

Abstract
Mechanisms linking ingested pollutants to increased incidence of allergy are poorly understood. We report that mice exposed to low doses of cadmium develop higher IgE responses following oral allergen sensitization and more severe allergic symptoms upon allergen challenge. The environmentally relevant doses of this pollutant also induced oxidative/inflammatory responses in the gut of SPF, but not germ-free mice. Interestingly, the increased IgE responses correlated with stimulation of the vitamin D3-metabolizing enzymes CYP27B1 and CYP24A1 in the gut, and increased luminal levels of oxidized vitamin D3 metabolites that are not ligands of the vitamin D receptor. Inhibition of CYP27B1 and CYP24A1 via oral administration of pharmacological inhibitors reduced IgE responses induced in mice orally exposed to cadmium. Our findings identify local alteration of vitamin D signaling as a new mechanism for induction of IgE responses by environmental pollutants. They also identify vitamin D3-metabolizing enzymes as therapeutic targets for treatment of allergy.
Keywords: Pollutant, gut, oxidative stress, vitamin D3 metabolism, IgE, cadmium
INTRODUCTION
Food allergies are IgE-mediated hypersensitivity reactions with clinical symptoms that are capable of affecting multiple tissues, including the skin, airways, and gastrointestinal tract. The increased incidence of allergic diseases over the last few decades demonstrates that both genetic predisposition (1) and nongenetic factors, including the excessive use of antibiotics, especially during early life, contribute to the development of allergies (2, 3). Environmental factors, such as pollutants and drugs, are also viewed as major contributors to the growing number of allergic individuals (4, 5); however, how environmental pollutants affect allergic sensitization and the development of allergen-specific IgE responses and/or immune mechanisms during the effector phase of allergic responses remains poorly understood.
Cadmium (Cd) is a cationic heavy metal that is commonly used for metal plating, or for making pigments and batteries. Cd can remain in the air, soil, and water (6), and can be ingested through contaminated water, leafy vegetables, fish, or grains (6, 7). Average daily intake of Cd through water and food has been estimated to be around 8–25 μg (7). Approximately 20% of arable land in China is reportedly contaminated with heavy metals (8). Furthermore, Cd content in contaminated rice paddy fields was found to range between 5 and 145 mg/kg, with Cd accumulation in brown rice between 1.9 and 9.4 mg/kg (9). Because Cd has a low rate of excretion and a half-life in the body greater than 15 years (6), the chronic ingestion of subtoxic doses may have important health implications. Exposure to toxic doses of Cd has been shown to induce inflammatory responses (10, 11) and impair immune cell function via mechanisms involving the production of reactive oxygen species (ROS) (12). With the exception of occupational exposure, the general adult population is more likely to be exposed to subtoxic doses of Cd. However, how Cd affects T helper cell differentiation, immunoglobulin (Ig) class switching, and production of IgE remain largely unknown.
Here, we report that chronic ingestion of subtoxic, and environmentally relevant, doses of Cd (6, 13, 14), enhances the production of IgE and the severity of subsequent allergic symptoms. We also show that Cd enhances the expression of the vitamin D3-oxidizing enzymes CYP27B1 and CYP24A1. The subsequent increased levels of oxidized vitamin D3 metabolites that are not ligands of vitamin D receptor drive IgE responses in hosts exposed to subtoxic doses of this heavy metal.
RESULTS
Chronic ingestion of subtoxic doses of Cd enhances IgE responses
The gut is the primary target of ingested and inhaled Cd (7). To mimic chronic exposure to subtoxic and environmentally relevant doses of Cd (13–15), groups of mice were given drinking water containing 10 μM CdCl2 (Cd10) or 25 μM CdCl2 (Cd25) for 28 days. Compared to controls (NoCd), mice exposed to Cd ingested the same amount of water and had the body weight. They also had similar numbers of T and B lymphocytes in tissues (Figure S1A), suggesting that the doses of Cd ingested had no major effect on the overall number of cells of adaptive immunity in Gut-Associated Lymphoid Tissues or in the spleen.
To determine whether such exposure to Cd could modulate allergic sensitization and promote IgE responses, age-matched controls and mice exposed to Cd for 28 days were sensitized by oral administration of ovalbumin (OVA) and the Th2-promoting adjuvant cholera toxin (Figure 1A). Cd-exposed mice produced higher levels of OVA-specific serum IgE antibody (Ab) responses than control mice (Figure 1B). However, antigen-specific IgG1 and IgA responses were unchanged in Cd-exposed mice, indicating that Ig classes were differentially affected by this pollutant (Figure 1B). We also found that a four weeks exposure of mice to Cd doses lower than Cd10 (i.e., Cd4) did not enhance IgE responses in this experimental model (Figure S1B). The allergen-specific Ab responses in Cd-treated mice were associated with elevated frequencies of IL-4+CD4+, IFNγ+CD4+, IL-17+, and IL-21+ CD4+ T cells in the spleens (Figure 1C and Figure S1C).
Figure 1. Prior chronic ingestion of subtoxic doses of Cd enhances IgE responses and allergy severity.
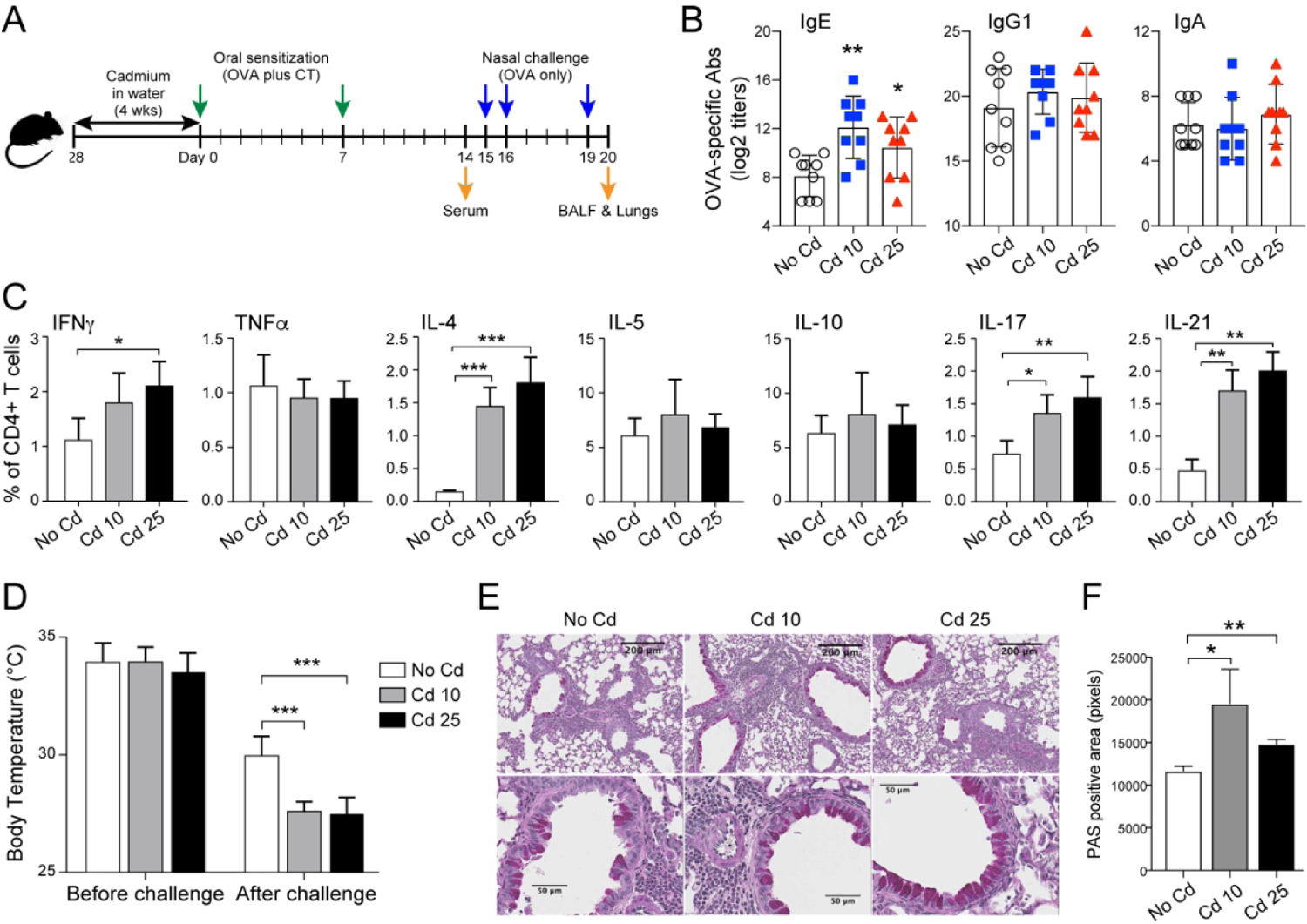
(A) Experimental scheme. Mice (n=5 per group) were provided CdCl2 [10 μM (Cd10) or 25 μM (Cd25)] in drinking water for 28 days and sensitized by the oral administration of OVA (1 mg) and cholera toxin (CT 15 μg). (B) OVA-specific serum Ab titers 1 week after the last sensitization (Day 14). (C) Allergen-specific CD4+ T cell responses. Spleens were collected on day 14 and restimulated in vitro with 1 mg/mL of OVA before flow cytometry analysis of CD4+ T cell cytokine responses. (D) Surface body temperatures of anesthetized mice before and 1 h after nasal allergen challenge with OVA (200 μg). (E) Lung inflammation and mucus production, and (F) quantification of mucus production. Data represent one of at least four independent experiments with five mice per group. Data are expressed as the mean ± SD. *p < 0.05; **p < 0.01 compared to No Cd.
