

Abstract
DNA polymerase theta (Polθ, encoded by POLQ gene) plays an essential role in Polθ-mediated end-joining (TMEJ) of DNA double-strand breaks (DSB). Inhibition of Polθ is synthetic lethal in homologous recombination (HR)-deficient tumor cells. However, DSBs can be also repaired by PARP1 and RAD52-mediated mechanisms. Because leukemia cells accumulate spontaneous DSBs, we tested if simultaneous targeting of Polθ and PARP1 or RAD52 enhance the synthetic lethal effect in HR-deficient leukemia cells. Transformation potential of the oncogenes inducing BRCA1/2-deficiency (BCR-ABL1 and AML1-ETO) was severely limited in Polq−/−;Parp1−/− and Polq−/−;Rad52−/− cells when compared with single knockouts, which was associated with accumulation of DSBs. Small-molecule inhibitor of Polθ (Polθi) when combined with PARP or RAD52 inhibitors (PARPi, RAD52i) caused accumulation of DSBs and exerted increased effect against HR-deficient leukemia and myeloproliferative neoplasm cells.
Implications:
In conclusion, we show that PARPi or RAD52i might improve therapeutic effect of Polθi against HR-deficient leukemias.
Graphical Abstract
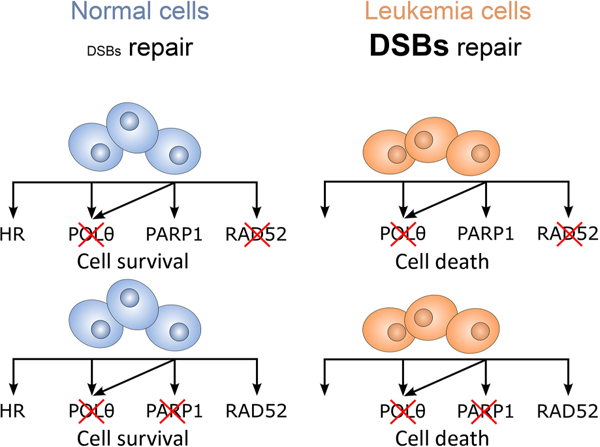
Introduction
Myeloid leukemia cells accumulate high levels of spontaneous and drug-induced DNA damage, but they survive because of enhanced/altered DNA repair activities (1). Thus, DNA damage response (DDR) activities could be targeted to eliminate leukemia cells. We reported that inhibition of RAD52 or PARP1 exerted synthetic lethal effect in HR-deficient hematologic malignancies (2, 3). Our recent report indicated that acute myeloid leukemias (AML) express the highest levels of DNA polymerase theta (Polθ, encoded by POLQ gene) among all tumors in The Cancer Genome Atlas (TCGA) database (4). In addition, Polθ protected homologous recombination (HR)-deficient leukemia cells from the lethal effect of DNA double-strand breaks (DSB) suggesting synthetic lethal effect. However, targeting RAD52, PARP1 or Polθ usually did not completely eradicate HR-deficient leukemia cells indicating that more aggressive approach is required to achieve stronger effect (2–4).
Polθ, PARP1, and RAD52 have mostly distinct and a few overlapping biological activities (5–8). Polθ exerts DNA polymerase and helicase activities and it is involved in Polθ-mediated end-joining (TMEJ) of DSBs, which acts independently of DNA-PK–mediated nonhomologous end-joining (D-NHEJ) and HR (9). In addition, Polθ seals post-replicative ssDNA gaps to maintain genome stability (10). PARP1 plays a key role in base excision repair (BER), single-strand break (SSB) repair, alternative NHEJ (Alt-NHEJ), and in MRE11-mediated recruitment of RAD51 to promote stalled replication fork restart (11). RAD52 forms multimeric ring structures with strong affinity to ssDNA and lesser affinity to dsDNA, and plays a key role in single-strand annealing (SSA), in transcription-coupled HR (TC-HR) in G0–G1 cells, and in transcription-activated HR (TA-HR) in S–G2 cells (12).
We reported that simultaneous targeting of two proteins working in independent DNA repair pathways exerted exceptionally strong dual synthetic lethality effect in HR-deficient tumor cells (13). Here, we examined if simultaneous targeting of Polθ + RAD52 and/or Polθ + PARP1 exert dual synthetic lethality HR-deficient leukemia cells.
