

Summary
Efficient sensory processing requires the nervous system to adjust to ongoing features of the environment. In primary visual cortex (V1), neuronal activity strongly depends on recent stimulus history. Existing models can explain effects of prolonged stimulus presentation, but remain insufficient for explaining effects observed after shorter durations commonly encountered under natural conditions. We investigated the mechanisms driving adaptation in response to brief (100 ms) stimuli in L2/3 V1 neurons by performing in vivo whole-cell recordings to measure membrane potential and synaptic inputs. We find that rapid adaptation is generated by stimulus-specific suppression of excitatory and inhibitory synaptic inputs. Targeted optogenetic experiments reveal that these synaptic effects are due to input-specific short-term depression of transmission between layers 4 and 2/3. Thus, brief stimulus presentation engages a distinct adaptation mechanism from that previously reported in response to prolonged stimuli, enabling flexible control of sensory encoding across a wide range of time scales.
Keywords: adaptation, stimulus-specific suppression, paired-pulse plasticity, vesicle depletion, intracellular electrophysiology, extracellular electrophysiology, layer 4, layer 2/3, pyramidal cell, excitation, inhibition
Graphical Abstract
eTOC
Sensory adaptation filters stimulus information by reducing responses to redundant inputs. Using a combination of in vivo and in vitro electrophysiological approaches, Li and Glickfeld find that short-term depression generates stimulus-specific adaptation in superficial layers of mouse primary visual cortex and shapes temporal integration at a seconds-long timescale.
Introduction
Adaptation plays a key role in dynamic regulation of sensory systems. A proposed function of sensory adaptation is to maximize stimulus information from the environment while minimizing metabolic cost of the nervous system1-3. This optimization is particularly important for naturalistic stimuli, which contain highly correlated temporal and spatial structure4-6. By reducing neuronal sensitivity to relatively constant stimulus features, adaptation can improve the efficiency of stimulus representation. To accomplish this, sensory systems must reference a stored memory of recent stimulus statistics, and modulate responses accordingly.
Notably, naturalistic stimuli fluctuate over a wide range of timescales, spanning milliseconds to many minutes, and these dynamics are further enriched by self-generated movements during active sensation7,8. Therefore, reducing redundant encoding requires sensory systems to concurrently store stimulus statistics across a wide range of timescales. Indeed, responses recorded from neurons in visual, auditory, and somatosensory cortices are best predicted by sets of temporal filters that encompass multiple timescales of stimulus history9,10. However, whether adaptation acts to improve encoding through a singular mechanism that acts on multiple timescales, or multiple mechanisms, is still unknown.
Part of this ambiguity arises from the complexity of biological processes related to adaptation. Ion channel kinetics and short-term synaptic plasticity can often be fit with multiple time constants that differ by orders of magnitude1,11-14. However, systematic measures of adaptation across multiple timescales provide strong evidence for contributions from distinct mechanisms. In primary visual cortex (V1), brief and prolonged visual stimulus presentation produce distinct effects on neurons’ orientation tuning curves16. Similar results are observed in human psychophysics, where the duration and dynamics of an adapter determine not only the magnitude, but also specific features of perceived visual aftereffects17,18. Altogether, both perceptual and neural effects of adaptation are consistent with multiple mechanisms that act across different timescales.
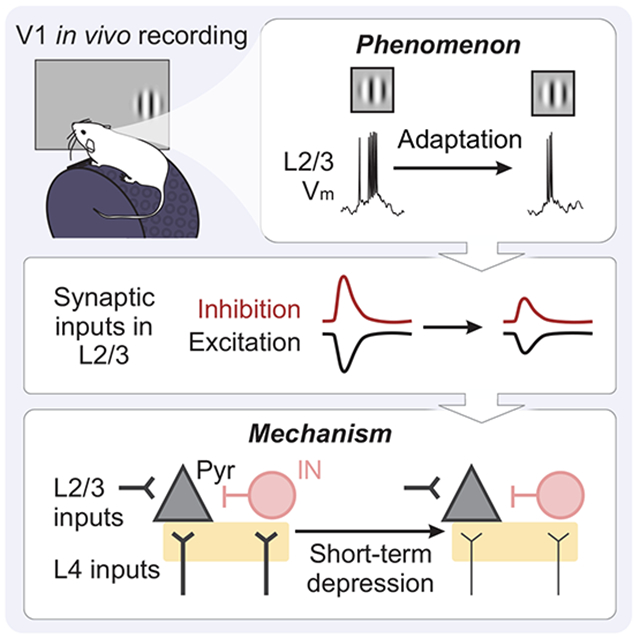
Here, we investigated the mechanism underlying adaptation in layer 2/3 (L2/3) neurons in V1 of alert mice. L2/3 neurons in V1 undergo a profound degree of adaptation to brief stimulus presentations (0.1 s; rapid adaptation)19,20. Consistent with an efficient coding model, visual responses to repeated stimuli are suppressed more than responses to novel stimuli. The majority of this effect appears to originate within cortex, as neurons in both the visual thalamus (lateral geniculate nucleus; LGN) and the thalamic input layer of cortex (layer 4; L4) show relatively little adaptation at this time scale20,21. Cell-intrinsic mechanisms identified within V1 can explain adaptation effects with prolonged stimulus presentation22,23, but are inconsistent with the brief time scale of induction and stimulus-selectivity of rapid adaptation. Instead, these properties have largely been attributed to synaptic mechanisms involving inhibition and short-term plasticity14,24-28.
Using a combination of in vivo and in vitro electrophysiological approaches, we measured the relative contribution of cell-intrinsic and synaptic mechanisms to rapid adaptation. We find that brief stimulus presentation does not hyperpolarize V1 neurons, but rather decreases subsequent stimulus-evoked excitatory and inhibitory synaptic inputs in a manner that can account for firing rate changes with rapid adaptation. Manipulations that directly activate L4, or decrease probability of release at L4 synapses, demonstrate that this site is both necessary and sufficient for rapid adaptation, and argue for a role of short-term depression at this synapse. Altogether, our results highlight a synaptic mechanism that could complement previously characterized cell-intrinsic mechanisms in order to generate sensory adaptation at multiple time scales.
Results
Rapid adaptation reduces stimulus-evoked synaptic inputs
Visual responses of neurons in L2/3 of V1 are reduced following brief (0.1 s) visual stimuli9,19,21. We first investigated whether this rapid adaptation is mediated by cell-intrinsic mechanisms by making intracellular membrane potential recordings of L2/3 V1 neurons in awake, head-fixed mice. Pairs of high-contrast, static gratings (0.1 s, 0.1 cycles per degree, 30° diameter) at the neuron's preferred orientation were presented at a range of inter-stimulus intervals (ISIs) to measure the magnitude and time course of recovery from rapid adaptation (Figure 1A). Presentation of the baseline stimulus induces a decrease in firing rate (FR) in response to the test, consistent with previous studies using calcium imaging and extracellular recordings19,20 (Figure 1B). At short ISIs, responses to the test stimulus are suppressed by 40% (n=13 cells; p<0.001, paired t-test; Figure 1C) and recover with a time constant of ~1 second.
Figure 1. Adaptation suppresses stimulus-evoked responses in L2/3 neurons without affecting cell-intrinsic properties.

A. Left: Recording setup and stimulus paradigm. Animals are head-fixed on a treadmill and membrane potential (Vm) of L2/3 neurons is recorded with a glass pipette. Two stimuli (baseline and test; 0.1 s) are separated by an inter-stimulus interval (ISI) varying from 0.25 to 4 s. Right: Membrane potential is separated into the stimulus-evoked firing rate (black) and stimulus-evoked post-synaptic potential (blue; PSP). B. Top: Membrane potential from an example cell during a 0.25 s ISI trial. Grey shading indicates stimulus presentation. Middle: Raster plot of spike output during 0.25 s (light purple) and 4 s (dark purple) ISI trials and binned peri-stimulus spike histogram (PSTH). Bottom: Same example cell’s trial-averaged subthreshold membrane potential during baseline (left) and test stimulus presentations at 0.25 s (middle) and 4 s (right) ISIs. C. Left: Average normalized firing rate (FR; test/baseline) as a function of ISI for individual cells (gray lines) and all cells (black circles; n=13). Error is SEM across cells. Black line is an exponential fit (τ=0.82 s). Right: Same as left, for average normalized PSP amplitude (τ=0.79 s). D. Average membrane potential preceding baseline and test stimuli for individual cells in 0.25 s ISI trials. Black dot is mean across cells. Error is SEM across cells. E-G. Same as D for spike threshold (E), membrane variance (F), and PSP amplitude (G). See also Figure S1.
To determine whether this decrease in firing rate could be explained by long-lasting hyperpolarization following the baseline stimulus, we compared the membrane potential preceding baseline and test stimuli. Despite the strong suppression of spike output, there is no significant hyperpolarization of membrane potential prior to the test stimulus (p=0.74, paired t-test; Figure 1D). Other properties of the recorded cell that could impact spike output are also unchanged, including the spike threshold (p=0.50, paired t-test; Figure 1E) and spontaneous membrane variance (p=0.85, paired t-test; Figure 1F). Additionally, most neurons have a positive correlation between the number of spikes in response to baseline and test stimuli on each trial, arguing against cell-intrinsic fatigue (Figure S1A). Although activity-dependent hyperpolarization can be generated in V1 L2/3 neurons, it arises only after prolonged periods of activity (Figure S1B-D).
Instead, adaptation in response to brief stimuli greatly reduces stimulus-evoked post-synaptic potentials (PSPs; p<0.001, paired t-test Figure 1B-C, G). Changes in PSPs are similar in both magnitude and time course of recovery to changes in spiking, reflecting the relatively linear transform between membrane potential and firing rate in these neurons (R2 of an exponential vs linear fit, p=0.79, paired t-test). Therefore, rapid adaptation engages a synaptic, rather than cell-intrinsic, mechanism to reduce stimulus-evoked responses to repeated stimuli.
Increases in inhibition, decreases in excitation, or decreases in total conductance could all lead to the reduced stimulus-evoked depolarization that we observed. To identify changes in stimulus-evoked synaptic excitation and inhibition, we made voltage clamp recordings from L2/3 neurons using the same stimulus paradigm (Figure 2A). We recorded excitatory currents (EPSCs) and inhibitory currents (IPSCs) within individual neurons by clamping the membrane potential near the reversal for inhibition (−70 mV) and excitation (+10 mV), respectively (n=10 cells; Figure 2A and S2A). Consistent with our current clamp recordings, there are no changes in either the mean or the standard deviation of the holding current in the time windows preceding baseline and test stimulus onset (paired t-test: p>0.05 for all comparisons), arguing against a role for long-lasting inhibition or changes in overall network excitability in rapid adaptation.
Figure 2. Adaptation drives a balanced reduction of intracortical stimulus-evoked excitation and inhibition.

A. Left: Schematic of recording setup for measuring excitatory and inhibitory currents (EPSCs and IPSCs) in L2/3 neurons. Right: Single trial voltage traces from an example cell held at −70 mV (black) and +10 mV (red), to measure EPSCs and IPSCs respectively. B. Stimulus-evoked EPSCs and IPSCs averaged across all cells (n=10) in response to baseline and test stimuli for all ISIs. Shaded error is SEM across cells. C. Average normalized current amplitudes (test/baseline) for EPSCs (black) and IPSCs (red) for individual cells (small dots) and across all cells (large dots). Curve is exponential fit to the average across cells for each current type (τEPSC=1.10 s; τIPSC=0.93 s). Error is SEM across cells. D. Ratio of excitation to inhibition (E/I) for the baseline and test stimulus in 0.25 s ISI trials. Grey lines are individual cells, black line is average across cells, error is SEM across cells. E. Comparison of visual adaptation in 0.25 s ISI trials in putative pyramidal cells (RS, black) and inhibitory interneurons (FS, gray), obtained from extracellular recordings. Error is SEM across units. F. Top: Schematic of cortical silencing for isolating thalamic excitatory input: photostimulation of ChR2 (green) in parvalbumin (PV)-expressing inhibitory interneurons. Bottom: Stimulus-evoked EPSCs recorded in an example neuron during control (EPSCTotal) and cortical silencing (EPSCThal.) trials. Thin lines are individual trials, thick lines are trial-averaged response. G. Fraction of remaining thalamic current (EPSCThal./EPSCTotal) as a function of cell depth from pia. Vertical dashed lines are depth boundaries used for layer identification. Filled and empty circles are cells with (n=21) and without (n=17) detectable thalamic input, respectively. H. Same as G, but normalized current amplitude of EPSCTotal (test/baseline). I. Same as G, but normalized current amplitude of EPSCThal. (test/baseline). Only cells with thalamic input are included. J. Comparison of normalized EPSC (test/baseline) measured from total versus thalamic input in L2/3 (left) and L4 (right) cells. Grey lines are individual cells and black line is average across cells. Error is SEM across cells. See also Figures S2-3.