Chronic ingestion of subtoxic doses of Cd enhances allergic responses
To determine whether ingestion of cadmium could affect the severity of allergic responses, mice orally-sensitized were nasally challenged with the allergen. This nasal allergen challenge triggered a greater drop in body temperature in Cd-exposed mice than in controls, indicating more severe signs of allergy (Figure 1D). Allergen challenge also increased the frequencies of CD4+ T cells, SSChiCD11cloGR1loCD3−CD19− eosinophils and macrophages, including CD38+ M1 macrophages, in the bronchoalveolar lavage fluid (BALF) of mice exposed to Cd (Figure S2A). The increased number of eosinophils in the lungs of Cd-treated mice was further confirmed by the levels of eosinophil-associated Siglec-F and peroxidase (Epx) mRNA (Figure S2B). When T cell collected from BALF were re-stimulated with allergen in vitro, they showed increased frequency of Th2 (IL-4), and Th1 (IFNγ) cells (Figure S2C). This profile was consistent with elevated levels of IL-4, IFNγ, and TNFα measured in the BALF of mice exposed to Cd10 (Figure S2D) which also exhibited lung inflammation as indicated by increased leukocyte infiltrates and mucus production after nasal allergen challenge (Figures 1E and 1F).
We addressed whether stimulation of IgE responses by ingested Cd was mediated by events that occurred in mucosal tissues of the gut or the systemic compartment. In contrast with mice sensitized orally, mice sensitized via the systemic route (i.e., intraperitoneal, (i.p.)) route by injection of OVA and cholera toxin as adjuvant showed no increase in OVA-specific IgE responses or other Ig isotype responses (Figure S2E). Together, these data indicate that chronic ingestion of subtoxic doses of Cd enhances the magnitude of IgE responses via events that occur locally in gut tissues.
Chronic ingestion of subtoxic doses of Cd promotes oxidative stress and inflammatory responses in the intestines
Consistent with reports that Cd induces reactive oxygen species (ROS) production and oxidative stress responses(12), intestines of mice chronically exposed to subtoxic doses of Cd had higher mRNA levels of dual oxidase 2 (Duox2) and dual oxidase maturation factor 2 (Duoxa2) than controls (Figure 2A). Antioxidants and phase II-detoxifying enzymes are under the control of the transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) (12), and dysregulation of the NRF2 pathway stimulates the expression of inflammatory cytokines. Accordingly, mice exposed to subtoxic doses of Cd exhibited a dose-dependent induction of NRF2 in intestinal epithelial cells (Figures 2B and 2C) and increases in proinflammatory (i.e., Ifnγ, Il-1β, Mip2, and Kc) and anti-inflammatory (i.e., Il-10 and Tgfβ mRNA responses (Figure 2D). Furthermore, mice exposed to Cd does not increase the expression of Th2 (i.e., IL-4 and IL-5) cytokines, but IL-33 and TSLP were elevated by Cd exposure (Figure 2D). Oxidative stress responses were recently shown to induce PGE2 (16), and PGE2 can promote IgE in vivo (17, 18). Thus, we examined PGE2 production in intestinal tissues by immunofluorescence staining (Figures 2E and 2F). The higher levels of PGE2 detected in tissues of mice treated with Cd10 and to a lesser extend Cd25 was confirmed by quantification of PGE2 in small intestinal tissues by ELISA (Figure 2G).
Figure 2. Chronic ingestion of subtoxic doses of Cd induces oxidative stress and inflammatory responses.
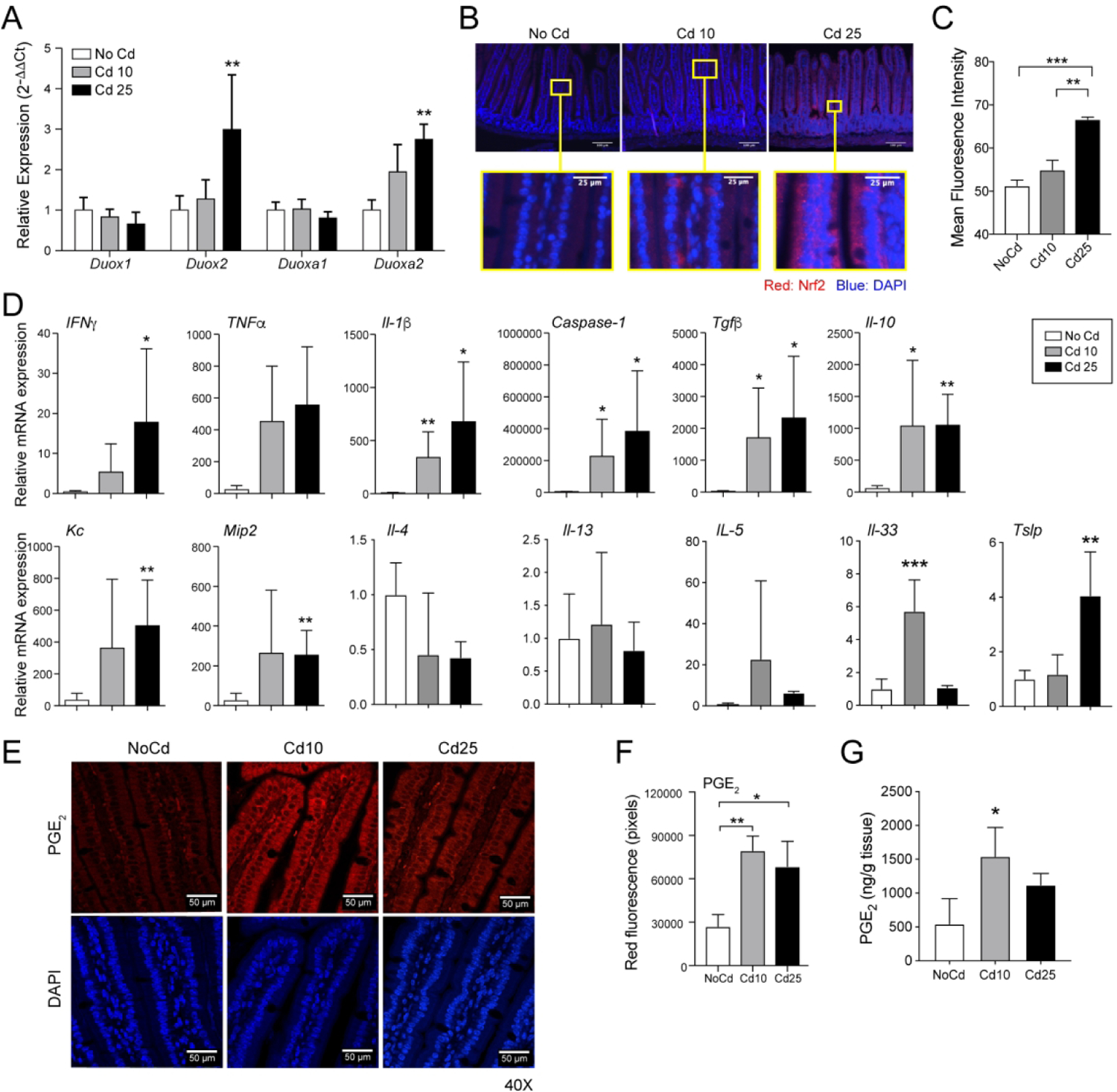
Mice (n=5 per group) were provided CdCl2 [10 μM (Cd10) or 25 μM (Cd25)] in drinking water for 28 days. (A) Expression of mRNA levels of the oxidase subunits Duox1, Duox2, Duoxa1, and Duoxa2. cDNAs were generated from small intestines, and mRNA responses were analyzed by qRT-PCR. (B and C) Expression of NRF2. Sections of small intestinal. (C) Quantification of NRF2 staining. (D) Cytokine mRNA responses in intestinal tissues. qRT-PCR data represent one of at least four independent experiments with five mice per group and are expressed as the mean ± SD. (E-G) Expression of PGE2 in small intestinal tissues. (E) Representative immunofluorescence staining. Sections of small intestines were stained with anti-PGE2 antibody and counterstained with DAPI to visualize nuclei. (F) Quantification of PGE2 staining. (G) Analysis of PGE2 levels in small intestinal tissues by ELISA (n=5 per group). *p < 0.05; **p < 0.01 compared to NoCd.
IgE-promoting effects of low doses of Cd are lost in the absence of gut microbiota
We next used germ-free (GF) mice to address whether the increased IgE responses observed in Cd-treated mice reflected direct host response to Cd or indirect effects mediated through the gut microbiota (Figure 3A). As observed in conventional specific pathogen-free (SPF) mice, chronic exposure to subtoxic doses of Cd did not change the frequency of T and B cells in lymphoid tissues of GF mice (Figures 3B). Interestingly, prior exposure to subtoxic doses of Cd did not enhance allergen-specific IgE responses in GF mice (Figure 3C). However, allergen-specific serum IgA (Figure 3D) and IgG subclass (Figure 3E) responses were increased in Cd-treated mice, suggesting that IgE responses are differentially regulated by Cd than other Ig isotypes. To better understand the inability of Cd to enhance IgE responses in germ-free mice, we examined oxidative stress and inflammatory responses in their gut tissues prior to allergic sensitization. NRF2 was not induced (Figure 3F) and we detected minimal levels of inflammatory and type 2 cytokine mRNA responses with the exception of Tnfα mRNA which were significantly enhanced (Figure 3G). Finally, both Cd-treated and control germ-free mice expressed similar levels of PGE2 in gut tissues (Figure 3H and 3I).
