

Abstract
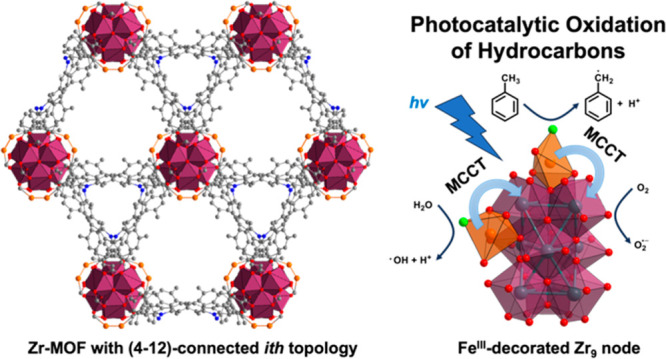
Heterogeneous catalysis plays an indispensable role in chemical production and energy conversion. Incorporation of transition metals into metal oxides and zeolites is a common strategy to fine-tune the activity and selectivity of the resulting solid catalysts, as either the active center or promotor. Studying the underlying mechanism is however challenging. Decorating the metal-oxo clusters with transition metals in metal–organic frameworks (MOFs) via postsynthetic modification offers a rational approach to construct well-defined structural models for better understanding of the reaction mechanism. Therefore, it is important to expand the materials scope beyond the currently widely studied zirconium MOFs consisting of Zr6 nodes. In this work, we report the design and synthesis of a new (4,12)-connected Zr-MOF with ith topology that consists of rare Zr9 nodes. FeIII was further incorporated onto the Zr9 nodes of the framework, and the resulting MOF material exhibits significantly enhanced activity and selectivity toward the photocatalytic oxidation of toluene. This work demonstrates a delicate ligand design strategy to control the nuclearity of Zr-oxo clusters, which further dictates the number and binding sites of transition metals and the overall photocatalytic activity toward C–H activation. Our work paves the way for future exploration of the structure–activity study of catalysts using MOFs as the model system.
Introduction
Heterogeneous catalysis typically involves reusable solid phase catalysts that are readily separated from the reaction mixture, which has laid the foundation for many applications in daily life ranging from fine chemical production to energy conversion.1 Typical heterogeneous catalysts are based on solid supports such as metal oxides and zeolites that incorporate active metal species.2,3 Indeed, the binding between a metal catalyst and its support significantly affects the selectivity and activity via the coordination mode, geometry, and electronic structures.4,5 However, elucidation of the structure–activity relationship in heterogeneous catalysts is a daunting task since it is challenging to identify the active species due to the amorphous nature of many solid supports and inhomogeneous binding sites. Moreover, the tunability of most common solid supports is quite limited, which also hampers the establishment of a systematic variable for the thorough investigation within a series of supports.
Metal–organic frameworks,6−11 a class of highly porous crystalline materials that are composed of organic linkers and inorganic nodes, represent an ideal platform to meet the needs as emerging heterogeneous catalysts.12−16 The periodic spacing of the metal nodes and ligands, as well as the well-ordered pore structures, enables the systematic study of the structure–activity relationships of the catalytic reactions.17−23 In addition, for metal–organic frameworks (MOFs) with the same topology, one can use reticular chemistry to finetune the pore size, an important feature for understanding the structure–activity relationship that is not attenable in other solid supports.24 Zr-based MOFs (Zr-MOFs) are an ideal choice of the solid support material due to their outstanding thermal and chemical stability.25 The terminal and bridging hydroxyl/aqua groups (−OH/H2O) on the Zr-oxo nodes can serve as the binding sites for extraneous metal, and their number and availability are controlled by ligand connectivity and Zr nuclearity. The uniform distribution of the metal species with atomic precision can not only avoid aggregation but also facilitate the structural determination by a variety of spectroscopy methods and single-crystal X-ray diffraction (sc-XRD), further underlining the importance of Zr-MOFs as the ideal model catalyst supports.26−32
To date, diverse MOFs featuring Zr6,33−39 Zr8,40 and Zr1241 nodes have been widely studied as porous supports for the facile construction of single-site catalysts in heterogeneous catalysis.42 We envision that the design space can be further expanded for Zr-MOFs with other types of nodes with different Zr nuclearity, such as the newly discovered Zr9 node.43−45 In particular, with the composition of Zr9O9(OH)6(H2O)6 in (4,12)-connected Zr-MOFs with ith topology, the terminal −OH/H2O groups, which are absent in the conventional 12-connected Zr6 node in the UiO series, offer new opportunities to introduce metal species onto the node. The distinct binding manner of terminal carboxylates with the Zr9 node is likely to provide a new local chemical environment that steers the reactivity. Unfortunately, the design insight for the synthesis of (4,12)-connected Zr-MOFs consisting of Zr9 nodes, besides a pseudotetrahedral ligand being used for the construction of such MOFs, needs further development.43 Herein, we screened several tetrahedral ligands based on tetraphenylmethane and N,N′-bicarbazole backbone together with synthesis condition optimization and successfully prepared a new (4,12)-connected Zr-MOF with ith topology that consists of Zr9 nodes, i.e., NPF-520 (NPF = Nebraska porous framework). We further incorporate FeIII ions into NPF-520 by base-promoted coordinative binding to the Zr9 nodes, resulting in Fe-doped NPF-520-FeIII. With three FeIII ions incorporated in each Zr9 node and near-visible absorption of the bicarbazole ligand, NPF-520-FeIII exhibits enhanced activity and selectivity toward the photocatalytic oxidation of hydrocarbons.
Results and Discussion
Ligand Steric Tuning
The screening of tetrahedral ligands starts with H4L0 (Figure 1), which has been used by Zhou and co-workers to construct a (4,8)-connected Zr-MOF (i.e., PCN-521) with the flu topology (Figure 1b).46 Previous work by our group showed that rigidification of the four phenyl rings in N,N′-bicarbazole-based ligand H4L1 does not affect the topology of the resulting (4,8)-connected Zr-MOF (i.e., NPF-500, Figure 1b).47 We reasoned that further tuning the sterics of terminal benzoates of these two ligands by functionalizing with a methyl group at the 3-position might lead to a proper ligand geometry that facilitates the formation of the targeted (4,12)-Zr-MOF. To our delight, although with the additional methyl groups H4L2 (Figure 1b) still results in a (4,8)-connected NPF-510 with the flu topology (Table S1), such a strategy proves to be successful for the modification of H4L1: with the two additional methyl groups, H4L3 forms a Zr-MOF named NPF-520, with the desired ith topology that is composed of Zr9O9(OH)6(H2O)6 nodes (Figure 1b, Table S2). Interestingly, further increase of the steric effect of H4L1 by replacing the terminal benzoate with naphthalene carboxylate in ligand H4L4 retains the topology of the resulting NPF-530 as flu (Figure 1b, Table S3), which underlines the delicate impact of ligand steric and consequent MOF topology. It should be noted here, however, that besides the choice of ligand, other synthetic parameters such as modulator also contribute to the formation of Zr-MOFs with a specific topology. Nevertheless, such steric tuning outlined in this work offers a solid strategy to targeted synthesis of MOFs with specific topological structures.
