

Abstract
Artificial intelligence (AI) has been used in many areas of medicine, and recently large language models (LLMs) have shown potential utility for clinical applications. However, since we do not know if the use of LLMs can accelerate the pace of genetic discovery, we used data generated from mouse genetic models to investigate this possibility. We examined whether a recently developed specialized LLM (Med-PaLM 2) could analyze sets of candidate genes generated from analysis of murine models of biomedical traits. In response to free-text input, Med-PaLM 2 correctly identified the murine genes that contained experimentally verified causative genetic factors for six biomedical traits, which included susceptibility to diabetes and cataracts. Med-PaLM 2 was also able to analyze a list of genes with high impact alleles, which were identified by comparative analysis of murine genomic sequence data, and it identified a causative murine genetic factor for spontaneous hearing loss. Based upon this Med-PaLM 2 finding, a novel bigenic model for susceptibility to spontaneous hearing loss was developed. These results demonstrate Med-PaLM 2 can analyze gene-phenotype relationships and generate novel hypotheses, which can facilitate genetic discovery.
1. Introduction
A major challenge in biomedical science is to identify genetic factors in a population that affect the properties (i.e., phenotypes or traits) of an individual, especially those for disease susceptibility. Many genetic discoveries have been made using genome wide association study (GWAS) methods, which compare the pattern of allelic differences in mouse or human populations with variation in phenotypic responses. Irrespective of whether the subjects are mouse or human, a major barrier for genetic discovery is that GWAS results will identify a true causative genetic variant along with multiple other false positive associations because allelic patterns within genomic regions that are commonly inherited from their ancestors can randomly correlate with any phenotypic response pattern in a population. To address this issue, we recently developed an artificial intelligence (AI)-based computational pipeline for mouse genetic discovery that could sift through a set of candidate genes emerging from a GWAS and identify those most likely to be causal based upon assessment of the candidate gene-phenotype relationships in the published literature and other factors [1]. This pipeline analyzed publicly available datasets of biomedical responses (or disease susceptibility) that were measured in panels of inbred strains and identified novel causative genetic factors for various disease models, which included diabetes/obesity and cataract formation. However, this pipeline could only analyze a specific dataset and the analysis required entry of its numeric label.
The utility of a genetic discovery AI would be greatly expanded if it could answer free-text queries. Consistent with this possibility, large language models (LLMs) have recently been developed, which are general-purpose AI systems that are commonly implemented with transformer based neural network architectures [2]. They are typically pretrained on internet scale text corpora [3] and have demonstrated a wide range of language understanding and processing capabilities including information extraction, reasoning, summarizing data, and entailment capabilities. While general-purpose LLMs encode knowledge and have demonstrated their capabilities across a wide range of applications, the uniqueness of the scientific and biomedical domain requires that they be further specialized and adapted. LLM task performance can be improved through fine-tuning, which uses high quality domain-specific texts (i.e., scientific and biomedical text, or clinical guidelines) and/or learning from expert feedback. For example, Med-PaLM 2 is a recently developed medically aligned LLM that was fine-tuned using high quality biomedical text corpora and was aligned using clinician feedback. It demonstrated significantly improved performance when answering medical questions relative to general-purpose LLMs, and its answers matched or exceeded those of physicians when controlled benchmarks were assessed [4, 5]. Despite these advances and the large volume of biomedical and scientific knowledge encoded within LLMs, it remains to be determined if LLMs can be used to generate novel hypotheses that facilitate genetic discovery. Here, we test the potential of the specialized Med-PaLM 2 model to uncover gene-phenotype associations (Figure 1). We found that Med-PaLM 2 correctly responded to free-text queries about potential sets of candidate genes and that it could identify a novel causative genetic factor for an important biomedical trait.
Figure 1 ∣. The Med-PaLM 2 pipeline for genetic discovery.
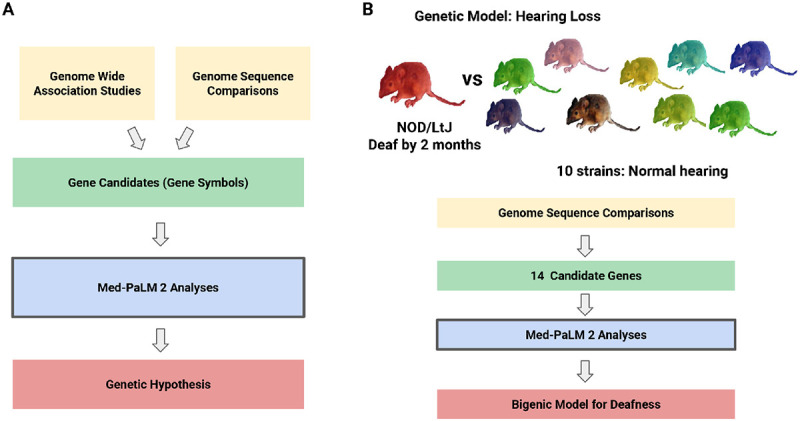
(A) Overview of the genetic analysis pipeline. A set of candidate genes are identified through analysis of the results obtained from either Genome Wide Association Study (GWAS) or genomic sequence comparison. Med-PaLM 2 evaluates the gene candidates (represented by their gene symbols) and it generates a genetic hypothesis by identifying those with the strongest association with a queried phenotype. (B) Overview of how Med-PaLM 2 generated a bigenic model for spontaneous hearing loss in a mouse strain. The genomic sequence of a mouse strain (NOD/LtJ) that spontaneously develops a hearing loss by 7 weeks of age was compared with that of 10 strains that maintain normal hearing during their lifetime. Fourteen genes with NOD/LtJ-specific high impact alleles were identified by this analysis. Med-PaLM 2 identified one gene as the most likely to contain the causative genetic factor for hearing loss. However, NOD/LtJ mice have another genetic factor, that is shared among multiple inbred strains with early onset hearing loss, which is necessary but it alone is not sufficient to cause their severe hearing loss. Therefore, based upon the Med-PaLM 2 results, the genetic hypothesis developed was that two genetic factors (i.e., a bigenic model) jointly contribute to the hearing loss of NOD/LtJ mice.
