

Abstract
Despite being collectively among the most frequent congenital developmental conditions worldwide, differences of sex development (DSD) lack recognition and research funding. As a result, what constitutes optimal management remains uncertain. Identification of the individual conditions under the DSD umbrella is challenging and molecular genetic diagnosis is frequently not achieved, which has psychosocial and health-related repercussions for patients and their families. New genomic approaches have the potential to resolve this impasse through better detection of protein-coding variants and ascertainment of under-recognized etiology, such as mosaic, structural, non-coding, or epigenetic variants. Ultimately, it is hoped that better outcomes data, improved understanding of the molecular causes, and greater public awareness will bring an end to the stigma often associated with DSD.
INTRODUCTION
Conditions affecting the development of chromosomal, gonadal, or anatomic sex, termed DSD (see Box 1), encompass a large spectrum of overlapping phenotypes and an equally wide array of etiologies1–3. They may present in isolated form, as an index feature of well-defined syndromes (Figure 1), or as an incompletely penetrant trait of many complex conditions3,4. They can have an origin in various developmental processes including gonadal formation, hormone biosynthesis or responses in target tissues, or signaling along the hypothalamus-pituitary-gonad axis. Phenotypes range from minor malformations (such as undescended testes or hypertrophy of the clitoris) to abnormal gonadal development leading to ambiguity of the genital anatomy or discordance between sex chromosomes and anatomy. The conditions under the DSD umbrella are each individually rare and multiple genetic etiologies have been demonstrated, ranging from missense single nucleotide variants (SNVs) to whole chromosome aneuploidies (Table 1). At least 75 genes have been associated with one occurrence of DSD in humans, with variable strength of evidence2. In addition, anatomical phenotypes and hormonal profiles frequently overlap between the various conditions under the DSD umbrella, making an accurate DSD diagnosis one of the most difficult in medicine.
Box 1. DSD nomenclature.
Patient-centered and multidisciplinary care, promoted by the Consensus Statement1, has forced clinical teams to reflect further on the nomenclature for the conditions they care for. The term ‘Disorders of Sex Development’ was coined at the 2005 Chicago Consensus Conference as an answer to several historical issues1. One was the inherently confusing meaning of the word ‘Intersex’, which has a strong significance for identity in addition to a medical meaning. ‘Intersex’ suggested that, from a clinical standpoint, there is a requirement for ambiguous genitalia to ‘qualify’ for this diagnosis. Conditions with no external ambiguity, such as complete androgen insensitivity syndrome (CAIS), would illogically be excluded from the Intersex category, while congenital adrenal hyperplasia (CAH), which is often accompanied with ambiguous external genitalia in XX individuals, would be considered by some advocacy groups, such as the CARES Foundation, as a ‘disorder affecting the adrenal glands’ without acknowledgement of the word ‘Intersex.’ It was also felt that a clear medical slant to naming was needed to ensure that all the tools of evidence-based medicine would be available to systematically evaluate the effects of clinical management on outcomes. Finally, it was proposed that the term should be as precise as possible and should reflect the genetic etiology when available.
The Global DSD Update7, a follow-up to the Chicago Consensus Conference, summarized the positive aspects of the DSD nomenclature, including facilitated access to health care and insurance, an umbrella term helping generate a comprehensive and integrated model of care, and distinction from other conditions such as gender dysphoria. It also warned against the negative aspects: stigma associated with the word ‘Disorder’, the perceived implication that ‘sex’ implies sexual behavior, and the possible risk of increasing participant refusal in research under the DSD heading.
Now that the medical and health insurance community are aware of the complexities of DSD, the role of the word ‘disorder’ as a catalyst is waning, and it is time to retire it. The word ‘Difference’ has emerged as a trending alternative, and is increasingly being used in publications over the past few years. ‘Difference’ does not carry the potential stigma of ‘Disorder’, has been used by advocacy organizations and the National Health Service in the UK, and allows the recognizable acronym DSD to be retained, which is widely used throughout the medical literature.
Figure 1: Disruption of processes in many different organs can result in isolated and syndromic forms of DSD.
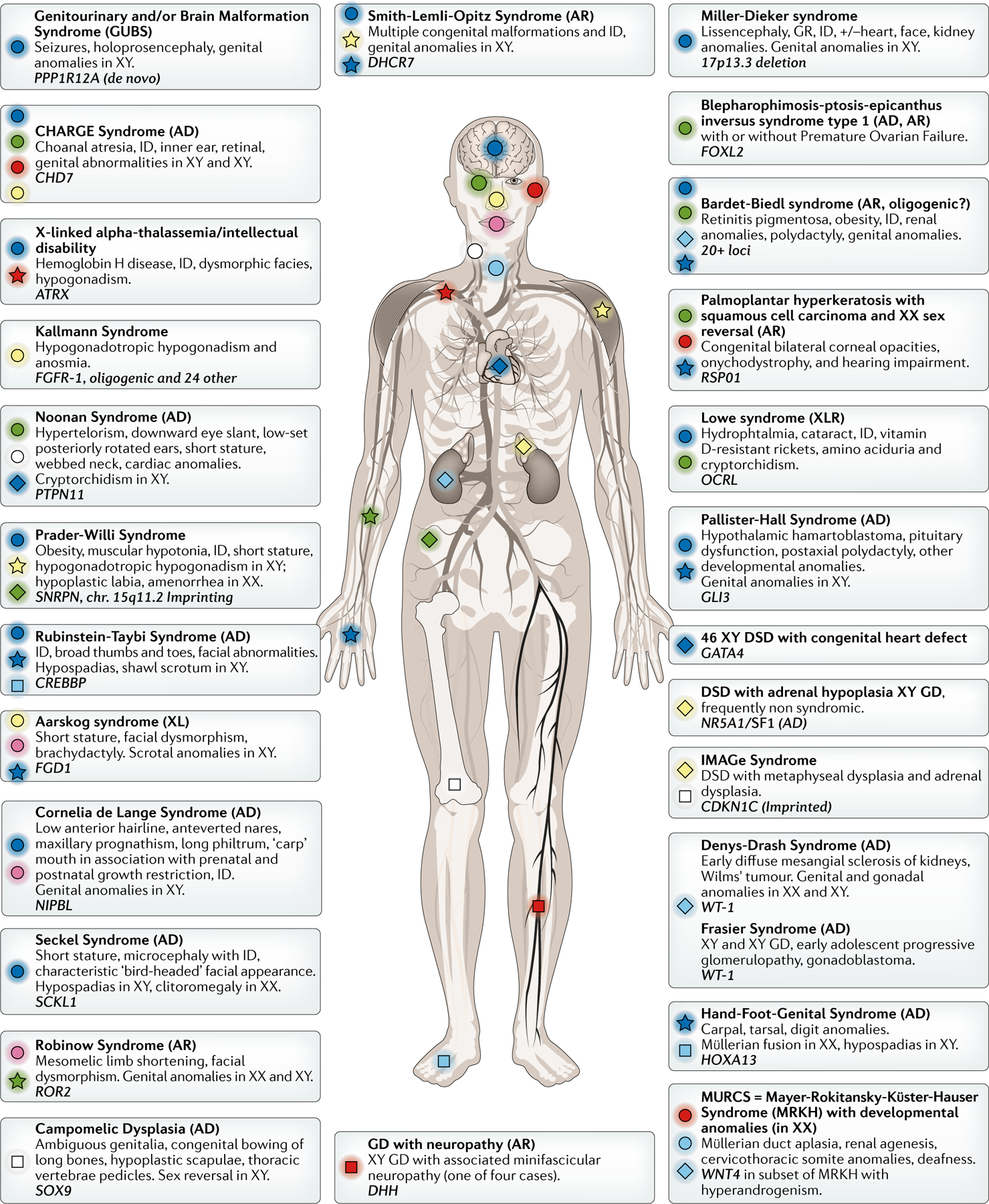
A selection of conditions is represented. The main organs affected (each represented by a unique combination of colour and symbol), mode of inheritance and the main genetic etiology are shown. Although not recommended nomenclature by the 2006 Consensus Statement1, the term ‘sex reversal’ is used when it is part of the OMIM description. AD: autosomal dominant; AR: autosomal recessive; XL(R): X-linked (recessive), GD: gonadal dysgenesis; GR: growth retardation; ID: intellectual disability.
Table 1: Major genetic etiology of 46,XX, 46, XY, and sex chromosome DSD.
Additional rare syndromic forms of DSD are shown in Fig. 1.
| DSD | Inheritance1 | Typical DSD genital phenotype | Genetic cause(s)2 | Available tests | ClinVar limitations | Comments |
|---|---|---|---|---|---|---|
| 46XX, DSD (~35 % of cases) | ||||||
| Disorders of androgen excess: CAH | AR | Clitoromegaly, urogenital sinus, labioscrotal fusion. Normal ovaries. | CYP21A2 in >90% | Newborn screening Gene panel for common variants Seq | Variant zygosity, sex chromosome complement, and genital phenotype are not indicated for these recessive conditions with sex-limited expression. Unclear which variants are linked to DSD. | Genetic diagnosis rarely pursued. Endocrine test: level of 17OHP |
| CYP11B1 | Seq | - | ||||
| CYP19A1 | Seq | P450 aromatase deficiency | ||||
| Testicular or ovotesticular DSD | De novo | Male or ambiguous genitalia. No uterus. | SRY translocation | FISH CMA Karyotype | Four pathogenic variants marked as copy number gain of Y including SRY. Whether patient is XX is not indicated. | Diagnosis of (ovo)testicular DSD requires histological examination of gonad. |
| SOX9 CNV | CMA | Five variants curated from original papers. | ||||
| SOX3 CNV | CMA | No SOX3 CNV in database. Six reported variants linked to a different condition. | ||||
| R92W variant in NR5A1 | Seq | Shown as “conflicting interpretations of pathogenicity” despite strong published evidence for pathogenicity. | ||||
| WT1 | Seq | None of 637 variants indicate 46,XX DSD | ||||
| Premature ovarian insufficiency (POI)* | AR, AD, XL | Primary or secondary amenorrhea in phenotypic females. | FSHR, BMP 15, FMR1, NR5A1 and 20 other genes | Seq | Variable. See examples in Supp. Table 1 | Each gene explains a few cases. Etiology overlaps with POF and ODG. |
| MRKH | Not known | Aplasia of Mllerian structures | Not known | — | — | Candidate loci only. Not clinically tested. |
| 46XY, DSD (~ 50% of cases) | ||||||
| Disorders of gonadal development | Y-linked | Pure, partial, or mixed gonadal dysgenesis. Uterus or urogenital sinus present. Female or ambiguous genitalia. | SRY SNV | Seq CMA | Strong evidence. 28 likely/pathog enic variants, three VUS, none benign. | Disorders of sex determination |
| AD | NR5A1 SNV | 14 variants (36%) deposited by Invitae indicate Oligosynaptic infertility (OMIM #258150), a condition linked to a different gene. | ||||
| AD | SOX9 CNV, SNV | Three variants curated from original papers. eight associated with campomelic dysplasia and sex reversal, but karyotype is not indicated. Only 17/160 variants associated with sex reversal. Most others do not indicate phenotype. | ||||
| XL | NROB1 dup | 13 variants deposited by Invitae do not distinguish between phenotypes of loss of function variants (causing a different condition, AHC) and duplications. | ||||
| AD | MAP3K1 SNV | Majority (21/30) of variants are reported as benign. Rapidly increasing numbers of benign, but not pathogenic, variants reported over time. | ||||
| De novo | 9p24 deletion | n/a | ||||
| AR | CBX2 SNV | Three variants including the two alleles of the original patient155; zygosity is not indicated for third variant. | ||||
| AR | DHH SNV | 35 variants, mostly VUS, with zygosity not indicated for this recessive condition. | ||||
| AD | WT1 SNV | 637 variants. None of the entries specify with which of the several WT1-linked conditions they are associated. | ||||
| AD | DHX37 SNV | Five, but not all, of the original published variants are in the database. | ||||
| Disorders of androgen action (AIS) | XL (~2/3), de novo (~1/3) | Complete AIS (CAIS): Female external genitalia, functional often inguinal testes, blind vagina, no uterus. Partial AIS (PAIS): Hypospadias, micropenis, gynecomastia. | AR SNV, deletion | Deletion by NGS or CMA (CAIS only) Seq | Only a fraction of known variants in the database. Most entries do not specify if variant is found in DSD condition. | The PAIS term should be reserved for cases in which a variant is found in the AR gene. |
| Disorders of androgen synthesis | AR | Hypospadias, micropenis/clitoromegaly, urogenital sinus, labia or bifid scrotum. No uterus. | SRD5A2 | Seq | Variant zygosity, sex chromosome complement, and genital phenotype are not indicated for these recessive conditions with sex-limited expression. Unclear which variants are Linked to DSD. | 5α reductase deficiency |
| CYP17A1 | CAH | |||||
| HSD3B2 | CAH | |||||
| StAR | Lipoid CAH | |||||
| HSD17B3 | See Box 2 | |||||
| POR | Cytochrome P450 oxidoreductase deficiency | |||||
| Persistent Müllerian Duct Syndrome | AR | Müllerian and Wolffian ducts coexist; inguinal hernias or undescended testes. | AMHR2, AMH SNV Indel | Seq | Historical variants are curated. For AMH, they are the only pathogenic variants in the database. For AMHR2, two other pathogenic variants are reported, but zygosity is not indicated. 13/14 variants from original published series are not in database. | - |
| Sex chromosome DSD (~15% of cases) | ||||||
| Klinefelter syndrome | De novo | Gynecomastia, small testes, Leydig cell hyperplasia. | 47,XXY | Karyotype | - | - |
| Turner syndrome variants | De novo | Various degrees of virilization. Mixed gonadal dysgenesis. | 45,X/45,XY mosaics | Karyotype | - | - |
| Chimerism or Mosaicism | De novo | Ovotesticular DSD | 46,XX/46,XY | Karyotype | - | - |
The most frequent mode inheritance is indicated; note that ‘AD’ may also include new mutations.