Rather, the main effect of adaptation is a robust decrease in the peak amplitude of stimulus-evoked excitation (p<0.001, paired t-test; Figure 2B-C) and inhibition (p<0.001, paired t-test) in response to the test stimulus. This reduces the overall conductance (baseline: 7.45±3.11 nS; test: 3.87±2.76 nS; p<0.001, paired t-test) while preserving the excitation/inhibition (E/I) ratio (p=0.16, paired t-test; Figure 2D). We sometimes observed an overshoot in the recovery of visually-evoked PSCs beyond baseline levels (Figure 2B). This was evident in both EPSCs and IPSCs, arguing against voltage clamp errors, and instead suggests a transient removal of both excitation and inhibition from the network29,30. However, since suppression of excitation recovers relatively quickly and does not vary significantly with ISI (p=0.22, one-way ANOVA), this network suppression cannot explain adaptation.
Our data reveal that adaptation is best explained by the combined decrease in excitatory and inhibitory currents in response to the test stimulus. Consistent with previous experiments and modeling, the decrease in inhibition is not sufficient to overcome the decrease in excitation, resulting in a decrease in PSP amplitude and firing rate31. Stimulus-evoked synaptic inputs are suppressed to a similar degree as the postsynaptic potentials (EPSC vs PSP: p=0.98; IPSC vs PSP: p=0.94; unpaired t-test) and firing rates (EPSC vs FR: p=0.19; IPSC vs FR: p=0.16; unpaired t-test), and recover at a similar time scale (Figure 2C). Thus, the decrease in synaptic drive can account for the magnitude and time course of the reduced visual response following rapid adaptation.
The similar magnitude and time course of changes in excitatory and inhibitory synaptic inputs suggests that these inputs may be yoked by a shared mechanism, such as a decrease in the excitation onto both excitatory and inhibitory cells. Indeed, we find a comparable decrease in FR of both putative excitatory (regular-spiking [RS], n=135 units) and inhibitory neurons (fast-spiking [FS], n=67 units; p=0.08, unpaired t-test; Figures 2E and S3A-B). Altogether, these observations suggest that adaptation arises through short-term depression of excitatory synapses onto both excitatory and inhibitory neurons.
Adaptation acts at specific, intracortical excitatory synapses
Previous studies have attributed the effects of adaptation to short-term depression at excitatory thalamocortical synapses25,27,32. Although thalamocortical input primarily ramifies in L4, L2/3 neurons can also receive direct thalamic input33,34. Thus, we investigated whether adaptation of synaptic inputs in L2/3 reflects short-term depression at thalamocortical synapses or instead arises at intracortical synapses. To isolate the excitatory thalamic input (EPSCThal.) onto neurons in L2/3 and L4, we optogenetically silenced the cortex by activating channelrhodopsin (ChR2) in parvalbumin-expressing inhibitory interneurons and measured visually-evoked activity in response to baseline and test stimuli (STAR Methods; Figure 2F).
Consistent with the anatomical distribution of thalamocortical inputs32,34, the relative proportion of EPSCThal. to EPSCTotal is strongly correlated with proximity to L4 (p=0.001, Pearson correlation; Figure 2G). Only ~50% of neurons in L2/3 receive excitatory input during cortical suppression (n=14/29 cells), whereas this is true for all neurons recorded in L4 (n=7/7 cells). In L2/3 cells with thalamic input, this excitation comprises only a small fraction of the total excitation evoked by the baseline stimulus, significantly less than the proportion in L4 cells (p<0.001, unpaired t-test). Importantly, the depth dependence of thalamic input is not simply due to decreased efficacy of cortical silencing since we reliably observe silencing of spiking across layers (Figure S2D-F) and find complete suppression of excitatory input onto neurons in L5 (Figure 2G). Thus, thalamic input to L2/3 is only a minority of the total input.
To test whether short-term depression of thalamic input can account for cortical adaptation, we measured the normalized EPSC (test/baseline) within interleaved control and cortical suppression trials. On control trials, adaptation of stimulus-evoked EPSCs is significantly stronger in L2/3 than in L4 (p<0.001, unpaired t-test; Figure 2H,J), consistent with extracellular recordings that find adaptation largely arises in L2/320. However, the contribution of thalamic inputs to this effect is minimal—in L2/3 neurons that receive significant thalamic input, this input undergoes significantly less adaptation than the total input (p<0.001, paired t-test; Figure 2I-J) and is similar in magnitude to the weak adaptation of thalamic input in L4 (p=0.71, unpaired t-test). Thus, regardless of layer, thalamic input is not strongly depressing, indicating that adaptation of excitatory inputs to L2/3 must arise at intracortical synapses downstream of thalamocortical input.
If reduction in excitation and inhibition in L2/3 neurons in vivo is generated by short-term synaptic depression of intracortical synapses, adaptation of synaptic inputs should reflect two key features of this type of plasticity. First, repeated visual stimulus presentations will drive increasing depression of visual responses that eventually saturate at a level determined by the balance between time constants of vesicle depletion and replenishment11,12,14. Second, effects will be restricted to the specific subset of synapses activated by features of the baseline stimulus (Figure 3A). To test these predictions, we measured EPSCs and IPSCs in response to static gratings of matched and orthogonal orientations.
Figure 3. Changes in synaptic input are selective to previously activated synapses.

A. Schematic of proposed model of synapse-specific effect of adaptation on excitatory inputs from L4 to L2/3. Color of axons correspond to L4 inputs to L2/3 synapses tuned to vertical (black) versus horizontal (blue) orientations. Line thickness represents strength of inputs. B. Top: Visual stimulus paradigm with repeated presentation of the same stimulus orientation (baseline and test 1-4) followed by an orthogonal orientation (test 5). Middle: Trial-averaged stimulus evoked EPSCs (black) and IPSCs (red) for an example cell. Bottom: Average normalized current (test/baseline) for all cells (n=8). Response to the orthogonal orientation is normalized to its own baseline. Error is SEM across cells. C. Left: Schematic of stimuli presented to measure the tuning width of adaptation. Test orientation was kept constant while the baseline orientation varied. Right: Average normalized current (test/baseline, where the baseline is the same orientation as the test) as a function of similarity between baseline and test stimuli for EPSCs and IPSCs for all cells (n=13). D. Average adaptation tuning curve fits from data in C. Shaded error is SEM across cells. Tuning width (TW) is half-width at half-max. E. Average orientation tuning curve fits from extracellular recording of RS (black) and FS (gray) units. See also Figure S3.
Our results confirm both predictions. First, we find that suppression of both EPSCs and IPSCs accumulate and saturate over repeated stimuli (n=8 cells; test 1 vs baseline 1, p=0.04 for EPSCs and p=0.009 for IPSCs; test 2-4 vs baseline, p<0.001 for EPSCs and IPSCs; all comparisons within test 2-4, p>0.05 for EPSCS and IPSCS; one-way ANOVA with post hoc Tukey test; Figure 3B). Second, excitation and inhibition evoked by a subsequent, orthogonal test stimulus are not significantly different from the baseline response at that orientation (test 5 vs baseline: EPSCs p=0.89; IPSCs p=0.98; paired t-test), consistent with a synapse-specific mechanism.
Across all stimuli presented, the relative ratio of excitation to inhibition remains the same following adaptation (two-way ANOVA: effect of current type, p=0.81). We attribute the maintenance of the excitation-inhibition ratio to a concomitant decrease in excitatory drive to neighboring pyramidal cells and interneurons, which receive divergent feedforward excitatory input from the same presynaptic population31,35. We further tested this by probing the orientation selectivity of adaptation of excitation and inhibition. If decreases in EPSCs and IPSCs are the result of short-term depression at excitatory synapses, the orientation selectivity of adaptation of excitation and inhibition should be matched. Additionally, this selectivity should reflect the tuning of spike output in pyramidal neurons, which are generally more narrowly tuned than interneurons36-38. To measure the tuning width of adaptation, we measured excitation and inhibition in response to pairs of stimuli with orientation differences between 0 and 90 degrees, sampled in 22.5 degree increments (0.25 s ISI only; Figure 3C). We find that the degree of adaptation depends on orientation difference (n=13 cells; two-way ANOVA: main effect of orientation, p=0.009) but not current type. EPSCs and IPSCs undergo a similar degree of suppression across all orientation differences (main effect of current type: p=0.32).
To compare the orientation selectivity of this suppression to orientation tuning of spike output in V1 neurons, we fit individual neurons’ normalized EPSCs and IPSCs in response to the test stimulus with a von Mises function (Figure 3D). We then compared these intracellular adaptation tuning curves to the orientation tuning curves of either RS or FS units obtained in extracellular recordings (Figure 3E and S3C). We find that the bandwidth of adaptation observed in EPSCs and IPSCs more closely matches the bandwidth of orientation tuning of RS units than FS units (tuning width (TW): RS = 21.31±1.25, FS = 26.78±1.91; EPSC = 19.15±3.36, IPSC = 21.57±3.44). This match between the orientation-selectivity of adaptation of IPSCs and RS tuning further supports the idea that changes in excitation and inhibition are yoked by a shared short-term depression mechanism that reduces excitation onto both classes of L2/3 neurons.
Activation of L4 depresses excitatory inputs in L2/3
Our data suggest that rapid adaptation is due to short-term depression of excitatory synapses in L2/3. If so, direct activation of synaptic inputs onto L2/3 neurons should induce short-term depression and mimic the effects of visual adaptation. To test this prediction, we optogenetically activated inputs to L2/3 in slices from mice expressing Channelrhodopsin-2 (ChR2) selectively in L4 pyramidal neurons (Scnn1a-Tg3-Cre x Ai32). To trigger vesicle release in response to somatic action potentials (as opposed to directly exciting axons in L2/3), we targeted a small spot of blue light to the somas of ChR2-expressing L4 neurons below the recorded cell (Figure 4A; STAR Methods). Optogenetic activation of cells in L4 for 0.1 s activates monosynaptic and polysynaptic excitatory inputs to L2/3 neurons (Figure 4B). Repeated stimulation of L4 reveals a history-dependent reduction of optogenetically-evoked EPSCs. As with our in vivo recordings, responses to the test stimulus are strongly suppressed at short ISIs (n=11 cells; p<0.001, paired t-test; Figure 4C) and recover with a time constant of nearly 1 second. This effect is not due to reduced efficacy of L4 activation since optogenetically-evoked activity in L4 neurons is stable across ISIs (p=0.09, one-way ANOVA, effect of ISI; Figure S4A-C). Therefore, the long-lasting suppression of excitatory input to L2/3 neurons observed with rapid adaptation in vivo can be reproduced by engaging a local, activity-dependent mechanism in V1.
Figure 4. Excitatory inputs to L2/3 neurons decrease with repeated stimulation in vitro.

A. Schematic of setup for recording EPSCs in L2/3 neurons during optogenetic stimulation of L4. Two 0.1 s square pulses of blue light (baseline and test) were used to activate L4 neurons. B. Traces during baseline (dark blue) and test (light blue) stimuli from an example cell during 0.25 s (left) versus 4 s (right) ISI trials. Thin lines are individual trials, thick lines are trial-averaged response. C. Average normalized EPSC amplitudes (test/baseline) as a function of ISI for each cell (gray) and the across all cells (blue). Blue line is exponential fit to the average across cells (τ=1.03 s). Dashed line is exponential fit from EPSCs recorded in vivo in Figure 2. Error is SEM across cells (n=11). D. Schematic for recording EPSCs from a L2/3 pyramidal cell while electrically stimulating L4 or L2/3 inputs on alternating trials. E. EPSCs from an example cell in response to stimulation of L2/3 (top; gray) or L4 (bottom; black). Thin lines are individual trials, thick lines are trial-averaged response. F. Average EPSC amplitudes normalized to the first stimulus in response to L2/3 (gray) and L4 (black) stimulation. Error is SEM across cells. See also Figure S4.
L4 stimulation in vitro could drive short-term depression at L4 to L2/3 synapses or at L2/3 to L2/3 synapses. To determine whether these synapses depress equally or in an input-specific manner39,40, we used low intensity electrical stimulation to selectively drive monosynaptic inputs from L4 or L2/3 onto L2/3 neurons (Figure 4D; STAR Methods). Repeated electrical stimulation of L4 in vitro at 4 Hz (0.25 s ISI) is sufficient to depress EPSCs recorded in L2/3 (n=14 cells; Figure 4E). Direct optogenetic activation of L4 somas at the same frequency depresses EPSCs to a similar extent (Figure S4D-F). In contrast, L2/3 excitatory inputs onto the same cells depress significantly less (two-way ANOVA: main effect of input layer, p<0.001; Figure 4F). Given the relatively weak depression observed at L4 to L2/3 inputs at low frequency, we tested whether more closely approximating the in vivo, visually-evoked activity of L4 neurons with high frequency bursts (33 Hz) could produce stronger short-term depression. Indeed, we find that this stimulation paradigm drives depression more comparable to that seen in vivo (Figure S4G-H). Thus, adaptation in V1 L2/3 neurons likely arises from short-term depression at specific excitatory synapses originating from L4 neurons.