2. Results
Assessing candidate gene sets.
To investigate whether Med-PaLM 2 could be used for genetic discovery in response to free-text input, it was asked to analyze sets of candidate genes, which were previously identified by analysis of mouse GWAS data for six previously studied biomedical traits and identify the most likely causative genetic factors. In each example, Med-PaLM 2 was asked to identify which gene among a set of input genes was most likely to be associated with a queried phenotype. The candidate gene sets for cataract and diabetes susceptibility were identified using the mouse genetic discovery AI [1]; and those for the albino skin color [6], warfarin metabolism [7], aromatic hydrocarbon response [6], and chronic pain [8] phenotypes output by the computational genetic mapping program after analysis of the data obtained from inbred mouse strain panels. For all six examples, Med-PaLM 2 correctly identified the gene with the experimentally verified causative factor (Table 1, Table ED.1 and Table ED.2).
Table 1 ∣.
This table shows the candidate genes sets analyzed by Med-PaLM 2. The murine candidate genes, which had allelic patterns that correlated with the pattern of phenotypic responses of the inbred strains for the indicated phenotype, for each referenced study are listed. The experimentally proven causative genetic factor, which was correctly identified by the Med-PaLM 2 response is in bold. The queries and responses are shown in Table ED.1 and Table ED.2.
| Phenotype | Gene Candidates | Correct Answer |
|---|---|---|
| Cataract | Miox, Nid1, Trappc1, Pi4ka, Adgrv1, Slc8b1 | Nid1 [1] |
| Diabetes | Krtap5-1, Act1, Act2, Act3, Actb, Abra, Tlr5, Myh1, Myh2, Myh8, Unc45a, Cdc102a | Tlr5 [1] |
| Chronic Pain | P2rx7, Arntl, Pttg1, Kcnk2, Clcn4-2, Mhrt, Mh7b, Cttnb1, Dact, Cdh2 | P2rx7 [8] |
| Albino Skin | Grm5, Fzdr, Dlg2, Tyr, Polc4, Sec11a, Ap3b2, Rab38, Tmem135, Cedc81, 17rn6, Fcd, Picalm, Cdc83, Nrs155 | Tyr [6] |
| Warfarin Metabolism | Igsf8, Kcnj9, Kcnj10, Slc30a1, Icnh1, Masp2, Itpr1, Grin2b, Ctsc, Art5, Trpc2, Rrm1, Adam9, Lpl, Mmp2, Nnmt, Acy1, Gabrg2, Ctsl, Adprtl2, Abhd4, Mmp14, Psmb5, cacna1i, Tmprss2, Sncaip, Aldh7a1, Cyp2c55, Abcc2, Gabre | Cyp2c55 [7] |
| Aromatic Hydrocarbon Response | Ahr, Dnpep, Dusp2, Vdr, Ren1, Cyp2b19, Pola1, Esr2 | Ahr [6] |
While Med-PaLM 2 correctly identified the experimentally verified gene (P2rx7) [8] in response to the chronic pain query, it also mentioned two other genes whose potential relationship to chronic pain we had not previously considered: (i) Kcnk2 is a potassium channel that affects multiple types of pain responses [9-11]; and (ii) Arntl, which is a major circadian clock regulator [12], because maladaptive pain is affected by circadian rhythm disruption [13]. Although the connection between Arntl and chronic pain is tenuous, the hypothesis that Kcnk2 alleles could impact chronic pain is a creative and intriguing one. These results indicate that Med-PaLM 2 can assess a list of genes and identify those most likely to contain genetic factors for a studied trait, and also that it could generate novel hypothesis about gene-phenotype relationships.
A bigenic model for hearing loss.
We next investigated whether Med-PaLM 2 could facilitate novel genetic discovery by determining if it could identify murine genes with genetic variants, which were identified by genomic sequence comparison, that were most likely to cause hearing loss. Sixteen inbred mouse strains (of 80 tested) spontaneously developed an age-related nonsyndromic hearing loss by 3 months of age [14]. A Cadherin 23 (Cdh23 G->A) frameshift allele [15] that reduced the stability of a cochlear sensory hair cell bundle protein was previously shown to contribute to hearing loss in mice [16, 17]. We examined our 25M SNP database with alleles covering 48 classical inbred strains [18, 19] and found an extremely strong correlation between strains with early onset hearing loss and Cdh23753A alleles (Table SI.1). However, since only a subset of the strains with Cdh23753A alleles developed early onset hearing loss, there must be other contributing genetic factors. The age-related hearing loss of NOD/LtJ mice was of interest because it occurs by 3 weeks of age [14] and we found that it is more severe than was previously known. NOD/LtJ cochlear sensitivity was dramatically reduced across all frequencies tested. C57BL/6 mice, which also have Cdh23753A alleles, develop a hearing loss that is much less severe at 7 weeks of age (see Figure 2) and it is more fully manifested at a later age [14]. These features confirm that genetic factors other than Cdh23753A alleles must contribute to the NOD/LtJ hearing loss, but none have yet been identified. To identify them, the genomic sequence of NOD/LtJ was compared with 10 other strains (A/HeJ, AKR, BALB/C, CBA, FVB, LG/J, PL/J, SJL, SM/J, SWR) that maintained normal hearing throughout much of their life [14], and variant alleles that were also present in any these other strains were removed. Among the high impact NOD/LtJ-specific SNP alleles (Table ED.4), those within genes that encoded polymorphic genomic markers, immunoglobulin variable region genes, or affected splice sites were removed. Med-PaLM 2 was then asked to analyze the remaining 14 genes using CoT prompting with self-consistency, and it identified Crystallin mu (Crym) as the gene that was most likely to be associated with hearing loss (Table ED.3). When Med-PaLM 2 analyses were repeated 100 times after the input gene order was randomized, Crym was identified as one of the top 5 candidates in 98 of the analyses (Table SI.2). NOD/LtJ mice have a 2-bp frameshift deletion allele (rs216145143) within the codon for amino acid 220 in Crym that was not present in 47 classic inbred strains [18, 20] including the closely related NOR/LtJ stain [21], which generates a truncated protein with a premature termination codon at position 230 (Figure 3A-C). Immunoblotting indicated that the CRYM protein is completely absent from NOD/LtJ tissue (Figure 3D), which results from degradation of mutated Crym protein or of its mRNA. Hence, the NOD/LtJ frameshift deletion makes NOD/LtJ mice the equivalent of a Crym knockout mouse.