Figure 1.

(a) Formation of (4,8)-connected flu and (4,12)-connected ith topologies from a tetrahedral ligand with Zr6 and Zr9 nodes, respectively. (b) Sequential rigidification and steric tuning of tetrahedral ligands to form NPF-520 with ith topology.
The synthesis and structural characterization of NPF-510 and NPF-530 are described in the Supporting Information, and here we focus on the discussion of NPF-520. Tetratopic ligand H4L3 was synthesized via Suzuki coupling of tetrabromo-N,N′-bicarbazole and hexyl 3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate followed by hydrolysis in a basic medium (see Supporting Information for detailed procedures). Colorless hexagonal crystals of NPF-520 were obtained by solvothermal reaction of ZrCl4 and H4L3 in the presence of acetic acid as the modulation agent at 120 °C for 48 h (Figure S7). sc-XRD studies at 273 K reveal that NPF-520 crystallizes in the trigonal crystal system, in chiral space group R32 with the lattice parameters a = b = 35.168 Å and c = 28.593 Å (Table S2). Close examination of the zirconium cluster reveals two crystallographically distinct Zr atoms (i.e., Zr1 and Zr2) in each asymmetric unit (Figure S8). Zr1 is bridged by eight oxygen atoms, derived from four μ3-O2–/OH– groups, as well as three L3 ligands and a capping H2O molecule. Zr2 is bridged by two oxygen atoms emanating from two L3 ligands and seven μ3-O2–/OH– groups. Grouped together, six Zr1 and three Zr2 atoms are bound by eight μ3-O2–/OH– groups, forming the rare Zr9 nodes. Topologically, each Zr9 cluster can be described as the face-sharing of two Zr6 clusters that links 12 tetrahedral ligands (Figure S8), and each tetrahedral ligand is bridged by four Zr9 clusters to give an extremely rare (4,12)-connected ith net (Figure 1b).43,48 Such interconnection between the ligand and Zr9 node results in a 3D framework with a charge-balanced formula of Zr9O9(OH)6(H2O)6(L4)3. The resulting 3D framework contains two types of triangular 1D channels with diameters of 1.2 and 0.4 nm along the c-axis (Figure 1b). PLATON calculations indicate the presence of 66.7% of void space accessible for guest molecules.49 Powder X-ray diffraction (PXRD) patterns of NPF-520 confirm the bulk phase purity of the as-synthesized sample when compared with the simulated patterns from its corresponding single-crystal structure (Figure 2a).
Figure 2.

(a) PXRD patterns of NPF-520 after acid, water, and base treatment. (b) N2 sorption isotherm at 77 K and pore size distribution of NPF-520.
Stability and Porosity of NPF-520
The thermal stability of NPF-520 was studied by thermogravimetric analysis (TGA) under N2. The initial weight loss in the temperature range of 150 to 250 °C is attributed to the removal of the solvent molecules, and NPF-520 exhibits excellent thermal stability exemplified by the thermal decomposition around 540 °C (Figure S12). In addition, the chemical stability of NPF-520 was examined by treating MOFs in H2O, basic (pH = 11 and 12), and acidic (pH = 1 and 0) conditions. The PXRD patterns after 24 h remained intact, suggesting good stability with no phase transition or framework collapse (Figure 2a). After activation using supercritical CO2 exchange, the permanent porosity of NPF-520 was measured by N2 adsorption isotherms at 77 K (Figure 2b). NPF-520 exhibits a typical type I isotherm with a saturated N2 uptake of 915 cm3 g–1. The Brunauer–Emmett–Teller surface area (SABET) was determined to be 3463 m2 g–1, consistent with the calculated accessible surface area of 3358 m2 g–1.
Postsynthetic Incorporation of Fe
Many studies of transition-metal-doped Zr-MOFs have been focused on chemical catalysis in gas and solution phases, and recently photocatalytic applications of these materials have also emerged.50−53 One interesting strategy reported by Jiang and co-workers involves the incorporation of FeIII onto the Zr-oxo clusters of UiO-66, which not only creates catalytic active center but also shifts the light absorbance that is attributed to metal-to-cluster or metal-to-metal charge transfer (MCCT or MMCT) from FeIII to the Zr6 node.54 It was proposed that the photoinduced charge separation originated from MCCT55−57 promotes the oxidation of H2O to hydroxyl radical (•OH), which subsequently activates C–H bonds of toluene in the presence of O2 and ultimately the formation of benzoic acid. We hypothesize that the different local chemical environment around the FeIII-decorated Zr9 node in NPF-520-FeIII and/or a photoactive bicarbazole-based ligand might induce a different photocatalytic activity or selectivity toward C–H activation.
In the past, the incorporation of FeIII into Zr-MOFs has been realized by microwave-54 and solvent-assisted58 synthesis, by which ∼1.2 Fe and up to 2.2 Fe atoms per Zr6 node were introduced on UiO-66 and NU-1000, respectively. Here, we adopted a deprotonation-assisted metalation strategy developed by Lin and co-workers.59 Briefly, NPF-520 was first deprotonated with trimethylsilylmethyllithium followed by reaction with anhydrous FeCl3 to afford the Fe-modified MOF NPF-520-FeIII. ICP-OES analysis indicates that 3.1 Fe atoms were incorporated onto each Zr9 node. PXRD patterns show that the framework structure of NPF-520 is retained after metalation (Figure S20a). Installation of Fe centers reduced SABET from 3463 m2/g to 2511 m2/g for NPF-520-FeIII, and the pore size also decreased slightly from 1.9/1.1 nm to 1.6/0.9 nm (Figure S20b). The hexagonal crystal shape of NPF-520 was retained with no additional particles forming, as seen by SEM images (Figure S20c,d). Energy dispersive X-ray (EDX) mapping analysis of NPF-520-FeIII also shows a similar distribution of Fe and Zr throughout the crystals (Figure S21), consistent with the even distribution of Fe on the Zr9 clusters. The 1H NMR spectrum of NPF-520-FeIII digested with sulfuric acid in D2O confirmed that the linker was intact after the chemical modification (Figure S22).
