
Figure 4.
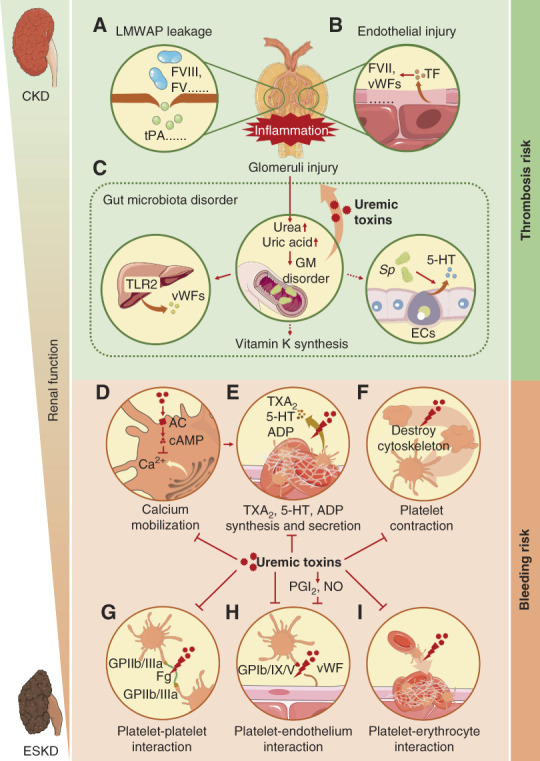
Mechanism of thrombosis in CKD and bleeding in ESKD. (A) The injuries in the glomeruli filtration barrier in CKD resulted in the leakage of LMWAPs and retention of HMWPPs, which promoted coagulation. (B) Glomeruli endothelial injuries in CKD led to the release of TF which triggered the extrinsic coagulation pathway. (C) Glomeruli injuries in CKD led to an accumulation of urea and uric acid in the gut, causing GM disorder, which on one hand enhanced the production of uremic toxins, resulting in glomeruli inflammation and injury; on the other hand, promoted vWFs, vitamin K, and 5-HT production. (D) Uremic toxins in ESKD activated AC and promoted cAMP production, which inhibited calcium mobilization, resulting in platelet dysfunction. (E) Uremic toxins and calcium mobilization abnormality inhibited the synthesis and secretion of TXA2, 5-HT, and ADP in platelets, leading to platelet dysfunction. (F) Uremic toxins destroyed the cytoskeleton of platelet, which disturbed platelet contraction, inhibiting platelet aggregation. (G) Uremic toxins inhibited platelet–platelet interaction by blocking the combination of fibrinogen (Fg) and GPIIb/IIIa, resulting in platelet aggregation dysfunction. (H) PGI2 and nitric oxide (NO) induced by uremic toxins and uremic toxins themselves inhibited platelet–endothelium interaction by blocking the combination of vWF and GPIb/IX/V, resulting in platelet aggregation dysfunction. (I) Uremic toxins inhibited platelet–erythrocyte interaction, resulting in platelet aggregation dysfunction. 5-HT, 5-hydroxytryptamine; AC, adenylate cyclase; GM, gut microbiota; HMWPP, high molecular weight procoagulant protein; LMWAP, low molecular weight anticoagulant protein; NO, nitric oxide; PGI2, prostacyclin; TF, tissue factor; TXA2, thromboxane A2; vWF, von Willebrand factor.