

Abstract
Alzheimer’s disease (AD) is the major form of dementia in the elderly population. The main neuropathological changes in AD patients are neuronal death, synaptic alterations, brain inflammation, and the presence of cerebral protein aggregates in the form of amyloid plaques and neurofibrillary tangles. Compelling evidence suggests that the misfolding, aggregation, and cerebral deposition of amyloid-beta (Aβ) plays a central role in the disease. Thus, prevention and removal of misfolded protein aggregates is considered a promising strategy to treat AD. In the present study, we describe that the development of cerebral amyloid plaques in a transgenic mice model of AD (Tg2576) was significantly reduced by 40–80% through exchanging whole blood with normal blood from wild type mice having the same genetic background. Importantly, such reduction resulted in improvement in spatial memory performance in aged Tg2576 mice. The exact mechanism by which blood exchange reduces amyloid pathology and improves memory is presently unknown, but measurements of Aβ in plasma soon after blood exchange suggest that mobilization of Aβ from the brain to blood may be implicated. Our results suggest that a target for AD therapy may exist in the peripheral circulation, which could open a novel disease-modifying intervention for AD.
INTRODUCTION
Alzheimer’s disease (AD) is a chronic, progressive brain disorder characterized by impairments of learning and memory. AD is the most common cause of dementia in the elderly population and one of the leading causes of death in the US. Despite decades of effort to understand the genetic and molecular bases of AD, there is not yet available a cure or disease-modifying intervention for this devastating disease. Much evidence has been reported suggesting that misfolding, oligomerization, and progressive accumulation of cerebral deposits of amyloid-beta (Aβ) are a central event in AD [1]. Thus, preventing and removing Aβ aggregates is considered one of the most promising strategies to treat the disease.
Brain Aβ levels seem to be strictly regulated and kept in balance homeostatically. Inhibiting only 20% of Aβ production in the brain has been shown to be sufficient to suppress the formation and growth of amyloid plaques in vivo [2]. In contrast, the total cerebral clearance rate of Aβ in AD patients show a 25–30% decrease compared to normal subjects [3, 4], indicating that impairment of Aβ clearance mechanisms may be critically important in the development of AD. It has been shown that Aβ is constitutively produced by many cells [5], circulates in the blood [6], and is even secreted in urine [7]. Moreover, there are some reports suggesting that plasma Aβ measures may be useful biomarkers for predicting cerebral Aβ and tau deposition [8] and may even enable to predict disease progression [9, 10].
Monomeric and oligomeric Aβ peptides can cross the blood-brain barrier in both directions [11, 12]. Indeed, intravenous administration of labeled Aβ into old non-human primates showed that the labeled peptide was integrated into virtually all amyloid deposits in the brain [13]. Compelling evidence indicates that there is a tight relationship between the pools of Aβ circulating in the periphery and accumulating in the brain [14]. Indeed, a series of experiments have demonstrated that there is a dynamic equilibrium between Aβ in brain and blood and that sequestration of Aβ in plasma leads to a rapid efflux from the brain [15, 16]. Recently, additional reports have provided further support for the important role of circulating Aβ on brain pathology. Bu et al. showed that wild-type mice develop brain amyloid deposition after parabiosis with transgenic mice over-expressing mutated versions of APP and presenilin-1 [17]. Conversely, parabiosis between transgenic and WT mice before and after Aβ deposition in the brain of transgenic mice significantly reduced brain Aβ burden without alterations in the expression of amyloid precursor protein, Aβ generating and degrading enzymes, Abeta transport receptors, or other type of AD abnormalities including hyperphosphorylated tau, neuroinflammation, neuronal degeneration in the brains of parabiotic transgenic mice [18]. Another report suggested that intra-venous (i.v.) injection of brain homogenates containing Aβ aggregates promote the appearance of cerebral amyloid angiopathy [19]. Finally, our own recent study showed that cerebral amyloidosis can be significantly accelerated by transfusion with whole blood or plasma from old animals with extensive Aβ deposition [20]. Moreover, intra-venous injection of purified Aβ aggregates accelerated amyloid pathology, supporting the concept that Aβ seeds present in blood can reach the brain to promote neuropathological alterations in the brain of treated animals [20]. Altogether, these findings suggest that Aβ circulating in blood may contribute to the pathology in the brain.
Considering the putative role of the peripheral circulation in the brain pathological abnormalities, in this study we investigated the possibility of reducing Alzheimer’s pathology by blood exchange. For this purpose, we subjected transgenic mice expressing human mutant APP (Tg2576) to a series of total blood exchange procedures to replace endogenous vascular components with that coming from non-transgenic animals of the same background. We found that blood exchange treatment significantly decreased brain plaque formation and growth and resulted in the amelioration of spatial memory impairment in aged Tg2576 mice. These results suggest an important role for circulating Aβ or other blood factors in the development of AD pathogenesis and that the disease may be targeted in the periphery, which represents a goal much easier to accomplish than intervening in the brain.
MATERIALS AND METHODS
Blood exchange treatment
Tg2576 mice having 129S genetic background were used as a model of amyloid pathology in AD. These mice express the human amyloid precursor protein containing the Swedish mutation and begin to exhibit amyloid deposits at 8–9 months of age [21]. Normal blood was obtained from 129S wild type mice under Avertin (2%) anesthesia by heart puncture with a heparin-coated syringe. For all experiments, blood donors were 5 weeks old female 129S mice. Blood (300 μL) was manually withdrawn from the jugular vein of Tg2576 mice by a heparin-coated syringe with a 30-gauge needle. Thereafter, 300 μL-aliquot of whole blood obtained separately from wild-type mice was slowly infused (100 μL/min) into the jugular vein of Tg2576 mice to compensate for the withdrawn volume (Supplementary Fig. 1A). This procedure was repeated multiple times in each surgery. The number of withdrawal/infusion in a single surgery was determined by the body weight (BW) of animals due to their growth during the course of this longitudinal experiment. The locations of needle insertion are schematically shown in Supplementary Fig. 1B. The withdrawal/infusion in the jugular vein was performed through a thin layer of connective tissue left on the blood vessel at the time of jugular vein exposure, and the needle insertion was initiated from the proximal region to the heart. When one blood exchange treatment was done, the location for the next needle insertion was slightly moved to the distal location to avoid a rupture of the blood vessel wall. The relationship between the blood exchange ratio and the BW of recipient mice over the periods of the experiment was estimated as shown in Supplementary Fig. 1C. We initiated whole blood exchange in Tg2576 mice at the age of 3 months and continued the treatment once a month for 10 or 14 consecutive months. The second cohort was initiated at the age of 13 months and the treatment finished at the age of 17 months. In the sham group, original blood withdrawn from Tg2576 mice was re-infused into the same animal (Supplementary Fig. 1A). Untreated Tg2576 mice and wild type littermates only received monthly anesthesia to negate the effect of anesthetics for the long-term study. Animals were sacrificed 2 weeks after the end of the treatment for histological and biochemical analyses. The sample size used for the different studies is indicated in the figure legends and was chosen considering our previous experience with this model and the methodologies used for analysis. Initial groups included a larger number of mice to offset expected mortality due to the recurrent procedures. In our experiments, survival rate to the end-point was between 31 and 87%. In average for the three experiments, the sham group showed a survival rate of 65.3% whereas the blood exchange group was 50.6%. No animals were excluded from the analysis. Animals were randomly assigned to each group. All animal procedures described in this article were approved by the Center of Laboratory Animal Medicine and Care and the Animal Welfare Committee of the McGovern Medical School, University of Texas Health Science Center at Houston. Animals were monitored daily after surgery and if any indication of excessive pain occurred, the affected animal was humanely sacrificed.