Single-crystal XRD analysis was conducted to obtain structural insights into the incorporated FeIII species in NPF-520-FeIII (Figure 3a and Table S4). After refinement of the parent framework structure, the residual electron density was calculated to assist in the identification of the location and occupancy of Fe. There are two crystallographically distinct Fe sites (i.e., Fe1 and Fe2) in each asymmetric unit (Figure 3b). Fe1 is located inside the two corners of the trigonal bipyramidal cage (Figure S23) and coordinated to the Zr9 node through μ3-bridging oxygen (O6, 2.63 Å) and carboxylate oxygens (O4/O5, 3.15 and 3.19 Å). Fe2 sits in the triangular pore and coordinates to the Zr9 node through terminal oxygen (O7, 2.84 Å), carboxylate oxygens (O1/O2, 2.56 and 2.45 Å), and μ3-bridging oxygens (O8, 3.02 and 2.92 Å). The total occupancies of the Fe sites are 1.15 (Fe1) and 1.66 (Fe2) per Zr9 node, which corresponds to a total iron content of 2.81 Fe/Zr9, consistent with the ICP-OES data (3.1 Fe/Zr9). The location of Cl– could not be determined, likely due to severe disorder and low occupancy. The relatively long Fe–O distances (2.5–3.2 Å) suggest a weak interaction between Fe and the Zr node at the solvated state during which diffraction data were collected. Thus, although sc-XRD is helpful to reveal the general location and occupancy, the local coordination of Fe needs further investigation.
Figure 3.
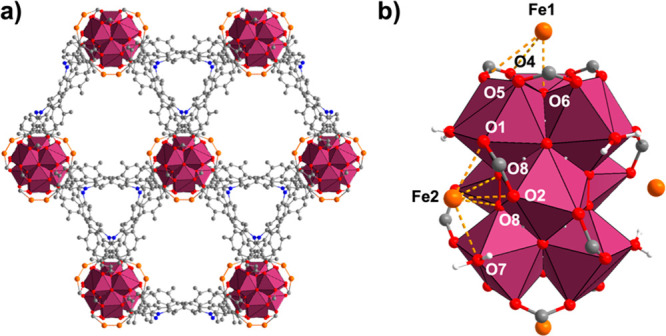
(a) Single-crystal structure of NPF-520-FeIII viewed down the c-axis. (b) Side view of the Zr9 node showing the two crystallographically distinct Fe sites.
Next, X-ray absorption spectroscopy (XAS) was performed to gain direct insights into the local electronic structure at the Fe center in NPF-520-FeIII. The X-ray absorption near edge (XANES) spectrum of the Fe foil reference exhibits the main feature at 7112 eV, corresponding to the Fe 1s–4p transition (Figure 4a). In contrast, an additional weak pre-edge feature corresponding to the quadrupole allowed 1s–3d transition was observed in the spectrum of NPF-520-FeIII, indicating the non-centrosymmetric geometry of the Fe center (Figure 4a). To quantitatively examine the local coordination environment, extended X-ray absorption fine structure (EXAFS) spectra at the Fe K-edge were fit using the Demeter X-ray absorption analysis package. The R-space spectrum with the best fit is shown in Figure 4a with fitting parameters. Compared to Fe reference foil (Figure S24), no prominent Fe–Fe bond was observed in NPF-520-FeIII, consistent with the fact that the multiple Fe2 sites are the result of symmetry generation instead of a real close Fe–Fe contact. The first shell was dominated by an Fe–O/Cl bond with an average coordination number of five. From the best fitting results, the bond distances of Fe–O and Fe–Cl were determined to be between 1.96 and 2.29 Å and were consistent with a structural model shown in Figure 4b, in which Fe1 is coordinated with two carboxylate oxygens, one μ3-bridging oxygen, one chloride, and one oxygen from water or hydroxide, and Fe2 is coordinated with one carboxylate oxygen, one terminal oxygen, one μ3-bridging oxygen, one chloride, and one oxygen from water or hydroxide. X-ray photoelectron spectroscopy (XPS) was further used to study the chemical states of Zr and Fe in NPF-520 and NPF-520-FeIII. The binding energy of 711.2 eV of Fe 2p3/2 confirms the FeIII state in NPF-520-FeIII (Figure S25a). Importantly, due to the stronger electron-withdrawing effect of FeIII, the peak of Zr 3d5/2 shifts from 181.78 eV in NPF-520 to 182.10 eV in NPF-520-FeIII (Figure S25b,c), consistent with the formation of ZrIV-O-FeIII in the latter.
Figure 4.

(a) Fe K-edge XANES of NPF-520-FeIII and Fe foil and fitting parameters (inset: EXAFS spectrum and the fit). (b) Structural model of the FeIII-decorated Zr9 node used for EXAFS fitting.
