

Abstract
Rab GTPases are important regulators of intracellular vesicular trafficking. RAB5C is a member of the Rab GTPase family that plays an important role in the endocytic pathway, membrane protein recycling and signaling. Here we report on 12 individuals with nine different heterozygous de novo variants in RAB5C. All but one patient with missense variants (n = 9) exhibited macrocephaly, combined with mild-to-moderate developmental delay. Patients with loss of function variants (n = 2) had an apparently more severe clinical phenotype with refractory epilepsy and intellectual disability but a normal head circumference. Four missense variants were investigated experimentally. In vitro biochemical studies revealed that all four variants were damaging, resulting in increased nucleotide exchange rate, attenuated responsivity to guanine exchange factors and heterogeneous effects on interactions with effector proteins. Studies in C. elegans confirmed that all four variants were damaging in vivo and showed defects in endocytic pathway function. The variant heterozygotes displayed phenotypes that were not observed in null heterozygotes, with two shown to be through a dominant negative mechanism. Expression of the human RAB5C variants in zebrafish embryos resulted in defective development, further underscoring the damaging effects of the RAB5C variants. Our combined bioinformatic, in vitro and in vivo experimental studies and clinical data support the association of RAB5C missense variants with a neurodevelopmental disorder characterized by macrocephaly and mild-to-moderate developmental delay through disruption of the endocytic pathway.
Introduction
The Rab family of GTPases is involved in regulation of intracellular membrane trafficking. About 70 members of the Rab family exist in humans, each with separate functions (1). Several of these have been associated with neurodevelopmental disorders (2–4), while other Rab GTPases have been studied in cell models and model organisms, without association with human disease (5). The Rab GTPases function as cellular switches, cycling between GTP-bound active and a GDP-bound inactive states. The balance between activation and inactivation is influenced by multiple factors, and the final effect depends on the timing of activation, as well as the amount and subcellular localization of the protein. To function properly, the timing, amount and localization must be in sync: normal development is the final result of a cellular balancing act.
Rab5, a subgroup of the Rab GTPase family, is a master regulator of the endocytic pathway, functioning in the uptake and utilization of extracellular materials, membrane protein recycling or degradation and signaling. Rab5 mediates heterotypic fusion of nascent cargo containing endocytic vesicles with early endosomes (EEs), homotypic fusion of EEs and the recruitment of Rab7 in the maturation of EEs to late endosomes (6,7). In mammals there are three paralogs, Rab5a, Rab5b and Rab5c, with Rab5c the focus of this study. Rab5c regulates the homeostasis of several membrane-bound proteins, including adhesion molecules and receptors. In addition, Rab5c has also been implicated in several (patho)physiological processes, including signal transduction through EGF and VEGF receptors (8–10), cell motility and cell adhesion (11,12). In previous studies, Rab5 was found to have a role in neuronal migration (13) and axonal transport (14). RAB5C has, up until now, not definitively been associated with human disease, although a link has been suggested based on the presence of loss of function variants in RAB5C in cohorts of patients with epilepsy (15,16).
Here we describe pediatric and adult patients with heterozygous de novo variants in RAB5C, along with in vitro biochemical characterization and in vivo studies of these variants in C. elegans and zebrafish.
Results
Patients
Using GeneMatcher (17), we identified 12 unrelated individuals with heterozygous variants in RAB5C (Table 1). Of these variants 11 were de novo, while inheritance was unknown in one patient. Nine variants were missense, one was an indel and two were loss of function (Table 1, Supplementary Material, Table S1).
Table 1.
Description of phenotypic characteristics of patients with missense (1–9), indel (10) and loss-of-function (11,12) variants
| Patient | Gene (NM_004583.3) | Protein (NP_004574.2) | Gender | Age (yrs) | Head circumference (SDS) | DD/ID | Epilepsy | Coarse facial features | Sleep disturbances | Hematologic abnormalities | Recurrent infections | Hypothyroidism | Other | MRI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | c.91G > C | p.Ala31Pro | M | 7 | 4 | + | + | Short 4th and 5th metacarpals. Umbilical hernia, cryptorchidism. | No specific findings | |||||
| 2 | c.104C > G | p.Ser35Cys | M | 16 | 4.8 | + | + | + | + | Macroglossia. High-arched palate, low- set ears. Knee valgus, flat feet. Hyperinsulinemia, hypercholesterolemia, hypertension. Panic attacks. | Agenesis of CC | |||
| 3 | c.104C > G | p.Ser35Cys | F | 14 | 4.6 | + | + | Slightly elevated regulatory T cells; persistent hypergammaglobulinemia (IgA and IgM). Short stature, normal GH but good response on GH therapy. Elevated CK levels. Hypermobility (BS 7/9). Mild DD (learning difficulties). | Periventricular leukomalacia; multiple cysts in lateral ventricle, cavum septum pellucidum | |||||
| 4 | c.239A > G | p.Gln80Arg | F | 27 | 4.9 | ++ | + | + | + | + | Mild hypertelorism, anteverted nares, small jaw, low-set ears. 5th digit clinodactyly. Angiokeratomas, lipogranulomas, cutaneous xanthomas. | Mesial temporal sclerosis, small prepontine cyst | ||
| 5 | c.239A > G | p.Gln80Arg | F | 2.5 | 5.7 | + | + | + | Ventriculo-peritoneal shunt placed at age 27 months. Thrombocytopenia and lympopenia, hypogammaglobulinemia. | Enlarged extra-axial space, ventriculomegaly | ||||
| 6 | c.386 T > A | p.Ile129Asn | M | 2 | 3 | + | + | Mild motor delay | No specific findings | |||||
| 7 | c.405G > C | p.Lys135Asn | M | 3 | 2.9 | + | + | Hypotonia. Prominent forehead. Hydrocephaly (shunted). Umbilical hernia. | DWM. Agenesis of CC. Enlarged extra-axial space, widely separated lateral ventricles. Mild-to- moderate confluent white matter disease. | |||||
| 8 | c.409G > A | p.Asp137Asn | F | 7 | 1.2 | ++ | + | + | Facial dysmorphism | N/E | ||||
| 9 | c.409G > A | p.Asp137Asn | F | 6 | 2.2 | + | + | + | Bulbous nasal tip. Mottled skin. | N/E | ||||
| 10 | c.95_97dup | p. Val32dup | F | 4 | 0.26 | + | + | Ptosis, epicanthal folds. Bulbous nasal tip. Tented upper lip, high-arched palate. Thickened and overfolded helices. Inverted nipples. Erythematous skin, dysplastic toenails. Cyanosis in extremities. Rhabdomyosarcoma. | Periventricular leukomalacia | |||||
| 11 | c.238C > T | p.Gln80 * | M | 18 | −0.9 | +++ | ++ | Asymmetric spastic diplegia, intellectual disability, seizure disorder with behavior disturbances, autism spectrum disorder | Periventricular leukomalacia, mild cerebral and cerebellar degenerative changes | |||||
| 12 | c.364C > T | p.Gln122 * | M | 17 | 1.5 | +++ | ++ | Early onset epilepsy, Lennox–Gastaut syndrome | No specific findings |
Note that several transcripts of RAB5C are known. We have used the canonical transcript NM_004583.3 in the table and text. Head circumference is given as standard deviation scores (SDSs). Macrocephaly is defined as a head circumference SDS > 2 SD. Developmental delay/intellectual disability is graded mild (+), moderate (++) or severe (+++) according to description of the clinician. Abbreviations: BS: Beighton score; CC: corpus callosum; DD/ID: developmental delay/intellectual disability; DWM: Dandy-Walker malformation; HC: head circumference GH: growth hormone; N/E: not evaluated.
Patients with de novo heterozygous missense variants presented with developmental delay, ranging from mild to moderate. Macrocephaly was a consistent feature, present in eight out of nine patients (range 1.2–5.7 standard deviations (SD)). Next to these highly prevalent features, a variety of additional phenotypic features were described. Facial dysmorphism, mostly described as coarse facial features, were present in six out of nine patients (Fig. 1). Other phenotypic features included sleep disturbances (4/9), hypothyroidism (3/9), epilepsy (2/9), hematologic abnormalities (2/9) and cardiac abnormalities (2/9). Several patients showed abnormalities on imaging of the brain (5/7), including periventricular leukomalacia in two, and absence of the corpus callosum in two patients (Table 1, Supplementary Material, Table S1).
Figure 1.

Photographs of patients with RAB5C variants, showing macrocephaly and (increase in) coarse facial features in several patients.
Recurrence was observed for three of the missense variants, with two patients for each of the recurrent variants. Similar macrocephaly (2/3 pairs) and mild-to-moderate developmental delay (2/3 pairs) was observed, while the additional less penetrant phenotypic features were not consistent among each pair.
One patient carrying a duplication of Val32 showed phenotypic overlap with patients with missense variants but did not have macrocephaly.
Two patients were heterozygous for loss of function variants and presented with potentially clinically distinct features from those with missense variants. Macrocephaly was absent in these patients. Instead, they exhibited severe refractory epilepsy, profound intellectual disability with autistic features.
Bioinformatic features of the missense variants
RAB5C is widely expressed in humans (https://gtexportal.org/, release V8) and thus may function in the tissues affected in the patient cohort. To assess pathogenicity of the RAB5C variants, we first evaluated the genetic data. Constraint data from gnomAD (v2.1.1) (18) for RAB5C were consistent with intolerance to loss of function heterozygosity [observed/expected ratios for loss of function variants (o/e) = 0.08 (0.03–0.36), probability of being loss-of-function intolerant (pLI) = 0.94], suggesting haploinsufficiency, while there is marginal intolerance for missense variation (Z score 1.42, o/e 0.67). Combined Annotation Dependent Depletion (CADD) scores of the missense variants in our patients were all above 24.6, where values >20 represent variants that are among the 1% most deleterious (19). CADD scores of patient variants tended to be higher than those of missense variants reported in gnomAD (mean 27.7 ± 2.7 versus 19.1 ± 8.2, p = 0.1). Similarly, REVEL (rare exome variant ensemble learner) scores of missense variants (range 0–1, with higher scores indicating greater likelihood of pathogenicity) were between 0.81 and 0.94 (mean 0.89) (20) (Supplementary Material, Table S3).