Materials and Methods
Primary leukemia cells
Human primary BRCA1/2 deficient and proficient AML samples and myeloproliferative neoplasm (MPN) samples were characterized before (3, 14). The investigators obtained informed written consent from the subjects. Lin−CD34+ cells were obtained from mononuclear fractions by magnetic sorting using the EasySep negative selection human progenitor cell enrichment cocktail followed by human CD34 positive selection cocktail (Stemcell Technologies) as described before (3). Lin−CD34+ cells were incubated in StemSpan SFEM supplemented with growth factors (AML, 100 ng/mL SCF; 20 ng/mL IL3, 100 ng/mL Flt-3 ligand, 20 ng/mL G-CSF, 20 ng/mL IL6; MPN, 100 ng/mL SCF’ 10 ng/mL Flt-3 ligand; 20 ng/mL IL3, IL6, G-CSF, and GM-CSF, 12 U/mL Epo, 2.5 ng/mL thrombopoietin) as described before (3, 14).
Primary cells from knockout mice
Parp1−/− and Rad52−/−- mice were described before (13). Polq−/− (B6.Cg-Polqtm1Jcs/J) mice were purchased from The Jackson Laboratories (#006194). Polq−/− mice were crossbred with Rad52−/− and Parp1−/− mice to generate Polq−/−;Rad52−/− and Polq−/−;Parp1−/− mice and individual knockouts and wild-type (WT) littermates. Knockout mice were identified by PCR of tail snip DNA. DNA isolation and purification from mice tails were performed using the REDExtract-N-Amp Tissue PCR Kit (Sigma-Aldrich). Genotyping was performed using knockout specific primers (Operon) and 2X DreamTaq polymerase Master Mix (Thermo Fisher Scientific). Parp1 specific primers were: forward: 5’-CATGTTCGATGGGAAAGTCCC-’3; WT reverse: 5’-CCAGCGCAGCTCAGAGAAGCCA-’3; mutant reverse: 5’-CATGTTCGATGGGAAAGTCCC-’3, which amplified a 112 base pair fragment if WT, a 350 base pair fragment if Parp1 null, and both 112 and 350 base pair fragments if heterozygous. Polq-specific primers were: WT forward: 5’-TGCAGTGTACAGATGTTACTTTT-’3; WT reverse: 5’-TGGAGGTAGCATTTCTTCTC-3’; mutant forward: 5’-TCACTAGGTTGGGGTTCTC-3’; mutant reverse: 5’-CATCAGAAGCTGACTCTAGAG-3’. The primers amplified a 190 base pair fragment if WT, a 300 base pair fragment if Polq null, and both 190 and 300 base pair fragments if heterozygous. The amplification conditions for both Parp1 and Polq-specific primers consisted of 10 cycles at 94°C for 30 seconds, 65°C for 30 seconds, and 68°C for 20 seconds, followed by 28 cycles at 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 20 seconds. Rad52-specific primers were: forward: 5’-AGCCAGTATACAGCGGATG-’3; WT reverse: 5’-CAACTAGATACATGCCCACG-’3; mutant reverse: 5’-CGCATCGCCTTCTATCGCCT-’3. The primers amplified a 120 base pair fragment if WT, a 320 base pair product if Rad52 null, and both 120 and 320 base pair fragments if heterozygous. The amplification conditions consist of 35 cycles at 93°C for 1 minute, 55°C for 1 minute, and 72°C for 3 minutes. All PCR products were run in a 1.5% agarose gel containing ethidium bromide and visualized using the Alpha Imager Mini Imaging System. Lin-cKit+ murine cells were obtained from mononuclear fractions by magnetic sorting using the EasySep Lin negative selection followed by cKit positive selection cocktail as described before (3). Lin-cKit+ cells were maintained in StemSpanSFEM medium supplemented with 100 ng/mL SCF, and 20 ng/mL IL3 as described before (3).
Viral infection
Retroviral infections were performed as described before (2, 3). Briefly, retroviruses were prepared by cotransfecting HEK 293T/17 cells in a 10-cm plate with 10 mg of packaging pCL-ECO plasmid (Addgene #12371) and 10 mg MIGR1 based vectors by using Lipofectamine 2000 Transfection Reagent (Invitrogen) according to manufacturer’s protocol. GFP+ cells were plated in in Methocult (Stemcell Technologies) as described before (3). Colonies were counted after 7 to 10 days.
Cell lines
Nalm6 and Nalm6-RAD54−/− cells were described before (13). JAK2(V617F)-positive HEL cells were described before (15) and cells with tetracycline-inducible IDH1wt or IDH1(R132H) mutant were obtained from Yuanbin Song and Stephanie Halene, Yale Cancer Center and DeLuca Center for Innovation in Hematology Research, Yale University School of Medicine (16). Cell lines were routinely tested for Mycoplasma and used within a month after thawing.