The electronic structure of NPF-520 before and after the incorporation of FeIII was also studied. UV–vis spectra show a red shift and enhanced visible light absorption after the incorporation of FeIII, consistent with its light-yellow color (Figure S26). The positive slopes of the linear Mott–Schottky plots of NPF-520 and NPF-520-FeIII at different frequencies indicate the nature of the n-type semiconductor (Figure S27). The LUMO energy levels were determined from the intersection with the values of −0.77 and −0.42 V vs Ag/AgCl (i.e., −0.58 and −0.23 V vs NHE) for NPF-520 and NPF-520-FeIII, respectively.50 Combined with the band gaps estimated by Tauc plots (Figure S26), the energy diagrams were determined (Figure S28), which suggests that the incorporation of FeIII lowers the LUMO energy level while it raises the HOMO energy level, resulting in the decrease of the band gap from 4.20 eV to 3.19 eV, a value comparable to that of the previously reported Fe-UiO-66 (i.e., 3.02 eV).54 From the thermodynamic perspective, similar to Fe-UiO-66, NPF-520-FeIII has the capability for water oxidation (E•OH/OH– = 1.89 V vs NHE; EO2/H2O = 1.23 V vs NHE) as well as O2 reduction (EO2/O2•– = −0.16 V).60
Photocatalytic Toluene Oxidation
In view of the appropriate energy levels of NPF-520-FeIII, we next used the toluene oxidation reaction to evaluate its photocatalytic performance in C–H activation. Under visible light irradiation from a blue LED photoreactor (λ = 395 nm), toluene oxidation was first carried out in the presence of 5 mol % photocatalyst and O2 (1 atm). To our delight, the reaction gives 100% conversion in 8 h with nearly exclusive selectivity to benzaldehyde (entry 1 and Table 1), which is consistent with the fact that the absence of water eliminates the production of a hydroxyl radical required for the complete oxidation to benzoic acid. Continuing to run the reaction for another 16 h still does not produce any detectable benzoic acid. Hot filtration experiments demonstrate that oxidation catalysis occurs heterogeneously (Figure S29). However, upon the introduction of 20 μL of water into the reaction mixture, benzoic acid started to emerge (41%, entry 3, Table 1, Figure S30). Complete benzoic acid selectivity was achieved within 6 h by increasing the amount of water to 100 μL (entry 4, Table 1 and Figure S30). As expected, the reaction does not proceed in the absence of O2 (entries 5 and 6, Table 1), and unmodified NPF-520 and FeCl3 show no catalytic activity under the same reaction conditions (entries 7–11, Table 1), confirming the photoactivity originating from MCCT due to Fe incorporation. NPF-520-FeIII exhibits good stability, suggested by similar PXRD patterns after catalysis (Figure S31). ICP-OES indicates negligible iron leaching (<1%) after the first run, and the iron content remains stable after the next four repetitions at room temperature (Table S5). The recyclability of the MOFs is also well maintained within five consecutive runs (Figure S32).
Table 1. Toluene Oxidation under Different Conditionsa.
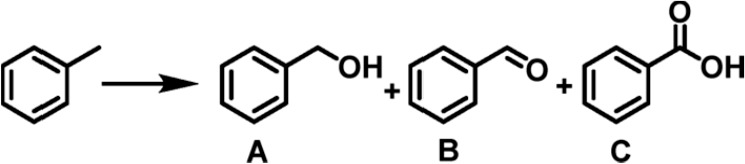
| entry | catalyst | H2O/μL | t/h | conversionb | selectivityb (A/B/C) |
|---|---|---|---|---|---|
| 1 | NPF-520-FeIII | 0 | 8 | 100 | 0/100/0 |
| 2 | NPF-520-FeIII | 0 | 24 | 100 | 0/100/0 |
| 3 | NPF-520-FeIII | 20 | 8 | 41 | 0/94/6 |
| 4 | NPF-520-FeIII | 100 | 6 | 100 | 0/0/100 |
| 5c | NPF-520-FeIII | 0 | 8 | 0 | |
| 6c | NPF-520-FeIII | 100 | 6 | 0 | |
| 7 | NPF-520 | 0 | 12 | 0 | |
| 8 | NPF-520 | 100 | 12 | 0 | |
| 9 | FeCl3 | 0 | 12 | 0 | |
| 10 | FeCl3 | 100 | 12 | 0 | |
| 11 | UiO-66-FeIII | 0 | 12 | 0 | |
| 12 | UiO-69-FeIII | 0 | 12 | 4 | 0/100/0 |
| 13 | UiO-66-FeIII | 100 | 12 | 6 | 0/36/64 |
| 14 | UiO-69-FeIII | 100 | 12 | 12 | 0/18/82 |
Reaction conditions: 5 μL of toluene, 1 mL of MeCN, 5 mol % catalyst (based on FeIII), 395 nm blue LED photoreactor, 1 atm O2.
Determined by GC/GC-MS.
Without O2.
To further benchmark the catalytic activity of NPF-520-FeIII, we synthesized two FeIII-doped Zr-MOFs, UiO-66-FeIII (1.1 Fe per Zr6 node) and UiO-69-FeIII (1.2 Fe per Zr6 node), as the comparison adopting a literature synthesis procedure (see Supporting Information for details).59 Both UiO materials exhibit inferior activities compared to NPF-520-FeIII under similar reaction conditions (entries 11–14, Table 1). Although the larger channel size of UiO-69-FeIII is likely responsible for its slightly higher activity compared with UiO-66-FeIII, the overall significantly lower activity of UiO materials suggests an enhanced energy utilization efficiency in NPF-520-FeIII. On one hand, the presence of near-visible absorption of the bicarbazole ligand in NPF-520 increases light harvesting, which likely undergoes energy transfer to the MMCT process. This is corroborated by the decreased fluorescence of NPF-520 upon FeIII doping (Figure S33). On the other hand, compared to the Zr6 nodes doped with a single FeIII ion, a more efficient charge separation process might occur in Zr9 nodes doped with ∼3 FeIII ions in NPF-520-FeIII. Further investigation is required to elucidate the dominant contribution; nevertheless, it is clear that modification of the tetrahedral ligand not only facilitates the formation of the Zr9 nodes but also enhances the light harvesting, contributing to both the enhanced activity and selectivity toward the photocatalytic oxidation of hydrocarbons.
Photocatalytic Mechanism
Our work demonstrates that, depending on the availability of water in the reaction system, the MCCT process can lead to a different product selectivity, which we attribute to the different reactive oxygen species (ROS) (i.e., hydroxyl radical •OH and superoxide O2•–) generated under the different conditions. We propose the mechanism for the photocatalytic oxidation of toluene as follows (Figure 5). In the presence of water, the photoexcited hole is transferred to H2O, resulting in the formation of the hydroxyl radical •OH and proton. The photoexcited electron then reacts with the O2 to form O2•–. From there, the generated ROS, •OH and O2•–, can react with toluene and its partially oxidized derivatives to form benzyl alcohol, benzaldehyde, and benzoic acid. In the absence of water, the photoexcited hole can only react with toluene to form the toluene radical and H+, which then combines with O2•– to form benzaldehyde. The superoxide does not further react with benzaldehyde to form benzoic acid, resulting in selectivity for the aldehyde product, which is observed in all cases when the photooxidation reaction is conducted under anhydrous conditions.
Figure 5.

(a) Schematic illustration of the FeIII to Zr-oxo cluster charge transfer process in the FeIII-decorated Zr9 node (Zr: violet, O: red, Fe: orange; Cl: green). (b) Generation of reaction intermediates in the presence of water and under anhydrous conditions. (c) Proposed mechanism for the photocatalytic oxidation of toluene.
