is usually sheared at the proximal poly(A) site, resulting in cancer tissues with multiple short 3′UTR events.15 Moreover, our previous two-stage case-control study found that the variant G allele of apaQTL-SNP rs10138506 in CHURC1 was significantly associated with an increased risk of LUAD.16 These pieces of evidence suggest that APA-mediated regulation of gene expression may play an essential role in the development of lung cancer.
Besides, the selection of APA sites is also closely related to the prognosis of cancer. Several studies have revealed the prognostic role of APA events in different cancers. Zhang et al. used the prognostic characteristics of associated APA events as a predictor of survival and treatment in rectal cancer patients,17 and Wang et al. developed an APA event-based model for predicting the efficacy of immunotherapy, demonstrating the clinical application of APA events as potential biomarkers in cancer immunotherapy.18 Therefore, APA may become a new and potential biomarker for disease prevention and treatment. However, relevant analyses addressing the prognosis of APA events in LUAD are still lacking, especially in East Asian populations.
In the present study, we aimed to identify novel prognostic biomarkers of LUAD using bioinformatics tools and data to deeply analyze specific APA events in LUAD. First, we downloaded LUAD genes associated with APA events from a publicly available database by Xiang et al.15 Subsequently, we screened for prognosis-related APA events and identified genes differentially expressed in tumor tissues and adjacent non-tumor tissues. Finally, these LUAD prognosis-related genes were validated and a prognostic signature based on APA in East Asian populations was constructed. Our research identifies prognostic genes associated with LUAD, advances our understanding of the molecular mechanisms underlying LUAD progression, unveils the potential role of APA in regulating LUAD prognosis.
Results
Integrated screening for LUAD prognosis-related genes
In this study, we identified candidate prognostic genes for LUAD associated with specific APA events by several computational methods. The study design is shown in Figure 1. The original gene data obtained from publicly available databases were screened for a total of 518 LUAD genes that were significantly associated with APA events (|Rs|>0.3 and PFDR<0.05). Each gene was subjected to survival analysis, first grouped according to PDUI values to plot Kaplan-Meier survival curves, and a total of 246 genes were screened (Log rank p < 0.05). Then, these genes were grouped by gene expression for survival analysis and survival curves were plotted. The significance of differences in OS between groups was tested by Log rank test, with a total of 143 genes associated with survival being identified (Log rank p < 0.05). The PDUI values of 143 genes are shown in Table S1. Next, the LUAD differentially expressed genes were screened. A total of 106 pairs of LUAD tumor tissues and adjacent non-tumor tissues were analyzed, including 57 pairs from the TCGA database and 49 pairs from the LUAD database of the Chinese population.19 By overlapping the genes screened in the two databases, 23 prognostic candidate genes for LUAD with APA events were selected.
Figure 1.

The screening process of LUAD prognostic genes associated with APA events
LUAD, lung adenocarcinoma; APA, alternative polyadenylation; RS, spearman correlation; FDR, false discovery rate; PDUI, the percentage of distal poly(A) site usage index; TCGA, The Cancer Genome Atlas; FC, fold change.
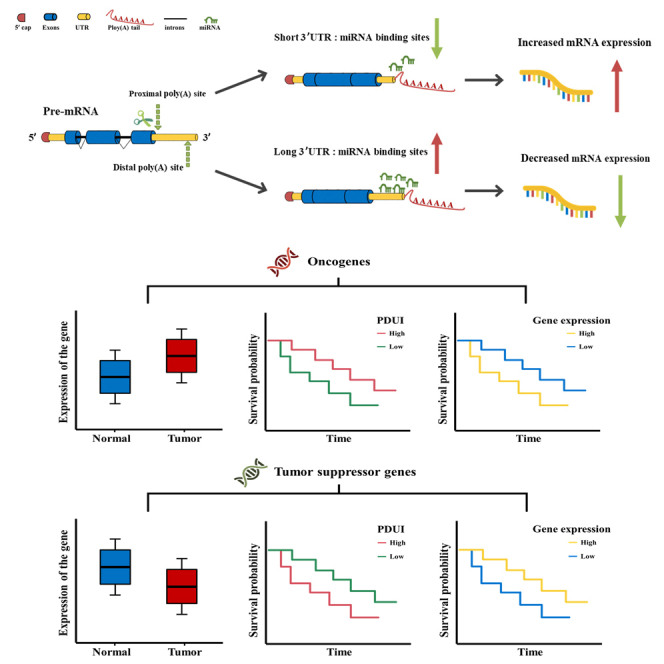
We performed a final screening step to ensure that candidate prognostic genes fit the regulation mechanism of APA events. The presence of the APA site allows mRNA to produce mRNA isoforms with different lengths of the 3′UTR, broadly classified into two isoforms: long and short. This can directly affect the stability and expression level of mRNA. If the gene tends to use the distal poly(A) site, a transcript with a longer 3′UTR will be produced. This makes it subject to more negative regulation by miRNAs. When the gene is an oncogene, miRNAs can bind to the longer 3′UTR of the oncogenes. As a result, miRNAs may inhibit translation of oncogenes or target mRNAs for degradation, resulting in reduced expression of oncogenes.