Calculation of the exchange rate
The theoretical percentage of elimination of vascular components by blood exchange treatment was estimated by the blood volume based on mice BW (90 ml/kg) [22] using the following equation; Cn = Cn-1–[0.3 mL/(BW/11.1)] × Cn–1, where C is the concentration, n is the number of repetitions of withdrawal/infusion procedure, BW represents the BW (g), and the conversion value 11.1 was obtained from the ratio of blood volume to mouse BW [22]. A conversion value of 11.1 (g/mL) was calculated from the average blood volumes per BW reported in previous studies. The estimated average blood volume in mice was 90.3 mL/kg which indicates the blood volume (mL) = BW (g)/11.1 (g/mL) [22, 23]. The rate of exchange was also experimentally calculated in mice. For this purpose, wild-type mice (28.6 ± 0.7 g) received i.v. injection of [125I]-albumin (1 × 106 cpm) prior to perform blood exchange treatment to create an equilibrated distribution of [125I]-albumin in the systemic circulation. Thereafter, blood exchange treatment was initiated with whole blood containing [131I]-albumin (3 × 105 cpm/300 μl) to replace systemic circulating components. Blood exchange volume was 2.4 ml which was achieved by repeating the procedure eight times at the basis of 300 μl replacement per time. Radioactivities from [125I]- and [131I]-albumin in withdrawn blood were determined by a gamma counter (Packard Cobra II) and plotted against the number of repetitions of blood exchange procedure. The percentages of radiolabeled albumin in blood with each isotope were estimated as % of total injected radioactivity (Supplementary Fig. 1D).
Histology
Animals receiving whole blood exchange treatments were analyzed at the ages of 13 and 17 months. Brains were dissected out after transcardiac brain perfusion with heparinized phosphate buffer (pH 7.4, 20 mL), and the right hemisphere of the brain was immersed into histology grade paraformaldehyde solution (10%). The brain was subsequently processed in ethanol and xylene and embedded into paraffin wax. Sagittal brain sections (10 μm thickness) were immunostained with the 4G8 anti-Aβ antibody (1:1000, Covance, Princeton, NJ, USA) and counter-stained with hematoxylin. Sections were also stained with thioflavin S (ThS) to identify amyloid plaques, using previously described procedures [24]. The number and burden of Aβ and ThS reactive deposits were quantified in 3–4 sagittal sections per brain hemisphere. Each section was separated by at least 40 μm. The number and burden of plaques were averaged from these 3–4 sagittal sections and employed as representative values per animal examined. The burden was defined as the labeled area of the brain per total area analyzed and was expressed as a percentage. The number of plaques and areas covered by plaques in the brain were quantified by densitometric analysis with NIH Image J software. Both the histological staining and image analyses were performed in a blinded fashion.
Biochemical determination of soluble and insoluble Aβ1–40 and Aβ1–42 by ELISA
Brain samples from 13- or 17- month-old animals were processed for the determination of the concentrations of Aβ1–40 and Aβ1–42. The left hemisphere of the brain after dissection was snap-frozen in liquid nitrogen and stored at −80 °C for further processing. The tissues were later used for sequential extraction in phosphate-buffered saline (PBS) and formic acid (FA) fractions. Briefly, the frozen tissue was thawed and weighed. A 10% brain homogenate (wt/vol) was prepared in PBS containing protease inhibitors (EDTA free, Protease inhibitor cocktail tablets, 1 tablet in 50 ml PBS; Roche Applied Sciences, Germany) using a homogenizer (5000 rpm, 15 s, Precellys 24 Lysis and Homogenization, Bertin Technologies, France). The brain homogenate was centrifuged at 36,200 rpm for 1 h at 4 °C. The PBS soluble fraction obtained after this step was immediately frozen using liquid nitrogen and stored at −80 °C until use. The resultant pellet was sonicated (20 s, five times at amplitude 90; S-4000, Misonix Inc., Farming-dale, NY) and dissolved in 100 μl of 70% FA solution. Then, the FA soluble fraction was separated by centrifugation at 36,200 rpm for 1 h at 4 °C, the supernatant collected in new tubes, and pH neutralized (1:20 dilution) with 1 M Tris (pH 10.8). The neutralized FA fractions (1:200 final dilution) were immediately frozen in liquid nitrogen and stored at −80 °C until use.
The levels of Aβ1–40 and Aβ1–42 were measured in the PBS and FA fractions using human Aβ40 and Aβ1–42 ELISA kits (Invitrogen, Carlsbad, CA). The PBS fractions without further dilution were used for the measurement of both Aβ1–40 and Aβ1–42 for 13- and 17- month-old animal brain extracts. The FA fraction was further diluted 100- or 1000- fold for 13 month-old animal brain extracts and 1000- to 10,000- folds for 17-month-old animal samples. The dilutions were prepared in the sample diluents provided with the ELISA kits and the protocol recommended by the manufacturer was used for the measurements of Aβ1–40 and Aβ1–42 levels.
Plasma was obtained by centrifuging whole blood immediately after sampling at 5000 rpm for 10 min at room temperature, and isolated plasma was stored at −80 °C until further analysis. Plasma concentrations of Aβ1–40 and Aβ1–42 were separately measured by ELISA according to the manufacturer’s protocol. In some experiments, the total amount of Aβ withdrawn by the treatment was estimated by total blood volume based on the body weight of animals.
Behavioral study
Animals of 12.5 or 17.5 months of age were subjected to the Barnes maze test which consists of placing the animals onto a flat round table with multiple holes and a secure hiding chamber underneath one of the holes. The maze was surrounded by visual cues and the position of the secure chamber with respect to these visual cues remained constant throughout the experiment. After placing a mouse in the center of the maze, a high-pitch buzzer-sound was played and the time taken to reach the secure chamber was measured. Animals were trained during 4 consecutive days. Spatial short- and long- term memory was tested on the day 5th and 12th, respectively. Each mouse only received one set of these trainings plus the short- and long-term prove trials. The cut-off time was set to 300 s per training or prove trials. The entire maze and the target tunnel were extensively cleaned with isopropyl alcohol (30%) to remove odor cues after each trial. The behavioral studies were performed in a blinded fashion. The Barnes maze test was always performed between 8 am and 11 am. Prior to the test, mice were acclimated overnight in the behavior test preparation room having the same light intensity, temperature, room color and size configuration to the test room. Mice were housed in their home cage in the preparation room with cage mates and brought to the Barnes maze apparatus in the test room right next to the preparation room.
Statistical analysis
Means are presented with their standard errors and compared by one-way ANOVA followed by Dunnet or Newman–Keuls multiple comparison tests, or by two-way ANOVA followed by Bonferroni test with the Prism 5.0 program (GraphPad, San Diego, CA). For some studies, we used unpaired t-test, as indicated in the figure legends. Significance levels were evaluated and represented as P value *,P < 0.05, **,P < 0.01, and ***,P < 0.001 or †,P < 0.05, ††,P < 0.01, and †††,P < 0.001.
RESULTS
We performed a series of whole blood exchange treatments to partially replace blood from Tg2576 mice with complete blood from healthy wild-type littermate mice having the same genetic background. We initiated whole blood exchange in Tg2576 at the age of 3 months and continued the treatment once a month for 10 or 14 consecutive months. For the procedure, 300 μL of total blood was manually withdrawn from the jugular vein by a heparin-coated syringe with a 30-gauge needle. Thereafter, 300 μL of whole blood obtained separately from wild-type mice was slowly infused (100 μL/min) into the jugular vein of Tg2576 mice to compensate for the withdrawn volume (Supplementary Fig. 1A). By repeating 6–8 times this withdrawal/infusion procedure of whole blood transfusion, we can achieve 1.8–2.4 mL of blood exchange per animal per month which corresponds to an estimated replacement of 40–60% of the original blood each time the procedure was performed (Supplementary Fig. 1C). This estimation was supported by experimental calculation of the actual replacement ratio of circulating components, using radiolabeled albumin as a marker of the vascular space (Supplementary Fig. 1D). The data showed that after replacement of 2.4 ml of whole blood by the treatment, we found 41.6 ± 1.2% of original blood components remaining in the circulation and 58.4 ± 1.2% of injected donor blood components accumulating in recipient mice (Supplementary Fig. 1D). As controls for our blood exchange procedure, we included a sham group, in which original blood withdrawn from Tg2576 mice was re-infused into the same animals (Supplementary Fig. 1A). Other controls included untreated Tg2576 mice and wild type littermates, which only received monthly anesthesia to negate the effect of anesthetics for the long-term study.
