

SUMMARY
Fungal infections are a global threat, yet there are no licensed vaccines to any fungal pathogens. Th17 cells mediate immunity to Candida albicans, particularly oropharyngeal candidiasis (OPC), but essential downstream mechanisms remain unclear. In murine model of OPC, IκBζ (Nfkbiz, a noncanonical NF-κB transcription factor) was upregulated in an IL-17-dependent manner and was essential to prevent candidiasis. Deletion of Nfkbiz rendered mice highly susceptible to OPC. IκBζ was dispensable in hematopoietic cells and acted partially in the suprabasal oral epithelium to control OPC. One prominent IκBζ-dependent gene target was β-defensin (BD)-3 (Defb3), an essential antimicrobial peptide. Human oral epithelial cells required IκBζ for IL-17-mediated induction of BD2 (DEFB4A, human orthologue of mouse Defb3) through binding to the DEFB4A promoter. Unexpectedly, IκBζ regulated the transcription factor EGR3, which was essential for C. albicans induction of BD2/IDEFB4A. Accordingly, IκBζ and EGR3 comprise an antifungal signaling hub mediating mucosal defense against oral candidiasis.
Keywords: Candida albicans, IL-17 signaling, oral epithelium, defensins, signal transduction, cytokines
Graphical Abstract
eTOC blurb
Fungal pathogens are of increasing medical concern. Oropharyngeal candidiasis (OPC) is the most common fungal infection in humans, but tissue-specific correlates of oral immunity are poorly understood. Taylor et al show that IL-17 prevents OPC through the non-canonical transcription factor, IκBζ, that regulates essential antimicrobial peptide effectors.
INTRODUCTION
Fungal infections are of increasing medical concern, especially among the immunocompromised. To date, there are only limited effective anti-fungal therapies and no licensed vaccines to any fungal species. Candida albicans is the most common causative agent of fungal infections in humans, with multiple manifestations including oral, vaginal and systemic disease 1. Oropharyngeal candidiasis (OPC, oral thrush) can range from a mild, self-limiting condition to a severe superficial infection linked to oral cancer, nutritional deficits, and failure to thrive in infants 2,3. OPC occurs as a result of systemic immune defects that occur in HIV infection, chemotherapy, antibiotic use or congenital immune defects, as well conditions that negatively impact oral-specific immunity such as head-neck irradiation and Sjögren’s syndrome 4.
Compared to many other tissues, site-specific immunity within the oral mucosa is still poorly understood 5,6. The cytokine IL-17 (IL-17A) has emerged as a vital mediator of immunity to candidiasis, especially in the oropharynx 7. Although produced predominantly by lymphocytes, IL-17 acts upon nonhematopoietic cells, particularly epithelial cells lining the gut, lung and mouth. C. albicans is dimorphic, and the conversion from a yeast form to an invasive hyphal morphotype is a key feature of pathogenesis. Oral epithelial cells (OECs) respond to pathogenic hyphae by upregulating an innate immune defense program composed of cytokines, chemokines, antimicrobial peptides (AMPs), and other antifungal effectors 8,9.
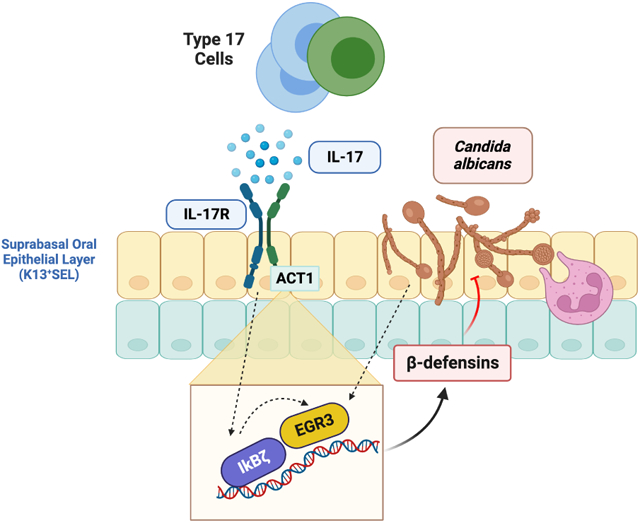
The stratified oral epithelium is comprised of multilayered cell subsets that provide the first line of pathogen recognition 10-12. Upon infection and damage caused by C. albicans hyphal invasion, the suprabasal epithelial layer (SEL) of the tongue and buccal mucosa is sloughed and swallowed, aiding clearance. The basal epithelial layer (BEL) responds to C. albicans-induced injury through a wound healing process that replaces the exfoliated SEL 13. OEC layers are characterized by distinct cytokeratin pairs, with the SEL expressing keratin 13 (K13) and K4, and the BEL is marked by K5 and K14 14. We showed that IL-17RA signaling is required in K13+ cells, whereas IL-22R acts dominantly in the K14+ cell layer 15. Thus, specific cellular subsets within the oral epithelium respond preferentially to inflammatory stimuli.
IL-17 signals through IL-17RA and IL-17RC via the essential adaptor Act1 16. Loss of either subunit or Act1 in mice or humans predisposes to oral candidiasis 17-23. IL-17 orchestrates essential antifungal events via complex downstream signaling pathways that converge on activation of transcription factors (TFs). Prominent IL-17-activated TFs include CCAAT/enhancer binding proteins (C/EBP)-β and -δ, MAPK-associated TFs (c-Fos, c-Jun), AT-rich interacting protein 5a (Arid5a), CUX1, and NF-κB family members 16,24,25. In addition, IL-17 promotes post-transcriptional pathways that stabilize or otherwise control target mRNAs, many of which include these TFs 16. Prior studies surprisingly ruled out roles for many IL-17-induced TFs in OPC, including C/EBPβ, c-Fos and Arid5a 26-28 and thus the essential signaling activators that underlie IL-17-driven immunity to OPC have been enigmatic.
Another IL-17-activated TF is IκBζ, encoded by Nfkbiz. IκBζ is an atypical member of the IκB family that, despite its name, usually acts as a transcriptional activator 29,30. Originally identified as a mediator of IL-1/TLR responses, IκBξ regulates genes characteristic of the IL-17 pathway through promoter activation and chromatin remodeling 25,31-35. In T cells, IκBζ promotes Th17 differentiation. Consequently, mice with a global Nfkbiz gene deletion are resistant to IL-17-driven conditions such as experimental autoimmune encephalomyelitis (EAE) and psoriasis-like skin inflammation 34,36. Despite such impairments, IκBζ-deficient mice develop spontaneous dermatitis and lacrimal inflammation that bears similarities to Sjögren’s syndrome 37.
Given these dichotomous physiologic impacts of IκBζ, it was not obvious whether IκBζ loss would enhance IL-17-driven inflammation and thus protect against OPC, or instead impair IL-17 signaling and thereby promote candidiasis. Here we report that mice lacking Nfkbiz in all tissues are highly sensitive to OPC. A portion of this susceptibility was mediated through suprabasal OECs, particularly via antifungal β-defensins. Moreover, IκBζ regulates the TF early growth factor 3 (Egr3) in OECs. Although not a direct target of IL-17, Egr3 drives production of β-defensins in response to C. albicans infection in a manner distinct from IκBζ. These findings revealing a regulatory TF circuit that coordinates the fungal- and host-driven pathways controlling oral mucosal candidiasis.
RESULTS
IκBζ is induced by IL-17 in oral candidiasis
IL-17 and IL-22 are nonredundant mediators of host defense against OPC. Whereas IL-17 acts dominantly on a K13+ suprabasal layer of the oral epithelium (SEL), IL-22 acts on the stem-like basal K14+ epithelial cell layer (BEL) 13,15 (Fig 1a). To probe mechanisms that drive effective antifungal immunity, we evaluated a previously-published RNASeq dataset 15 for TFs induced in the oral mucosa in an IL-17-dependent manner 38. We specifically focused on genes induced at day 2, as this coincides with peak expression of Type 17 cytokines (Il17a, Il22) and essential antifungal genes (Deft3, CXC chemokines) (Fig 1b) 15,17.
Figure 1. IκBζ is induced by IL-17 in OPC.

(A) The stratified oral epithelium and cytokine receptor localization. (B) Timeline of oral C. albicans infection procedure. (C) Heat map of TF mRNAs upregulated in tongues from WT mice (Nfkbiz+/+ animals obtained from breeding colonies) or Il17ra−/− mice on day 2 p.i. Data derived from published RNA-Seq data 15 (n= 2 or 3) (D) The indicated mice were subjected to OPC and RNA extracted from tongue at day 2. Nfkbiz levels were assessed by qPCR normalized to Gapdh. Data are mean ± SEM, analyzed by ANOVA with Tukey’s multiple comparisons test. (E) Top: Tongue homogenates from WT and Il17ra−/− mice isolated 2 days p.i. were immunoblotted for IκBζ and β-actin. Size markers are shown. Bottom: Densitometry analysis normalized to β-actin (n=3). Data are mean ± SEM, analyzed by 1-way ANOVA with Tukey’s multiple comparisons test. (F) Top: TR146 cells were treated ± IL-17 (100 ng/ml) for the indicated times and immunoblotted for IκBξ and β-actin. Bottom: Densitometry analysis normalized to β-actin (n=3). Data are from 3 independent experiments. Data are mean ± SEM, analyzed by 1-way ANOVA with Tukey’s multiple comparisons test.
Multiple TF classes were upregulated in the tongue during OPC. CCAAT/Enhancer binding proteins C/EBPβ and C/EBPδ are instrumental in regulating many downstream IL-17 target genes 39,40 and were upregulated in OPC, though surprisingly expression was not strongly IL-17-dependent in this setting (Fig 1c). C/EBPβ contributes to OPC immunity in only a limited way 27, but the contribution of C/EBPd is unknown. Cebpd−/− mice 41 were infected orally with C. albicans and fungal loads assessed at 5 days p.i., the time point at which control WT mice efficiently cleared the infection. Mice lacking C/EBPδ cleared the fungus completely, with no weight changes or detectable fungal loads after 5 days of infection (Supplementary Fig S1a, b). Therefore, IL-17-associated C/EBPβ and C/EBPδ are dispensable to control OPC.
In addition to C/EBPs, several members of the NF-κB family were elevated during OPC, including Nfkbiz, encoding IκBζ (Fig 1c, d). IκBζ protein was significantly increased in WT tongue homogenates following oral C. albicans infection and was largely absent in tongues from Il17ra−/− mice (Fig 1e). In keeping with findings in mouse, a prominent band corresponding to IκBζ was evident in immunoblots of lysates from a human OEC line (TR146) following IL-17 treatment, though densitometric measurements showed quite a bit of variablity (Fig 1f). Induction of IκBζ was cyclical, with prominent induction consistently seen at 1 hour, reduced expression at 1-3 hours and then rebound at 6 hours. This likely reflects the dependence of IκBζ on the canonical NF-κB pathway, which undergoes dynamic oscillations due to a complex network of transcriptional and post-transcriptional feedback regulators 29,42. Upon IL-17 stimulation, a prominent, slower-migrating band was observed in immunoblots, which likely corresponds to a post-transcriptionally modified form of IκBζ (e.g., a phosphorylated isoform 43). Hence, IκBζ is elevated in OPC in a manner requiring IL-17 signaling.