In contrast, APA-driven shortening of the 3′UTR can eliminate the target of the miRNA, which may lead to increased oncogene mRNA expression; when the gene is an oncogene, the higher survival rate in the high PDUI group in the survival curve graph. This is because, with a PDUI value close to 1, the gene tends to utilize the distal poly(A) site, producing a longer 3′UTR transcript and reducing mRNA expression. Conversely, a PDUI value close to 0 means that the gene utilizes the proximal poly(A) site, resulting in a lower survival rate for the low PDUI group of oncogenes in the survival curve. Therefore, candidate prognostic genes should fit the specific regulatory pattern of APA events. For example, suppose this gene is up-regulated in cancerous tissue. In that case, it will have a higher survival rate among the group with high PDUI in the survival curve and a lower survival rate among the group with high expression. Finally, a total of eleven candidate genes were screened. The differential expression of these genes in the tumor tissues and adjacent non-tumor tissues is shown in Table 1. Nine genes were up-regulated compared to the adjacent non-tumor tissues, and two were down-regulated in the tumor tissues.
Table 1.
Specific information on PDUI values and differential expression of eleven candidate genes
| Gene | PDUI value |
TCGA database |
LUAD database in Chinese population (GSE140343) |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tumor | Normal | Expressiona |
Pb | Regulated direction | Expressiona |
Pb | Regulated direction | |||
| Tumor | Normal | Tumor | Normal | |||||||
| C1GALT1 | 0.272 ± 0.089 | 0.420 ± 0.088 | 0.533 ± 0.200 | 0.392 ± 0.109 | 6.39 × 10−6 | UP | 0.235 ± 0.181 | 0.119 ± 0.081 | 1.54 × 10−4 | UP |
| CISD2 | 0.022 ± 0.008 | 0.043 ± 0.011 | 0.497 ± 0.199 | 0.264 ± 0.150 | 2.07 × 10−12 | UP | 0.228 ± 0.155 | 0.142 ± 0.119 | 4.30 × 10−3 | UP |
| DCUN1D5 | 0.017 ± 0.012 | 0.055 ± 0.027 | 0.489 ± 0.207 | 0.258 ± 0.145 | 1.48 × 10−11 | UP | 0.443 ± 0.198 | 0.156 ± 0.069 | 1.99 × 10−12 | UP |
| NAA50 | 0.056 ± 0.015 | 0.068 ± 0.014 | 0.418 ± 0.221 | 0.256 ± 0.137 | 3.86 × 10−7 | UP | 0.367 ± 0.193 | 0.212 ± 0.073 | 9.18 × 10−6 | UP |
| PSMC4 | 0.081 ± 0.030 | 0.119 ± 0.032 | 0.428 ± 0.164 | 0.232 ± 0.125 | 8.76 × 10−12 | UP | 0.394 ± 0.203 | 0.165 ± 0.069 | 5.27 × 10−9 | UP |
| PSMD11 | 0.134 ± 0.059 | 0.280 ± 0.063 | 0.499 ± 0.198 | 0.270 ± 0.134 | 1.76 × 10−10 | UP | 0.286 ± 0.142 | 0.124 ± 0.044 | 4.55 × 10−10 | UP |
| RAN | 0.099 ± 0.034 | 0.177 ± 0.041 | 0.452 ± 0.205 | 0.286 ± 0.138 | 8.17 × 10−8 | UP | 0.232 ± 0.202 | 0.106 ± 0.070 | 1.60 × 10−4 | UP |
| RPF2 | 0.183 ± 0.090 | 0.248 ± 0.096 | 0.375 ± 0.185 | 0.241 ± 0.126 | 5.41 × 10−7 | UP | 0.319 ± 0.203 | 0.204 ± 0.080 | 5.64 × 10−4 | UP |
| TFAM | 0.302 ± 0.058 | 0.361 ± 0.046 | 0.409 ± 0.207 | 0.251 ± 0.126 | 8.28 × 10−7 | UP | 0.444 ± 0.197 | 0.256 ± 0.078 | 1.08 × 10−7 | UP |
| THRA | 0.791 ± 0.115 | 0.779 ± 0.107 | 0.397 ± 0.184 | 0.632 ± 0.154 | 2.36 × 10−11 | Down | 0.267 ± 0.167 | 0.669 ± 0.192 | 4.33 × 10−15 | Down |
| TMEM100 | 0.677 ± 0.118 | 0.673 ± 0.082 | 0.402 ± 0.171 | 0.767 ± 0.124 | 6.16 × 10−21 | Down | 0.047 ± 0.143 | 0.184 ± 0.173 | 1.85 × 10−4 | Down |
Expression: Gene expression after normalization. Data are represented as mean ± SD.
P: Paired Student’s t test (Comparison of gene expression levels in paired tumor and normal samples).
Moreover, their differential expression in the TCGA and LUAD databases of the Chinese population was consistent (Figure 2; Figure 3). Kaplan-Meier survival curves for the eleven genes plotted based on the optimal cut-off values of PDUI values dividing LUAD patients into high and low groups are shown in Figure 4. The Kaplan-Meier survival curves for the eleven genes plotted for LUAD patients divided into high and low groups based on the optimal cut-off values for gene expression are shown in Figure 5. The differences in survival time distributions between the two groups were statistically significant and consistent with a specific regulation pattern of APA events. It is worth noting that eight of the candidate genes also differed statistically significantly (p < 0.05) in the Chinese population for expression of the associated proteins (Figure 6).