Next, we evaluated the amino acid location of the patient variants compared to gnomAD variants in the linear gene sequence (Fig. 2). Most patient missense variants localized to stretches of the gene that were devoid of gnomAD missense variants, which are thought to be largely benign. These ‘desert stretches’ (21) corresponded to the G-motifs, which are involved in nucleotide binding and are highly conserved in all G-protein, Rab and Ras family members (Fig. 2). Variants A31P, V32dup, S35C and Q80R were all located in regions of the gene that were previously found to be constrained coding regions (22). Interestingly, patient #6, with the only variant (I129N) outside of the G-motifs, exhibited a similar but milder clinical phenotype.
Figure 2.
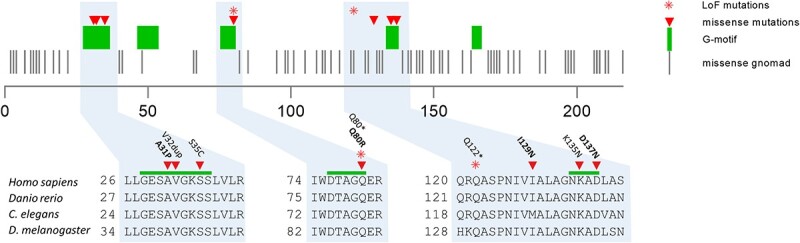
RAB5C is depicted along with known G-motifs (green bars). Missense mutations as retrieved from gnomAD are depicted with gray bars. Rab5c variants found in patients are depicted in triangles (missense) and asterisk (frameshift). Note that most missense mutations localize to a G-motif that is essential in nucleotide binding, except for one patient (I129N) that had a relatively mild phenotype. Variants used for further functional characterization in bold.
Available structural data confirmed that most patient variants affect residues involved in nucleotide binding (Fig. 3A–C). The exception is I129, which is localized to the hydrophobic core of the protein (Fig. 3D).
Figure 3.
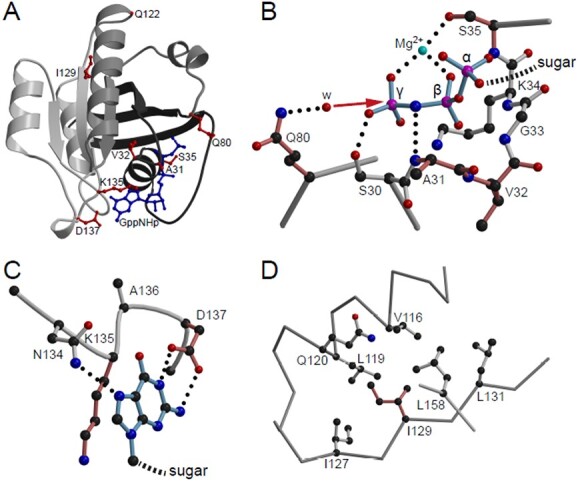
Structural visualization of Rab5c variants. (A) Crystal structure of Rab5c bound to the hydrolysis-resistant GTP analogue GppNHp (pdb-entry 1HUQ). Tints of gray are used to illustrate the incompatibility of the non-sense variants with protein folding; dark gray, N-terminal part till residue 80; light gray C-terminal part from residue 122 onward. GppNHp (blue) and residues affected by point mutations (red) are shown in ball-and-stick representation. (B) Detailed view of the phosphate binding cavity. For clarity, only the three phosphates of the nucleotide are shown and labeled α, β and γ. The direction of the nucleophilic attack of a water molecule (w) to the γ-phosphate in course of the non-GAP catalyzed hydrolysis reaction is indicated by a red arrow. Residues affected by point mutations are colored in a tint of red. Dotted lines, hydrogen bonds. (C) Interaction of the base with the NKxD motif. For clarity of representation, only the base of the nucleotide is shown. (D) Hydrophobic environment of Ile129. Figures were generated by use of the programs molscript (55) and Raster3D (56).
Below we describe in vitro and in vivo functional studies of four of the missense variants. We chose three variants that reside in different nucleotide binding G-motifs (RAB5C p.A31P, p.Q80R and p.D137N), that are predicted to be damaging, and the single missense variant that is not in the nucleotide binding motif (RAB5C p.I129N), as the functional effect of the variant in the hydrophobic core was unclear (Supplementary Material, Fig. S1).
Biochemical characterization of Rab5c variants in vitro
The activation status of Rab proteins is dependent on nucleotide exchange, which, in turn, is influenced by guanine exchange factors (GEFs). The eventual downstream effect is mediated through effector proteins. We hypothesized that missense variants could impact cellular function either through a change in nucleotide exchange, altered responsivity to GEFs or a change in effector protein interaction.
Wild-type recombinant Rab5c protein displays a high nucleotide affinity, as reflected by a slow nucleotide exchange rate, and is very responsive to the GEF Rin1 (Fig. 4A). In contrast, Rab5c variants A31P, Q80R, I129N or D137N exhibited increased nucleotide exchange rates (Fig. 4B–F). The change in nucleotide exchange was different among the variants: the increase in exchange rate of Rab5c Q80R and I129N was mild, that of A31P was pronounced (Fig. 4E), while the nucleotide exchange of D137N was even about one order of magnitude faster than that of A31P (Fig. 4E and F). All variants were still responsive to GEF Rin1, although the GEF effect was attenuated compared to wild type and partially masked by the increased nucleotide exchange rate (Fig. 4A–F).
Figure 4.

Intrinsic and GEF Rin1 catalyzed nucleotide exchange. (A)–(D) Nucleotide exchange rates of 200 nM Rab5c wt and Rab variants in the absence or presence of different concentrations of Rin1 (0–100 nM) as indicated at 20°C. The vertical grey line in (A), (C) and (D) corresponds to the length of the x-axis in (B). (E) Intrinsic nucleotide exchange rates of Rab5C wt and Rab5c variants at 30°C. (F) 1300 nM Rab5c D137N and subsequently 10 mm GPD were added to 200 nM mGDP at 20°C. (G). GTPγS-loaded Rab5c proteins, together with seven different unrelated reference baits (cb01 to cb07), were immobilized and used for precipitation of interacting proteins from HeLa cell lysates, in duplicate (49). The precipitation proteins were identified by mass spectroscopy. The summed peptide intensities of individual identified proteins were logarithmical-transformed to the basis of 10 and are represented by shades of gray. Note, intensities smaller than 104 are not obtained due to technical constraints. The 19 identified known Rab binding proteins (in bold, y-axis) and randomly selected ‘background’ proteins are shown.
While a change in nucleotide exchange rate and GEF-responsivity results in differences in activation status of Rab5c, the eventual function of Rab5c is mediated through effector proteins. Of interest in the context of this study were known Rab5 interacting proteins of which in total 19 were identified (Fig. 4G). Among these proteins are effector proteins as well as GEFs. The effector protein interaction was altered in all four variants, with qualitative differences between different variants. A decrease in effector protein interaction was observed for I129N and D137N. Especially for the D137N variant, many effectors found with wild-type Rab5 or other Rab5c variants were absent, although the interaction with GEFs like Rin1 and RabGEF1 was maintained. In contrast, a general increase in effector protein interaction was observed for A31P and Q80R. For example, OCLR1, OSBPL11 and PI3KR3 were found in particular with Rab5c A31P (Fig. 4G).
Variants affect Rab-5 function in C. elegans
We exploited gene editing and phenotypic analysis including established assays for the endocytic pathway in C. elegans to assess the in vivo functional consequences of four RAB5C missense variants A31P, Q80R, I129N or D137N (corresponding to rab-5 A29P, Q78R, M127N, D135N) (Supplementary Material, Fig. S1) (23,24). The goals of the experimental studies were to determine if each variant was damaging in vivo, to provide information on the genetic mechanism of the de novo dominant presentation of the cases and to ascertain if there were alterations in endocytic pathway function. The results are described below and summarized in Supplementary Material, Table S1A and B.
Control edited worms (A29A, Q78Q, D135D and M127M) were not phenotypically different from the wild-type VC2010 strain that was edited, indicating that the synonymous changes do not alter rab-5 gene function. Furthermore, M127I humanized edited worms were not different from control edited worms or wild type, indicating that both methionine (C. elegans sequence) and isoleucine (human sequence) are interchangeable in RAB-5.
First, we assessed the consequences of the variants when present in the homozygous state. Worms homozygous for A29P, Q78R and D135N were lethal at the first larval (L1) stage, indistinguishable from the L1 lethal phenotype of the rab-5 homozygous deletion allele ok2605 (25). Interestingly, when modeling the mildest patient variant, rab-5 M127N homozygotes were viable and fertile but exhibited cold-sensitive phenotypes including slower growth, reduced adult body length and reduced crawling speed (Supplementary Material, Fig. S3).
Given the dominant presentation in affected individuals, we then examined worms heterozygous for the different variants. We found that rab-5 M127N heterozygotes had reduced crawling speed (Supplementary Material, Fig. S4), while rab-5 D135N heterozygotes displayed cold-sensitive phenotypes including reduced length and reduced crawling speed (Supplementary Material, Fig. S5). In contrast, rab-5 Q78R and A29P heterozygotes did not display temperature sensitivity, adult size or crawling speed phenotypes. Based on the clinical observation of disrupted sleep in patient 4 harboring RAB5C Q80R, we next assessed sleep phenotypes for rab-5 Q78R, as well as for A29P, making use of established assays for sleep in C. elegans (26). Indeed, rab-5 Q78R heterozygotes, but not A29P heterozygotes, displayed a significant decrease in stress-induced sleep (Supplementary Material, Fig. S6A).
rab-5 is not a haploinsufficient gene in C. elegans. rab-5(ok2605) heterozygotes, which produce ~50% of the wild-type protein level, were viable and fertile, indistinguishable from wild-type worms in adult size and crawling speed, in endocytic uptake of secreted soluble GFP (ssGFP) and in the formation of large EEs in coelomocytes (25). Thus, rab-5 D135N, M127N and Q78R heterozygotes displayed phenotypes not observed in the deletion null heterozygote, suggesting that these missense variants result in a change of protein function.