Several sets of experiments were conducted to provide further support for the proposed mechanism. The presence of these produced ROS can be detected using certain agents, such as terephthalic acid for •OH and luminol for O2•–.61 As expected, under anhydrous conditions, no •OH was produced, as no fluorescence signal was observed around 430 nm due to the oxidation of terephthalic acid (Figure S34). This is consistent with the proposed reaction mechanism where water reacts with the photogenerated hole to produce •OH. However, the luminol-amplified chemiluminescence test indicates that O2•– is produced regardless of the presence of water (Figure S35), confirming that O2 gas is indeed the electron acceptor. Moreover, quenchers such as 2-methylfuran (for singlet oxygen), 1,4-diazabicyclo[2.2.2]octane (DABCO, for singlet oxygen), and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO, for all radicals) were also used to help identify ROS involved in the reaction.61 According to Table S6, the addition of 2-methylfuran and DABCO did not affect the photocatalytic reaction at all, suggesting that singlet oxygen is not involved in the oxidation reaction. On the other hand, TEMPO completely quenched the reaction, highlighting the importance of radical species in the photocatalysis. Besides the radical quenchers, sacrificial electron or hole donors were also added to the reaction mixture to confirm if the oxidation was hole or electron driven. While the sacrificial electron donor triethylamine completely quenched the reaction, the reaction proceeded in the presence of (NH4)2Ce(NO3)6, suggesting that the photooxidation is hole-driven (Table S6). Finally, hydrogen peroxide (H2O2) affords only minimal conversion, suggesting the Fenton reaction is not the dominant reaction driving the toluene oxidation (Table S6).
The kinetic isotope effect (KIE) was studied to determine the rate-limiting step in the photo-oxidation of toluene (Figure S36). Although a minimal KIE (1.06) was observed when D2O was used instead of H2O, switching toluene to toluene-d8 demonstrated the largest KIE (i.e., 3.23 in the presence of water and 6.67 in the absence of water), indicating that C–H bond activation is the rate-limiting step under both reaction conditions. In the absence of water, hole-to-toluene oxidation instead occurs to produce the toluene radical, which then reacts with ROS such as superoxide to form benzaldehyde. Under anhydrous conditions, the rate-limiting effect of toluene is even greater than with water present. Finally, although O182 shows a KIE of more than 1 for both reactions (Figure S36), indicating that the breaking of the O–O bond is crucial to the reaction, it is not the predominant rate-limiting step.
Conclusions
In summary, we have demonstrated a rational strategy to construct a Zr-MOF consisting of nonanuclear nodes that undergoes a postsynthetic modification to afford a highly active photocatalyst for aerobic C–H activation. Using ligand rigidification and steric tuning, we successfully modified an N,N′-bicarbazole-based tetrahedral ligand and synthesized a highly stable Zr-MOF named NPF-520 that features the (4,12)-connected ith topology. The unique metal binding sites on the Zr9 node in NPF-520 facilitate the addition of up to three FeIII atoms to the nonanuclear Zr-oxo clusters. The resulting NPF-520-FeIII exhibits significantly enhanced photocatalytic activity toward toluene oxidation. Moreover, the reaction selectivity can be controlled by varying the water content to afford exclusively either aldehyde or benzoic acid as the oxidation product via an MCCT process that likely involves the enhanced light harvesting via the near-visible absorption of ligand. This work highlights the importance of the metal-binding mode of a metal-oxo cluster to modulate the catalytic activity of MOFs for heterogeneous catalysis and paves the road for building model systems to understand the structure–activity relationship for broader application avenues of MOFs for both chemical and photochemical catalysis.
Acknowledgments
Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy, and by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under U.S. Department of Energy contract no. DE-AC02-05CH11231. The use of X-ray absorption spectroscopy at Advanced Photon Source, Beamline 12-BM, in Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award No. DE-AC02-06CH11357.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c07237.
Synthesis, experimental data and procedures, single-crystal X-ray diffraction, TGA, PXRD, N2 sorption isotherms, postsynthetic modification, and photocatalysis data (PDF)
Author Contributions
R.S.H.K. and C.F. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Schlogl R. Heterogeneous catalysis. Angew. Chem., Int. Ed. 2015, 54, 3465–3520. 10.1002/anie.201410738. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Zaera F.. Heterogeneous Catalysis by Metals; John Wiley & Sons, Ltd, 2006. [Google Scholar]
- Rothenberg G.Catalysis: Concepts and Green Applications; Wiley-VCH, 2008. [Google Scholar]