Figure 3. IgE-promoting effects of low doses of Cd are lost in the absence of gut microbiota.
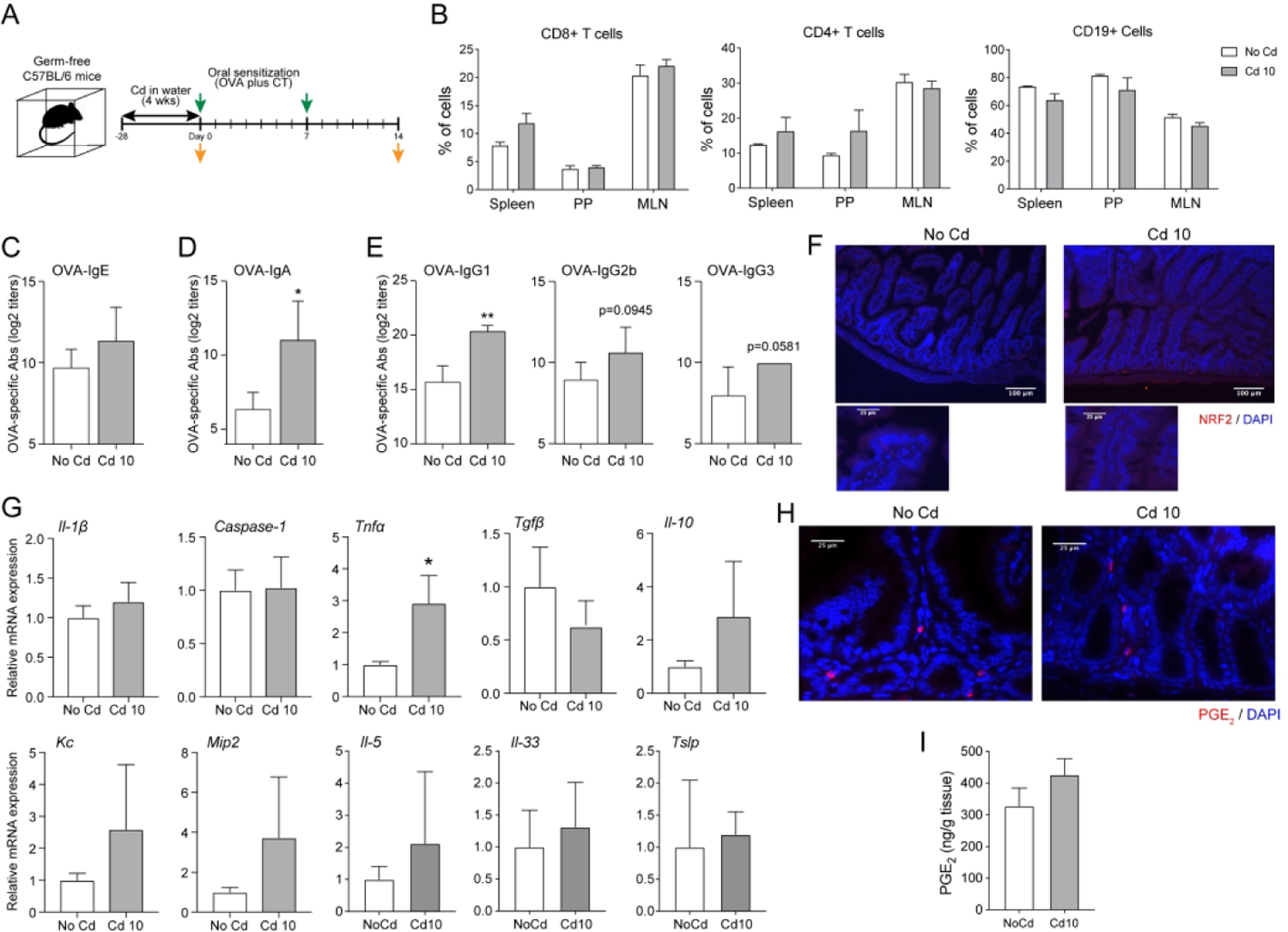
(A) Scheme of experiments. Germ-free mice (n=5 per group) were provided CdCl2 [10 μM (Cd10) or 25 μM (Cd25)] in drinking water for 28 days. (B) Frequency of B and T lymphocytes in mucosal (Peyer’s patches (PP); mesenteric lymph node (MLN)) and peripheral (spleen) lymphoid tissues from germ-free mice exposed to subtoxic doses of Cd. (C-E) Allergen-specific antibody responses after oral sensitization. (C) Allergen-specific serum IgE responses. (D) Allergen-specific serum IgA responses. (E) Allergen-specific serum IgG subclass responses. (F-I) Gut innate responses before sensitization. (F) Expression of NRF2. Sections of small intestines were stained with anti-NRF2 antibody and counterstained with DAPI to visualize nuclei. (G) Cytokine mRNA responses in intestinal tissues. (H and I) Expression of PGE2. (H) Sections of small intestines were stained with anti-PGE2 antibody and DAPI. (I) Quantification of PGE2 levels in small intestinal tissues by ELISA. Data are expressed as mean ± SD. *p < 0.05; **p < 0.01 and represent one of at least four independent experiments with five mice per group. Immunofluorescence images are representative of five mice per group.
Chronic ingestion of subtoxic doses of Cd alters the gut microbiome
Increased intestinal permeability is known to facilitate allergic sensitization. We found no difference in the absorption of orally administrated FITC-dextran by gut tissues of control and Cd-treated mice either at the basal level (groups no CT, Figure 4A). Furthermore, Cd-treated mice did not show higher intestinal permeability than control mice after oral administration of the adjuvant cholera toxin (CT) (group CT 16h, Figure 4A). These results indicate that the ingestion of Cd at these levels does not increase intestinal permeability. Although Cd has no known nutritional role in bacterial growth, it may interfere with divalent cationic metal ions that limit the growth of certain bacteria. Microbial colonization is the single most important event that controls intestinal secretory IgA (SIgA) levels (19, 20), and reduction of SIgA levels is a potential marker of dysbiosis (21, 22). Intestinal SIgA levels were reduced after 2 weeks of ingestion of subtoxic doses of Cd and remained low after 28 days of exposure (Figure 4B). Proteomic analyses of intestinal contents showed that mice treated with low doses of Cd (Cd10) expressed higher levels of the antimicrobial peptide regenerating islet-derived protein 3 beta (Figure S3A). Other luminal proteins increased after Cd10 ingestion, including ferritin, laminin, and histone (Figures 4C and S3A). In contrast, Cd exposure reduced overall levels of mucins, kallikrein-1, and proteins associated with the digestive function of epithelial cells (e.g., aminopeptidase, alkaline phosphatase, sucrose isomaltase) (Figures 4C and S3A and S3B). Proteomics data also showed that Cd stimulates antimicrobial responses and impairs the function of goblet cells. Reduced levels of mucus may facilitate sensitization, since mucus has been reported to deliver regulatory signals that enhance oral tolerance (23). Kallikrein-1 cleaves kininogens into kinins that enhance the permeability of capillaries and epithelial cell layers (24). Thus, reduced levels of kallikrein-1 in fecal samples of mice exposed to Cd support the conclusions our FITC-dextran absorption studies (Figure 4A), that intestinal permeability was not increased in these mice.
Figure 4. Chronic ingestion of subtoxic doses of Cd alters the gut microbiome, and luminal metabolite and proteomic profiles.