Figure 2.

Expression levels of the eleven prognostic candidate genes in LUAD tumor tissues and adjacent non-tumor tissues based on the TCGA database
(A–K) (A) C1GALT1; (B) CISD2; (C) DCUN1D5; (D) NAA50; (E) PSMC4; (F) PSMD11; (G) RAN; (H) RPF2; (I) TFAM; (J) THRA; (K) TMEM100.
Figure 3.

Expression levels of the eleven prognostic candidate genes in LUAD tumor tissues and adjacent non-tumor tissues based on LUAD databases of the Chinese population
(A–K) (A) C1GALT1; (B) CISD2; (C) DCUN1D5; (D) NAA50; (E) PSMC4; (F) PSMD11; (G) RAN; (H) RPF2; (I) TFAM; (J) THRA; (K) TMEM100.
Figure 4.

Kaplan-Meier survival curves for the eleven prognostic candidate genes were plotted by dividing LUAD patients into high and low PDUI value groups based on the optimal cut-off values for PDUI value
(A–K) (A) C1GALT1; (B) CISD2; (C) DCUN1D5; (D) NAA50; (E) PSMC4; (F) PSMD11; (G) RAN; (H) RPF2; (I) TFAM; (J) THRA; (K) TMEM100; PDUI, the percentage of distal poly(A) site usage index.
Figure 5.

Kaplan-Meier survival curves for the eleven prognostic candidate genes were plotted by dividing LUAD patients into high and low gene expression groups based on the optimal cut-off values for gene expression
(A–K) (A) C1GALT1; (B) CISD2; (C) DCUN1D5; (D) NAA50; (E) PSMC4; (F) PSMD11; (G) RAN; (H) RPF2; (I) TFAM; (J) THRA; (K) TMEM100.
Figure 6.

In the Chinese population, eight of the eleven candidate genes were differentially expressed in the associated proteins. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Validation of candidate prognostic genes
To verify whether the eleven candidate genes could also have prognostic value in other population cohorts, we downloaded gene expression profiles and clinical survival information from GEO for the East Asian cohort20 and the USA cohort.21 In the East Asian population, we selected 204 LUAD patients with reliable prognostic information and validated them by COX regression analysis. The results showed that there were seven LUAD prognosis-related genes (p < 0.05). Of these, five genes were poor prognostic factors (HR > 1) and two genes were favorable prognostic factors (HR < 1). In the USA population, we selected 331 LUAD patients with complete clinical information for validation, and the results showed a total of eight LUAD prognosis-associated genes (p < 0.05). Of these, six genes were poor prognostic factors (HR > 1) and two genes were favorable prognostic factors (HR < 1). A comprehensive analysis of the validation results showed that five genes (DCUN1D5, PSMC4, TFAM, THRA, TMEM100) had prognostic significance in both populations (Figure 7).
Figure 7.

Prognostic validation of 11 candidate genes in East Asian and USA cohorts
GSE31210 for East Asia cohort; GSE72094 for USA cohort;a P-value: adjusted for age, sex, race, and smoking status in the COX regression analysis.
Construction and evaluation of a prognostic signature based on APA
To construct a prognostic profile of APA based on East Asian populations, we retained genes with survival significance in East Asian populations during the validation phase. The seven genes with independent prognostic APA events were further analyzed using LASSO Cox regression to screen for the strongest predictive power to construct the prognostic risk score model. Selection of the tuning parameter in the LASSO model by 10-fold cross-validation based on minimum criteria (Figures 8A and 8B), optimal lambda.min = 0.016. A total of six prognostic genes (DCUN1D5, PSMC4, RPF2, TFAM, THRA, and TMEM100) were identified and APA-based prognostic signature were established. The following formula was utilized for calculating the risk scores: Risk score = (0.4222×PSMC4 expression level) + (0.1542×DCUN1D5 expression level) + (0.1769×RPF2 expression level) + (0.5343×TFAM expression level) + (−0.5567×THRA expression level) + (−0.0474×TMEM100 expression level). We divided patients into high and low-risk groups according to the median of the prognostic model risk scores and built Kaplan-Meier survival curves (Figure 8C). The OS in the high-risk group was significantly lower (Log rank p = 0.00023). Risk curves with associated scatterplots for constructing prognostic model survival are shown in Figures 8D and 8E. The prognosis of the low-risk group was better than that of the high-risk, with lower survival time and increased number of deaths as the risk score increased. Meanwhile, the risk heatmap of the expression distribution (Figure 8F) shows that the expression of high-risk genes (DCUN1D5, PSMC4, RPF2, TFAM) increased and the expression of low-risk genes (THRA, TMEM100) decreased as the risk value increased. The predictive accuracy of the prognostic signature was further evaluated by the time-dependent ROC in this cohort, with 1-, 2-, and 3-year AUCs of 0.86, 0.81, and 0.71, respectively (Figure 8G). The results indicate that this prognostic signature has promising predictive power in East Asian populations.