IκBζ in the nonhematopoietic compartment is required for immunity to OPC
Although numerous TFs are upregulated during OPC in an IL-17-dependent manner, surprisingly few evaluated thus far are needed for fungal clearance 26-28. Studies of IκBζ in oral settings have been confounded by the fact that Nfkbiz−/− mice show major baseline defects, including a Sjögren’s-like syndrome with oral manifestations 35,37. Therefore, to avoid developmental or early-life influences of IκBζ deficiency, we crossed Nfkbizfl/fl to Rosa26CreERT2 mice, which express Cre under control of a ubiquitous but tamoxifen (TAM)-inducible promoter, permitting timed but permanent deletion of the Nfkbiz gene in all tissues. NfkbizR26ERT2 mice were administered TAM for 5 days and rested for 7-9 days (Fig 1b). Unlike mice lacking IκBξ from birth 37, NfkbizR26ERT2 mice given TAM and subject to sham infections (PBS) did not show apparent inflammatory lesions or other deficits. However, NfkbizR26ERT2 mice subjected to OPC exhibited high fungal loads after 5 days, at levels similar to mice lacking the IL-17 receptor (Il17ra−/−) or its essential adaptor Act1 (Act1−/−) (Fig 2a). All infected NfkbizR26ERT2 mice had detectable fungal loads and showed progressive weight loss throughout the infection (Fig 2a). Therefore, IκBζ is vital for immunity to OPC.
Figure 2. IκBζ in non-hematopoietic cells protects against OPC.

(A) The indicated mice were treated with TAM on days −12 to −8, rested for 7-9 d, and infected orally with C. albicans. Fungal loads were assessed on days 4-5. Data show geometric mean of CFU/g tongue homogenates, analyzed by ANOVA with Dunn’s multiple comparisons test. The percentage of mice per cohort with a detectable fungal load is shown. Weight loss is shown as percentage of starting weight, analyzed by 2-way ANOVA with Tukey’s multiple comparisons test. Pooled from 2 independent experiments. (B, C) Expression of indicated genes in tongue was assessed by qPCR normalized to Gapdh. Data are ± SEM relative to WT untreated mice, analyzed by ANOVA with Tukey’s multiple comparisons test. (D) The indicated mice were subjected to OPC and cells from tongue harvested at day 2 were analyzed by flow cytometry. Left: Representative plot showing percentage of CD11b+ Ly6G+ cells in neutrophils (gated on live, CD45+ cells). Right: Data compiled from two independent experiments. (E) Bone marrow from indicated donors was transferred into irradiated recipients (KO = NfkbizR26ERT2). After 6 weeks, recipients were given TAM on days −12 to −8, subjected to OPC, and fungal burden assessed on day 5. Data show geometric mean, analyzed by ANOVA with Dunn’s multiple comparisons test. Data pooled from two independent experiments, analyzed by ANOVA test with Dunn’s multiple comparison test.
IL-17 mediates immunity to oral C. albicans through antimicrobial peptides (AMPs) such as β-defensin 3 (BD3, encoded by Defb3) and through neutrophil recruitment, mediated indirectly by epithelial expression of CXC chemokines 15,17,44. Loss of Nfkbiz impaired Defb3 expression in tongue during OPC (Fig 2b). Expression of Cxcl1 and Cxcl5 both trended downward in NfkbizR26ERT2 mice, and neutrophil recruitment to the oral mucosa was concomitantly reduced (Fig 2c, d). These defects likely explain the susceptibility of Iκζ-deficient mice to oral candidiasis.
IL-17 and IL-22 act dominantly in nonhematopoietic cells due to restricted expression of their respective receptors, but Nfkbiz is ubiquitously expressed in oral cell types in both hematopoietic and stromal cell types (Supplementary Fig S2) 45. To determine whether IκBζ functions in hematopoietic cells to during OPC, WT recipient mice were lethally irradiated and adoptively transferred with bone marrow from WT or NfkbizR26ERT2 donors. After 6 weeks, chimeric mice and controls were treated with TAM (to delete Nfkbiz), subjected to OPC, and fungal loads evaluated at day 5. WT recipients cleared the fungus, regardless of whether they were given WT or NfkbizR26ERT2 BM (Fig 2e), indicating that IκBζ is, rather surprisingly, not required in hematopoietic cells to drive immunity to OPC.
IκBζ functions in the oral epithelium
The above data pointed to essential IκBζ activities within the nonhematopoietic compartment, and we have shown that IL-17 antifungal signaling occurs dominantly in the SEL (see Fig 1a). To determine if IκBζ was similarly required for antifungal activities in the suprabasal oral epithelia, we crossed Nfkbizfl/fl to mice bearing a K13Cre cassette 15. At 5 days p.i. NfkbizK13 mice consistently had detectable fungal loads, whereas WT and Nfkbizfl/fl littermate controls cleared the infection (Fig 3a). Nearly all mice showed detectable fungal loads, whereas only 2/10 control mice showed any evidence of colonization. Moreover, fungal burdens in NfkbizK13 mice were reproducibly lower than in mice with a complete Nfkbiz deletion (compare to Fig 2a). NfkbizK13 mice fully regained body weight, commensurate with the modest oral fungal loads. At 10 days p.i., half the NfkbizK13 mice still bore fungal loads (Fig 3b). Thus, IκBζ exerts some of its antifungal activities in K13+SELs, notably the cell type that is most responsive to IL-17, yet also clearly acts in additional stromal cell types.
Figure 3. IκBζ functions within the oral epithelium.

(A, B) The indicated mice were infected orally with C. albicans. Fungal loads, percentage of mice with detectable fungal loads, and weight loss were assessed on day 5 (A) or day 10 (B). Data were pooled from 3 independent experiments. (C) The indicated mice were treated with TAM on days −12 to −8, rested for 7 d, and infected orally with C. albicans. Fungal loads, percent of mice with detectable fungal loads, and weight loss were assessed on day 5. Throughout: Bars show geometric mean. Data were analyzed by ANOVA with Dunn’s multiple comparison test. Weight loss was analyzed by 2-way ANOVA with Tukey’s multiple comparison test.
Since only part of the nonhematopoietic IκBζ-dependent response could be attributed to K13+ cells, we interrogated its role in the K14+ BEL (see Fig 1a) 13. Nfkbizfl/fl mice were crossed to mice bearing a TAM-inducible K14CreERT2 cassette, administered TAM as described above, and subjected to OPC. NfkbizK14ERT2 mice showed a very modest susceptibility to OPC, with only 35% (9/26) mice having detectable fungal burdens (Fig 3c). Even when considering only mice with a detectable infectious load, the mean fungal burden in NfkbizK14ERT2 mice was low, averaging 196 CFU/g. Collectively, these data suggest that IκBζ is an essential mediator of immunity to OPC, acting partially in OECs but evidently in additional cell types as well.
IκBζ regulates oral β-defensins
To understand mechanisms by which IκBζ drives host defense within oral epithelium, we performed transcriptomic profiling of tongues from NfkbizK13 mice or Nfkbizfl/fl (WT) littermates. This was assessed at day 2 p.i., when fungal loads were not yet different (Fig 4a). Partek Pathway analysis identified ‘Cytokine-cytokine receptor interactions’ and the ‘IL-17 signaling pathway’ as the top pathways altered by an SEL-specific Nfkbiz deletion (Fig 4b). Consistent with this, characteristic IL-17-target genes known to be required for immunity to OPC were impaired in NfkbizK13 tongue tissue, including Defb3 and neutrophil-attracting chemokines (Cxcl1, Cxcl2, Cxcl5) (Fig 4c). Verification by qPCR confirmed that Defb3 mRNA was reduced in NfkbizK13 mice (Fig 4d).
Figure 4. IκBζ in the SEL regulates β-defensins.

(A) WT and NfkbizK13 were infected orally with C. albicans, and fungal loads assessed on day 2. Data show geometric mean, analyzed by ANOVA with Tukey’s multiple comparison test. Data pooled from 3 independent experiments. (B) RNA-seq data (n=3) from C. albicans-infected tongues of WT and NfkbizK13 mice on day 2 was analyzed by Partek pathway analysis. (C) Comparison of transcriptional responses induced in tongue during OPC from WT and NfkbizK13 mice on day 2. (D) Defb3 was assessed by qPCR normalized to Gapdh. Data are mean ± SEM relative to WT untreated mice, analyzed by ANOVA with Tukey’s multiple comparisons test. (E) TR146 cells transfected with indicated siRNAs targeting were treated with IL-17 and DEFB4A was assessed by qPCR normalized to GAPDH. Data are as fold-increase compared to untreated (time 0), analyzed by ANOVA with Dunn’s multiple comparison test. (F) Supernatants from TR146 samples from panel D were analyzed for human BD2 by ELISA. Data are mean ± SEM, analyzed by ANOVA with Holm-Sidak’s multiple comparisons test. (G) Top: Diagram of predicted TF binding sites in DEFB4A proximal promoter. Bottom: TR146 cells were treated ± IL-17 (100 ng/ml) for 4 h and subjected to ChIP with anti-IκBξ Abs or IgG control. Indicated promoter regions were analyzed by PCR, normalized to input. Data are mean ± SEM of three independent experiments. Data analyzed pairwise by Student’s t test. (H) Comparison of transcriptional responses induced in tongue during OPC from WT and Defb3−/− mice on day 1. Data are from 3 mice.
The human orthologue of murine BD3 is human β-defensin 2 (BD2, encoded by DEFB4A) 46. BD2 is upregulated in the OEC cell line TR146 upon IL-17 stimulation or C. albicans infection in vitro 15,47. RNA silencing of NFKBIZ in TR146 cells abrogated IL-17 induction of DEFB4A mRNA and BD2 secretion (Fig 4e, f). To ascertain whether IκBζ occupied the DEFB4A proximal promoter, we performed ChIP-qPCR using primers spanning four predicted TF binding site regions upstream of the DEFB4A transcriptional start site (TSS) (Fig 4g). There was strong enrichment of DEFB4A in IκBξ-pulldowns within region 1, a distal site encompassing a C/EBP recognition element (also known as NFIL6) and region 4, a proximal site encompassing a C/EBP and NF-κB sites. Thus, IκBζ regulates DEFB4A/BD2 but not via canonical NF-κB binding sites.