We employed classic gene dosage studies (27) to better understand the genetic nature of the change of function activity for two of the variants, rab-5 D135N and Q78R. If a variant is dominant negative, then adding wild-type copies will suppress the phenotype, while in a hypermorphic/hyperactive variant, adding wild-type copies will enhance the phenotype. The endogenous rab-5 gene resides on chromosome I and we employed a transgenic wild-type single copy of rab-5(+), on chromosome II, which is expressed at the same level as the endogenous rab-5(+) gene (Supplementary Material, Fig. S5) (25). For the rab-5 D135N heterozygous length phenotype, addition of one transgenic copy of rab-5(+) (a total of two wild-type copies) fully suppressed the defect, with no additional change with two transgenic copies (Supplementary Material, Fig. S5B). For the crawling speed phenotype, one transgenic copy of rab-5(+) partially suppressed and two transgenic copies (a total of three wild-type copies) fully suppressed the rab-5 D135N phenotype (Supplementary Material, Fig. S5C). Similarly, the addition of one transgenic copy of rab-5(+) almost fully suppressed the stress-induced sleep phenotype of the Q78R heterozygotes (Supplementary Material, Fig. S6B). The finding that the addition of a wild-type gene copy suppressed the phenotype indicates that the rab-5 D135N and Q78R variants result in a dominant negative effect. By extension, human RAB5C p.D137N and p.Q80R, which were recurrent in our patient cohort (Table 1), are likely dominant negative variants.
Variants affect endocytic pathways in C. elegans
Rab5 functions in endocytosis by mediating heterotypic fusion of cargo-containing nascent endocytic vesicles with EEs and homotypic fusion of EEs to become large EEs, which have a ring-shaped appearance in cross section. We assessed the effect of the four missense variants on endocytic uptake, measuring fluid phase endocytic uptake by coelomocytes, which are scavenger cells in C. elegans that nonspecifically endocytose fluid from the body cavity. In the endocytosis assay at a steady state, ssGFP constitutively secreted by muscle cells (myo-3p::ssGFP) is efficiently cleared from the body cavity by endocytosis into the coelomocytes and subsequently degraded in wild-type (24) and in the control edits. In contrast, worms homozygous for the M127N variant were defective in ssGFP clearance (Fig. 5). Similarly, the D135N heterozygotes had a more than 3-fold reduction in ssGFP fluorescence in the coelomocytes, indicating decreased ssGFP endocytosis (Fig. 6). The rab-5 Q78R and A29P heterozygotes did not display an obvious defect in the steady-state ssGFP endocytosis assay. Employing a more sensitive assay determining the rate of endocytic uptake, following a pulse of ssGFP production, revealed that Q78R heterozygotes, but not A29P heterozygotes, were significantly defective in clearing ssGFP (Supplementary Material, Fig. S7). Taken together, rab-5 M127N, D135N and Q78R variants displayed defects in endocytic uptake.
Figure 5.
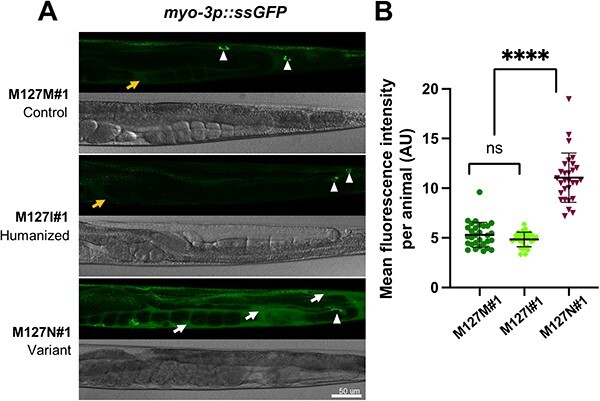
rab-5 M127N homozygotes are defective in endocytic uptake. (A) Steady-state levels of ssGFP (arIs37[myo-3p::ssGFP]) in rab-5 M127M#1 control edit homozygotes (top), rab-5 M127I#1 humanized edit homozygotes (middle) and rab-5 M127N#1 variant homozygotes (bottom). Representative GFP (above) and bright-field (below) images of the posterior half of worms are shown. In M127M#1 control edit and M127I#1 humanized edit homozygotes, there is rapid endocytic uptake of ssGFP from the body cavity (weak signal, orange arrow) into coelomocytes (white arrowheads). In M127N#1 variant homozygotes, ssGFP accumulates in the body cavity (strong signal, white arrows) with limited uptake in coelomocytes (white arrowhead), although this is difficult to visualize with the large amount of ssGFP in the surrounding body cavity. Scale bar, 50 μm. Full genotypes of strains are listed in Supplementary Material, Table S2. (B) Quantification of mean GFP fluorescence intensity per animal, in arbitrary units (AU) (assessed with ImageJ) for rab-5 M127M#1 control edit homozygotes (dark green), rab-5 M127I#1 humanized edit homozygotes (light green) and rab-5 M127N#1 variant homozygotes (dark red). Three independent biological replicates were combined for each genotype. n = 10 per condition. Scatter plots indicate the median with 95% confidence interval. Differences between groups were determined using ordinary one-way ANOVA followed by a Dunnett multiple-comparisons test. ns, not significant; ****P < 0.0001.
Figure 6.
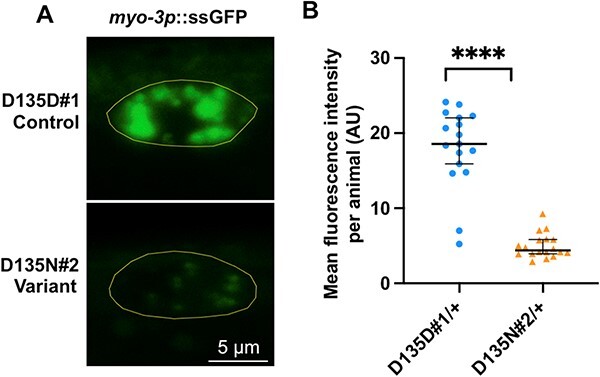
rab-5 D135N heterozygotes are defective in endocytic uptake. (A) Confocal images showing the steady-state ssGFP level in coelomocytes (yellow outline) of a rab-5 D135D#1 control edit heterozygote (top) and a D135N#2 variant edit heterozygote (bottom). Scale bar, 5 μm. All genotypes were generated by crosses with wild-type males; the full genotypes of the parental strains are listed in Supplementary Material, Table S2. (B) Quantification of the mean GFP fluorescent intensity from rab-5 D135D#1 (circles) (n = 17) and D135N#2 (triangles) (n = 18) heterozygous coelomocytes. The scatter plot shows the mean and standard deviation. Differences between groups were determined by Student’s t-test. ****P < 0.0001.
For the rab-5 M127N variant, which showed a strong endocytic defect in the homozygous state, we also assessed if there was an effect on EE fusion. We visualized the size of EE in coelomocytes employing the 2xFYVE::GFP marker, which binds to PI (3) P-modified lipids on EE (28,29). The coelomocytes of rab-5 M127M control edit and rab-5 M127I humanized edit worms contained EE of different sizes, from small 2xFYVE::GFP puncta to numerous large, ring-like EE viewed in cross section (Supplementary Material, Fig. S8), consistent with continuous endocytic trafficking and increase in size from heterotypic to homotypic EE fusion events. By contrast, in coelomocytes of M127N homozygous worms, there was a 5-fold reduction in the formation of large 2xFYVE::GFP ring-shaped EE and a more than 10-fold increase in the number, as well as an increase in staining intensity, of small and medium sized puncta. The failure to form large ring-like EE and the increase in number of small puncta suggest that the M127N variant results in a defect in EE fusion.
In the heterozygous state, rab-5 Q78R, a change from a polar to a positively charged amino acid, resulted in a small, but significant defect in endocytic uptake. In contrast, a different missense change at the corresponding residue, RAB5A p.Q79L in humans (polar to hydrophobic amino acid), resulted in constitutive activation and increased endocytosis (30). A C. elegans rab-5 Q78L overexpression transgenic model recapitulates the constitutive activation phenotype and resulted in the formation of massively enlarged RAB-7 ring-like late endosomes (31). To test whether rab-5 Q78R variant heterozygotes also form enlarged RAB-7 ring-like late endosomes, we examined late endosomes using the GFP::RAB-7 intestinal marker. We also examined rab-5 A29P heterozygotes as we had not thus far observed an endocytic pathway phenotype with this variant. For rab-5 Q78R, as well as A29P, variant heterozygotes did not display enlarged GFP::RAB-7 ring-like late endosomes (Fig. 7), indicating that the variants behave differently from the constitutively active rab-5 Q78L. Instead, we found that the number of GFP::RAB-7 ring-like structures differed between the respective variants and the control edits: Q78R heterozygote intestines contained significantly fewer RAB-7 ring-like structures compared to the control Q78Q, while the A29P heterozygote intestines contained significantly more RAB-7 ring-like structures compared to A29A (Fig. 7). Currently, it is unclear how to interpret changes in number, rather than size, of RAB-7 late endosomes. Nevertheless, the RAB-7::GFP late endosome number phenotype indicates that the rab-5 A29P variant disrupts endocytic pathway function.
Figure 7.
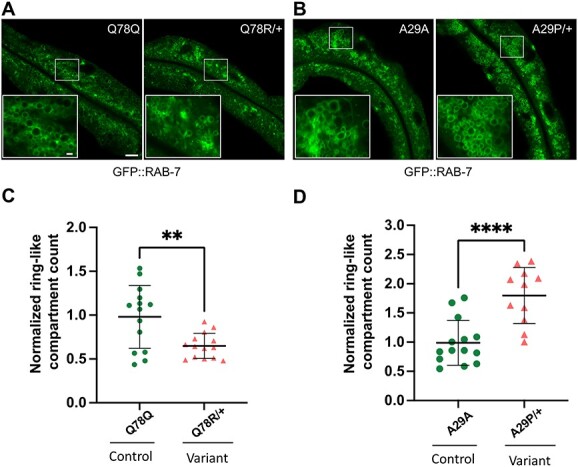
rab-5 Q78R and A29P heterozygotes contain altered numbers of normal size GFP::RAB-7 late endosomes. Assessment of late endosome formation with marker GFP::RAB-7 in Q78R and A29P variant heterozygotes. (A) and (B) Low-magnification GFP::RAB-7 fluorescent images of a middle plane from the worm intestine, with the lumen running along the length and insets (lower left) showing high-magnification confocal views of late endosome ring-like structures, in cross section. Images from (A) the Q78Q control edit (left) and Q78R variant edit (right) heterozygotes and (B) the A29A control edit (left) and A29P variant edit (right) heterozygotes. (C) and (D) Scatter plot of the normalized number of ring-like compartments viewed in cross section for (C) Q78Q control edit (green circles) (n = 15) and Q78R variant edit (orange triangles) (n = 14) heterozygotes and (D) A29A control edit (green circles) (n = 14) and A29P variant edit (orange triangles) (n = 11) heterozygotes. The scatter plot shows the mean and standard deviation. Differences between groups were determined by Student’s t-test. **P < 0.01, and ***P < 0.001. Whole image scale bar, 10 μm. The panel at the bottom left inset shows a zoomed-in region represented by the white rectangle in the main image, inset scale bar, 1 μm.