Figure 8.

Establishment of APA-based prognostic signature
(A) LASSO coefficients profiles of the seven prognostic genes associated with APA events.
(B) LASSO regression analysis obtained six prognostic genes with APA events.
(C) Kaplan–Meier curves for OS in the high-risk and low-risk groups stratified by the prognostic model risk scores.
(D) Risk curves based on risk scores for each sample, with yellow indicating high risk and blue indicating low risk.
(E) Scatterplot based on the survival status of each sample, with yellow indicates death, blue indicates alive.
(F) The heatmap shows the expression of six prognostic genes in the high-risk and low-risk groups.
(G) The predictive accuracy of the prognostic signature was evaluated by the time-dependent ROC, with 1-, 2-, and 3-year AUCs of 0.86, 0.81, and 0.71, respectively.
Discussion
In this study, we combined APA events with TCGA and Chinese databases to systematically analyze survival information of LUAD patients, resulting in the candidate of eleven APA event-related genes as potential prognostic modifiers of their presence in LUAD. Worth mentioning, in the Chinese population, we found that eight of the eleven candidate genes also showed statistically significant differences in the expression of the corresponding encoded proteins. As we know, APA may alter the binding of miRNAs or RBPs to target genes by regulating the length of the 3′UTR, which in turn affects the stability, expression level and translation ability of the target genes. And recent studies have shown that the shift of 3′UTRs to shorter isoforms may lead to higher gene translation efficiency.22 While the above eight proteins with differential expression levels had lower PDUI values in tumor tissues than in adjacent non-tumor tissues, and the expression of the genes encoding seven of these proteins was upregulated in tumors in concert with the proteins. Moreover, survival analysis revealed that patients in their low PDUI group had a poorer prognosis. Therefore, we hypothesized that mRNAs with shorter 3′UTRs in tumor tissues might have higher translational efficiency, which leads to differences in protein expression levels and impacts a range of cellular malignant phenotypic processes, leading to poor prognosis. However, we found that DCUN1D5, although having a shorter 3′UTR in tumor tissues and more gene expression relative to adjacent non-tumor tissues, in yet showed downregulation in proteins. This inconsistency between gene and protein expression may be due to post-transcriptional translational diversity. It is important to know that protein translation itself is a complex multi-step process that is extensively regulated at the level of initiation, elongation, localization and ribosome composition, and the concordance rate between differential gene and protein expression in tissues is only 40%.23 Therefore, we speculate that DCUN1D5 may be influenced by other complex mechanisms besides splicing and polyadenylation regulation in LUAD and deserves further investigation.
In the validation phase, we performed validation analyses of eleven candidate genes using the East Asian population cohort and the USA population, finding that five of these genes had prognostic significance in both populations. These five genes screened for the prognosis of LUAD patients in different populations, indicating they have good diagnostic power. It is well known that the occurrence and progression of cancer is a complex multi-stage process, and the prognosis of patients may not be accurately predicted by a single biomarker alone. However, the prognosis of such cancers has been relatively less studied in Asian populations, especially in East Asian populations, than in Western countries. Therefore, to construct an APA prognostic signature for the East Asian population, we retained genes with survival significance in the East Asian population in the validation phase and performed LASSO regression on these seven genes to construct the prognostic signature. This model assessed the risk score of each patient, and LUAD patients were divided into high-risk and low-risk groups with significant differences. And the AUCs for the prognostic signature were 0.86, 0.81 and 0.71 at 1, 2, and 3 years, respectively. This suggests that this prognostic signature has good predictive power. The six prognostic genes with APA events that constructed this model could provide alternative molecular markers for basic research related to prognosis in LUAD.
Among these APA genes in the signature, most are associated with tumorigenesis and progression. Research has shown that DCUN1D5 has oncogenic potential. In oral and lung squamous cell carcinomas, mRNA levels of DCUN1D5 corresponded to protein levels, and upregulation of expression was associated with decreased disease-specific survival.24 PSMC4 belongs to the PSMC family, and most genes of the PSMC family are up-regulated in many cancers. For example, in LUAD tissues, there was a significant correlation between overexpression of PSMCs and poorer overall and recurrence-free survival in LUAD patients, demonstrating that PSMCs are ideal targets for LUAD diagnosis.25 Furthermore, PSMC4 is not only involved in prostate tumorigenesis but is also considered one of the best biomarkers for endometrial cancer.26,27 RPF2 is not only overexpressed in colorectal cancer tissues but also associated with colorectal cancer cell proliferation. It was suggested that RPF2 could activate the AKT/GSK-3β signaling pathway through direct interaction with CARM1 and promote epithelial-mesenchymal transition, thereby enhancing the migration and invasion of colorectal cancer cells.28 TFAM is a key molecule in carcinogenesis and its protein is the only nuclear-associated protein that functions as a histidine-like factor. TFAM expression levels are up-regulated in various cancers, such as gastric cancer29 and breast cancer.30 Compared with normal tissues, tumor cells produce more TFAM to adequately compact mtDNA in tumors. The mtDNA in cancer cells is tightly wrapped by the increased TFAM, resulting in reduced expression of the relevant mtDNA-encoding genes and promoting the utilization of aerobic glycolysis by tumor cells.31 The thyroid hormone receptor (THR) is encoded by THRA, which is known to have tumor suppressive effects, and loss of functional THR in mice leads to follicular thyroid cancer.32 Loss of THR expression in clinical samples of hepatocellular carcinoma,33 along with a negative correlation of THRA expression in clinical liver cancer specimens.34 The TMEM100 gene has previously been down-regulated in tumor tissues of LUAD patients. Meanwhile, in vitro functional studies have shown that TMEM100 inhibits colony formation when overexpressed in transfected lung cancer cell lines, suggesting that suppression of TMEM100 expression is an essential factor contributing to the development of lung cancer.35,36 Although these six genes with APA events may influence the development and prognosis of some cancers, biological functions played by these APA events in cancer are not yet precise and deserve further investigation.