While β-defensins exert direct candidacidal activity and are critical to prevent OPC 15,48, they are also reported to have additional immune properties. For example, many defensins have chemotactic properties, engaging the chemokine receptor CCR6 that is expressed on Type 17 cells and thus serving to recruit cells to the inflammatory milieu 49-52. To explore the possibility that BD3 might more broadly influence the antifungal immune landscape, we subjected Defb3−/− mice to OPC or sham infections and analyzed the transcriptome from tongues harvested at 24 h p.i. by RNA-Seq (Fig 4h). Remarkably few genes apart from Defb3 itself were differentially expressed in Defb3−/− mice upon OPC compared to sham-treated controls. The absence of any genes implicated in immune cell mobilization or immune pathways contrasts with the marked differential gene expression changes seen in the absence of the IL-17 signaling system or IκBζ 15,17. Hence, β-defensins appear to be a functional endpoint of antifungal signals, rather than amplifying any kind of feed-forward inflammatory circuit through chemotactic recruitment of immune cells.
We next evaluated the impact of IκBζ in the SEL on neutrophil recruitment. NfkbizK13 mice showed reduced expression of the chemokines Cxcl1 and Cxcl5 (Fig 5a). However, neutrophil recruitment to tongue was not impaired in NfkbizK13 mice (Fig 5b). NfkbizK14ERT2 mice did not show defects in Defb3 or neutrophil recruitment (Supplementary Fig S3), commensurate with their mild susceptibility to OPC. This observation is reminiscent of other knockout settings where disease is modest (e.g., IL-17F-deficiency 53,54), and explains why the oral fungal loads in NfkbizK13 mice are less than those with a complete IL-17 signaling defect.
Figure 5. IκBζ in the SEL does is not required for neutrophil recruitment in OPC.

(A) WT and NfkbizK13 mice were subjected to OPC. On day 2, Cxcl1 and Cxcl5 were assessed by qPCR normalized to Gapdh. Data are presented as mean ± SEM normalized to WT untreated mice, analyzed by ANOVA with Tukey’s multiple comparisons test. (B) Cells from tongue harvested at day 2 p.i. were stained with the indicated Abs and analyzed by flow cytometry. Left: Representative plot showing percentage of CD11b+ Ly6G+ cells in neutrophils (gated on live, CD45+ cells). Right: Data pooled from two independent experiments, analyzed by ANOVA with Tukey’s multiple comparisons test.
Egr3 regulates β-defensins in OECs
RNA-Seq analysis of NfkbizK13 tongue showed that IκBζ-deficiency in K13+ OECs reduced expression of multiple TFs during OPC (Fig 6a). Egr3 (early growth factor 3) stood out because it has been previously shown to be upregulated in response to C. albicans hyphae in macrophages and OECs 55-57, though Egr3 was not IL-17-inducible. Unlike IκBζ, total Egr3 protein levels were not altered in tongue upon OPC (Fig 6b), nor was EGR3 mRNA induced in TR146 cells in response to IL-17 (Fig 6c). However, C. albicans infection of TR146 cells in culture upregulated EGR3 mRNA, which was blocked by NFKBIZ siRNA (Fig 6d). Though not IL-17-inducible, EGR3 silencing in TR146 cells diminished IL-17 induction of DEFB4A mRNA and BD2 secretion (Fig 6e, f). Although there were no predicted Egr3 recognition elements in the DEFB4A promoter, Egr3 can form complexes with NF-κB p50 and thereby bind DNA indirectly 58. ChIP indicated that Egr3 occupancy of the DEFB4A promoter was elevated in the vicinity of regions 2, 3 and 4 (Fig 6g), which notably differs from the occupancy site identified for IκBζ (see Fig 4g). These data suggest that Egr3 participates in the oral host response to C. albicans by regulating key β-defensins.
Figure 6. Egr3 regulates β-defensins in OECs.

(A) Heatmap of differentially expressed transcription factor genes in WT (Nfkbizfl/fl littermates) or NfkbizK13 mice (n=3) on day 2. Red asterisk highlights TFs examined in this study. (B) Top: Tongue homogenates from infected WT and Il17ra−/− mice on day 2 were probed for EGR3 or β-actin. Bottom: Densitometry analysis normalized to β-actin, analyzed using ANOVA with Tukey’s multiple comparisons test (Note: these data are from same experiment as Figure 1e, so the β-actin loading control is repeated). (C) TR146 cells were transfected with the indicated siRNAs, treated ± IL-17 and EGR3 assessed by qPCR normalized to GAPDH. Data are fold-increase compared to untreated (time 0), analyzed by ANOVA with Dunn’s multiple comparisons test. (D) TR146 cells were co-cultured with PBS or C. albicans (MOI = 10) for the indicated times. EGR3 was assessed by qPCR normalized to GAPDH and presented as fold-increase compared to untreated cells (time 0) and analyzed by ANOVA. (E) TR146 cells were transfected with the indicated siRNAs, treated ± IL-17 and DEFB4A assessed by qPCR normalized to GAPDH. Data are fold-increase compared to untreated (time 0), analyzed by ANOVA with Dunn’s multiple comparisons test. (F) Supernatants from samples in panel E were analyzed for human BD2 by ELISA and analyzed by ANOVA with Holm-Šídák's multiple comparisons test (G) Top: Diagram of predicted TF binding sites in DEFB4A proximal promoter. Bottom: TR146 cells were treated ± IL-17 (100 ng/ml) for 4 h and subjected to ChIP with anti-EGR3 Abs or IgG control. Indicated promoter regions were analyzed by PCR, normalized to input. Data are mean ± SEM of three independent experiments. Data analyzed pairwise by Student’s t test.
Egr3 is induced by live C. albicans or zymosan in vitro, but how this occurs is not well understood 55,56. Multiple fungal properties are required for virulence in OECs (Fig 7a). To determine requirements for EGR3 induction, TR146 cells were infected with mutant, avirulent C. albicans strains defective in (i) cellular adhesion (als3Δ), (ii) hyphal formation (a ‘yeast-locked’ strain, efgΔ/cph1Δ), and (iii) candidalysin, a secreted pore-forming peptide needed to drive innate gene expression in OECs (ClysΔ) 12,59. As controls, we used C. albicans strains CAF2-1, SC5314, and the autotrophic strain BWP17 + CIp30 (parental control for ClysΔ). After infection in vitro, EGR3 was induced at 4 h p.i by the virulent controls but not by the avirulent efgΔ/cph1Δ, als3Δ or the ClysΔ strains (Fig 7a). Although the yeast-locked efgΔ/cph1Δ strain failed to induce EGR3, hyphal formation per se appears not to be a prerequisite for this response, as als3Δ and ClysΔ can form filaments normally. Rather, upregulation of EGR3 requires adherence to target cells and secretion of the candidalysin peptide is required.
Figure 7. EGR3 responses in oral candidiasis.

(A) TR146 cells were infected with the indicated strains of C. albicans (MOI = 10) for 4 h and EGR3 levels were assessed by qPCR, normalized to GAPDH, analyzed by ANOVA with Tukey’s multiple comparisons test. Representative of three independent experiments. Diagram of C. albicans virulence components and interactions with oral epithelium. (B, C) TR146 cells were transfected with indicated siRNAs and infected with C. albicans for 4 h. Indicated mRNAs were analyzed by qPCR normalized to GAPDH, analyzed by Mann Whitney U. Data were pooled from four independent experiments.
We next investigated how IκBζ and EGR3 regulate anti-fungal cytokine and chemokine genes in oral epithelial cells in response to C.albicans. Accordingly, we silenced NFKBIZ or EGR3 in TR146 cells, which impaired induction of IL1F9 (IL-36γ, required for immunity to OPC 60) and the chemokine CCL20 (Fig 7b). Silencing EGR3 IL1F9 and IL6 with a trend to impaired IL8 and CCL20 (Fig 7c). Hence, NFKBIZ and EGR3 both promote OEC expression of genes involved in immunity to C. albicans.
DISCUSSION
The data described here reveal that IκBζ is a central TF mediating oral defense to C. albicans. IκBζ works jointly with other TFs such as Egr3 to regulate production of β-defensins, essential for effective clearance of C. albicans from the oral mucosa. IκBζ is induced by many cytokines that participate in immunity to candidiasis, so it is likely that IκBζ is a convergence point downstream of many cytokine receptors that function to limit fungal colonization.
Despite high microbial loads, infections of the oral mucosa are uncommon, implying unique oral immune defense mechanisms 5,6,45. IL-17 and IL-22 are potent antifungal cytokines operative in the oral cavity, in large part because of their capacity to drive expression of β-defensins 13,15. Though produced by the same Type 17 cells, IL-17 and IL-22 function in distinct layers of the stratified oral epithelium, with each cell subset mounting separate and distinctive responses to C. albicans infection. Even so, their actions are interconnected, because IL-22 drives proliferation of BELs that replenish the SEL, thus restoring expression of IL-17R and licensing responsiveness to this cytokine 13. Both IL-17 and IL-22 upregulate IκBζ, in OECs, though this is just one of many TFs known to be IL-17-inducible. Here we show empirically that IκBζ, but not C/EBPδ, is required for immunity to oral candidiasis, and that in the oral epithelium IκBζ acts within the post-mitotic SEL. This is consistent with a model in which IκBζ acts downstream of the IL-17R within the SEL. Still, IκBζ likely acts in other cytokine pathways (e.g., IL-1 or IL-36) that also participate in OPC immunity 60-62. Related to this, the relatively modest phenotype observed in mice lacking IκBζ in the K14+ BEL (NfkbizK14) implies that IκBζ is not a major component of IL-22 antifungal signaling, which rather is the province of STAT3 13.
In the context of oral candidiasis, antimicrobial peptides (AMPs) are a particularly important yet relative understudied component of immune defense 63. According to the APD3 database, there are 3569 AMPs, including 152 human host defense peptides, 1288 antifungal peptides, and 737 anti-Candida peptides 64. In the mouth, AMPs are produced by OECs and neutrophils and found in high concentrations in saliva 65-68. AMPs function in diverse ways, by targeting and disrupting microbial cell membranes, mediating apoptosis, or by inhibiting protein and nucleic acid biosynthesis, protease activity and cell division 69. AMPs can also serve as chemoattractants to recruit immune cells to the local microenvironment and promote wound healing 49,70. β-defensins are primarily expressed in mucosal and epithelial cells 63. In humans, BD1 is constitutively expressed, whereas BD2 and BD3 are induced by inflammatory stimuli, including C. albicans, IL-17 and IL-22 8,13,17. Human BD1, BD2 and BD3 exert direct antifungal activity against C. albicans 65,71,72. Though evolutionarily divergent, murine β-defensins have similar activities and are nonredundant for immunity to OPC 15,48. Human BD2 and mouse BD3 were reported to bind to CCR6, a receptor for CCL20 found on Th17 cells 49,51,52, though this relationship is not universally accepted 50. Loss of BD3 in mice surprisingly did not result in detectable gene disruption during the peak stages of immunity to OPC, contrasting with the substantial changes in mice lacking IL-17RA or IκBζ. The only altered gene in Defb3−/− mice was Sycp1, which is not part of the classic IL-17 or Candida albicans gene signatures. Consequently, these findings imply that BD3 acts at the “end of the line” to clear the fungus without further amplifying the immune response.