Variants affect zebrafish development in a dose-dependent manner
We used a zebrafish model to evaluate whether the Rab5c variants were damaging in a vertebrate model. Zebrafish embryos in which human RAB5C variants were overexpressed exhibited a disturbed development, varying from subtle changes in yolk extension to severe shortening of anterior–posterior axis, caudal edema and tissue necrosis (Fig. 8A). The edema formation seen (Fig. 8A, class III and IV) points to disturbed establishment of vasculature. The spectrum of severity depended on the mRNA dose as well as the variant injected: the phenotype of zebrafish injected with the previously studied S35N variant (with a predicted dominant negative effect (32)) and Q80R variant showed more severely disturbed development even at the lowest dose used as compared to WT (Fig. 8B). Conversely, the dose needed to induce abnormalities using variants A31P and I129N was higher.
Figure 8.
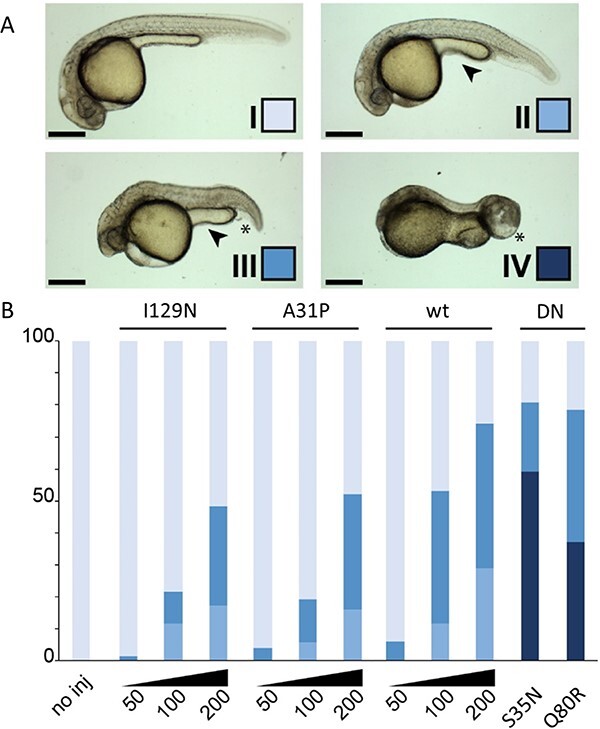
Rab5c variants and dose disturb zebrafish development. Expression of the Rab5c variants was achieved by mRNA microinjection in zebrafish embryos. The different classes and color scheme (A) are defined on developmental parameters such as cephalic development, anterior–posterior axis, caudal edema formation and yolk extension shape. Tissue necrosis and abnormal tail growth are also observed. Class I depicts normal development. Class II displays mild developmental defects observable in the reduced size of the eyes and slight shortening of the AP axis. The yolk extension is short and thick (arrowhead). In class III, the AP axis is visibly shortened, the yolk extension is further shortened and caudal edema may be observed (asterisk). These phenotypes are further exacerbated in class IV; here, head necrosis is also visible. (B) Quantification of the phenotypical classification as described in (A) for the tested Rab5c variants. The y-axis is the percentage of embryos showing each of the four classes. Note the dose–response effect obtained with wt, A31P and I129N variants with increasing mRNA quantities (from 50 pg/embryo to 200 pg/embryo). Injections with S35N and Q80R were carried out at 50 pg/embryo. Scale bars: 100 μm. DN: dominant negative.
Discussion
The Ras-related protein Rab5c is a small GTPase that was first discovered as a regulator of the endocytic pathway (33,34). We provide evidence that RAB5C missense variants can cause human disease based on combined bioinformatic, in vitro biochemical, in vivo model organism and patient data. Individuals harboring missense variants exhibited macrocephaly and varying degrees of developmental delay. In vitro and in vivo experimental studies performed for four of the missense variants demonstrated that variants changed the proteins biochemical characteristics, were damaging in model organisms and caused disruption of the endocytic pathway. This supports that the de novo heterozygous RAB5C missense variants were likely responsible for the more penetrant phenotypes observed in the patients.
Several additional, less penetrant phenotypic characteristics (six phenotypes from 6/9 to 2/9 patients) were noted in individuals with missense variants. The broad tissue expression of RAB5C and the fact that the individual phenotypes were observed in patients with different variants suggests that RAB5C plays a causal role for at least some of the less penetrant phenotypes. Factors such as segregating modifiers, environmental and/or stochastic events may contribute to the incomplete penetrance.
Two patients with truncating variants displayed therapy resistant epilepsy and profound developmental delay, but not macrocephaly, suggesting that RAB5C loss of function and missense variants may contribute to distinct clinical conditions. However, given the missense variant patient phenotypic heterogeneity and the lack of biological material for molecular evaluation of the consequences of the truncating variants, no conclusions can be drawn on potential phenotypic differences between the dominant missense variants and loss of function variants. Expanding the cohort of patients with loss of function and with missense variants will be important for informing on the range of phenotypes that can be attributed to disruption of RAB5C function.
Variants disrupt Rab5c GTPase activity and effector interactions, in diverse ways
The Rab GTPases are a family of proteins that are involved in regulation of intracellular membrane transport. The proteins switch between a GTP-bound ‘active’ and GDP-bound ‘inactive’ state. The intracellular concentration of GTP exceeds that of GDP, favoring a GTP- bound activated state. Endogenous GTPase activity results in a GDP-bound state and inactivation. The cycle completes upon dissociation of GDP and binding of a new GTP. The eventual effect depends on the interaction of the activated protein with membrane-bound effector proteins (35). Interaction with effector proteins is possible in the GTP-bound active conformation. Influencing the activation/inactivation cycle are guanine exchange factors (GEFs) that stimulate nucleotide exchange and thus favor activation; GTPase activating proteins (GAPs) that stimulate GTP hydrolysis, favoring inactivation; and GDP dissociation inhibitors that keep Rab in its GDP-bound inactive state (1,5,36). This extensive regulation of Rab GTPases also implies that its function can be disturbed in multiple ways.
Our in vitro studies indicate that several of the factors that determine the activation/inactivation balance are disturbed in RAB5C variants. For most variants, especially those with missense changes in nucleotide binding motifs, nucleotide exchange was increased. A slight increase in nucleotide exchange would lead to more GTP-bound active Rab5c. However, a strong decrease in nucleotide affinity, as was observed for Rab5c D137N, would results in Rab5c that is mainly nucleotide-free. Nucleotide-free Rab would bind to GEFs with high affinity and thereby trap GEFs in an unproductive complex. In this way, the variant would have a dominant negative effect, as it would prevent the GEFs from acting on wild-type Rab5c. As GEFs for Rab5a are to some extent promiscuous and act on related Rab proteins as well, those pathways might also be affected. Furthermore, studies of effector protein interaction showed that the analyzed variants influenced effector protein interaction in diverse ways. Thus, although the data clearly demonstrate that the biochemical properties of the variants are altered, it remains difficult to predict which consequences of these alterations would dominate in context of cellular signaling. We therefore studied the cellular effects of variants in model organisms.
Variants are damaging in vivo, in diverse ways
Our in vivo functional studies modeling RAB5C variants p.A31P, p.Q80R, p.I129N and p.D137N, in C. elegans demonstrated that the missense changes were damaging, but with diverse characteristics. Homozygous A29P, Q78R and D135N worms were L1 larval lethal, while the consequences of M127N homozygosity was less detrimental, resulting in cold-sensitivity, slow growth, shorter adult length and defective in motility. The M127N variant retained a significant level of wild-type activity, consistent with the milder clinical phenotype of the patient with the p.I129N variant (Table 1). All four tested missense variants, unlike the rab-5 gene deletion allele, showed dominant phenotypes in C. elegans, consistent with the dominant presentation observed in our patients. Gene dosage studies demonstrated that Q78R and D135N variants could be outcompeted by wild-type RAB-5, indicating that the rab-5 variants likely have a dominant negative effect. Previous studies found that a different missense change, rab-5 Q78L and RAB5A Q79L, when overexpressed, result in a constitutively active variant (30). From our C. elegans experiments, rab-5 Q78R appears to act as a dominant negative variant. Also, in the zebrafish experiments, expression of the Q80R variant resulted in developmental defects comparable to the known dominant negative Rab5c S35N variant (32).
Experiments directed at examining functional effects of the variants on the endocytic pathway revealed that all variants altered the endocytic pathway, but again in diverse ways. D135N, M127N and Q78R resulted in defects in endocytic uptake, while A29P increased the steady-state number of normal size RAB-7 late endosomes.
The expression of human Rab5c variants in zebrafish resulted in a clear disturbance of development for the A31P and I129N variants. Previous studies found that Rab5c influences vascular development in zebrafish, through regulation of VEGFR2 (9), Notch and AKT (10) signaling. The edema formation seen in zebrafish embryos injected with human Rab5c variants is likely related to disturbed vascular development. Heng et al. (10) evaluated the effect of Rab5c modulation on endocytic trafficking in hematopoietic stem cells in zebrafish. They found that both inhibition of Rab5c, expression of dominant negative as well as constitutively active variants of Rab5c all resulted in disturbed hematopoietic stem cell development. Strikingly, in our studies, overexpression of WT human RAB5C in zebrafish also resulted in a dose-dependent disruption of development. This underscores the concept that the effects of Rab5c do not depend on its mere presence but rather on the balance in quantity or activation status.
While these studies illustrated the detrimental effects of altered Rab5c expression, our zebrafish model did not mimic the human phenotype in terms of macrocephaly. Obviously, injecting human RAB5C mRNA in zebrafish embryos does not take into account the intricate spatiotemporal regulation that characterizes most Rab proteins (1). More sophisticated genetic manipulation might help understand the exact way in which genetic variations result in the observed phenotype.
Together, the experimental studies demonstrate that the four missense variants interfere with properly controlled Rab5c function, but the biochemical, organismal and endocytic pathway phenotypes can differ between variants (Supplementary Material, Fig. S9, Table S1B). Correspondingly, there is phenotypic heterogeneity between patients with different variants and even with the same variant. Comparisons of the ‘strength’ of the experimental phenotypes and patient phenotypes are complicated by this heterogeneity. For the penetrant missense phenotypes macrocephaly and developmental delay, the strength of phenotypic defect is not consistent among variants across phenotypic features. For example, variant D137N is strongest for developmental delay but weakest for macrocephaly/head circumference. Similarly, in the experimental studies, D137N/D135N tended to have the strongest defects but was wild type for the stress-induced sleep phenotype in C. elegans. The heterogeneity suggests that the requirement for RAB5C function and/or the variant effects can differ between cell types. To correlate phenotypic differences unambiguously with the different biochemical defects in the Rab5c variants more patients with the same variants would be required.
