
Figure 5.
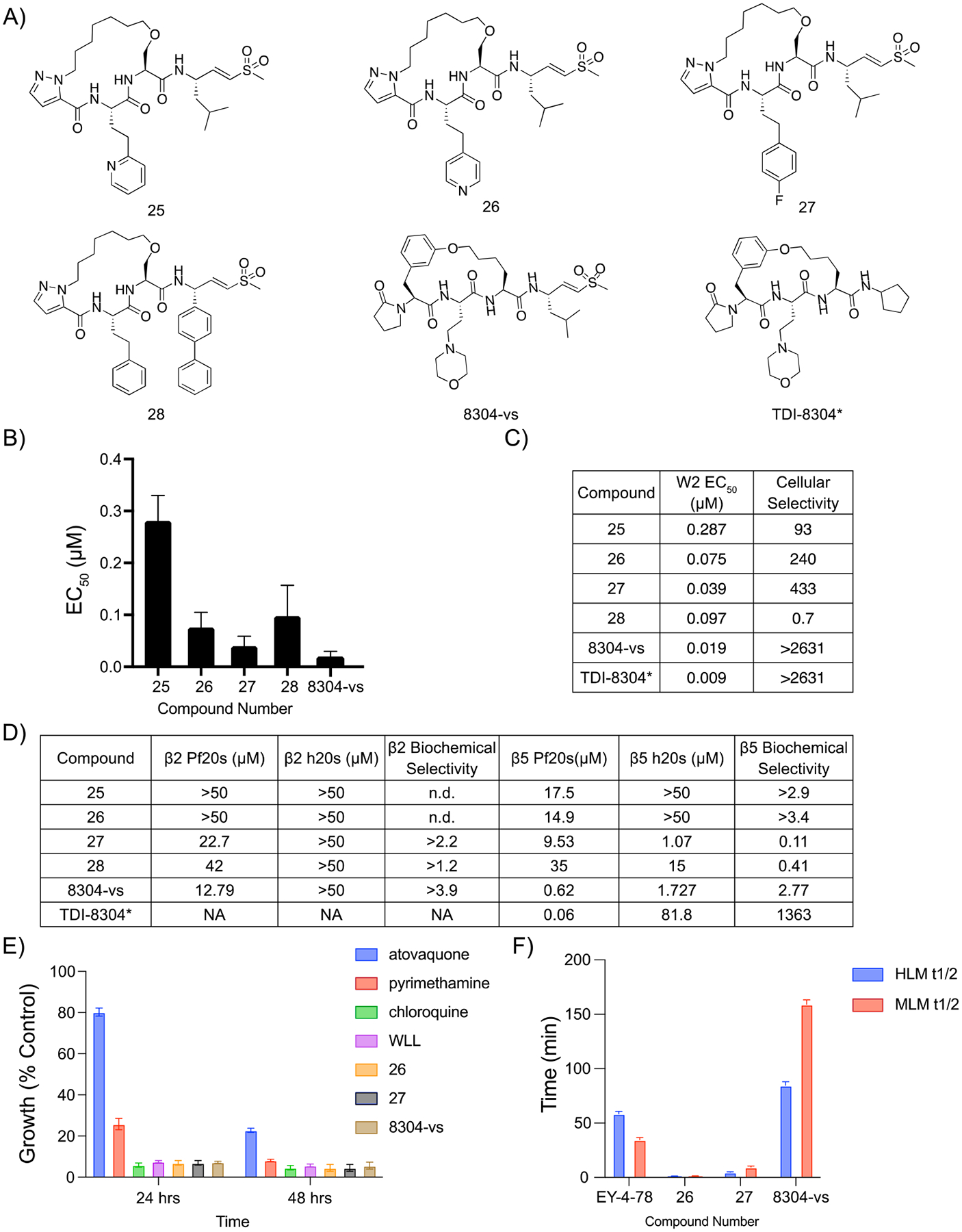
Covalent irreversible proteasome inhibitor 8304-vs is a potent, selective, and stable inhibitor of the P. falciparum proteasome. (A) Structures of pyrazole capped inhibitors with varying P3 or P1 groups and 8304-vs, a covalent version of a previously described noncovalent macrocyclic inhibitor of the proteasome. (B) Compound mean ± SEM EC50 values for each inhibitor assayed against P. falciparum asexual blood stage W2 parasites. Data were generated using 72 h dose–response assays (N, n = 2,2). (C) EC50 values against W2 parasites and cellular indexes comparing P. falciparum parasites to mammalian HFF cells (listed as the ratio of HFF EC50 to parasite EC50 values). (D) Potencies (IC50) of compounds assayed either the β2 or the β5 subunits of the purified Plasmodium or human 20s proteasomes in 1 h inhibition assays. Biochemical selectivity was calculated as the ratio of potency for the two proteasomes (h20s/Pf20s). (E) Plot of rate of kill assay, in which compounds were dosed at their EC50 and parasite viability was monitored over time and compared against known fast (chloroquine), medium (pyrimethamine), and slow (atovaquone) acting inhibitors of parasite growth ± SEM. N,n = 6,2. (F) Bar graphs depicting the half-life of compounds in either human (blue) or mouse (red) microsomes. Compound metabolism was measured by LC/MS/MS. *TDI-8304 data were previously reported.17