For the first experiment, group of animals were subjected to blood exchange once a month between the ages of 3 and 13 months (Fig. 1A). Figure 1 shows representative sagittal cross-sections of the brain from blood exchanged, sham-operated and untreated transgenic mice at the age of 13 months. While abundant cerebral amyloid plaques identified by 4G8 anti-Aβ antibody and ThS staining were observed in both sham and untreated groups, predominantly in the cerebral cortex and hippocampus areas (Fig. 1B and D), only few and small plaques were observed in age-matched blood exchanged Tg2576 mice (Fig. 1C). The number and the area covered by amyloid plaques in these regions were significantly reduced in blood exchanged Tg2576 mice. Indeed, the number of deposits were 50.0% and 64.2% less in the cerebral cortex (Fig. 1E) and hippocampus (Fig. 1F), respectively, compared with the average observed in sham controls and untreated mice. As expected, no amyloid lesions were observed in age-matched wild type mice subjected to the same regimen of anesthesia than experimental groups (Supplementary Fig. 2A). The reduction in amyloid deposition by blood exchange was even higher when we estimated the amyloid burden, i.e., the area of the brain occupied by ThS-positive amyloid plaques. Indeed, a 75.6% and 75.9% reduction of the amyloid area in the cerebral cortex (Fig. 1E) and hippocampus (Fig. 1F), respectively, was observed between the blood exchange group and the average between sham-operated and untreated transgenic mice. These findings suggest that blood exchange effectively decreased cerebral plaque formation.
Fig. 1. Blood exchange treatment decreases the accumulation of cerebral amyloid plaques.

Tg2576 mice receiving whole blood exchange treatment during 10 consecutive months were analyzed at the age of 13 months (A). Sagittal cross-sections of sham-operated (B), blood exchanged (C), and untreated (D) Tg2576 mouse brains were stained with the 4G8 anti-Aβ antibody (a–d) and ThS (e–h). a and e In each treatment group show a low magnification image. Hippocampal areas depicted by dashed squares in (a and e) were further magnified and shown in (b and f), respectively. Similarly, cortical areas indicated by dashed squares in (a and e) are showed with higher magnification in (c and g), respectively. Representative plaque morphology in the cortex in broken squares in (c), and (g) were further magnified and shown in (d), and (h), respectively. Scale bars represent 500 μm (a and e), 100 μm (b, c, f and g) and 25 μm (d and h). The number of amyloid deposits and the plaque burden in the cerebral cortex (E) and hippocampus (F) were measured from ThS-stained sections. Each bar represents the mean ± S.E. of four to six mice. Asterisks indicate significant differences in the respective groups, which were evaluated by one-way ANOVA followed by Newman–Keuls multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ND: not detected. BE: blood exchanged Tg2576 (n = 4, 3 males, 1 female), Sham: Sham-operated Tg2576 (n = 4, 2 males and 2 females), uTg: untreated Tg2576 mice (n = 4, 1 males and 3 females) and WT: untreated wild-type littermates (n = 6, 4 males, 2 females). WT group was not considered in the statistical analysis because of no detectable levels. Following are the individual results for statistical analysis. E Number of plaques One-way ANOVA [F(3, 14) = 22.25, P < 0.0001], BE vs Sham [q = 3.841, p < 0.05], BE vs uTg [q = 5.760, p < 0.05], Sham vs uTg [q = 1.919, ns]; Plaque burden One-way ANOVA [F(3, 14) = 19.89, p < 0.0001], Post-hoc BE vs Sham [q = 6.766, p < 0.001], BE vs uTg [q = 4.624, p < 0.01], Sham vs uTg [q = 2.142, ns]. F Number of plaques One-way ANOVA [F(3, 14) = 27.98, P < 0.0001], Post-hoc BE vs Sham [q = 6.897, p < 0.001], BE vs uTg [q = 4.631, p < 0.01], Sham vs uTg [q = 2.266, ns]; Plaque burden One-way ANOVA [F(3, 14) = 9.851, p = 0.0009], Post-hoc BE vs Sham [q = 4.524, p < 0.05], BE vs uTg [q = 3.797, p < 0.05], Sham vs uTg [q = 0.7265, ns].
To study whether blood exchange resulted in functional changes in spatial memory, we performed Barnes’ maze test starting 2 weeks before sacrificing animals. Figure 2 shows the learning curve, short- and long-term memory in Tg2576 mice (12.5-month-old at the time of the test) receiving multiple blood exchange treatments for 10 consecutive months. There were no differences in learning curves among the groups regardless of the treatment (Fig. 2A), indicating that the animals effectively learned the task. Analysis of short-term memory (Fig. 2B) showed that blood exchanged mice spent a significantly shorter time seeking the destination than sham-operated animals or untreated Tg2576 mice. A long-term memory study (Fig. 2C) revealed that blood exchanged mice retained significantly higher performance compared to sham and untreated Tg2576. Interestingly, both short- and long-term memory in the blood exchange group were not significantly different from untreated wild-type mice. These results suggest that spatial memory performance in Tg2576 mice was improved by multiple blood exchange treatments to the level observed in wild type animals.
Fig. 2. Blood exchange treatment improves spatial memory.

Learning curve (A), short- (B) and long- term (C) memory were investigated. The graphs show the primary latency, which corresponds to the time taken for the animals to reach the hiding secure chamber. Short-term memory was evaluated at day 5, after 4 consecutive training days and long-term memory at day 12. Each point or bar represents the mean ± S.E. of eight to twelve mice. BE: blood exchanged Tg2576 (n = 9, 5 males and 4 females), Sham: Sham-operated Tg2576 (n = 11, 6 males and 5 females), uTg: untreated Tg2576 mice (n = 8, 2 males and 6 females) and WT: untreated wild-type mice (n = 12, 8 males and 4 females). Data in (A) was evaluated by Two-ways ANOVA using days of training and treatment as the variables (Interaction [F(9, 148) = 1.441, P = 0.1758], Treatment group [F(3, 148) = 45.74, p < 0.0001], Training [F(3, 148) = 3.718, P = 0.0129]). In (B and C), data were evaluated by one-way ANOVA followed by Newman–Keuls multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Whereas there were no significant differences in learning the task among the groups, short- and long- term memories in blood exchanged Tg2576 showed significant improvement showing a performance similar to wild-type animals. Following are the individual results for statistical analysis. B One-way ANOVA [F(3, 36) = 5.379, p = 0.0037], Post-hoc BE vs Sham [q = 2.563, ns], BE vs uTg [q = 3.846, p < 0.05], BE vs WT [q = 0.9870, ns], Sham vs uTg [q = 1.543, ns], Sham vs WT [q = 3.803, p < 0.05], uTg vs WT [q = 5.048, p < 0.01]. C One-way ANOVA [F(3, 36) = 11.42, p < 0.0001], Post-hoc BE vs Sham [q = 4.597, p < 0.01], BE vs uTg [q = 6.123, p < 0.001], BE vs WT [q = 0.3749, ns], Sham vs uTg [q = 1.957, ns], Sham vs WT [q = 5.346, p < 0.01], uTg vs WT [q = 6.881, p < 0.001].
To investigate whether blood exchange treatment persistently reduces cerebral amyloid formation and growth, we extended the period of blood exchange treatment for an additional 4 months (Fig. 3A), until a time in which untreated Tg2576 mice exhibit abundant and large plaques (Fig. 3D). In 17-month-old Tg2576 mice receiving blood exchange treatment for 14 consecutive months (Fig. 3C), the magnitude of staining for amyloid plaques, both using anti-Aβ antibody and ThS, was substantially lower than in age-matched animals receiving sham treatment or no treatment at all (Fig. 3B and D). The number and burden of plaques in blood exchanged Tg2576 mice compared to controls in the cerebral cortex (Fig. 3E) were reduced by 43.7% and 58.9%, respectively, and in the hippocampus (Fig. 3F) by 52.3% and 82.0%, respectively. Interestingly, comparison of data in Figs. 1E, F and 3E, F, showed that the plaque number and burden in blood exchanged Tg2576 mice at 17 months of age were in a similar range seen in 13-month-old sham controls or untreated mice. These results suggest that blood exchange reduced plaque deposition in the brain of 17 months old animals to those expected in untreated animals at 13 months old, indicating that blood exchange treatment persistently reduced cerebral plaque development in aged Tg2576 mice. As before, no amyloid lesions were detected in 17-month-old wild type mice (Supplementary Fig. 2B). To study the effect of blood exchange treatment on the growth of brain plaques, we plotted the amyloid burden observed in the different groups at 13 and 17 months of age and estimated the rate of plaque growth (Supplementary Fig. 3A). Two-way ANOVA revealed that blood exchange treatment has a significant effect in reducing the plaque burden in the cerebral cortex (P < 0.01) and hippocampus (P < 0.05) over the entire study period. The rates of plaque growth in the cerebral cortex (0.075%/month) and hippocampus (0.037%/month) in blood exchanged group were markedly smaller than the growth rates in sham controls (0.158 and 0.214%/month in the cortex and hippocampus, respectively) (Supplementary Fig. 3A, inset). The mean growth rates of plaques in untreated Tg2576 mice were similar to those seen in sham controls (Supplementary Fig. 3B). These results suggest that whole blood exchange not only decreases the net accumulation of Aβ aggregates, but also reduces the rates of plaque growth over time.