Only the main genes involved are included. Where ‘SNV’ is indicated, most conditions can also be caused by small indels or splice variants, which are also detectable by sequencing.
POI includes premature ovarian failure (POF) and hypergonadotropic ovarian failure (ODG) conditions. Those cases for which causative genes are known are named ODG1–8 and POF1–16 in the Online Mendelian Inheritance in Man (OMIM, omim.org) classification.
AR: autosomal recessive, AD: autosomal dominant, XL: X-linked, Seq: sequencing, FISH: fluorescence in situ hybridization, dup: duplication, CMA: chromosome microarray, CAH: congenital adrenal hyperplasia, 17OHP: 17-hydroxyprogesterone, CNV: copy number variant, MRKH: Mayer Rokitansky Küster Heuser syndrome, AHC: adrenal hypoplasia congenita, AIS: androgen insensitivity syndrome, VUS: variant of uncertain significance.
However, the 2005 Consensus Conference and recent efforts towards standardization of care, such as those spearheaded by the DSD-Translational Research Network (DSD-TRN5) in the US or the International DSD (I-DSD) registry (initiated in Europe6), have emphasized the need to reach an early diagnosis for optimal outcomes7,8. In particular, potential impaired fertility9,10, gender identity uncertainty, and self-image issues all contribute to poor psychosocial and medical quality-of-life outcomes8. An accurate diagnosis is critical as risk of gonadal cancer11–13, comorbidities (Fig. 1), and associated risks for variant-carrying family members vary greatly among conditions. It is critical to refer patients to a specialized multidisciplinary team as soon as DSD is suspected. At birth, it is urgent to initiate treatment to avoid life-threatening crises in the salt-wasting forms of Congenital Adrenal Hyperplasia (CAH), as well as to accurately distinguish between conditions with overlapping phenotypes but different outcomes to optimally decide on management and sex of rearing. Finally, benefits of having a diagnosis for individuals living with a rare disorder, even if no treatment is available, have been well documented14,15.
Yet, despite the high number of genes reported since the discovery of the first sex-determining gene, SRY, 30 years ago16–20, the molecular mechanisms underlying human sex development remain far from understood, and a majority of patients do not receive a diagnosis. Even with massively parallel sequencing technology (also known as next-generation sequencing, NGS), causative variants are reported in only 35%−45% of XY DSD in research series21,22, with even lower diagnostic rates in the clinical setting23. An inability to discover and interpret variants, especially beyond protein-coding regions, and poor delivery of care have been major limitations to diagnostic efficacy.
In this review, we discuss approaches and hurdles to diagnosis, challenges with the interpretation of variants, pre-clinical genomic technologies bringing promise of improvement, and how genetic information for individuals with a DSD is interpreted at a societal level. We refer to recent expert reviews for description and management of individual DSD conditions and known underlying molecular mechanisms2,3,9,24–28.
GENETIC TESTS IN CLINICAL USE
The traditional approach to DSD diagnosis has been step-wise stratification starting with clinical phenotyping and karyotyping to orient subsequent endocrine analysis, followed by genetic testing (which is often limited to individual candidate genes) to resolve intractable cases. Many algorithms have been put forth over the years to try and streamline the diagnostic process16,25,29,30, but progress is needed on the genetic diagnostic front. Apart from CAH, where hormonal markers orient the diagnosis, and chromosomal DSD (such as 45,X and 47,XXY and variants), clinically available genetic testing techniques fail to make a molecular diagnosis in more than half of cases. Diagnostic yield is particularly low for 46, XY DSD with gonadal dysgenesis.
Newborn screening for CAH
The vast majority (90–95%) of DSD in 46,XX individuals with genital atypia is associated with CAH due to CYP21A2 deficiency (~1:15,000 (ref31)). Because the life-threatening salt-wasting crises associated with this condition can be treated, CYP21A2 deficiency is now part of newborn screening panels in many parts of the world. However, a recent survey of protocols used across US states showed that criteria and technology are not standardized, resulting in poor test positive predictive value (PPV) [G], ranging from 0.7% to 50%32. This test is purely biochemical, as genetic testing for this gene is complex and not adapted to a screen. Most cases of CAH are diagnosed by endocrine testing alone and, as a result, our understanding of the correlation between genotype and outcomes remains insufficient to adapt treatment throughout life to specific genotypes33.
Sex chromosome assessment by karyotype
For newborns with atypical genitalia, a karyotype is still the test of reference (Table 2). However, interphase fluorescent in situ hybridization (FISH) for X and Y chromosomes and SRY is recommended as it is faster (24 hrs versus 1–2 weeks). About ~15% of DSD are thought to be chromosomal DSD, including Klinefelter syndrome, Turner syndrome variants with Y chromosome contribution (typically mosaic 45,X/46XY, with or without isodicentric Y [G]), segmental deletions of the X, or translocations of an SRY-containing Y fragment to the X5,23. With non-invasive prenatal screening (NIPS) using cell-free fetal DNA [G] to detect aneuploidies, many parents may now learn the sex chromosomes of their child around 10 weeks of gestation and DSD are increasingly ascertained this way34–36. Accuracy of this screen is lower for sex chromosomes than for Down syndrome (where sensitivity and specificity are close to 100%37). PPV of NIPS for sex chromosome aneuploidies has been reported at 32%−57.6%38–40, with very low PPVs of 18–21% for 45,X (Turner syndrome), and PPVs ranging from 39%38 to 90%40 for Klinefelter syndrome (47,XXY).
Table 2:
Clinical and preclinical tests for DSD.
| Technique | Time taken1 | Variants detected | Resolution | Diagnostic yield | ||||
|---|---|---|---|---|---|---|---|---|
| A/M | CNV | cSNV | ncSNV | SV | ||||
| Clinically available methods | ||||||||
| Karyotype | 1–2 weeks | ✓ | ✗ | ✗ | ✗ | ✓ | >5 Mb | 15% (mostly mosaics) |
| Interphase FISH (X, Y and SRY markers) | <3 days | ✓ | ✗ | ✗ | ✗ | ✗ | N/A | Rapid sex determination |
| Microarray | 2–3 weeks | ✓ | ✓ | ✗ | ✗ | ✗ | >50 kb | 15% |
| Single gene test or gene panel2 | Up to 6 weeks | ✗ | ✗ | ✓ | (✓) | ✗ | SNV: 1 nt Indels: <50 bp | Panel-dependent |
| Exome sequencing | Up to 12 weeks (<1 week possible if urgent) | ✗ | ✗ | ✓ | ✗ | ✗ | SNV: 1 nt Indels: <50 bp | 30–45% |
| Whole genome sequencing | Up to 16 weeks | ✗ | ✓ (requires validation) | ✓ | ✓ | ✓ (requires validation) | SNV: 1 nt | At least 30–45% |
| Pre-clinical methods | ||||||||
| Optical genome mapping3 | Up to 12 weeks | ✓ | ✓ | ✗ | ✗ | ✓ | SV: >500 bp | Not known |
| Long-read sequencing4 | Not known | ✗ | ✓ | ✗ (not yet) | ✗ (not yet) | ✓ | SV: 50 bp – 5 kb? | Not known |
Availability of test and time to results is highly dependent on location. Time estimates do not include time to obtain insurance pre-authorization to perform a test.
The nature of the single nucleotide variants (SNVs) detected will depend on sequencing primer design (for single gene tests) or the variants included in the gene panel.
Optical Genome Mapping is currently clinically approved in the USA only for FSHD (facioscapulohumeral muscular dystrophy), and has yet to be validated specifically in DSD. OGM is not designed for base-pair resolution and therefore cannot detect SNVs.
Long-range sequencing is not in current clinical use. Although it does not yet have base-pair accuracy (the current error rate is ~5% at each nucleotide position), improved algorithms and detection methods should allow reliable detection of SNVs in the future. A/M: aneuplodies and/or mosaics, SV: structural variants, CNV: copy number variants, cSNV: coding SNV. ncSNV: non-coding SNV, N/A: not applicable.
If NIPS suggests a normal sex chromosome complement, subsequent ultrasounds showing genitalia typical of the other sex (reported in 1/14,300 pregnancies36) may indicate a DSD. In a series, ascertainment errors were reported in ~1/3 of cases (tube mislabeling or incorrect identification of sex on ultrasound) and a DSD actually diagnosed at birth in 36%36. A vanishing twin (of the opposite sex) is a frequent cause of apparent discrepancy in the remaining 30%35. As NIPS is only a screen, follow up with confirmatory genetic testing (with karyotype or microarray) is needed and protocols have been proposed for this35.
Chromosomal microarrays
The advent of chromosomal microarrays revealed small, cryptic copy number variants [G] (CNVs) as an overlooked, albeit rare, DSD etiology41–43. Sex chromosome aneuploidies, and deletions or duplications of not only coding but also regulatory regions of SOX9, WNT4, or NR0B1 have been shown to cause DSD (genes, conditions, and research-based methods of detection are reviewed in41). Gene expression dosage threshold effects may also explain the incomplete penetrance observed in several conditions and pedigrees, which brings an element of extreme complexity to diagnosis. CNVs may also underlie the elusive etiology of developmental forms of DSD, such as cloacal malformations44 or Mayer Rokitansky Küster Heuser (MRKH) syndrome45,46.
Chromosomal microarrays are the reference method for detection of CNV, which can be identified even in mosaic form (if the CNV is present in more than 20–30% of cells). They often provide answers in syndromic cases of DSD and occasionally in isolated cases (for example 14% of cases in the DSD-TRN registry were diagnosed by chromosomal microarray 5). Clinical laboratories typically report CNVs larger than 25–50 kb (Table 2), which is larger than some DSD genes, although the current high density of probes on the arrays could allow detection of much smaller CNVs. A research bioinformatics tool was designed specifically for detection and functional annotation of smaller (> 1 kb) CNVs in DSD47. In 52 cases it identified >300 CNVs overlapping 68 DSD genes. Validation of the pathogenicity of these variants awaits replication in other cohorts and in vitro validation experiments prior to clinical deployment of the tool.