Figure 2 ∣. NOD/LtJ mice have a severe hearing loss.

Auditory brainstem responses (ABR) and distortion product otoacoustic emissions (DPOAE) were measured in 7-week-old CBA/J (n=10, red), NOD/LtJ (n=7, light gray) and C57BL/6J (n=14, dark gray) mice. Each bar represents the mean ± standard error of the mean, and each dot represents the sound pressure level (SPL) in decibels measured for one mouse. The ABR threshold levels for NOD/LtJ mice demonstrate that they have a profound hearing loss compared to that of CBA/J and C57BL/6J mice of the same age. The DPOAE thresholds in NOD/LtJ mice are substantially elevated across all frequencies tested in comparison to that of CBA/J and C57BL/6 mice. Interestingly, C57BL/6 have a hearing defect that is less severe than that of NOD/LtJ mice, and they show significantly higher DPOAE thresholds in mid-to-higher frequency range than CBA/J mice. The p-values for the CBA/J vs. NOD/LtJ comparisons are represented by asterisks: *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001. Similarly, the p-values for C57BL/6J vs. NOD/LtJ comparisons represented by #, and + are used to represent the p-values for CBA/J vs. C57BL/6J comparisons.
Figure 3 ∣.
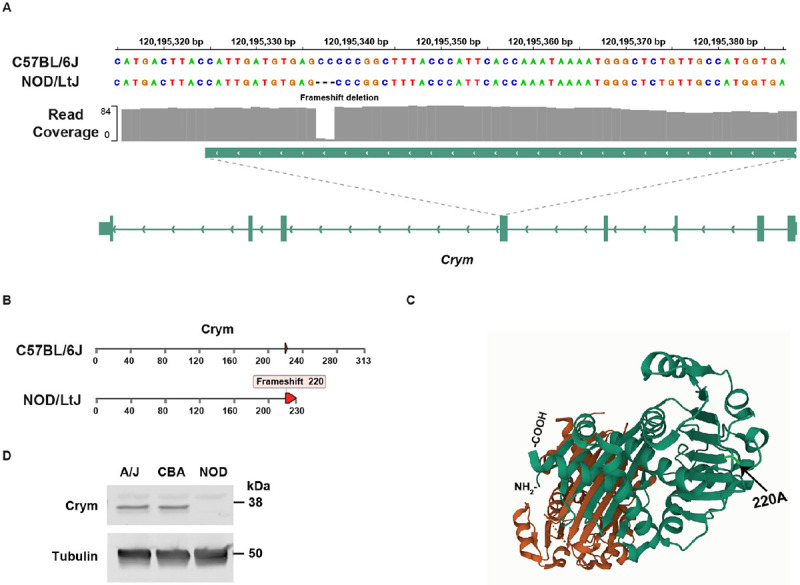
(A) NOD/LtJ mice have a 2 bp frameshift deletion in exon 4 of Crym, which is not present in 42 other strains. (B) The full length Crym protein has 313 amino acids, but the NOD/LTJ frameshift deletion within the codon for amino acid 220, which generates a premature termination codon at amino acid 230. (C) The Crym protein structure (PDB: 4BVA) is shown; and the position of NOD/LtJ-unique frameshift mutation that disrupts the COOH-terminal region of Crym is highlighted along with the location of the NH2- and COOH-terminal amino acids. (D) Crym protein is not present in NOD/LtJ mice. The proteins in lysates prepared from brain tissue obtained from a 5-week-old male A/J, CBA/J or NOD/LtJ mice were separated by SDS-PAGE and immunoblotted with mouse monoclonal anti-Crym or anti-tubulin antibodies. The blots were scanned after incubation with dye-labelled goat anti-mouse IgG. While the lysates have comparable amounts of tubulin, Crym is not present in the NOD/LtJ lysates.
Crym was identified by Med-PaLM 2 because it is highly expressed in a gradient (apex higher than base) along the length of the mouse cochlea [22], and two point mutations in human CRYM (X315Y or K314T) cause autosomal dominant non-syndromic deafness (DFNA40) [23, 24]. Thyroid hormone activity is essential for auditory function [25, 26]; CRYM is a triiodothyronine (T3) binding protein that regulates the intracellular level of T3 in cochlear cells [24] and the K314T mutation in human CRYM was shown to prevent thyroid hormone binding to CRYM [24]. Moreover, analysis of the crystal structure of Crym reveals that T3 binding is highly dependent upon the COOH-terminal domain of Crym: T3 binds to the aliphatic side chain of Arg229, Ser228 is one of two amino acids that hydrogen bonds with T3, and Ser228 and Leu292 interact with T3 through water molecules [27]. Since point mutations within the COOH terminal region of human CRYM cause early onset hearing loss, a NOD/LtJ Crym mutation that abolishes Crym protein expression is highly likely to contribute to its hearing loss.