Activation of L4, but not L2/3, is sufficient to drive adaptation in vivo
To test whether activation of L4 is sufficient to drive adaptation in vivo, we made extracellular recordings from transgenic mice expressing ChR2 in L4 (Figure 5A). Units were identified as L4 or L2/3 neurons based on their waveform position relative to layer boundaries determined by the visually-evoked current source density (Figure S5). In agreement with the in vitro results, 0.1 s of repeated optogenetic activation of L4 neurons significantly decreases responses in L2/3 neurons, but not L4 neurons (Figure S6A-C). To investigate the interaction between optogenetic activation and subsequent visually-driven responses, we used a 0.5 s sinusoidal light stimulus (Figure S6D-F) and randomly interleaved trials with visual stimulation alone and visual stimulation preceded by optogenetic stimulation of L4 (Figure 5B). We then compared adaptation induced by visual stimulation (Testcontrol/Baselinecontrol: “Visual adapt”; Figure 5C) and adaptation induced by optogenetic stimulation (Baselineopto/Baselinecontrol: “Opto. adapt” Figure 5C).
Figure 5. Activation of L4 neurons is sufficient to recapitulate the effects of visual adaptation.

A. Schematic of in vivo extracellular recording setup with optrode. B. Structure of control trials (black) and ChR2 activation trials (blue). On control trials, baseline and test stimuli are presented with varying ISI. On ChR2 activation trials, 0.5 s of sinusoidal (20 Hz) blue light is used to activate L4 neurons optogenetically at varying intervals prior to baseline visual stimulus presentation. C. Visual adaptation is quantified as the response to the test divided by the response to the baseline stimulus (gray shaded box). Optogenetic adaptation is quantified as the response to the baseline stimulus in ChR2 activation trials divided by the response to the baseline stimulus in control trials (blue arrow). Optogenetic visual adaptation is quantified as the response to the test stimulus divided by the baseline stimulus on ChR2 activation trials (blue shaded box). D. Average z-scored PSTH for L2/3 units during baseline (black) and test (gray) stimuli in control trials and baseline stimulus in ChR2 activation trials (blue; n=34 units). Black line indicates stimulus presentation. Shaded error is SEM across units. E. Comparison of visual adaptation (black) and optogenetic adaptation (blue) in L2/3 (left) and L4 (right) units. Green fill indicates optogenetic stimulation of L4. F. Average z-scored PSTH for L2/3 units during baseline (blue) and test (light blue) stimuli in L4 ChR2 activation trials. G. Visual adaptation (black) and Optogenetic visual adaptation (blue) with 0.25 s ISI at increasing intervals after L4 stimulation (0.25 s, 1 s, 4 s). Normalized firing rate is calculated relative to baseline visual response in control trials (horizontal dashed line). Error is SEM across units. See also Figures S5-6.
Consistent with previous work, V1 neurons in L2/3 are suppressed by visual adaptation at short ISIs (n=34 units; Figure 5D-E) while neurons in L4 undergo significantly less adaptation20 (n=47 units; p<0.001, unpaired t-test). Adaptation induced by optogenetic activation of L4 neurons generates layer-specific effects that are similar to visual adaptation: baseline visual responses are more strongly reduced in L2/3 than in L4 (p<0.001, unpaired t-test; Figure 5D-E). The time scale of recovery from optogenetic adaptation is also similar to recovery from visual adaptation. Consequently, across all ISIs, optogenetic adaptation is indistinguishable from visually-evoked adaptation (two-way ANOVA, effect of stimulation type: L2/3- p=0.93, L4- p=0.47). Notably, in a subset of L2/3 neurons that are not activated by L4 optogenetic stimulation, visual responses are unaffected even shortly after ChR2 activation (n=26 laser-activated and 8 not laser-activated units, p<0.001; Figure S6G). L4 neurons showed a similar, but not significant trend (n=39 laser-activated and 8 not laser-activated units, p=0.11; unpaired t-test; Figure S6H). The restriction of this effect to laser-activated units suggests a role for specific L4 to L2/3 synapses, rather than recruitment of broad, network-level effects. To ensure that these effects are not due to cell-intrinsic fatigue in ChR2-expressing L4 neurons, we monitored L4 in vitro following optogenetic activation and find no significant impact on membrane potential or firing rate (Vm: p=0.95; FR: p=0.10; paired t-test; Figure S6I-L). Thus, activation of L4 is sufficient to reproduce the magnitude, recovery, and layer-specific effects of visual adaptation.
Although optogenetic stimulation of L4 is sufficient to drive adaptation, it is possible that these effects are produced through a different mechanism than visual adaption. Because the effects of visual adaptation saturate quickly with additional stimulus presentations (Figure 3B, 4F and 5C), we reasoned that if optogenetic stimulation and visual adaptation act through the same mechanism, stimulation of L4 should also reduce subsequent visual adaptation20. Conversely, persistence of strong visual adaptation would indicate engagement of distinct mechanisms. To test this, we compared the magnitude of visual adaptation at short (0.25 s) ISIs in control trials and after optogenetic stimulation (Testopto/Baselineopto: “Opto. Visual adapt”; Figure 5C). Following optogenetic stimulation of L4, responses in L2/3 to the test stimulus undergo little visual adaptation (n=24 units; p=0.49) and visual adaptation is significantly reduced (p<0.001, paired t-test; Figure 5F-G). Occlusion of adaptation in L2/3 by stimulation of L4 indicates that adaptation evoked by visual and optogenetic stimulation likely act through the same mechanism.
Optogenetic stimulation of L4 activates both feedforward and recurrent inputs within L2/3. To test whether activation of feedforward inputs from L4 are necessary for visual adaptation, we used in utero electroporation to selectively express ChR2 in L2/3 pyramidal cells (Figure 6A-B). Firing rates in L2/3 are not reduced following L2/3 stimulation (n=27 units; p=0.94, paired t-test; Figure 6C-D), and the magnitude of this effect is significantly smaller than in response to both visually evoked adaptation (p<0.001; paired t-test) and L4 stimulation (p=0.007; unpaired t-test). In addition, unlike L4, L2/3 stimulation does not occlude visual adaptation (n=27 units; p=0.002, paired t-test; Figure 6E-F). Consistent with the preferential short-term depression at L4 inputs to L2/3 observed in vitro, activation of L4, but not L2/3, can recapitulate the effects of visual adaptation. Thus, activation of the L4 to L2/3 synapse is both necessary and sufficient for visual adaptation in L2/3.
Figure 6. Activation of L2/3 neurons does not recapitulate the effects of visual adaptation.

A. Left: Schematic of in vivo extracellular recording setup in mice expressing ChR2 in L2/3 neurons. Right: expression of ChR2-mRuby in L2/3 neurons following in utero electroporation. Scale bar is 100 μm. B. Structure of control trials (black) and ChR2 activation trials (blue). C. Average z-scored PSTH for L2/3 units during baseline (black) and test (gray) stimuli in control trials and baseline stimulus in ChR2 activation trials (blue; n=27 units). Black line indicates stimulus presentation. Shaded error is SEM across units. D. Comparison of visual adaptation (black) and optogenetic adaptation (blue) in L2/3 (left) and L4 (right) units. Red fill indicates optogenetic stimulation of L2/3. E. Average z-scored PSTH for L2/3 units during baseline (blue) and test (light blue) stimuli in L2/3 ChR2 activation trials. F. Visual adaptation (black) and Optogenetic visual adaptation (blue) with 0.25 s ISI at increasing intervals after L2/3 stimulation (0.25 s, 1 s, 4 s). Normalized firing rate is calculated relative to baseline visual response in control trials (horizontal dashed line). Error is SEM across units. See also Figure S5.
Rapid adaptation results from short-term depression at L4 to L2/3 synapses
Short-term depression is associated with activity-dependent depletion of readily releasable vesicles at high release probability (Pr) synapses11. To test whether short-term depression at L4 synapses is necessary for rapid adaptation, we optogenetically manipulated Pr using the modified mosquito opsin, eOPN341. With green light exposure, eOPN3 activates a Gi/o pathway to inhibit calcium channels and SNARE complex formation, reducing vesicle release and decreasing depletion11,41. Thus, we can use eOPN3 to decrease Pr selectively at L4 synapses and test whether this also decreases short-term depression and rapid adaptation.
We expressed eOPN3 in L4 neurons by injecting a Cre-dependent viral construct in Scnn1a-Tg3-Cre mice and confirmed its effects using in vitro whole-cell recordings of EPSCs in L2/3 neurons (Figure 7A). We evoked EPSCs with 4 Hz electrical stimulation in L4 and used a small spot of green light positioned over the recorded cell to activate eOPN3 expressed at L4 axon terminals (Figure 7B). To ensure the effects were specific to eOPN3 activation in L4 axons, we recorded EPSCs evoked with a second stimulation electrode in L2/3 on alternating trials. Due to the relatively slow off-kinetics of eOPN3, we performed these experiments in a block-wise structure (Figure 7A).
Figure 7. Activation of eOPN3 in L4 terminals reduces probability of release at inputs onto L2/3 neurons.

A. Left: Schematic of in vitro recording setup for recording EPSCs in L2/3 neurons while electrically stimulating L4 or L2/3. eOPN3 expressed in L4 neurons is activated with green light over L4 terminals in L2/3. Right: Example image of viral expression pattern. Scale bar is 100 μm. B. EPSCs from an example cell in response to L4 stimulation during first (P1) and second (P2) stimuli in a train (4 Hz), either before (black) or after eOPN3 activation (green). Thin lines are individual trials, thick lines are trial-averaged response. C. Average time course of normalized P1 EPSC amplitudes following L4 (left) or L2/3 (right) stimulation aligned to the time of eOPN3 activation (n=14 cells). Vertical green lines indicate eOPN3 activation trials: induction of 10 s of pulsed green light prior to visual stimulus presentation, followed by a top-up of 0.5 s of pulsed green light prior to each trial. Black curve is exponential fit to recovery (τ=3.34 min). Shaded error is SEM across cells. D. Paired pulse ratio (PPR) during L4 or L2/3 stimulation for individual cells (gray lines) and the average of all cells (black) in control (white) and after eOPN activation (green). Error is SEM across cells. E. Same as D, for coefficient of variation.
Consistent with a reduction in Pr, activation of eOPN3 significantly reduces the amplitude of EPSCs elicited by L4 electrical stimulation (p<0.001, paired t-test; Figure 7C), increases the paired-pulse ratio (p=0.02; Figure 7D), and increases the coefficient of variation (p=0.02; Figure 7E). Activation of eOPN3 also significantly reduces short-term depression of excitation during L4 electrical stimulation matched to in vivo visual responses (p=0.001, paired t-test; Figure S7A-B). In contrast, EPSCs evoked by L2/3 electrical stimulation are significantly less suppressed than L4 stimulation (p=0.003, paired-t-test; Figure 7C) and have no significant change in paired-pulse ratio (p=0.69, paired t-test; Figure 7D) or coefficient of variation (p=0.32; Figure 7E). Following the eOPN3 activation block, the amplitude of evoked L4 EPSCs recover over minutes (Figure 7C). Thus, optogenetic inhibition of L4 terminals can reversibly reduce short-term depression and vesicle depletion in a pathway-specific manner.
We next determined whether decreasing Pr and short-term depression at L4 synapses prevents rapid adaptation of visual responses in vivo. To test this, we recorded V1 neurons extracellularly while presenting pairs of static gratings (0.25 s ISI) and activated eOPN3 using the same block-wise paradigm validated in vitro (L2/3- n=105 units, L4- n=61 units). Green light was delivered via an optic fiber outside the brain to preferentially activate eOPN3 in L4 axons arborizing in L2/3 (Figure 8A-B). To quantify the effect of this manipulation on visual responses, we compared responses to the baseline stimulus on control and eOPN3 activation trials (Figure 8C and S7C). Neurons with less than a 20% change in firing rate were categorized as stable, while neurons that decreased or increased by more than 20% as inhibited or facilitated, respectively (Figure 8D). Consistent with our manipulation largely targeting L4 to L2/3 synapses, most neurons in L2/3 are inhibited following eOPN3 activation (p<0.001, Chi-squared test; Figure 8E), whereas most neurons in L4 are stable (p=0.003, Chi-squared test;).
Figure 8. Decreasing probability of release at L4 terminals decreases visual adaptation in vivo.