Single gene testing
Single gene testing is recommended when CAH is suspected clinically, as some degree of phenotype-genotype correlation may exist to distinguish the salt-wasting, simple virilizing, and non-classical forms48,49. For this and other conditions, targeted Sanger sequencing is useful once a variant has been identified in a proband to ascertain the phase of compound heterozygous variants and inheritance or de novo status. Because of the phenotypic overlap between conditions and the large number of candidate genes, single candidate gene testing (whether by NGS or Sanger sequencing) is not typically efficient in other cases. Indeed, analysis of the two most frequently ordered single gene tests for 46,XY DSD (AR and SRD5A2) in the DSD-TRN Registry showed that negative results were returned for ~40% of AR and ~ 55% of SRD5A2 tests5.
Exome sequencing
To identify SNVs or small INDELs in the dozens of known DSD genes, the method of choice is now massively parallel sequencing using short (~150 bp) reads covering the protein-coding regions of genes (clinical exome sequencing methods and best practices were recently reviewed in50). It can be performed with enrichment (targeted capture of a panel of known causative genes) or without enrichment (‘whole’ exome capture, with reporting limited to selected genes). Expertly curated enriched panels (from 30 to 180 DSD genes) have shown great diagnostic rates22,51–57, as well as time- and cost-efficiency53 in a research setting, but those are not typically commercially available. Clinical whole exome sequencing (WES), interpreted by an expert board, resulted in 35% firm diagnoses in 46,XY DSD21.
WES and panels for DSD testing have respective advantages (reviewed in2,55,58). While panels need to be re-engineered when a new causative gene is discovered, and patients retested, WES allows both easy reanalysis and gene discovery. However capture kits have evolved dramatically, and some DSD genes were poorly captured in older exome tests5, and variants in un-captured regions would be missed even upon reanalysis. For WES, whether targeted (to known clinical genes, covering ~30Mb of the genome), whole exome (45Mb), or expanded (into adjacent non-coding regions, 60Mb) capture is performed could greatly influence the number of discoverable variants. Sequencing platforms and analysis pipelines also evolve rapidly, and it is increasingly difficult for patients and providers alike to understand what the results of a test mean, as the ‘same’ test a few years later may result in a different outcome.
Whole genome sequencing
Whole genome sequencing (WGS), while based on the same short-read sequencing (SRS) principle, avoids the capture limitation of exome or panels. WGS has been shown to expand diagnostic utility and improve clinical management in rare pediatric disorders 59,60. WES misses about 650 true variants (3% of coding variants) per individual, which WGS is able to detect thanks to more even coverage of GC-rich and hard-to-capture exons61. Also, the proportion of false-positive SNVs is much higher for WES than for WGS (78%versus 17% (ref61)). Thus, WGS alone should increase diagnostic yield in the protein-coding regions of known DSD genes. Early indications suggest that this may be the case, but supporting evidence has yet to be published.
In addition, WGS allows detection of variants outside of exons, for example in regulatory elements found in promoters, introns or enhancers, a known source of DSD variants. Currently, 89% of pathogenic variants reported in the ClinVar database are in coding sequences (99% if adjacent regions are included)62. However, the exome represents only 1–2% of the genome and many causal variants are expected to be missed using exome sequencing alone. In the USA, Children’s National Hospital and Rady Institute have recently pioneered shifting their accredited pipeline to WGS as the primary technology for clinical genetic testing, but DSD panels are not yet available. With a cost still much higher than WES, and limited interpretability of variants in non-coding regions, the current appeal of WGS as a clinical test remains limited for providers in spite of the clear technical advantages.
IMPROVING DETECTION OF DSD VARIANTS
Current clinical technology detects mostly coding SNVs and large CNVs in the heterozygous or homozygous state. Detection yield should increase when methods become clinically available for detecting other types of variants and etiology, such as variants in mosaic states, oligogenic etiology, complex genomic variants and epigenetic variants—all known but under-ascertained causes of DSD. Emerging, pre-clinical approaches for improving detection of these classes of variants are discussed below.
Mosaicism
Chimerism [G] 63,64 and mosaicism [G] 65 have been reported as a DSD etiology and possibly the cause of missing diagnoses, phenotypic variability, and reduced penetrance. Turner syndrome variants with mosaic 45,X/46,XY typically present with mixed gonadal dysgenesis, with different tissue anatomy in the two gonads. Mosaic X-inactivation explained phenotypic variability in 46,XX individuals with a Y-to-X translocation66. A mosaic frameshift mutation in SRY in a man with testicular dysgenesis presented as a typical female phenotype in two 46,XY daughters, who inherited the mutation in a non-mosaic state67. Mosaicism for variants in other DSD genes may be under-recognized because of the lack of appropriate clinically available technology. In particular, better detection of mosaicism is expected to improve the diagnostic yield in ovotesticular DSD and other conditions resulting in asymmetric genital phenotypes, cases where the phenotype is at the mild end of the spectrum for a known condition; or in genes known for syndromic DSD when DSD condition presents in isolated form.
Next-generation sequencing was shown to be more sensitive than karyotype or microarray for detection of Y material in Turner syndrome variants68, but standard variant calling pipelines are not adequate to detect low level mosaicism or chimerism. They assume a diploid genome, and the depth of coverage (~100x for WES; ~30X for WGS) is too low for robust mosaic variant calling. Targeted short-read sequencing applied to high read depth (1000x) should allow robust detection of these otherwise cryptic DSD etiologies69.
Oligogenic etiology
Involvement of variants in multiple genes has been hypothesized to explain phenotypic variability in individuals sharing genetic etiology. For example, variants in NR5A1/SF-1 are associated with phenotypes ranging from isolated adrenal insufficiency, premature ovarian insufficiency, or ovotesticular DSD in 46,XX individuals, to undescended testes or infertility in typical males, and ambiguous genitalia or gonadal dysgenesis with female genitalia in 46,XY individuals. An oligogenic mode of inheritance, with variants in other genes explaining the variability in phenotype, has been proposed in this70,71 and other forms of DSD9,72,73. However, current bioinformatics pipelines are not designed for efficient identification of oligogeny. Publications have started sharing lists of other variants found even in resolved cases74 to help identify contribution of other genes to DSD phenotypic variability and pathogenicity69.
Structural variation
Structural variants [G] (SVs) interrupting the coding regions of DSD genes or modifying the genomic architecture around regulatory regions can result in DSD. While microarrays reliably identify large gains or losses of genetic material, they are blind to balanced rearrangements, inversions, or translocations. The short-read sequencing methodology common to both WES and WGS does not accurately detect large or complex SVs75. Several technologies that can detect complex SVs have been developed, with complementary strengths, and their combined use will be necessary to recognize the full extent of the currently under-ascertained role of SVs in DSD. Below, we discuss optical genome mapping (OGM) and long-read sequencing (LRS), and the current barriers to their clinical use (for details about techniques and algorithms to identify SVs in SRS, LRS and OGM see76,77).
Optical Genome Mapping detects variants 500 bp to Mbp in size.
OGM (supported by the Bionano Genomics platform) can identify large and/or complex SVs with high specificity and sensitivity78–80. OGM images megabase-size DNA molecules fluorescently labeled at specific sequence motifs throughout the genome. The resultant pattern of fluorescent tags within long DNA molecules is used for de novo assembly of each allele of the sample genome81. Because genomic architecture is preserved in the ultralong DNA molecules, comparison of the sample-specific maps with reference maps allows for identification of large deletions, insertions, inversions, translocations and complex rearrangements, including those not associated with copy number variation. The resolution limit of the method is dictated by both the spacing of the fluorescent tags along the genome and the pixel resolution of the camera capturing the signal. OGM can currently resolve SVs starting at about 500 bp in size, but this could evolve in the future with different labeling enzymes and improved sensors. The ability of OGM to identify clinically relevant SVs has been shown in cancer82–84 and Duchenne muscular dystrophy85. The first clinical application of OGM was approved in 201986 and it is being adopted in place of Southern blotting to identify the 3.3 kb D4Z4 repeats in facioscapulohumeral muscular dystrophy87, even in a low-level mosaic state.
Because OGM does not provide base-to-base information, a novel software was developed and integrated into the Genoox sequence analysis platform. It uses an advanced reference graph structure and short read WGS data from the same sample to refine breakpoint localization of OGM-based SV calls (from multiple kbp to ~200 bp). Integrated datasets enable more precise identification of breakpoints that might interrupt genes and facilitate interpretation of SVs. Identification of high-quality, clinically relevant SVs in OGM datasets is facilitated by nanotatoR88, a newly developed annotation tool that provides a variety of filtration options, information on the identity of and distance to overlapping genes and parental zygosity, and enables integration with gene expression (RNA-Seq) data if available. A limitation is that the Bionano Genomics Database contains data for only ~230 healthy individuals. Although other databases, such as the Database of Structural Variants (DGV), have larger numbers (~55,000), most are aggregated from different studies and techniques, which makes it difficult to achieve a harmonized dataset wherein frequency values translate to true population frequency. In addition, comparison of SVs to reported variants is more complex than for SNVs, as SVs of different sizes that interrupt a gene in the same region may have a similar effect but will be categorized as different. Just as the exponential expansion of public databases of control variants (such as gnomAD) and disease variants (such as ClinVar) has revolutionized interpretation of SRS data, interpretation of non-coding and structural variants will become more accurate as more samples are analyzed and databases expand.
Long-read sequencing excels at detection of SVs in the 50 bp-5 kbp range.
LRS, sometimes called third-generation sequencing, produces reads over 10 kb and up to megabases in length (compared to the typical 150 bp of clinical SRS), greatly facilitating mapping of sequences in areas of the genome difficult to sequence because of repetitive elements or pseudogenes. The role of LRS in medical genetics is emerging89. Two main technologies currently dominate: nanopore-based sequencing (developed by Oxford Nanopore Technologies) and single-molecule real-time (SMRT) sequencing (developed by Pacific Biosciences). Because long stretches of DNA are sequenced from a single molecule, LRS allows phasing of variants along a haplotype, variants to be resolved in low complexity regions, and detection of SVs missed by SRS90. In comparisons of SMRT, OGM and WGS, OGM shone at detecting SVs in the 1kb-1Mb range but only SMRT was able to detect ~90% of insertions and >75% of deletions in the 50 bp to 1 kbp range 75. Bioinformatics tools to identify more complex SVs and annotate variants are still being developed (for methods to identify SVs in LRS and SRS, see76,91).
Nanopore sequencing recently enabled telomere-to-telomere assembly of the X chromosome92, which may be useful for X-linked DSD (assembly of the Y chromosome is still to be tackled). Because of its high per-base error rate, LRS cannot currently be used to detect SNVs but should be very helpful to identify SVs too small to be accurately ascertained with OGM and CNVs smaller than the clinical threshold for microarrays. However, clinical applicability of LRS is still a few years away because of its current very high compute requirements and cost.
Epigenetic variation
Genital atypia is part of several imprinting disorders such as Prader Willi (OMIM 176270), Beckwith-Wiedemann (OMIM 130650), and IMAGe93 (OMIM 614732) syndromes. Changes in methylation in the promoters of DSD genes and histone modifications have been shown to affect mammalian sex determination (reviewed in2,94) and at least one case has been reported of differential methylation upstream of SRY in a 46,XY woman compared to her father95.