3. Discussion
Based upon Med-PaLM 2 output, we propose that the spontaneous hearing loss of NOD/LtJ mice is likely to have (at minimum) a bigenic origin: the NOD/LtJ Cdh23 and Crym frameshift alleles jointly contribute to it. This bigenic model explains four features of the hearing loss that develops in NOD/LtJ and in several other inbred strains. (i) Some inbred strains with Cdh23753A alleles do not develop a hearing loss. (ii) The time of onset and the severity of the hearing loss developing in strains with Cdh23753A alleles is variable. For example, the NOD/LtJ hearing loss occurs earlier and is more severe than in C57BL/6 mice, which have the Cdh23753A allele but do not have the NOD/LtJ Crym frameshift allele. (iii) Reversion of the Cdh23753A allele to wildtype only partially suppressed the hearing loss that develops in NOD mice [28]. (iv) Crym knockout mice were reported to have normal hearing [29], but this is a result of the Crym knockout being bred onto the 129Sv background: 129Sv substrains maintain normal hearing [14] throughout their life, and they have the Cdh23753G that is associated with normal hearing. Since hearing loss does not develop in all strains with Cdh23753A susceptibility alleles, the variability in the severity of the hearing loss in the strains with the Cdh23753A susceptibility alleles, the inability of the reversion of the Cdh23753A allele to wildtype in NOD mice to completely reverse the hearing defect, and the absence of a hearing loss in Crym knockout mice indicate that at least two genetic factors contribute to the NOD/LtJ hearing loss.
The Med-PaLM 2 analyses for hearing loss in a mouse is analogous to the methods used to identify causal genetic factors in newborns presenting with clinical abnormalities. Every year, around 7M newborns are affected by severe genetic diseases [30] and many are monogenetic diseases (MGD). Despite the increased use of genome sequencing, identification of MGD-causing variants is difficult because hundreds of rare variants of unknown significance (VUS) are present within many genes in any individual’s genome [31-35]. As in the mouse, the large number of VUS make it difficult to identify the true causative variant for a phenotype in a human subject’s genome. The magnitude of this problem is compounded by the fact that ~ 60M suspected MGD patients will be sequenced by 2025. Numerous computational tools that automate some aspects of the MGD analysis pipeline have been developed to assess the potential pathogenicity of VUS [36-42]. Nevertheless, the rate of identifying disease-causing genes for suspected MGD (~ 30%) is only modestly better than that achieved a decade ago [35]. Since Med-PaLM 2 can help identify disease-causing genetic variants in mice, it is likely that it can similarly be used to analyze VUS in human subjects.
Our findings also indicate how LLM genetic discovery performance can be improved. First, by analyzing the output, we identified at least two factors that contribute to the false associations generated by an LLM (which are referred to as ‘hallucinations’). (i) The tokenizer (i.e., the program that breaks down a piece of text into tokens) has difficulty analyzing gene symbols, which consist of letters (that are often acronyms) and numbers. Tokenizers can incorrectly assign gene symbols to other genes (or words), which generate false associations based upon analysis of the related genes (or words). For example, one input gene (Med1) that was not associated with hearing loss was associated with hearing loss because mutations in another gene (MED12) cause congenital hearing impairment [43]. (ii) The next word prediction objective used to train the LLM sometimes leads to spurious associations between genes and phenotypes, which do not have causal relationships, due to their co-occurrence within the same context window. In one case, a spurious association was caused by a series of >40 abstracts, each covering a different topic, that were published together as a conference summary. In another case, a diverse set of phenotypes exhibited by an inbred strain (which included hearing loss) were summarized in the introduction to a paper, which characterized the effect of a gene knockout on an unrelated phenotype [44]. Secondly, a scalable LLM based AI system for genetic discovery can be designed to overcome the current limitations of the knowledge encoded in LLMs. For example, such an AI system could be taught to use external tools, which include: advanced variant calling tools like AlphaMissense [45] to identify the more probable disease-causing alleles; or search based retrieval augmentation to provide supporting evidence (from other genetic data or the published literature) and remove false positive associations. Also, by using high-quality domain specific data, process supervision and providing granular feedback to the AI system, we can further enable the AI system to analyze regulatory elements, protein-protein interactions, or non-coding RNA functions which in turn will further expand the range of genetic hypothesis that these models can develop, and therefore lead to a deeper understanding of genetic diseases.
Methods
Animals and Experimental Design.
All mouse procedures were approved by the Institutional Animal Care and Use Committee at the Stanford University Medical School; they were conducted in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals, Eighth Edition; and the results are reported according to the ARRIVE guidelines [46]. Male NOD/LtJ, C57BL/6 and CBA/J mice were purchased from Jackson Laboratories (Sacramento, CA, United States). CBA/J mice were used as a control since they maintain good cochlear sensitivity through most of their adulthood. Cochlear function was examined by measuring auditory brainstem responses (ABR) and distortion product otoacoustic emissions (DPOAE) at 7 weeks of age (N=7 for NOD/LtJ, N=14 for C57BL/6, and N=10 for CBA/J).
Cochlear function testing.
Cochlear function testing was performed using previously described methods [47, 48]. In brief, mice were anesthetized with ketamine (120 mg/kg) and xylazine (12 mg/kg) that was administered intraperitoneally. A custom acoustic system consisting of two speakers (MF1, TDT, Alachua, FL, United States) and a microphone (ER-10B+, Ethymotic Research, Elk Grove Village, IL, United States) coupled to a probe tube was used to measure pressure near the mouse eardrum. DPOAEs were measured as ear canal pressure in response to two tones presented into the ear canal (f1 and f2, with f2/f1=1.2) at half-octave steps, from f2 = 5.6 to 45.2 kHz, and in 5 dB intensity increments from 10 to 80 dB sound pressure level (SPL). ABR responses to 5-ms tone pips were measured between subdermal electrodes (placed adjacent to ipsilateral pinna, at the vertex, and near the tail), amplified 10,000 times through an amplifier (Medusa4Z, TDT, Alachua, FL, United States) and filtered (0.3-3.0 kHz). For each frequency and sound level assessed, 512 responses were recorded and averaged using the BioSigRZ software run on a RZ6 Multi-I/O Processor (TDT, Alachua, FL, United States). ABR waveforms stacked from lowest to highest SPL were visually inspected to define the threshold as the first level at which a repeatable wave I was detected. In the absence of an ABR or DPOAE response, 85 dB SPL was chosen as a threshold because it was 5 dB above the highest sound pressure level tested [49].
