
Fig. 1 |. Overview of MAGICAL for mapping disease-associated regulatory circuits from scRNA-seq and scATAC-seq data.
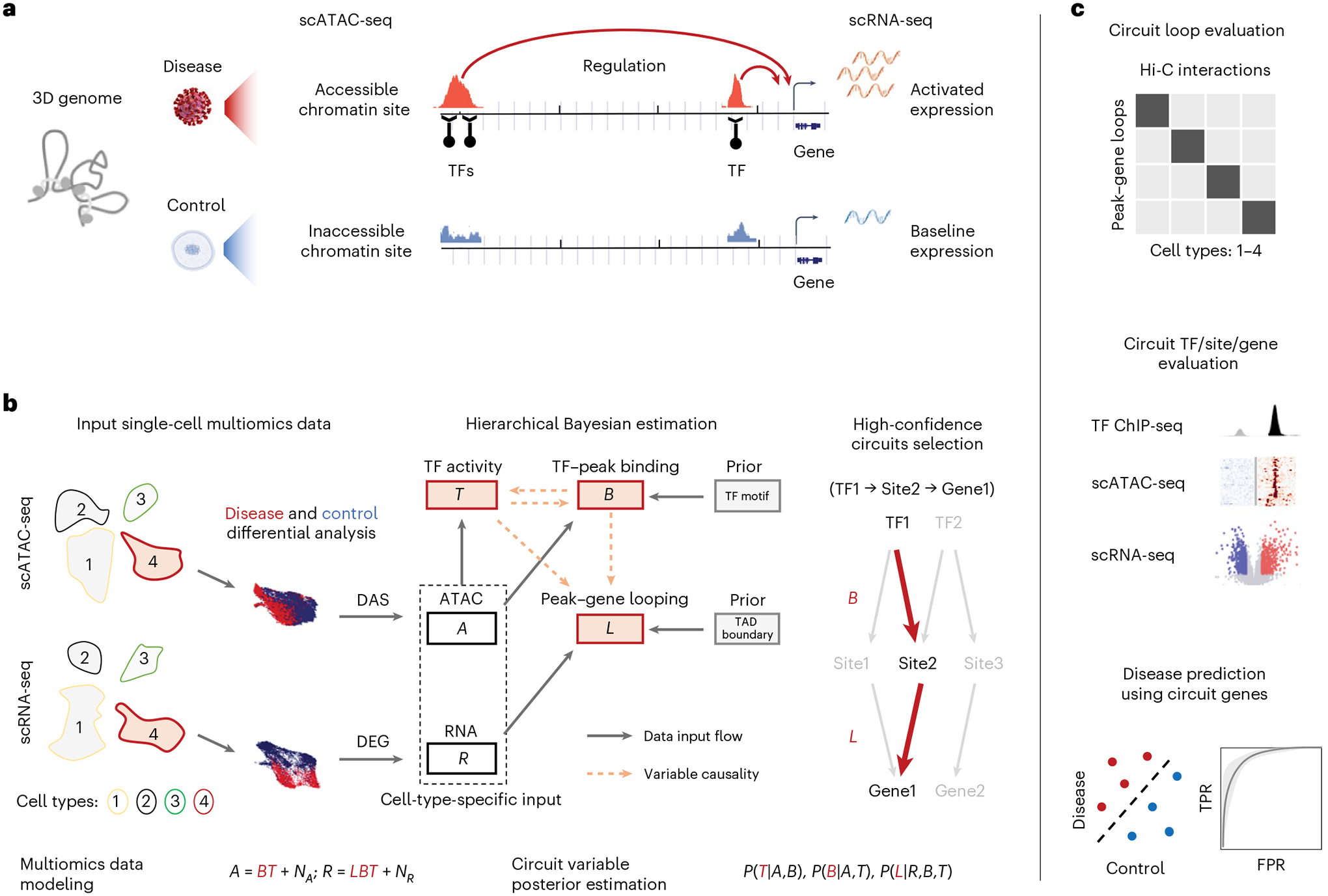
a, Disease-modulated regulatory circutis. In the 3D genome, the altered gene expression in cells between disease and control conditions can be attributed to the chromatin accessibility changes of proximal and distal chromatin sites regulated by TFs. b, MAGICAL framework. To identify disease-associated regulatory circuits in a selected cell type (including ATAC assay cells and RNA assay cells from samples being compared), MAGICAL selects DAS as candidate chromatin sites (peaks) and DEG as candidate genes. Then, the filtered ATAC data and RNA data of DAS and DEG are used as input to a hierarchical Bayesian framework pre-embedded with the prior TF motifs and TAD boundaries. The chromatin activity A is modeled as a linear combination of TF–peak binding confidence B and the hidden TF activity T, with data noise contamination NA. The gene expression R is modeled as a linear combination of B, T, and peak–gene looping confidence L, with data noise contamination NR. MAGICAL estimates the posterior probabilities P(B|A,T), P(T|A,B), and P(L|R,B,T) by iteratively sampling variables B, T, and L to optimize against the data noise NA and NR in both modalities. Finally, regulatory circuits with high posterior probabilities of B and L (for example, a high confidence circuit with inferred interactions between TF1, Site2, and Gene1) are selected. c, Results validation. We evaluate the accuracy and cell-type specificity of the inferred peak–gene looping interactions by checking their enrichment with cell-type-matched chromatin interactions in Hi-C experiments. For the identified TFs, chromatin sites, and genes in circuits, we checked the accuracy of each using independent ChIP-seq, scATAC-seq, and scRNA-seq data. Finally, as a demonstration of the utility of MAGICAL, we used the circuit target genes as features to predict disease states.