The diagnostic utility of genome-wide DNA methylation analysis has been recently demonstrated96 including in syndromes with genital involvement (Cornelia de Lange, Kabuki, genitopatellar, ATRX, and CHARGE syndromes). Systematic analysis of epimutations [G] (single locus) and episignatures [G] (across multiple loci) is likely to both identify new DSD etiology and help resolve variants of unknown significance (VUS). In addition to existing technology using bisulfite conversion followed by SRS or SNP arrays, LRS via nanopore-based sequencing97 or SMRT98,99 technology and OGM86 all hold the promise of detecting epigenetic and genetic variation on the same long DNA molecule100,101. Improving integration of epigenetic data (DNA methylation, histone modifications, imprinted regions) into sequence and SV analysis algorithms will be critical to increase DSD diagnostic yield, as was shown for microarrays47.
IMPROVING VALIDATION OF DSD VARIANTS
Typically, WES and WGS identify ~21,000 and ~3 million variants per genome respectively, with WGS variants largely found in non-coding regions, where interpretability is even lower than in coding sequence. Variant filtration using a pre-established list of genes relevant to the phenotype, population frequency, predicted protein-damaging effects, and previously reported occurrence in the same condition helps prioritize potentially pathogenic variants. However, the yield of likely pathogenic variants that can be confidently called is low because evidence of causation is very low for many of the dozens of DSD genes. Many genes have been published once, with limited in vitro or in vivo evidence of their deleteriousness. Existing guidelines (for example, ref 103 and the SVI General Recommendations for Using ACMG/AMP Criteria to incorporate results of validation studies into variant interpretation algorithms have focused on traditional, low throughput assays, which cannot keep pace with the amount of data produced by NGS. Multiplexed, high-throughput assays are necessary102, but designing assays specific for DSD is complex as they need to be relevant to each gene and affected tissue type.
Improving reference database curation
The 2015 publication of the 28 ACMG/APA criteria103 for classification of variants (from pathogenic to benign) was followed by the creation of tools to automate classification, such as Varsome104, Franklin and Sherloc105. These, and clinical platforms, use the ClinVar and/or UniProt variant repositories to score the ‘reputable source’ criteria (PP5 and BP6103 ). However, a recent detailed analysis showed that annotation of DSD genes in ClinVar is grossly insufficient2. Many published pathogenic variants, supported by in vitro validation, have not yet been curated and included in the database. As a result, evidence of pathogenicity may be over- or under-estimated, even for well-known DSD genes (Table 1 and Supplementary Table 1). For data on more genes, types of variants, and associated references, see2.) For example, ClinVar contained only 109 variants in the androgen receptor gene (AR)2, while the AR mutation database (maintained until 2014) annotated >550 variants106. For Persistent Müllerian Duct Syndrome (PMDS), caused by variants in either the Anti-Müllerian Hormone (AMH) or its receptor (AMHR2), ClinVar contains 4 AMH and 5 AMHR2 pathogenic variants. They were curated from historical publications and deposited by OMIM, but another 20+ variants from the largest published series107,108 were not.
Furthermore, phenotypic description associated with the deposited variant is often absent or sparse. For syndromes where the genital phenotype is incompletely penetrant or for genes where different types of mutations cause different conditions, it is critical to know the phenotypes associated with reported variants. For example, AR variants can cause X-linked spinobulbar atrophy (not a DSD) or androgen insensitivity syndrome (AIS). One of the largest depositors into ClinVar (Invitae) does not specify which of the conditions is linked to variants and many other depositors do not indicate a phenotype at all. As a result, out of the ~400 AR variants in Clinvar, 190 provide a phenotypic description that includes DSD but only ~100 seem to be specifically linked to an actual DSD case.
Another problem is that the sex chromosome complement associated with the reported variant typically is not provided. Finding a variant associated with DSD in an XX individual may not be relevant to explaining an XY individual’s condition, as some genes only affect genital phenotype (either in syndromic or isolated forms) in an XX or XY genetic background3. For example, AMHR2 variants cause PMDS in men but not in XX women, who do not express AMH during fetal development. Reference databases for control, non-pathogenic variants (such as gnomAD) have started systematically reporting XX or XY status, and this needs to be adopted in databases aggregating disease variants as well. While the Leiden Open Variation Database reports zygosity and sex chromosome complement, when known, it is not routinely used by clinical algorithms. For these databases to realize their full potential, genotype-phenotype correlation, when known, needs to be curated, with an indication of known inheritance models. New, unpublished variants should be associated with a phenotypic description (for example. Did the individual have the condition for which this gene is known? In syndromic conditions, which of the incompletely penetrant features were present?). This will require a massive effort by expert curators. Unfortunately, unlike other conditions, there is no current working group for curation of DSD variants funded by the ClinGen consortium. For example, the only annotation currently available in ClinGen for SRY is “sufficient evidence for haploinsufficiency”, without specification that this is true only in XY, and without mention of DSD caused by abnormal presence of SRY in XX individuals or by SNVs in SRY in XY individuals. A PanelApp effort sponsored by Genomics England is underway to crowd-source evaluation of existing clinical panels, including for DSD. For any such effort, specific functional protein regions and variants will need to be curated in addition to gene names, and the curated database integrated into analytic pipelines. We have started a special interest group on the Franklin platform to curate DSD variants, which will be open to interested contributors worldwide and would have the advantage of being directly integrated into the sequence analysis platform.
Validation of VUS
The emergence of databases reporting population frequency has helped prioritize variants tremendously, but new approaches need to be developed and brought to the clinical testing realm to enable classification of the thousands of variants uncovered by next-generation genome sequencing and mapping.
Integration with transcriptome analysis.
RNA-seq profiles can be utilized for both assessing functional consequences of VUSs and molecular phenotyping of a disease state. RNA-seq data can be interrogated directly if a variant is expected to lead to changes in splicing, allelic skewing, RNA instability, or expression levels. Alternatively, RNA-Seq datasets from multiple DSD samples can be grouped based on expression profile and those groups investigated to search for common causative variants. To facilitate these analyses, platforms integrating RNA Seq profiles with WGS and microarray data need to be made user-friendly and publicly available for wider use. However, the utility of transcriptome-based VUS validation is limited by the availability of a relevant tissue on which to perform RNA-seq. Only about half of known DSD genes are expressed in blood cells, and a slightly higher percentage in skin fibroblasts109. For many DSD, whose origin is in the developing embryo, the relevant tissue is not available. Even a close proxy, such as adult genital or gonad tissue, is rarely available.
Interpretation of non-coding variants.
Pathogenic non-coding variants in known DSD genes have been reported (reviewed in9,110) and causative SVs have been identified outside of coding regions, in promoters and enhancers111. For example, duplication of an enhancer upstream of the AR gene was shown to increase expression76 and a recurrent variant in the promoter region resulted in aberrant translation and complete AIS112. A 3-bp deletion in the promoter of SRY that partially deletes a binding site for the SP1 transcription factor resulted in 46,XY complete gonadal dysgenesis. Other examples include variants in regulatory regions for SOX3, DMRT1, or NR0B1/DAX-1. The best studied example is the amazingly complex SOX9 regulatory region110. Variants affecting non-coding regions of SOX9 lead to at least 4 different conditions, including 46,XX testicular DSD and long-bone disorder campomelic dysplasia, which is associated with DSD in 75% of 46,XY individuals. Non-conservation between mouse and human genomes complicates the study of DSD variants in animal models.
Non-coding variants are currently under-ascertained (only a few hundred regulatory variants have been classified as pathogenic in human disease113–115), and validation of such variants is difficult. Integration of regulatory region annotation into WGS, OGM, or LRS analysis pipelines is still being developed. High-throughput tests of chromatin accessibility, configuration and binding site occupancy need to be applied to DSD datasets and tissues to prioritize variants that may affect functional regions of the genome, such as enhancers, relevant transcription factor binding sites, or regions of open chromatin116,117,118. Focusing variant searches to the few percent of the genome with open chromatin and potential regulatory function vastly reduces the logistical complexity of WGS interpretation. High-throughput cis-regulatory element sequencing (CRE-seq) reporter assays to test the impact of regulatory variants on gene expression119 should also be useful to classify VUS identified in DSD datasets.
DSD-specific cellular models.
For many DSD genes, such as SOX9, non-coding variants cannot be tested in animal models because of lack of evolutionary sequence conservation. Cellular models could be powerful tools to evaluate VUS and candidate genes affecting the molecular pathways present in the developing gonad120. The pluripotent human testicular embryonal carcinoma NT2/D1 cell line121–125 expresses the regulatory pathways downstream of SRY and SOX9 and responds to SOX9 perturbation similarly to what is observed during early sex determination. SOX9 is key to maintaining the integrity of Sertoli cells126,127 and controls a vast number of biological processes in gonadal development128. Over- or under-expression of SOX9 occurs in many XX and XY DSD. NT2/D1is therefore a powerful screening tool to detect patterns of gene network perturbation caused by DSD variants.
Attempts to reprogram skin fibroblasts into Sertoli cells, either via iPS cells or by direct transformation, have also emerged 129,130. These would allow the generation of case-specific cell models for high-throughput testing of the downstream effects of VUSs on molecular pathways and cellular phenotype (cell adhesion, division, response to hormones, etc.).
IMPROVING CLINICAL PRACTICE
The aspirational goals set by the International Rare Diseases Research Consortium (IRDiRC) include a call for reaching an accurate diagnosis within one year of an individual coming to medical attention. Yet many individuals with DSD undergo years of endocrine testing, imaging, and diagnostic surgeries without reaching a diagnosis. Hormone assays may not reliably distinguish between conditions (Box 2), and practice varies considerably131. A European effort is under way to standardize laboratory assessment in DSD131,132. But beyond endocrine testing, the consensus statements of 2006 and 20161,7 emphasized the need for interdisciplinary care, which needs to include expert competence for ordering appropriate genetic tests and interpreting their results. DSD-TRN registry data showed that 97% of undiagnosed patients at centers in the network had not exhausted clinically available genetic tests5. Causes are likely to include reluctance by insurance companies or health authorities to authorize testing, limited access to testing, and discomfort toward genetic testing, but may also reflect clinician habits or the lack of a genetics provider on the team altogether133,134.
Box 2: A case for early comprehensive NGS testing in DSD diagnosis.
Here, we outline the diagnostic process and timeline (see figure) for an individual taken from the DSD-TRN registry.
Family history.
Ultrasound during the sixth pregnancy of second cousins identified female genitalia in the fetus while Non-Invasive Prenatal Screening (NIPS) suggested the presence of a Y chromosome. An older brother had been born with ambiguous genitalia, but no cause had been established. An ultrasound at birth showed testes in the labioscrotal folds.
Endocrine testing.
While 21-hydroxylase deficiency does not cause genital atypia in XY individuals, measurement of electrolytes and 17-OH progesterone was performed on day 2 and showed normal levels. A second blood draw at about five weeks of age showed normal gonadotropins, cortisol, Anti-Müllerian hormone, very low testosterone (T), and a dihydrotestosterone (DHT)-to-testosterone ratio not diagnostic for SRD5A2 deficiency. A confirmatory DHT test was normal. The child was subjected to a human chorionic gonadotropin (hCG) stimulation test in pursuit of a HSD17B3 deficiency diagnosis. This rare recessive condition results in ambiguous genitalia in XY individuals owing to impaired testosterone synthesis. Stimulated testosterone was borderline low, but the test was not diagnostic.
Genetic testing.
The 46,XY karyotype was confirmed at birth. A microarray identified regions of homozygosity, as expected in this consanguineous family, but no copy number variants. An 11-gene NGS panel reported a variant of uncertain significance in ATRX; testing of the mother and affected brother ruled out this etiology. Finally, a firm diagnosis was made at age 71/2 months when another NGS panel (of 19 genes) identified a homozygous variant in HSD17B3.
Conclusion.