Consequences of RAB5 variants differ between paralogs and transcripts
In humans, there are three paralogs, RAB5A, RAB5B and RAB5C. The underrepresentation of loss of function variants (gnomAD v2.1.1, probability of being loss-of-function intolerant (pLI) =0.94, o/e = 0.08) suggests that RAB5C is a haploinsufficient gene. In contrast, gene constraint data suggest that RAB5A and RAB5B are not haploinsufficient genes. These differences in intolerance to loss-of-function are consistent with RAB5C having functions and expression level requirements that are unique among the RAB5 paralogs. Therefore, RAB5A and RAB5B are not able to compensate for disruptions of at least some RAB5C functions for the missense and loss of function variants. Recently, the RAB5B variant p.D136H has been implicated in a patient without macrocephaly, but with interstitial lung disease due to surfactant dysfunction disorder (25). The absence of interstitial lung disease in the RAB5C patient cohort and the absence of macrocephaly in the RAB5B patient support the concept that human RAB5 paralogs can have specific functions and may cause distinct pathophysiology when mutated. The absence of haploinsufficiency for C. elegans rab-5 (in contrast to human RAB5C) is not unexpected given that different species titrate gene expression differently and that haploinsufficiency is thought to be rare in C. elegans (37).
We are aware of one patient with a nonsense variant within an alternative RAB5C transcript (NM_001252039.2), which has low expression according to the Genotype-Tissue Expression (GTEx) database. This patient had a normal cognitive development and no dysmorphism, but presented with longstanding T-cell lymphocytosis and periodic fever.
Consequences of disturbed endocytosis
Rab5 is involved in regulation of early endocytosis (34). Through endocytosis, membrane-associated receptors and signaling molecules are targeted to other cellular compartments or for degradation, effectively providing a means of signal transduction regulation (38). Thus, Rab5c has previously been found to be important in hematopoietic stem cell development through regulation of Notch signaling (10). Interestingly, the two patients with the Q80R variant showed hematologic abnormalities, especially neutropenia.
Cell surface accumulation of the EGF receptor is also regulated by Rab5-mediated endocytosis and recycling (12,39–41). One of the patients in our cohort developed a rhabdomyosarcoma. Although this event in a single patient does not prove a relation with the Rab5c variant, neither can we exclude the possibility that disturbed regulation of endocytosis may induce a higher risk of cancer. In support, previous studies have shown a role for Rab5c in EGF-induced invasive behavior of breast cancer cells (8) and implicated Rab5-mediated processes in tumor progression (42,43).
Rab GTPases and disease
A growing number of Rab GTPases have been linked to neuronal development and neurologic disease (5,35). This may help provide support for pathogenicity of variants and provide possibilities to search for shared pathophysiological mechanisms. For example, Lamers et al. (2) describe mutations in the GTPase domain of RAB11B that resulted in a clinical phenotype in patients with developmental delay, microcephaly and epilepsy. The RAB5C variant of patient #10 in our study affects the same amino acid as the variant described in three patients with RAB11B mutations. Although this supports pathogenicity of this variant, the eventual effect is difficult to predict, as RAB11B and RAB5C have a role in different cellular compartments.
RAB39B has also been associated with developmental delay and macrocephaly (4), the latter explained by the interaction between Rab39b and the mTOR pathway (44). It is tempting to speculate that the known interaction of Rab5 with PI3-kinase subunits (45) might similarly lead to increased mTOR pathway activity and, consequently, to macrocephaly in our patients. Several Rab-interacting proteins have also been associated with neurodevelopmental syndromes. GDI1, encoding GDP dissociation inhibitor-1, is associated with X-linked mental retardation (46), RAB18, RAB3GAP1 and RAB3GAP2 mutations cause Warburg micro syndrome (47). Similarly, we would predict that changes in proteins implicated in Rab5c function, for example, GEFs or effector proteins, would also result in comparable phenotypes. Interestingly, macrocephaly and coarse facial features, but not developmental delay, have been reported in mutations in RIN2, one of the Rab5-interacting proteins (48). This may provide a first step to more precisely delineate the biological mechanisms that lead to the different phenotypic characteristics.
Conclusion
Taken together, we found de novo missense variants in RAB5C in nine patients with variable degrees of developmental delay and macrocephaly. In vitro and in vivo functional characterization suggests that these variants disturb Rab5c function. Although our studies do not permit a clear delineation of the pathophysiological mechanism that led to clinical characteristics of our patients, we do demonstrate that variants in RAB5C disrupt cellular processes that are essential for normal development and demonstrate the power of multimodal approaches to understand human gene variants of unknown significance.
Materials and Methods
Editorial policies and ethical considerations
Informed consent was obtained from each participant or legal representative for diagnostic procedures that led to the discovery of RAB5C variants and inclusion in this study.
Patients
Individual patients with variants in RAB5C were identified through diagnostic procedures. Using GeneMatcher (17), we identified a total of 12 patients with RAB5C variants and compiled the clinical, biochemical and imaging data that were available.
We started functional studies to further characterize RAB5C variants. While these studies were ongoing, further patients were identified. All patients are included in this paper, but we have not functionally characterized all RAB5C variants. We included variants from different G-motifs (see below and Fig. 2 and Supplementary Material, Fig. S1) in the functional studies.
Genotype analyses
Exome analyses was performed as part of routine diagnostic assessment using the regular procedures in different centers. We retrieved known RAB5C variants from gnomAD (gnomad.broadinstitute.org, v2.1.1 (18)). CADD and REVEL scores were obtained for all missense variants (Supplementary Material, Table S3) (19,20).
RAB5C has several overlapping transcripts and protein isoforms. We used the canonical transcript (RefSeq NM_004583.3) and protein isoform (NP_004574.2, Uniprot P51148–1).
In vitro studies
Nucleotide exchange of Rab5c and variants A31P, Q80R, I129N and D137N was analyzed as described previously for the small GTPase Ras (49). Briefly, recombinant Rab5c was expressed as GST-fusion proteins in E. coli strain CK600K and purified via affinity chromatography. Variants were introduced in the plasmid DNA by site direct mutagenesis and sequence verified. The GST tag was cleaved off with human rhinovirus type 14 3C protease and removed from the Rab5c proteins by size exclusion chromatography. Amino acid residues 320–540 of Rin1 (Mus musculus) were expressed as GST-fusion from pGEX4T3 in E. coli strain CK600K essentially as described for Epac (50).
Nucleotide affinity and nucleotide exchange is important for proper signaling by small GTPases and can be analyzed in a fluorescence-based assay. Recombinant Rab5c was loaded with the fluorescent GDP analogue mGDP. The fluorescence intensity of this analogue depends on the local environment of the fluorophore and is approximately twice as high if bound to the protein than if it is free in solution. In the presence of excess unlabeled nucleotides, fluorescently labeled nucleotides that dissociate from Rab are replaced with unlabeled nucleotides. Therefore, nucleotide release can be monitored as a decay in fluorescence. Nucleotide exchange was monitored in the presence of different concentrations of the GEF Rin1 at temperatures as indicated in buffer containing 50 mm Tris HCl pH 7,5, 50 mm NaCl, 5 mm MgCl2, 5 mM dithiothreitol and 5% glycerol. The D137N variant was prone to precipitation, possibly because the reduced nucleotide affinity of this variant caused a loss of nucleotide during the purification resulting in a nucleotide free state. Consequently, it was not possible to subject Rab5c D137N to nucleotide loading procedures. As an alternative approach, Rab5c D137N was added to mGDP, followed by addition of excess GDP.
Downstream signaling of Rab5c is mediated through effector proteins. Protein interactions were analyzed as described previously for the GTPase Ras (49). Rab5c proteins were expressed with an avi-tag in bacteria, purified, loaded with the hydrolysis resistant GTP analogue GTPγS and immobilized on streptavidin beads. These beads were incubated with HeLa cell lysates to precipitate Rab5c-interacting proteins. To discriminate against unspecific protein binding seven unrelated proteins were immobilized in parallel as control. The precipitated proteins were subjected for identification by mass spectroscopy, and the summed intensities of the identified peptides were used as a measure for the amount of precipitated protein.
Model organisms
C. elegans
Strains and culture conditions
C. elegans strains were cultured on nematode growth medium agar plates at 20°C, with E. coli strain OP50 as the food source. Strains containing variants rab-5 D135N and M127N were cold-sensitive. Therefore, D135N strains, and corresponding controls, were maintained at 25°C, while M127N strains and corresponding controls, were maintained at 22°C. C. elegans strains used in this study are listed in Supplementary Material, Table S2.
CRISPR/Cas9 genome editing
C. elegans contains a single RAB5 ortholog, rab-5, while humans have three paralogs, RAB5A, RAB5B and RAB5C. Human Rab5c and C. elegans RAB-5 proteins are largely identical in amino acid sequence (75% identical and 83% conserved), except at the very N- and C-termini (Supplementary Material, Fig. S2). To model the RAB5C missense changes p.D137N, p.Q80R and p.A31P, each variant was knocked-in to the corresponding residue of the endogenous rab-5 gene (D135N, Q78R and A29P, respectively, Supplementary Material, Fig. S1) of the VC2010 wild-type strain, using the dpy-10 co-conversion method of CRISPR/Cas9 gene editing (51) as described in Huang et al. (25). The editing strategy employed synonymous changes to generate a restriction enzyme cleavage site used for allele genotyping and to block Cas9 re-cleavage following homology-directed repair. Therefore, as controls, we generated lines that contained only the synonymous and restriction enzyme changes, but not the missense variants. For RAB5C p.I129N, rab-5 contains a conservative substitution (M127) at the corresponding position. Therefore, in addition to the variant, we generated a humanized edit, M127I, to assess whether isoleucine would be tolerated at residue 127 in rab-5. Independent lines were obtained from different injected P0 worms. The full rab-5 gene was Sanger-sequenced and confirmed to have all designed variants, but no extraneous changes in the gene. All strains were backcrossed with VC2010 at least twice to remove unlinked random variation introduced during editing or homozygousing. The guide RNAs, repair templates and PCR primers used for genotyping are found in Supplementary Material, Table S3.
