

Abbreviations
- 5‐Aza
5‐Aza‐2’‐deoxycytidine
- CCDC80
coiled‐coil domain containing 80
- COL12A1
collagen type XII alpha 1 chain
- CRC
colorectal cancer
- DDR2
discoidin domain receptor tyrosine kinase 2
- DEGs
differenital expressed genes
- DFS
disease‐free survival
- DNA
desoxyribonucleic acid
- ECs
endothelial cells
- GBP
guanylate binding protein
- HLA‐DRA/‐DPB1/‐DQB1/‐DQA1
major histocompatibility complex, class II‐DR alpha/DP beta 1/DQ beta 1/DQ alpha 1
- IFN
interferon
- LIFR
leukemia inhibitory factor receptor
- LRMP
lymphoid restricted membrane protein
- NECs
normal endothelial cells
- MERTK
MER proto‐oncogene, tyrosine kinase
- Pat
patient
- PBMCs
peripheral blood mononuclear cells
- PDGFRB
platelet derived growth factor receptor beta
- SNX10
sorting nexin 10
- STC2
stanniocalcin 2
- SPARCL1
secreted protein acidic and cysteine rich like 1
- TECs
tumor endothelial cells
- TGFB2
transforming growth factor beta 2
- Th
T helper
- TME
tumor microenvironment
- VE‐cadherin
vascular endothelial‐cadherin
Dear Editor,
Tumor microenvironment (TME)‐dependent stromal cell plasticity governs tumor development and therapy response. Tumor endothelial cells (TECs) are a major cellular component in this context [1]. In colorectal carcinoma (CRC), the stromal cell‐dependent impact of the TME is illustrated by an improved survival depending on an interferon (IFN)‐γ‐dominated Th1‐like TME associated with high T‐cell density [2] and suppressed angiogenesis [3, 4]. Cellular transcriptional memory is reported in cell lines after repeated exposure to IFN‐γ in vitro [5], suggesting that a Th1‐like TME may also exert stable imprinting effects in vivo. Here, we investigated whether TME‐dependent transcriptional imprinting in TECs from CRC patients can be exploited to retrieve clinically relevant signatures predicting outcomes.
TECs, corresponding normal endothelial cells (NECs), peripheral blood mononuclear cells (PBMCs) and tumor cells were isolated from CRC patients with a favorable Th1‐ or a worse Control‐(non‐Th‐1)‐TME and analyzed by multi‐omics analyses to identify differentially expressed genes (DEGs) with parallel epigenetic and/or genomic alterations (methods given in Supplementary Material). Respective DEGs were validated, and their clinical impact was analyzed (Figure 1A). Guanylate‐binding protein (GBP)‐1, an established marker of an angiostatic Th1‐TME in CRC [3, 4], was used to categorize Th1‐ and Control‐CRC patients (Supplementary Figures S1 and S2A) based on a positive correlation with high IFN‐γ levels and CD3 T cell density (Supplementary Figure S2B‐C), that exhibited similar clinical characteristics (Supplementary Table S1). ECs were isolated using triple‐positive‐FACS‐sorting [6] with a mean of 15,100 cells/sample (Figure 1A). EC purity (Supplementary Figures S2E and S3) and phenotype (Supplementary Figure S2F) were confirmed, and a population doubling time of up to 35 days was identified (Supplementary Figure S2G‐H).
FIGURE 1.