Whole genome variant calling and annotation.
The pipeline used to identify and annotate the SNPs and structural variants in the database was previously described [1]. In summary, the raw reads of the genomic sequence for 53 inbred strains was aligned to the reference genome (GRCm38) using bwa [50] and variants were discovered using BCFtools [51]. Each variant was annotated using Ensemble Variant Effect Predictor (VEP) [52]. For this analysis, a variant was retained if it had a high-impact consequence on any transcript, as determined by VEP. To identify unique high-impact variants in the NOD/LtJ strain, we subtracted variant alleles that were also present in any of 10 other strains (A/HeJ, AKR, BALB/C, CBA, FVB, LG/J, PL/J, SJL, SM/J, SWR) that maintained normal hearing throughout much of their life [14]. VEP defines high-impact variants as those causing transcript ablation, splice acceptor/donor variants, stop gain or stop loss, frameshifts, start lost, or transcript amplification (Table ED.4). Alleles within genes encoding polymorphic genomic markers or immunoglobulin variable regions, or within splice sites were removed. Then, the 14 genes with alleles that caused a frameshift, stop loss, or stop gain were selected for analysis by Med-PaLM 2.
Prompting strategies for Med-PaLM 2.
To assess the capability of Med-PaLM 2 to identify a causative genetic factor for previously studied biomedical traits, Med-PaLM 2 was zero-shot prompted to select the most likely causative gene from a set of genes that were identified previously through computational genetic analysis of mouse GWAS data. We evaluated the model’s answers for the six biomedical traits because the correct causative gene for each phenotype was validated in previous studies. The prompts and model outputs for all 6 test examples are shown in Table ED.1 and Table ED.2.
To assess the ability of Med-PaLM 2 to facilitate novel genetic discovery, Med-PaLM 2 was used to identify murine genes with genetic variants that contribute to hearing loss. A set of 14 high impact NOD/LtJ-specific SNP alleles were identified as described above. Chain-of-Thought (CoT) and self-consistency prompting strategies were used to ask Med-PaLM 2 to output the top 5 most likely genes that could be associated with hearing loss. CoT involves augmenting a one-shot example in a prompt with a step-by-step explanation of how to move towards a final answer [53]. CoT prompts were crafted to provide clear demonstrations on how to appropriately answer the gene-phenotype association questions shown in Table ED.3. Since the problem was formulated as a multiple-choice question, a self-consistency strategy was used to determine the most consistent answer [54]. Specifically, 100 decoding outputs from the model were sampled by randomizing the input order of the candidate genes. At each inference time, Med-PaLM 2 was instructed to select the top 5 genes associated with hearing loss. The total count for the number of times that each gene was selected across 100 decodes was aggregated.
Immunoblotting.
Brain tissues obtained from a 5-week-old male A/J, CBA/J or NOD/LtJ mice were homogenized in RIPA buffer supplemented with a protease inhibitor cocktail (Sigma P8340, 1 to 100) using a Precellys tissue homogenizer. Thirty micrograms each of protein lysate were resolved on a 4-15% gradient SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. The membranes were incubated with a 1:1000 dilution of mouse monoclonal anti-μ-crystallin antibody (F-11) (Santa Cruz Biotechnology) or with a 1:4000 dilution of a mouse monoclonal anti-α-tubulin antibody (clone B 5-1-2) (Sigma Aldrich). The anti-Crym antibody is specific for an epitope that is located between amino acids 45 and 85 of the Crym protein. The membranes were then incubated with IRDye® 800CW goat anti-mouse IgG (Licor Bioscience) as the labeled secondary antibody, and were scanned using a Licor Odyssey imaging system.
Extended Data
Table ED.1 ∣.
Med-PaLM 2 answers to questions about phenotypes cataract, diabetes, and chronic pain.
|
Candidate gene set 1: Cataract Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Cataract. Gene candidates: Miox, Nid1, Trappc1, Pi4ka, Adgrv1, Slc8b1 |
| Med-PaLM 2 output: Of the genes listed, the most likely candidate for association with cataract is **Nid1**. Nid1 encodes nidogen-1, a protein that is a major component of the extracellular matrix in the eye. Mutations in Nid1 have been shown to cause a variety of eye disorders, including cataracts. For example, a study of families with autosomal dominant cataracts found that mutations in Nid1 were responsible for the condition in some of the families. The other genes listed are not known to be associated with cataracts. Miox encodes a protein that is involved in the metabolism of iodine. Trappc1 encodes a protein that is involved in the transport of proteins. Pi4ka encodes a protein that is involved in the production of phosphatidylinositol 4-phosphate. Adgrv1 encodes a protein that is involved in the development of blood vessels. Slc8b1 encodes a protein that is involved in the transport of calcium. While it is possible that mutations in any of these genes could contribute to the development of cataracts, there is no evidence to suggest that they are directly involved in the condition. Therefore, the most likely candidate for association with cataract is Nid1. |
|
Candidate gene set 2: Diabetes Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Diabetes. Gene candidates: Krtap5-1, Act1, Act2, Act3, Actb, Abra, Tlr5, Myh1, Myh2, Myh8, Unc45a, Cdc102a |
| Med-PaLM 2 output: Of the genes listed, the most likely candidate for association with diabetes is **Tlr5**. Tlr5 is a Toll-like receptor that is involved in the innate immune system. It is known to play a role in the recognition of bacterial flagellin, a component of bacterial flagella. Activation of Tlr5 can lead to the production of inflammatory cytokines, which can contribute to the development of insulin resistance. Insulin resistance is a condition in which the body’s cells become less responsive to insulin. This can lead to high blood sugar levels, which is a hallmark of diabetes. Studies have shown that mice lacking Tlr5 are protected from developing insulin resistance. Additionally, polymorphisms in the Tlr5 gene have been associated with an increased risk of developing type 2 diabetes in humans. The other genes listed are not known to be directly involved in the development of diabetes. However, it is possible that they could play a role in the development of complications associated with diabetes. For example, Krtap5-1 is a gene that is involved in the formation of keratin, a protein that is found in hair, skin, and nails. People with diabetes are at increased risk of developing skin problems, so it is possible that Krtap5-1 could play a role in these complications. Overall, the evidence suggests that Tlr5 is the most likely candidate gene for association with diabetes. However, it is important to note that genetic factors are only one part of the puzzle when it comes to understanding the development of diabetes. Environmental factors, such as diet and lifestyle, also play a role. |
|
Candidate Gene Set 3: Chronic Pain Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Chronic pain. Gene candidates: P2rx7, Arntl, Pttg1, Kcnk2, Clcn4-2, Mhrt, Mh7b, Cttnb1, Dact, Cdh2 |
| Med-PaLM 2 output: Of the genes listed, P2rx7 is the most likely to be associated with chronic pain. P2rx7 is a gene that encodes for a receptor protein that is involved in the transmission of pain signals. Studies have shown that mutations in the P2rx7 gene can lead to increased pain sensitivity. Additionally, P2rx7 has been shown to be upregulated in the spinal cord of patients with chronic pain. The other genes listed are not as well-studied in the context of chronic pain. However, some of them have been implicated in other pain-related conditions. For example, Arntl has been linked to migraine headaches, and Kcnk2 has been linked to neuropathic pain. Overall, the evidence suggests that P2rx7 is the most likely of the listed genes to be associated with chronic pain. However, it is important to note that chronic pain is a complex condition that is likely influenced by multiple genes. Therefore, it is possible that other genes on the list may also play a role in chronic pain. If you are interested in learning more about the genetics of chronic pain, I would be happy to provide additional information. |
Table ED.2 ∣.