- Comotti M.; Li W. C.; Spliethoff B.; Schuth F. Support effect in high activity gold catalysts for CO oxidation. J. Am. Chem. Soc. 2006, 128, 917–924. 10.1021/ja0561441. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Chen L.; Qi Z.; Zhuo L.; Chen J. L.; Pao C. W.; Su J.; Somorjai G. A. Insights into the Mechanism of n-Hexane Reforming over a Single-Site Platinum Catalyst. J. Am. Chem. Soc. 2020, 142, 16533–16537. 10.1021/jacs.0c07911. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Cordova K. E.; O’Keeffe M.; Yaghi O. M. The chemistry and applications of metal-organic frameworks. Science 2013, 341, 1230444. 10.1126/science.1230444. [DOI] [PubMed] [Google Scholar]
- Cook T. R.; Zheng Y. R.; Stang P. J. Metal-organic frameworks and self-assembled supramolecular coordination complexes: comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem. Rev. 2013, 113, 734–777. 10.1021/cr3002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. C.; Kitagawa S. Metal-organic frameworks (MOFs). Chem. Soc. Rev. 2014, 43, 5415–5418. 10.1039/C4CS90059F. [DOI] [PubMed] [Google Scholar]
- Li B.; Wen H. M.; Cui Y.; Zhou W.; Qian G.; Chen B. Emerging Multifunctional Metal-Organic Framework Materials. Adv. Mater. 2016, 28, 8819–8860. 10.1002/adma.201601133. [DOI] [PubMed] [Google Scholar]
- Islamoglu T.; Goswami S.; Li Z.; Howarth A. J.; Farha O. K.; Hupp J. T. Postsynthetic Tuning of Metal-Organic Frameworks for Targeted Applications. Acc. Chem. Res. 2017, 50, 805–813. 10.1021/acs.accounts.6b00577. [DOI] [PubMed] [Google Scholar]
- Xie F.; Yu L.; Wang H.; Li J. Metal-Organic Frameworks for C6 Alkane Separation. Angew. Chem., Int. Ed. 2023, 62, 202300722. 10.1002/anie.202300722. [DOI] [PubMed] [Google Scholar]
- Zhu L.; Liu X. Q.; Jiang H. L.; Sun L. B. Metal-Organic Frameworks for Heterogeneous Basic Catalysis. Chem. Rev. 2017, 117, 8129–8176. 10.1021/acs.chemrev.7b00091. [DOI] [PubMed] [Google Scholar]
- Wang C.; An B.; Lin W. Metal–Organic Frameworks in Solid–Gas Phase Catalysis. ACS Catal. 2019, 9, 130–146. 10.1021/acscatal.8b04055. [DOI] [Google Scholar]
- Dhakshinamoorthy A.; Li Z.; Garcia H. Catalysis and photocatalysis by metal organic frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. 10.1039/C8CS00256H. [DOI] [PubMed] [Google Scholar]
- Bavykina A.; Kolobov N.; Khan I. S.; Bau J. A.; Ramirez A.; Gascon J. Metal-Organic Frameworks in Heterogeneous Catalysis: Recent Progress, New Trends, and Future Perspectives. Chem. Rev. 2020, 120, 8468–8535. 10.1021/acs.chemrev.9b00685. [DOI] [PubMed] [Google Scholar]
- Liu J.; Goetjen T. A.; Wang Q.; Knapp J. G.; Wasson M. C.; Yang Y.; Syed Z. H.; Delferro M.; Notestein J. M.; Farha O. K.; Hupp J. T. MOF-enabled confinement and related effects for chemical catalyst presentation and utilization. Chem. Soc. Rev. 2022, 51, 1045–1097. 10.1039/D1CS00968K. [DOI] [PubMed] [Google Scholar]
- Pattengale B.; Yang S.; Ludwig J.; Huang Z.; Zhang X.; Huang J. Exceptionally Long-Lived Charge Separated State in Zeolitic Imidazolate Framework: Implication for Photocatalytic Applications. J. Am. Chem. Soc. 2016, 138, 8072–8075. 10.1021/jacs.6b04615. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhang X.; Li P.; Otake K. I.; Cui Y.; Lyu J.; Krzyaniak M. D.; Zhang Y.; Li Z.; Liu J.; Buru C. T.; Islamoglu T.; Wasielewski M. R.; Li Z.; Farha O. K. Vanadium Catalyst on Isostructural Transition Metal, Lanthanide, and Actinide Based Metal-Organic Frameworks for Alcohol Oxidation. J. Am. Chem. Soc. 2019, 141, 8306–8314. 10.1021/jacs.9b02603. [DOI] [PubMed] [Google Scholar]
- Babucci M.; Guntida A.; Gates B. C. Atomically Dispersed Metals on Well-Defined Supports including Zeolites and Metal-Organic Frameworks: Structure, Bonding, Reactivity, and Catalysis. Chem. Rev. 2020, 120, 11956–11985. 10.1021/acs.chemrev.0c00864. [DOI] [PubMed] [Google Scholar]
- He X.; Looker B. G.; Dinh K. T.; Stubbs A. W.; Chen T.; Meyer R. J.; Serna P.; Román-Leshkov Y.; Lancaster K. M.; Dincă M. Cerium(IV) Enhances the Catalytic Oxidation Activity of Single-Site Cu Active Sites in MOFs. ACS Catal. 2020, 10, 7820–7825. 10.1021/acscatal.0c02493. [DOI] [Google Scholar]
- Chen C.; Alalouni M. R.; Dong X.; Cao Z.; Cheng Q.; Zheng L.; Meng L.; Guan C.; Liu L.; Abou-Hamad E.; Wang J.; Shi Z.; Huang K. W.; Cavallo L.; Han Y. Highly Active Heterogeneous Catalyst for Ethylene Dimerization Prepared by Selectively Doping Ni on the Surface of a Zeolitic Imidazolate Framework. J. Am. Chem. Soc. 2021, 143, 7144–7153. 10.1021/jacs.1c02272. [DOI] [PubMed] [Google Scholar]
- Liu C.; Luo T. Y.; Feura E. S.; Zhang C.; Rosi N. L. Orthogonal Ternary Functionalization of a Mesoporous Metal-Organic Framework via Sequential Postsynthetic Ligand Exchange. J. Am. Chem. Soc. 2015, 137, 10508–10511. 10.1021/jacs.5b06780. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Chen Y.; Wang W.; Yang H.; Hong A. N.; Bu X.; Feng P. Simultaneous Control of Flexibility and Rigidity in Pore-Space-Partitioned Metal-Organic Frameworks. J. Am. Chem. Soc. 2023, 145, 10980–10986. 10.1021/jacs.3c03130. [DOI] [PubMed] [Google Scholar]
- Ockwig N. W.; Delgado-Friedrichs O.; O’Keeffe M.; Yaghi O. M. Reticular chemistry: occurrence and taxonomy of nets and grammar for the design of frameworks. Acc. Chem. Res. 2005, 38, 176–182. 10.1021/ar020022l. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Dou Y.; Xie L. H.; Rutledge W.; Li J. R.; Zhou H. C. Zr-based metal-organic frameworks: design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. 10.1039/C5CS00837A. [DOI] [PubMed] [Google Scholar]