In vitro sensitivity assays
Novobiocin, olaparib, and talazoparib were purchased from Selleckchem, ART558 and RP6685 were from MedChemExpress. Inhibitors were added for 3 days following by plating in Methocult (Stemcell Technologies) when indicated; colonies were scored after 5 to 7 days. Cells were also incubated with the inhibitors for 14 days and cell viability was assessed by trypan blue exclusion test.
Western blotting
The nuclear and cytosolic protein lysates were prepared as described before (2, 3), separated in 4% to 15% SDS-PAGE (Bio-Rad), and transferred onto a nitrocellulose membrane. Indicated proteins were detected by Western blotting analysis using primary antibodies recognizing Polθ (Invitrogen PA5–69577, MyBioSource MBS9612322), RAD54 (Santa Cruz Biotechnology, sc-374598), RAD52 (Santa Cruz Biotechnology, sc-365341), PARP1 (Santa Cruz Biotechnology, sc-74470), IDH1(R132H) (OriGene CF190113), histone H3 (Invitrogen, AHO1432), actin (Santa Cruz Biotechnology, sc-47778) and Flag (Cell Signaling Technology, #2368).
Comet assay
DSBs were detected by neutral comet assay as described before (4) with modifications. Briefly, Oxiselect Comet Assay Kit (Cell Biolabs #STA-355) was used according to the manufacturer’s instructions. Images were acquired by an inverted Olympus IX70 fluorescence microscope using a FITC filter, and the percentage of tail DNA of individual cells was calculated using the OpenComet plugin of ImageJ.
HR activity
HR activity was measured by using DR-GFP reporter cassette as well as endonuclease I-SceI (Thermo Fisher Scientific) to generate a DSB in GFP cassette as described before (2). RFP plasmid was used as control for transfection efficiency. GFP+ and RFP+ cells were analyzed by flow cytometer (FACScanto, BD Biosciences).
In vivo treatment
NRGS mice (The Jackson Laboratories) were total body irradiated (600 cGy) and injected intravenously with 1 × 106 Lin-CD34+ HR-deficient AML primary cells described before (3). Treatment started when approximately 50% of human hCD45+ leukemia cells were detected in peripheral blood. Mice were treated daily for up to 14 consecutive days with vehicle (control), Polθ inhibitor RP6685 (80 mg/kg oral gavage; ref. 17), PARP1 inhibitor olaparib (50 mg/kg, i.p; ref. 3), and the combination. Median survival time was determined.
Statistical analysis
Data are expressed as mean SD from at least three independent experiments unless stated otherwise; #, +, *, P < 0.05; ##, ++, **, P < 0.01; ###, +++, ***, P < 0.001 and #####, ++++, ****, P < 0.0001. Comparisons between two groups were done via student t test. Multigroup comparisons used one-way Anova to find the initial F and Pr(>F) values, followed by Tukey honestly significant difference (HSD) post hoc test to discover the P values intergroup comparisons. Median survival time of the mice SE was calculated by Kaplan–Meier and compared by log-rank test.
Study approval
Studies involving primary AML and MPN samples were approved by the ethics committee of Temple University Lewis Katz School of Medicine (Philadelphia, PA) and met all requirements of the Declaration of Helsinki. Animal studies were approved by the Temple University Institutional Animal Care and Use Committee.
Data availability
Data and protocols will be shared upon request to the corresponding author.
Results
Genetic targeting of Polu and PARP1 or RAD52 prevented malignant transformation by AML1-ETO and BCR-ABL1
Beat-AML database analysis revealed that mutations in POLQ are extremely rare (POLQ) and mutations in RAD52, PARP1 were not detected (Supplementary Fig. S1A). In addition, patient-to patient differences in mRNA expression of these genes were not interdependent (Supplementary Fig. S1B).
To test the effect of simultaneous inhibition of Polθ + PARP1 and Polθ + RAD52, first we generated Polq−/−;Parp1−/− and Polq−/−;Rad52−/− mice. These double knockout mice, as well as their single knockout counterparts did not display any detectable organic defects, including bone marrow, based on blood cell counts, phenotypic blood and marrow cell analysis as well as a comprehensive macro and microscopic tissue examination (Supplementary Fig. S2).