Fig. 3. Sustained reduction in the cerebral amyloid deposition by blood exchange treatment.
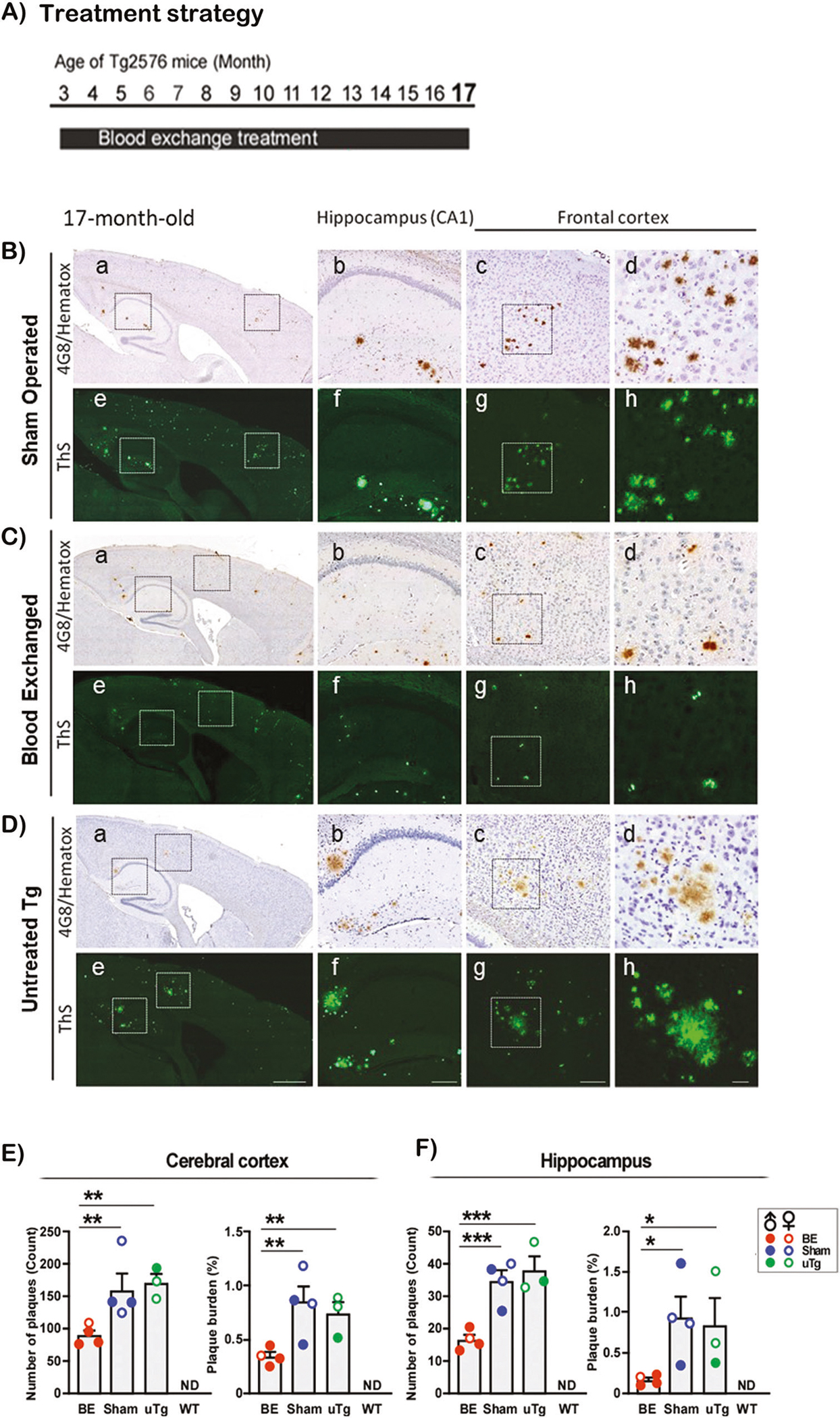
To evaluate whether the decrease of amyloid deposition was maintained at more severe stages of pathology, Tg2576 mice receiving blood exchange treatments during 14 consecutive months were analyzed at the age of 17 months (A). Sagittal cross-sections of sham-operated (B), blood exchanged (C), and untreated (D) Tg2576 mouse brains were stained with the 4G8 antibody (a–d) and ThS (e–h). As indicated in Fig. 1, the different panels correspond to distinct magnifications of the entire brain section (a and e), the hippocampal (b and f) and cerebral cortex (c, d, g and h) areas. Scale bars represent 500 μm (a and e), 100 μm (b, c, f and g) and 25 μm (d and h). The number of amyloid deposits and the plaque burden in the cerebral cortex (E) and hippocampus (F) were measured from ThS-stained sections. Each bar represents the mean ± S.E. of four to six mice. Asterisks indicate significant differences in the respective groups, which were evaluated by one-way ANOVA followed by Newman–Keuls multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ND: not detected. BE: blood exchanged Tg2576 (n = 4, 3 males and 1 female), Sham: Sham-operated Tg2576 (n = 4, 2 males and 2 females), uTg: untreated Tg2576 mice (n = 3, 1 males and 2 females) and WT: untreated wild-type littermates (n = 6, 3 males and 3 females). The WT group was not included in the statistical analysis because it showed no detectable levels of staining. Following are the individual results for statistical analysis. E Number of plaques One-way ANOVA [F(3, 13) = 41.22, P < 0.0001], BE vs Sham [q = 5.255, p < 0.01], BE vs uTg [q = 5.579, p < 0.01], Sham vs uTg [q = 0.7138, ns]; Plaque burden One-way ANOVA [F(3, 12) = 25.07, p < 0.0001], Post-hoc BE vs Sham [q = 5.824, p < 0.01], BE vs uTg [q = 4.440, p < 0.01], Sham vs uTg [q = 1.294, ns]. F Number of plaques One-way ANOVA [F(3, 12) = 59.69, P < 0.0001], Post-hoc BE vs Sham [q = 7.117, p < 0.001], BE vs uTg [q = 8.423, p < 0.001], Sham vs uTg [q = 1.221, ns]; Plaque burden One-way ANOVA [F(3, 12) = 8.032, p = 0.0033], Post-hoc BE vs Sham [q = 4.326, p < 0.05], BE vs uTg [q = 3.772, p < 0.05], Sham vs uTg [q = 0.5180, ns].
To confirm the reduction of cerebral amyloid burden found by histological analyses, we employed an enzyme-linked immunosorbent assay (ELISA) for quantifying the concentrations of the soluble and insoluble forms of Aβ1–40 and Aβ1–42 in the brain (Fig. 4). After sequential fractionation of brain Aβ with PBS and FA to separate the soluble and insoluble pools of the peptide, immunoreactive Aβ1–40 and Aβ1–42 were separately measured by ELISA (Invitrogen, Carlsbad, CA). Soluble Aβ1–40 and Aβ1–42 in the brain showed significant reductions in blood exchanged Tg2576 mice at the age of 13 months, to values that were 55.1% and 50.3%, respectively, of those found in control groups (sham controls and untreated mice) (Fig. 4A). Conversely, no significant decrease in soluble Aβ was observed in blood exchanged 17-month-old mice. Importantly, the levels of insoluble Aβ1–40 and Aβ1–42 extracted with FA, which correspond to the abnormally aggregated peptide, significantly decreased in blood exchanged Tg2576 mice at both 13 and 17 months of age. The values of insoluble Aβ1–40 and Aβ1–42 were reduced by 79.6% and 62.9%, respectively at 13 months in blood exchanged animals compared with the average obtained in the two control groups (Fig. 4B). Such remarkable reduction was also seen in older Tg2576 mice (17-month-old) receiving blood exchange treatment, even though the levels of insoluble Aβ in the brain increased with age (Fig. 4B). While there was a significant decrease in the plaque burden in 17-month-old Tg2576 mice receiving the BE treatment from 3 to 17 months of age, the soluble Aβ content in the brain remained unchanged among the groups. At present, we do not know the reason for these results. However, considering the age-associated reduction in Aβ clearance in AD [25], it is possible that multiple alternative mechanisms may be involved in the reduction of insoluble Aβ components in the brain such as immunomodulatory changes and/or alteration in blood-brain barrier functions.