A large candidate gene panel or whole exome sequencing, ordered as an emergency test at birth, would have reassured the family within two weeks, as this condition has no known comorbidities. It would also have been very cost-effective, avoiding 7 blood draws from 3 individuals, 7 endocrine tests, an hCG stimulation, an inadequate NGS panel, and a microarray. Understanding the genetic diagnosis will orient management. Prenatal testing can be offered for future pregnancies. Early treatment may result in more typical male anatomy in adulthood. Sisters of prepubertal age should be tested: while XX individuals are asymptomatic, some XY individuals are born with typical female anatomy but develop signs of virilization and hirsutism at puberty, leading to a change to male gender in about 50% of cases. Alternatively, removal of the (infertile) gonads prior to puberty prevents the phenotypic changes (for complexities of management and gender outcomes in this condition, see156–159).
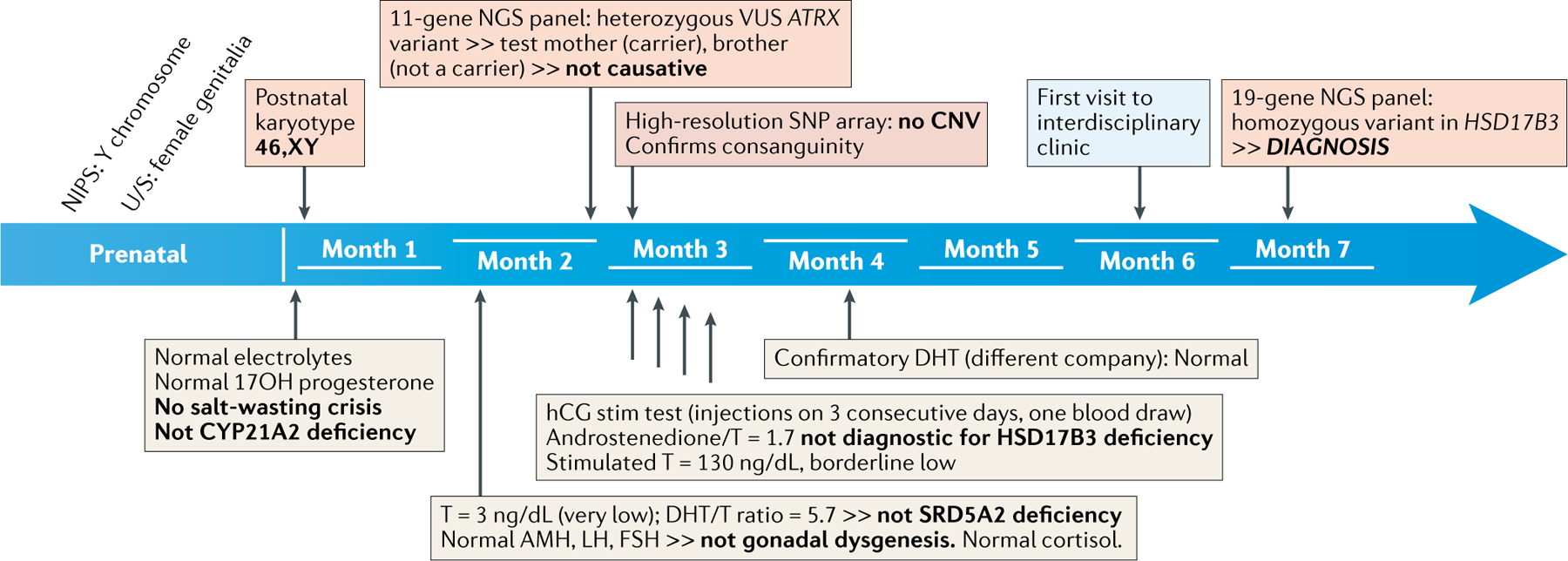
Prioritizing genetic tests
Prioritizing genetics as part of an integrated approach with parallel anatomical and biochemical assessment prior to interdisciplinary decision-making is recommended5,26,135 and should be considered standard of care. Cost-effectiveness of early genetic testing has been reported for DSD and other conditions22,51,54,56,136 (Box 2). Results of WES and gene panels are now available in a few weeks, on par with some endocrine tests, allowing genetic testing to become a front-line approach that can be ordered early in the diagnostic process to orient and focus expensive, and possibly invasive, phenotypic exploration and discovery of comorbidities. The feasibility and usefulness of stat exomes (results within 2 days) for diagnosis of critically ill newborns (including syndromic DSD, such as CHARGE, Kabuki, or Noonan syndromes) has been demonstrated and implemented in various settings137–141. For DSD gene panels, most facilities offer 2–3 week turnarounds, which would be sufficient once life-threatening adrenal insufficiency has been ruled out.
A major difficulty is choosing the list of candidate genes when interpreting WES data or designing an enriched panel. In published research studies, the number of genes included in targeted panels ranged from 30 to 18022,52–54,56. The lack of consensus is also evidenced by the extremely diverse panels offered by companies for clinical testing. In the US, several facilities use NGS to support their panels: some offer true panels, with capture enriched for a chosen list of genes; and some offer in silico panels with exome-wide capture and interpretation limited to the panel genes. Enrichment technology is proprietary and not disclosed and the actual number of bases covered or depth of coverage for individual genes is not described. The number of genes varies even for similar test descriptions. The ‘abnormal genitalia’ panels offered by Blueprint Genetics and the University of Chicago have 62 and 72 genes respectively. The ambiguously named ‘Disorder of male sex development’ (is it appropriate for males or for XY individuals?) panel from Invitae has 8 genes, the ‘neonatal 46,XY DSD panel’ from GeneDx has 19 genes, and the ‘46,XY DSD/complete gonadal dysgenesis’ panel from the University of Chicago has 26 genes. Average turnaround time ranges from 2 to 6 weeks and quoted price varies from $250 to $4,000. For facilities offering WES it is unclear what genes would be included in the analysis or if competency exists to specifically interpret DSD variants. In the Netherlands, Radboud-UMC offers a DSD/Primary adrenal deficiency WES-based test using a 143-gene panel with a 3–4 month turnaround for 900 euros in or a 15 working day turnaround for 4000 euros. Variability in practice is likely to be as great in other countries, but information is typically not made publicly available. Thus, establishing standardized testing protocols should be a priority of the field to improve test uptake and diagnostic yields.
Using diagnoses to improve care
A critical aspect of patient-centered care is diagnostic accuracy, which provides the means to avoid inappropriate treatments, adjust interventions to the specific condition, and improve outcomes142. For example, many individuals receive a working diagnosis of partial androgen insensitivity (PAIS), whereby an impaired tissue response to testosterone during development results in atypical external genitalia in 46,XY individuals. However, in only a minority of suspected de novo PAIS cases is a mutation actually found in the Androgen Receptor (AR) gene106,143,144. Those misdiagnosed may carry mutations in SRD5A2, NR5A1/SF-1, or LHCGR that can partially mimic PAIS. It is therefore critical to obtain a molecular diagnosis prior to deciding on a course of treatment, as response to androgen therapy could be very different depending on the gene mutated.
A precise genetic diagnosis is also needed to predict associated conditions. For example, WT1 heterozygous mutations can present as isolated DSD in XY individuals, but patients are at risk of tumors and kidney failure. Different types of variants in WT1 result in Denys-Drash syndrome (SNVs in exons 8/9) and Frasier syndrome (splice variants, reviewed in145). Wilms’ tumor is almost always associated with Denys-Drash, whereas risk of gonadoblastoma (but not Wilms’ tumor) is high in Frasier syndrome but kidney disease may be less severe 146. Other congenital malformations, in particular cardiovascular and skeletal abnormalities, are frequently associated with DSD and must be ascertained and monitored3,23,147.
Finally, it is important to determine the mode of inheritance not only for genetic counseling but also for carrier risk assessment. For example, many NR5A1/SF-1 mutations are inherited from seemingly unaffected parents148,149 who are at risk of early ovarian or testicular failure, information important to their family and life planning.
Need for large registries
Because each individual DSD condition is rare, and best practices often unclear, large registries are needed to capture longitudinal clinical data, in association with molecular variants when known. These registries need to include presentations and natural history, as well as the effects of hormonal or surgical interventions (or non-intervention) on gender identity, sexual function, psychosocial outcomes and quality of life.
The DSD-TRN has launched an effort to standardize practice and improve the standard of care at its member sites. It uses newly-designed, DSD-specific standardized forms to support all aspects of care (endocrine, genetic, anatomical, and psychosocial)5,8. The DSD-TRN registry collects this standardized information longitudinally to allow for investigation into long-term outcomes. The I-DSD registry6 has also recently started to collect longitudinal data in a standardized fashion150. DSD-Life , a large European effort to recruit participants to measure psychosocial adaptation, psychosexual health and mental health, reported that participation varied by condition151. It is critical that clinicians and families of all ethnic and socio-economic backgrounds are invited to actively participate in these registries so that an accurate, global picture of lives with DSD can be captured and serve all equitably.
CONCLUSIONS
DSD is a hyper-specialized subspecialty of pediatric endocrinology and urology, but awareness is growing, fueled by highly successful advocacy efforts which place front and center the issues faced by individuals with a DSD. Yet, the global impact of DSD remains under-recognized, and individuals with DSD continue to face stigma and societal attempts at discrimination (Box 3).
Box 3: Societal Challenges: DSD, genetic diagnosis, and sports.
Athletic authorities have attempted to prevent women athletes with a DSD from competing in women’s events for years, often using genetic data as justification. In 1968, genetic screening was imposed for all women athletes, initially as a sex chromatin test and later as a molecular genetic test. It is estimated that women were banned from events at almost every Olympic games between 1968 and 1984, while others likely ‘self-disqualified’160. Women who were removed had no proven advantage (such as women with complete androgen insensitivity), yet this genetically-based policy would potentially let 46,XX men compete. Under pressure from sports leaders and geneticists (such as Arne Ljungvist and Albert de la Chapelle, respectively) the history of discriminatory rulings targeting women with a Y chromosome concluded in 1999, with the declaration that the presence of a Y chromosome no longer precluded eligibility for women’s competitions (see161 for a detailed account).
However, this progress was undermined in 2018 when the International Association of Athletic Federation (IAAF, now World Athletics) introduced a new set of rules that lowered the testosterone limit to 5 nmol/L for eligibility to compete in the women’s category of a small number of events, namely distance running from 400m to the mile162. No clear rationale was provided for the rule change but it affected only those events in which Caster Semenya competed, a multiple women’s 800m champion whose biological sex had become a source of speculation. The controversy became an international outcry when, on May 1, 2019, the Court of Arbitration of Sports (CAS) sided with the IAAF, preventing Ms. Semenya and other women with naturally high testosterone levels from competing with other women unless they modified their natural biology.
The ruling is worrying for a number of scientific reasons. Only athletes with a DSD who carry a Y chromosome are targeted: a woman with an XX karyotype and a condition resulting in a testosterone level above 5 nmol/L (which has been reported, albeit rarely163,164) will remain eligible. (For a recent history of testosterone regulations in sports, refer to165). To take into account DSD mutations that cause complete or partial androgen sensitivity, only women athletes who are “sufficiently androgen‐sensitive that their elevated testosterone levels have a material androgenising effect”162 are affected by the ruling. However, there is currently no reliable diagnostic test to measure androgen sensitivity of muscles and no tests are performed on the athletes, which highlights the limitations of current genetic tests and their potential discriminatory consequences: although they can identify a genetic variant, physiological effects are not always clear. Finally, the new regulations are based on the unproven claim in the CAS Executive summary that “female athletes with 46 XY DSD enjoy a significant performance advantage over other female athletes without such DSD”. The departure by CAS from the expectation that guidelines should rely on evidence is disturbing. CAS had suspended similar guidelines in 2016, arguing that the IAAF could not demonstrate that testosterone accounts for the entirety of the 10%−12% difference between male and female performance and requesting the IAAF to produce additional data to justify the guidelines. The 2019 reversal of the 2016 ruling occurred without the production of such evidence.