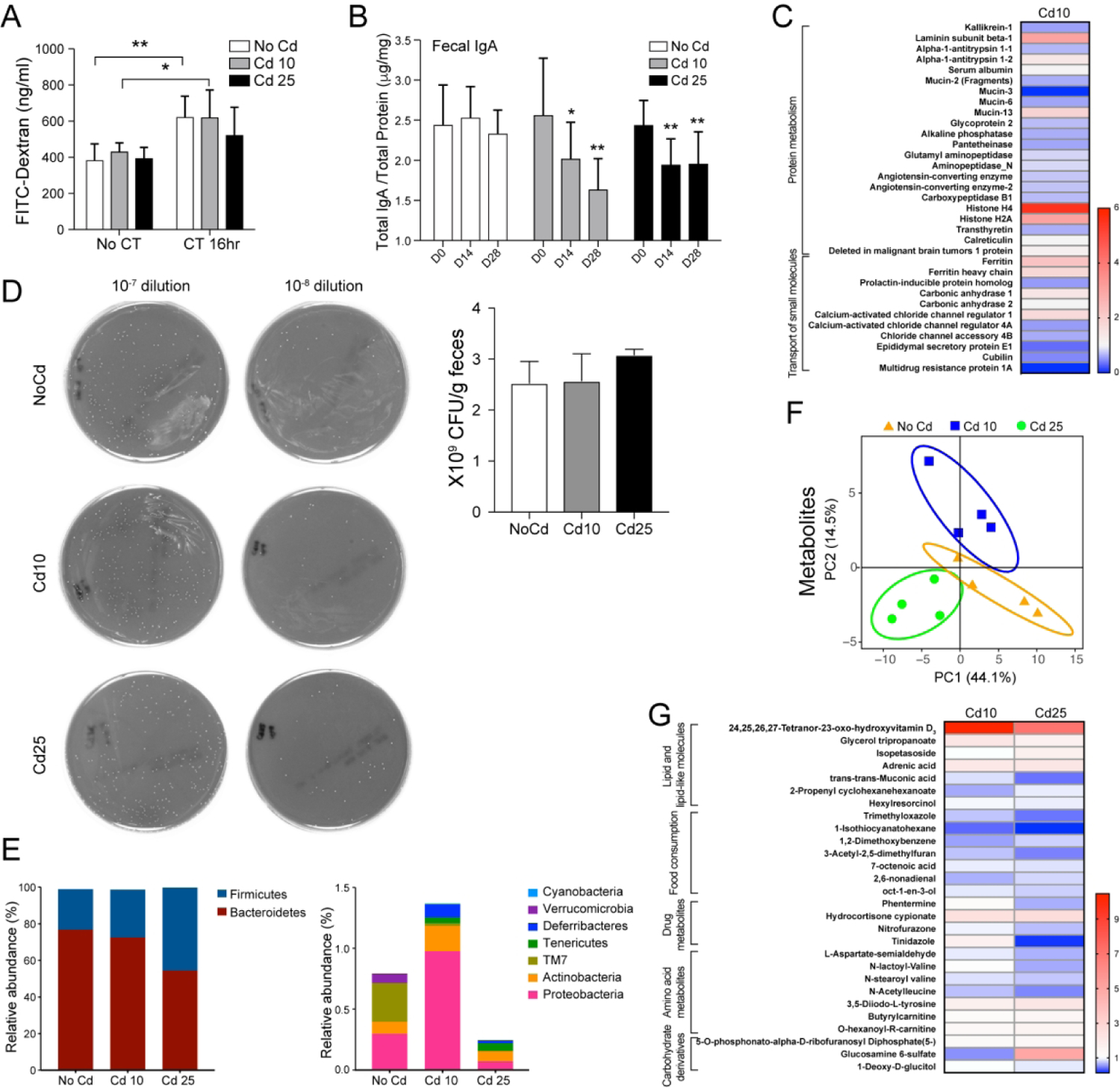
Mice (n=5 per group) were provided CdCl2 [10 μM (Cd10) or 25 μM (Cd25)] in drinking water for 28 days. (A) Intestinal permeability was analyzed by measuring serum FITC-dextran (4 kDa). (B) Fecal IgA levels. Total IgA levels in freshly emitted feces were analyzed by ELISA. The amounts of IgA were normalized by total protein content. (C) Relative proteins expression in mice treated with Cd10 vs. NoCd mice. (D) Bacterial load in fecal extracts. Dilution of fecal pellets were normalized by mass (g) of feces and plated on blood agar, and bacteria (CFU) were counted after 48 h of culture. (E) Relative abundance of Firmicutes to Bacteroidetes (upper) and other less dominant phyla (lower). (F and G) Metabolites in fecal extracts. (F) Principal component analysis of metabolite profiles. (G) Heat map of relative expression of metabolites with significantly different concentrations in samples from Cd-treated mice vs. NoCd mice (*p < 0.05). Data are expressed as the mean ± SD. *p < 0.05; **p < 0.01 (4–5 mice per group).
Ingestion of Cd did not affect the number of cultivable bacteria in the gastrointestinal tract (Figure 4D) but did induce dysbiosis (Figure 4E). The relative abundance of gut microbiota (Figures 4E and S3C and S3D) and a linear discriminant analyses (LDA) (Figure S3E) showed that Cd most significantly reduced bacteria of the genus Prevotella, whereas bacteria in genera Tannerella and Turicibacter increased the most in mice exposed to Cd10 and Cd25, respectively.
Chronic ingestion of subtoxic doses of Cd alters metabolite composition and protein expression in the gut
Consistent with the notion that dysbiosis is generally associated with changes in the composition of metabolites in the gut, Cd-treated mice exhibited profiles of metabolites that differed from control mice (Figures 4F and G). Statistical differences in metabolite composition between NoCd mice and at least one of the Cd-treated groups are depicted in Figure 4G. In general, samples from Cd-treated mice exhibited reduced levels of metabolites related to food consumption and amino acid metabolism. A main feature of Cd-treated mice was a major increase in the levels of lipids and lipid-like molecules (Figure 4G), principally, the vitamin D3 metabolite 24,25,26,27-tetranor-23-oxo-hydroxyvitamin D3 (Figure 4G and Table S2). This finding was of high significance, since mice exposed to Cd10, which contain the highest levels of 24,25,26,27-tetranor-23-oxo-hydroxyvitamin D3 (oxo-VD3) in the gut, were also those that exhibited the highest IgE responses upon oral sensitization (Figure 2B). In this regard, vitamin D3 metabolites were shown to be associated with increased production of PGE2 (25), and PGE2 promotes IgE production (17, 18).
IgE-enhancing effects of Cd can be transferred by fecal transplantation
To address the role of Cd-induced luminal metabolites/proteins and/or bacteria in increasing allergic sensitization, fecal materials from Cd-treated mice were transferred to groups of SPF mice, which were subsequently orally sensitized by the administration of OVA and cholera toxin as adjuvant (Figure 5A). OVA-specific serum IgE responses (Figure 5B), but not OVA-specific IgG subclasses or IgA (Figure 5C and S4A), were enhanced in recipients of fecal material transplantation (FMT) from mice exposed to Cd. Furthermore, subsequent nasal allergen challenge resulted in higher lung inflammatory responses and mucus production in recipients of FMT from mice chronically exposed to Cd (Figure S4B). In the reverse experiment (Figure S4C–H), fecal material from control untreated mice reduced allergen-specific IgE responses in recipient Cd-treated mice (Figure S4D) but had no effect on the other Ig isotypes (Figure S4E). In addition, Cd-treated mice, which received fecal material from untreated mice, developed fewer signs of allergy after nasal allergen challenge, as manifested by a less significant drop in body temperature (Figure S4F) and lower numbers of IFNγ+CD4+T and IL-4+CD4+T cells in the BALF (Figures S4G and H). Taken together, these results show that fecal materials of Cd-treated mice contain factors that regulate IgE responses.
Figure 5. Cd-induced gut microenvironment enhances IgE responses via stimulation of PGE2.
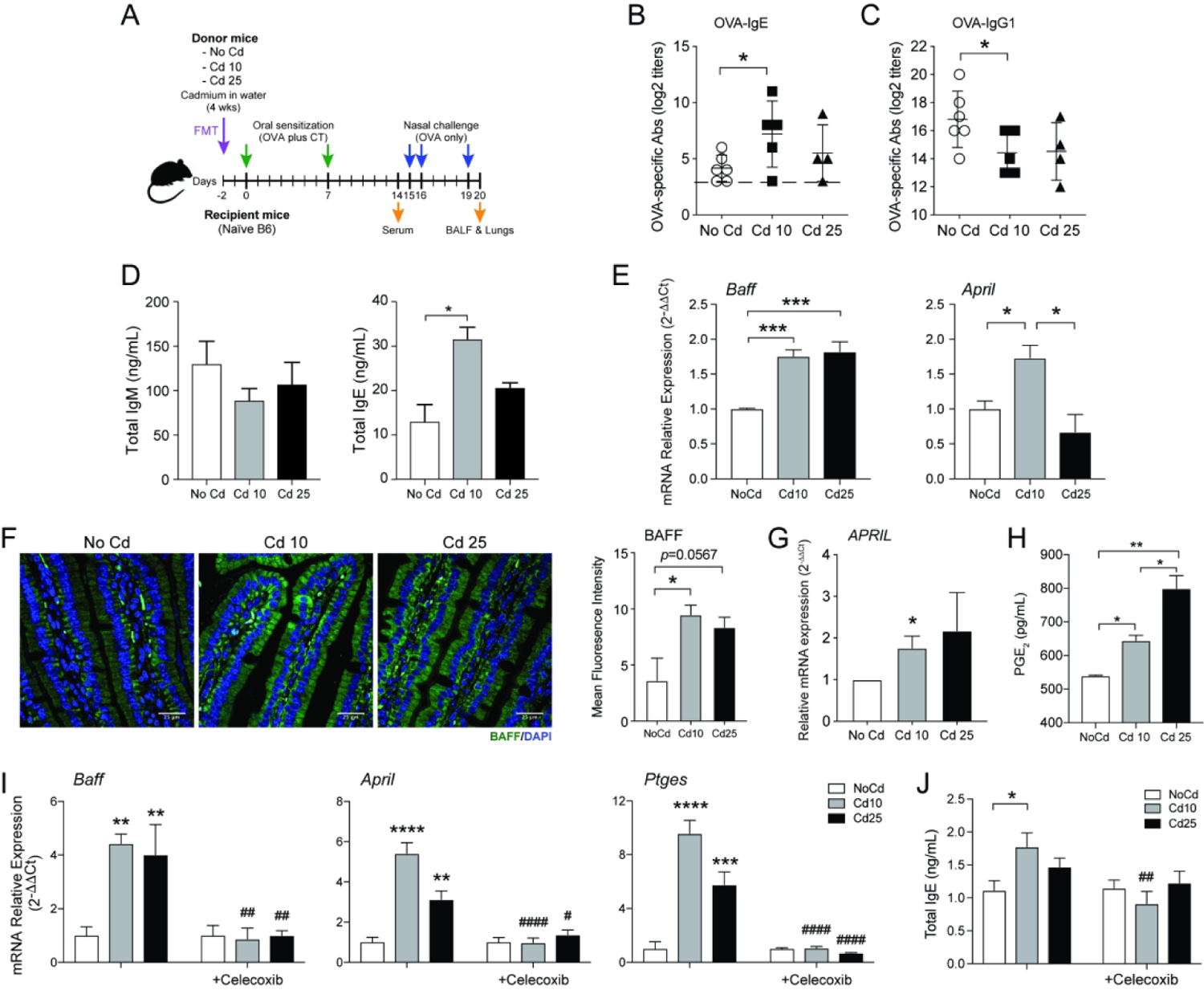
(A) Experimental scheme for the transfer of fecal materials from Cd-treated mice. Allergen-specific serum IgE (B) and IgG1 (C) responses after oral sensitization of recipient mice not exposed to Cd. (D) IgM and IgE production in culture supernatants of naïve murine spleen cells cultured for 4 days in the presence of IL-4 (10 ng/ml), anti-CD40 (1μg/ml), and bacteria-free fecal extracts (n = 4). (E) Baff and April mRNA responses in Myd88 KO macrophages cultured 24 h in the presence of bacteria-free fecal extracts (n=5). (F) Expression of BAFF in small intestinal tissues of naïve mice or mice exposed to subtoxic doses of Cd for 28 days (n=5). (G) PGE2 secretion from HT-29 cells 4 h after the addition of bacteria-free fecal extracts (n=5). (H) Baff, April, and Ptges mRNA responses by MLN lymphocytes (2×106 cells/mL) stimulated for 24 h with anti-CD40 and IL-4 in the presence of bacteria-free fecal extracts only, or together with the COX2 inhibitor celecoxib (10 μM) (n=3). (I) Effects of celecoxib on IgE production by MLN lymphocytes stimulated as described in (H) (n=3). Data are from at least four independent experiments and are expressed as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 compared to NoCd. ## p < 0.01; ##p < 0.001; ####p < 0.0001 compared to no celecoxib.