Our study has several strengths. First, we used PDUI values to quantify APA events, which clarified the complicated relationship between APA events and LUAD prognosis more clearly. Second, based on APA regulatory mechanisms, we explored the impact of 3′UTR length and gene expression values on survival. We used a rigorous logical framework to screen candidate genes associated with prognosis, strengthening our predictions. Finally, we validated the ability of candidate genes to identify patient prognosis using dual cohorts and constructed an APA-based prognostic signature for East Asian populations, enriching the understanding of the prognostic role of APA events in LUAD.
In summary, this study investigated APA events in LUAD patients and screened for prognostically relevant APA events. More importantly, we constructed a prognostic signature based on APA, which can well assess the OS of LUAD patients. These results not only enrich our understanding of the role of APA events in LUAD prognosis, but also promise to offer new markers for individualized diagnosis and prognostic assessment of LUAD, and provide precise treatment for potential targets.
Limitations of the study
However, there were some limitations in this study. The prognostic model we established was based on a dataset from Japan, representing only a portion of the population in East Asia. Furthermore, the model requires validation through clinical trials to enhance its clinical value.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| TCGA-LUAD gene expression data and clinical information | NIH GDC data portal | https://portal.gdc.cancer.gov/ |
| LUAD genes related to APA events | Xiang et al.,201815 | https://doi.org/10.1093/jnci/djx223 |
| Gene expression profiles and clinical information of the Chinese LUAD cohort | Xu et al.,202019 | GSE140343 |
| Proteomics data of the Chinese LUAD cohort | Xu et al.,202019 | iProx: IPX0001804000 |
| Gene expression profiles and clinical information of the Japanese LUAD cohort | Okayama et al., 201220 | GSE31210 |
| Gene expression profiles and clinical information of the USA LUAD cohort | Schabath et al.,201621 | GSE72094 |
| Software and algorithms | ||
| R (version 4.2.0) | The R Foundation | https://www.r-project.org |
| ggplot2 | R package | N/A |
| survival | R package | N/A |
| glment | R package | N/A |
| survminer | R package | N/A |
| timeROC | R package | N/A |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Minjie Chu (chuminjie@ntu.edu.cn).
Materials availability
The study did not generate any new materials.
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
The data analyzed in this study were obtained from The Cancer Genome Atlas (TCGA) database and the Gene Expression Omnibus (GEO) database. The characteristics of all patients in this study are summarized in Table S2. RNA-seq data and clinical survival information for LUAD samples were downloaded from the TCGA database and matched with PDUI values to create a reliable APA dataset. Gene expression profiles and clinical survival information for three independent LUAD cohorts were obtained from the GEO database.
Method details
Data collection and processing
LUAD genes associated with APA events were obtained from the study published by Xiang et al.,15 containing 13,737 genes. The study was dynamically analyzed for data using the DaPars algorithm (https://github.com/ZhengXia/DaPars) and against standard RNA-seq from The Cancer Genome Atlas (TCGA) database to identify APA events. The DaPars algorithm identifies dynamic APAs from standard RNA-seq to determine alternative proximal poly(A) sites and calculates the percentage of distal poly(A) site usage index (PDUI) for each transcript. The PDUI value enables the quantification of APA events into a more intuitive ratio, which is calculated by dividing the isoform expression level at the distal poly(A) locus by the total expression level of isoforms at the distal and proximal poly(A) loci. The value of PDUI ranges from 0 to 1. If PDUI is close to 1, the gene has a longer 3′UTR with more distal polyadenylation sites in its transcript and vice versa. To explore the correlation between APA factor expression and PDUI for each transcript in the tumor samples, linear regression modeling was performed using Spearman correlations (Rs). The Wilcoxon test was used and adjusted by the false discovery rate (FDR) using Benjamini & Hochberg to consider the PFDR<0.05 as statistically significant. |Rs|>0.3 and PFDR<0.05 were defined as significant correlations between APA factors and transcript PDUI. Finally, 518 LUAD genes significantly associated with APA events were screened. Relevant guidelines and regulations are carried out for all methods.