A. Schematic of recording setup and eOPN3 expression. B. Block-wise trial structure for measuring effects of eOPN3 activation on visual adaptation. Visual stimuli are always presented with 0.25 s ISI. C. PSTHs for two example units in control (black) and eOPN3 activation (green) trials. ΔFR is calculated as the change in peak stimulus-evoked response. D. Distribution of change in visually-evoked responses to the baseline stimulus in L4 (pink; n=61) and L2/3 (gray; n=105) units. Vertical solid lines indicate thresholds for categorization as inhibited (<0.8), stable (≥0.8 and ≤1.2), or facilitated (>1.2). E. Percent of units categorized as inhibited, stable, or facilitated in L2/3 and L4. F. Average z-scored PSTH of inhibited L2/3 units (n=65) in response to baseline (dark) and test (light) stimuli during control trials (left, black) and during eOPN3 activation trials (right, green). Black line indicates stimulus presentation. Shaded error is SEM across units. G. Same as F, for stable L2/3 units (n=28). H. Comparison of normalized response (test/baseline) in control and eOPN3 activation trials, for all L2/3 units colored by categorization in E. I. Average normalized response for inhibited (dark green) and stable (light green) units in L2/3. Error is SEM across units. See also Figures S5, 7 and 8.
If suppression of neurons in L2/3 is indicative of decreased Pr at L4 inputs to those neurons, visual adaptation should also be most affected in neurons L2/3 neurons inhibited by eOPN3 activation. Indeed, inhibited neurons in L2/3 undergo significantly less visual adaptation after eOPN3 activation (p<0.001, paired t-test; Figure 8F, H-I). In comparison, adaptation is unaffected in stable neurons in L2/3 (p=0.99; Figure 8G-I), inhibited neurons in L4 (p=0.21; Figure S7D-E), and stable neurons in L4 (p=0.45). The overall effect of eOPN3 activation on L2/3 neurons is similar to the effect we observed with eOPN3 activation with high frequency stimulation in vitro (norm. FR all L2/3, control: 0.61±0.02, +eOPN3: 0.87±0.06; Figure S7A-B). Thus, the effects observed in vivo are consistent with the changes observed in vitro, considering the neurons recorded in vitro are a random sample of neurons in L2/3.
These effects cannot be explained by non-specific effects of the laser, as green light has no significant effect on responses of L2/3 neurons to the baseline stimulus (n=35 units; p=0.75; paired t-test; Figure S7F-G), or the degree of adaptation (p=0.34), when L4 neurons express only a fluorophore. Nor could these effects be explained solely by reduced visual responses in L2/3 induced by eOPN3, as L2/3 neurons exhibit a comparable reduction of visually-evoked firing with a decrease in stimulus contrast (n=29 units; p<0.001; paired t-test; Figure S8) with no significant effect on adaptation (p=0.45; two-way ANOVA, effect of baseline contrast). Together, our results indicate that short-term depression at high Pr L4 to L2/3 synapses in V1 is necessary for the effects of visual adaptation.
Discussion
We show that synaptic depression at feedforward synapses within primary visual cortex can explain stimulus-specific adaptation of visually-evoked responses. Thus, features of rapidly changing visual stimuli are stored through activity-dependent modulation of synaptic efficacy. Moreover, the effects of this modulation reduce sensitivity to repeated stimulus features, potentially serving to improve efficiency of stimulus encoding.
Direct evidence for a synaptic depression mechanism in adaptation
Short-term plasticity is a fundamental feature of synaptic transmission that transforms physically static synapses into dynamic filters11,42,43. In vitro results from electrical stimulation12, two-photon optogenetic input mapping39, and paired recordings40,44 have found robust short-term depression at L4 to L2/3 synapses in V1. Using whole-cell recordings of L2/3 neurons in vitro and in vivo we demonstrate that short-term plasticity of L4 output can explain the long-lasting and stimulus-specific effects of rapid adaptation on spike output.
Many studies have hinted that synaptic depression plays a role in generating sensory adaptation in vivo10,24,25,27,35,45-49. In cat visual cortex, repeated electrical stimulation of LGN neurons decreased both excitation and inhibition in V127. Similarly, repeated whisker stimulation and auditory clicks drive a balanced reduction in excitation and inhibition in L4 of somatosensory and auditory cortex24,45. Although these findings are consistent with synaptic depression, these experiments did not directly test the role of short-term plasticity. Here, we leverage cell-type specific in vitro and in vivo optogenetic manipulations to reveal that activation of L4 inputs to L2/3 is both necessary and sufficient for producing the effects of visual adaptation. Notably, our manipulation of probability of release at L4 synapses (using the newly developed inhibitory opsin eOPN3 to inhibit calcium channel activation41) enabled us to directly test the role of synaptic depression in vivo in the awake animal. Although eOPN3 activation could also engage additional signaling pathways via cAMP to alter synaptic transmission50, our findings strongly argue that L4 synapses play a critical role in temporal integration in L2/3 neurons.
Synaptic depression has previously been reported at both retinogeniculate and thalamocortical synapses35,51-54. In our preparation, we found very little adaptation of thalamocortical inputs to V1. This is consistent with recent work in awake mice finding only weak adaptation of LGN neurons and thalamic input to V120,21,55. These results highlight how differences in spontaneous activity across state (awake vs anesthetized) and preparation (in vivo vs in vitro) alter the levels of vesicle depletion and therefore the degree synaptic depression at each site51,56-58. Thus, we view our in vitro results as a careful dissection of the dynamics of synaptic inputs to L2/3 rather than a quantitative recapitulation of adaptation in vivo, and our in vivo results as revealing the relevant mechanisms that govern visual processing in the alert animal.
Rapid adaptation is not associated with increased inhibition
Increased inhibition has also been proposed to mediate stimulus-specific adaptation59-63. Facilitation of excitatory inputs onto somatostatin-expressing interneurons is thought to sensitize them to repeated or prolonged stimulus presentations64-66. Furthermore, manipulation of these interneurons selectively affects responses to frequent, but not rare, stimuli in visual and auditory cortex26,67,68.
Contrary to this model, we do not find either long-lasting inhibition in response to the adapter or an increase in inhibition in response to the test. Effects of adaptation on excitation and inhibition are tightly linked, similar to adaptation of FS interneurons and neighboring RS cells. Thus, adaptation of inhibition is likely driven by short-term depression of excitatory inputs onto L2/3 interneurons rather than short-term dynamics of their output inhibitory synapses. We did not find evidence for additional depression of inhibition, which is somewhat surprising considering in vitro studies documenting robust depression of inhibitory synapses59,69-71. We propose that the high firing rates of interneurons in vivo may put their synapses in a tonically depressed state, rendering them stable across a range of stimulus intervals51,72.
While whole-cell recordings in vivo have limited space clamp and likely underestimate dendritic inhibition, we saw no dependence of adaptation of either excitatory or inhibitory currents on series resistance (Figure S2B-C). Thus, while space clamp errors may bias our estimates of the absolute E/I ratio, they do not influence our comparison of the E/I ratio before and after adaptation. Our finding that the E/I ratio is maintained following adaptation thus argues against a role for facilitating dendritic inhibition. Furthermore, since decreases in total synaptic drive are sufficient to explain the changes observed in spike output, increased inhibition is unlikely to explain adaptation at this time scale of induction and recovery.
Distinct time scales and perceptual effects of adaptation
Short-term depression yields a distinct set of computational capacities from cell-intrinsic fatigue. One key difference is the ability to store stimulus history via modifications to individual synapses. Independent gain changes at each of a neurons’ thousands of inputs can greatly increase the specificity and complexity of adaptation73-75. Our results indicate that adaptation selectively regulates L4 to L2/3 inputs, a key cortical, feedforward synapse in visual processing. Input-specific depression at L4 but not L2/3 inputs could shift the relative balance of information flow from feedforward to recurrent connections. Moreover, a cortical site of depression (as opposed to thalamocortical) enables adaptation to be specific to cortically-computed stimulus features (e.g. orientation, phase and spatial frequency)76,77. This stimulus-specificity is a necessary component for adaptation to reduce spatiotemporal correlations in visual input. Therefore, our findings could provide the mechanistic basis for increased efficiency of encoding following adaptation1,46,78,79.
Additionally, synaptic depression has been proposed to act as a filter for cortical processing1,12,42,80. Modeling studies predict that short-term depression normalizes each input to its own firing rate to optimize detection of changes in weaker presynaptic inputs43,75. Frequency-dependent depression also implements a low-pass temporal filter that has been hypothesized to set the threshold for perceptual flicker-fusion55. Thus, this form of adaptation may also shape temporal integration by limiting the rates at which cortical neurons can follow fluctuations in visual inputs43,81.
Moving forward, we can begin to connect the diversity of perceptual effects of adaptation to the diversity of underlying biological mechanisms. Perceptual effects of adaptation depend on stimulus duration17,18. Our data indicate that this could arise through complementary mechanisms that ebb and flow on different time scales within the same neurons. This is consistent with studies that have identified multiple timescales of adaptation within single neurons that vary by orders of magnitude9,10. As a result, visual perception is shaped by concurrent dependencies on stimulus history that vary in their computational capacities. Another interpretation of these multiple forms of adaptation is as a series of mechanisms that work together to reduce activity in stages—during periods of elevated excitation, if synaptic depression has saturated and firing rates remain high, cell-intrinsic hyperpolarization can be engaged to reduce activity. Although this comes at the expense of stimulus specificity, it may be necessary to maintain cortical homeostasis. Indeed, prior studies using a greater number of stimulus presentations have identified both orientation specific and nonspecific components of adaptation10,20. Thus, future work will be important for understanding the interactions between distinct mechanisms in individual neurons.
In summary, we have linked a well-studied synaptic mechanism to the in vivo phenomenon of adaptation at rapid timescales in V1. Synaptic and cell-intrinsic mechanisms likely co-exist within single neurons but are engaged at different time scales9,10,46. Our findings provide long sought-after evidence for a synaptic depression mechanism that generates sensory adaptation and sparsens representations. Release probability regulates short-term depression11; thus, molecular machinery and neuromodulators that affect Pr are widespread means of specifying temporal integration. Given the similarities between sensory cortical circuits, it is possible this mechanism is shared across other stimulus modalities9,82-84. Input-specific release dynamics could further specialize transmission by tuning the degree of adaptation within an area or cell type. The excitatory neurons we studied in L4 of mouse V1 are primarily pyramidal neurons, whereas L4 of somatosensory cortex in mice and L4 of V1 in other species are primarily comprised of stellate cells85. Differences in cell type identity may translate to area-specific or species- specific short-term plasticity, and consequently, adaptation. Therefore, studies of short-term plasticity at synapses further along the visual hierarchy20, in different behavioral contexts86, sensory areas9, or species16, could all generate insights into how fundamental attributes of synapses shape the neural code.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to Lindsey Glickfeld (glickfeld@neuro.duke.edu).
Materials availability
No new reagents were generated as a result of this study.
Data and code availability
All electrophysiology data included in the manuscript figures is available on Figshare. A link is provided in the Key resources table.
All original code needed to generate the manuscript figures is available on Figshare. A link is provided in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| rAAV2/1&2.hSyn.SIO-eOPN3-mScarlet | Addgene | 125713 |
| AAV1.CAG.Flex.tdTomato.WPRE.bGH | Addgene | 51503 |
| Deposited data | ||
| Data and code for analysis | This paper | https://doi.org/10.6084/m9.figshare.23519394.v1 |
| Experimental models: Organisms/strains | ||
| Scnn1a-Tg3-Cre | Jackson Labs | 009613 |
| Pvalb-Cre | Jackson Labs | 008069 |
| Ai32 | Jackson Labs | 012569 |
| CBA | Jackson Labs | 000654 |
| Recombinant DNA | ||
| pCAG-ChR2-mRuby | Addgene | 109125 |
| Software and algorithms | ||
| ImageJ | NIH | https://micro-manager.org |
| Micromanager | NIH | https://imagej.nih.gov/ij/ |
| MWorks | MWorks | http://mworks-project.org |
| pClamp 10 Software Suite | Molecular Devices | N/A |
| MATLAB | Mathworks | https://www.mathworks.com |
| Chemicals | ||
| Fast Green | Sigma Aldrich | F7252 |
| Fluoromount G | Invitrogen | 00-4959-52 |
| DiO | Invitrogen | V22886 |
| NBQX | Tocris Bioscience | Cat #: 1044; CAS: 479347-86-9 |
| D-APV | Tocris Bioscience | Cat #: 0106; CAS: 79055-68-8 |
| Dental Cement | C&B Metabond | S380 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals.