In the acute OPC infection model used here, IκBζ is not essential in hematopoietic cells to prevent infection. However, IκBζ can play T cell- and DC-intrinsic roles in driving Th17 cell differentiation 36,73. In this regard, the 5-day OPC model represents an innate response to C. albicans, since this fungus is not part of the commensal flora in mice 2,7,61,74,77. IL-17 and IL-22 in are produced mainly in γδ-T cells and CD4+TCRβ+ ‘natural’ Th17 cells 44,61,74,78,79; only after exposure to C. albicans do mice mount an antigen-specific conventional Th17 adaptive response 61,75,77,80. Hence, it will be informative to determine the role of IκBζ in recall settings.
IκBζ was originally termed ‘IL-1-inducible nuclear ankyrin-repeat protein’ (INAP) or ‘molecule-possessing ankyrin repeats induced by lipopolysaccharide,’ (MAIL) 81-83. Unlike canonical NF-kB, IκBζ is regulated by expression rather than subcellular localization 30, and its expression is quite dynamic. The oscillatory nature of IκBξ expression is not a unique observation 29,43, and the underlying mechanisms governing this are likely to be a result of intersecting feedback regulatory systems affecting transcription, mRNA stability and protein degradation84. IκBζ is transcriptionally controlled by classical NF-κB, which itself exhibits cyclical expression due to feedback inhibitors such as IκB and the ubiquitin editing enzyme A20 42. The 3’ untranslated region (UTR) of the Nfkbiz gene harbors AU-rich elements recognized by RNA binding proteins (RBP) that control translation efficiency and/or mRNA stability 85-87, including the IL-17-induced inhibitor/endoribonuclease Regnase-1 (MCPIP1)that degrades Nfkbiz. Consequently, loss of Regnase-1 improves systemic responses to candidiasis by releasing restraints on IL-17 signaling 87. Another IL-17-activated RBP, Arid5a, stabilizes Nfkbiz mRNA and augments RNA translation of IκBζ 86, although surprisingly Arid5a is dispensable for immunity to oral and systemic candidiasis 28. The ability of Nfkbiz to be regulated at both transcriptional and post-transcriptional levels indicates complex nodes of IκBζ regulation, the nuances of which have yet to be elucidated in full.
IκBζ contains 7 ankyrin repeat motifs that facilitate association with other TFs, such as NF-κB p65 or p50 subunits or ROR nuclear receptors, interactions important for its capacity to transactivate gene expression 25,88,94. ChIP demonstrated association of IκBζ with NF-κB and C/EBP (NFIL6) recognition elements. Since we have ruled out essential roles for C/EBPβ and C/EBPδ in immunity to OPC, the nature of other proteins interacting with IκBζ in this context remains unknown and will be interesting to pursue.
Egr3 is a zinc-finger TF important for cell growth and development 95. Egr3 is linked to IL-17 production in γδ-T cells and directs expression of IL-17 target genes such as IL6 and IL8 96,97. C. albicans induces Egr3 in reconstituted human oral epithelium 55. Likewise, Egr3 is induced in a human OEC line by zymosan and live pathogenic C. albicans, dependent on the Dectin-1 receptor 56. Here we connect IκBζ to Egr3 in vivo based on altered expression in NfkbizK13 mice, and show that this TF regulates IL-17 induction of BD2 in an oral epithelial cell line. An acknowledged limitation of this study is that we were not able to evaluate OPC in Egr3-deficient mice. Exactly how EGR3 participates in the response to OPC will require further study but is likely to be revealing.
IκBζ has emerged as a central signaling mediator in several IL-17-mediated inflammatory diseases. We first connected IκBξ to IL-17 in mesenchymal cells 49,103, and multiple studies have since linked this TF to downstream IL-17 signaling 27,37. IκBζ also mediates signals by IL-1 89-91 and IL-36 40,104, both of which help control candidiasis 60-62. There is no information regarding IκBζ in other fungal infections, to our knowledge, nor are there genetic associations with NFKBIZ and human candidiasis. GWAS studies link this factor to psoriasis vulgaris, psoriasis arthritis, and ulcerative colitis 98-100. Furthermore, Nfkbiz-deficient mice are resistant to experimental autoimmune encephalomyelitis (EAE), an IL-17-dependent model of multiple sclerosis 36. IκBζ in intestinal epithelial cells is essential for promoting hemostasis in the gut and skin 35. Blockade of IL-17 has been very successful clinically, and therefore IκBζ has potential as a therapeutic target for similar conditions. However, the present studies suggest that targeting IκBζ is likely to be come with similar risks for mucosal candidiasis 101.
Mucosal candidiasis is associated with clinical IL-17 blockade 101. IL-17 integrates multiple TFs in its downstream signaling cascades 16,24, but the majority evaluated so far are surprisingly dispensable for immunity to OPC, even though many drive IL-17-dependent autoimmunity 26,28,102,103. Accordingly, the pathways vital for host defense are not always those that promote pathogenic autoimmunity, and therefore defining the relevant molecular players in each will be valuable for pursuing clinical strategies to spare host defense while intervening in autoimmune disease.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sarah Gaffen (sarah.gaffen@pitt.edu).
Materials availability
All unique/stable reagents generated in this study will be available from the Lead Contact with a completed Materials Transfer Agreement. The following mice are under MTA from other institutions: Nfkbizfl/fl (RIKEN #RBRC06410) 104, Act1−/− (NCH). Cebpd−/− (NCI) 105,106, Il17ra−/− (Amgen) and Il22−/− (Genentech). K14CreERT2, Rosa26CreERT2, Defb3−/− mice were from the MMMRC (stock # 011694). K13Cre 15 were from The Jackson Laboratory. Candida albicans strain SC5314 is available at ATCC. Other fungal strains (CAF2-1, BWP17+CIp30, als3Δ, efgΔ/cph1Δ, ClysΔ) will be made available upon request.
Data and Code Availability
RNA-seq data have been deposited at GEO and will be publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data (GSE164241, SRP075350, also listed in the key resources table. Uncropped original immunoblot images are available at the Mendeley Data repository (https://data.mendeley.com/datasets/jxpw9krpz2/1). Diagrams were created on Biorender.com.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGR3 | Cell Signaling Technology | Cat # 2559; AB_11142006 |
| EGR3 | Santa Cruz Biotechnology | Sc-390967 |
| IkappaBzeta | Cell Signaling Technology | Cat # 9244; AB_2151602 |
| IkappaBzeta | Cell Signaling Technology | Cat # 76041; AB_2799879 |
| Rabbit IgG | Cell Signaling Technology | Cat #2729; AB_1031062 |
| IgG Isotype | Cell Signaling Technology | Cat # 3900; AB_1550038 |
| Histone H3 | Cell Signaling Technology | Cat # 4620 AB_1904005 |
| IgG-Light chain- HRP | Cell Signaling Technology | Cat # 93702; AB_2800208 |
| Human BD2 | Peprotech | 500-P161G |
| Biotinylated Human BD2 | Peprotech | 500-P161GBt |
| beta actin | Abcam | Cat# ab49900; AB_867494 |
| rabbit HRP | Thermo Fisher Scientific | Cat# A-11008; AB_143165 |
| mouse HRP | Thermo Fisher Scientific | Cat# 31430; AB_228307 |
| CD45 | Thermo Fisher Scientific | Cat# 48-0451-82; AB_1518806 |
| Ly6G | BD Biosciences | Cat# 551461; AB_394208 |
| CD11b | BioLegend | Cat# 101222; AB_493705 |
| Ghost Dye BV510 | TONBO Biosciences | Cat # 13-870-T100 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IL-17A | Peprotech | Cat# 200-17 |
| Human beta defensin 2 | Peprotech | Cat # 300-49 |
| DharmaFect Transfection Reagent | Dharmacon | T-2001-03 |
| Opti-MEM I reduced serum medium | Gibco | Cat# 31985-070 |
| Collagenase IV | Gibco | Cat# 17104-019 |
| Streptavidin HRP | R&D systems | DY998 |
| Protease inhibitor cocktail | Thermo Fisher Scientific | Cat# 11697498001 |
| DNase | Qiagen | Cat# 79256 |
| Cell lysis buffer II | Thermo Fisher Scientific | Cat # FNN0021 |
| gentleMACS Dissociator C tubes | Miltenyi Biotec | CAt# 130096334 |
| gentleMACS Dissociator M tubes | Miltenyi Biotec | Cat # 130096335 |
| Critical Commercial Assays | ||
| PerfeCTa SYBR Green FastMix, ROX | Quantabio | Cat# 84071 |
| SsoAdvanced University Syber | Quantabio | Cat # 1725270 |
| SuperScript III First Strand Kits | Thermo Fisher Scientific | Cat# 18080-044 |
| iScript cDNA Synthesis kit | BioRad | Cat# 1708890 |
| West Pico PLUSChemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23228 |
| Deposited Data | ||
| Human oral mucosa single cell atlas | Williams, D.W., Greenwell-Wild, T., Brenchley, L., Dutzan, N., Overmiller, A., Sawaya, A.P., Webb, S., Martin, D., Genomics, N.N., Computational Biology, C., et al. (2021). Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell 184, 4090-4104 e4015. 10.1016/j.cell.2021.05.013. | GSE164241 |
| NfkbizK13 2 day OPC RNASeq | PRJNA955230 | |
| Defb3−/− 1 day OPC RNASeq | SUB13072303 | |
| Il17ra−/− 2 day OPC RNASeq | Conti, H., Bruno, V., Childs, E., Daugherty, S., Hunter, J., Mengesha, B., Saevig, D., Hendricks, M., Coleman, B.M., Brane, L., et al. (2016). IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606-617. | SRP075350 |
| Uncropped original immunoblots | This study | https://data.mendeley.com/datasets/jxpw9krpz2/1 |
| Experimental Models: Cell Lines | ||
| TR146 cells (human oral squamous epithelium, female) | ECACC | Cat # 10032305 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | The Jackson Laboratory (JAX) | Cat #000664 |
| C57BL/6 mice | Taconic Farms | B6NTac |
| Nfkbizfl/fl | RIKEN | #RBRC06410 (under MTA) |
| Act1−/− | NIH (Ulrich Siebenlist, deceased) | under MTA |
| Cebpd−/− | NCI (Esta Sterneck) | under MTA |
| Il17ra−/− | Amgen | under MTA |
| Il22−/− | Genentech | under MTA |
| Defb3−/− | MMMRC | under MTA |
| K13Cre | JAX, Conti, H., Bruno, V., Childs, E., Daugherty, S., Hunter, J., Mengesha, B., Saevig, D., Hendricks, M., Coleman, B.M., Brane, L., et al. (2016). IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606-617. | Cat # 034382 |
| Rosa26CreERT2 | JAX | Cat # 008463 |
| K14CreERT2 | JAX | Cat # 005107 |
| Candida albicans SC5314 | ATCC | MYA-2786 |
| Candida albicans CAF2-1 | M. Edgerton, University at Buffalo | N/a |
| Candida albicans BWP17+CIp30 | J. Naglik, King’s College London | N/a |
| Candida albicans als3Δ | S. Filler, Lundquist Inst. | N/a |
| Candida albicans efgΔ/cph1Δ | A Mitchell, University of Georgia | N/a |
| Candida albicans ClysΔ | J. Naglik, King’s College London | N/a |
| Oligonucleotides | ||
| see Table S2 | ||
| Software and Algorithms | ||
| De-Seq2 | Bioconductor | doi:10.1186/s13059-014-0550-8 |
| Prism | Graphpad | https://www.graphpad.com/ |
| Biorender | N/a | https://www.biorender.com/ |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper will be made available from the lead contact upon request.