Fig. 4. Reduction in insoluble Aβ levels in Tg2576 mice receiving blood exchange treatment.

Animals were sacrificed at the ages of 13 or 17 months and half the brain was frozen for biochemical studies. The brain was homogenized and the concentration of soluble Aβ after extraction in PBS (A) as well as insoluble Aβ (formic acid soluble) (B) was measured by ELISA. The concentrations of Aβ1–40 and Aβ1–42 were measured by C-terminal specific antibodies, using the Invitrogen kit. Each bar represents the mean ± S.E. of three to five mice. Asterisks indicate significant differences in the respective groups, which were evaluated by one-way ANOVA followed by Newman–Keuls multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ND: not detected. BE: blood exchanged Tg2576 (n = 5, 3 males and 2 females), Sham: Sham-operated Tg2576 (n = 4, 2 males and 2 females), uTg: untreated Tg2576 mice (n = 4, 2 males and 2 females) and WT: untreated wild-type littermates (n = 5, 3 males and 2 females). The WT group was not considered in the statistical analysis since all values were zero. Following are all the individual values for statistical analysis. A 13-month-old Aβ40, One-way ANOVA [F(3, 14) = 27.25, p < 0.0001], Post-hoc BE vs Sham [q = 4.906, p < 0.01], BE vs uTg [q = 4.730, p < 0.01], Sham vs uTg [q = 0.1669, ns]. 13-month-old Aβ42, One-way ANOVA [F(3, 14) = 81.77, p < 0.0001], Post-hoc BE vs Sham [q = 10.49, p < 0.001], BE vs uTg [q = 4.691, p < 0.01], Sham vs uTg [q = 0.5.502, p < 0.01]. 17-month-old Aβ40, One-way ANOVA [F(3, 10) = 24.90, p < 0.0001]. Post-hoc comparisons were not significant in all groups [q < 0.001, ns]. 17-month-old Aβ42, One-way ANOVA [F(3, 9) = 28.35, p < 0.0001]. Post-hoc comparisons were not significant in all groups [q < 0.001, ns]. B 13-month-old Aβ40, One-way ANOVA [F(3, 14) = 8.080, p = 0.0023], Post-hoc BE vs Sham [q = 4.959, p < 0.01], BE vs uTg [q = 3.097, p < 0.05], Sham vs uTg [q = 1.767, ns]. 13-month-old Aβ42, One-way ANOVA [F(3, 14) = 17.72, p < 0.0001], Post-hoc BE vs Sham [q = 6.041, p < 0.01], BE vs uTg [q = 3.520, p < 0.05], Sham vs uTg [q = 2.391, ns]. 17-month-old Aβ40, One-way ANOVA [F(3, 10) = 15.69, p = 0.0004], Post-hoc BE vs Sham [q = 3.886, p < 0.05], BE vs uTg [q = 4.008, p < 0.05], Sham vs uTg [q = 0.1145, ns]. 17-month-old Aβ42, One-way ANOVA [F(3, 10) = 59.79, p < 0.0001], Post-hoc BE vs Sham [q = 5.015, p < 0.01], BE vs uTg [q = 5.521, p < 0.01], Sham vs uTg [q = 0.4739, ns].
A possible interpretation of the findings that whole blood exchange resulted in a substantial reduction in amyloid pathology in the brain is that the procedure altered the dynamic equilibrium between the brain and blood pools of Aβ. This model is consistent with our results showing a reduction in soluble Aβ in the brain of blood exchanged mice at 13 months (Fig. 4A). To further study this issue, we measured plasma Aβ levels at various times after blood exchange treatment. For these measurements, we employed a simplified setting in which Tg2576 mice received the treatment only one time to avoid possible complications of multiple treatments on the interpretations of the results. Groups of experimentally naïve Tg2576 mice at the ages of 5 and 13 months were used for single blood exchange treatment. The reason to measure the levels of plasma Aβ at 5 months of age was to evaluate how whole blood exchange affect the levels of circulating Aβ ‘before’ the onset of cerebral amyloidosis in the Tg2576 mouse model. On the other hand, the measurement at 13 months of age was done to assess if whole blood exchange treatment does the same ‘after’ the onset of cerebral amyloidosis. During the treatment, 50 μL aliquots of whole blood were kept for measuring Aβ levels in plasma. At designated time points after the treatment, 50 μL of whole blood was withdrawn and after separation of plasma, Aβ concentrations were measured by ELISA (Fig. 5). During the blood exchange treatment in 13-month-old Tg2576 mice, the concentrations of Aβ1–40 and Aβ1–42 in plasma were estimated to be 1511.7 and 103.0 pg/ml, respectively, and did not change significantly during the procedure. In 5-month-old Tg2576 mice, similar plasma concentrations of Aβ1–40 and Aβ1–42 were obtained (1095.0 and 89.4 pg/ml). Surprisingly, plasma Aβ concentration starts increasing 1 h after the treatment and reached the highest peak between 3 and 6 h after the blood exchange procedure was completed (Fig. 5). At these times, the concentrations of both Aβ1–40 and Aβ1–42 in plasma increased 2–3 folds respect to the basal levels. The higher concentration of Aβ in plasma remained for several hours, returning to basal levels by 3 days post-treatment. This behavior did not depend on the peptide measured (Aβ1–40 or Aβ1–42) or the age of the mice. Since no differences were observed between the 5 months and 13-months groups, we expect that age and presence of cerebral amyloidosis do not change this behavior. The mechanism responsible for the increase in plasma Aβ is not known, however taking into account that blood exchange reduced the levels of Aβ in the brain, we could speculate that relocation of Aβ from the brain to blood may explain in part our results.
Fig. 5. Plasma concentrations of total Aβ increased soon after blood exchange.

Changes in plasma Aβ levels in 5- and 13- month-old Tg2576 mice were measured at different times during and after a single blood exchange procedure. Groups of 7 Tg2576 female mice were subjected to the same regimen of blood withdrawals and replacement with WT blood as in the experiments described in Fig. 1. Blood (50 μL) was collected at 1, 3, 6, 12, 24, and 72 h after blood exchange procedure. Whole blood was immediately mixed with 0.1% heparin, the plasma separated by centrifugation for 15 min at 5000 × g at 4 °C, and used to measure Aβ levels. Immunoreactive Aβ1–40 and Aβ1–42 were separately measured by ELISA kit. There was no cross-reactivity in determining the Aβ1–40 and Aβ1–42 by ELISA. Each point represents the mean ± S.E. of four to seven plasma samples per point. Asterisks and daggers indicate significant differences from respective baselines concentration of total Aβ in plasma estimated during the blood exchange treatment (gray shaded period), which were evaluated by one-way ANOVA followed by Dunnett’s multiple comparison test. **, P < 0.01 for 13-month-old mice and †, P < 0.05, ††, P < 0.01, and †††, P < 0.001 for 5-month-old mice. Following are the individual values from the statistical analysis. 13-month-old Aβ40 One-way ANOVA [F(6, 69) = 3.233, P = 0.0074], Post-hoc comparison at 3 h [q = 3.257, p < 0.01]. All other time points were not significant [q < 0.001, ns]. 5-month-old Aβ40 One-way ANOVA [F(6, 71) = 13.99, P < 0.0001], Post-hoc comparison at 1 h [q = 4.395, p < 0.001], 3 h [q = 4.887, p < 0.001], 6 h [q = 6.765, p < 0.001], 12 h [q = 3.642, p < 0.01], 24 h [q = 4.183, p < 0.001], and 72 h [q = 2.864, p < 0.05]. 13-month-old Aβ42 One-way ANOVA [F(6, 69) = 3.285, P = 0.0067], Post-hoc comparison at 3 h [q = 3.851, p < 0.01]. All other time points were not significant [q < 0.001, ns]. 5-month-old Aβ42 One-way ANOVA [F(6, 70) = 20.68, P < 0.0001], Post-hoc comparison at 1 h [q = 5.580, p < 0.001], 3 h [q = 5.736, p < 0.001], 6 h [q = 8.879, p < 0.001], 12 h [q = 4.023, p < 0.001], 24 h [q = 1.629, ns], and 72 h [q = 0.5152, ns].