We are now back to the pre-1999 position, whereby chromosomes define who is a man and who is a woman – a position that excluded countless athletes with 46, XY DSD conditions who were born, raised and self-identified continuously as women. With an increasing number of countries (including top Olympic countries such as Germany and Australia) allowing for non-binary gender identification on legal documents, the regulations are disconnected from societal trends and from the principles of the Olympic Charter: “Every individual must have the possibility of practicing sport without discrimination of any kind and in the Olympic spirit”166. The long journey of athletes with a DSD, and the discriminatory rulings they face, should inspire us to consider the complexity of interpreting genetic tests and cherish our most treasured value in science: evidence.
Estimates of DSD incidence vary widely, depending on the conditions included; even expert providers disagree on what should be included, and the conditions referred to specialized centers vary widely133. When mild malformations are included, estimates can reach 1.7% live births152; isolated hypospadias alone has been calculated to affect about 1 in 125 boys153. For severe conditions under the DSD umbrella, recent studies cite aggregated incidences between 0.2% and 0.5%26,110, making DSD one of the most frequent birth defects. An incidence of 0.5% for DSD equates to ~39 million affected families worldwide, which is similar to the number of people living with HIV/AIDS (according to HIV.gov) and 3–4 times more than the number of people with Parkinson’s disease (according to the Parkinson’s Foundation). As a comparison, arguably the best known rare genetic disease, cystic fibrosis, affects an estimated 70–100,000 people globally (according to the Cystic Fibrosis Foundation, Cystic Fibrosis Worldwide and ref. 154.
While a multidisciplinary approach to care is becoming the norm, it requires important resources and competencies not currently available in many settings. Major fundraising initiatives for medical research and infrastructure development will be necessary to increase the global awareness of DSD, support systematic large-scale variant validation studies and design of condition-specific bioinformatic analysis tools, and deploy evidence-based best practices in all aspects of clinical practice to benefit the DSD community at large, improve outcomes, and reduce stigma.
Supplementary Material
Acknowledgements
The authors thank Dr. P. Speiser for helpful discussions while preparing the case report for Box 2 and Mr. M. Almalvez for assistance with references and figures.
GLOSSARY
- Positive predictive value (PPV) [G]:
The PPV of a clinical test is the ratio of the number of individuals confirmed to have the condition being tested for to those who tested positive with the test, irrespective of disease status. It predicts the likelihood of someone who tests positive of having the condition.
- Isodicentric Y [G]:
Abnormal Y chromosome resulting in two centromeres and two identical arms (Yp or Yq). Breakpoints vary, but individuals with 2 short (Yp) arms may have two copies of the testis-determining gene SRY (located in Yp11.2), while those with 2 long arms typically do not carry SRY.
- Cell-free fetal DNA [G]:
Fragments of DNA of fetal origin circulating in the maternal blood during pregnancy, which can be tested to screen for aneuploidies such as trisomy 21 or to ascertain the sex chromosome complement of the fetus.
- Copy number variants [G]:
Variants, typically larger than 50 bp, that result in increased or decreased ploidy, such as a deletion on an autosome resulting in a single copy of the region instead of two. Copy number variants are a type of structural variant.
- Chimerism [G]:
a condition whereby two different genomes are found in a single individual, usually as a result of fusion of two zygotes during a twin pregnancy. DSD can arise when the two genomes have a different sex chromosome complement (XX and XY).
- Mosaicism [G]:
a condition whereby two different genomes are found in a single individual, typically resulting from a post fertilization mutation that is found only in the daughter cells of a subset of embryonic cells and thereby results in different phenotypic expression in different tissues. A frequent type of mosaicism associated with DSD is Turner syndrome variants with 45,X/46,XY mosaic karyotypes.
- Structural variants [G]:
Structural variants include copy number variants, insertions, translocations or inversions. They can be balanced (when the rearrangement does not result in loss or gain of genomic material) or unbalanced.
- Epimutation [G]:
A heritable variant that modifies gene expression through gain or loss of DNA methylation or other modification of chromatin without affecting the underlying DNA sequence.
- Episignature [G]:
A unique pattern of epigenetic variation (typically DNA methylation) occurring at multiple nucleotide locations throughout the genome. Episignatures have shown potential diagnostic value in syndromic conditions where no underlying genetic etiology is found.
Footnotes
Competing interests
E. V. has scientifically advised Bionano Genomics. E. D. declares no competing interests.
Peer review information
Nature Reviews Genetics thanks [Referee#1 name], [Referee#2 name] and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Related links
DSD-TRN https://dsdtrn.org
I-DSD https://home.i-dsd.org
Genoox sequence analysis platform https://www.genoox.com/genoox-integrated-omsv-platform/
SVI General Recommendations for Using ACMG/AMP Criteria https://clinicalgenome.org/working-groups/sequence-variant-interpretation/
Franklin https://franklin.genoox.com/clinical-db/home
Leiden Open Variation Database www.lovd.nl
ClinGen consortium, http://clinicalgenome.org/
PanelApp https://panelapp.genomicsengland.co.uk/panels/9/
IRDiRC https://irdirc.org/about-us/vision-goals/
DSD/Primary adrenal deficiency WES-based test https://www.radboudumc.nl/getmedia/ea3f05c7-2d81-4127-9d15-66aee97aff3e/DSDPRIMARYADRENALINSUFFICIENCY_DG300.aspx
DSD-Life https://www.dsd-life.eu/home/index.html
HIV.gov https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics
Parkinson’s Foundation https://www.parkinson.org/Understanding-Parkinsons/Statistics
Cystic Fibrosis Foundation https://www.cff.org/What-is-CF/About-Cystic-Fibrosis/
Cystic Fibrosis Worldwide https://www.cfww.org/
CAS Executive summary https://www.tas-cas.org/fileadmin/user_upload/CAS_Executive_Summary_5794_.pdf
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s415XX-XXX-XXXX-X
Contributor Information
Emmanuèle C. Délot, Center for Genetic Medicine Research, Children’s Research Institute, Children’s National Hospital, Washington DC 20010, USA
Eric Vilain, Department of Genomics and Precision Medicine, School of Medicine and Health Sciences, George Washington University, Washington DC 20052, USA.
REFERENCES
- 1. Lee PA et al. Consensus statement on management of intersex disorders. Pediatrics 118, e488–e500 (2006). This seminal publication of the 2005 international Consensus Conference on Management of Intersex Disorders, convened by the Lawson Wilkins Pediatric Endocrine Society (LWPES) and the European Society for Paediatric Endocrinology (ESPE), coined the term DSD and laid out the current framework for nomenclature, diagnosis, and interdisciplinary management of DSD.
- 2. Parivesh A, Barseghyan H, Délot E & Vilain E Translating genomics to the clinical diagnosis of disorders/differences of sex development. in Current Topics in Developmental Biology vol. 134 317–375 (2019). This review of the genetic etiology and testing of DSD provides a comprehensive analysis of the available evidence of pathogenicity, and the current limitations of the ClinVar database, for 69 DSD genes.
- 3. Délot EC & Vilain E Disorders of Sex Development. Yen & Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management: Eighth Edition (Elsevier Inc., 2018). doi: 10.1016/B978-0-323-47912-7.00016-0. This textbook chapter details clinical DSD categories, etiology, testing, and management approaches.
- 4.Hutson JM & Kearsey I Disorders of sex development (DSD): not only babies with ambiguous genitalia. A practical guide for surgeons. Pediatr. Surg. Int 33, 355–361 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Délot EC et al. Genetics of Disorders of Sex Development: The DSD-TRN Experience. Endocrinol. Metab. Clin. North Am 46, 519–537 (2017). This publication lays out the principles of the practice standardization effort undertaken by the DSD-TRN, and includes a report of early genetics data from its registry.
- 6.Ahmed SF, Bryce J & Hiort O International networks for supporting research and clinical care in the field of disorders of sex development. Endocr. Dev 27, 284–292 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Lee PA et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr 85, 158–180 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Sandberg DE, Gardner M, Callens N & Mazur T Interdisciplinary care in disorders/differences of sex development (DSD): The psychosocial component of the DSD—Translational research network. Am. J. Med. Genet. Part C Semin. Med. Genet 175, 279–292 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomes NL, Chetty T, Jorgensen A & Mitchell RT Disorders of Sex Development-Novel Regulators, Impacts on Fertility, and Options for Fertility Preservation. Int. J. Mol. Sci 21, 2282 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson EK et al. Gonadal Tissue Cryopreservation for Children with Differences of Sex Development. Horm. Res. Paediatr 92, 84–91 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Looijenga LHJ, Kao CS & Idrees MT Predicting gonadal germ cell cancer in people with disorders of sex development; insights from developmental biology. Int. J. Mol. Sci 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morin J et al. Oncologic outcomes of pre-malignant and invasive germ cell tumors in patients with differences in sex development – A systematic review. J. Pediatr. Urol 16, 576–582 (2020). [DOI] [PubMed] [Google Scholar]
- 13.Adam MP & Vilain E Emerging issues in disorders/differences of sex development (DSD). Am. J. Med. Genet. Part C Semin. Med. Genet 175, 249–252 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Gainotti S et al. Meeting Patients’ Right to the Correct Diagnosis: Ongoing International Initiatives on Undiagnosed Rare Diseases and Ethical and Social Issues. Int. J. Environ. Res. Public Health 15, 2072 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Austin CP et al. Future of Rare Diseases Research 2017–2027: An IRDiRC Perspective. Clin. Transl. Sci 11, 21–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croft B, Ayers K, Sinclair A & Ohnesorg T Review disorders of sex development: The evolving role of genomics in diagnosis and gene discovery. Birth Defects Res. Part C - Embryo Today Rev 108, 337–350 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Ahmed F et al. Disorders of sex development: advances in genetic diagnosis and challenges in management. Adv. Genomics Genet 2015, (2015). [Google Scholar]
- 18.Ohnesorg T, Vilain E & Sinclair AH The Genetics of Disorders of Sex Development in Humans 8, 262–272 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Ono M & Harley VR Disorders of sex development: new genes, new concepts 9, 79–91 (2013). [DOI] [PubMed] [Google Scholar]
- 20. Koopman P, Sinclair A & Lovell-Badge R Of sex and determination: marking 25 years of Randy, the sex-reversed mouse. Development 143, 1633–1637 (2016). This article provides a lively account of the characterization of SRY as the key mammalian gonadal sex determination gene, and is told by three of the main protagonists.
- 21. Baxter RM et al. Exome sequencing for the diagnosis of 46, XY disorders of sex development. J. Clin. Endocrinol. Metab 100, E333–E344 (2015). This article reports an early use of clinical exome sequencing for DSD diagnosis on a cohort of 46,XY individuals with various DSD conditions.
- 22.Eggers S et al. Disorders of sex development: Insights from targeted gene sequencing of a large international patient cohort. Genome Biol 17, 243 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heeley JM et al. Risk association of congenital anomalies in patients with ambiguous genitalia: A 22-year single-center experience. J. Pediatr. Urol 14, 153.e1–153.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Koopman P The Curious World of Gonadal Development in Mammals. Curr. Top. Dev. Biol 116, 537–45 (2016). [DOI] [PubMed] [Google Scholar]
- 25. León NY, Reyes AP & Harley VR A clinical algorithm to diagnose differences of sex development. Lancet Diabetes Endocrinol 7, 560–574 (2019). This article describes a recent comprehensive clinical algorithm that guides diagnosis and management of newborns with ambiguous genitalia.