The Cd-induced gut microenvironment enhances IgE responses via stimulation of PGE2
The FMT experiments described above did not elucidate whether bacteria, bacteria product, or host molecule mediated the IgE-promoting effects. To elucidate the mechanism(s) driving IgE responses in Cd-treated mice, bacteria-free fecal extracts were added to spleen cells stimulated in vitro with anti-CD40 in the presence of IL-4, and secretion of IgE was assessed in cell culture supernatants. Cells cultured in the presence of fecal extracts from Cd-treated mice produced more IgE than cells exposed to fecal extracts from mice that did not receive Cd (Figure 5D). This effect was unlikely due to cytokines which were either unchanged or reduced (IL-6 and GM-CSF) in the fecal extracts from Cd-treated mice (Figure S5A). Fecal extracts from Cd-treated mice increased the mRNA levels of April and Baff members of the TNF family that support Ig class switching (26–28), in macrophages from Myd88 KO mice (Figure 5E) and enhanced APRIL and C4BP mRNA responses in human epithelial HT-29 cells (Figure S5B). These findings indicate that the luminal contents of Cd-treated mice stimulate APRIL/BAFF expression via molecules distinct from microbial PAMPs and can induce similar responses in human epithelial cells. Finally, BAFF expression was upregulated in vivo in small intestinal tissues of naïve mice exposed for 28 days to subtoxic doses of Cd (Figure 5F).
Gut expansion of fungi was reported to stimulate PGE2 and promote allergic inflammation (29) and PGE2 supports the production of IgE but not IgG1 (18). Control and Cd-treated mice had similar levels of PGE2 in their feces (Figure S5C), and showed no evidence of Candida overgrowth (Figure S5D). On the other hand, bacteria-free fecal extracts from Cd-treated mice enhanced PGE2 production in both human HT-29 cells (Figure 5G) and in LPS-unresponsive Trif/Tram double-KO murine macrophages (Figure S5E). To determine the link between PGE2, BAFF/APRIL, and IgE production, we added the specific COX2 inhibitor celecoxib to cultures of spleen and mesenteric lymph node cells (MLN) stimulated with anti-CD40 in the presence of IL-4 and bacteria-free fecal extracts from Cd-treated mice. Celecoxib suppressed the transcription of PGE2 synthase (Ptges), Baff, and April (Figures 5H, S5F and S5G) and the production of IgE by these cells (Figure 5I). Together, these results indicate that a product(s) found in fecal extracts stimulated the production of PGE2 in vivo by intestinal epithelial cells and myeloid cells, and that inhibition of PGE2 response by COX-2 inhibitor limits the production of BAFF and APRIL and associated high IgE responses
Host vitamin D3-metabolizing enzymes play key roles in the Cd-mediated increase in IgE responses
The main feature of fecal samples obtained from Cd-treated mice was the presence of high levels of oxo-VD3, an oxidized vitamin D3 metabolite. Oxidized vitamin D3 metabolites can be induced by oxidation of active vitamin D3 by 25-hydroxyvitamin D3 1-alpha-hydroxylase (CYP27B1) and the steroid 21-hydroxylase (CYP24A1) (Figure S5H) (30, 31). Interestingly, Cyp27b1 mRNA levels were significantly enhanced in the intestines of mice chronically exposed to subtoxic doses of Cd (Figure 6A). An inverse relationship has been reported between vitamin D3 receptor (VDR) signaling and PGE2 production (32). Consistent with the fact that oxo-VD3 is not a ligand of the VDR, and that expression of VDR is stimulated by its ligands (32), the addition of bacteria-free fecal extracts from Cd-treated mice to J774 macrophages cultures reduced Vdr mRNA levels (Figure S5I).
Figure 6. Vitamin D3-metabolizing enzymes and oxidized vitamin D3 metabolites regulate IgE production in mice exposed to Cd.
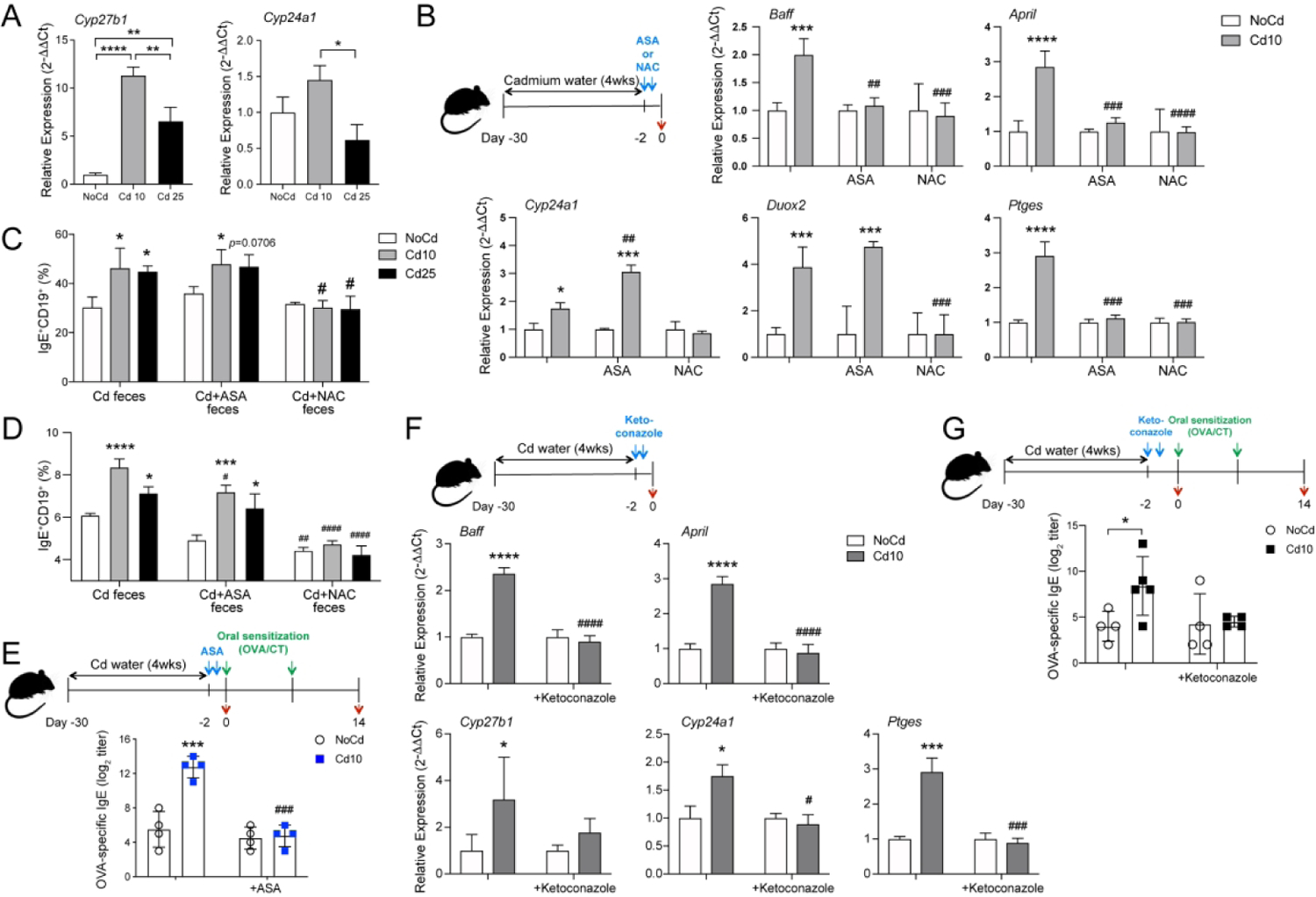
(A) Cyp27b1 and Cyp24a1 mRNA responses in small intestines from control mice or mice exposed to Cd for 28 days (n=5 per group). (B) COX2 inhibitor (ASA; aspirin)- or antioxidant (NAC; N-acetyl cysteine)-mediated regulation of cytokine (Baff and April) and vitamin D3-metabolizing enzyme (Cyp24a1) mRNAs in mice exposed to subtoxic doses of Cd. Ptges and Duox2 mRNAs were measured to confirm inhibitor specificity. (C and D) Numbers of IgE+CD19+ cells in cultures of cells from spleen (C) and mesenteric lymph node (D) from IgE Verigem mice stimulated in vitro with anti-CD40 and IL-4 in the presence of bacteria-free fecal extracts from mice exposed to Cd, mice exposed to Cd and treated with a COX2 inhibitor (Cd+ASA), or mice exposed to Cd and treated with an antioxidant (Cd+NAC). (E) Antigen-specific IgE responses in Cd-treated mice treated with a COX2 inhibitor before allergic sensitization (n=4). (F) In vivo regulation of Baff, April, and Ptges mRNA responses in mice exposed to subtoxic doses of Cd by ketoconazole, an inhibitor of vitamin D3 metabolism (n=5). (G) Antigen-specific IgE responses in Cd-treated mice treated with an inhibitor of vitamin D3-metabolizing enzymes. Data are from at least four independent experiments and are expressed as mean ± SD. *p < 0.05; **p < 0.01 compared to control NoCd mice. ##P < 0.001 compared to Cd alone.
