

Abstract
Usher syndrome (USH) is a clinically heterogeneous condition characterized by sensorineural hearing loss, progressive retinal degeneration, and vestibular dysfunction. There are two phenotypically recognizable types of Usher syndrome described in the literature. Usher type 1 individual have no vestibular function and profound sensorineural hearing loss. Usher type 2 individuals have a normal vestibular function and mild-to-severe hearing loss with visual impairment that is presented later in life. We are reporting a case of 35 years old gentleman with hearing loss and visual impairment presented to the ENT clinic at the tertiary care center. Clinical evaluations as well as comprehensive testing of hearing, vestibular function, and visual function have confirmed USH. It’s a rare but serious cause of hearing loss that requires comprehensive multidisciplinary evaluation in conjunction with an ophthalmology team. Further genetic, audiological, and vestibular assessments are required to help diagnose and management of specific subtypes of this syndrome.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12070-023-03970-4.
Keywords: Usher’s syndrome, Retinitis pigmentosa, Autosomal recessive, Congo democratic
Background
Usher syndrome (USH) is a major cause of genetic deafness and blindness, inherited as an autosomal recessive mode with dual sensory impairments. At least 3 distinct USHl loci and 2 distinct USH2 loci exist. The 2 phenotypically recognizable types of USH are characterized by PPR. USH1 individuals have no vestibular function and a profound sensorineural hearing loss. USH2 individuals have normal vestibular function and a mild-to-severe hearing loss. The minimal test battery reliably identifies individuals with USHl and USH2. The comprehensive test battery is recommended in the evaluation and discrimination of patients with USHl and USH2 [1].
The incidence of Usher Syndrome is of 1.8 to 4.4 cases per 100,000 births. In America, it affects about 45,000 resident population. According to a National Institute survey on Deafness and Other Communication Disorders, half of the patients with deafness disorders are patients with Usher Syndrome. In the severe subtype, hearing loss developed since birth, with an onset of retinitis pigmentosa at the age of 10 years and blindness at the age of 20 years [2]. Studies performed in Denmark, Sweden, Switzerland and Finland have found the prevalence to vary from 1.8 to 3.5 per 100,000 population [3].
Types 1 and 2 account for the majority of USH in many countries, while only approximately 2% of all USH is type 3. The Finish population is an exception where USH3 accounts for about 40% of all USH cases [4]. Clinically USH has been divided into three clinical subtypes, USH1, USH2 and USH3, all manifesting combined sensorineural hearing loss and visual loss due to a progressive retinal degeneration termed retinitis pigmentosa. Vestibular dysfunction may also be a feature. Molecular definition Further subdivision of Usher syndrome has been possible due to the discovery of distinct genomic loci and causative genes. Since the first gene for USH, discovered in1995, a total of 11 loci including nine genes have been identified as causing various clinical subtypes of USH [5]. Mutations in five of these genes can also give rise to isolated (nonsyndromic) sensorineural hearing loss, and mutations in USH2A can produce isolated (nonsyndromic) autosomal recessive retinitis pigmentosa without hearing loss. For three of the USH1 genes, USH1C, CDH23 and PCDH15, a genotype–phenotype correlation exists with truncating mutations causing USH, whereas missense/inframe alterations result in nonsyndromic deafness [5].
Case Presentation
This article illustrates a case of usher syndrome. A 35 years old man patient presented to ENT clinic at tertiary care center in East congo with progressive hearing loss, first noted at 21years old. His right and left ear hearing declined slowly over the past six years. He is the second of three siblings, has a positive family history of hearing loss explained by the fact that his old brother got a total hearing loss at age of 5. No tinnitus or disschraging ear symptoms or complaints of dysequilibrium or dizziness, or past medical history of head or ear trauma, noise exposure or surgery, he never used hearing aids. no history of mumps, measles, and chickenpox, or systemic problems like hypertension or diabetes. Patient was born full term through normal vaginal delivery, assisted by midwives with no postnatal issues.
Regarding his growth and development, there was no delay in walking, he has normal speech development, patient has a good educational and academic performance background. Currently, the patient has a family. a wife with two kids, the first is 10 years old, and the second is 6 years old. Till now, the children are in good health and there are no signs of hearing or visual impairment.
His ENT clinical exam showed a normal otoscope, Tunning fork tests showed central weber and positive Rinne test over both sides. The pure tone audiometry has shown a bilateral moderate to severe degree of sensorineural hearing loss. Down-sloping configuration audiogram (Fig. 1). Tympanometry examination was normal with present stapedial reflex, normal vestibular assessment. The neurological exam found no abnormalities.
Fig. 1.