Phenotypic assays
Phenotypes were scored 24 h after the mid-L4 stage. The exception was rab-5 D135N/+ adults, which were sickly and sterile; phenotypes were scored at the L4 stage where worms appeared healthier. To propagate rab-5 D135N/+, we employed a transgenic wild-type extra copy of rab-5, on chromosome II (25), that suppresses the sickly and sterile phenotypes. To generate D135N heterozygotes for phenotypic analysis, wild-type males were mated to a strain with rab-5 D135N variant or control edits over balancer tmC18 (mCherry, chromosome I) and the transgenic extra copy of rab-5(+) over the balancer mnC1 (GFP, chromosome II) and green, non-red cross progeny analyzed. Viability, body length and locomotion (using the Wormlab system, MBF Biosciences) was assessed as previously described (25,52). Stress and developmental sleep assays were performed as described in Churgin et al. (26).
Endocytic pathway analysis
Measurement of endocytic uptake of constitutive muscle synthesized ssGFP from the body cavity by the coelomocytes was as previously described (25). For the rab-5 D135D control heterozygotes, there were low and variable levels of ssGFP in the body cavity, possibly because the ssGFP transgene was heterozygous and the L4 worms examined were grown at 25°C. Therefore, accumulation of ssGFP within coelomocytes was measured at steady state for the rab-5 D135N variant and control heterozygotes. For variants Q78R and A29P, where no defect was observed at steady state, a more sensitive assay of the rate of ssGFP uptake by the coelomocytes during the chase period, following a heat shock pulse of synthesis (hsp::ssGFP), was employed, as described in Fares and Greenwald (53). Assessment of early endosome fusion in coelomocytes, using the 2xFYVE::GFP marker, for rab-5 M127N variant and control edits was as previously described (25). The size and number of late endosomes was measured in rab-5 Q78R, A29P and respective control edit strains, employing the late endosome marker GFP::RAB-7.
Imaging
For the ssGFP imaging, worms were placed in 5 μl of 100 mm NaN3 in PBS, in a 35 mm cover-glass bottom dish (MatTek). Worms with the 2xFYVE::GFP early endosome marker were immobilized with levamisole and transferred to a 35 mm cover-glass bottom dish and covered with a 12 mm circular coverslip and then a 25 mm square coverslip. Confocal images were taken with a Lecia SP8X tandem scanning confocal microscope with a white light laser using either a 40× 1.3 NA oil PlanApo objective over ≥20 z-planes and a pinhole size of 1.00 (Leica Microsystems). Images for ssGFP and 2xFYVE::GFP were displayed as maximum intensity projections. Images for Phsp::ssGFP were displayed as single planes. Images were rendered and analyzed using LASX (Leica Microsystems) and ImageJ software.
For analysis of late endosomes with GFP::RAB-7, worms were imaged with a Zeiss LSM800 point scanning confocal microscope equipped with an Airyscan module, using the 488 nm laser line. A 63× 1.4 NA objective was used for image acquisition. Data were captured as a single image in the middle plane of the C. elegans intestine using ZEISS ZEN 2.3 imaging software. By selecting the middle plane of the worm intestine, the apical membranes and the basolateral membranes are visible. The auto-fluorescence from the C. elegans intestine was removed by the Linear Unmixing function in ZEN. The images were exported as TIF files for analysis in Metamorph version 7.7 software. For each animal ring-like compartments where manually counted and graphed using GraphPad Prism 7.04 software.
Statistics
Statistical analyses were performed using GraphPad Prism (Version 9). Student’s t-test was used to compare the difference between two groups. One-way ANOVA followed by Tukey’s HSD was used to compare the difference among more than two groups, followed by a Dunnett multiple-comparisons test. Dot plots indicate the median, and whiskers extend to the 5th and 95th percentile confident interval. The F test was used to compare the slopes generated by a linear regression model, which represent the ssGFP signal changes per unit time. A P-value <0.05 was considered statistically significant.
Zebrafish
Fish lines and husbandry
Zebrafish (Danio rerio) of the Tübingen longfin strain were kept in standard laboratory conditions (54). Animal experiments were approved by the Animal Experimentation Committee of the Royal Netherlands Academy of Arts and Sciences.
Expression assay in zebrafish embryos
Template human cDNA was used to generate novel cDNA encoding the following RAB5C variants: wild type, S35N (32), A31P, Q80R and I129N. After cloning into pCS2GW by Gateway cloning (Life Technologies), the resulting template constructs were linearized with NotI-HF (New England BioLabs) and used for in vitro synthesis of capped mRNA with MESSAGE mMACHINE SP6 Ultra kit (Life Technologies). Microinjections in 1-cell-stage embryos were carried out with 50, 100 or 200 pg mRNA per embryo. After microinjection, embryos were kept at 28.5°C in E3 medium and development was assessed at ~28 h post-fertilization.
Imaging
Live phenotypical assessment of 28 hpf zebrafish embryos was carried out on a Zeiss StemiSV6 stereomicroscope (Carl Zeiss AG, Oberkochen, Germany). Pictures were captured with a Leica DFC420C digital microscope camera (Leica Microsystems, Wetzlar, Germany) mounted on a Zeiss Axioplan brightfield microscope (Carl Zeiss AG).
Consortia (Members of the Undiagnosed Diseases Network)
Maria T. Acosta, Margaret Adam, David R. Adams, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Guney Bademci, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Hugo Bellen, Jimmy Bennet, Beverly Berg-Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Thomas Cassini, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D’Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Matthew Deardorff, Esteban C. Dell’Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Elizabeth L. Fieg, Laurie C. Findley, Paul G. Fisher, Brent L. Fogel, Irman Forghani, William A. Gahl, Ian Glass, Bernadette Gochuico, Rena A. Godfrey, Katie Golden-Grant, Madison P. Goldrich, Alana Grajewski, Irma Gutierrez, Don Hadley, Sihoun Hahn, Rizwan Hamid, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Wendy Introne, Rosario Isasi, Kosuke Izumi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Orpa Jean-Marie, Vaidehi Jobanputra, Lefkothea Karaviti, Jennifer Kennedy, Shamika Ketkar, Dana Kiley, Gonench Kilich, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Seema R. Lalani, Byron Lam, Christina Lam, Grace L. LaMoure, Brendan C. Lanpher, Ian R. Lanza, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Roy Levitt, Richard A. Lewis, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Rachel Mahoney, Bryan C. Mak, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M. Moretti, John J. Mulvihill, Mariko Nakano-Okuno, Stan F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen C. Pak, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Barbara N. Pusey, Aaron Quinlan, Wendy Raskind, Archana N. Raja, Deepak A. Rao, Anna Raper, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Mario Saporta, C. Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Vandana Shashi, Jimann Shin, Rebecca Signer, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica-Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Jennifer A. Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Amelia L. M. Tan, Queenie K.-G. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Brianna M. Tucker, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Stephanie Wallace, Nicole M. Walley, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Monika Weisz-Hubshman, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Jeremy D. Woods, Kim Worley, Changrui Xiao, Shinya Yamamoto, John Yang, Diane B. Zastrow, Zhe Zhang, Chunli Zhao, Stephan Zuchner.
Conflict of Interest statement. The authors declare no competing interests.
Supplementary Material
Contributor Information
Klaas Koop, Department of Pediatrics, University Medical Center Utrecht, Utrecht, 3584 EA, The Netherlands.
Weimin Yuan, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Federico Tessadori, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht, 3584 CT, The Netherlands.
Wilmer R Rodriguez-Polanco, Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, Piscataway, NJ, 08854, USA.
Jeremy Grubbs, Department of Neurology and the Chronobiology and Sleep Institute, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Bo Zhang, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Matt Osmond, Children's Hospital of Eastern Ontario Research Institute, University of Ottawa, Ottawa, Ontario, K1H 8L1, Canada.
Gail Graham, Children's Hospital of Eastern Ontario Research Institute, University of Ottawa, Ottawa, Ontario, K1H 8L1, Canada.
Sarah Sawyer, Department of Pediatrics, Children's Hospital of Eastern Ontario, University of Ottawa, Ottawa, Ontario, K1H 8L1, Canada.
Erin Conboy, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, 46202, USA.
Francesco Vetrini, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, 46202, USA.
Kayla Treat, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, 46202, USA.
Rafal Płoski, Department of Medical Genetics, Medical University of Warsaw, Warsaw, 02-106, Poland.
Victor Murcia Pienkowski, Department of Medical Genetics, Medical University of Warsaw, Warsaw, 02-106, Poland; Marseille Medical Genetics U1251, Aix Marseille University, Marseille, 13005, France.
Anna Kłosowska, Department of Pediatrics, Hematology and Oncology, Medical University of Gdańsk, Gdańsk, 80-210, Poland.
Elizabeth Fieg, Brigham and Women's Hospital, Boston, MA, 02115, USA; Harvard Medical School, Boston, MA, 02115, USA.
Joel Krier, Brigham and Women's Hospital, Boston, MA, 02115, USA; Harvard Medical School, Boston, MA, 02115, USA.
Coralie Mallebranche, Unité d'Onco-Hémato-Immunologie pédiatrique, CHU d’Angers, Angers, 49933, France.
Ziegler Alban, Service de génétique, CHU d’Angers, Angers, 49933, France.
Kimberly A Aldinger, Center for Integrative Brain Research, Seattle Children's Research Institute, Seattle, WA, 98195, USA; Division of Genetic Medicine, Department of Pediatrics, University of Washington, Seattle, WA, 98195, USA.
Deborah Ritter, Department of Pediatrics, Oncology Section, Baylor College of Medicine, Houston, TX, 77030, USA.
Ellen Macnamara, Undiagnosed Diseases Program Translational Laboratory, NHGRI, National Institutes of Health, Bethesda, MD, 20892, USA.
Bonnie Sullivan, Division of Clinical Genetics, Department of Pediatrics, Children's Mercy-Kansas City, Kansas City, MO, 64108, USA.
John Herriges, Department of Pathology and Laboratory Medicine, Children's Mercy-Kansas City, Kansas City, MO, 64108, USA.
Joseph T Alaimo, Department of Pathology and Laboratory Medicine, Children's Mercy-Kansas City, Kansas City, MO, 64108, USA.
Catherine Helbig, The Epilepsy Neurogenetics Initiative, Division of Neurology, Department of Pediatrics, The Children's Hospital of Philadelphia, Philadelphia, PA, 19104, USA.