Identification of the Feature genes associated with LUAD prognosis
The RNA-seq data and clinical survival information of LUAD samples were downloaded from the TCGA database (https://portal.gdc.cancer.gov/) and matched with PDUI values to generate a reliable APA dataset. In order to evaluate the association between APA events and overall survival (OS), survival analysis was performed on the dataset to identify survival-related APA events. The optimal cut-off value for the PDUI value was determined using the "surv_cutpoint" function of the R package "survminer", which determines the most significant cut-off value for survival by using the maximum selected rank statistic information from the "maxstat" package (http://r-addict.com/2016/11/21/Optimal-Cutpoint-maxstat.html). PDUI values were cut by the optimal cut-off value and all patients were divided into a low PDUI group and a high PDUI group. Survival curves were plotted by the Kaplan-Meier method and compared between the two groups using the Log rank test. Survival analysis of gene expression data was performed to screen for further prognosis-related genes. The expression was divided into high and low groups based on the optimal cut-off values. Survival curves were also plotted using the Kaplan-Meier method. Only Log rank p < 0.05 were selected for further analysis to obtain APA events with a good prognosis.
Differential profiling of mRNA expression levels between tumor tissues and adjacent non-tumor tissues was performed to obtain differentially expressed genes. A total of 106 pairs of LUAD tumor tissues and adjacent non-tumor tissues were analyzed, including 57 pairs from the TCGA database and 49 pairs from the LUAD database of the Chinese population.19 Due to the different databases of genes with different expression ranges, in order to make reliable and meaningful comparisons of gene expression. Therefore, we balance these values by the min-max normalization procedure, which converts them to the range (0, 1). Using p < 0.05 and fold change (FC) > 3/2(cases/controls:> 3/2-fold upregulated or<2/3-fold downregulated) as screening criteria. Candidate LUAD prognostic genes with APA events were obtained by overlapping the genes screened from the two databases. In addition, the proteomic data of 103 pairs from the LUAD database of the Chinese population19 were analyzed to investigate the expression of candidate gene-related proteins.
Validation of candidate prognostic genes
We downloaded the gene expression profiles and clinical survival information from the East Asian population cohort study20 and the USA population cohort study21 from GEO. The East Asian population cohort included 226 patients from Japan with prognostic information for LUAD, 22 of whom were excluded because they received chemotherapy and/or radiation therapy, resulting in 204 patients being included in this study. A total of 331 patient samples were included in the USA population cohort after removing samples with missing clinical information. The COX regression analysis was used to assess the correlation between each candidate prognostic gene in the GEO cohort and the survival status of LUAD patients, adjusting for confounding variables that may affect survival outcomes, namely age, sex, race, and smoking status. Finally, prognostic genes based on APA events were validated.
Establishment of APA-based prognostic signature
The least absolute shrinkage and selection operator (LASSO) regression was constructed to select the most valuable prognostic factors. The patient’s risk score was calculated based on the expression level of the gene and the corresponding lasso factor. The risk score was calculated as: risk score = ∑exp(i) × coef(i). After obtaining the risk score for the prognostic model, patients in the dataset were divided into high and low-risk groups by median and Kaplan-Meier survival curves were plotted. The area under the curve (AUC) of the receiver operating characteristic (ROC) was calculated to assess the predictive power of the risk score for 1-, 2- and 3-year survival.
Quantification and statistical analysis
All statistical analyses in this research were performed using R software (version 4.2.0). Kaplan-Meier survival curves were plotted using the "survival" package in the R and the Log rank test was used to compare OS between the two groups. Paired Student’s t test were used to compare mRNA expression levels in tumor tissue and adjacent non-tumor tissue between groups. The "Coxph" function in the "survival" package was used for COX regression analysis, and the LASSO regression was constructed using the "glment" package. The packages "survminer" and "timeROC" were used to calculate AUC for the ROC. All statistical tests were considered statistically significant at p < 0.05.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (82273715, 82203771), the National Key Research and Development Program of China (2022YFC2503202), the Science and Technology Program of Nantong City (MS22022062, JC22022002), and the Postgraduate Research and Practice Innovation Program of Jiangsu Province (KYCX23-3436).
Author contributions
Conceptualization, W.Z., H.X., and A.Q.; Data curation, L.M., Y.Y., and W.Z.; Formal analysis, L.M., Y.Y., and Y.L.; Funding acquisition, M.C.; Methodology, A.Q., W.Z., and M.C.; Software, L.M., X.T., and Y.W.; Supervision, M.C., J.C., and L.Z.; Validation, L.Z., Y.D., and M.C.; Visualization, Y.Z., Y.L., and Q.C.; Writing – original draft, W.Z., T.T., and M.C.; Writing – review and editing, W.Z., Y.H., and M.Q.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: September 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108068.
Contributor Information
Lei Zhang, Email: zhanglei94@ntu.edu.cn.
Jiahua Cui, Email: cuijiahua@ntu.edu.cn.
Minjie Chu, Email: chuminjie@ntu.edu.cn.