In concordance with previous reports (3, 18, 19) BCR-ABL1 and AML1-ETO downregulated BRCA1 and/or BRCA2 to induce BRCA-deficient phenotype of leukemia cells (Fig. 1A and B). Therefore, bone marrow cells from WT, single knockout (Polq−/−, Parp1−/− and Rad52−/−) and double knockout (Polq−/−;Parp1−/− and Polq−/−;Rad52−/−) mice were infected with retroviral particles encoding for GFP (control), GFP and BCR-ABL1, or GFP and AML1-ETO. As expected, expression of AML1-ETO and BCR-ABL1 inhibited colony formation capacity of GFP+Lin-Sca1+c-Kit+ stem cell-enriched Polq−/−, Parp1−/− and Rad52−/− cells by 2 to 3-fold, respectively, when compared with WT counterparts (Fig. 1C). Remarkably, AML1-ETO and BCR-ABL1 reduced clonogenic potential of Polq−/−;Parp1−/− and Parp1−/−;Rad52−/− cells by 4 to 8-fold when compared with single knockout counterparts. Nontransformed GFP+Lin-Sca1+c-Kit+ stem cell-enriched cells from WT, single and double knockout mice displayed similar clonogenic activity.
Figure 1.
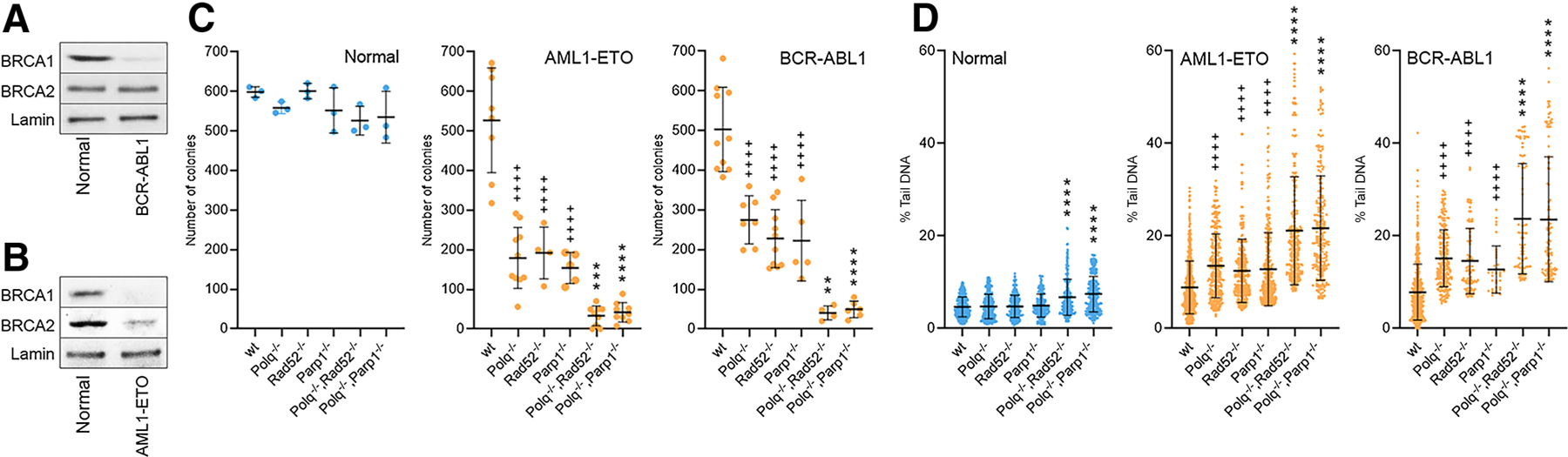
Simultaneous deletion of Polq and Parp1 or Rad52 impaired transforming activity of BCR-ABL1 and AML1-ETO. A and B, Western blot analysis showing the expression of indicated proteins in the nuclear fractions of Lin- mBMCs expressing indicated oncogene or GFP (normal). Clonogenic activity (C) and neutral comet assay (D) in Lin−Sca1+c-Kit+ (LSK) murine bone marrow cells of the indicated genotypes expressing GFP (Normal) and BCR-ABL1 or AML1-ETO. Results represent mean number SD of colonies (C) and mean % of tail DNA SD (D). * and + indicate statistical significance when compared with corresponding individual knockouts and to WT, respectively.
A neutral comet assay was performed to determine the impact of the absence of Polθ and PARP1 or Polθ and RAD52 on accumulation of DSBs in GFP+Lin-Sca1+c-Kit+ cells transduced with BCR-ABL1 and AML1-ETO. As expected, BCR-ABL1 and AML1-ETO enhanced DSBs accumulation in WT cells, which was further increased in Polq−/−, Parp1−/−, and Rad52−/− single knockout cells (Fig. 1D). However, the highest levels of DSBs were detected in Polq−/−;Parp1−/− and Polq−/−;Rad52−/− double knockout cells transduced with these oncogenes. Nontransformed Polq−/−;Parp1−/− and Parp1−/−; Rad52−/− GFP+Lin-Sca1+c-Kit+ cells displayed very modest, but statistically significant increase of DSBs when compared with the single knockout counterparts.