- 26.Audi L et al. GENETICS IN ENDOCRINOLOGY: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 ‘DSDnet’. Eur. J. Endocrinol 179, R197–R206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Délot EC & Vilain EJ Nonsyndromic 46,XX Testicular Disorders of Sex Development. GeneReviews® (1993). [PubMed]
- 28.Mohnach L, Fechner PY & Keegan CE Nonsyndromic Disorders of Testicular Development. GeneReviews® (1993). [PubMed]
- 29.Alhomaidah D, McGowan R & Ahmed SF The current state of diagnostic genetics for conditions affecting sex development. Clin. Genet 91, 157–162 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Cools M et al. Caring for individuals with a difference of sex development (DSD): A Consensus Statement. Nat. Rev. Endocrinol 14, 415–429 (2018). This consensus statement by European workgroups details approaches and data collection processes for management of individuals with DSD throughout their life span.
- 31.Speiser PW et al. Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab 103, 4043–4088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Speiser PW et al. Newborn Screening Protocols and Positive Predictive Value for Congenital Adrenal Hyperplasia Vary across the United States. Int. J. Neonatal Screen 6, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krone N et al. Genotype-phenotype correlation in 153 adult patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: Analysis of the United Kingdom congenital adrenal hyperplasia adult study executive (CaHASE) cohort. J. Clin. Endocrinol. Metab 98, E346–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smet ME, Scott FP & McLennan AC Discordant fetal sex on NIPT and ultrasound. Prenat. Diagn 40, 1353–1365 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Byers HM et al. Discordant sex between fetal screening and postnatal phenotype requires evaluation. J. Perinatol 39, 28–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhamankar R, DiNonno W, Martin KA, Demko ZP & Gomez-Lobo V Fetal Sex Results of Noninvasive Prenatal Testing and Differences With Ultrasonography. Obstet. Gynecol 135, 1198–1206 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dey M, Sharma S & Aggarwal S Prenatal screening methods for aneuploidies. N. Am. J. Med. Sci 5, 182–190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng C et al. Clinical application of noninvasive prenatal screening for sex chromosome aneuploidies in 50,301 pregnancies: initial experience in a Chinese hospital. Sci. Rep 9, 7767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neufeld-Kaiser WA, Cheng EY & Liu YJ Positive predictive value of non-invasive prenatal screening for fetal chromosome disorders using cell-free DNA in maternal serum: independent clinical experience of a tertiary referral center. BMC Med 13, 129 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y et al. Cell-free DNA screening for sex chromosome aneuploidies by non-invasive prenatal testing in maternal plasma. Mol. Cytogenet 13, 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Croft B, Ohnesorg T & Sinclair AH The Role of Copy Number Variants in Disorders of Sex Development. Sex. Dev 12, 19–29 (2018). This article provides a comprehensive literature review of the impact of structural variants in DSD etiology.
- 42.Ledig S et al. Array-CGH analysis in patients with syndromic and non-syndromic XY gonadal dysgenesis: Evaluation of array CGH as diagnostic tool and search for new candidate loci. Hum. Reprod 25, 2637–2646 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Tannour-Louet M et al. Identification of De Novo copy number variants associated with human disorders of sexual development. PLoS One 5, e15392 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harrison SM, Seideman C & Baker LA DNA copy number variations in patients with persistent cloaca. J. Urol 191, 1543–1546 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Backhouse B et al. Identification of Candidate Genes for Mayer-Rokitansky-Küster-Hauser Syndrome Using Genomic Approaches. Sex. Dev 13, 26–34 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Takahashi K et al. Exome and copy number variation analyses of Mayer–Rokitansky–Küster–Hauser syndrome. Hum. Genome Var 5, 27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amarillo IE et al. Integrated small copy number variations and epigenome maps of disorders of sex development. Hum. Genome Var 3, 16012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.New MI et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. U. S. A 110, 2611–2616 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narasimhan ML & Khattab A Genetics of congenital adrenal hyperplasia and genotype-phenotype correlation. Fertil. Steril 111, 24–29 (2019). [DOI] [PubMed] [Google Scholar]
- 50.Koboldt DC Best practices for variant calling in clinical sequencing. Genome Med 12, 91 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arboleda VA et al. Targeted massively parallel sequencing provides comprehensive genetic diagnosis for patients with disorders of sex development. Clin. Genet 83, 35–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim JH et al. Diagnostic yield of targeted gene panel sequencing to identify the genetic etiology of disorders of sex development. Mol. Cell. Endocrinol 444, 19–25 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Özen S et al. Rapid Molecular Genetic Diagnosis with Next-Generation Sequencing in 46,XY Disorders of Sex Development Cases: Efficiency and Cost Assessment. Horm. Res. Paediatr 87, 81–87 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Fan Y et al. Diagnostic application of targeted next-generation sequencing of 80 genes associated with disorders of sexual development. Sci. Rep 7, 44536 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barseghyan H, Delot EC & Vilain E New technologies to uncover the molecular basis of disorders of sex development. Mol. Cell. Endocrinol 468, 60–69 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hughes LA et al. Next generation sequencing (NGS) to improve the diagnosis and management of patients with disorders of sex development (DSD). Endocr. Connect 8, 100–110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buonocore F & Achermann JC Primary adrenal insufficiency: New genetic causes and their long-term consequences. Clin. Endocrinol. (Oxf) 92, 11–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barseghyan H, Delot E & Vilain E New genomic technologies: an aid for diagnosis of disorders of sex development. Horm Metab Res 47, 312–320 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Stavropoulos DJ et al. Whole-genome sequencing expands diagnostic utility and improves clinical management in paediatric medicine. npj Genomic Med 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wright CF, FitzPatrick DR & Firth HV Paediatric genomics: Diagnosing rare disease in children. Nat. Rev. Genet 19, 253–268 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Belkadi A et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. U. S. A 112, 5473–5478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barbitoff YA et al. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep 10, 2057 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ravishankar A, Derraik JGB, Mathai S, Cutfield WS & Hofman PL Karyotypes, confined blood chimerism, and confusion: A case of genetic sex mislabelling and its potential consequences. N. Z. Med. J 128, 62–65 (2015). [PubMed] [Google Scholar]
- 64.Strain L, Dean JCS, Hamilton MPR & Bonthron DT A True Hermaphrodite Chimera Resulting from Embryo Amalgamation after in Vitro Fertilization. N. Engl. J. Med 338, 166–169 (1998). [DOI] [PubMed] [Google Scholar]
- 65.Hatano M, Fukuzawa R & Hasegawa Y The Mosaicism Ratio of 45,X May Explain the Phenotype in a Case of Mixed Gonadal Dysgenesis. Sex. Dev 12, 175–179 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Boucekkine C et al. The sole presence of the testis-determining region of the Y chromosome (SRY) in 46,XX patients is associated with phenotypic variability. Horm. Res. Paediatr 37, 236–240 (1992). [DOI] [PubMed] [Google Scholar]
- 67.Isidor B et al. Familial Frameshift SRY Mutation Inherited from a Mosaic Father with Testicular Dysgenesis Syndrome. J. Clin. Endocrinol. Metab 94, 3467–3471 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Murdock DR et al. Whole-Exome sequencing for diagnosis of turner syndrome: Toward next-generation sequencing and newborn screening. J. Clin. Endocrinol. Metab 102, 1529–1537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hu P et al. Low-level parental mosaicism affects the recurrence risk of holoprosencephaly. Genet. Med 21, 1015–1020 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Camats N, Fernández-Cancio M, Audí L, Schaller A & Flück CE Broad phenotypes in heterozygous NR5A1 46,XY patients with a disorder of sex development: an oligogenic origin? Eur. J. Hum. Genet 26, 1329–1338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Werner R et al. New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PLoS One 12, e0176720 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Flück CE et al. Broad Phenotypes of Disorders/Differences of Sex Development in MAMLD1 Patients Through Oligogenic Disease. Front. Genet 10, 746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindstrand A et al. Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome. Am. J. Hum. Genet 99, 318–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baetens D et al. NR5A1 is a novel disease gene for 46,XX testicular and ovotesticular disorders of sex development. Genet. Med 19, 367–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chaisson MJP et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 (2019). This landmark benchmarking study compares the respective outputs of LRS, SRS, and OGM datasets for detection of SVs of various sizes and types on the same genome.
- 76.Ho SS, Urban AE & Mills RE Structural variation in the sequencing era. Nat. Rev. Genet 21, 171–189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sedlazeck FJ, Lee H, Darby CA & Schatz MC Piercing the dark matter: Bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19, 329–346 (2018). [DOI] [PubMed] [Google Scholar]
- 78.Chan S et al. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol 1833, 193–203 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Mak ACY et al. Genome-wide structural variation detection by genome mapping on nanochannel arrays. Genetics 202, 351–362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levy-Sakin M et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun 10, 1025 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bocklandt S, Hastie A & Cao H Bionano Genome Mapping: High-Throughput, Ultra-Long Molecule Genome Analysis System for Precision Genome Assembly and Haploid-Resolved Structural Variation Discovery. Adv. Exp. Med. Biol 1129, 97–118 (2019). [DOI] [PubMed] [Google Scholar]
- 82.Jaratlerdsiri W et al. Next generation mapping reveals novel large genomic rearrangements in prostate cancer. Oncotarget 8, 23588–23602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Du C et al. A tandem duplication of BRCA1 exons 1–19 through DHX8 exon 2 in four families with hereditary breast and ovarian cancer syndrome. Breast Cancer Res. Treat 172, 561–569 (2018). [DOI] [PubMed] [Google Scholar]
- 84.Dixon JR et al. Integrative detection and analysis of structural variation in cancer genomes. Nat. Genet 50, 1388–1398 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Barseghyan H et al. Next-generation mapping: A novel approach for detection of pathogenic structural variants with a potential utility in clinical diagnosis. Genome Med 9, 1–11 (2017). This paper explains the OGM technology and demonstrates its clinical utility for clinical diagnosis in human disease on a cohort of individuals with Duchenne muscular dystrophy.
- 86.Sharim H et al. Long-read single-molecule maps of the functional methylome. Genome Res 29, 646–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rieken A, Bossler AD, Mathews KD & Moore SA CLIA Laboratory Testing for Facioscapulohumeral Dystrophy (FSHD): A Retrospective Analysis. Neurology 96, 10.1212/WNL.0000000000011412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bhattacharya S, Barseghyan H, Délot EC & Vilain E nanotatoR: a tool for enhanced annotation of genomic structural variants. BMC Genomics 22, 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mantere T, Kersten S & Hoischen A Long-Read Sequencing Emerging in Medical Genetics. Front. Genet 10, 426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amarasinghe SL et al. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21, 30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mahmoud M et al. Structural variant calling: The long and the short of it. Genome Biol 20, 246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arboleda VA et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet 44, 788–792 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.García-Acero M, Moreno O, Suárez F & Rojas A Disorders of Sexual Development: Current Status and Progress in the Diagnostic Approach. Curr. Urol 13, 169–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gimelli G, Giorda R, Beri S, Ginielli S & Zuffardi OA 46,X,inv(Y) young woman with gonadal dysgenesis and gonadoblastoma: Cytogenetics, molecular, and methylation studies. Am. J. Med. Genet 140 A, 40–45 (2006). [DOI] [PubMed] [Google Scholar]
- 96.Aref-Eshghi E et al. Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions. Am. J. Hum. Genet 104, 685–700 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Q et al. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun 10, 2449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang Y & Scott SA DNA methylation profiling using long-read single molecule real-time bisulfite sequencing (SMRT-BS). in Methods in Molecular Biology (eds. Kaufmann M, Klinger C & Savelsbergh A) vol. 1654 125–134 (Springer; New York, 2017). [DOI] [PubMed] [Google Scholar]
- 99.Beaulaurier J et al. Single molecule-level detection and long read-based phasing of epigenetic variations in bacterial methylomes. Nat. Commun 6, 7438 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gouil Q & Keniry A Latest techniques to study DNA methylation. Essays Biochem 63, 639–648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y et al. Accurate targeted long-read DNA methylation and hydroxymethylation sequencing with TAPS. Genome Biol 21, 54 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gelman H et al. Recommendations for the collection and use of multiplexed functional data for clinical variant interpretation. Genome Med 11, 85 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Richards S et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med 17, 405–424 (2015). This joint report from the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology established the criteria and guidelines currently used by all accredited clinical sequencing facilities and research laboratories to interpret and classify sequence variants (often called the ACMG criteria).