EXPERIMENTAL MODEL DETAILS
Mice
WT mice were from The Jackson Laboratory, Taconic Farms or generated in-house as littermates from breeding. All mice were on the C57BL/6 background and housed in groups in SPF conditions. Both sexes were used and were assigned randomly to experimental groups.
Cell lines
TR146 cells (female) were purchased from ECACC (European Collection of Authenticated Cell Cultures) and cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM-F12; Gibco) with Pen/Strep and 15% FBS. Aliquots from original purchase were stored as early passage stocks. Aliquots were used for a limited number of passages. Cells were authenticated by visual assessment and distinctive culture characteristics.
Candida albicans
C. albicans were stored as frozen aliquots, streaked on YPD/Amp to generate single colonies, and cultured to log phase in YPD at 30°C.
METHOD DETAILS
Oropharyngeal candidiasis
Mice were maintained in specific pathogen free conditions with food and water ad libitum and a 12 hour dark-light cycle. Mice of both sexes ages 6-12 weeks were used. OPC was induced by sublingual inoculation with 0.0025g cotton balls saturated with 107 CFU C. albicans yeast (strain CAF2-1, cultured to log phase at 30°C) or PBS (uninfected controls) for 75 mins under general anesthesia 38. Weight was tracked daily. Tongue homogenates were prepared using gentleMACS (Miltenyi Biotec) with C-tubes. Fungal burdens were determined by plating serial dilutions in PBS plated on YPD agar with ampicillin and cultured for 48 h at 30°C. Limit of detection is ~30 CFU/g. Where indicated, tamoxifen (TAM, 20 mg/ml in sesame or corn oil) was administered i.p. prior to infection starting on day −12 for 5 days, then rested for 7-9 days. Mice were euthanized if they lost more than 25% weight. Animalsexperiments were conducted according to national and international guidelines and approved by the University of Pittsburgh IACUC.
Cell Culture and RNA silencing
Recombinant mouse or human IL-17 (PeproTech) was used at 100 ng/mL. SiRNAs were from Dharmacon (SMARTpool ON-TARGET plus). See Table S1 for detailed information. TR146 cells were seeded overnight in antibiotic-free media and transfected 18-24 h later with 50 nM siRNA in DharmaFect Reagent. Culture media was replaced after 24 h and stimulations or infections occurred 24 h later. TR146 cells were infected with an MOI of 10 C. albicans yeast 15.
Radiation Chimeras
On day −1, recipient mice (CD45.1 wild type) were given sulfamethoxazole and trimethoprim in the drinking water for 10 days. On day 0, mice were irradiated (900 rad). One day post irradiation, 7-9 x106 cells donor femoral BM cells from WT or NfkbizR26ERT2 were injected i.v. into recipients. After 6 weeks, reconstitution was assessed by flow cytometry for CD45 markers. Reconstituted mice were treated with TAM (20 mg/ml in sesame or corn oil) to induce Cre-mediated deletion, then mice were subjected to OPC.
Flow Cytometry
Tongue tissue was digested with collagenase IV (0.7 mg/mL) in HBSS. Filtered cell suspensions were separated by Percoll gradient centrifugation. Antibodies: anti-CD45 (Invitrogen; #48-0451-82), anti-CD11b (BioLegend; #101222) and anti-Ly6G (BD Biosciences; #551461). Dead cells were excluded using Ghost Dye (Tonbo Biosciences; #13-0870-T100). Data were acquired with an LSRFortessa and analyzed using FlowJo software (Tree Star).
qPCR, RNA Seq
RNA from tongue homogenates was extracted with RNeasy kits (Qiagen). cDNA was synthesized by superscript III Frist Strand Kit (Thermo Fisher) or iScript cDNA synthesis kit (Biorad). Real time PCR was performed using SYBER Green FastMix ROX (Quanta Biosciences) or iTaq Universal Syber Supermix (BioRad) on a CFX Real time Detection System (Biorad). Data were normalized to Gapdh. Primers were from QuantiTect Primer Assays (Qiagen) and see Table S1 for detailed information. For RNA Seq, cDNA libraries were prepared from tongue RNA (Nextera XT Kit) harvested at day 2 p.i. and RNASeq was performed on the Illumina NextSeq 500 platform by the Health Sciences Sequencing Core at the University of Pittsburgh. For Defb3−/− RNASeq reads were annotated and aligned to the UCSC mouse refence genome (mm10, GRCm38.75) using HISAT2. Read counts were generated using subreads FeatureCounts and differential gene expression was performed using De-Seq2 package. Statistical analysis was calculated using R. Il17ra−/− RNASeq data was downloaded from Ref 15 and analyzed using CLC genomics Workbench. NfkbizK13 RNASeq data was analyzed using Partek Flow Software. Sequencing reads were annotated and aligned to UCSC mm10 using STAR. STAR alignment files were used to generate read counts for each gene and differentially expressed genes was performed using Gene Specific Analysis (GSA). RNA-seq raw data files are available at the NCBI Sequence Read Archive (BioProject ID PRJNA955230 and SUB13072303).
Chromatin Immunoprecipitation
ChIPs were performed with the SimpleChIP Plus sonication Kit (Cell Signaling Technology). Cells were crosslinked using disuccinimidyl glutarate (ThermoFisher), followed by formaldehyde cross-linking. Proteins were immunoprecipitated using the following Abs: anti- IκBζ (Cell Signaling; #9244), anti-Egr3 (Cell Signaling; #2559) or IgG (Cell Signaling; #2729). The following primer pairs spanning NF-kB and C/EBP (NFIL6) sites of the DEFB4A proximal promoter were used for PCR: Region 1: (5’-CAGCCCCTCACTCCATTCAC-3’; 5’-TGGTGAGTCAGAGAATGGTCC-3’) Region 2: (5’-GAGGAAGGAAGTGGGCATCC-3’; 5’-ATACAGGGCTGGCTCAAACC-3’), Region 3: (5’-CCATCACCAACAGGGAGACC-3’; 5’-CTACCACCCGCACTTGAGTT-3’) Region 4: (5’-CAGCCCCTCACTCCATTCAC-3’; 5’-TGGTGAGTCAGAGAATGGTCC-3’). Delta Ct value was calculated for each sample from the Ct value obtained for the input.
Immunoblotting and ELISA
TR146 cells were seeded in DMEM-F12 18-24 hours before transfection, infection, or cytokine treatment. Standard denaturing SDS-PAGE and wet transfer were performed. Abs used for immunoblotting: IκBζ (Cell Signaling; #9244, #76041), Egr3 (Cell Signaling; #2559), β-actin (Abcam; #49900). Human BD2 (hBD2) ELISA: plates were coated with 100 uL of anti-hBD2 antibody (Peprotech; #6500P16) overnight. Standard curves were generated with recombinant hBD2 (Peprotech; #300-49). Biotinylated goat anti-hBD2 (Peprotech; #500-P161Gbt) was secondary Ab followed by detection with streptavidin-HRP and absorbance at 450 and 570 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Fungal loads were analyzed by ANOVA with Dunn’s multiple comparisons test. Weight loss was analyzed by two-way ANOVA. qPCR was analyzed by one-way ANOVA, Student’s t-test and indicated post hoc analyses described in Figure Legends. Normality and lognormality tests were performed for each dataset. Data were analyzed on GraphPad Prism and a P value of <0.05 was considered significant. Each symbol represents one mouse or sample. *P <0.05, ** <0.01, ***<0.001, and ****< 0.0001.
Supplementary Material
Highlights.
During candidiasis, IκBζ (Nfkbiz) is upregulated in the oral mucosa via IL-17
Deletion of Nfkbiz renders mice highly susceptible to oropharyngeal candidiasis
IκBζ acts partially in the suprabasal oral epithelium
IκBζ and EGR3 regulate β-defensins, nonredundant antifungal effectors
ACKNOWLEDGMENTS
This work was supported by NIH grants to SLG (DE022550), HRC (DE026898). TCT was supported by T32-AI089443. Nfkbiz−/− mice were provided by RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. We thank Amgen for Il17ra−/− mice, Genentech for Il22−/− mice, U. Siebenlist (NIH, deceased) for Act1−/− mice and Esta Sterneck (NCI) for Cebpd−/− mice. We are grateful to A. Mitchell (University of Georgia), B. Hube (University of Jena), J. Naglik (King’s College London) and S. Filler (Lundquist Institute) for C. albicans strains. L. D’Cruz and N. Moustopoulos provided helpful input. Diagrams were created on Biorender.com.