Chemical targeting of Polu and PARP1 or RAD52 exerted dual synthetic lethality effect in HR-deficient leukemias
Polθ is a novel and very promising target in HR-deficient tumors (9). The antibiotic novobiocin (NVB) was identified as an inhibitor of Polθ ATPase (helicase) activity, and ART558 inhibits Polθ DNA polymerase activity (9). Both inhibitors, when used individually, inhibit the growth of HR-deficient tumor cells.
We reported that simultaneous targeting of different functions of PARP1 by two inhibitors exerted increased effect against BRCA1-deficient cells (20). Therefore, we also tested the combinatorial effect of ART558 and NVB. NVB + ART558 exerted increased effect against Nalm6(RAD54−/−) cells and a less profound effect against Nalm6 cells (Supplementary Fig. S3A and S3B). NVB was identified through a small screen and has not undergone medicinal chemistry optimization to improve its potency/selectivity, thus we cannot exclude that off-target effect(s) of NVB might contribute to this result. On the other-hand, ART558 has been thoroughly developed, and likely has significantly less off-target effects than NVB. Therefore, ART558 was applied in further experiments.
The effectiveness of simultaneous inhibition of Polθ + PARP1 and Polθ + RAD52 by small molecule inhibitors was tested using four clinically relevant HR-deficient hematological malignancy models: Nalm6(RAD54−/−) cells, JAK2(V617F);IDH1(R132H) HEL cells, HR-deficient AML primary cells, and MPL(W515L)-positive MPN cells (2, 3, 14, 16). Polθ, PARP, and RAD52 inhibitors were validated and applied following the protocols described before (2–4).
HR-deficient status of Nalm6(RAD54−/−) cells was confirmed by loss of expression of RAD54 protein (Fig. 2A) and abrogation of HR activity when compared with isogenic parental cells (Fig. 2B). Trypan blue exclusion test confirmed that Nalm6(RAD54−/−) cells were more sensitive to Polθi (ART558), PARPi (talazoparib), and RAD52i (6-hydroxy-DL–dopa) when compared to Nalm6 parental cells (Supplementary Fig. S3C). Remarkably, ART558 IC50 was increased 4 to 6 times in Nalm6(RAD54−/−) cells in the presence of talazoparib and 6-hydroxy-DL–dopa (Supplementary Fig. S3C and S3D).
Figure 2.
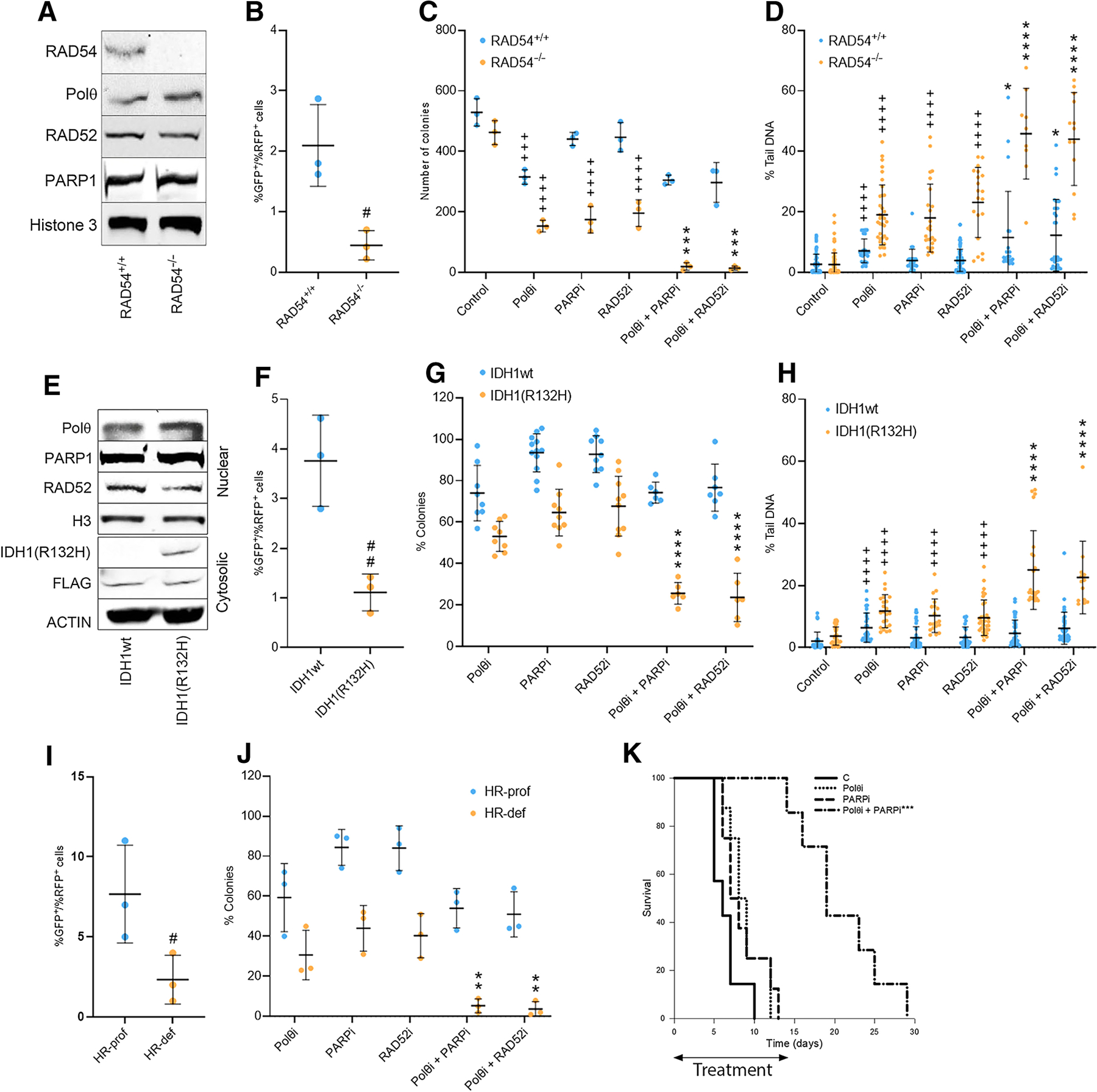