Colin A Ellis, Department of Neurology, University of Pennsylvania Perelman School of Medicine, Philadelphia PA, 19104, USA.
Clare van Eyk, Robinson Research Institute, Faculty of Health and Medical Sciences, University of Adelaide, Adelaide, SA, 5006, Australia.
Jozef Gecz, Robinson Research Institute, Faculty of Health and Medical Sciences, University of Adelaide, Adelaide, SA, 5006, Australia.
Daniel Farrugia, Haematology, Mater Dei Hospital, Msida, MSD2090, Malta.
Ikeoluwa Osei-Owusu, Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, MA, 02142, USA.
Lesley Adès, Department of Clinical Genetics, The Children’s Hospital at Westmead Clinical School, Faculty of Medicine and Health, University of Sydney, Sydney, 2145, Australia.
Marie-Jose van den Boogaard, Department of Genetics, Center for Molecular Medicine, University Medical Center Utrecht, Utrecht University, Utrecht, 3584EA, The Netherlands.
Sabine Fuchs, Department of Pediatrics, University Medical Center Utrecht, Utrecht, 3584 EA, The Netherlands.
Jeroen Bakker, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht, 3584 CT, The Netherlands.
Karen Duran, Hubrecht Institute-KNAW and University Medical Center Utrecht, Utrecht, 3584 CT, The Netherlands.
Zachary D Dawson, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Anika Lindsey, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Huiyan Huang, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Dustin Baldridge, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Gary A Silverman, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Barth D Grant, Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, Piscataway, NJ, 08854, USA.
David Raizen, Department of Neurology and the Chronobiology and Sleep Institute, University of Pennsylvania, Philadelphia, PA, 19104, USA.
Gijs van Haaften, Department of Genetics, Center for Molecular Medicine, University Medical Center Utrecht, Utrecht University, Utrecht, 3584EA, The Netherlands.
Stephen C Pak, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Holger Rehmann, Department of Energy and Biotechnology, Flensburg University of Applied Sciences, 24943, Flensburg, Germany.
Tim Schedl, Departments of Pediatrics and Genetics, C. elegans Model Organism Screening Center, Washington University in St Louis School of Medicine, St Louis, MO 63110, USA.
Peter van Hasselt, Department of Pediatrics, University Medical Center Utrecht, Utrecht, 3584 EA, The Netherlands.
Undiagnosed Diseases Network:
Maria T Acosta, Margaret Adam, David R Adams, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A Ashley, Mahshid S Azamian, Carlos A Bacino, Guney Bademci, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H Beggs, Edward Behrens, Gill Bejerano, Hugo Bellen, Jimmy Bennet, Beverly Berg-Rood, Jonathan A Bernstein, Gerard T Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A Burke, Lindsay C Burrage, Manish J Butte, Peter Byers, William E Byrd, John Carey, Olveen Carrasquillo, Thomas Cassini, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D Clark, Terra R Coakley, Laurel A Cobban, Joy D Cogan, Matthew Coggins, F Sessions Cole, Heather A Colley, Cynthia M Cooper, Heidi Cope, William J Craigen, Andrew B Crouse, Michael Cunningham, Precilla D’Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G Dayal, Matthew Deardorff, Esteban C Dell’Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L Doss, Emilie D Douine, Laura Duncan, Dawn Earl, David J Eckstein, Lisa T Emrick, Christine M Eng, Cecilia Esteves, Marni Falk, Liliana Fernandez, Elizabeth L Fieg, Laurie C Findley, Paul G Fisher, Brent L Fogel, Irman Forghani, William A Gahl, Ian Glass, Bernadette Gochuico, Rena A Godfrey, Katie Golden-Grant, Madison P Goldrich, Alana Grajewski, Irma Gutierrez, Don Hadley, Sihoun Hahn, Rizwan Hamid, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M Hisama, Ingrid A Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yong Huang, Wendy Introne, Rosario Isasi, Kosuke Izumi, Fariha Jamal, Gail P Jarvik, Jeffrey Jarvik, Suman Jayadev, Orpa Jean-Marie, Vaidehi Jobanputra, Lefkothea Karaviti, Jennifer Kennedy, Shamika Ketkar, Dana Kiley, Gonench Kilich, Shilpa N Kobren, Isaac S Kohane, Jennefer N Kohler, Deborah Krakow, Donna M Krasnewich, Elijah Kravets, Susan Korrick, Mary Koziura, Seema R Lalani, Byron Lam, Christina Lam, Grace L LaMoure, Brendan C Lanpher, Ian R Lanza, Kimberly LeBlanc, Brendan H Lee, Hane Lee, Roy Levitt, Richard A Lewis, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K Loo, Joseph Loscalzo, Richard L Maas, Ellen F Macnamara, Calum A MacRae, Valerie V Maduro, Rachel Mahoney, Bryan C Mak, May Christine V Malicdan, Laura A Mamounas, Teri A Manolio, Rong Mao, Kenneth Maravilla, Ronit Marom, Gabor Marth, Beth A Martin, Martin G Martin, Julian A Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Alexa T McCray, Elisabeth McGee, Heather Mefford, J Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo M Moretti, John J Mulvihill, Mariko Nakano-Okuno, Stan F Nelson, John H Newman, Sarah K Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P Orengo, Laura Pace, Stephen C Pak, J Carl Pallais, Christina G S Palmer, Jeanette C Papp, Neil H Parker, John A Phillips III, Jennifer E Posey, Lorraine Potocki, Barbara N Pusey, Aaron Quinlan, Wendy Raskind, Archana N Raja, Deepak A Rao, Anna Raper, Genecee Renteria, Chloe M Reuter, Lynette Rives, Amy K Robertson, Lance H Rodan, Jill A Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B Sampson, Mario Saporta, C Ron Scott, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A Scott, Vandana Shashi, Jimann Shin, Rebecca Signer, Edwin K Silverman, Janet S Sinsheimer, Kathy Sisco, Edward C Smith, Kevin S Smith, Emily Solem, Lilianna Solnica-Krezel, Ben Solomon, Rebecca C Spillmann, Joan M Stoler, Jennifer A Sullivan, Kathleen Sullivan, Angela Sun, Shirley Sutton, David A Sweetser, Virginia Sybert, Holly K Tabor, Amelia L M Tan, Queenie K-G Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J Tifft, Camilo Toro, Alyssa A Tran, Brianna M Tucker, Tiina K Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P Vogel, Colleen E Wahl, Stephanie Wallace, Nicole M Walley, Melissa Walker, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F Wangler, Patricia A Ward, Daniel Wegner, Monika Weisz-Hubshman, Mark Wener, Tara Wenger, Katherine Wesseling Perry, Monte Westerfield, Matthew T Wheeler, Jordan Whitlock, Lynne A Wolfe, Jeremy D Woods, Kim Worley, Changrui Xiao, Shinya Yamamoto, John Yang, Diane B Zastrow, Zhe Zhang, Chunli Zhao, and Stephan Zuchner
Funding
Research reported in this manuscript was supported by the NIH Common Fund through Office of Strategic Coordination/Office of the NIH Director Award U54 NS108251 (T.S. and Lila Solnica-Krezel), Grant U01 HG007690 and Grant U01 HG007709. Funding was also provided by the St. Louis Children’s Hospital Foundation (G.A.S. and S.C.P.) 5R01GM135326 to B.D.G and R01NS107969 and R01NS122779 to D.R. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs Grant P40 OD010440. The work on patient 1 was supported under the Care4Rare Canada Consortium funded by Genome Canada and the Ontario Genomics Institute (OGI-147), the Canadian Institutes of Health Research, the Ontario Research Fund, Genome Alberta, Genome British Columbia, Genome Quebec and the Children’s Hospital of Eastern Ontario Foundation.