As prophylactic treatments done at the ages between 3 and 17 months in Tg2576 mice showed the reduction in cerebral amyloid burden and the restoration in spatial memory, we wanted to evaluate the efficiency of the treatment in a more therapeutic setting. For this purpose, we examined the effect of whole blood exchange in aged Tg2576 mice with already established amyloid pathology on their cerebral amyloid burden and the spatial memory function. We have characterized that untreated Tg2576 mice at the age of 13 months showed abundant cerebral amyloid burden (Fig. 1) as well as abnormalities in spatial memory (Fig. 2). To test the therapeutic effectivity of whole blood exchange treatment in the symptomatic phase in aged Tg2576 mice, we initiated the blood exchange treatment at 13 months of age and evaluated the brain histology and behavior memory at 17 months (Fig. 6A). We increased the number of blood exchanges in the therapeutic setting, to compensate for the changes on the animal weight. As shown in Supplementary Fig. 1C, the proportion of blood exchanged depend on the weight of the mice. Since weight increased with time, we performed additional number of exchanges in the therapeutic experiment to reach a similar estimated extent of exchange.
Fig. 6. Changes in the cerebral amyloid burden by blood exchange treatment in aged Tg2576 mice after the onset of cerebral plaque pathology.

Tg2576 mice receiving whole blood exchange treatment during 5 consecutive months were analyzed at the age of 17 months (A). Sagittal cross-sections of sham-operated (B), and blood exchanged (C) were stained with the 4G8 anti-Aβ antibody (a–d) and ThS (e–h). a and e In each treatment group show a low magnification image. Hippocampal areas depicted by dashed squares in (a and e) were further magnified and shown in (b and f), respectively. Similarly, cortical areas indicated by dashed squares in (a and e) are showed with higher magnification in (c and g), respectively. Representative plaque morphology in the cortex in broken squares in (c and g) were further magnified and shown in (d and h), respectively. Scale bars represent 500 μm (a and e), 100 μm (b, c, f and g) and 25 μm (d and h). The number of amyloid deposits and the plaque burden in the cerebral cortex (D) and hippocampus (E) were measured from ThS-stained sections. Each bar represents the mean ± S.E. of seven to eleven mice. Asterisks indicate significant differences in the respective groups, which were evaluated by unpaired t-test with Welch’s correction. *, P < 0.05; **, P < 0.01; BE: blood exchanged Tg2576 (n = 11, 6 males and 5 females), and Sham: Sham-operated Tg2576 (n = 7, 4 males and 3 females). Following are the individual values for statistical analysis. D Number of plaques F-test to compare variances [F(6, 10) = 5.385, p = 0.02], unpaired t-test [p = 0.0059]. Plaque burden [F(6, 10) = 2.657, p = 0.166, ns], unpaired t-test [p = 0.0081]. E Number of plaques F-test to compare variances [F(10, 6) = 3.473, p = 0.141, ns] unpaired t-test [p = 0.106, ns]. Plaque burden F-test to compare variances [F(10, 6) = 2.419, p = 0.2917, ns], unpaired t-test [p = 0.0152].
Figure 6 shows the brain amyloid burden in Tg2576 mice receiving the blood exchange treatment from 13 months to 17 months of age. The number of plaques and the amyloid plaque burden in both the cerebral cortex and hippocampus were significantly reduced in the blood exchanged Tg2576 mice compared to age-matched sham Tg2576 mice (Fig. 6B–E). The plaque growth rate in this cohort also showed a significant reduction in these brain regions (Supplementary Fig. 3C). Interestingly, the plaque load of these 17 months old animals treated since month 13, was not significantly different as Tg2576 animals sacrificed without treatment at 13 months of age. This data indicates that blood exchange keep the extent of Aβ deposition constant during the treatment. The spatial memory acquisition in aged Tg2576 mice (Fig. 7A) showed impairment in Sham and untreated Tg2576 mice at the age of 17 months, compared to age-matched wild type mice, which was improved by the blood exchanged Tg2576 mice. Both short- and long-term memories (Fig. 7B and C) were significantly improved in aged Tg2576 mice receiving the blood exchange treatment to the levels seen in age-matched wild type control, while sham and untreated Tg2576 mice showed identical primary latency in the Barnes’ maze trial. These data suggest that whole blood exchange treatment was effective in keeping pre-existing cerebral amyloid burden without further significant grow between the ages of 13 and 17 months in Tg2576 mice. The behavioral data showed that improvement of spatial memory function is possible even after the onset of cerebral plaque development.
Fig. 7. Changes in the spatial memory by blood exchange treatment after the onset of cerebral plaque pathology.
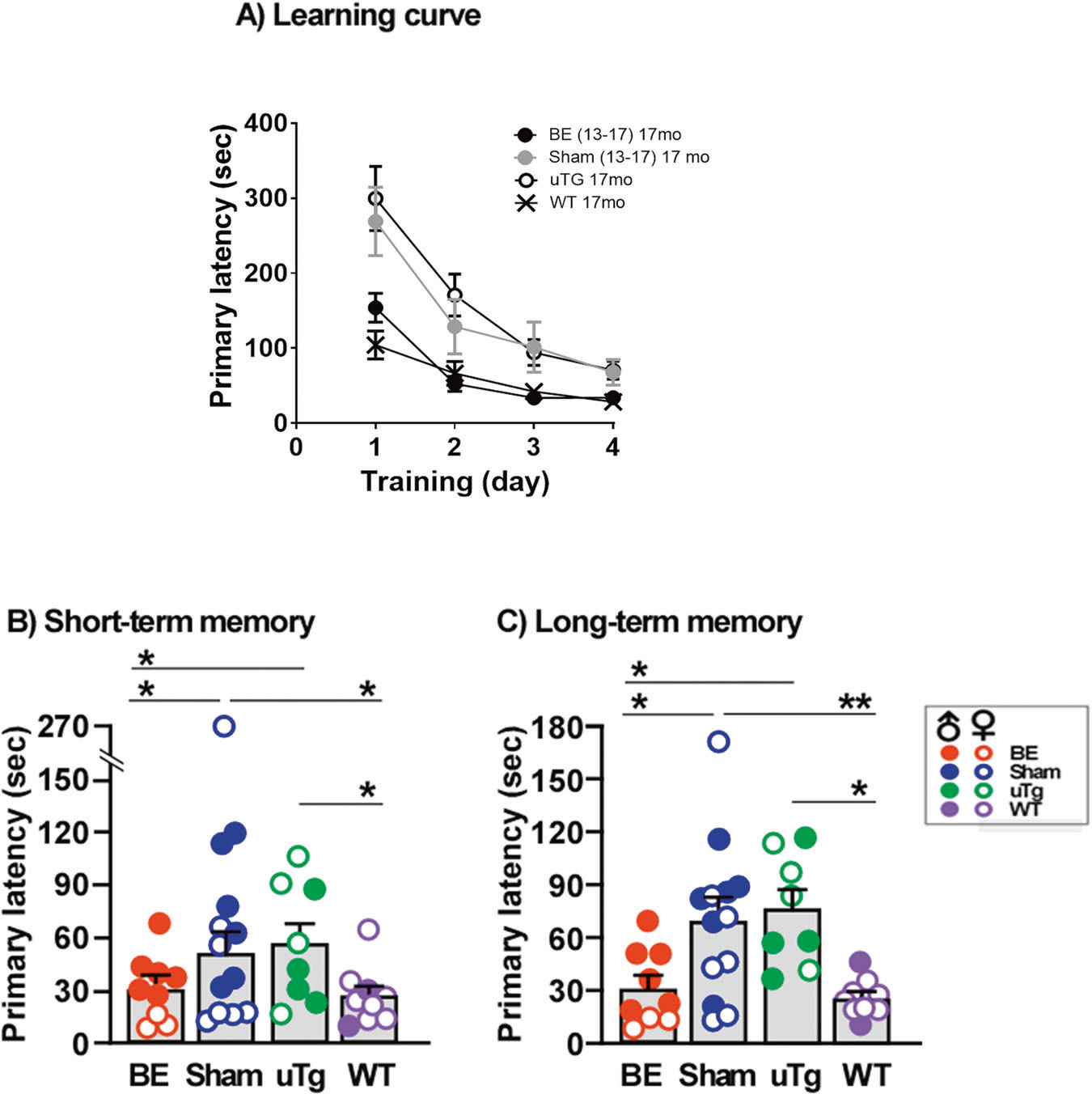
Learning curve (A), short- (B) and long- term (C) memory were investigated. The graphs show the primary latency, which corresponds to the time taken for the animals to reach the hiding secure chamber. Short-term memory was evaluated at day 5, after 4 consecutive training days and long-term memory at day 12. Each point or bar represents the mean ± S.E. of eight to thirteen mice. BE: blood exchanged Tg2576 (n = 9, 6 males and 3 females), Sham: Sham-operated Tg2576 (n = 13, 6 males and 7 females), uTg: untreated Tg2576 mice (n = 8, 4 males and 4 females) and WT: untreated wild-type mice (n = 9, 2 males and 7 females). Asterisks indicate statistically significant differences in the respective groups, *, P < 0.05; **, P < 0.01. Following are the individual values from statistical analysis. A Two-way ANOVA Interaction [F(9, 140) = 1.44, p = 0.1786], Treatment group [F(3, 140) = 13.19, p < 0.0001], Training [F(3, 140) = 25.65, p < 0.001. Data in (B and C) were evaluated by one-way ANOVA followed by Newman–Keuls multiple comparison test. B One-way ANOVA [F(3, 35) = 5.046, p = 0.0052], Post-hoc comparison BE vs Sham [q = 4.011, p < 0.05], BE vs uTg [q = 3.425, p < 0.05], BE vs WT [q = 0.2306, ns], Sham vs uTg [q = 0.1668, ns], Sham vs WT [q = 4.262, p < 0.05], uTg vs WT [q = 3.649, p < 0.05]. C One-way ANOVA [F(3, 34) = 5.602, p = 0.0031], Post-hoc comparison BE vs Sham [q = 3.725, p < 0.05], BE vs uTg [q = 3.612, p < 0.05], BE vs WT [q = 0.7555, ns], Sham vs uTg [q = 0.2468, ns], Sham vs WT [q = 4.533, p < 0.01], uTg vs WT [q = 4.345, p < 0.05].
DISCUSSION
