
Figure 3: Identifying disease variants affecting cell type regulatory activity.
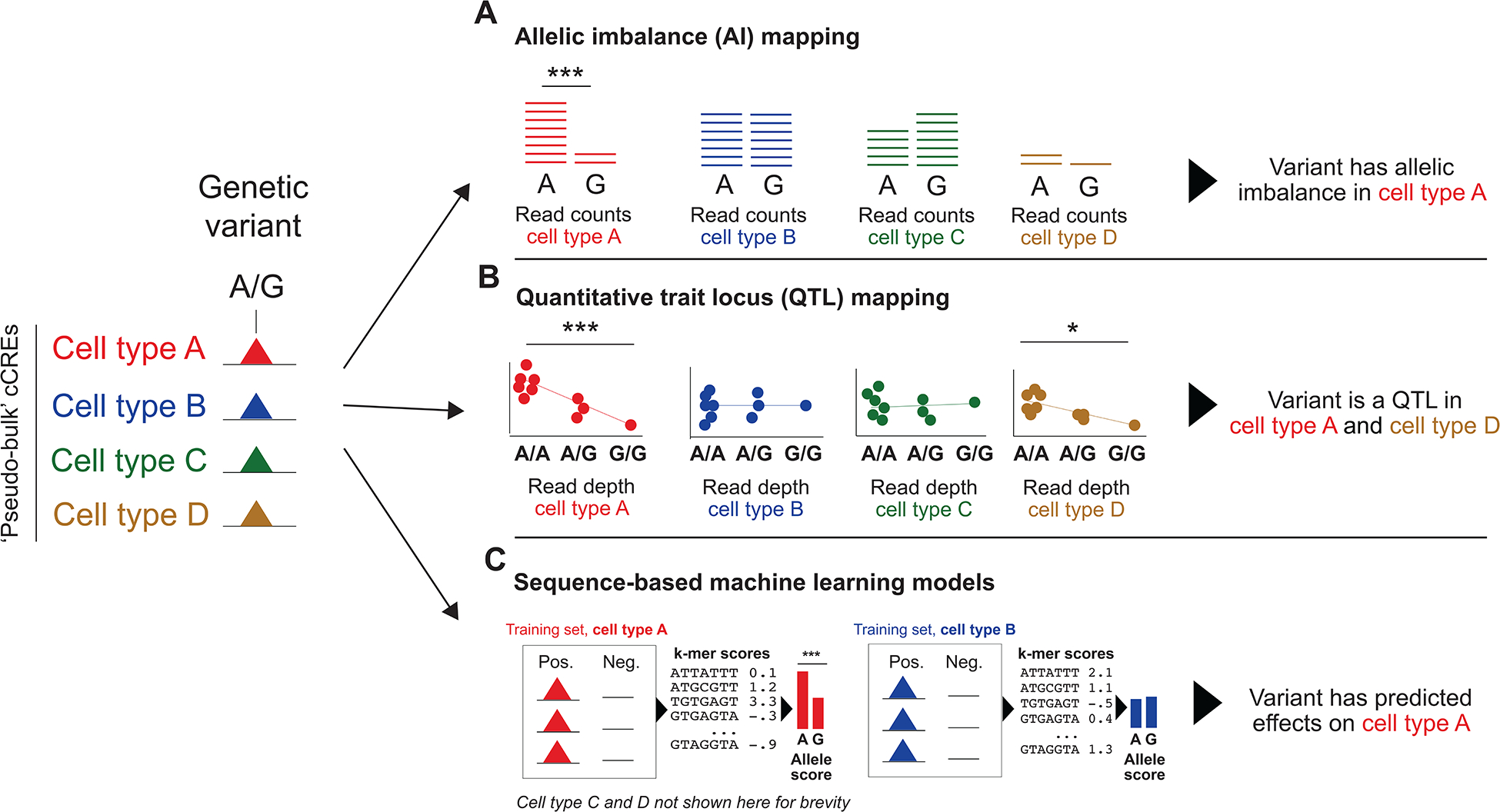
Overview of approaches for determining the functional effects of disease variants in specific cell types using single-cell epigenomics. All examples in this figure use the same genetic variant with alleles A and G overlapping a cCRE derived from pseudo-bulk profiles of cell types A, B, C, and D. Many of the methods currently used to determine variant effects on cCRE activity using pseudo-bulk profiles have been co-opted from previous studies using conventional bulk genomics. a, In allelic imbalance (AI) mapping, read counts for variant alleles in heterozygous samples are compared for each cell type. In this example, the variant has significant imbalance in allele read counts for cell type A but not for cell types B, C and D. b, In quantitative trait locus (QTL) mapping, the cell type read depth for a cCRE is compared across samples with different genotypes. In this example, the variant is a QTL in cell types A and D but not in cell types B and C. c, In sequence-based machine learning using gapped k-mers, a model is trained using cCREs active and not active in each cell type and sequence k-mers are scored based on this model. In this example the models for each cell type are trained separately, although many models can jointly consider data from many cell types. The k-mer scores for the sequence around each variant allele are compared to identify variants with large allelic differences. In this example, the genetic variant has significant allelic differences in cell type A and therefore is predicted to affect cell type A function, but does not have corresponding allelic differences in cell types B-D.