All procedures conformed to standards set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Duke University's Animal Care and Use Committee. Mice were housed on a normal 12:12 light-dark cycle. Data in this study were collected from 82 mice (39 female). For experiments involving selective expression in layer 4 V1 neurons, we used Cre-positive offspring from Scnn1a-Tg3-Cre mice (Jackson Labs #009613) crossed with either Ai32 (Jackson Labs #012569, n = 15), or CBA (Jackson Labs, #000654, n = 20). We also used offspring from Scnn1a-Tg3-Cre and CBA mice for in utero electroporation (n = 11) but did not select for Cre expression. For cortical silencing experiments, we used offspring from Pvalb-Cre mice (Jackson Labs, #008069) crossed with Ai32 mice (n = 6) or CBA mice that were injected in V1 to express ChR2 virally (n = 2). All other experiments did not require cell-type specific expression; thus, mice were a mix of genotypes (n = 32). Transgenic mice were heterozygous and bred on a C57/B6J background (Jackson Labs #000664) with up to 50% CBA/CaJ (Jackson Labs #000654). In vivo electrophysiology experiments used mice 6-22 weeks old and in vitro electrophysiology experiments used mice 4-12 weeks old. At the time of viral injection, mice were at least 4 weeks old.
METHOD DETAILS
Surgical Procedures
Intracranial viral injections.
Burrhole injections of viral constructs [rAAV2/1&2.hSyn.SIO-eOPN3-mScarlet (Addgene 125713 diluted to 6 x 1012 viral genomes/mL) or AAV1.CAG.Flex.tdTomato.WPRE.bGH (Addgene 51503; diluted to 3 x 1012 viral genomes/mL)] were used to selectively express opsins and control fluorophores in layer 4. This approach was also used to express ChR2 in two of the Pvalb-Cre mice used in cortical silencing experiments (AAV5-EF1a-double floxed-hChR2(H134R)-EYFP-WPRE-HGHpA [Addgene 20298-AAV5 diluted to 4 x 1011 viral genomes/mL]). Mice were anesthetized with isoflurane and positioned in a stereotax (Kopf Instruments). Meloxicam (5 mg/kg) was administered subcutaneously and bupivacaine (5 mg/kg) was administered locally prior to incision. After the skull was exposed, a small hole was drilled −2.6 mm lateral from lambda and directly anterior to the lambdoid suture targeting the posterior and medial aspect of the primary visual cortex (V1). Injection micropipettes were pulled from glass capillary tubes (1B100F-4, World Precision Instruments) and backfilled with virus and then mineral oil and mounted on a Hamilton syringe. The pipette was lowered into the brain and pressure injected at multiple depths using an UltraMicroPump (World Precisions Instruments; 2 x 100 nL; −350 μm and −450 μm from the surface for L4; −550 μm and −250 μm from the surface for ChR2 expression in PV interneurons). We waited between 4.5-7 weeks for viral expression of eOPN3 (for both in vitro and in vivo electrophysiology) and 2-3 weeks for viral expression of ChR2 in PV-expressing interneurons. Viral expression was confirmed expression post hoc.
In utero electroporation.
Embryos from timed-pregnant CBA female mice (E15.5-16.5) mated to Scnn1a-Tg3-Cre males were used to obtain expression in layer 2/3. Meloxicam was administered pre-operatively (1 mg/mL, 5 mg/kg; subcutaneous). Animals were maintained under anesthesia (2.5% isoflurane), the abdomen was cleaned with ethanol and then swabbed with iodine. An incision was made in the skin and then in the abdominal wall, then covered in a drape made with sterile surgical gauze. Uterine horns were carefully removed and kept moist with warm PBS throughout the surgery. Embryos were injected with plasmid mixture (1.5 μg/uL pCAG-ChR2-mRuby-ST in 0.5% Fast Green in UltraPure water, Addgene 109125) in the left ventricle using a glass micropipette pulled to a 70 μm beveled tip. After injection, a series of voltage steps (five voltage pulses of 50 V at 1 Hz with each pulse lasting 50 ms) was applied to each embryo using 5 mm round tweezertrodes (BTX, BTX ECM 830 ElectroSquarePorator). Paddles were oriented to target V1. Embryos were gently returned to the abdomen in the same side that they were removed from. The abdominal wall was sutured before applying bupivicane (5 mg/kg) and then suturing the skin. Animals were allowed to recover on a heating pad until mobile. Strength and location of expression was screened with trans-cranial fluorescence of mRuby following headpost implantation.
Headpost implantation.
Mice were anesthetized with a mixture of ketamine/xylazine (ketamine: 50 mg/kg, xylazine: 5 mg/kg; intraperitoneal) and isoflurane (1.2–2% in 100% O2). Meloxicam was administered pre-operatively (1 mg/mL, 5 mg/kg; subcutaneous). Using aseptic technique, a custom-made titanium headpost was secured over V1 using clear dental cement (C&B Metabond, Parkell). Buprenex (0.05 mg/kg) and cefazolin (50 mg/kg) were administered post-operatively. Animals were allowed to recover for at least 1 week prior to experiments.
Visual and optogenetic stimulus presentation.
Visual stimuli were presented on a 144-Hz (Asus) LCD monitor, calibrated with an i1 Display Pro (X-rite). The monitor was positioned 21 cm from the contralateral eye. Visual stimuli were controlled with MWorks (http://mworks-project.org). Circular gabor patches containing sine-wave gratings (30° diameter; 0.1 cycles per degree; 80% contrast) alternated with periods of uniform mean luminance (60 cd/m2). Timing of visual stimulus onset was measured for aligning neural data via a photodiode that directly measured output from the LCD. All baseline and test stimuli were presented for 0.1 s, with inter-stimulus intervals (ISIs) ranging from 0.25 s to 4 s and inter-trial interval of 8 s to allow for adequate recovery.
PV-ChR2 cortical silencing.
Control and cortical suppression trials were randomly interleaved. All trials consisted of two vertically oriented static gratings separated by a 0.25 s ISI. On cortical suppression trials, PV interneurons were activated with a square laser step with a tapered offset (to prevent rebound effects). Suppression started 0.3 s prior to the onset of the first visual stimulus and ended 0.75 s after the offset of the second visual stimulus.
ChR2 activation of L4 and L2/3.
Control and ChR2 activation trials were randomly interleaved. Control trials consisted of two vertically oriented static gratings separated by a 0.25, 1, or 4 s ISI. ChR2 activation trials consisted of a sine-wave laser pulse (0.5 s, 20 Hz, 450 nm, Optoengine) followed by a grating (0.25, 1, or 4 s ISI). In a subset of experiments, two static gratings (0.25 s ISI) were presented following ChR2 activation to measure the effect on visual adaptation. The effect of serial ChR2 activation was also tested using brief (0.1 s) square-wave pulses (Figure S6A-C).
Stimulus specificity of adaptation.
Two protocols were used to test the stimulus specificity of adaptation. 1) Five repeated presentations of a static grating (baseline and test 1-4; 0.25 s ISI) followed by a presentation of the orthogonal orientation (test 5). On randomly interleaved trials, the repeated and orthogonal orientation were switched to obtain the baseline amplitude of both orientations. 2) Two oriented gratings were presented with an ISI of 0.25 s. The test stimulus was the same across trials while the baseline stimulus was varied from 0 to 90 degrees from the test in 22.5 degree increments.
Orientation tuning.
Drifting gratings (2 Hz) moving in 16 directions (22.5 degree increments) were presented for 1 s with an 8 s inter-trial interval to measure the orientation tuning of neurons.
eOPN3 activation.
All trials consisted of two vertically oriented baseline and test stimuli separated by 0.25 s ISI. After 20 control trials, eOPN3 was activated with a square-wave laser pulse (10 s, 10 Hz, 530 nm, Optoengine). We then tested the effect of eOPN3 over 20 trials with top-up activation (0.5 s, 10 Hz) preceding visual stimulation on each trial. Recovery was measured during a subsequent 90-100 trials. Each experiment contained 1-3 repeats of eOPN3 activation blocks.
Contrast dependence of adaptation.
Two vertically oriented gratings were presented with an ISI of 0.25 s. The test stimulus was the same (80% contrast) across trials while the baseline stimulus was either 40 or 80% contrast.
Experimental Procedures
In vivo retinotopic mapping.
For all in vivo electrophysiological recordings, V1 boundaries were first identified with retinotopic mapping with intrinsic signal imaging through the skull. The skull was illuminated with orange light (590 nm LED, Thorlabs), and unfiltered emitted light was collected using a CCD camera (Rolera EMC-2, Q Imaging) at 2 Hz through a 5x air immersion objective (0.14 numerical aperture (NA), Mitutoyo), using Micromanager acquisition software87. Drifting gratings (80% contrast, 2 Hz, 0.1 cpd) were presented for 2 s at 3 positions with a 4 s interstimulus interval. Collected images were analyzed in ImageJ to measure changes in reflectance at each position (dR/R; with R being the average of all frames) to identify V1.
Preparation for in vivo electrophysiology.
Animals were habituated to head-fixation for 1-3 days prior to surgery. On the day of recording, animals were anesthetized with isoflurane and a small craniotomy (< 1 mm diameter) was made over a V1 location identified by intrinsic signal imaging. For extracellular recordings, a gold ground pin was inserted in an anterior portion (outside of visual areas) within the headpost and secured with dental cement. Damage to superficial cortex was minimized by drilling in brief bouts (< 1 s) and alternating drilling and cooling with chilled glucose-free HEPES-based artificial cerebral spinal fluid (ACSF; in mM: 141 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2 1.3 MgCl2). A slit was made in the dura with a syringe and the craniotomy was kept covered with ACSF for the remainder of the experiment. For laser stimulation during in vivo whole-cell recordings, an optic fiber (Thorlabs) was attached to the headpost with dental cement on the day of recording. The optic fiber was positioned at an angle with the tip terminating ~500-700 um from the craniotomy. Animals were allowed to recover on the running wheel for at least 45 minutes before recording. In a subset of experiments, recording was performed the day after the craniotomy or animals were used for up to 2 consecutive recording days. In these cases, the craniotomy was protected overnight with Dura-Gel (Cambridge NeuroTech) and dental cement, which were removed and replaced with ACSF prior to recording.
In vivo whole-cell recordings.
Whole-cell recordings were performed using blind patch technique. A silver chloride ground pellet was placed in the recording well outside of the brain. Recording ACSF was wicked away from the craniotomy and a 3-5 MOhm glass micropipette with high positive pressure (~120 mbar) was lowered until the pipette tip touched the brain (confirmed by appearance of a square pulse on the membrane test); this position was zeroed and the well was refilled with recording ACSF. All recordings were documented relative to this depth. If the dura was removed well, pipettes showed minimal resistance increase (<0.1 MOhm) when lowering in first 50 μm into the brain. This observation was critical to successful recordings. Once in the brain, the pressure was lowered to ~50 mbar. The pipette was lowered to ~100 μm depth and positive pressure was lowered again to ~30 mbar and stepped in 1-2 μm/second increments until a bounce-like increase in resistance was observed and pressure was released to form a GΩ seal. Cells recorded at 180-350 μm depths were considered to be within L2/3. For current clamp recordings, internal solution contained (in mM): 142 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 5 phosphocreatine-di(tris), 5 phosphocreatine-Na2, 3 Mg-ATP, 0.5 GTP; for voltage clamp recordings, internal solution contained (in mM): 125 Cs-methanesulfonate, 5 TEA-Cl, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na3GTP, 8 phosphocreatine-di(tris), 3 NaCl. For voltage clamp recordings, EPSCs were recorded at −70 mV and IPSCs were recorded at +10 mV based on previous literature and our own calibration with ChR2 activation of interneurons in vivo (Figure S2A). Series resistance was monitored using −5 mV steps preceding each stimulus; recordings that reached >35 MΩ resistance or >20% change from baseline were discarded. The order of recording EPSCs and IPSCs was varied across experiments, and there was no relationship between the series resistance and the normalized current for either holding potential (Figure S2B-C; EPSCs p = 0.21, IPSCs p = 0.57).
In a subset of recordings, a low resistance pipette (1 MΩ) was filled with 3 M NaCl and lowered ~200 μm to measure local field potential and determine optimal stimulus position. Otherwise, optimal stimulus position was guided by retinotopic mapping and then determined separately for azimuth and elevation by observing spikes or EPSCs in response to a flashing white bar (0.1 s on, 1 s off, 5 degree width). For current clamp and voltage clamp experiments (Figures 1-3), following optimization of stimulus position, spikes or EPSCs were analyzed online to determine a stimulus orientation that reliably evoked the largest response. For EPSCs, effort was made to select a neuron’s preferred orientation, however given the broad tuning of synaptic inputs relative to spiking88-90, as well as the strong influence of spontaneous activity on single trial responses, if multiple orientations elicited a strong synaptic response during online optimization, we proceeded with any of those orientations to maximize time for data collection. Depending on the stimulus protocol, average recording duration ranged from around 5 minutes (Figure 2F, Figure 3) to 30 minutes if multiple inter-stimulus intervals were tested (Figure 1, Figure 2A).