- Otake K. I.; Cui Y.; Buru C. T.; Li Z.; Hupp J. T.; Farha O. K. Single-Atom-Based Vanadium Oxide Catalysts Supported on Metal-Organic Frameworks: Selective Alcohol Oxidation and Structure-Activity Relationship. J. Am. Chem. Soc. 2018, 140, 8652–8656. 10.1021/jacs.8b05107. [DOI] [PubMed] [Google Scholar]
- Hackler R. A.; Pandharkar R.; Ferrandon M. S.; Kim I. S.; Vermeulen N. A.; Gallington L. C.; Chapman K. W.; Farha O. K.; Cramer C. J.; Sauer J.; Gagliardi L.; Martinson A. B. F.; Delferro M. Isomerization and Selective Hydrogenation of Propyne: Screening of Metal-Organic Frameworks Modified by Atomic Layer Deposition. J. Am. Chem. Soc. 2020, 142, 20380–20389. 10.1021/jacs.0c08641. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Zhang X.; Kanchanakungwankul S.; Lu Z.; Noh H.; Syed Z. H.; Farha O. K.; Truhlar D. G.; Hupp J. T. Unexpected ″Spontaneous″ Evolution of Catalytic, MOF-Supported Single Cu(II) Cations to Catalytic, MOF-Supported Cu(0) Nanoparticles. J. Am. Chem. Soc. 2020, 142, 21169–21177. 10.1021/jacs.0c10367. [DOI] [PubMed] [Google Scholar]
- Ma X.; Liu H.; Yang W.; Mao G.; Zheng L.; Jiang H. L. Modulating Coordination Environment of Single-Atom Catalysts and Their Proximity to Photosensitive Units for Boosting MOF Photocatalysis. J. Am. Chem. Soc. 2021, 143, 12220–12229. 10.1021/jacs.1c05032. [DOI] [PubMed] [Google Scholar]
- Ye G.; Wang H.; Chen W.; Chu H.; Wei J.; Wang D.; Wang J.; Li Y. In Situ Implanting of Single Tungsten Sites into Defective UiO-66(Zr) by Solvent-Free Route for Efficient Oxidative Desulfurization at Room Temperature. Angew. Chem., Int. Ed. 2021, 60, 20318–20324. 10.1002/anie.202107018. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Shi Y.; Jiang Y.; Zhang X.; Long C.; An P.; Zhu Y.; Shao S.; Yan Z.; Li G.; Tang Z. Fe-O Clusters Anchored on Nodes of Metal-Organic Frameworks for Direct Methane Oxidation. Angew. Chem., Int. Ed. 2021, 60, 5811–5815. 10.1002/anie.202013807. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Ahn S.; Mian M. R.; Wang X.; Ma Q.; Son F. A.; Yang L.; Ma K.; Zhang X.; Notestein J. M.; Farha O. K. Modulating Chemical Environments of Metal-Organic Framework-Supported Molybdenum(VI) Catalysts for Insights into the Structure-Activity Relationship in Cyclohexene Epoxidation. J. Am. Chem. Soc. 2022, 144, 3554–3563. 10.1021/jacs.1c12421. [DOI] [PubMed] [Google Scholar]
- Nguyen H. G. T.; Schweitzer N. M.; Chang C.-Y.; Drake T. L.; So M. C.; Stair P. C.; Farha O. K.; Hupp J. T.; Nguyen S. T. Vanadium-Node-Functionalized UiO-66: A Thermally Stable MOF-Supported Catalyst for the Gas-Phase Oxidative Dehydrogenation of Cyclohexene. ACS Catal. 2014, 4, 2496–2500. 10.1021/cs5001448. [DOI] [Google Scholar]
- Yuan S.; Chen Y. P.; Qin J.; Lu W.; Wang X.; Zhang Q.; Bosch M.; Liu T. F.; Lian X.; Zhou H. C. Cooperative Cluster Metalation and Ligand Migration in Zirconium Metal-Organic Frameworks. Angew. Chem., Int. Ed. 2015, 54, 14696–14700. 10.1002/anie.201505625. [DOI] [PubMed] [Google Scholar]
- Yang D.; Odoh S. O.; Borycz J.; Wang T. C.; Farha O. K.; Hupp J. T.; Cramer C. J.; Gagliardi L.; Gates B. C. Tuning Zr6 Metal–Organic Framework (MOF) Nodes as Catalyst Supports: Site Densities and Electron-Donor Properties Influence Molecular Iridium Complexes as Ethylene Conversion Catalysts. ACS Catal. 2016, 6, 235–247. 10.1021/acscatal.5b02243. [DOI] [Google Scholar]
- Manna K.; Ji P.; Greene F. X.; Lin W. Metal-Organic Framework Nodes Support Single-Site Magnesium-Alkyl Catalysts for Hydroboration and Hydroamination Reactions. J. Am. Chem. Soc. 2016, 138, 7488–7491. 10.1021/jacs.6b03689. [DOI] [PubMed] [Google Scholar]
- Li Z.; Peters A. W.; Bernales V.; Ortuno M. A.; Schweitzer N. M.; DeStefano M. R.; Gallington L. C.; Platero-Prats A. E.; Chapman K. W.; Cramer C. J.; Gagliardi L.; Hupp J. T.; Farha O. K. Metal-Organic Framework Supported Cobalt Catalysts for the Oxidative Dehydrogenation of Propane at Low Temperature. ACS Cent. Sci. 2017, 3, 31–38. 10.1021/acscentsci.6b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Ye J.; Ortuno M. A.; Fulton J. L.; Gutierrez O. Y.; Camaioni D. M.; Motkuri R. K.; Li Z.; Webber T. E.; Mehdi B. L.; Browning N. D.; Penn R. L.; Farha O. K.; Hupp J. T.; Truhlar D. G.; Cramer C. J.; Lercher J. A. Selective Methane Oxidation to Methanol on Cu-Oxo Dimers Stabilized by Zirconia Nodes of an NU-1000 Metal-Organic Framework. J. Am. Chem. Soc. 2019, 141, 9292–9304. 10.1021/jacs.9b02902. [DOI] [PubMed] [Google Scholar]
- Platero-Prats A. E.; Mavrandonakis A.; Liu J.; Chen Z.; Chen Z.; Li Z.; Yakovenko A. A.; Gallington L. C.; Hupp J. T.; Farha O. K.; Cramer C. J.; Chapman K. W. The Molecular Path Approaching the Active Site in Catalytic Metal-Organic Frameworks. J. Am. Chem. Soc. 2021, 143, 20090–20094. 10.1021/jacs.1c11213. [DOI] [PubMed] [Google Scholar]
- Ji P.; Manna K.; Lin Z.; Urban A.; Greene F. X.; Lan G.; Lin W. Single-Site Cobalt Catalysts at New Zr8(μ2-O)8(μ2-OH)4 Metal-Organic Framework Nodes for Highly Active Hydrogenation of Alkenes, Imines, Carbonyls, and Heterocycles. J. Am. Chem. Soc. 2016, 138, 12234–12242. 10.1021/jacs.6b06759. [DOI] [PubMed] [Google Scholar]
- An B.; Li Z.; Song Y.; Zhang J.; Zeng L.; Wang C.; Lin W. Cooperative copper centres in a metal–organic framework for selective conversion of CO2 to ethanol. Nat. Catal 2019, 2, 709–717. 10.1038/s41929-019-0308-5. [DOI] [Google Scholar]