Supplemental information
References
- 1.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 2.Barta J.A., Powell C.A., Wisnivesky J.P. Global Epidemiology of Lung Cancer. Ann. Glob. Health. 2019;85:8. doi: 10.5334/aogh.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Succony L., Rassl D.M., Barker A.P., McCaughan F.M., Rintoul R.C. Adenocarcinoma spectrum lesions of the lung: Detection, pathology and treatment strategies. Cancer Treat Rev. 2021;99:102237. doi: 10.1016/j.ctrv.2021.102237. [DOI] [PubMed] [Google Scholar]
- 4.Lin J.J., Cardarella S., Lydon C.A., Dahlberg S.E., Jackman D.M., Jänne P.A., Johnson B.E. Five-Year Survival in EGFR-Mutant Metastatic Lung Adenocarcinoma Treated with EGFR-TKIs. J. Thorac. Oncol. 2016;11:556–565. doi: 10.1016/j.jtho.2015.12.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstraw P., Chansky K., Crowley J., Rami-Porta R., Asamura H., Eberhardt W.E.E., Nicholson A.G., Groome P., Mitchell A., Bolejack V., et al. The IASLC Lung Cancer Staging Project: Proposals for Revision of the TNM Stage Groupings in the Forthcoming (Eighth) Edition of the TNM Classification for Lung Cancer. J. Thorac. Oncol. 2016;11:39–51. doi: 10.1016/j.jtho.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Oskarsdottir G.N., Bjornsson J., Jonsson S., Isaksson H.J., Gudbjartsson T. Primary adenocarcinoma of the lung--histological subtypes and outcome after surgery, using the IASLC/ATS/ERS classification of lung adenocarcinoma. APMIS. 2016;124:384–392. doi: 10.1111/apm.12522. [DOI] [PubMed] [Google Scholar]
- 7.Li L., Huang K.L., Gao Y., Cui Y., Wang G., Elrod N.D., Li Y., Chen Y.E., Ji P., Peng F., et al. An atlas of alternative polyadenylation quantitative trait loci contributing to complex trait and disease heritability. Nat. Genet. 2021;53:994–1005. doi: 10.1038/s41588-021-00864-5. [DOI] [PubMed] [Google Scholar]
- 8.Tian B., Manley J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017;18:18–30. doi: 10.1038/nrm.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardy J.G., Norbury C.J. Cleavage factor Im (CFIm) as a regulator of alternative polyadenylation. Biochem. Soc. Trans. 2016;44:1051–1057. doi: 10.1042/BST20160078. [DOI] [PubMed] [Google Scholar]
- 10.Masamha C.P., Wagner E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis. 2018;39:2–10. doi: 10.1093/carcin/bgx096. [DOI] [PubMed] [Google Scholar]
- 11.Gruber A.J., Zavolan M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 2019;20:599–614. doi: 10.1038/s41576-019-0145-z. [DOI] [PubMed] [Google Scholar]
- 12.Lin Y., Li Z., Ozsolak F., Kim S.W., Arango-Argoty G., Liu T.T., Tenenbaum S.A., Bailey T., Monaghan A.P., Milos P.M., John B. An in-depth map of polyadenylation sites in cancer. Nucleic Acids Res. 2012;40:8460–8471. doi: 10.1093/nar/gks637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xia Z., Donehower L.A., Cooper T.A., Neilson J.R., Wheeler D.A., Wagner E.J., Li W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types. Nat. Commun. 2014;5:5274. doi: 10.1038/ncomms6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S., Zhang X., Lei W., Liang J., Xu Y., Liu H., Ma S. Genome-wide profiling reveals alternative polyadenylation of mRNA in human non-small cell lung cancer. J. Transl. Med. 2019;17:257. doi: 10.1186/s12967-019-1986-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang Y., Ye Y., Lou Y., Yang Y., Cai C., Zhang Z., Mills T., Chen N.Y., Kim Y., Muge Ozguc F., et al. Comprehensive Characterization of Alternative Polyadenylation in Human Cancer. J. Natl. Cancer Inst. 2018;110:379–389. doi: 10.1093/jnci/djx223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiu A., Xu H., Mao L., Xu B., Fu X., Cheng J., Zhao R., Cheng Z., Liu X., Xu J., et al. A Novel apaQTL-SNP for the Modification of Non-Small-Cell Lung Cancer Susceptibility across Histological Subtypes. Cancers. 2022;14:5309. doi: 10.3390/cancers14215309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Wang Y., Li C., Jiang T. Systemic Analysis of the Prognosis-Associated Alternative Polyadenylation Events in Breast Cancer. Front. Genet. 2020;11:590770. doi: 10.3389/fgene.2020.590770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G., Xie Z., Su J., Chen M., Du Y., Gao Q., Zhang G., Zhang H., Chen X., Liu H., et al. Characterization of Immune-Related Alternative Polyadenylation Events in Cancer Immunotherapy. Cancer Res. 2022;82:3474–3485. doi: 10.1158/0008-5472.CAN-22-1417. [DOI] [PubMed] [Google Scholar]
- 19.Xu J.Y., Zhang C., Wang X., Zhai L., Ma Y., Mao Y., Qian K., Sun C., Liu Z., Jiang S., et al. Integrative Proteomic Characterization of Human Lung Adenocarcinoma. Cell. 2020;182:245–261.e17. doi: 10.1016/j.cell.2020.05.043. [DOI] [PubMed] [Google Scholar]