- 104.Kopanos C et al. VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nykamp K et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet. Med 19, 1105–1117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gulía C et al. Androgen Insensitivity Syndrome. in European Review for Medical and Pharmacological Sciences (eds. Adam MP et al.) vol. 22 3873–3887 (University of Washington, Seattle, 2018). [DOI] [PubMed] [Google Scholar]
- 107.Imbeaud S et al. Molecular genetics of the persistent müllerian duct syndrome: A study of 19 families. Hum. Mol. Genet 3, 125–131 (1994). [DOI] [PubMed] [Google Scholar]
- 108.Imbeaud S et al. A 27 base-pair deletion of the anti-Mullerian type II receptor gene is the most common cause of the persistent Mullerian duct syndrome. Hum. Mol. Genet 5, 1269–1277 (1996). [DOI] [PubMed] [Google Scholar]
- 109.Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baetens D, Mendonça BB, Verdin H, Cools M & De Baere E Non-coding variation in disorders of sex development. Clin. Genet 91, 163–172 (2017). [DOI] [PubMed] [Google Scholar]
- 111.Croft B et al. Human sex reversal is caused by duplication or deletion of core enhancers upstream of SOX9. Nat. Commun 9, 5319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hornig NC et al. A recurrent germline mutation in the 5’UTR of the androgen receptor causes complete androgen insensitivity by activating aberrant uORF translation. PLoS One 11, e0154158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smedley D et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet 99, 595–606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Caron B, Luo Y & Rausell A NCBoost classifies pathogenic non-coding variants in Mendelian diseases through supervised learning on purifying selection signals in humans. Genome Biol 20, 32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kwasnieski JC, Fiore C, Chaudhari HG & Cohen BA High-throughput functional testing of ENCODE segmentation predictions. Genome Res 24, 1595–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.ENCODE Project Consortium et al. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pranzatelli TJF, Michael DG & Chiorini JA ATAC2GRN: Optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference. BMC Genomics (2018) doi: 10.1186/s12864-018-4943-z. [DOI] [PMC free article] [PubMed]
- 119.Shen SQ et al. Massively parallel cis-regulatory analysis in the mammalian central nervous system. Genome Res 26, 238–255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sharpe RM, McKinnell C, Kivlin C & Fisher JS Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction 125, 769–784 (2003). [DOI] [PubMed] [Google Scholar]
- 121.Bernard P, Sim H, Knower K, Vilain E & Harley V Human SRY inhibits β-catenin-mediated transcription. Int. J. Biochem. Cell Biol 40, 2889–2900 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bernard P et al. Wnt signaling in ovarian development inhibits Sf1 activation of Sox9 via the Tesco enhancer. Endocrinology 153, 901–912 (2012). [DOI] [PubMed] [Google Scholar]
- 123.Alankarage D et al. SOX9 regulates expression of the male fertility gene Ets variant factor 5 ( ETV5 ) during mammalian sex development. Int. J. Biochem. Cell Biol 79, 41–51 (2016). [DOI] [PubMed] [Google Scholar]
- 124.Lee J et al. Sex-specific neuroprotection by inhibition of the Y-chromosome gene, SRY, in experimental Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A 116, 16577–16582 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Knower KC et al. Failure of SOX9 Regulation in 46XY Disorders of Sex Development with SRY, SOX9 and SF1 Mutations. PLoS One 6, e17751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Matson CK et al. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 476, 101–105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Uhlenhaut NH et al. Somatic Sex Reprogramming of Adult Ovaries to Testes by FOXL2 Ablation. Cell 139, 1130–1142 (2009). [DOI] [PubMed] [Google Scholar]
- 128.Rahmoun M et al. In mammalian foetal testes, SOX9 regulates expression of its target genes by binding to genomic regions with conserved signatures. Nucleic Acids Res 45, 7191–7211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liang J et al. Induction of Sertoli-like cells from human fibroblasts by NR5A1 and GATA4. Elife 8, e48767 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rodríguez Gutiérrez D, Eid W & Biason-Lauber A A Human Gonadal Cell Model From Induced Pluripotent Stem Cells. Front. Genet 9, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hannema SE & De Rijke YB Improving Laboratory Assessment in Disorders of Sex Development through a Multidisciplinary Network. Sex. Dev 12, 135–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kulle A et al. Steroid hormone analysis in diagnosis and treatment of DSD: position paper of EU COST Action BM 1303 ‘DSDnet’. Eur. J. Endocrinol 176, P1–P9 (2017). This position paper reports the recommendations developed by the European Cooperation in Science and Technology (EU-COST) workgroup, DSDnet, for harmonization of steroid hormone analysis in diagnosis and treatment of DSD.
- 133.Rolston AM et al. Disorders of sex development (DSD): Clinical service delivery in the United States. Am. J. Med. Genet. Part C Semin. Med. Genet 175, 268–278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kyriakou A et al. Current models of care for disorders of sex development – results from an International survey of specialist centres. Orphanet J. Rare Dis 11, 155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vora KA & Srinivasan S A guide to differences/disorders of sex development/intersex in children and adolescents. Aust. J. Gen. Pract 49, 417–422 (2020). [DOI] [PubMed] [Google Scholar]
- 136.Farnaes L et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. npj Genomic Med 3, 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Meng L et al. Use of exome sequencing for infants in intensive care units ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr 171, e173438–e173438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smith LD, Willig LK & Kingsmore SF Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb. Perspect. Med 6, a023168–a023168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chung CCY et al. Rapid whole-exome sequencing facilitates precision medicine in paediatric rare disease patients and reduces healthcare costs. Lancet Reg. Heal. - West. Pacific 1, 100001 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mestek-Boukhibar L et al. Rapid Paediatric Sequencing (RaPS): Comprehensive real-life workflow for rapid diagnosis of critically ill children. J. Med. Genet 55, 721–728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lunke S et al. Feasibility of Ultra-Rapid Exome Sequencing in Critically Ill Infants and Children with Suspected Monogenic Conditions in the Australian Public Health Care System. JAMA - J. Am. Med. Assoc 323, 2503–2511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Balogh EP, Miller BT, Ball JR & National Academies of Sciences Medicine and Engineering. Overview of diagnostic error in health care. in Improving Diagnosis in Health Care (National Academies Press (US), 2015). [PubMed] [Google Scholar]
- 143.Sultan C et al. Disorders of Androgen Action. Semin Reprod Med 20, 217–228 (2002). [DOI] [PubMed] [Google Scholar]
- 144.Lek N et al. Low frequency of androgen receptor gene mutations in 46 XY DSD, and fetal growth restriction. Arch. Dis. Child 99, 358–361 (2014). [DOI] [PubMed] [Google Scholar]
- 145.Eozenou C et al. Testis formation in XX individuals resulting from novel pathogenic variants in Wilms’ tumor 1 (WT1) gene. Proc. Natl. Acad. Sci. U. S. A 117, 13680–13688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hutson JM, Grover SR, O’Connell M & Pennell SD Malformation syndromes associated with disorders of sex development. Nat. Rev. Endocrinol 10, 476–487 (2014). [DOI] [PubMed] [Google Scholar]
- 147.Cox K et al. Novel associations in disorders of sex development: findings from the I-DSD Registry. J. Clin. Endocrinol. Metab 99, E348–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ferraz-de-Souza B, Lin L & Achermann JC Steroidogenic factor-1 (SF-1, NR5A1) and human disease. Mol. Cell. Endocrinol 336, 198–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Philibert P et al. Predominant Sertoli cell deficiency in a 46,XY disorders of sex development patient with a new NR5A1/SF-1 mutation transmitted by his unaffected father. Fertil. Steril 95, 1788.e5–1788.e9 (2011). [DOI] [PubMed] [Google Scholar]
- 150.Flück C et al. Standardised data collection for clinical follow-up and assessment of outcomes in differences of sex development (DSD): Recommendations from the COST action DSDnet. Eur. J. Endocrinol 181, 545–564 (2019). [DOI] [PubMed] [Google Scholar]
- 151.Röhle R et al. Participation of adults with disorders/differences of sex development (DSD) in the clinical study dsd-LIFE: design, methodology, recruitment, data quality and study population. BMC Endocr. Disord 17, 52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fausto-Sterling A Sexing the Body: Gender Politics and the Construction of Sexuality (Basic Books, 2000). [Google Scholar]
- 153.Pohl HG, Joyce GF, Wise M & Cilento BG Cryptorchidism and Hypospadias. J. Urol 177, 1646–1651 (2007). [DOI] [PubMed] [Google Scholar]
- 154.Kelly J Environmental scan of cystic fibrosis research worldwide. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc 16, 367–370 (2017). [DOI] [PubMed] [Google Scholar]
- 155.Biason-Lauber A, Konrad D, Meyer M, DeBeaufort C & Schoenle EJ Ovaries and Female Phenotype in a Girl with 46,XY Karyotype and Mutations in the CBX2 Gene. Am. J. Hum. Genet 84, 658–663 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Khattab A et al. Pitfalls in hormonal diagnosis of 17-beta hydroxysteroid dehydrogenase III deficiency. J. Pediatr. Endocrinol. Metab 28, 623–628 (2015). [DOI] [PubMed] [Google Scholar]
- 157.BALDUCCI R et al. Familial Male Pseudohermaphroditism With Gynaecomastia Due To 17Β‐Hydroxysteroid Dehydrogenase Deficiency. a Report of 3 Cases. Clin. Endocrinol. (Oxf) 23, 439–444 (1985). [DOI] [PubMed] [Google Scholar]
- 158.Boehmer ALM et al. 17β-Hydroxysteroid dehydrogenase-3 deficiency: Diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J. Clin. Endocrinol. Metab 84, 4713–4721 (1999). [DOI] [PubMed] [Google Scholar]
- 159.Chuang J et al. Complexities of gender assignment in 17β-hydroxysteroid dehydrogenase type 3 deficiency: is there a role for early orchiectomy? Int. J. Pediatr. Endocrinol 2013, 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Elsas LJ et al. Gender verification of female athletes. Genet. Med 2, 249–254 (2000). [DOI] [PubMed] [Google Scholar]
- 161.Ha NQ et al. Hurdling Over Sex? Sport, Science, and Equity. Arch. Sex. Behav 43, 1035–1042 (2014). [DOI] [PubMed] [Google Scholar]
- 162.IAAF. Eligibility Regulations for the Female Classification (Athletes With Differences of Sex Development) 1–22 https://www.iaaf.org/download/download?filename=0c7ef23c-10e1-4025-bd0c-e9f3b8f9b158.pdf&urlslug=IAAF Eligibility Regulations for the Female Classification %5BAthletes with Differences of Sex Development%5D in force as from 1st November 2018 (2018).
- 163.Bartolone L, Smedile G, Arcoraci V, Trimarchi F & Benvenga S Extremely high levels of estradiol and testosterone in a case of polycystic ovarian syndrome. Hormone and clinical similarities with the phenotype of the α estrogen receptor null mice. J. Endocrinol. Invest 23, 467–472 (2000). [DOI] [PubMed] [Google Scholar]
- 164.Legro RS et al. Total Testosterone Assays in Women with Polycystic Ovary Syndrome: Precision and Correlation with Hirsutism. J. Clin. Endocrinol. Metab 95, 5305–5313 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vilain E & Martinez-Patiño MJ Science’s place in shaping gender-based policies in athletics. Lancet (London, England) vol. 393 1504 (2019). [DOI] [PubMed] [Google Scholar]
- 166.International Olympic Committee. 2011 Olympic charter 1–95 https://stillmed.olympic.org/media/Document Library/OlympicOrg/General/EN-Olympic-Charter.pdf (2011).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.