ABBREVIATIONS
- AMP
antimicrobial peptide
- ARID
AT-rich interacting domain
- BD
β-defensin
- BEL
basal epithelial layer
- C/EBP
CCAAT Enhancer binding protein
- ChIP
chromatin immunoprecipitation
- Clys
candidalysin
- IκBζ
Inhibitor of NF-κB zeta
- MOI
multiplicity of infection
- OEC
oral epithelial cell
- OPC
oropharyngeal candidiasis
- RBP
RNA binding protein
- SEL
suprabasal epithelial layer
- TAM
tamoxifen
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. We unequivocally support inclusive, diverse and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci Transl Med 4, 165rv113. 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Lionakis MS, and Levitz SM (2018). Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annual Rev Immunol 36, 157–191. 10.1146/annurev-immunol-042617-053318. [DOI] [PubMed] [Google Scholar]
- 3.Becker KL, Ifrim DC, Quintin J, Netea MG, and van de Veerdonk FL (2015). Antifungal innate immunity: recognition and inflammatory networks. Semin Immunopathol 37, 107–116. 10.1007/s00281-014-0467-z. [DOI] [PubMed] [Google Scholar]
- 4.Fidel PL Jr. (2011). Candida-Host Interactions in HIV Disease: Implications for Oropharyngeal Candidiasis. Adv Dent Res 23, 45–49. 23/1/45 [pii] 10.1177/0022034511399284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaffen SL, and Moutsopoulos N (2020). Regulation of host-microbe interactions at oral mucosal barriers by type 17 immunity. Sci Immunol 5, eaau4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moutsopoulos NM, and Konkel JE (2018). Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends Immunol 39, 276–287. 10.1016/j.it.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conti HR, and Gaffen SL (2015). IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J Immunol 195, 780–788. 10.4049/jimmunol.1500909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moyes D, Wilson D, Richardson J, S M, Tang S, J W, Hofs S, Gratacap R, Robbins J, Manohursingh R, et al. (2016). Candidalysin: A fungal peptide toxin critical for mucosal infection. Nature 532, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Naglik JR, Konig A, Hube B, and Gaffen SL (2017). Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr Opin Microbiol 40, 104–112. 10.1016/j.mib.2017.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao R, Han W, Tang K, Shao R, Zhu P, Zhang S, Xu P, and He Y (2022). Function of normal oral mucosa revealed by single-cell RNA sequencing. J Cell Biochem 123, 1481–1494. 10.1002/jcb.30307. [DOI] [PubMed] [Google Scholar]
- 11.Presland R, and Dale B (2000). Epithelial structural proteins of the skin and oral cavity: Function in health and disease. Crit Rev Oral Biol Med 11, 383–408. [DOI] [PubMed] [Google Scholar]
- 12.Swidergall M, and Filler SG (2017). Oropharyngeal Candidiasis: Fungal Invasion and Epithelial Cell Responses. PLoS Pathog 13, e1006056. 10.1371/journal.ppat.1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aggor FEY, Break TJ, Trevejo-Nunez G, Whibley N, Coleman BM, Bailey RD, Kaplan H, Naglik JR, Shan W, Shetty AC, et al. (2020). Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis. Sci Immunol 5, eaba0570. 10.1126/sciimmunol.aba0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Presland R, and Jurevic R (2002). Making sense of the epithelial barrier: What molecular biology and genetics tell us about the functions of the oral mucosal and epidermal tissues. J Dent Educ 66, 564–574. [PubMed] [Google Scholar]
- 15.Conti H, Bruno V, Childs E, Daugherty S, Hunter J, Mengesha B, Saevig D, Hendricks M, Coleman BM, Brane L, et al. (2016). IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Bechara R, Zhao J, McGeachy MJ, and Gaffen SL (2019). Interleukin 17 receptor-based signaling and implications for disease. Nat Immunol 20, 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Conti H, Shen F, Nayyar N, Stocum E, JN S, Lindemann M, Ho A, Hai J, Yu J, Jung J, et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho AW, Shen F, Conti HR, Patel N, Childs EE, Peterson AC, Hernandez-Santos N, Kolls JK, Kane LP, Ouyang W, and Gaffen SL (2010). IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol 185, 1063–1070. 10.4049/jimmunol.0903739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, et al. (2015). Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med 212, 619–631. 10.1084/jem.20141065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Puel A, Cypowji S, Bustamante J, Wright J, Liu L, Lim H, Migaud M, Israel L, Chrabieh M, Audry M, et al. (2011). Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332, 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, et al. (2013). A biallelic ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira MC, Whibley N, Mamo AJ, Siebenlist U, Chan YR, and Gaffen SL (2014). Interleukin-17-induced protein lipocalin 2 is dispensable for immunity to oral candidiasis. Infect Immun 82, 1030–1035. 10.1128/IAI.01389-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Vinh DC, Casanova JL, and Puel A (2017). Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr Opin Microbiol 40, 46–57. 10.1016/j.mib.2017.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Draberova H, Janusova S, Knizkova D, Semberova T, Pribikova M, Ujevic A, Harant K, Knapkova S, Hrdinka M, Fanfani V, et al. (2020). Systematic analysis of the IL-17 receptor signalosome reveals a robust regulatory feedback loop. EMBO J 39, e104202. 10.15252/embj.2019104202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slowikowski K, Nguyen HN, Noss EH, Simmons DP, Mizoguchi F, Watts GFM, Gurish MF, Brenner MB, and Raychaudhuri S (2020). CUX1 and IκBζ (NFKBIZ) mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts. Proc Natl Acad Sci U S A 117, 5532–5541. 10.1073/pnas.1912702117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikou SA, Zhou C, Griffiths J, Kotowicz N, Green M, Moyes D, Gaffen SL, Naglik JR, and PJ P (2022). Candidalysin triggers p38 independent of EGFR by two parallel pathways with distinct outputs during oral Candida albicans infection Sci Signal 15, eabj6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simpson-Abelson MR, Childs EE, Ferreira MC, Bishu S, Conti HR, and Gaffen SL (2015). C/EBPβ Promotes Immunity to Oral Candidiasis through Regulation of β-Defensins. PLoS One 10, e0136538. 10.1371/journal.pone.0136538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor TC, Li Y, Li D-D, Majumder S, McGeachy MJ, Biswas PS, Gingras S, and Gaffen SL (2022). Arid5a mediates an IL-17-dependent pathway that drives autoimmunity but not antifungal host defense. J Immunol 209, 1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willems M, Dubois N, Musumeci L, Bours V, and Robe PA (2016). IκBζ: an emerging player in cancer. Oncotarget 7, 66310–66322. 10.18632/oncotarget.11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Annemann M, Plaza-Sirvent C, Schuster M, Katsoulis-Dimitriou K, Kliche S, Schraven B, and Schmitz I (2016). Atypical IκB proteins in immune cell differentiation and function. Immunol Lett 171, 26–35. 10.1016/j.imlet.2016.01.006. [DOI] [PubMed] [Google Scholar]
- 31.Johansen C, Mose M, Ommen P, Bertelsen T, Vinter H, Hailfinger S, Lorscheid S, Schulze-Osthoff K, and Iversen L (2015). IκBζ is a key driver in the development of psoriasis. Proc Natl Acad Sci U S A 112, E5825–5833. 10.1073/pnas.1509971112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kayama H, Ramirez-Carrozzi VR, Yamamoto M, Mizutani T, Kuwata H, Iba H, Matsumoto M, Honda K, Smale ST, and Takeda K (2008). Class-specific regulation of proinflammatory genes by MyD88 pathways and IκBζ. J Biol Chem 283, 12468–12477. 10.1074/jbc.M709965200. [DOI] [PubMed] [Google Scholar]
- 33.Lorscheid S, Muller A, Loffler J, Resch C, Bucher P, Kurschus FC, Waisman A, Schakel K, Hailfinger S, Schulze-Osthoff K, and Kramer D (2019). Keratinocyte-derived IκBζ drives psoriasis and associated systemic inflammation. JCI Insight 4. 10.1172/jci.insight.130835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, and Kramer D (2018). IκBζ is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci U S A 115, 10088–10093. 10.1073/pnas.1801377115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamazaki S, Inohara N, Ohmuraya M, Tsuneoka Y, Yagita H, Katagiri T, Nishina T, Mikami T, Funato H, Araki K, and Nakano H (2022). IκBζ controls IL-17-triggered gene expression program in intestinal epithelial cells that restricts colonization of SFB and prevents Th17-associated pathologies. Mucosal Immunol 15, 1321–1337. 10.1038/s41385-022-00554-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, and Takayanagi H (2010). IκBζ regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 464, 1381–1385. 10.1038/nature08922. [DOI] [PubMed] [Google Scholar]
- 37.Shiina T, Konno A, Oonuma T, Kitamura H, Imaoka K, Takeda N, Todokoro K, and Morimatsu M (2004). Targeted disruption of MAIL, a nuclear IκB protein, leads to severe atopic dermatitis-like disease. J Biol Chem 279, 55493–55498. 10.1074/jbc.M409770200. [DOI] [PubMed] [Google Scholar]
- 38.Solis NV, and Filler SG (2012). Mouse model of oropharyngeal candidiasis. Nat Protoc 7, 637–642. nprot.2012.011 [pii] 10.1038/nprot.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL, and Gaffen SL (2004). Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer binding protein family members. J Biol Chem 279, 2559–2567. [DOI] [PubMed] [Google Scholar]
- 40.Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, Woodgett JR, Wood TD, and Gaffen SL (2009). IL-17 Receptor Signaling Inhibits C/EBPβ by Sequential Phosphorylation of the Regulatory 2 Domain. Sci Signal 2, ra8. 2/59/ra8 [pii] 10.1126/scisignal.2000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawar SA, Sarkar TR, Balamurugan K, Sharan S, Wang J, Zhang Y, Dowdy SF, Huang AM, and Sterneck E (2010). C/EBPδ targets cyclin D1 for proteasome-mediated degradation via induction of CDC27/APC3 expression. Proc Natl Acad Sci U S A 107, 9210–9215. 10.1073/pnas.0913813107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffmann A, Levchenko A, Scott ML, and Baltimore D (2002). The IκB-NF-κB signaling module: temporal control and selective gene activation. Science 298, 1241–1245. 10.1126/science.1071914 298/5596/1241 [pii]. [DOI] [PubMed] [Google Scholar]
- 43.Grondona P, Bucher P, Schmitt A, Schonfeld C, Streibl B, Muller A, Essmann F, Liberatori S, Mohammed S, Hennig A, et al. (2020). Threonine Phosphorylation of IκBζ Mediates Inhibition of Selective Proinflammatory Target Genes. J Invest Derm 140, 1805–1814 e1806. 10.1016/j.jid.2019.12.036. [DOI] [PubMed] [Google Scholar]