In vivo extracellular recordings.
Extracellular recordings were performed with a 32-site acute probe (A1x32-Poly2–5mm-50s-177-A32, NeuroNexus or H4, Cambridge NeuroTech). Probes were connected through an A32-OM32 adapter to a Cereplex Mu digital headstage (Blackrock Microsystems). Signals were digitized at 30 kHz and recorded by a Cerebus multichannel data acquisition system (Blackrock Microsystems). Probes were slowly lowered into the brain until all sites were inserted and allowed to stabilize for 40–50 min before recording. Recordings typically lasted 1-2 hours. For experiments involving localized viral expression, the probe was painted with DiO (Thermo Fisher) to confirm with post hoc histology that the electrode tract was within the expression region.
For optogenetic experiments, we used either a 450 nm or 532 nm laser (Optoengine) to activate ChR2-expressing neurons or inhibit L4 terminals with eOPN3, respectively. Lasers were coupled to an optic shutter and patch cable terminating in an optic fiber. For L2/3 ChR2 activation and eOPN3 inhibition experiments, probes had attached optic fibers (200 μm core, 0.22 NA) that terminated 100 μm above the surface of the brain. For L4 ChR2 stimulation, a tapered lambda fiber (100 μm core with 0.9 mm taper, 0.22 NA, Optogenix) was inserted in the brain aligned to the tip of the probe for enhanced light transmission deeper in the brain. Laser power was calibrated to deliver 1 mW power at fiber tip for ChR2 activation and 1.2 mW power at the fiber tip for eOPN3 inhibition.
On a subset of recordings, putative ChR2-expressing units were identified by blocking excitatory transmission with a mix of AMPAR and NMDAR blockers (3 mM NBQX and 6 mM APV, respectively) diluted in 100 μL of recording ACSF91. At the end of the recording, ACSF was wicked away from the recording well and the drug mixture was dripped onto the craniotomy. After at least 20 minutes (up to 45 minutes, based on visually-evoked responses at the deepest electrode sites) ~50 pulses of 450 nm laser (10 ms, 0.1 Hz) were presented to activate ChR2-expressing cells.
In vitro slice preparation.
Mice were deeply anesthetized with isoflurane, the brain was removed and then transferred to oxygenated (95% O2 and 5% CO2), ice-cold artificial cerebrospinal fluid (ACSF, in mM: NaCl 126, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, glucose 20, CaCl2 2, MgCl2 1.3). Coronal brain slices (300 μm thickness) were prepared using a vibrating microtome (VT1200S, Leica) and transferred to a holding solution (at 34° C) for 12 minutes, and then transferred to storage solution for 30 min before being brought to room temperature. The holding solution contained (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 2 CaCl2, 2 MgSO4. The storage solution contained (in mM): 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 thiourea, 5 Na-ascorbate, 3 Na-pyruvate, 0.5 CaCl2, 10 MgSO4. Micropipettes pulled from borosilicate glass (1B150F-4, World Precision Instruments) were filled with internal solution containing (in mM): 142 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 5 phosphocreatine-tris, 5 phosphocreatine-Na2, 3 Mg-ATP, 0.5 GTP. Recording pipettes had resistances of 3-5 MΩ.
In vitro slice recordings.
Recordings occurred between 1.5 and 5 hours after the animal was sacrificed. Brain slices were transferred to a recording chamber and maintained at 34° C in oxygenated ACSF (containing, in mM: NaCl 126, KCl 2.5, NaHCO3 26, NaH2PO4 1.25, glucose 20, CaCl2 2, MgCl2 1.3, bubbled with 95% O2 and 5% CO2) perfused at 2 mL/min. Electrophysiological recordings were restricted to layer 2/3 and V1 was identified either by reference atlas alignment or visualization of fluorescence expression at the viral injection site. Neural signals were recorded using a MultiClamp 700B and digitized with a Digidata 1550 (Axon Instruments) with a 20 kHz sample rate. Data acquisition and stimulus presentation was controlled using the Clampex software package (pClamp 10.5, Axon Instruments).
In current-clamp recordings, a constant positive current was injected to maintain membrane potential near resting membrane potential measured in vivo. To test effects of depolarization on membrane potential, positive current was injected for a duration that varied between 0.1 and 5 s. Current level was calibrated with 0.1 s current injections to elicit a similar firing rate (~30 Hz) across cells, but generally ranged between 400-600 pA.
In voltage-clamp recordings, series resistance was monitored using −5 mV steps preceding each trial. At least 10 sweeps were collected for each recording condition. Only cells that had < 20 MΩ series resistance, < 20% series resistance change, and stable holding current (<100 pA baseline variation) were included for analysis. EPSCs were evoked by either electrical stimulation with a steel monopolar electrode placed in L4 or L2/3 (100 μs pulses) or optical activation of ChR2 over cell bodies in L4 (light power 0.5-1.5 mW/mm2, 470 nm LED, 100 ms square pulse). For optical activation, light pulses from a 4-color LED controller (ThorLabs) were coupled to the epifluorescence path (Olympus BX-RFA) and projected through a 40x water immersion lens (Olympus, 0.8 NA).
For electrical stimulation of L4 and L2/3 (Figures 4 and S4D-H), stimulation location and intensity were adjusted prior to data collection to minimize polysynaptic activation (assessed with online observation of EPSCs). Based on our previous data silencing local action potentials with muscimol, we considered monosynaptic responses to be short-latency (< 5 ms) EPSCs92. Since electrical stimulation activates axons non-selectively, in Scnn1a-Cre x Ai32 animals we compared EPSCs in L2/3 neurons in response to electrical and optogenetic activation in L4. In these experiments, we performed a similar optimization for the optogenetic stimulation site. After patching a L2/3 neuron, a small spot (50 μm, 1 ms) of 470 nm light was moved around within L4 to search for a position that elicited short-latency EPSCs. The intensity of optogenetic stimulation was then calibrated to reliably elicit EPSCs with as little polysynaptic activity as possible (although this was more difficult than with electrical stimulation). To best match the subset of L4 neurons projecting to the recorded neuron, the stimulation electrode was placed in the center of this spot. Optogenetic stimulation produced EPSCs with significantly longer latency than electrical stimulation (4.16 ± 0.14 ms vs 2.22 ± 0.21 ms; p < 0.001, paired t-test), suggesting that optogenetic EPSCs reflect propagation of spikes generated in L4 somas, whereas electrical stimulation activates axons directly. However, EPSCs recorded in L2/3 neurons displayed the same depression for electrical and optogenetic stimulation, indicating that L4 electrical stimulation is sufficient to reveal the dynamics of L4-L2/3 synapses (Figure S4D-F; two-way ANOVA, effect of stimulation type, p = 0.11).
For 0.1 s paired pulse optogenetic stimulation of L4, the same optimization (with 1 ms stimulus duration) was performed prior to data collection to identify an optimal stimulation position. The intensity of the optogenetic stimulation was then reduced for the 0.1 s stimulus duration to elicit EPSCs with ~100-250 pA amplitude.
eOPN3 in L4 terminals was activated by illuminating a small area (100 μm diameter) around the recorded neuron with green light (0.8 mW/mm2, 530 nm LED) for 10 s, followed by a 0.5 s top-up preceding each trial.
Post hoc histology.
After recording in virally injected or electroporated animals, brains were imaged to confirm viral expression in the recorded area. For in vitro recordings, slices were incubated 12-16 hours in 4% paraformaldehyde (PFA) in PBS, washed 3x with PBS and mounted. For in vivo recordings, the probe tract was visualized with DiI or DiO painted on the probe prior to insertion (Invitrogen V22889). After recording, animals were anesthetized with an overdose of ketamine/xylazine and perfused with PBS followed by 4% PFA in PBS. Brains were dissected and incubated in 4% PFA overnight, rinsed 3x with PBS, then sliced in 100 μm sections and mounted on glass slides. Slides were mounted with Fluoromount G with DAPI (Invitrogen) and imaged using a Zeiss inverted microscope (Zeiss Axiovert).
QUANTIFICATION AND STATISTICAL ANALYSIS
All analyses were performed in custom code written in either MATLAB or Python. All data are presented as mean ± SEM. N values refer to number of cells or units isolated. Sample sizes were not predetermined but were collected to be comparable to published literature for each type of experiment. For similar types of whole-cell recordings, studies typically report a minimum of 6-10 cells24,25,29,30,93,94; our experiments had a minimum of 8 cells for recordings in vivo and 10 for recordings in vitro. For in vivo extracellular recordings, we had a minimum number of 15 well-isolated single units. For all experiments adaptation is quantified as the normalized response:
Where the Baseline response is the response to the first visual stimulus (or electrical or optical stimulus in vitro) on a trial and the Test is the response to the same stimulus following a visual, electrical, or optical adapter.
Analysis of in vivo whole-cell recordings
Current clamp recordings.
Raw membrane potential was separated into firing rate and subthreshold membrane potential. Firing rate was obtained by setting a voltage threshold on a cell-by-cell basis for detecting spikes. Subthreshold membrane potential was obtained by using a median filter to clip spikes. For each ISI, pre-stimulus mean membrane potential and variance were measured from subthreshold membrane potential in a 0.1 s window prior to stimulus onset. Spike threshold was measured from spikes detected in a 0.4 s window around stimulus onset (0.1 s before and 0.3 s after stimulus onset). Spike threshold was calculated by averaging over the membrane potential at the time of the peak of the second derivative for all spikes within this time window. PSP amplitude was measured in a 20 ms window around the peak of the trial-averaged response during the stimulus-evoked response window (0-0.25 s after stimulus onset), relative to the baseline window (0.1 s before stimulus onset). Each condition contained at least 8 trials.
Voltage clamp recordings.
EPSC or IPSC stimulus-evoked amplitude was quantified by averaging trials in each condition (minimum 8) and measuring the current amplitude within a 20 ms window around the peak of the response 0-0.25 s after stimulus onset. Mean and standard deviation of the holding current was quantified in a 0.1 s window prior to stimulus onset. Recovery time constants were fit for EPSCs and IPSCs using a single exponential from the normalized current amplitude averaged across cells. For all stimulus specificity experiments, current amplitudes were normalized to the baseline stimulus of the same orientation. Adaptation tuning width was measured by fitting the normalized responses with a von Mises function.
Analysis of in vitro whole-cell recordings
Amplitudes of EPSCs in response to electrical stimulation or 1 ms ChR2 activation were calculated as the mean of the trial-averaged response in a 2 ms window around the peak of the response. Some recordings displayed late-onset polysynaptic EPSCs. To specifically measure the early-onset, presumably monosynaptic, amplitude we limited the search window for the peak response to the first 5 ms after stimulation. Each condition contained at least 10 trials. Amplitudes of EPSCs in response to 0.1 s ChR2 activation were calculated in a 20 ms window around the peak of the response. Recovery of optogenetically evoked EPSCs from adaptation with 0.1 s ChR2 activation was fit with a single exponential. For recordings of EPSCs in response to bursts of electrical stimulation, the normalized EPSC was quantified by normalizing the total charge on the second burst to the charge on the first burst. Charge was quantified for each electrical stimulation pulse by summing the current in a 10 ms window post-stimulation and then summed for each stimulus in the burst.
Analysis of extracellular recordings
Spike sorting.
Single units were isolated with KiloSort 2.5 (https://github.com/MouseLand/Kilosort) using refractory period violations and steepness of the autocorrelogram as criteria for isolation. We then manually curated these units in Phy (https://github.com/cortex-lab/phy) such that only units that were detected throughout the entire recording are included for subsequent analysis. Depth of the unit was assigned based on their waveforms’ center-of-mass across sites. Fast-spiking (FS) and regular-spiking (RS) units were separated within recordings according to peak-to-trough time of the maximum amplitude waveform across all contact sites (Figure S3A-B).
Layer identification.
To functionally identify cortical layers, we used the local field potential (LFP) obtained from filtering the raw data (downsampled to 10 kHz) from 1 to 200 Hz. The trial-averaged, stimulus-evoked LFP during a 1-second drifting grating presentation was converted to a current source density (CSD) plot by taking the discrete second derivative across the electrode sites and interpolated. Layer bounds were assigned relative to an initial sink in layer 4, followed by a sink in layer 2/3 and a sustained sink in layer 5 (Figure S5).
To confirm layer identification, in a subset of experiments ChR2-activated units were identified in the presence of excitatory synaptic blockers to identify ChR2-expressing units. Each unit’s distance in depth from the L23-L4 boundary was measured to compare depth of L4 versus L2/3 ChR2-expressing neurons across the two experiment types (Figure S5).