To determine the sequence of events that led to the increase in IgE responses in mice exposed to Cd, control and Cd-treated mice were orally treated with inhibitors of oxidative stress (N-acetyl cysteine, NAC) or PGE2 (the COX2 inhibitor aspirin, ASA). Treatment with NAC reduced the mRNA levels of Ptges, Baff, April, and Cyp24a1 in Cd-treated mice (Figure 6B). The COX2 inhibitor downregulated the mRNA levels of Ptges, Baff, and April in the intestines of Cd-treated mice but failed to reduce the expression of Cyp24a1 (Figure 6B). We next used in vitro cultures of spleen, mesenteric lymph node or Peyer’s patch cells from IgE Veregem mice to determine whether treatment with these inhibitors affect the IgE-promoting effects of the gut luminal microenvironment of Cd-treated mice. As depicted in Figure 6C, NAC completely suppressed the IgE-promoting effects of fecal materials from Cd-treated mice while Cd-treated mice that had been administered ASA orally retained their IgE-promoting capabilities in vitro. These findings indicate that products of the vitamin D3-metabolizing enzymes that were present in the fecal materials of mice given ASA drove IgE responses. Furthermore, oral sensitization with OVA in the presence of cholera toxin induced the same levels of IgE in control and Cd-exposed mice that were treated with aspirin prior to sensitization (Figure 6E). These results demonstrate that PGE2 is a central check point, downstream of vitamin D3-metabolizing enzymes, in the increase in IgE responses in vivo in hosts exposed to subtoxic doses of Cd.
To conclusively demonstrate the role of vitamin D3-metabolizing enzymes, mice exposed to Cd were treated orally with ketoconazole, a pharmacological inhibitor of CYP27B1 and CYP24A1 (Figure 6F). This treatment suppressed the mRNA expression of Cyp27b1, Cyp24a1, Ptges, Baff, and April in intestinal tissues of Cd-treated mice (Figure 6F). It also reduced the levels of allergen-specific IgE induced after oral sensitization to levels seen in control mice that were not exposed to Cd (Figure 6G). These results show that vitamin D3-metabolizing enzymes are previously unknown regulators of allergic sensitization in hosts chronically exposed to low, but environmentally relevant, doses of this heavy metal pollutant (Figure 7).
Figure 7. Mechanisms of enhanced IgE sensitization by ingested cadmium.
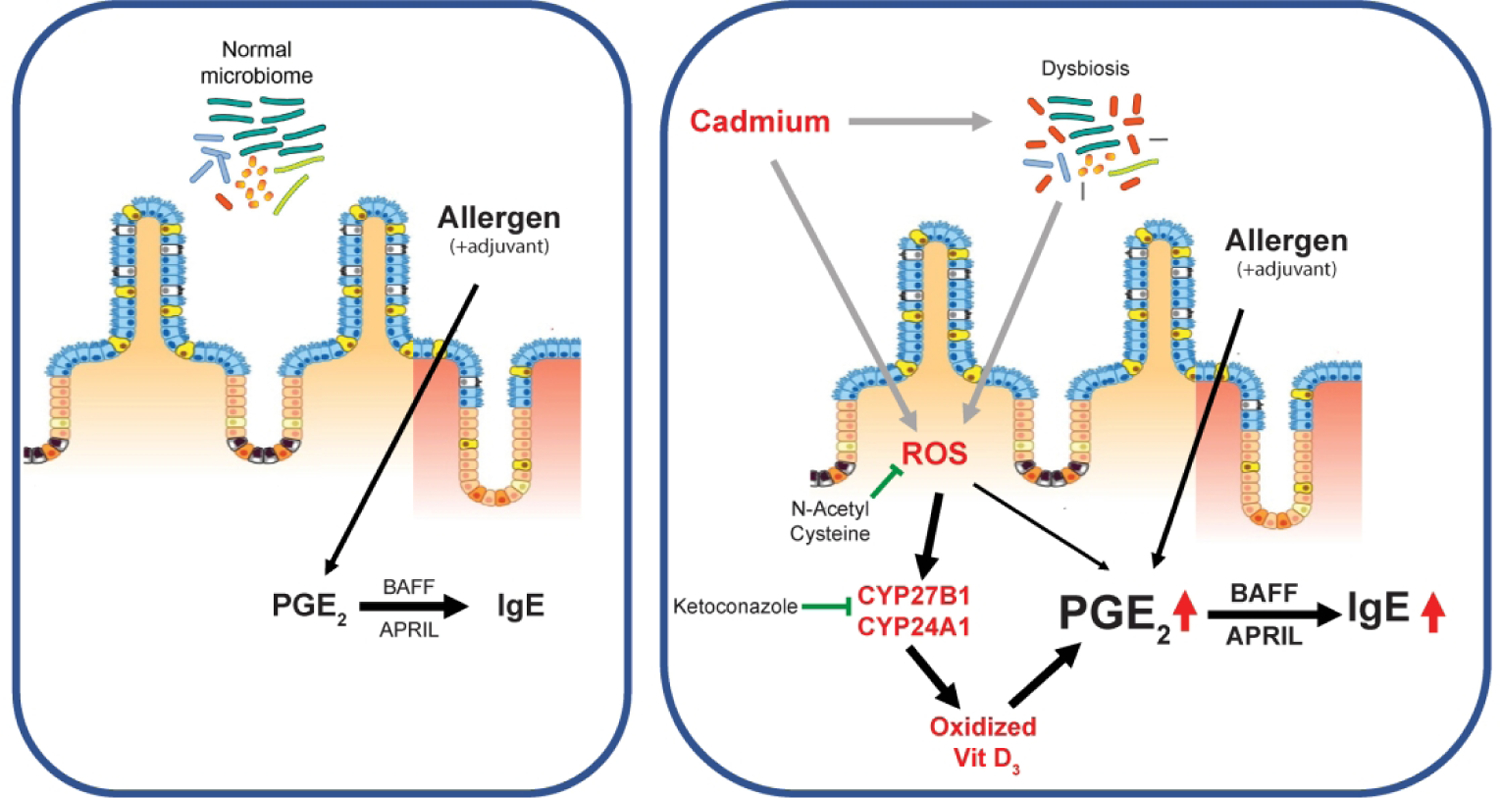
Chronic ingestion of subtoxic doses of Cd stimulate expression of the Vitamin D metabolizing enzymes CYP27B1 and CYP24A1 leading to increased levels of oxidized Vit D3 metabolites. This results in enhanced PGE2 responses to allergic sensitization and higher IgE responses.
DISCUSSION
Extensive use of antibiotics early in life is often mentioned as a leading factor driving the increased prevalence of allergic diseases. But other factors including diet and environmental pollutants are also believed to play roles in steady increase of allergic diseases worldwide (3, 4, 33). Results summarized herein reveal a previously unknown mechanism for increased allergic sensitization in the gut and possibly other mucosal tissues. More specifically, using chronic exposure to subtoxic dose of Cd as an experimental system that induces oxidative responses, we have identified the induction of vitamin D3-metabolizing enzymes in gut tissues and the subsequent increased luminal levels of vitamin D3-oxidized metabolites which are not ligands of the VDR as central mechanisms for selective enhancement of IgE responses.
Production of IgE is initiated by IL-4 and IL-13, which provide the signals for the transcription of the IgE (ε) germline (34). Engagement of CD40 on the B cell surface by CD40L expressed on activated T cells further enables Ig isotype switching to IgE and subsequent IgE production. In vitro studies have shown that exposure of B cells to low doses of Cd (0.1 μM) inhibits IgE production (35). We show that oral allergen sensitization of mice exposed to environmentally relevant doses of Cd actually increased IgE responses. Cd-treated mice also had increases allergen-specific Th1 and Th17 cells, which further enhance histological and clinical signs of allergy after allergen challenge. It was reported that splenocytes of the offspring from C57BL/6 mice exposed to 10 ppm Cd, produced lower amounts of IFNγ upon in vitro stimulation than cells from control mice (36). Nonetheless, upon vaccination, these offspring developed higher levels of both T-dependent and -independent serum antibodies compared to control animals (36). Although, other Ig isotypes can contribute to allergic responses, in our study, Cd did not alter IgG1 or IgA responses. This suggests that IgE-promoting signals are selectively targeted/stimulated by this heavy metal in the gastrointestinal tract.