- 44.Break TJ, Oikonomou V, Dutzan N, Desai JV, Swidergall M, Freiwald T, Chauss D, Harrison OJ, Alejo J, Williams DW, et al. (2021). Aberrant type 1 immunity drives susceptibility to mucosal fungal infections. Science 371. 10.1126/science.aay5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams DW, Greenwell-Wild T, Brenchley L, Dutzan N, Overmiller A, Sawaya AP, Webb S, Martin D, Genomics NN, Computational Biology C, et al. (2021). Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell 184, 4090–4104 e4015. 10.1016/j.cell.2021.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganz T (2003). Defensins: Antimicrobial peptides of innate immunity. Nat Rev Immunol 3, 710–720. [DOI] [PubMed] [Google Scholar]
- 47.Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, Kohli A, Islam A, Mora-Montes H, Challacombe SJ, and Naglik JR (2010). A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8, 225–235. 10.1016/j.chom.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tomalka J, Azodi E, Narra HP, Patel K, O'Neill S, Cardwell C, Hall BA, Wilson JM, and Hise AG (2015). β-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J Immunol 194, 1788–1795. 10.4049/jimmunol.1203239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schröder JM, Wang JM, Howard OMZ, and Oppenheim JJ (1999). β-Defensins: Linking innate immunity and adaptive immunity through dendritic and T cell CCR6. Science 286, 525–528. [DOI] [PubMed] [Google Scholar]
- 50.Soruri A, Grigat J, Forssmann U, Riggert J, and Zwimer J (2007). β-Defensins chemoattract macrophages and mast cells but not lymphocytes and dendritic cells: CCR6 is not involved. Eur J Immunol 37, 2474–2486. 10.1002/eji.200737292. [DOI] [PubMed] [Google Scholar]
- 51.Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, Martin-Orozco N, Kang HS, Ma L, Panopoulos AD, et al. (2008). CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol 181, 8391–8401. 181/12/8391 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee AY, Phan TK, Hulett MD, and Korner H (2015). The relationship between CCR6 and its binding partners: does the CCR6-CCL20 axis have to be extended? Cytokine 72, 97–101. 10.1016/j.cyto.2014.11.029. [DOI] [PubMed] [Google Scholar]
- 53.Whibley N, Tritto E, Traggiai E, Kolbinger F, Moulin P, Brees D, Coleman BM, Mamo A, Garg A, Jaycox JR, et al. (2016). Antibody blockade of IL-17-family cytokines in immunity to acute murine oral mucosal candidiasis. J Leukoc Biol 99, 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou C, Monin L, Gordon R, Aggor FEY, Bechara R, Edwards TN, Kaplan DH, Gingras S, and Gaffen SL (2020). An IL-17F.S65L Knock-In Mouse Reveals Similarities and Differences in IL-17F Function in Oral Candidiasis: A New Tool to Understand IL-17F. J Immunol 205, 720–730. 10.4049/jimmunol.2000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moyes DL, Shen C, Murciano C, Runglall M, Richardson JP, Arno M, Aldecoa-Otalora E, and Naglik JR (2014). Protection against epithelial damage during Candida albicans infection is mediated by PI3K/Akt and mammalian target of rapamycin signaling. J Infect Dis 209, 1816–1826. 10.1093/infdis/jit824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goodridge HS, Simmons RM, and Underhill DM (2007). Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol 178, 3107–3115. [DOI] [PubMed] [Google Scholar]
- 57.Barker KS, Liu T, and Rogers PD (2005). Coculture of THP-1 human mononuclear cells with Candida albicans results in pronounced changes in host gene expression. J Infect Dis 192, 901–912. 10.1086/432487. [DOI] [PubMed] [Google Scholar]
- 58.Wieland GD, Nehmann N, Muller D, Eibel H, Siebenlist U, Suhnel J, Zipfel PF, and Skerka C (2005). Early growth response proteins EGR-4 and EGR-3 interact with immune inflammatory mediators NF-κB p50 and p65. J Cell Sci 118, 3203–3212. 10.1242/jcs.02445. [DOI] [PubMed] [Google Scholar]
- 59.Naglik JR, Gaffen SL, and Hube B (2019). Candidalysin: discovery and function in Candida albicans infections. Curr Opin Microbiol 52, 100–109. 10.1016/j.mib.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor FEY, Ainscough JS, Ho J, Richardson JP, Coleman BM, et al. (2018). IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J Immunol 201, 627–634. 10.4049/jimmunol.1800515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verma A, Richardson J, Zhou C, Coleman BM, Moyes D, Ho J, Huppler AR, Ramani K, McGeachy MJ, Mufazalov IA, et al. (2017). Oral epithelial cells orchestrate innate Type 17 responses to Candida albicans through the virulence factor Candidalysin. Sci Immunol 2, eeam8834. 10.1126/sciimmunol.aam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Altmeier S, Toska A, Sparber F, Teijeira A, Halin C, and LeibundGut-Landmann S (2016). IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa. PLoS Pathog 12, e1005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khurshid Z, Naseem M, Sheikh Z, Najeeb S, Shahab S, and Zafar MS (2016). Oral antimicrobial peptides: Types and role in the oral cavity. Saudi Pharm J 24, 515–524. 10.1016/j.jsps.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang G, Li X, and Wang Z (2016). APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44, D1087–1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vylkova S, Nayyar N, Li W, and Edgerton M (2007). Human β-defensins kill Candida albicans in an energy-dependent and salt-sensitive manner without causing membrane disruption. Antimicrob Agents Chemother 51, 154–161. AAC.00478-06 [pii] 10.1128/AAC.00478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bishu S, Su E, Wilkerson E, Reckley K, Jones D, McGeachy MJ, Gaffen SL, and M L (2014). RA patients exhibit impaired Candida albicans-specific Th17 responses but preserved protective oral immunity. Arth Res Ther 16, R50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Conti H, Baker O, Freeman A, Jang W, Li R, Holland S, Edgerton M, and Gaffen S (2011). New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol 4, 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Salvatori O, Puri S, Tati S, and Edgerton M (2016). Innate Immunity and Saliva in Candida albicans-mediated Oral Diseases. J Dent Res 95, 365–371. 10.1177/0022034515625222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Huan Y, Kong Q, Mou H, and Yi H (2020). Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front Microbiol 11, 582779. 10.3389/fmicb.2020.582779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dale BA, and Fredericks LP (2005). Antimicrobial peptides in the oral environment: expression and function in health and disease. Curr Issues Mol Biol 7, 119–133. 10.1093/jac/dki103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vylkova S, Li XS, Berner JC, and Edgerton M (2006). Distinct antifungal mechanisms: β-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob Agents Chemother 50, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joly S, Maze C, McCray PB Jr., and Guthmiller JM (2004). Human β-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J Clin Microbiol 42, 1024–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Michaelis L, Tress M, Low HC, Klees J, Klameth C, Lange A, Griesshammer A, Schafer A, Menz S, Steimle A, et al. (2020). Gut Commensal-Induced IκBξ Expression in Dendritic Cells Influences the Th17 Response. Front Immunol 11, 612336. 10.3389/fimmu.2020.612336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Conti H, Peterson A, Huppler A, Brane L, Hernaádez-Santos N, Whibley N, Garg A, Simpson-Abelson M, Gibson G, Mamo A, et al. (2014). Oral-resident ‘natural’ Th17 cells and γδ-T cells control opportunistic Candida albicans infections. J Exp Med 211, 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hernández-Santos N, Huppler AR, Peterson AC, Khader SA, KC M, and Gaffen SL (2013). Th17 cells confer long term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol 6, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pandiyan P, Conti H, Zheng L, Peterson A, Mathern D, Hernandez-Santos N, Edgerton M, Gaffen S, and Lenardo M (2011). CD4+CD25+Foxp3+ regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 infection model. Immunity 34, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bär E, Gladiator A, Bastidas S, Roschitzki B, Acha-Orbea H, Oxenius A, and LeibundGut-Landmann S (2012). A novel Th cell epitope of Candida albicans mediates protection from fungal infection. J Immunol 188, 5636–5643. jimmunol.1200594 [pii] 10.4049/jimmunol.1200594. [DOI] [PubMed] [Google Scholar]
- 78.Verma A, Gaffen SL, and Swidergall M (2017). Innate Immunity to Mucosal Candida Infections. J Fungi 3. 10.3390/jof3040060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sparber F, Dolowschiak T, Mertens S, Lauener L, Clausen BE, Joller N, Stoitzner P, Tussiwand R, and LeibundGut-Landmann S (2018). Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog 14, e1007069. 10.1371/journal.ppat.1007069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, and Huehn J (2019). Microbiome Dependent Regulation of Tregs and Th17 Cells in Mucosa. Front Immunol 10, 426. 10.3389/fimmu.2019.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Haruta H, Kato A, and Todokoro K (2001). Isolation of a novel interleukin-1-inducible nuclear protein bearing ankyrin-repeat motifs. J Biol Chem 276, 12485–12488. 10.1074/jbc.C100075200. [DOI] [PubMed] [Google Scholar]
- 82.Yamazaki S, Muta T, and Takeshige K (2001). A novel IκB protein, IκBζ, induced by proinflammatory stimuli, negatively regulates NF-κB in the nuclei. J Biol Chem 276, 27657–27662. 10.1074/jbc.M103426200. [DOI] [PubMed] [Google Scholar]
- 83.Kitamura H, Kanehira K, Okita K, Morimatsu M, and Saito M (2000). MAIL, a novel nuclear IκB protein that potentiates LPS-induced IL-6 production. FEBS Lett 485, 53–56. 10.1016/s0014-5793(00)02185-2. [DOI] [PubMed] [Google Scholar]
- 84.Dhamija S, Winzen R, Doerrie A, Behrens G, Kuehne N, Schauerte C, Neumann E, Dittrich-Breiholz O, Kracht M, and Holtmann H (2013). Interleukin-17 (IL-17) and IL-1 activate translation of overlapping sets of mRNAs, including that of the negative regulator of inflammation, MCPIP1. J Biol Chem 288, 19250–19259. 10.1074/jbc.M113.452649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamazaki S, Muta T, Matsuo S, and Takeshige K (2005). Stimulus-specific induction of a novel NF-κB regulator, IκBξ, via Toll/Interleukin-1 receptor is mediated by mRNA stabilization. J Biol Chem 280, 1678–1687. [DOI] [PubMed] [Google Scholar]