References
- 1. Zhen, Y. and Stenmark, H. (2015) Cellular functions of Rab GTPases at a glance. J. Cell Sci., 128, 3171–3176. [DOI] [PubMed] [Google Scholar]
- 2. Lamers, I.J.C., Reijnders, M.R.F., Venselaar, H., Kraus, A., Study, D.D.D., Jansen, S., deVries, B.B.A., Houge, G., Gradek, G.A., Seo, J.et al. (2017) Recurrent De novo mutations disturbing the GTP/GDP binding pocket of RAB11B cause intellectual disability and a distinctive brain phenotype. Am. J. Hum. Genet., 101, 824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bem, D., Yoshimura, S.-I., Nunes-Bastos, R., Bond, F.C., Bond, F.F., Kurian, M.A., Rahman, F., Handley, M.T.W., Hadzhiev, Y., Masood, I.et al. (2011) Loss-of-function mutations in RAB18 cause Warburg micro syndrome. Am. J. Hum. Genet., 88, 499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giannandrea, M., Bianchi, V., Mignogna, M.L., Sirri, A., Carrabino, S., D’Elia, E., Vecellio, M., Russo, S., Cogliati, F., Larizza, L.et al. (2010) Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am. J. Hum. Genet., 86, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Veleri, S., Punnakkal, P., Dunbar, G.L. and Maiti, P. (2018) Molecular insights into the roles of Rab proteins in intracellular dynamics and neurodegenerative diseases. NeuroMolecular Med., 20, 18–36. [DOI] [PubMed] [Google Scholar]
- 6. Langemeyer, L., Fröhlich, F. and Ungermann, C. (2018) Rab GTPase function in endosome and lysosome biogenesis. Trends Cell Biol., 28, 957–970. [DOI] [PubMed] [Google Scholar]
- 7. Stenmark, H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol, 10, 513–525. [DOI] [PubMed] [Google Scholar]
- 8. Onodera, Y., Nam, J.-M., Hashimoto, A., Norman, J.C., Shirato, H., Hashimoto, S. and Sabe, H. (2012) Rab5c promotes AMAP1-PRKD2 complex formation to enhance β1 integrin recycling in EGF-induced cancer invasion. J. Cell Biol., 197, 983–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kempers, L., Wakayama, Y., van derBijl, I., Furumaya, C., De Cuyper, I.M., Jongejan, A., Kat, M., vanStalborch, A.-M.D., vanBoxtel, A.L., Hubert, M.et al. (2021) The endosomal RIN2/Rab5C machinery prevents VEGFR2 degradation to control gene expression and tip cell identity during angiogenesis. Angiogenesis, 24, 695–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heng, J., Lv, P., Zhang, Y., Cheng, X., Wang, L., Ma, D. and Liu, F. (2020) Rab5c-mediated endocytic trafficking regulates hematopoietic stem and progenitor cell development via notch and AKT signaling. PLoS Biol., 18, e3000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barbera, S., Nardi, F., Elia, I., Realini, G., Lugano, R., Santucci, A., Tosi, G.M., Dimberg, A., Galvagni, F. and Orlandini, M. (2019) The small GTPase Rab5c is a key regulator of trafficking of the CD93/Multimerin-2/β1 integrin complex in endothelial cell adhesion and migration. Cell Commun Signal, 17, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen, P.-I., Schauer, K., Kong, C., Harding, A.R., Goud, B. and Stahl, P.D. (2014) Rab5 isoforms orchestrate a “division of labor” in the endocytic network; Rab5C modulates Rac-mediated cell motility. PLoS One, 9, e90384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawauchi, T., Sekine, K., Shikanai, M., Chihama, K., Tomita, K., Kubo, K.-I., Nakajima, K., Nabeshima, Y.-I. and Hoshino, M. (2010) Rab GTPases-dependent endocytic pathways regulate neuronal migration and maturation through N-cadherin trafficking. Neuron, 67, 588–602. [DOI] [PubMed] [Google Scholar]
- 14. Deinhardt, K., Salinas, S., Verastegui, C., Watson, R., Worth, D., Hanrahan, S., Bucci, C. and Schiavo, G. (2006) Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron, 52, 293–305. [DOI] [PubMed] [Google Scholar]
- 15. de Kovel, C.G.F., Brilstra, E.H., vanKempen, M.J.A., Van’t Slot, R., Nijman, I.J., Afawi, Z., De Jonghe, P., Djémié, T., Guerrini, R., Hardies, K.et al. (2016) Targeted sequencing of 351 candidate genes for epileptic encephalopathy in a large cohort of patients. Mol Genet Genomic Med, 4, 568–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen, A.S., Berkovic, S.F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E.E., Epstein, M.P., Glauser, T. et al. (2013) De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karczewski, K.J., Francioli, L.C., Tiao, G., Cummings, B.B., Alföldi, J., Wang, Q., Collins, R.L., Laricchia, K.M., Ganna, A., Birnbaum, D.P.et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ioannidis, N.M., Rothstein, J.H., Pejaver, V., Middha, S., McDonnell, S.K., Baheti, S., Musolf, A., Li, Q., Holzinger, E., Karyadi, D.et al. (2016) REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet., 99, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haijes, H.A., Koster, M.J.E., Rehmann, H., Li, D., Hakonarson, H., Cappuccio, G., Hancarova, M., Lehalle, D., Reardon, W., Schaefer, G.B.et al. (2019) De novo heterozygous POLR2A variants cause a neurodevelopmental syndrome with profound infantile-onset Hypotonia. Am. J. Hum. Genet., 105, 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Havrilla, J.M., Pedersen, B.S., Layer, R.M. and Quinlan, A.R. (2019) A map of constrained coding regions in the human genome. Nat. Genet., 51, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dickinson, D.J. and Goldstein, B. (2016) CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics, 202, 885–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fares, H. and Greenwald, I. (2001) Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics, 159, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang, H., Pan, J., Spielberg, D.R., Hanchard, N.A., Scott, D.A., Burrage, L.C., Dai, H., Murdock, D., Rosenfeld, J.A., Mohammad, A.et al. (2022) A dominant negative variant of RAB5B disrupts maturation of surfactant protein B and surfactant protein C. Proc. Natl. Acad. Sci. U. S. A., 119, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Churgin, M.A., Szuperak, M., Davis, K.C., Raizen, D.M., Fang-Yen, C. and Kayser, M.S. (2019) Quantitative imaging of sleep behavior in Caenorhabditis elegans and larval Drosophila melanogaster. Nat. Protoc., 14, 1455–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muller, H.J. (1932) Further studies of the nature and causes of gene mutations. In Jones, D.F. (ed), Proceedings of the 6th International Congress on Genetics. Brooklyn Botanic Garden, Menasha, WI: 1932, Vol. 1, pp. 213–255. [Google Scholar]
- 28. Gillooly, D.J., Raiborg, C. and Stenmark, H. (2003) Phosphatidylinositol 3-phosphate is found in microdomains of early endosomes. Histochem. Cell Biol., 120, 445–453. [DOI] [PubMed] [Google Scholar]
- 29. Dang, H., Li, Z., Skolnik, E.Y. and Fares, H. (2004) Disease-related myotubularins function in endocytic traffic in Caenorhabditis elegans. Mol. Biol. Cell, 15, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stenmark, H., Parton, R.G., Steele-Mortimer, O., Lütcke, A., Gruenberg, J. and Zerial, M. (1994) Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. EMBO J., 13, 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chotard, L., Mishra, A.K., Sylvain, M.-A., Tuck, S., Lambright, D.G. and Rocheleau, C.E. (2010) TBC-2 regulates RAB-5/RAB-7-mediated endosomal trafficking in Caenorhabditis elegans. Mol. Biol. Cell, 21, 2285–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clark, B.S., Winter, M., Cohen, A.R. and Link, B.A. (2011) Generation of Rab-based transgenic lines for in vivo studies of endosome biology in zebrafish. Dev. Dyn., 240, 2452–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gorvel, J.P., Chavrier, P., Zerial, M. and Gruenberg, J. (1991) rab5 controls early endosome fusion in vitro. Cell, 64, 915–925. [DOI] [PubMed] [Google Scholar]
- 34. Bucci, C., Lütcke, A., Steele-Mortimer, O., Olkkonen, V.M., Dupree, P., Chiariello, M., Bruni, C.B., Simons, K. and Zerial, M. (1995) Co-operative regulation of endocytosis by three Rab5 isoforms. FEBS Lett., 366, 65–71. [DOI] [PubMed] [Google Scholar]
- 35. Guadagno, N.A. and Progida, C. (2019) Rab GTPases: switching to human diseases. Cell, 8, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gabe Lee, M.T., Mishra, A. and Lambright, D.G. (2009) Structural mechanisms for regulation of membrane traffic by Rab GTPases. Traffic, 10, 1377–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hodgkin, J. (2005) Karyotype, ploidy, and gene dosage. WormBook, 1–9. 10.1895/wormbook.1.3.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan, W. and Song, C. (2020) The emerging role of Rab5 in membrane receptor trafficking and signaling pathways. Biochem. Res. Int., 2020, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen, P.-I., Kong, C., Su, X. and Stahl, P.D. (2009) Rab5 isoforms differentially regulate the trafficking and degradation of epidermal growth factor receptors. J. Biol. Chem., 284, 30328–30338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Barbieri, M.A., Fernandez-Pol, S., Hunker, C., Horazdovsky, B.H. and Stahl, P.D. (2004) Role of rab5 in EGF receptor-mediated signal transduction. Eur. J. Cell Biol., 83, 305–314. [DOI] [PubMed] [Google Scholar]
- 41. Ceresa, B.P. (2006) Regulation of EGFR endocytic trafficking by Rab proteins. Histol. Histopathol., 21, 987–993. [DOI] [PubMed] [Google Scholar]
- 42. Kiral, F.R., Kohrs, F.E., Jin, E.J. and Hiesinger, P.R. (2018) Rab GTPases and membrane trafficking in neurodegeneration. Curr. Biol., 28, R471–R486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Igarashi, T., Araki, K., Yokobori, T., Altan, B., Yamanaka, T., Ishii, N., Tsukagoshi, M., Watanabe, A., Kubo, N., Handa, T.et al. (2017) Association of RAB5 overexpression in pancreatic cancer with cancer progression and poor prognosis via E-cadherin suppression. Oncotarget, 8, 12290–12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang, W., Ma, L., Yang, M., Shao, Q., Xu, J., Lu, Z., Zhao, Z., Chen, R., Chai, Y. and Chen, J.F. (2020) Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev., 34, 580–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Whitecross, D.E. and Anderson, D.H. (2017) Identification of the binding sites on Rab5 and p110beta phosphatidylinositol 3-kinase. Sci. Rep., 7, 16194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. D’adamo, P., Menegon, A., Lo Nigro, C., Grasso, M., Gulisano, M., Tamanini, F., Bienvenu, T., Gedeon, A.K., Oostra, B., Wu, S.-K.et al. (1998, 1998) Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat. Genet., 19, 134–139. [DOI] [PubMed] [Google Scholar]
- 47. Handley, M.T., Morris-Rosendahl, D.J., Brown, S., Macdonald, F., Hardy, C., Bem, D., Carpanini, S.M., Borck, G., Martorell, L., Izzi, C.et al. (2013) Mutation Spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype–phenotype correlations in Warburg micro syndrome and Martsolf syndrome. Hum. Mutat., 34, 686–696. [DOI] [PubMed] [Google Scholar]
- 48. Basel-Vanagaite, L., Sarig, O., Hershkovitz, D., Fuchs-Telem, D., Rapaport, D., Gat, A., Isman, G., Shirazi, I., Shohat, M., Enk, C.D.et al. (2009) RIN2 deficiency results in macrocephaly, alopecia, cutis laxa, and scoliosis: MACS syndrome. Am. J. Hum. Genet., 85, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eijkelenboom, A., vanSchaik, F.M.A., vanEs, R.M., Ten Broek, R.W., Rinne, T., van derVleuten, C., Flucke, U., Ligtenberg, M.J.L. and Rehmann, H. (2019) Functional characterisation of a novel class of in-frame insertion variants of KRAS and HRAS. Sci. Rep., 9, 8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rehmann, H. (2006) Characterization of the activation of the rap-specific exchange factor Epac by cyclic nucleotides. Methods Enzymol, 407, 159–173. [DOI] [PubMed] [Google Scholar]
- 51. Arribere, J.A., Bell, R.T., Fu, B.X.H., Artiles, K.L., Hartman, P.S. and Fire, A.Z. (2014) Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics, 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Corsi, A.K., Wightman, B. and Chalfie, M. (2015) A transparent window into biology: a primer on Caenorhabditis elegans. Genetics, 200, 387–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fares, H. and Greenwald, I. (2001) Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat. Genet., 28, 64–68. [DOI] [PubMed] [Google Scholar]
- 54. Aleström, P., D’Angelo, L., Midtlyng, P.J., Schorderet, D.F., Schulte-Merker, S., Sohm, F. and Warner, S. (2020) Zebrafish: housing and husbandry recommendations. Lab. Anim., 54, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kraulis, P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- 56. Merritt, E.A. and Murphy, M.E. (1994) Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr D Biol Crystallogr, 50, 869–873. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.