Data inclusion and analysis.
For all recordings, only cells that were visually responsive, according to a paired t-test in a 0.15 s window before and after stimulus onset, were included. In ChR2 activation experiments, “laser active” units were defined as units significantly driven by ChR2 activation in this same time window. For eOPN experiments, inhibited and facilitated units were defined as having >20% decrease or increase, respectively, in visually-evoked responses during eOPN3 activation compared to control trials. Categorization of units with significant increase or decrease defined by paired t-test yielded similar results. Neurons that were classified as inhibited in L2/3 and L4 were monitored for recovery of visually-evoked responses following eOPN3 activation and the recovery time constant was fit with a single exponential from the start of eOPN3 induction.
PSTHs were generated by binning spiking activity in 0.01 s windows across all trials of each type, aligned to stimulus onset. Each stimulus condition contained at least 20 repeats. Maximum firing rate was measured as the average firing rate in a 20 ms window around the peak of the PSTH. For plots visualizing average stimulus-evoked responses across units, firing rates were z-scored prior to averaging. Orientation tuning was measured using the mean firing rate in a 20 ms window around the peak PSTH for each stimulus direction, collapsed by orientation and fit with a von Mises function.
Supplementary Material
Highlights.
Brief visual experience induces long-lasting adaptation of L2/3 neurons in mouse V1
Adaptation decreases both stimulus-evoked synaptic excitation and inhibition
Activation of L4 is necessary and sufficient to induce effects of adaptation
Adaptation reflects short-term depression of L4 excitatory inputs to L2/3 neurons
Acknowledgements
We thank Gloria Kim for surgical assistance, TJ Wagner and Wenjuan Kong for husbandry assistance, Drs. Debra Silver and LJ Pilaz for equipment and instruction for performing in utero electroporation, and Dr. Hiroyuki Kato for advice in optimizing in vivo whole-cell recordings. We thank Drs. Celine Cammarata, Court Hull and Nicholas Priebe for comments on the manuscript, and members of the Hull and Glickfeld labs for insight throughout the project. This work was supported by grants from the National Eye Institute (R01-EY031328 to L.L.G. and F31-EY031941 to J.Y.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests:
The authors declare no competing interests.
References
- 1.Weber AI, Krishnamurthy K, and Fairhall AL (2019). Coding Principles in Adaptation. Annu. Rev. Vis. Sci 5, 427–449. 10.1146/annurev-vision-091718-014818. [DOI] [PubMed] [Google Scholar]
- 2.Barlow HBH (1961). Possible principles underlying the transformation of sensory messages. In Sensory Communication, pp. 217–234. 10.1080/15459620490885644. [DOI] [Google Scholar]
- 3.Kohn A. (2007). Visual adaptation: Physiology, mechanisms, and functional benefits. J. Neurophysiol 97, 3155–3164. 10.1152/jn.00086.2007. [DOI] [PubMed] [Google Scholar]
- 4.Vinje WE, and Gallant JL (2000). Natural Vision Sparse Coding and Decorrelation in Primary Visual Cortex During Sparse Coding and Decorrelation in Primary Visual Cortex During Natural Vision. Science (80-. ) 287, 1273–1276. 10.1126/science.287.5456.1273. [DOI] [PubMed] [Google Scholar]
- 5.Simoncelli EP, and Olshausen BA (2001). Natural Image Statistics and Neural Representation. Annu. Rev. Neurosci 24, 1193–1216. 10.1146/annurev.neuro.24.1.1193. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz O, Hsu A, and Dayan P (2007). Space and time in visual context. Nat. Rev. Neurosci 8, 522–535. 10.1038/nrn2155. [DOI] [PubMed] [Google Scholar]
- 7.Parker PRL, Abe ETT, Leonard ESP, Martins DM, and Niell CM (2022). Joint coding of visual input and eye/head position in V1 of freely moving mice. Neuron, 1–10. 10.1016/j.neuron.2022.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nigam S, Milton R, Pojoga S, and Dragoi V (2023). Adaptive coding across visual features during free-viewing and fixation conditions. Nat. Commun 14, 87. 10.1038/s41467-022-35656-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latimer KW, Barbera D, Sokoletsky M, Awwad B, Katz Y, Nelken I, Lampl I, Fairhall AL, and Priebe NJ (2019). Multiple timescales account for adaptive responses across sensory cortices. J. Neurosci 39, 10019–10033. 10.1523/JNEUROSCI.1642-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ulanovsky N, Las L, Farkas D, and Nelken I (2004). Multiple time scales of adaptation in auditory cortex neurons. J. Neurosci 24, 10440–10453. 10.1523/JNEUROSCI.1905-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zucker RS, and Regehr WG (2002). Short-term synaptic plasticity. Annu. Rev. Physiol 64, 355–405. 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 12.Varela JA, Sen K, Gibson J, Fost J, Abbott LF, and Nelson SB (1997). A quantitative description of short-term plasticity at excitatory synapses in layer 2/3 of rat primary visual cortex. J. Neurosci 17, 7926–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abolafia JM, Vergara R, Arnold MM, Reig R, and Sanchez-Vives MV (2011). Cortical auditory adaptation in the awake rat and the role of potassium currents. Cereb. Cortex 21, 977–990. 10.1093/cercor/bhq163. [DOI] [PubMed] [Google Scholar]
- 14.Beck O, Chistiakova M, Obermayer K, and Volgushev M (2005). Adaptation at synaptic connections to layer 2/3 pyramidal cells in rat visual cortex. J. Neurophysiol 94, 363–376. 10.1152/jn.01287.2004. [DOI] [PubMed] [Google Scholar]
- 15.Baccus SA, and Meister M (2002). Fast and slow contrast adaptation in retinal circuitry. Neuron 36, 909–919. 10.1016/S0896-6273(02)01050-4. [DOI] [PubMed] [Google Scholar]
- 16.Patterson CA, Wissig SC, and Kohn A (2013). Distinct Effects of Brief and Prolonged Adaptation on Orientation Tuning in Primary Visual Cortex. J. Neurosci 33, 532–543. 10.1523/JNEUROSCI.3345-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolfe JM (1984). Short test flashes produce large tilt aftereffects. Vision Res. 24, 1959–1964. 10.1016/0042-6989(84)90030-0. [DOI] [PubMed] [Google Scholar]
- 18.Harris JP, and Calvert JE (1989). Contrast, spatial frequency and test duration effects on the tilt aftereffect: Implications for underlying mechanisms. Vision Res. 29, 129–135. 10.1016/0042-6989(89)90179-X. [DOI] [PubMed] [Google Scholar]
- 19.Jin M, Beck JM, and Glickfeld LL (2019). Neuronal adaptation reveals a suboptimal decoding of orientation tuned populations in the mouse visual cortex. J. Neurosci, 3172–18. 10.1523/JNEUROSCI.3172-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin M, and Glickfeld LL (2020). Magnitude, time course, and specificity of rapid adaptation across mouse visual areas. J. Neurophysiol 124, 245–258. 10.1152/jn.00758.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fritsche M, Solomon SG, and de Lange FP (2022). Brief Stimuli Cast a Persistent Long-Term Trace in Visual Cortex. J. Neurosci 42, 1999–2010. 10.1523/jneurosci.1350-21.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Vives MV, Nowak LG, and McCormick DA (2000). Membrane mechanisms underlying contrast adaptation in cat area 17 in vivo. J Neurosci 20, 4267–4285. 20/11/4267 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carandini M, and Ferster D (1997). A Tonic Hyperpolarization Underlying Contrast A Tonic Hyperpolarization Underlying Contrast Adaptation in Cat Visual Cortex. 276, 949–952. 10.1126/science.276.5314.949. [DOI] [PubMed] [Google Scholar]
- 24.Wehr M, and Zador AM (2005). Synaptic mechanisms of forward suppression in rat auditory cortex. Neuron 47, 437–445. 10.1016/j.neuron.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 25.Chung S, Li X, and Nelson SB (2002). Short-term depression at thalamocortical synapses contributes to rapid adaptation of cortical sensory responses in vivo. Neuron 34, 437–446. 10.1016/S0896-6273(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 26.Natan RG, Briguglio JJ, Mwilambwe-Tshilobo L, Jones SI, Aizenberg M, Goldberg EM, and Geffen MN (2015). Complementary control of sensory adaptation by two types of cortical interneurons. Elife 4, 1–27. 10.7554/elife.09868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boudreau CE, and Ferster D (2005). Short-term depression in thalamocortical synapses of cat primary visual cortex. J. Neurosci 25, 7179–7190. 10.1523/JNEUROSCI.1445-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu B, Garrett ME, Groblewski PA, Ollerenshaw DR, Shang J, Roll K, Manavi S, Koch C, Olsen SR, and Mihalas S (2021). Adaptation supports short-term memory in a visual change detection task. PLoS Comput. Biol 17, 1–22. 10.1371/journal.pcbi.1009246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kato HK, Asinof SK, and Isaacson JS (2017). Network-Level Control of Frequency Tuning in Auditory Cortex. Neuron 95, 412–423.e4. 10.1016/j.neuron.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato TK, Haider B, Häusser M, and Carandini M (2016). An excitatory basis for divisive normalization in visual cortex. Nat. Neurosci 19, 568–570. 10.1038/nn.4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.House DRC, Elstrott J, Koh E, Chung J, and Feldman DE (2011). Parallel regulation of feedforward inhibition and excitation during whisker map plasticity. Neuron 72, 819–831. 10.1016/j.neuron.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kloc M, and Maffei A (2014). Target-specific properties of thalamocortical synapses onto layer 4 of mouse primary visual cortex. J. Neurosci 34, 15455–15465. 10.1523/JNEUROSCI.2595-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balcioglu A, Gillani R, Doron M, Burnell K, Ku T, Erisir A, Chung K, Segev I, and Nedivi E (2023). Mapping thalamic innervation to individual L2/3 pyramidal neurons and modeling their ‘readout’ of visual input. Nat. Neurosci 26, 470–480. 10.1038/s41593-022-01253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun W, Tan Z, Mensh BD, and Ji N (2016). Thalamus provides layer 4 of primary visual cortex with orientation- and direction-tuned inputs. Nat. Neurosci 19, 308–315. 10.1038/nn.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabernet L, Jadhav SP, Feldman DE, Carandini M, and Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron 48, 315–327. 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 36.Sohya K, Kameyama K, Yanagawa Y, Obata K, and Tsumoto T (2007). GABAergic neurons are less selective to stimulus orientation than excitatory neurons in layer II/III of visual cortex, as revealed by in vivo functional Ca2+ imaging in transgenic mice. J. Neurosci 27, 2145–2149. 10.1523/JNEUROSCI.4641-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niell C, and Stryker M (2008). Highly Selective Receptive Fields in Mouse Visual Cortex. J. Neurosci 28, 7520–7536. 10.1523/JNEUROSCI.0623-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerlin AM, Andermann ML, Berezovskii VK, and Reid RC (2010). Broadly Tuned Response Properties of Diverse Inhibitory Neuron Subtypes in Mouse Visual Cortex. Neuron 67, 858–871. 10.1016/j.neuron.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hage TA, Bosma-Moody A, Baker CA, Kratz MB, Campagnola L, Jarsky T, Zeng H, and Murphy GJ (2022). Synaptic connectivity to L2/3 of primary visual cortex measured by two-photon optogenetic stimulation. Elife 11, 1–46. 10.7554/elife.71103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lefort S, and Petersen CCH (2017). Layer-Dependent Short-Term Synaptic Plasticity between Excitatory Neurons in the C2 Barrel Column of Mouse Primary Somatosensory Cortex. Cereb. Cortex 27, 3869–3878. 10.1093/cercor/bhx094. [DOI] [PubMed] [Google Scholar]