Targeting Polθ + PARP and Polθ + RAD52 induced dual synthetic lethality against HR-deficient leukemia cells. A, Western blot analysis showing the expression of indicated proteins in the nuclear fractions of Nalm6(RAD54+/+) and Nalm6(RAD54−/−) isogenic cell lines. B, HR activity in Nalm6(RAD54+/+) and Nalm6(RAD54−/−) cells: results represent mean % SD of GFP+ cells in RFP+ cells. Nalm6(RAD54+/+) and Nalm6(RAD54−/−) cells were treated for 72 hours (C) and 12 hours (D) with 25 mmol/L Polθi ART558, 2.5 nmol/L PARPi talazoparib, 10 mmol/L RAD52i 6-hydroxy-DL-dopa and the indicated combinations. Results show mean number SD of colony numbers (C) and mean % of tail DNA SD (D). E, Western blot analysis showing the expression of indicated proteins in the nuclear and cytosolic fractions of HEL cells with tet-inducible IDH1wt or IDH1(R132H) mutant. F, HR activity in HEL cells expressing IDH1wt or IDH1(R132H) mutant: results represent mean % SD of GFP+ cells in RFP+ cells. HEL cells expressing IDH1wt or IDH1(R132H) mutant were treated for 72 hours (G) and 12 hours (H) with 25 mmol/L Polθi ART558, 1 mmol/L PARPi olaparib, 2.5 mmol/L RAD52i 6-hydroxy-DL-dopa and the indicated combinations. Results show mean % SD of colony numbers when compared with untreated cells (G) and mean % of tail DNA SD (H). I, HR activity in Lin-CD34+ cells from HR-proficient and HR-deficient AMLs (n = 3 of each). Results represent mean % SD of GFP+ cells in RFP+ cells. J, Lin-CD34+ cells from HR-proficient and HR-deficient AMLs (n = 3 of each) were treated with 25 mmol/L Polθi ART558, 1.25 mmol/L PARPi olaparib, 2.5 mmol/L RAD52i 6-hydroxy-DL–dopa and the indicated combinations. Results represent mean % SD of colonies in comparison with untreated cells. K, Survival curves of the AML mice treated with vehicle (C), Polθi RP6685, PARPi olaparib, and the combination. (B–D and F–J) Statistical significance when compared with: # another group, + control, and * corresponding individual treatments. K, ***, P < 0.001 in comparison to other groups.
In concordance, combinations of Polθi (ART558) + PARPi (talazoparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) caused increased inhibition of colony formation capacity of RAD54−/− Nalm6 cells with modest effect on Nalm6 parental cells (Fig. 2C). The inhibitory effect of ART558 + talazoparib and ART558 + 6-hydroxy-DL–dopa on clonogenic activity was accompanied by proportional increase of DSBs detected by neutral comet assay (Fig. 2D).