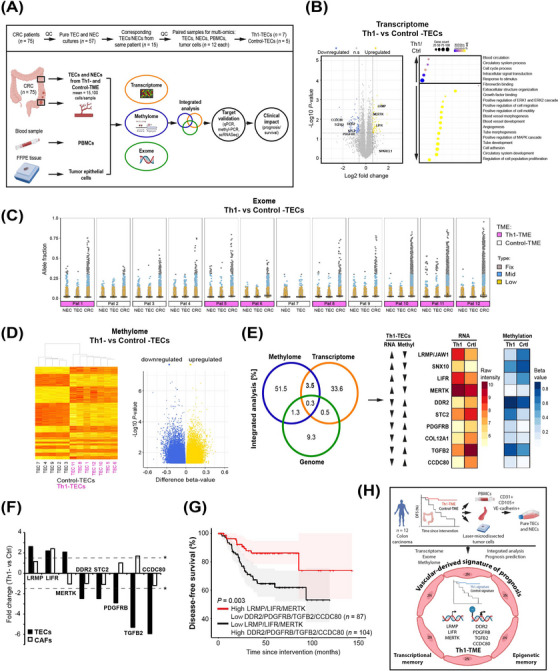
Tumor microenvironment‐dependent epigenetic imprinting in the vasculature predicts colon cancer outcome. (A) Pure and viable tumor endothelial cells (TECs) and corresponding normal endothelial cells (NECs) were isolated from human CRC patients (mean 15,100 cells/sample) with different TMEs (Th1‐ vs Control‐TME). To exploit tumor vessel‐derived TME‐dependent transcriptional imprinting and mechanisms of its manifestation, the cells were expanded in culture and compared by a multi‐omics analysis of the transcriptome, methylome and exome. DNA extracted from fresh PBMCs and laser‐microdissected tumor cells from FFPE‐blocks of the same patients was used as a control in the exome analysis. The obtained results were subjected to integrated bioinformatical analyses and independently experimentally validated. The prognostic value of the extracted signature was analyzed in CRC patients. (B) Transcriptome analyses of Th1‐ vs Control‐TECs to identify DEGs (left, P < 0.05, log2FC > 0.585/FC > 1.5‐fold) and functional differences (right). n.s. = not significant. (C) Exome analysis of Th1‐ vs Control‐TECs as depicted by scatter plots of the PBMC‐corrected allele fractions with somatic variants in corresponding TECs, NECs, and tumor cells (CRC) for each patient depicted individually. Detailed subgroup analyses are shown in Supplementary Figure S6B. (D) Methylome analysis of Th1‐ vs Control‐TECs conducted by EPIC methylation chip analyses and depicted after hierarchical clustering (left, y‐axis corresponds to individual probe sets) and by a volcano plot (right, P < 0.05). (E) Integrated analysis (gene overlap percentage) of the transcriptome, methylome and genome reveals an increased methylome/transcriptome overlap as compared to the genome/transcriptome (left). The top 10 DEGs in Th1‐TECs and Control‐TECs with inverse relation of transcription and methylation are given (right). (F) TEC imprinting genes show a different expression pattern in cancer associated fibroblasts (CAFs) isolated from a Th1‐ vs. Control‐TME (n = 3 each) compared to TECs (FC > 1.5, *P < 0.05 is indicated by dashed lines). (G) Survival analysis as depicted by a Kaplan‐Meier curve of the 7 independently validated vascular imprinting genes associated with a Th1‐TME (high LRMP, LIFR and MERTK expression in combination with low DDR2, PDGFRB, TGFB2 and CCDC80 expression) predicted significantly improved disease‐free survival in CRC patients (GEO database GSE161158). (H) In summary, ultrapure viable TECs from 12 human CRC patients were isolated by FACS (CD31‐, CD105‐, and VE‐cadherin‐positive cells) and systematically compared at the genomic, transcriptional and epigenomic levels with corresponding normal endothelial cells (NECs), PBMCs and laser‐microdissected tumor cells. These analyses identified TME‐imprinted transcriptional imprinting in TECs maintained in culture by epigenetic mechanisms rather than somatic variants. Integrative bioinformatics retrieved a seven gene imprinting signature capable to predict patient's prognosis. Illustrations created using BioRender.com.
Cellular transcriptional memory in vitro is defined by an enhanced expression of marker genes upon repeated exposure to a stimulus (priming and re‐stimulation). GBP‐2/‐3/‐4/‐5/‐7, HLA‐DRA/‐DPB1/‐DQB1, and ‐DQA1 are IFN‐γ‐dependent memory genes long‐term activated by chromatin reorganization identified in HeLa cells [5]. The expression levels of these IFN‐γ‐memory markers were not different between Th1‐ versus Control‐TECs, neither at basal untreated levels (Supplementary Figure S4A) nor after re‐stimulation by IFN‐γ (Supplementary Figure S4B). However, ECs could elicit IFN‐γ‐dependent cellular memory in vitro after repeated exposure to IFN‐γ similar to HeLa (Supplementary Figure S4C). This suggested that Th1‐TME‐driven transcriptional imprinting of TECs in vivo is more complex and/or mechanistically different from solely IFN‐γ‐driven transcriptional memory in vitro.