Med-PaLM 2 answers to questions about phenotypes albino skin, warfarin metabolism, and aromatic hydrocarbon response.
|
Candidate Gene Set 4: Albino Skin Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Albino skin. Gene candidates: Grm5, Fzdr, Dlg2, Tyr, Polc4, Sec11a, Ap3b2, Rab38, Tmem135, Cedc81, 17rn6, Fcd, Picalm, Cdc83, Nrs155 |
| Med-PaLM 2 output: The gene most likely associated with the albino skin phenotype is **Tyr**. The Tyr gene encodes the tyrosinase enzyme, which is responsible for the production of melanin. Melanin is the pigment that gives skin, hair, and eyes their color. Mutations in the Tyr gene can lead to a lack of melanin production, resulting in albinism. The other genes listed are not known to be associated with albinism. However, some of them may play a role in other aspects of skin pigmentation. For example, the Grm5 gene is involved in the production of melanocytes, which are the cells that produce melanin. Mutations in the Grm5 gene can lead to a condition called piebaldism, which is characterized by patches of white skin. If you are concerned about a genetic condition, it is important to speak with a doctor or genetic counselor. They can provide you with more information about the condition and its inheritance pattern. They can also order genetic testing to confirm a diagnosis. |
|
Candidate Gene Set 5: Warfarin Metabolism Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Warfarin metabolism. Gene candidates: Igsf8, Kcnj9, Kcnj10, Slc30a1, Icnh1, Masp2, Itpr1, Grin2b, Ctsc, Art5, Trpc2, Rrm1, Adam9, Lpl, Mmp2, Nnmt, Acy1, Gabrg2, Ctsl, Adprtl2, Abhd4, Mmp14, Psmb5, cacna1i, Tmprss2, Sncaip, Aldh7a1, Cyp2c55, Abcc2, Gabre |
|
Med-PaLM 2 output: Out of the genes you listed, the most likely gene associated with warfarin metabolism is **CYP2C9**. This gene encodes an enzyme that is responsible for the metabolism of warfarin. Variations in the CYP2C9 gene can affect how quickly or slowly a person metabolizes warfarin. This can impact the dose of warfarin that a person needs to take in order to achieve the desired effect. Other genes that may be associated with warfarin metabolism include VKORC1, CYP4F2, and CYP2C19. However, CYP2C9 is the most well-studied and has the strongest association with warfarin metabolism. It is important to note that genetic factors are not the only factors that can affect warfarin metabolism. Other factors, such as age, diet, and medications, can also play a role. Therefore, it is important for people taking warfarin to be monitored closely by their healthcare provider. I hope this information is helpful. Please let me know if you have any other questions. Comment: Cyp2c55 is located within a haplotype block that contains murine Cyp29, which is the homolog of human CYP2C29. |
|
Candidate Gene Set 6: Aromatic hydrocarbon response Input prompt: You are a helpful genetic assistant. The following is a question about genetic discovery. Which of the following genes is most likely associated with phenotype Aromatic hydrocarbon response. Gene candidates: Ahr, Dnpep, Dusp2, Vdr, Ren1, Cyp2b19, Pola1, Esr2 |
| Med-PaLM 2 output: The gene most likely associated with the phenotype Aromatic hydrocarbon response is **Ahr**. The Ahr gene encodes the aryl hydrocarbon receptor, which is a transcription factor that regulates the expression of genes involved in the metabolism of aromatic hydrocarbons. Aromatic hydrocarbons are a class of chemicals that are found in many environmental pollutants, such as cigarette smoke and car exhaust fumes. When Ahr is activated by binding to an aromatic hydrocarbon, it can induce the expression of genes that help to detoxify these chemicals. Studies have shown that mutations in the Ahr gene can lead to altered responses to aromatic hydrocarbons. For example, people with certain mutations in the Ahr gene may be more susceptible to the harmful effects of exposure to these chemicals. The other genes listed are not as likely to be associated with the phenotype Aromatic hydrocarbon response. However, some of these genes may play a role in the metabolism of aromatic hydrocarbons or in the response to these chemicals. For example, the Cyp2b19 gene encodes an enzyme that is involved in the metabolism of some aromatic hydrocarbons. Overall, the Ahr gene is the most likely candidate gene for the phenotype Aromatic hydrocarbon response. |
Table ED.3 ∣.