- 41.Mahn M, Saraf-Sinik I, Patil P, Pulin M, Bitton E, Karalis N, Bruentgens F, Palgi S, Gat A, Dine J, et al. (2021). Efficient optogenetic silencing of neurotransmitter release with a mosquito rhodopsin. Neuron 109, 1621–1635.e8. 10.1016/j.neuron.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chance FS, Nelson SB, and Abbott LF (1998). Synaptic depression and the temporal response characteristics of V1 cells. J. Neurosci 18, 4785–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abbott LF, Varela JA, Sen K, and Nelson SB (1997). Synaptic depression and cortical gain control. Science (80-. ) 275, 220–224. 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- 44.Feldmeyer D, Lübke J, Silver RA, and Sakmann B (2002). Synaptic connections between layer 4 spiny neurone-layer 2/3 pyramidal cell pairs in juvenile rat barrel cortex: Physiology and anatomy of interlaminar signalling within a cortical column. J. Physiol 538, 803–822. 10.1113/jphysiol.2001.012959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higley MJ, and Contreras D (2006). Balanced Excitation and Inhibition Determine Spike Timing during Frequency Adaptation. J. Neurosci 26, 448–457. 10.1523/JNEUROSCI.3506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitmire CJ, and Stanley GB (2016). Rapid Sensory Adaptation Redux: A Circuit Perspective. Neuron 92, 298–315. 10.1016/j.neuron.2016.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heiss JE, Katz Y, Ganmor E, and Lampl I (2008). Shift in the balance between excitation and inhibition during sensory adaptation of S1 neurons. J. Neurosci 28, 13320–13330. 10.1523/JNEUROSCI.2646-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carandini M, Heeger DJ, and Senn W (2002). A synaptic explanation of suppression in visual cortex. J. Neurosci 22, 10053–10065. 10.1523/jneurosci.22-22-10053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crowder NA, Price NSC, Hietanen MA, Dreher B, Clifford CWG, and Ibbotson MR (2006). Relationship between contrast adaptation and orientation tuning in V1 and V2 of cat visual cortex. J. Neurophysiol 95, 271–283. 10.1152/jn.00871.2005. [DOI] [PubMed] [Google Scholar]
- 50.Rosenbaum DM, Rasmussen SGF, and Kobilka BK (2009). The structure and function of G-protein-coupled receptors. Nature 459, 356–363. 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borst JGG (2010). The low synaptic release probability in vivo. Trends Neurosci. 33, 259–266. 10.1016/j.tins.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 52.Chen C, and Regehr WG (2003). Presynaptic modulation of the retinogeniculate synapse. J. Neurosci 23, 3130–3135. 10.1523/jneurosci.23-08-03130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stratford KJ, Tarczy-Hornoch K, Martin KAC, Bannister NJ, and Jack JJB (1996). Excitatory synaptic inputs to spiny stellate cells in cat visual cortex. Nature 382, 258–261. 10.1038/382258a0. [DOI] [PubMed] [Google Scholar]
- 54.Gil Z, Connors BW, and Amitai Y (1997). Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19, 679–686. 10.1016/S0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 55.Reinhold K, Lien AD, and Scanziani M (2015). Distinct recurrent versus afferent dynamics in cortical visual processing. Nat. Neurosci 18, 1789–1797. 10.1038/nn.4153. [DOI] [PubMed] [Google Scholar]
- 56.Litvina EY, and Chen C (2017). An evolving view of retinogeniculate transmission. Vis. Neurosci 34. 10.1017/S0952523817000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hirsch JA, Martinez LM, Alonso JM, Desai K, Pillai C, and Pierre C (2002). Synaptic physiology of the flow of information in the cat’s visual cortex in vivo. J. Physiol 540, 335–350. 10.1113/jphysiol.2001.012777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Crochet S, Chauvette S, Boucetta S, and Timofeev I (2005). Modulation of synaptic transmission in neocortex by network activities. Eur. J. Neurosci 21, 1030–1044. 10.1111/j.1460-9568.2005.03932.x. [DOI] [PubMed] [Google Scholar]
- 59.Nelson SB, and Turrigiano GG (1998). Synaptic depression: A key player in the cortical balancing act. Nat. Neurosci 1, 539–541. 10.1038/2775. [DOI] [PubMed] [Google Scholar]
- 60.Seay MJ, Natan RG, Geffen MN, and Buonomano DV (2020). Differential short-term plasticity of PV and SST neurons accounts for adaptation and facilitation of cortical neurons to auditory tones. J. Neurosci 40, 9224–9235. 10.1523/JNEUROSCI.0686-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu H, and Agmon A (2016). Differential excitation of distally versus proximally targeting cortical interneurons by unitary thalamocortical bursts. J. Neurosci 36, 6906–6916. 10.1523/JNEUROSCI.0739-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wright NC, Borden PY, Liew YJ, Bolus MF, Stoy WM, Forest CR, and Stanley GB (2021). Rapid Cortical Adaptation and the Role of Thalamic Synchrony during Wakefulness. J. Neurosci 41, 5421–5439. 10.1523/JNEUROSCI.3018-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yarden TS, Mizrahi A, and Nelken I (2022). Context-Dependent Inhibitory Control of Stimulus-Specific Adaptation Context-Dependent Inhibitory Control of Stimulus-Specific Adaptation Abbreviated Title: 4 Inhibitory Control of Stimulus-Specific Adaptation. 42, 4629–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yavorska I, and Wehr M (2016). Somatostatin-expressing inhibitory interneurons in cortical circuits. Front. Neural Circuits 10, 1–18. 10.3389/fncir.2016.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beierlein M, Gibson JR, and Connors BW (2003). Two Dynamically Distinct Inhibitory Networks in Layer 4 of the Neocortex. J. Neurophysiol 90, 2987–3000. 10.1152/jn.00283.2003. [DOI] [PubMed] [Google Scholar]
- 66.Heintz TG, Hinojosa AJ, Dominiak SE, and Lagnado L (2022). Opposite forms of adaptation in mouse visual cortex are controlled by distinct inhibitory microcircuits. Nat. Commun 13, 1–14. 10.1038/s41467-022-28635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Phillips EAK, Schreiner CE, and Hasenstaub AR (2017). Cortical Interneurons Differentially Regulate the Effects of Acoustic Context. Cell Rep. 20, 771–778. 10.1016/j.celrep.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamm JP, and Yuste R (2016). Somatostatin Interneurons Control a Key Component of Mismatch Negativity in Mouse Visual Cortex. Cell Rep. 16, 597–604. 10.1016/j.celrep.2016.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Varela JA, Song S, Turrigiano GG, and Nelson SB (1999). Differential depression at excitatory and inhibitory synapses in visual cortex. J. Neurosci 19, 4293–4304. 10.1523/jneurosci.19-11-04293.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kapfer C, Glickfeld LL, Atallah BV, and Scanziani M (2007). Supralinear increase of recurrent inhibition during sparse activity in the somatosensory cortex. Nat. Neurosci 10, 743–753. 10.1016/j.immuni.2010.12.017.Two-stage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, and Sakmann B (1998). Target-cell-specific facilitation and depression in neocortical circuits. Nat. Neurosci 1, 279–284. 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- 72.Pala A, and Petersen CCH (2015). InVivo Measurement of Cell-Type-Specific Synaptic Connectivity and Synaptic Transmission in Layer 2/3 Mouse Barrel Cortex. Neuron 85, 68–75. 10.1016/j.neuron.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scholl B, Thomas CI, Ryan MA, Kamasawa N, and Fitzpatrick D (2020). Cortical response selectivity derives from strength in numbers of synapses. Nature 12. 10.1038/s41586-020-03044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Petersen CCH, and Crochet S (2013). Synaptic Computation and Sensory Processing in Neocortical Layer 2/3. Neuron 78, 28–48. 10.1016/j.neuron.2013.03.020. [DOI] [PubMed] [Google Scholar]
- 75.Tsodyks MV, and Markram H (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc. Natl. Acad. Sci. U. S. A 94, 719–723. 10.1073/pnas.94.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chance FS, and Abbott LF (2001). Input-specific adaptation in complex cells through synaptic depression. Neurocomputing 38–40, 141–146. 10.1016/S0925-2312(01)00550-1. [DOI] [Google Scholar]
- 77.Foley JM, and Boynton GM (1993). Forward pattern masking and adaptation: Effects of duration, interstimulus interval, contrast, and spatial and temporal frequency. Vision Res. 33, 959–980. 10.1016/0042-6989(93)90079-C. [DOI] [PubMed] [Google Scholar]
- 78.Ulanovsky N, Las L, and Nelken I (2003). Processing of low-probability sounds by cortical neurons. Nat. Neurosci 6, 391–398. 10.1038/nn1032. [DOI] [PubMed] [Google Scholar]
- 79.Solomon SG, and Kohn A (2014). Moving sensory adaptation beyond suppressive effects in single neurons. Curr. Biol 24, R1012–R1022. 10.1016/j.cub.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nagel KI, Hong EJ, and Wilson RI (2015). Synaptic and circuit mechanisms promoting broadband transmission of olfactory stimulus dynamics. Nat. Neurosci 18, 56–65. 10.1038/nn.3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Horwitz GD (2020). Temporal information loss in the macaque early visual system 10.1371/journal.pbio.3000570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Douglas RJ, and Martin KAC (2004). Neuronal Circuits of the Neocortex. Annu. Rev. Neurosci 27, 419–451. 10.1146/annurev.neuro.27.070203.144152. [DOI] [PubMed] [Google Scholar]
- 83.Cheetham CEJ, and Fox K (2010). Presynaptic development at L4 to L2/3 excitatory synapses follows different time courses in visual and somatosensory cortex. J. Neurosci 30, 12566–12571. 10.1523/JNEUROSCI.2544-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voelcker B, Pancholi R, and Peron S (2022). Transformation of primary sensory cortical representations from layer 4 to layer 2. Nat. Commun 13, 5484. 10.1038/s41467-022-33249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Scala F, Kobak D, Shan S, Bernaerts Y, Laturnus S, Cadwell CR, Hartmanis L, Froudarakis E, Castro JR, Tan ZH, et al. (2019). Layer 4 of mouse neocortex differs in cell types and circuit organization between sensory areas. Nat. Commun 10, 1–12. 10.1038/s41467-019-12058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Castro-Alamancos MA (2004). Absence of Rapid Sensory Adaptation in Neocortex during Information Processing States. Neuron 41, 455–464. 10.1016/S0896-6273(03)00853-5. [DOI] [PubMed] [Google Scholar]
- 87.Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using μManager software. J. Biol. Methods 1, e10. 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu BH, Li YT, Ma WP, Pan CJ, Zhang LI, and Tao HW (2011). Broad inhibition sharpens orientation selectivity by expanding input dynamic range in mouse simple cells. Neuron 71, 542–554. 10.1016/j.neuron.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Priebe NJ, and Ferster D (2008). Inhibition, Spike Threshold, and Stimulus Selectivity in Primary Visual Cortex. Neuron 57, 482–497. 10.1016/j.neuron.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 90.Wilson DE, Whitney DE, Scholl B, and Fitzpatrick D (2016). Orientation selectivity and the functional clustering of synaptic inputs in primary visual cortex. Nat. Neurosci 19, 1003–1009. 10.1038/nn.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanzeni A, Akitake B, Goldbach HC, Leedy CE, Brunel N, and Histed MH (2020). Inhibition stabilization is a widespread property of cortical networks. Elife 9, 1–39. 10.7554/eLife.54875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li JY, Hass CA, Matthews I, Kristl AC, and Glickfeld LL (2021). Distinct recruitment of feedforward and recurrent pathways across higher-order areas of mouse visual cortex. Curr. Biol, 1–13. 10.1016/j.cub.2021.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu GK, Li P, Tao HW, and Zhang LI (2006). Nonmonotonic Synaptic Excitation and Imbalanced Inhibition Underlying Cortical Intensity Tuning. Neuron 52, 705–715. 10.1016/j.neuron.2006.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Adesnik H, Bruns W, Taniguchi H, Huang ZJ, and Scanziani M (2012). A neural circuit for spatial summation in visual cortex. Nature 490, 226–230. 10.1038/nature11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All electrophysiology data included in the manuscript figures is available on Figshare. A link is provided in the Key resources table.
All original code needed to generate the manuscript figures is available on Figshare. A link is provided in the Key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| rAAV2/1&2.hSyn.SIO-eOPN3-mScarlet | Addgene | 125713 |
| AAV1.CAG.Flex.tdTomato.WPRE.bGH | Addgene | 51503 |
| Deposited data | ||
| Data and code for analysis | This paper | https://doi.org/10.6084/m9.figshare.23519394.v1 |
| Experimental models: Organisms/strains | ||
| Scnn1a-Tg3-Cre | Jackson Labs | 009613 |
| Pvalb-Cre | Jackson Labs | 008069 |
| Ai32 | Jackson Labs | 012569 |
| CBA | Jackson Labs | 000654 |
| Recombinant DNA | ||
| pCAG-ChR2-mRuby | Addgene | 109125 |
| Software and algorithms | ||
| ImageJ | NIH | https://micro-manager.org |
| Micromanager | NIH | https://imagej.nih.gov/ij/ |
| MWorks | MWorks | http://mworks-project.org |
| pClamp 10 Software Suite | Molecular Devices | N/A |
| MATLAB | Mathworks | https://www.mathworks.com |
| Chemicals | ||
| Fast Green | Sigma Aldrich | F7252 |
| Fluoromount G | Invitrogen | 00-4959-52 |
| DiO | Invitrogen | V22886 |
| NBQX | Tocris Bioscience | Cat #: 1044; CAS: 479347-86-9 |
| D-APV | Tocris Bioscience | Cat #: 0106; CAS: 79055-68-8 |
| Dental Cement | C&B Metabond | S380 |