To retrieve marker genes imprinted by a Th1‐TME in vivo, the transcriptomes of Th1‐ versus Control‐TECs were compared (Figure 1B). Multiple DEGs (n = 1,381, P < 0.05) were detected with 147 genes above fold change > 1.5 (Supplementary Table S2). Among these, the major upregulated DEGs were LIFR, LRMP and MERTK and among the downregulated they were CCDC80, DDR2, PDGFRB, STC2 and TGFB2 (Figure 1B, Supplementary Table S2). Increased SPARCL1 expression, previously detected in Th1‐TECs [4], was also confirmed here (Figure 1B, Supplementary Figure S2D). A TME‐dependent DEG pattern was absent in corresponding NECs (Supplementary Figure S5B). The relationship of the Th1‐associated DEGs in cultivated TECs with the immuno‐angiostatic Th1‐TME was supported by functional enrichment analysis identifying downregulated terms, including “positive regulation of cell migration/motility” and “angiogenesis” (Figure 1B). Consistent with published work [7], transcriptional profiling of all TECs versus NECs revealed DEGs (n = 26) (Supplementary Table S3) associated with metabolism such as “prostaglandin metabolic process” and “benzo(a)pyrene metabolism” (Supplementary Figure S5A).
Transcriptional imprinting in Th1‐TECs may be manifested by genomic alterations [8] and/or epigenetic regulation [9]. Accordingly, ECs were subjected to exome sequencing compared to PBMCs and tumor cells as references to exclude patient‐ and to identify tumor‐specific alterations. The tumor cells exhibited the highest number of somatic variants, while all ECs exhibited only a few and comparable variants independent of Th1‐ or Control‐TME (Figure 1C, Supplementary Figure S6B, Supplementary Table S4). Patients 10‐12 displayed increased somatic variants in tumor cells and were identified as microsatellite instable (Figure 1C), indicating that a higher tumor cell mutation frequency did not increase somatic variants in ECs per se. Only a few tumor cell‐specific somatic variants were simultaneously present in ECs with no differences between TECs and NECs (Supplementary Figure S6A, Supplementary Table S5), rejecting the idea of genetic drift from tumor cells to ECs. This indicated that somatic variability is similarly low in TECs and NECs, irrespective of the TME, demonstrating that transcriptional imprinting is not manifested at the level of genomic somatic variants.
Next, the methylome was compared between Th1‐ and Control‐TECs and identified to be different (Figure 1D). All Th1‐TECs clustered together (Figure 1D), including multiple hits at single genes (Supplementary Table S6). Clustering was less stringent between TECs and NECs, indicated by a lower log2‐FC range and two TEC samples clustering with NECs (Supplementary Figure S7A) in contrast to the analysis between Th1‐ and Control‐TECs. This suggested that differential methylomes between Th1‐ and Control‐TECs may contribute to the regulation of TEC transcriptional imprinting.
Next, the potential of genomic versus methylation alterations to regulate TME‐dependent transcriptional imprinting was analyzed. Changes in genome structure occurred at 0.5%, and methylation changes occurred 7‐fold more frequently at 3.5% (Figure 1E), suggesting a superior role of methylation. Methylation is usually associated with suppression of gene expression and its absence with activation. Analyses of an inverse relation of methylation and gene expression identified the genes LIFR, LRMP/JAW1, MERTK, and SNX10 with increased transcription and reduced methylation, as well as CCDC80, COL12A1, DDR2, PDGFRB, STC2 and TGFB2 in the opposite direction in Th1‐TEC DEGs (Figure 1E, Supplementary Table S6). Differential RNA expression could be independently validated for 8 of the 10 genes (Supplementary Figure S5C) and the differential methylation pattern for all genes except STC2 (Supplementary Figure S7B‐C). Treatment of Th1‐TECs with the DNA methylation inhibitor decitabine restored the expression of methylated targets such as TGFB2 or CCDC80 (Supplementary Figure S7D), confirming their regulation by methylation. Accordingly, the multi‐omics analyses finally retrieved a seven‐gene imprinting signature with inverse relation of transcription and methylation.