Chain-of-Thought prompt and Med-PaLM 2 answer to the question of identifying 5 genes for hearing loss.
|
CoT prompt: You are a helpful genetic assistant. You need to identify genes with causal mutations for chronic pain. You are provided with a set of genes that contain causal genetic factors in mice that might cause chronic pain. You need to determine which of the following genes have causal mutations that are directly associated with chronic pain in mice or humans. Output top 3 options in the final answer. (1) P2rx7 (2) Arntl (3) Pttg1 (4) Kcnk2 (5) Clcn4-2 (6) Mhrt (7) Mh7b (8) Cttnb1 (9) Dact (10) Cdh2 (11) Gabrg2 (12) Slco3a1 (13) Scn8a (14) Clbp (15) Ctgf. |
| Based on the available evidence, the majority of the genes listed are not directly associated with chronic pain in mice or humans. However, the top 3 most likely genes in the list to have causal mutations that are directly associated with chronic pain are: **P2rx7**: P2rx7 is a gene that encodes for a receptor protein that is involved in the transmission of pain signals. Studies have shown that mutations in the P2rx7 gene can lead to increased pain sensitivity. Additionally, P2rx7 has been shown to be upregulated in the spinal cord of patients with chronic pain. P2X7 receptor antagonists have been considered as potential treatments for chronic pain. **Kcnk2**: Also known as TREK-1, this gene encodes a member of the superfamily of potassium channel proteins containing two pore-forming P domains. Changes in its activity have been associated with pain perception. TREK-1 channel modulators have been studied as potential analgesic drugs. **Arntl**: Also known as BMAL1, is mainly known for its role in the circadian rhythm. There is indirect evidence suggesting that disruptions in circadian rhythms can influence pain processing, but the direct causal relationship of Arntl mutations to chronic pain isn’t well-established. However, it is important to note that chronic pain is a complex condition that is likely influenced by multiple genes. Therefore, it is possible that other genes on the list may also play a role in chronic pain. Newer studies might have identified more direct links or clarified associations for the genes mentioned. Always refer to recent literature for the most up-to-date scientific evidence. |
| You are a helpful genetic assistant. You need to identify genes with causal mutations for hearing loss or deafness. You are provided with a set of genes that contain causal genetic factors in mice that might cause hearing loss or deafness. You need to determine which of the following genes have causal mutations that are directly associated with hearing loss or deafness in mice or humans. Output top 5 options in the final answer. (1) Zfp629 (2) Sdc2 (3) Gtpbp10 (4) Adgrb1 (5) Mettl7a1 (6) Il23r (7) Crym (8) Med1 (9) Stk38 (10) Fcgr1 (11) Armt1 (12) Mast4 (13) Spertl (14) Ifi27l2b (15) Tmem242 (16) Stxbp4 (17) Eppk1. |
| Med-PaLM 2 output: Based on the available evidence, the top 5 genes in the list that are most likely to have causal mutations that are directly associated with hearing loss or deafness in mice or humans are: **Crym**: Crym encodes a protein called α-crystallin, which is a major structural component of the lens. Mutations in Crym can lead to cataracts, which can cause hearing loss. **Med1**: Med1 encodes a protein that is involved in the development of the inner ear. Mutations in Med1 can lead to sensorineural hearing loss. **Mast4**: Mast4 encodes a protein that is involved in the transmission of sound signals from the inner ear to the brain. Mutations in Mast4 can lead to nonsyndromic hearing loss. **Tmem242**: Tmem242 encodes a protein that is involved in the development of the inner ear. Mutations in Tmem242 can lead to sensorineural hearing loss. **Stk38**: Stk38 encodes a protein that is involved in the development of the inner ear. Mutations in Stk38 can lead to sensorineural hearing loss. It is important to note that hearing loss is a complex condition that is likely influenced by multiple genes. Therefore, it is possible that other genes on the list may also play a role in hearing loss. Newer studies might have identified more direct links or clarified associations for the genes mentioned. Always refer to recent literature for the most up-to-date scientific evidence. |
Table ED.4 ∣.
High impact NOD/LtJ-specific SNP alleles.
| SYMBOL | Variant | Location | Allele | Consequence | HGVSp |
|---|---|---|---|---|---|
| Armt1 | 10_4450853_G/GG | 10:4450853 | GG | frameshift_variant | ENSMUSP00000114073.1:p.Val201CysfsTer15 |
| 4930505A04Rik | 11_30426247_TTTTTT/TTTTTTT | 11:30426247-30426252 | TTTTTTT | frameshift_variant | ENSMUSP00000045288.7:p.Thr207AsnfsTer73 |
| Stxbp4 | 11_90604823_C/T | 11:90604823 | T | stop_gained | ENSMUSP00000103504.1:p.Trp212Ter |