Tetracycline-inducible expression of IDH1(R132H) mutant and inhibition of HR activity was detected in JAK2(V617F);IDH1 (R132H) HEL cell line when compared to JAK2(V617F);IDH1wt HEL cells (Fig. 2E and F, respectively). Clonogenic test revealed that combinations of Polθi (ART558) + PARPi (olaparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) exerted stronger inhibitory effect against JAK2(V617F);IDH1(R132H) HEL cells when compared with JAK2(V617F);IDH1wt counterparts (Fig. 2G), which was associated with abundant accumulation of DSBs detected by neutral comet assay in the former cells (Fig. 2H).
Moreover, the antileukemia potential of combinations of Polθi (ART558) + PARPi (olaparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) were evaluated in Lin-CD34+ stem/progenitor enriched primary AML cells. HR-deficient and HR-proficient status of AML samples was validated by measuring HR activity (Fig. 2I). Combinations of Polθi (ART558) + PARPi (olaparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) increased the reduction of clonogenic potential of HR-deficient AMLs (Fig. 2J). HR-proficient AMLs did not respond to PARPi and RAD52i and were modestly affected by Polθi.
Finally, the antileukemia effect of the combination of Polθi and PARPi was tested in humanized immunodeficient NRGS mice bearing HR-deficient primary AML xenograft. Mice with advanced leukemia (approximately 50% of the peripheral blood leukocytes were hCD45+ cells) were treated with orally active Polθi RP6685, PARPi olaparib, and RP6685 + olaparib. While single treatment did not prolong the survival of AML mice (MST of the mice treated with vehicle = 6.5 0.7, RP6685 = 8.8 0.8, and olaparib = 8.5 0.9 days after starting the treatment), combination of RP6685 + olaparib exerted statistically significant antileukemia effect (MST = 20.7 2.0; Fig. 2K). No obvious overall toxicity (>10% body weight loss, reduced food/water intake, changes in hair/skin, changes in overall behavior) was attributed to the combinatorial treatment.
The combinations of Polθi (ART558) + PARPi (olaparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) were also tested in HR-deficient MPL(W515L) MPN cells (Supplementary Fig. S4A). Increased effect of dual synthetic lethality of Polθi (ART558) + PARPi (olaparib) and Polθi (ART558) + RAD52i (6-hydroxy-DL–dopa) was detected in clonogenic assay of Lin-CD34+ MPN primary cells (Supplementary Fig. S4B).
Discussion
Inactivation of PARP1 and RAD52 triggered synthetic lethality in HR-deficient leukemias (2, 3). Moreover, our recent report unraveled a pivotal role of Polθ in promoting proliferation and protecting against apoptosis induced by spontaneous DNA damage in myeloid malignancies carrying mutations causing HR deficiency such as AML1-ETO, IDH2(R140Q), and BCR-ABL1 (4). We show here that the antileukemia effect of Polθi could be greatly enhanced by simultaneous targeting of PARP1 or RAD52, likely triggering highly effective dual synthetic lethality (ref. 13; Supplementary Fig. S5). Despite recent advances the mechanisms of dual synthetic lethality by simultaneous targeting of Polθ+PARP1 and Polθ+RAD52 are not fully understood.
PARP1 is thought to promote recruitment of Polθ to DNA damage (21). Thus, the beneficiary effect of PARPi + Polθi may reflect reduced recruitment of Polθ to DSBs and inhibited Polθ activity, respectively. In addition, the fact that PARPi-resistant cells may be sensitive to Polθi (22) suggests that Polθ and PARP1 work also in distinct DNA repair pathways and/or different cell cycle phases, which may explain dual synthetic lethality effect. This hypothesis is further supported by recent discovery that Polθ plays a critical role in sealing ssDNA post-replicative gaps (10) and in TMEJ in mitosis (23).
Polθ and RAD52 have distinct roles in genome maintenance, e.g., in chromosomal break repair and replication stress response (5). Therefore, targeting two distinct DNA repair pathway may likely produce dual synthetic lethality in HR and/or D-NHEJ deficient cells. RAD52 also blocks premature TMEJ to prevent chromosomal aberrations (6). Thus, inhibition of RAD52 may cause premature usage of TMEJ and accumulation of lethal chromosomal aberrations.
Simultaneous inactivation of Polθ and PARP1 or Polθ and RAD52 did not significantly affect various tissues in mice, implicating potential clinical application of dual targeting of Polθ and PARP1 or RAD52. While RAD52 inhibitor still awaits clinical development, PARP1 inhibitors have been widely used to treat patients with HR-deficient tumors (11). In addition, Polθ inhibitor ART4215 and PARP1 inhibitor talazoparib are being explored in a clinical trial in patients with HR-deficient solid tumors (NCT04991480).
In conclusion, combination of Polθi with PARPi or RAD52i exerted remarkably strong dual synthetic lethality effect implicating clinical applications in growing cohorts of patients with leukemias displaying accumulation of DNA damage concomitant with DNA repair deficiencies (1).