This signature was identified to be EC‐specific due to lacking coherent expression in cancer‐associated fibroblasts (CAFs) from Th1‐ vs Control‐CRC for all genes, except LIFR (Figure 1F). TEC‐associated imprinting was validated using an independent scRNAseq data set in CRC. ECs were composed of lymphatic, proliferative, stalk‐like and tip‐like ECs, with proliferative EC only present in TECs (Supplementary Figure S8A). All imprinted genes were expressed in at least one EC population (Supplementary Figure S8C). Consistent with our previous findings, SPARCL1, a marker of ECs in an angiostatic Th1‐TME [4], was more highly expressed in NECs compared to TECs (Supplementary Figure S8C) and, within these, most highly in quiescent, nonproliferating stalk‐like TECs (Supplementary Figure S8B). In the Th1‐TEC population, 5/7 marker genes were coregulated in the expected direction (Supplementary Figure S8C), confirming TEC‐associated expression of the imprinted genes. Moreover, expression of the DEGs in tumor vessels was also confirmed by IHC, for example, for DDR2, MERTK and PDGFRB (Supplementary Figure S8D).
The clinical impact of the Th1‐TEC imprinting signature was analyzed in an independent CRC cohort (n = 191). Analyses of the seven individual genes revealed a prognostic value for DDR2, PDGFRB and MERTK only (Supplementary Figure S9). Remarkably, the combined imprinting signature was found to be associated with an overall improved disease‐free survival rate in accordance with the respective TME (Figure 1G), confirming its clinical relevance.
In conclusion, TME‐dependent transcriptional imprinting specifically induced in TECs and stably maintained by epigenetic DNA methylation was identified. A corresponding imprinting signature allowed tumor vessel‐based prognoses prediction (Figure 1H). Considering known epigenomic subtypes of CRC [10], this provides novel perspectives for corresponding targeted therapies and highlights the contribution of vascular cells within the TME.
AUTHOR CONTRIBUTIONS
Elisabeth Naschberger and Michael Stürzl designed the experiments. Elisabeth Naschberger, Richard Demmler, Yanmin Lyu and Christian Flierl contributed to data acquisition. Katja Petter and Tobias Gass helped in conducting the experiments. Maximilian Fuchs, Nicholas Dickel, Meik Kunz, Charles Gwellem Anchang, Bernd Popp and Steffen Uebe contributed to bioinformatical and statistical analysis. Elisabeth Naschberger, Richard Demmler, Arif Bülent Ekici, Carol Immanuel Geppert, Andreas Ramming, Claudia Günther, Susanne Merkel, Vera Simone Schellerer, Simon Völkl, Michael Scharl and Michael Stürzl interpreted and analyzed the data; Vera Simone Schellerer, Carol Immanuel Geppert and Arndt Hartmann provided key reagents; Elisabeth Naschberger and Michael Stürzl wrote the manuscript. All authors reviewed and edited the manuscript. All authors read and approved the final manuscript. The corresponding author is responsible for all aspects of the work.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
FUNDING INFORMATION
The work of the authors was supported by grants from the German Research Foundation (DFG) FOR 2438, subproject 2 (project ID 280163318) to Elisabeth Naschberger and Michael Stürzl; by the DFG SFB/TRR 241, subproject A06 (project ID 375876048) to Michael Stürzl; by the DFG STU 238/10‐1 (project ID 437201724) to Michael Stürzl and Michael Scharl; by the DFG TRR 305, subproject B08 (project ID 429280966) to Elisabeth Naschberger and Claudia Günther; by the DFG‐NOTICE program (project ID 493624887) to Elisabeth Naschberger; by the Interdisciplinary Center for Clinical Research (IZKF) of the Clinical Center Erlangen to Michael Stürzl; by the Programm zur Förderung von Corona‐Forschungsprojekten, StMWK München to Michael Stürzl; by the W. Lutz Stiftung to Michael Stürzl; by the Forschungsstiftung Medizin am Universitätsklinikum Erlangen to Michael Stürzl; by the German Federal Ministry of Education and Research (BMBF) Era‐Net grant 01KT1801 to Maximilian Fuchs and Meik Kunz; the CompLS program grant 031L0262C to Meik Kunz and Nicholas Dickel, and the Chinese scholarship program to Yanmin Lyu.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the ethics committee of the Friedrich‐Alexander‐Universität Erlangen‐Nürnberg (AZ 159_15B, TuMiC Study), and all patients provided written informed consent.