Patient’s audiogram, a bilateral moderate to severe degree of sensoneural hearing loss. with the typical “down-sloping” configuration
Patient reported a history of blurred vision since the age of 23 that got worse at night, with no history of trauma or surgery to the eye. Ophthalmologic examination has been requested and showed that best corrected visual acuity OD 6/60 OS count fingers 3 m, anterior segment was free. Fundus exam revealed a waxy pallor of the optic nerve head, arteriolar narrowing, small vessels, bone-spicule shaped pigment deposits, abnormal color vision and tubular visual field, all are consistent features of retinitis pigmentosa (Fig. 2).
Fig. 2.

The patient’s Fundus photograph. (A) pale image of the optic nerve (red arrow), the fundus centered on the posterior pole showing a decrease in vessels size (black arrow). (B) retinal periphery with pigmented migrations having the appearance of bone spicules (red arrows)
The diagnosis of Usher syndrome type 2 was made. Vision correction, with blue light filtering glasses (OD -1 -1 90 degrees, OS -1.5 -1 80 degrees), hearing aid were then prescribed. Genetic and daily life counseling also close 3 months follow up were advised.
Discussion
Differences in auditory acuity and vestibular function are the distinguishing characteristics of USHl and USH2. Among USH patients, the respective proportion of USHl and USH2 has been estimated at 90% and nearly 10%. Persons with USHl are born with a profound sensorineural hearing loss and absent vestibular function. Residual low frequency hearing may be detectable at 90–100 dB (i.e., a “corner” audiogram) but “islands” of hearing between 250 and 8,000 Hz hearing do not exist. These persons typically do not find traditional hearing aids beneficial [1].
Persons with USHl lack vestibular function, eventually they cannot perform these tests. Persons with USHl also fail to respond to icewater caloric stimulation of the horizontal semi-circular canal.
Persons with USH2, by contrast, have typically mild to severe hearing loss. The audiogram has a down-sloping configuration, with a mild to moderate loss in the low frequencies and a severe-to-profound loss in the high frequencies (Fig. 3). Vestibular function is normal. Some investigators have proposed the existence of an additional USH phenotype, Usher syndrome type III (USH3) [Davenport and Omenn, 19,771]. USH3 is similar to USH2, except that hearing loss is progressive and vestibular function deteriorates. However, more clinical information is required before USH3 can be recognized as a distinct USH phenotype [1].
Fig. 3.
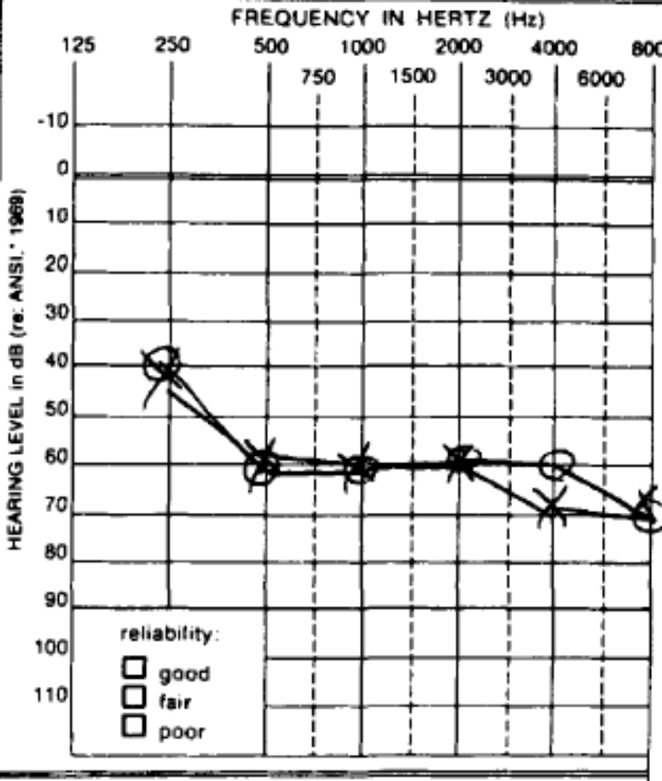
Audiograms characteristic of USH2 demonstrating the typical “down-sloping”
Our presented case had a progressive bilateral hearing impairment discovered around 21 years old; his audiogram shows a typical down sloping configuration, with moderate loss in low frequencies and severe loss in high frequencies as shown before. Vestibular function was normal. The results of sensorineural deafness examination were in accordance with the pathophysiology of deafness in patients with Usher Syndrome, which is generally caused by damage and degeneration of hair cells in organ of corti. Some authors have suggested that persons with USH2 may have vestibular dysfunction [Montandon and Spitzer, 1961; Stenger, 1961] [6, 7]. However, the nature and frequency of this finding is poorly documented. Noted that patient’s vestibular function was normal.