Supplementary Material
Acknowledgments
This work was funded by R01 CA186238, R01 CA244179, R01 CA237286, R01 CA247707, and the Leukemia and Lymphoma Society TRP 6628 (all to T. Skorski). K. Sullivan-Reed was supported by F31 CA203161.
Footnotes
Authors’ Disclosures
No disclosures were reported.
Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Esposito MT, So CW. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma 2014;123:545–61. [DOI] [PubMed] [Google Scholar]
- 2.Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, et al. Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood 2013;122: 1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nieborowska-Skorska M, Sullivan K, Dasgupta Y, Podszywalow-Bartnicka P, Hoser G, Maifrede S, et al. Gene expression and mutation-guided synthetic lethality eradicates proliferating and quiescent leukemia cells. J Clin Invest 2017; 127:2392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vekariya U, Toma M, Nieborowska-Skorska M, Le BV, Caron MC, Kukuyan AM, et al. DNA polymerase theta protects leukemia cells from metabolically induced DNA damage. Blood 2023;141:2372–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kelso AA, Lopezcolorado FW, Bhargava R, Stark JM. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet 2019;15:e1008319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llorens-Agost M, Ensminger M, Le HP, Gawai A, Liu J, Cruz-García A, et al. Polθ-mediated end joining is restricted by RAD52 and BRCA2 until the onset of mitosis. Nat Cell Biol 2021;23:1095–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman T, Bercovich A, Moskovitz Y, Chapal-Ilani N, Mitchell A, Medeiros JJF, et al. Recurrent deletions in clonal hematopoiesis are driven by microhomology-mediated end joining. Nat Commun 2021;12:2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luedeman ME, Stroik S, Feng W, Luthman AJ, Gupta GP, Ramsden DA. Poly (ADP) ribose polymerase promotes DNA polymerase theta-mediated end joining by activation of end resection. Nat Commun 2022;13:4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drzewiecka M, Barszczewska-Pietraszek G, Czarny P, Skorski T, Śliwiński T. Synthetic lethality targeting Polθ. Genes 2022;13:1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belan O, Sebald M, Adamowicz M, Anand R, Vancevska A, Neves J, et al. POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol Cell 2022;82:4664–80. [DOI] [PubMed] [Google Scholar]
- 11.Le BV, Podszywałow-Bartnicka P, Piwocka K, Skorski T. Pre-existing and acquired resistance to PARP inhibitor-induced synthetic lethality. Cancers 2022;14:5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottifredi V, Wiesmüller L. Current understanding of RAD52 functions: fundamental and therapeutic insights. Cancers (Basel) 2020;12:705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan-Reed K, Bolton-Gillespie E, Dasgupta Y, Langer S, Siciliano M, Nieborowska-Skorska M, et al. Simultaneous targeting of PARP1 and RAD52 triggers dual synthetic lethality in BRCA-deficient tumor cells. Cell Rep 2018; 23:3127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nieborowska-Skorska M, Maifrede S, Dasgupta Y, Sullivan K, Flis S, Le BV, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017;130:2848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quentmeier H, MacLeod RA, Zaborski M, Drexler HG. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia 2006;20:471–6. [DOI] [PubMed] [Google Scholar]
- 16.Sulkowski PL, Corso CD, Robinson ND, Scanlon SE, Purshouse KR, Bai H, et al. 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 2017;9:eaal2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bubenik M, Mader P, Mochirian P, Vallée F, Clark J, Truchon JF, et al. Identification of RP-6685, an orally bioavailable compound that inhibits the DNA polymerase activity of Polθ. J Med Chem 2022;65:13198–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med 2015;21:1481–90. [DOI] [PubMed] [Google Scholar]
- 19.Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, Wolanin K, Skowronek K, Nieborowska-Skorska M, et al. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle 2014;13:3727–41. [DOI] [PubMed] [Google Scholar]
- 20.Nieborowska-Skorska M, Maifrede S, Ye M, Toma M, Hewlett E, Gordon J, et al. Non-NAD-like PARP1 inhibitor enhanced synthetic lethal effect of NAD-like PARP inhibitors against BRCA1-deficient leukemia. Leuk Lymphoma 2019;60: 1098–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mateos-Gomez PA, Gong F, Nair N, Miller KM, Lazzerini-Denchi E, Sfeir A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015;518:254–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun 2021;12:3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brambati A, Sacco O, Porcella S, Heyza J, Kareh M, Schmidt JC, et al. RHINO restricts MMEJ activity to mitosis. Biorxiv 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data and protocols will be shared upon request to the corresponding author.