The main goal of the present study was to investigate whether peripheral circulation plays a role in the development of AD pathology in the brain and whether blood exchange might be an alternative treatment for AD pathology. Our results demonstrate that whole blood exchange treatment significantly reduced cerebral plaque burden which resulted in improvement of spatial memory performance in aged Tg2576 mice. Our findings provide a proof-of-concept for establishing a possible disease-modifying treatment for AD based on targeting the peripheral circulation. Although, periodic whole blood exchange is likely not feasible for treatment in humans, there are two procedures that are routinely used in the clinic that can be utilized to “clean” blood of Aβ aggregates: plasmapheresis and blood dialysis. Plasmapheresis is a process in which plasma is separated from the blood cells and is replaced with another solution such as saline or albumin, or the plasma is treated and then returned to the body [26]. Hemodialysis is a treatment to filter wastes and water from the blood used in cases of kidney dysfunction. Hemodialysis helps control blood pressure and balance important minerals, such as potassium, sodium, and calcium, in blood. For application to AD, dialysis could be done using a filter that specifically retains and removes Aβ from the blood. Interestingly, a small clinical study to evaluate the effect of plasmapheresis in patients affected by AD showed a clear difference between the treated and the control groups with regard to the levels of Aβ, both in plasma and in cerebrospinal fluid, and a small improvement in memory performance tests (MMSE and ADAS-Cog) [27]. Clinical studies have shown that regular dialysis reduces Aβ plasma levels in humans [28, 29] and attenuates AD-associated phenotypes in an APP/PS1 mouse model [29].
The most probable explanation for the reduction of cerebral pathology after blood exchange was that a factor present in the transgenic blood that contributes to promoting brain alterations was partially removed during the exchange procedure. We previously showed that transfusion of blood from mice having established cerebral amyloid pathology into young transgenic mice, significantly increase Aβ plaque deposition [20]. The increase in cerebral amyloid pathology is likely due to the presence of Aβ oligomers in blood, since adding synthetic Aβ aggregates in blood also increases plaque deposition. Thus, it is likely that removal of oligomeric Aβ from blood by blood exchange was responsible for the effect observed in the current study. However, since in our studies we did not specifically remove Aβ from the blood, but rather exchanged the whole blood, we cannot rule out the possibility that the effect observed is not due to changes in Aβ. It is possible that the mice have differences in the levels or activity of other proteins, such as cytokines, pro- or anti-inflammatory components, growth factors, etc. Developing a dialysis-like system which specifically removes monomeric and/or oligomeric Aβ species from the circulation would enable to better understand the contribution of peripheral Aβ to the development of cerebral pathology and may provide a more practical approach to translate the current results into a novel potential therapeutic approach for AD. In the framework that the beneficial effect of blood exchange was due to Aβ removal, there are at least 3 different mechanisms by which reduction of circulating Aβ may contribute to decreasing cerebral amyloid pathology: First, lowering blood-borne Aβ may promote the redistribution of the peptide from the brain to the periphery. This mechanism is well substantiated by our data showing a decrease in brain Aβ and the increase in plasma concentrations of Aβ1–40 and Aβ1–42 after blood exchange treatment. In this model, lowering Aβ in the bloodstream facilitates the relocation of exchangeable Aβ pools from the brain to the circulation, which results in the reduction of cerebral Aβ with the consequent decrease of plaque pathology. These findings suggest that there might be an equilibrium in the concentration of Aβ between the CNS and systemic circulation, supporting the peripheral sink hypothesis [15, 30]. However, the mechanism responsible for the peripheral sink model remain unclear. Alternative possibilities to the sink hypothesis are that blood exchange somehow prevent Aβ influx or inhibits re-uptake of cleared Aβ. Second, reduction in circulating Aβ oligomers may effectively inhibit the dissemination of Aβ oligomeric seeds from the vasculature to the CNS. This model is supported by our recent results showing that blood from AD transgenic mice carries Aβ aggregates that can seed amyloid deposition in the brain after blood transfusion [20]. Third, the blood exchange procedure may also contribute to sequester Aβ by providing more Aβ-free carriers for the peptide. It has been shown that in blood Aβ does not exist in a free form, but over 90% of Aβ is bound to serum albumin and apolipoproteins [6, 31]. Thus, blood proteins from normal animals may sequester circulating Aβ in blood, thereby suppressing amyloidogenic seeding activity of Aβ in the periphery. However, this last model seems less likely since Aβ carriers in plasma appear to be in large excess over Aβ concentration. It is important to highlight that the models of Aβ relocation from the brain to blood, the removal of Aβ seeds, and the peripheral capture of Aβ are not mutually exclusive, but these mechanisms may be operating simultaneously to explain the reduction in cerebral pathology after blood exchange. Finally, it is also possible that the effect observed is the result of an alteration in more general processes, such as brain health or aging. Indeed, a series of recent studies showed that exposing old mice to blood from young mice either by heterochronic parabiosis or direct injection of young plasma, leads to a reduction of some aging markers [32, 33]. Another study found that the small volume (~13% of total plasma volume) injection of young plasma and short-term heterochronic parabiosis in disease-laden human APP transgenic mice improved cognitive function, but the effect of young plasma on the brain Aβ levels was limited [34]. Young plasma may also influence the blood-brain barrier. Recent brain capillary blood vessel studies suggested that infusing young plasma into aged mice rejuvenated the transcriptome in capillary endothelia which raised a possibility for the existence of relay signals between blood and brain cells [35]. Very recently, Yang et al [36] suggested that the brain parenchyma in young mice received circulating proteins via a receptor-mediated transport process which decreases with aging. Considering the treatment regimen in the present study consisting of longitudinal (up to 15 months) high blood volume (~60%) exchange, the contribution of blood components to the brain function may be considerably different.