- 86.Amatya N, EE C, Cruz JA, Aggor F, Garg A, Berman A, Gudjonsson JE, Atasoy U, and Gaffen SL (2018). IL-17 integrates multiple self-reinforcing, feed-forward mechanisms through the RNA-binding protein Arid5a. Science Signaling 11, eaat4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, Conti HR, Hernandez Mir G, Sirakova T, Childs EC, et al. (2015). MCPIP1 Endoribonuclease Activity Negatively Regulates Interleukin-17-Mediated Signaling and Inflammation. Immunity 43, 475–487. 10.1016/j.immuni.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kohda A, Yamazaki S, and Sumimoto H (2016). The Nuclear Protein IκBζ Forms a Transcriptionally Active Complex with Nuclear Factor-κB (NF-κB) p50 and the Lcn2 Promoter via the N- and C-terminal Ankyrin Repeat Motifs. J Biol Chem 291, 20739–20752. 10.1074/jbc.M116.719302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trinh DV, Zhu N, Farhang G, Kim BJ, and Huxford T (2008). The nuclear IκB protein IκBζ specifically binds NF-κB p50 homodimers and forms a ternary complex on κB DNA. J Mol Biol 379, 122–135. 10.1016/j.jmb.2008.03.060. [DOI] [PubMed] [Google Scholar]
- 90.Kao CY, Kim C, Huang F, and Wu R (2008). Requirements for two proximal NF-κB binding sites and IκBζ in IL-17A-induced human β-defensin 2 expression by conducting airway epithelium. J Biol Chem 283, 15309–15318. M708289200 [pii] 10.1074/jbc.M708289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cowland JB, Sorensen OE, Sehested M, and Borregaard N (2003). Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1β, but not by TNF-α. J Immunol 171, 6630–6639. 10.4049/jimmunol.171.12.6630. [DOI] [PubMed] [Google Scholar]
- 92.Sorensen OE, Cowland JB, Theilgaard-Monch K, Liu L, Ganz T, and Borregaard N (2003). Wound healing and expression of antimicrobial peptides/polypeptides in human keratinocytes, a consequence of common growth factors. J Immunol 170, 5583–5589. 10.4049/jimmunol.170.11.5583. [DOI] [PubMed] [Google Scholar]
- 93.Karlsen JR, Borregaard N, and Cowland JB (2010). Induction of neutrophil gelatinase-associated lipocalin expression by co-stimulation with interleukin-17 and tumor necrosis factor-αis controlled by IκBζ but neither by C/EBP-β nor C/EBP-δ. J Biol Chem 285, 14088–14100. M109.017129 [pii] 10.1074/jbc.M109.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muromoto R, Hirao T, Tawa K, Hirashima K, Kon S, Kitai Y, and Matsuda T (2016). IL-17A plays a central role in the expression of psoriasis signature genes through the induction of IκBξ in keratinocytes. Int Immunol 28, 443–452. 10.1093/intimm/dxw011. [DOI] [PubMed] [Google Scholar]
- 95.Taefehshokr S, Key YA, Khakpour M, Dadebighlu P, and Oveisi A (2017). Early growth response 2 and Egr3 are unique regulators in immune system. Cent Eur J Immunol 42, 205–209. 10.5114/ceji.2017.69363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Parkinson RM, Collins SL, Horton MR, and Powell JD (2014). Egr3 induces a Th17 response by promoting the development of γδ T cells. PLoS One 9, e87265. 10.1371/journal.pone.0087265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baron VT, Pio R, Jia Z, and Mercola D (2015). Early Growth Response 3 regulates genes of inflammation and directly activates IL6 and IL8 expression in prostate cancer. Br J Cancer 112, 755–764. 10.1038/bjc.2014.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. (2015). Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47, 979–986. 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stuart PE, Nair RP, Tsoi LC, Tejasvi T, Das S, Kang HM, Ellinghaus E, Chandran V, Callis-Duffm K, Ike R, et al. (2015). Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture. Am J Hum Genet 97, 816–836. 10.1016/j.ajhg.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sakaue S, Kanai M, Tanigawa Y, Karjalainen J, Kurki M, Koshiba S, Narita A, Konuma T, Yamamoto K, Akiyama M, et al. (2021). A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet 53, 1415–1424. 10.1038/s41588-021-00931-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Davidson L, van den Reek J, Bruno M, van Hunsel F, Herings RMC, Matzaraki V, Boahen CK, Kumar V, Groenewoud HMM, van de Veerdonk FL, et al. (2022). Risk of candidiasis associated with interleukin-17 inhibitors: A real-world observational study of multiple independent sources. Lancet Reg Health Eur 13, 100266. 10.1016/j.lanepe.2021.100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simpson-Abelson MR, Hernandez-Mir G, Childs EE, Cruz JA, Poholek AC, Chattopadhyay A, Gaffen SL, and McGeachy MJ (2017). CCAAT/Enhancer-binding protein β promotes pathogenesis of EAE. Cytokine 92, 24–32. 10.1016/j.cyto.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balamurugan K, and Sterneck E (2013). The many faces of C/EBPδ and their relevance for inflammation and cancer. Int J Biol Sci 9, 917–933. 10.7150/ijbs.7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Okuma A, Hoshino K, Ohba T, Fukushi S, Aiba S, Akira S, Ono M, Kaisho T, and Muta T (2013). Enhanced apoptosis by disruption of the STAT3- IκBξ signaling pathway in epithelial cells induces Sjogren's syndrome-like autoimmune disease. Immunity 38, 450–460. 10.1016/j.immuni.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 105.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, et al. (2009). The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol 182, 1617–1630. 182/3/1617 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Banerjee S, Fu Q, Shah SK, Melnyk SB, Sterneck E, Hauer-Jensen M, and Pawar SA (2019). C/EBPδ protects from radiation-induced intestinal injury and sepsis by suppression of inflammatory and nitrosative stress. Scientific reports 9, 13953. 10.1038/s41598-019-49437-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO and will be publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data (GSE164241, SRP075350, also listed in the key resources table. Uncropped original immunoblot images are available at the Mendeley Data repository (https://data.mendeley.com/datasets/jxpw9krpz2/1). Diagrams were created on Biorender.com.
KEY RESOURCES TABLE
| REAGENT or RESOURCE |
SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EGR3 | Cell Signaling Technology | Cat # 2559; AB_11142006 |
| EGR3 | Santa Cruz Biotechnology | Sc-390967 |
| IkappaBzeta | Cell Signaling Technology | Cat # 9244; AB_2151602 |
| IkappaBzeta | Cell Signaling Technology | Cat # 76041; AB_2799879 |
| Rabbit IgG | Cell Signaling Technology | Cat #2729; AB_1031062 |
| IgG Isotype | Cell Signaling Technology | Cat # 3900; AB_1550038 |
| Histone H3 | Cell Signaling Technology | Cat # 4620 AB_1904005 |
| IgG-Light chain- HRP | Cell Signaling Technology | Cat # 93702; AB_2800208 |
| Human BD2 | Peprotech | 500-P161G |
| Biotinylated Human BD2 | Peprotech | 500-P161GBt |
| beta actin | Abcam | Cat# ab49900; AB_867494 |
| rabbit HRP | Thermo Fisher Scientific | Cat# A-11008; AB_143165 |
| mouse HRP | Thermo Fisher Scientific | Cat# 31430; AB_228307 |
| CD45 | Thermo Fisher Scientific | Cat# 48-0451-82; AB_1518806 |
| Ly6G | BD Biosciences | Cat# 551461; AB_394208 |
| CD11b | BioLegend | Cat# 101222; AB_493705 |
| Ghost Dye BV510 | TONBO Biosciences | Cat # 13-870-T100 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IL-17A | Peprotech | Cat# 200-17 |
| Human beta defensin 2 | Peprotech | Cat # 300-49 |
| DharmaFect Transfection Reagent | Dharmacon | T-2001-03 |
| Opti-MEM I reduced serum medium | Gibco | Cat# 31985-070 |
| Collagenase IV | Gibco | Cat# 17104-019 |
| Streptavidin HRP | R&D systems | DY998 |
| Protease inhibitor cocktail | Thermo Fisher Scientific | Cat# 11697498001 |
| DNase | Qiagen | Cat# 79256 |
| Cell lysis buffer II | Thermo Fisher Scientific | Cat # FNN0021 |
| gentleMACS Dissociator C tubes | Miltenyi Biotec | CAt# 130096334 |
| gentleMACS Dissociator M tubes | Miltenyi Biotec | Cat # 130096335 |
| Critical Commercial Assays | ||
| PerfeCTa SYBR Green FastMix, ROX | Quantabio | Cat# 84071 |
| SsoAdvanced University Syber | Quantabio | Cat # 1725270 |
| SuperScript III First Strand Kits | Thermo Fisher Scientific | Cat# 18080-044 |
| iScript cDNA Synthesis kit | BioRad | Cat# 1708890 |
| West Pico PLUSChemiluminescent Substrate | Thermo Fisher Scientific | Cat# 34580 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23228 |
| Deposited Data | ||
| Human oral mucosa single cell atlas | Williams, D.W., Greenwell-Wild, T., Brenchley, L., Dutzan, N., Overmiller, A., Sawaya, A.P., Webb, S., Martin, D., Genomics, N.N., Computational Biology, C., et al. (2021). Human oral mucosa cell atlas reveals a stromal-neutrophil axis regulating tissue immunity. Cell 184, 4090-4104 e4015. 10.1016/j.cell.2021.05.013. | GSE164241 |
| NfkbizK13 2 day OPC RNASeq | PRJNA955230 | |
| Defb3−/− 1 day OPC RNASeq | SUB13072303 | |
| Il17ra−/− 2 day OPC RNASeq | Conti, H., Bruno, V., Childs, E., Daugherty, S., Hunter, J., Mengesha, B., Saevig, D., Hendricks, M., Coleman, B.M., Brane, L., et al. (2016). IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606-617. | SRP075350 |
| Uncropped original immunoblots | This study | https://data.mendeley.com/datasets/jxpw9krpz2/1 |
| Experimental Models: Cell Lines | ||
| TR146 cells (human oral squamous epithelium, female) | ECACC | Cat # 10032305 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | The Jackson Laboratory (JAX) | Cat #000664 |
| C57BL/6 mice | Taconic Farms | B6NTac |
| Nfkbizfl/fl | RIKEN | #RBRC06410 (under MTA) |
| Act1−/− | NIH (Ulrich Siebenlist, deceased) | under MTA |
| Cebpd−/− | NCI (Esta Sterneck) | under MTA |
| Il17ra−/− | Amgen | under MTA |
| Il22−/− | Genentech | under MTA |
| Defb3−/− | MMMRC | under MTA |
| K13Cre | JAX, Conti, H., Bruno, V., Childs, E., Daugherty, S., Hunter, J., Mengesha, B., Saevig, D., Hendricks, M., Coleman, B.M., Brane, L., et al. (2016). IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20, 606-617. | Cat # 034382 |
| Rosa26CreERT2 | JAX | Cat # 008463 |
| K14CreERT2 | JAX | Cat # 005107 |
| Candida albicans SC5314 | ATCC | MYA-2786 |
| Candida albicans CAF2-1 | M. Edgerton, University at Buffalo | N/a |
| Candida albicans BWP17+CIp30 | J. Naglik, King’s College London | N/a |
| Candida albicans als3Δ | S. Filler, Lundquist Inst. | N/a |
| Candida albicans efgΔ/cph1Δ | A Mitchell, University of Georgia | N/a |
| Candida albicans ClysΔ | J. Naglik, King’s College London | N/a |
| Oligonucleotides | ||
| see Table S2 | ||
| Software and Algorithms | ||
| De-Seq2 | Bioconductor | doi:10.1186/s13059-014-0550-8 |
| Prism | Graphpad | https://www.graphpad.com/ |
| Biorender | N/a | https://www.biorender.com/ |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper will be made available from the lead contact upon request.