Diagnosis of Usher syndrome can be clinical posed; however today, molecular diagnosis is important but its expensiveness and inaccessibility make it impossible in most developing countries such as in our institution.
Although retinal damage cannot be prevented and treated until now, for this case counseling regarding diseases, low vision therapy, and instructions will help patient to be more independent in carrying out daily activities. Visual rehabilitation that was given is only supportive, so compatible glasses and eye drops were prescribed, educating patients about the use of sunglasses while outdoors, and providing vitamin and antioxidant supplements. Hearing aid rehabilitation was offered for the patient as well.
Studies on genetic therapy are ongoing. Finally, genetic counseling is necessary to assess and reduce the risk of transmission [7]. Many of the therapeutic possibilities, such as changes in the diet or the use of blue and ultraviolet light filtering glasses, are well suited to children and may prove beneficial in slowing the progression of the visual loss. If proven effective, such a simple therapy would give time for more effective gene-based therapies to be developed and to be proven safe for children. While the deafblindness that is the result of the progressive retinitis pigmentosa and occurs in the adult, it is none-the-less a pediatric disorder since, diagnosed early, intervention is predicted to be most effective [8].
Conclusion
Usher syndrome is a rare but serious cause of hearing loss that requires comprehensive multidisciplinary evaluation in conjunction with ophthalmology team. Further genetic, audiological and vestibular assessments are required to help diagnosis and management of specific subtype of this syndrome.
Counseling regarding diseases, low vision therapy, and instructions will help patient to be more independent in carrying out daily activities. Visual rehabilitation that was given is only supportive, so compatible glasses and eye drops were prescribed, educating patients about the use of sunglasses while outdoors, and providing vitamin and antioxidant supplements. Hearing aid rehabilitation was offered for the patient as well.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Acknowledgements
All authors are acknowledging the departments of Otolaryngology and Ophtalmology of the Hopital Provincial General de Reference de Bukavu, the Universite Catholique de Bukavu. Ahmed Youseef Sobhi of Alexandria University.
Authors’ Contributions
Amani Mudekereza Edouard, Nshokano Simba Gloria, Ahmad Yousef Sobhi, Amani Muzindusi, Kabego Fidele, Murhula Fabrice, Mulinganya Christian, Ngoma Basedeke Deogratias, Balungwe Birindwa Patrick.
Funding
Not applicable.
Data Availability
Available.
Declarations
Ethics Approval and Consent to Participate
This work has received the ethical approval from the Ethical committee of the Faculty of Medicine of the Alexandria University on 15/04/2021, under serial number: 0106767
Consent for Publication
Verbal informed consent was obtained from the patient
Competing Interest
The authors declare non conflict of interest
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Smith RJH, Berlin CI, Hejtmancik JF, Keats BJB, Kimberling WJ, Lewis RA, et al. Clinical diagnosis of the Usher syndromes. Am J Med Genet. 1994;50(1):32–38. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- 2.Kristiyan T, Dewi NA, Refa S. Usher Syndrome in two siblings, a Case Report. Int J Retina. 2019;2(1):42–47. doi: 10.35479/ijretina.2019.vol002.iss001.66. [DOI] [Google Scholar]
- 3.Samuelson S, Zahn J. Usher’s syndrome. Ophthalmic Genet. 1990;11(1):71–76. doi: 10.3109/13816819009012950. [DOI] [PubMed] [Google Scholar]
- 4.Friedman TB, Schultz JM, Ahmed ZM, Tsilou ET, Brewer CC (2011) Adv Otorhinolaryngol. Basel, Karger. Available from: http://www.ncbi.nlm.nih.gov/ [DOI] [PubMed]
- 5.Saihan Z, Webster AR, Luxon L, Bitner-Glindzicz M. Update on Usher syndrome. Curr Opin Neurol. 2009;22(1):19–27. doi: 10.1097/WCO.0b013e3283218807. [DOI] [PubMed] [Google Scholar]
- 6.Théra JP, Tiama JM, Konipo A, ,Dakouo PS. Retinitis Pigmentosa: Case Report of Usher Syndrome in Bamako. Acta Sci Ophthalmol. 2020;3(12):02–4. [Google Scholar]
- 7.Calvet C, Lahlou G, Safieddine S. Gene therapy progress: hopes for Usher syndrome. Medecine/Sciences: Editions EDK; 2018. pp. 842–848. [DOI] [PubMed] [Google Scholar]
- 8.Kimberling WJ, Hildebrand MS, Shearer AE, Jensen ML, Halder JA, Trzupek K et al (2010 Aug) Frequency of Usher syndrome in two pediatric populations: implications for genetic screening of deaf and hard of hearing children. Genet Sci 12(8):512–516 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Available.