It is important that Cd did not alter intestinal barrier function or increased intestinal permeability as indicated by absorption of FITC-dextran and reduced levels of kallikrein-1in the intestinal contents of mice exposed to Cd. On the other hand, subtoxic doses of Cd induced oxidative stress and increased the levels of the oxidative stress-sensing transcription factor NRF2 in the small intestinal tissues. These oxidative stress responses are associated with the stimulation of inflammatory cytokines and PGE2 production. These findings are consistent with the reported ability of Cd to induce oxidative stress (12), and that oral exposure to high doses of Cd through drinking water in early life promotes systemic inflammation and subsequent fat accumulation in male mice (37). Interestingly, the effects of Cd on oxidative stress, inflammatory responses, and PGE2 were not observed in germ-free mice. Accordingly, exposure to Cd failed to enhance the IgE responses in germ-free mice. In this regard, Duox2 expression in the intestine was shown to depend on microbial colonization (38, 39) and to be partially induced by TRIF and canonical NF-κB signaling (39). The cationic metal manganese was recently shown to increase the sensitivity of cGAS to dsDNA and STING activation (40). Thus, Cd, which is also a cationic metal that can accumulate in the cytosol, likely enhanced the reactivity of intestinal epithelial cells to TLR ligands from commensal microbes.
It was previously reported that PGE2 produced by Candida overgrowth in the gut tissues of mice treated with antibiotics increases allergic airway inflammation via the induction of M2 macrophages (29). The low doses of Cd used in our study did not affect the bacterial load, or result in Candida overgrowth or PGE2 secretion in fecal materials. On the other hand, overall fecal IgA levels were reduced by Cd ingestion. The two main events that regulate the levels of fecal IgA are the bacterial load in the intestine and the composition of the microbiome (19, 21, 22). Accordingly, Cd-induced dysbiosis was characterized by increased levels of the genera Tannerella and Turicibacter in mice exposed to Cd10 and Cd25, respectively. Our findings are consistent with previous reports that oral exposure to 100 ppm of Cd reduces the proportion of Bifibacterium and Lactobacillus (11) and increases the numbers of Lactobacillaceae and Erysipelotrichaceae, especially Turicibacter in the Erysipelotrichaceae family (41). However, it is unlikely that these changes alone could selectively enhance IgE responses.
The most striking and unexpected findings from this work were that Cd (a) stimulates the expression of the vitamin D3-metabolizing enzymes CYP27B1 and CYP24A1 in the gut and (b) enhances the levels of oxidized vitamin D3 metabolites, that are not ligands of the VDR. Since bacteria-free fecal extracts from Cd-treated mice downregulate Vdr mRNA expression by macrophage, collectively our data suggest that exposure to subtoxic doses of Cd mimics vitamin D deficiency. In this regard, epidemiologic data and animal studies have linked vitamin D deficiency to atopy, asthma, and food allergy (42–44) and vitamin D is believed to protect against allergic diseases, via inhibition of T cell proliferation, IFNγ and Th17 responses (45), and ILC2 (46), or stimulation of Treg (47). Vitamin D supplementation has been investigated as a strategy to mitigate or prevent allergic diseases. The timing and dose of Vitamin D used for supplementation were found to be crucial. Thus, vitamin D only protected when it was given before the allergy develops (i.e., before or early after birth) (42). Furthermore, high-dose vitamin D supplementation failed to prevent allergic sensitization of infants (48).
Our study suggests that activation of vitamin D-metabolizing enzymes is perhaps the missing link in our understanding of allergic sensitization and the efficacy of vitamin D supplementation therapy. In fact, we show that conditions of vitamin D deficiency and enhanced allergic sensitization can be induced in vitamin D-sufficient hosts by the activation of vitamin D-metabolizing enzymes and thus, depletion of functional vitamin D. The fact that these enzymes are inducible and that their expression is regulated by the concentration of vitamin D could explain the reported failure of high-dose vitamin D supplementation to prevent allergic sensitization (48). It is also important to note that Cd-induced oxidative stress responses were the main drivers of CYP27B1 and CYP24A1 expression in gut tissues. Diesel exhaust particles, which contain heavy metals and induce oxidative stress responses in the airways (49), are known to promote Th2-mediated allergic asthma (50, 51). Thus, induction CYP27B1 and CYP24A1 could represent a common mechanism to increase of allergic sensitization after oxidative stress responses in mucosal tissues. Taken together, our findings reveal a new mechanism for the promotion of IgE responses by environmental pollutants (Figure 7). These insights also have important implications for the prevention and treatment of allergic diseases since they identify CYP27B1 and CYP24A1 as potential targets for the prevention of allergic sensitization.
MATERIALS AND METHODS
Mice.
Specific pathogen-free (SPF) C57BL/6 mice (Jackson Laboratory (Bar Harbor, ME) were maintained at the Ohio State University animal care facility. Germ-free C57BL/6 mice were obtained by cesarean derivation and maintained in sterile isolators. The Verigem IgE reporter (IgE Verigem) mice were obtained from Dr. Christopher Allen (University of California at San Francisco). All animal experiments were approved by the OSU Animal Care and Use Committee.
Exposure of mice to subtoxic doses of Cd in vivo.
Mice aged 8–12 weeks received cadmium chloride (CdCl2, MW = 183.3; Sigma-Aldrich, St. Louis, MO) in drinking water for 4 weeks. Cd was given at the environmentally relevant doses of 10 μM (Cd10) or 25 μM (Cd25) (equivalent to 2 or 5 ppm (μg/L), based on previous studies (10).
Allergen sensitization and allergen challenge.
Mice were sensitized orally on days 0 and 7 by intragastric gavage of 1 mg of ovalbumin (OVA) and 15 μg cholera toxin in 250 μL of phosphate-buffered saline (PBS). Blood samples were collected on day 14 for analysis of serum IgE and other immunoglobulin isotypes. Nasal antigen challenges (200 μg of OVA in PBS 100 μL) were performed on days 15, 16, and 19 on mice anesthetized by intraperitoneal injection of ketamine/xylazine. The hypothermia associated with allergic responses was measured on the skin with a digital thermometer (Heat Spy infrared thermal imaging camera, Wahl, Culver City, CA).
In vivo treatment with pharmacological inhibitors of specific pathways.
To address the role of oxidative stress, PGE2 and Vitamin D3-metabolizing enzymes, mice received orally by intragastic gavage 250 uL of saline containing 250 mg/kg of N-acetyl L-cysteine, 25 mg/kg of aspirin, or 10 mg/kg of ketoconazole (Sigma-Aldritch, Saint Louis, MO).
Statistical analyses.
Results are expressed as mean ± SD. Statistical significance was determined by one- or two-way ANOVA, followed by the Tukey’s multiple range test. All statistical analyses were performed with the StataSE 12.0 software (StataCorp LLC, College Station, TX) and Prism 7 software (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
This work was supported by NIH grants AI18958, DK101323, and R01AI145144, and the UL1TR001070 award from the National Center for Advancing Translational Sciences. The authors thank Arpad Somogyi and Matthew Bernier of the OSU Center for Clinical and Translational Science Mass Spectrometry & Proteomics Core for assistance with metabolomics and proteomics studies. The Mass Spectrometry & Proteomics Core was supported by NIH grant P30 CA016058 and the Fusion Orbitrap instrument was supported by NIH grant S10 OD018056.
Footnotes
Conflict of interest: The authors declare no conflict of interest
Additional methods: are in the Supplemental Information which includes 2 tables and 5 figures.