CONSENT FOR PUBLICATION
Not applicable.
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Karin Ziegler (Department of Surgery, Universitätsklinikum Erlangen), the Central Biobank Erlangen (CeBE) of the Universitätsklinikum Erlangen, the Genomic Analysis Center (GAC) of the Helmholtz Center Munich, the BioChip Core Unit of the University of Essen (Prof. Ludger Klein‐Hitpass), the FACS Sorting Core Unit Erlangen (Uwe Appelt, Markus Mroz, Friedrich‐Alexander Universität Erlangen‐Nürnberg). Finally, we would like to apologize to authors whose important work was not cited due to referencing limitations.
DATA AVAILABILITY STATEMENT
Access to the multi‐omics data sets is available through ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) using the accession codes: E‐MTAB‐10465 (transcriptome), E‐MTAB‐10521 (exome) and E‐MTAB‐10467 (methylome). All other data presented in this study are available on request from the corresponding authors.
REFERENCES
- 1. Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, et al. An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell. 2020;37(3):421. [DOI] [PubMed] [Google Scholar]
- 2. Galon J, Costes A, Sanchez‐Cabo F, Kirilovsky A, Mlecnik B, Lagorce‐Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. [DOI] [PubMed] [Google Scholar]
- 3. Naschberger E, Croner RS, Merkel S, Dimmler A, Tripal P, Amann KU, et al. Angiostatic immune reaction in colorectal carcinoma: Impact on survival and perspectives for antiangiogenic therapy. Int J Cancer. 2008;123(9):2120–2129. [DOI] [PubMed] [Google Scholar]
- 4. Naschberger E, Liebl A, Schellerer VS, Schütz M, Britzen‐Laurent N, Kölbel P, et al. Matricellular protein SPARCL1 regulates tumor microenvironment‐dependent endothelial cell heterogeneity in colorectal carcinoma. J Clin Invest. 2016;126(11):4187–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siwek W, Tehrani SSH, Mata JF, Jansen LET. Activation of clustered ifngamma target genes drives cohesin‐controlled transcriptional memory. Mol Cell. 2020;80(3):396–409 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Naschberger E, Regensburger D, Tenkerian C, Langheinrich M, Engel FB, Geppert C, et al. Isolation of human endothelial cells from normal colon and colorectal carcinoma ‐ An improved protocol. J Vis Exp. 2018(134):57400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zecchin A, Kalucka J, Dubois C, Carmeliet P. How endothelial cells adapt their metabolism to form vessels in tumors. Front Immunol. 2017;8:1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Streubel B, Chott A, Huber D, Exner M, Jager U, Wagner O, et al. Lymphoma‐specific genetic aberrations in microvascular endothelial cells in B‐cell lymphomas. N Engl J Med. 2004;351(3):250–259. [DOI] [PubMed] [Google Scholar]
- 9. Nunez JK, Chen J, Pommier GC, Cogan JZ, Replogle JM, Adriaens C, et al. Genome‐wide programmable transcriptional memory by CRISPR‐based epigenome editing. Cell. 2021;184(9):2503–2519 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orouji E, Raman AT, Singh AK, Sorokin A, Arslan E, Ghosh AK, et al. Chromatin state dynamics confers specific therapeutic strategies in enhancer subtypes of colorectal cancer. Gut. 2022;71(5):938–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Access to the multi‐omics data sets is available through ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) using the accession codes: E‐MTAB‐10465 (transcriptome), E‐MTAB‐10521 (exome) and E‐MTAB‐10467 (methylome). All other data presented in this study are available on request from the corresponding authors.