- Feng X.; Song Y.; Lin W. Transforming Hydroxide-Containing Metal–Organic Framework Nodes for Transition Metal Catalysis. Trends in Chemistry 2020, 2, 965–979. 10.1016/j.trechm.2020.08.009. [DOI] [Google Scholar]
- Jiang H.; Zhang W.; Kang X.; Cao Z.; Chen X.; Liu Y.; Cui Y. Topology-Based Functionalization of Robust Chiral Zr-Based Metal-Organic Frameworks for Catalytic Enantioselective Hydrogenation. J. Am. Chem. Soc. 2020, 142, 9642–9652. 10.1021/jacs.0c00637. [DOI] [PubMed] [Google Scholar]
- Gong W.; Arman H.; Chen Z.; Xie Y.; Son F. A.; Cui H.; Chen X.; Shi Y.; Liu Y.; Chen B.; Farha O. K.; Cui Y. Highly Specific Coordination-Driven Self-Assembly of 2D Heterometallic Metal-Organic Frameworks with Unprecedented Johnson-type (J51) Nonanuclear Zr-Oxocarboxylate Clusters. J. Am. Chem. Soc. 2021, 143, 657–663. 10.1021/jacs.0c11881. [DOI] [PubMed] [Google Scholar]
- Gong W.; Chen X.; Fahy K. M.; Dong J.; Liu Y.; Farha O. K.; Cui Y. Reticular Chemistry in Its Chiral Form: Axially Chiral Zr(IV)-Spiro Metal-Organic Framework as a Case Study. J. Am. Chem. Soc. 2023, 145, 13869–13878. 10.1021/jacs.3c03036. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Chen Y. P.; Bosch M.; Gentle T. 3rd; Wang K.; Feng D.; Wang Z. U.; Zhou H. C. Symmetry-guided synthesis of highly porous metal-organic frameworks with fluorite topology. Angew. Chem., Int. Ed. 2014, 53, 815–818. 10.1002/anie.201307340. [DOI] [PubMed] [Google Scholar]
- Fiankor C.; Nyakuchena J.; Khoo R. S. H.; Zhang X.; Hu Y.; Yang S.; Huang J.; Zhang J. Symmetry-Guided Synthesis of N,N′-Bicarbazole and Porphyrin-Based Mixed-Ligand Metal-Organic Frameworks: Light Harvesting and Energy Transfer. J. Am. Chem. Soc. 2021, 143, 20411–20418. 10.1021/jacs.1c10291. [DOI] [PubMed] [Google Scholar]
- Furukawa H.; Gandara F.; Zhang Y. B.; Jiang J.; Queen W. L.; Hudson M. R.; Yaghi O. M. Water adsorption in porous metal-organic frameworks and related materials. J. Am. Chem. Soc. 2014, 136, 4369–4381. 10.1021/ja500330a. [DOI] [PubMed] [Google Scholar]
- Spek A. L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- Yuan S.; Qin J. S.; Xu H. Q.; Su J.; Rossi D.; Chen Y.; Zhang L.; Lollar C.; Wang Q.; Jiang H. L.; Son D. H.; Xu H.; Huang Z.; Zou X.; Zhou H. C. [Ti8Zr2O12(COO)16] Cluster: An Ideal Inorganic Building Unit for Photoactive Metal-Organic Frameworks. ACS Cent. Sci. 2018, 4, 105–111. 10.1021/acscentsci.7b00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi Y.; Feng X.; Song Y.; Xu Z.; Li Z.; Lin W. Metal-Organic Frameworks Integrate Cu Photosensitizers and Secondary Building Unit-Supported Fe Catalysts for Photocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2020, 142, 10302–10307. 10.1021/jacs.0c03906. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Abazari R.; Yao J.; Gao J. Recent strategies to improve the photoactivity of metal-organic frameworks. Dalton Trans. 2021, 50, 2342–2349. 10.1039/D0DT03844J. [DOI] [PubMed] [Google Scholar]
- Lin H.; Xu Y.; Wang B.; Li D.-S.; Zhou T.; Zhang J. Postsynthetic Modification of Metal–Organic Frameworks for Photocatalytic Applications. Small Struct. 2022, 3, 2100176. 10.1002/sstr.202100176. [DOI] [Google Scholar]
- Xu C.; Pan Y.; Wan G.; Liu H.; Wang L.; Zhou H.; Yu S. H.; Jiang H. L. Turning on Visible-Light Photocatalytic C-H Oxidation over Metal-Organic Frameworks by Introducing Metal-to-Cluster Charge Transfer. J. Am. Chem. Soc. 2019, 141, 19110–19117. 10.1021/jacs.9b09954. [DOI] [PubMed] [Google Scholar]
- Lin W.; Frei H. Photochemical CO2 splitting by metal-to-metal charge-transfer excitation in mesoporous ZrCu(I)-MCM-41 silicate sieve. J. Am. Chem. Soc. 2005, 127, 1610–1611. 10.1021/ja040162l. [DOI] [PubMed] [Google Scholar]
- Nakamura R.; Okamoto A.; Osawa H.; Irie H.; Hashimoto K. Design of all-inorganic molecular-based photocatalysts sensitive to visible light: Ti(IV)-O-Ce(III) bimetallic assemblies on mesoporous silica. J. Am. Chem. Soc. 2007, 129, 9596–9597. 10.1021/ja073668n. [DOI] [PubMed] [Google Scholar]
- Kramar B. V.; Phelan B. T.; Sprague-Klein E. A.; Diroll B. T.; Lee S.; Otake K.-i.; Palmer R.; Mara M. W.; Farha O. K.; Hupp J. T.; Chen L. X. Single-Atom Metal Oxide Sites as Traps for Charge Separation in the Zirconium-Based Metal–Organic Framework NDC–NU-1000. Energy Fuels 2021, 35, 19081–19095. 10.1021/acs.energyfuels.1c02623. [DOI] [Google Scholar]
- Otake K. I.; Ahn S.; Knapp J.; Hupp J. T.; Notestein J. M.; Farha O. K. Vapor-Phase Cyclohexene Epoxidation by Single-Ion Fe(III) Sites in Metal-Organic Frameworks. Inorg. Chem. 2021, 60, 2457–2463. 10.1021/acs.inorgchem.0c03364. [DOI] [PubMed] [Google Scholar]
- Manna K.; Ji P.; Lin Z.; Greene F. X.; Urban A.; Thacker N. C.; Lin W. Chemoselective single-site Earth-abundant metal catalysts at metal-organic framework nodes. Nat. Commun. 2016, 7, 12610. 10.1038/ncomms12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz H. A.; Dodson R. W. Equilibrium between hydroxyl radicals and thallium(II) and the oxidation potential of hydroxyl(aq). J. Phys. Chem. 1984, 88, 3643–3647. 10.1021/j150660a053. [DOI] [Google Scholar]
- Nosaka Y.; Nosaka A. Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. 10.1021/acs.chemrev.7b00161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.