References
- 1.Hong X, Tsai HJ, and Wang X. Genetics of food allergy. Curr Opin Pediatr. 2009;21(6):770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu AH. Revisiting the hygiene hypothesis for allergy and asthma. J Allergy Clin Immunol. 2015;136(4):860–5. [DOI] [PubMed] [Google Scholar]
- 3.Wesemann DR, and Nagler CR. The Microbiome, Timing, and Barrier Function in the Context of Allergic Disease. Immunity. 2016;44(4):728–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park S, Lee EH, and Kho Y. The association of asthma, total IgE, and blood lead and cadmium levels. J Allergy Clin Immunol. 2016;138(6):1701–3 e6. [DOI] [PubMed] [Google Scholar]
- 5.Yang SN, Hsieh CC, Kuo HF, Lee MS, Huang MY, Kuo CH, et al. The effects of environmental toxins on allergic inflammation. Allergy Asthma Immunol Res. 2014;6(6):478–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faroon O, Ashizawa A, Wright S, Tucker P, Jenkins K, Ingerman L, et al. Toxicological Profile for Cadmium. Atlanta (GA); 2012. [PubMed] [Google Scholar]
- 7.Jarup L, and Akesson A. Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol. 2009;238(3):201–8. [DOI] [PubMed] [Google Scholar]
- 8.Wei B, and Yang L. A review of heavy metal contamination in urban roads dusts and agricultural soils from China. Microchemical Journal. 2010;94(2):99–107. [Google Scholar]
- 9.Wang S, Huang DY, Zhu QH, Zhu HH, Liu SL, Luo ZC, et al. Speciation and phytoavailability of cadmium in soil treated with cadmium-contaminated rice straw. Environ Sci Pollut Res Int. 2015;22(4):2679–86. [DOI] [PubMed] [Google Scholar]
- 10.Kundu S, Sengupta S, Chatterjee S, Mitra S, and Bhattacharyya A. Cadmium induces lung inflammation independent of lung cell proliferation: a molecular approach. J Inflamm (Lond). 2009;6:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Li Y, Liu K, and Shen J. Exposing to cadmium stress cause profound toxic effect on microbiota of the mice intestinal tract. PLoS One. 2014;9(2):e85323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu KC, Liu JJ, and Klaassen CD. Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol Appl Pharmacol. 2012;263(1):14–20. [DOI] [PubMed] [Google Scholar]
- 13.Hogervorst J, Plusquin M, Vangronsveld J, Nawrot T, Cuypers A, Van Hecke E, et al. House dust as possible route of environmental exposure to cadmium and lead in the adult general population. Environ Res. 2007;103(1):30–7. [DOI] [PubMed] [Google Scholar]
- 14.Nawrot T, Plusquin M, Hogervorst J, Roels HA, Celis H, Thijs L, et al. Environmental exposure to cadmium and risk of cancer: a prospective population-based study. Lancet Oncol. 2006;7(2):119–26. [DOI] [PubMed] [Google Scholar]
- 15.Ke S, Cheng XY, Zhang JY, Jia WJ, Li H, Luo HF, et al. Estimation of the benchmark dose of urinary cadmium as the reference level for renal dysfunction: a large sample study in five cadmium polluted areas in China. BMC Public Health. 2015;15:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu YP, Peng YB, Zhang YF, Wang Y, Yu WR, Yao M, et al. Reactive Oxygen Species Mediated Prostaglandin E2 Contributes to Acute Response of Epithelial Injury. Oxid Med Cell Longev. 2017;2017:4123854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao Y, Zhao C, Wang W, Jin R, Li Q, Ge Q, et al. Prostaglandins E2 signal mediated by receptor subtype EP2 promotes IgE production in vivo and contributes to asthma development. Sci Rep. 2016;6:20505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuroda E, Ishii KJ, Uematsu S, Ohata K, Coban C, Akira S, et al. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity. 2011;34(4):514–26. [DOI] [PubMed] [Google Scholar]
- 19.Hapfelmeier S, Lawson MA, Slack E, Kirundi JK, Stoel M, Heikenwalder M, et al. Reversible microbial colonization of germ-free mice reveals the dynamics of IgA immune responses. Science. 2010;328(5986):1705–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, et al. Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature. 2016;534(7606):263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu W, Sun M, Chen F, Cao AT, Liu H, Zhao Y, et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2017;10(4):946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang XY, Liu ZM, Zhang HF, Li YS, Wen SH, Shen JT, et al. TGF-beta1 improves mucosal IgA dysfunction and dysbiosis following intestinal ischaemia-reperfusion in mice. J Cell Mol Med. 2016;20(6):1014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. 2013;342(6157):447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Mello RJ, Caldwell JM, Azouz NP, Wen T, Sherrill JD, Hogan SP, et al. LRRC31 is induced by IL-13 and regulates kallikrein expression and barrier function in the esophageal epithelium. Mucosal Immunol. 2016;9(3):744–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones SA, Schneider H, and Challis JR. Vitamin D3 metabolites stimulate prostaglandin production by human fetal membranes and placenta in vitro. Am J Perinatol. 1989;6(2):138–41. [DOI] [PubMed] [Google Scholar]
- 26.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, et al. TACI and BAFF-R mediate isotype switching in B cells. The Journal of experimental medicine. 2005;201(1):35–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castigli E, Scott S, Dedeoglu F, Bryce P, Jabara H, Bhan AK, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A. 2004;101(11):3903–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11(9):836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim YG, Udayanga KG, Totsuka N, Weinberg JB, Nunez G, and Shibuya A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell host & microbe. 2014;15(1):95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs ET, Van Pelt C, Forster RE, Zaidi W, Hibler EA, Galligan MA, et al. CYP24A1 and CYP27B1 polymorphisms modulate vitamin D metabolism in colon cancer cells. Cancer Res. 2013;73(8):2563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kundu R, Chain BM, Coussens AK, Khoo B, and Noursadeghi M. Regulation of CYP27B1 and CYP24A1 hydroxylases limits cell-autonomous activation of vitamin D in dendritic cells. Eur J Immunol. 2014;44(6):1781–90. [DOI] [PubMed] [Google Scholar]
- 32.Liu X, Nelson A, Wang X, Farid M, Gunji Y, Ikari J, et al. Vitamin D modulates prostaglandin E2 synthesis and degradation in human lung fibroblasts. Am J Respir Cell Mol Biol. 2014;50(1):40–50. [DOI] [PubMed] [Google Scholar]
- 33.Greer FR, Sicherer SH, Burks AW, American Academy of Pediatrics Committee on N, American Academy of Pediatrics Section on A, and Immunology. Effects of early nutritional interventions on the development of atopic disease in infants and children: the role of maternal dietary restriction, breastfeeding, timing of introduction of complementary foods, and hydrolyzed formulas. Pediatrics. 2008;121(1):183–91. [DOI] [PubMed] [Google Scholar]
- 34.Geha RS, Jabara HH, and Brodeur SR. The regulation of immunoglobulin E class-switch recombination. Nat Rev Immunol. 2003;3(9):721–32. [DOI] [PubMed] [Google Scholar]
- 35.Jelovcan S, Gutschi A, Kleinhappl B, Sedlmayr P, Barth S, and Marth E. Effects of low concentrations of cadmium on immunoglobulin E production by human B lymphocytes in vitro. Toxicology. 2003;188(1):35–48. [DOI] [PubMed] [Google Scholar]
- 36.Hanson ML, Holaskova I, Elliott M, Brundage KM, Schafer R, and Barnett JB. Prenatal cadmium exposure alters postnatal immune cell development and function. Toxicol Appl Pharmacol. 2012;261(2):196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ba Q, Li M, Chen P, Huang C, Duan X, Lu L, et al. Gender-Dependent Effects of Cadmium Exposure in Early Life on Gut Microbiota and Fat Accumulation in Mice. Environ Health Perspect. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grasberger H, Gao J, Nagao-Kitamoto H, Kitamoto S, Zhang M, Kamada N, et al. Increased Expression of DUOX2 Is an Epithelial Response to Mucosal Dysbiosis Required for Immune Homeostasis in Mouse Intestine. Gastroenterology. 2015;149(7):1849–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sommer F, and Backhed F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 2015;8(2):372–9. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Guan Y, Lv M, Zhang R, Guo Z, Wei X, et al. Manganese Increases the Sensitivity of the cGAS-STING Pathway for Double-Stranded DNA and Is Required for the Host Defense against DNA Viruses. Immunity. 2018;48(4):675–87 e7. [DOI] [PubMed] [Google Scholar]
- 41.Breton J, Massart S, Vandamme P, De Brandt E, Pot B, and Foligne B. Ecotoxicology inside the gut: impact of heavy metals on the mouse microbiome. BMC pharmacology & toxicology. 2013;14:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolcas PE, Brandt EB, Zhang Z, Biagini Myers JM, Ruff BP, and Khurana Hershey GK. Vitamin D supplementation attenuates asthma development following traffic-related particulate matter exposure. The Journal of allergy and clinical immunology. 2019;143(1):386–94 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hollams EM, Hart PH, Holt BJ, Serralha M, Parsons F, de Klerk NH, et al. Vitamin D and atopy and asthma phenotypes in children: a longitudinal cohort study. The European respiratory journal. 2011;38(6):1320–7. [DOI] [PubMed] [Google Scholar]
- 44.Hufnagl K, and Jensen-Jarolim E. Vitamin A and D in allergy: from experimental animal models and cellular studies to human disease. Allergo J Int. 2018;27(3):72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cantorna MT, Snyder L, Lin YD, and Yang L. Vitamin D and 1,25(OH)2D regulation of T cells. Nutrients. 2015;7(4):3011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ruiter B, Patil SU, and Shreffler WG. Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clin Exp Allergy. 2015;45(7):1214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chambers ES, Suwannasaen D, Mann EH, Urry Z, Richards DF, Lertmemongkolchai G, et al. 1alpha,25-dihydroxyvitamin D3 in combination with transforming growth factor-beta increases the frequency of Foxp3(+) regulatory T cells through preferential expansion and usage of interleukin-2. Immunology. 2014;143(1):52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rosendahl J, Pelkonen AS, Helve O, Hauta-Alus H, Holmlund-Suila E, Valkama S, et al. High-Dose Vitamin D Supplementation Does Not Prevent Allergic Sensitization of Infants. J Pediatr. 2019;209:139–45 e1. [DOI] [PubMed] [Google Scholar]
- 49.Lodovici M, and Bigagli E. Oxidative stress and air pollution exposure. J Toxicol. 2011;2011:487074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brandt EB, Biagini Myers JM, Acciani TH, Ryan PH, Sivaprasad U, Ruff B, et al. Exposure to allergen and diesel exhaust particles potentiates secondary allergen-specific memory responses, promoting asthma susceptibility. The Journal of allergy and clinical immunology. 2015;136(2):295–303 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Grove KC, Provoost S, Hendriks RW, McKenzie ANJ, Seys LJM, Kumar S, et al. Dysregulation of type 2 innate lymphoid cells and TH2 cells impairs pollutant-induced allergic airway responses. The Journal of allergy and clinical immunology. 2017;139(1):246–57 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.