Regardless of the mechanisms of action associated with blood exchange treatment, the present study indicates that a target for AD therapy may exist in the periphery. Treatment of CNS diseases, including AD, has long been complicated, due to the difficulty in delivering therapeutic agents across the blood-brain barrier [37]. Our results showing that exchanging blood significantly delayed brain plaque development indicate that AD brain reflects the changes in the systemic environment. Thus, manipulating circulating components in AD could be a novel disease-modifying therapeutic approach. Interestingly, there are already some examples of this approach in the literature. For example, inhibition of presenilin 2, expressing only in peripheral tissues and not in the brain, by pharmacological manipulation, resulted in about 50% reduction of cerebral plaque load [38]. Administration of anti-Aβ antibodies that do not cross the blood-brain barrier reduced brain amyloid burden in animals [15] and possibly in humans. An albumin replacement clinical trial is ongoing in multiple international locations to attempt scavenging of albumin for circulating Aβ [39]. Nevertheless, the advantage of our approach and others involving blood exchange is that there is no need to introduce any compound that could be potentially toxic into the body. More studies need to be performed to investigate the precise mechanism by which blood exchange produces therapeutic benefit in transgenic mice and to develop more specific and practical strategies to produce the same effect which could be tested in humans affected by AD.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by the Mitchell Foundation and R01AG059321 to CS.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41380-022-01679-4.
Reprints and permission information is available at http://www.nature.com/reprints
REFERENCES
- 1.Mucke L, Selkoe DJ. Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, et al. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009;29:10706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Potter R, Patterson BW, Elbert DL, Ovod V, Kasten T, Sigurdson W, et al. Increased in vivo amyloid-beta42 production, exchange, and loss in presenilin mutation carriers. Sci Transl Med. 2013;5:189ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selkoe DJ. Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol. 1994;10:373–403. [DOI] [PubMed] [Google Scholar]
- 6.Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916–22. [DOI] [PubMed] [Google Scholar]
- 7.Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, et al. Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett. 1997;408:105–8. [DOI] [PubMed] [Google Scholar]
- 8.Risacher SL, Fandos N, Romero J, Sherriff I, Pesini P, Saykin AJ, et al. Plasma amyloid beta levels are associated with cerebral amyloid and tau deposition. Alzheimers Dement (Amst). 2019;11:510–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koyama A, Okereke OI, Yang T, Blacker D, Selkoe DJ, Grodstein F. Plasma amyloid-beta as a predictor of dementia and cognitive decline: a systematic review and meta-analysis. Arch Neurol. 2012;69:824–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luis CA, Abdullah L, Paris D, Quadros A, Mullan M, Mouzon B, et al. Serum beta-amyloid correlates with neuropsychological impairment. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn. 2009;16:203–18. [DOI] [PubMed] [Google Scholar]
- 11.Zlokovic BV, Ghiso J, Mackic JB, McComb JG, Weiss MH, Frangione B. Blood-brain barrier transport of circulating Alzheimer’s amyloid beta. Biochem Biophys Res Commun. 1993;197:1034–40. [DOI] [PubMed] [Google Scholar]
- 12.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Investig. 2000;106:1489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mackic JB, Bading J, Ghiso J, Walker L, Wisniewski T, Frangione B, et al. Circulating amyloid-beta peptide crosses the blood-brain barrier in aged monkeys and contributes to Alzheimer’s disease lesions. Vasc Pharm. 2002;38:303–13. [DOI] [PubMed] [Google Scholar]
- 14.DeMattos RB, Bales KR, Parsadanian M, O’Dell MA, Foss EM, Paul SM, et al. Plaque-associated disruption of CSF and plasma amyloid-beta (Abeta) equilibrium in a mouse model of Alzheimer’s disease. J Neurochem. 2002;81:229–36. [DOI] [PubMed] [Google Scholar]
- 15.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:8850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, et al. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–8. [DOI] [PubMed] [Google Scholar]
- 17.Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, et al. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 2018;23:1948–56. [DOI] [PubMed] [Google Scholar]
- 18.Xiang Y, Bu XL, Liu YH, Zhu C, Shen LL, Jiao SS, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015;130:487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burwinkel M, Lutzenberger M, Heppner FL, Schulz-Schaeffer W, Baier M. Intra-venous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA). Acta Neuropathol Commun. 2018;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morales R, Duran-Aniotz C, Bravo-Alegria J, Estrada LD, Shahnawaz M, Hu PP, et al. Infusion of blood from mice displaying cerebral amyloidosis accelerates amyloid pathology in animal models of Alzheimer’s disease. Acta Neuropathol Commun. 2020;8:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. [DOI] [PubMed] [Google Scholar]
- 22.Riches AC, Sharp JG, Thomas DB, Smith SV. Blood volume determination in the mouse. J Physiol. 1973;228:279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093–5. [DOI] [PubMed] [Google Scholar]
- 24.Morales R, Estrada LD, Diaz-Espinoza R, Morales-Scheihing D, Jara MC, Castilla J, et al. Molecular cross talk between misfolded proteins in animal models of Alzheimer’s and prion diseases. J Neurosci. 2010;30:4528–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdo F, Krajcsi P. Age-Related Functional and Expressional Changes in Efflux Pathways at the Blood-Brain Barrier. Front Aging Neurosci. 2019;11:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen TC, Kiss JE, Goldman JR, Carcillo JA. The role of plasmapheresis in critical illness. Crit Care Clin. 2012;28:453–68. vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boada M, Ortiz P, Anaya F, Hernandez I, Munoz J, Nunez L, et al. Amyloid-targetedtherapeutics in Alzheimer’s disease: use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug N Perspect. 2009;22:325–39. [DOI] [PubMed] [Google Scholar]
- 28.Tholen S, Schmaderer C, Chmielewski S, Forstl H, Heemann U, Baumann M, et al. Reduction of Amyloid-beta Plasma Levels by Hemodialysis: An Anti-Amyloid Treatment Strategy? J Alzheimers Dis. 2016;50:791–6. [DOI] [PubMed] [Google Scholar]
- 29.Jin WS, Shen LL, Bu XL, Zhang WW, Chen SH, Huang ZL, et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017;134:207–20. [DOI] [PubMed] [Google Scholar]
- 30.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science. 2002;295:2264–7. [DOI] [PubMed] [Google Scholar]
- 31.Koudinov A, Matsubara E, Frangione B, Ghiso J. The soluble form of Alzheimer’s amyloid beta protein is complexed to high density lipoprotein 3 and very high density lipoprotein in normal human plasma. Biochem Biophys Res Commun. 1994;205:1164–71. [DOI] [PubMed] [Google Scholar]
- 32.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med. 2014;20:659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Middeldorp J, Lehallier B, Villeda SA, Miedema SS, Evans E, Czirr E, et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol. 2016;73:1325–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu CC, Fu SY, Chen YX, Li L, Mao RL, Wang JZ, et al. Advances in Drug Therapy for Alzheimer’s Disease. Curr Med Sci. 2020;40:999–1008. [DOI] [PubMed] [Google Scholar]
- 36.Yang AC, Stevens MY, Chen MB, Lee DP, Stahli D, Gate D, et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature. 2020;583:425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008;7:84–96. [DOI] [PubMed] [Google Scholar]
- 38.Sutcliffe JG, Hedlund PB, Thomas EA, Bloom FE, Hilbush BS. Peripheral reduction of beta-amyloid is sufficient to reduce brain beta-amyloid: implications for Alzheimer’s disease. J Neurosci Res. 2011;89:808–14. [DOI] [PubMed] [Google Scholar]
- 39.Boada M, Lopez OL, Olazaran J, Nunez L, Pfeffer M, Paricio M, et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: primary results of the AMBAR Study. Alzheimers Dement. 2020;16:1412–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.