- 20.Okayama H., Kohno T., Ishii Y., Shimada Y., Shiraishi K., Iwakawa R., Furuta K., Tsuta K., Shibata T., Yamamoto S., et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 21.Schabath M.B., Welsh E.A., Fulp W.J., Chen L., Teer J.K., Thompson Z.J., Engel B.E., Xie M., Berglund A.E., Creelan B.C., et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene. 2016;35:3209–3216. doi: 10.1038/onc.2015.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu Y., Chen L., Chen C., Ge Y., Kang M., Song Z., Li J., Feng Y., Huo Z., He G., et al. Crosstalk between alternative polyadenylation and miRNAs in the regulation of protein translational efficiency. Genome Res. 2018;28:1656–1663. doi: 10.1101/gr.231506.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buccitelli C., Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020;21:630–644. doi: 10.1038/s41576-020-0258-4. [DOI] [PubMed] [Google Scholar]
- 24.Bommeljé C.C., Weeda V.B., Huang G., Shah K., Bains S., Buss E., Shaha M., Gönen M., Ghossein R., Ramanathan S.Y., Singh B. Oncogenic function of SCCRO5/DCUN1D5 requires its Neddylation E3 activity and nuclear localization. Clin. Cancer Res. 2014;20:372–381. doi: 10.1158/1078-0432.CCR-13-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ullah M.A., Islam N.N., Moin A.T., Park S.H., Kim B. Evaluating the Prognostic and Therapeutic Potentials of the Proteasome 26S Subunit, ATPase (PSMC) Family of Genes in Lung Adenocarcinoma: A Database Mining Approach. Front. Genet. 2022;13:935286. doi: 10.3389/fgene.2022.935286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hellwinkel O.J.C., Asong L.E., Rogmann J.P., Sültmann H., Wagner C., Schlomm T., Eichelberg C. Transcription alterations of members of the ubiquitin-proteasome network in prostate carcinoma. Prostate Cancer Prostatic Dis. 2011;14:38–45. doi: 10.1038/pcan.2010.48. [DOI] [PubMed] [Google Scholar]
- 27.Ayakannu T., Taylor A.H., Konje J.C. Selection of Endogenous Control Reference Genes for Studies on Type 1 or Type 2 Endometrial Cancer. Sci. Rep. 2020;10:8468. doi: 10.1038/s41598-020-64663-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Hu X., Cheng C., Lu M., Huang L., Dou H., Zhang Y., Wang T. Ribosome production factor 2 homolog promotes migration and invasion of colorectal cancer cells by inducing epithelial-mesenchymal transition via AKT/Gsk-3beta signaling pathway. Biochem. Biophys. Res. Commun. 2022;597:52–57. doi: 10.1016/j.bbrc.2022.01.090. [DOI] [PubMed] [Google Scholar]
- 29.Lee W.R., Na H., Lee S.W., Lim W.J., Kim N., Lee J.E., Kang C. Transcriptomic analysis of mitochondrial TFAM depletion changing cell morphology and proliferation. Sci. Rep. 2017;7:17841. doi: 10.1038/s41598-017-18064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan X., Zhou S., Zheng M., Deng X., Yi Y., Huang T. MiR-199a-3p enhances breast cancer cell sensitivity to cisplatin by downregulating TFAM (TFAM) Biomed. Pharmacother. 2017;88:507–514. doi: 10.1016/j.biopha.2017.01.058. [DOI] [PubMed] [Google Scholar]
- 31.Sun X., Zhan L., Chen Y., Wang G., He L., Wang Q., Zhou F., Yang F., Wu J., Wu Y., et al. Increased mtDNA copy number promotes cancer progression by enhancing mitochondrial oxidative phosphorylation in microsatellite-stable colorectal cancer. Signal Transduct. Target. Ther. 2018;3:8. doi: 10.1038/s41392-018-0011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu X.G., Zhao L., Willingham M.C., Cheng S.Y. Thyroid hormone receptors are tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Oncogene. 2010;29:1909–1919. doi: 10.1038/onc.2009.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frau C., Loi R., Petrelli A., Perra A., Menegon S., Kowalik M.A., Pinna S., Leoni V.P., Fornari F., Gramantieri L., et al. Local hypothyroidism favors the progression of preneoplastic lesions to hepatocellular carcinoma in rats. Hepatology. 2015;61:249–259. doi: 10.1002/hep.27399. [DOI] [PubMed] [Google Scholar]
- 34.Tseng Y.H., Huang Y.H., Lin T.K., Wu S.M., Chi H.C., Tsai C.Y., Tsai M.M., Lin Y.H., Chang W.C., Chang Y.T., et al. Thyroid hormone suppresses expression of stathmin and associated tumor growth in hepatocellular carcinoma. Sci. Rep. 2016;6:38756. doi: 10.1038/srep38756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu X., Zhang W., Hu Y., Yi X. Bioinformatics approach reveals systematic mechanism underlying lung adenocarcinoma. Tumori. 2015;101:281–286. doi: 10.5301/tj.5000278. [DOI] [PubMed] [Google Scholar]
- 36.Frullanti E., Colombo F., Falvella F.S., Galvan A., Noci S., De Cecco L., Incarbone M., Alloisio M., Santambrogio L., Nosotti M., et al. Association of lung adenocarcinoma clinical stage with gene expression pattern in noninvolved lung tissue. Int. J. Cancer. 2012;131:E643–E648. doi: 10.1002/ijc.27426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.