| Med1 | 11_98152811_AAAAAAAAAA/AAAAAAAAA | 11:98152811-98152820 | AAAAAAAAA | frameshift_variant | ENSMUSP00000090411.5:p.Phe598SerfsTer4 |
| Ifi27l2b | 12_103451875_ACA/ACACA | 12:103451875-103451877 | ACACA | frameshift_variant | ENSMUSP00000041712.6:p.Ser169CysfsTer18 |
| Mast4 | 13_102734852_TTTTTTTTTTT/TTTTTTTTTTTTTTT | 13:102734852-102734862 | TTTTTTTTTTTTTTT | frameshift_variant | ENSMUSP00000132263.1:p.Gly1202LysfsTer20 |
| Vmn1r222 | 13_23233007_AAAAAAAA/AAAAAAA | 13:23233007-23233014 | AAAAAAA | frameshift_variant | ENSMUSP00000076365.2:p.Phe12SerfsTer3 |
| Vmn1r222 | 13_23233007_AAAAAAAA/AAAAAAA | 13:23233007-23233014 | AAAAAAA | frameshift_variant | ENSMUSP00000157644.1:p.Phe12SerfsTer3 |
| Vmn1r222 | 13_23233007_AAAAAAAA/AAAAAAA | 13:23233007-23233014 | AAAAAAA | frameshift_variant | ENSMUSP00000157499.1:p.Phe12SerfsTer3 |
| Spertl | 14_95881801_AAAAAAAAA/AAAAAAAAAA | 14:95881801-95881809 | AAAAAAAAAA | start_lost,start_retained_variant,5_prime_UTR_variant | - |
| Spertl | 14_95881801_AAAAAAAAA/AAAAAAAAAA | 14:95881801-95881809 | AAAAAAAAAA | start_lost,start_retained_variant,5_prime_UTR_variant | - |
| Mettl7a1 | 15_100328283_C/A | 15:100328283 | A | stop_gained | ENSMUSP00000155112.1:p.Tyr188Ter |
| Sdc2 | 15_32921251_GCGCGCG/GCGCG | 15:32921251-32921257 | GCGCG | frameshift_variant | ENSMUSP00000022871.5:p.Ala4ValfsTer24 |
| Adgrb1 | 15_74563913_A/G | 15:74563913 | G | splice_donor_variant | - |
| Eppk1 | 15_76103351_TG/AAGAGACTG | 15:76103351-76103352 | AAGAGACTG | frameshift_variant | ENSMUSP00000154609.1:p.Gln3111SerfsTer64 |
| Eppk1 | 15_76104896_-/AAGAGAC | 15:76104895-76104896 | AAGAGAC | frameshift_variant | ENSMUSP00000154609.1:p.Gln2596SerfsTer64 |
| Stk38 | 17_28991283_TTTTTTTTTTTTT/TTTTTTTTTTTTTT | 17:28991283-28991295 | TTTTTTTTTTTTTT | splice_donor_variant | - |
| Stk38 | 17_28991283_TTTTTTTTTTTTT/TTTTTTTTTTTTTT | 17:28991283-28991295 | TTTTTTTTTTTTTT | splice_donor_variant | - |
| Stk38 | 17_28991283_TTTTTTTTTTTTT/TTTTTTTTTTTTTT | 17:28991283-28991295 | TTTTTTTTTTTTTT | frameshift_variant | ENSMUSP00000156597.1:p.Ser108IlefsTer8 |
| Stk38 | 17_28991283_TTTTTTTTTTTTT/TTTTTTTTTTTTTT | 17:28991283-28991295 | TTTTTTTTTTTTTT | splice_donor_variant | - |
| Arid1b | 17_5321261_TTTTTTT/TTTTTTTTTTT | 17:5321261-5321267 | TTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Arid1b | 17_5321261_TTTTTTT/TTTTTTTTTTT | 17:5321261-5321267 | TTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Arid1b | 17_5321261_TTTTTTT/TTTTTTTTTTT | 17:5321261-5321267 | TTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Arid1b | 17_5321261_TTTTTTT/TTTTTTTTTTT | 17:5321261-5321267 | TTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Tmem242 | 17_5432735_G/A | 17:5432735 | A | stop_gained | ENSMUSP00000139629.1:p.Arg114Ter |
| F830016B08Rik | 18_60299849_G/A | 18:60299849 | A | start_lost | ENSMUSP00000131437.1:p.Met1? |
| As3mt | 19_46712200_TTTTTTTTTTTTTTTT/TTTTTTTTTTTTTTTTTTTTT | 19:46712200-46712215 | TTTTTTTTTTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| As3mt | 19_46712200_TTTTTTTTTTTTTTTT/TTTTTTTTTTTTTTTTTTTTT | 19:46712200-46712215 | TTTTTTTTTTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Morrbid | 2_128336371_C/G | 2:128336371 | G | splice_donor_variant,non_coding_transcript_variant | - |
| Morrbid | 2_128336371_C/G | 2:128336371 | G | splice_donor_variant,non_coding_transcript_variant | - |
| Cst8 | 2_148799222_G/A | 2:148799222 | A | splice_donor_variant | - |
| Pygb | 2_150808785_TTTTTTTTT/TTTTTTTTTTTTT | 2:150808785-150808793 | TTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Vtcn1 | 3_100892537_TTTTTTTT/TTTTTTTTTTTTTTTTTTTT | 3:100892537-100892544 | TTTTTTTTTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Amy2-ps1 | 3_113192372_C/T | 3:113192372 | T | splice_acceptor_variant,non_coding_transcript_variant | - |
| Cfi | 3_129850678_TTTTTTTTT/TTTTTTTTTTTTTTT | 3:129850678-129850686 | TTTTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Adam15 | 3_89340118_T/C | 3:89340118 | C | splice_acceptor_variant | - |
| Fcgr1 | 3_96284506_TCTCTCTCT/TCTCT | 3:96284506-96284514 | TCTCT | frameshift_variant | ENSMUSP00000029748.3:p.Glu328ArgfsTer5 |
| Zfp697 | 3_98431002_T/A | 3:98431002 | A | splice_donor_variant | - |
| Zmym4 | 4_126904386_TTTTTTTTT/TTTTTTTTTT | 4:126904386-126904394 | TTTTTTTTTT | splice_donor_variant | - |
| Zfp985 | 4_147562959_G/T | 4:147562959 | T | splice_donor_variant | - |
| Gtpbp10 | 5_5559360_-/G | 5:5559359-5559360 | G | frameshift_variant | ENSMUSP00000143003.1:p.Val36AlafsTer13 |
| Lrmp | 6_145160524_CCCCC/CCCCCCCCC | 6:145160524-145160528 | CCCCCCCCC | splice_acceptor_variant,intron_variant | - |
| Il23r | 6_67491779_A/G | 6:67491779 | G | start_lost | ENSMUSP00000113342.1:p.Met1? |
| Igkv17-121 | 6_68037077_G/T | 6:68037077 | T | stop_gained | ENSMUSP00000100116.2:p.Gly37Ter |
| Igkv17-121 | 6_68037212_C/T | 6:68037212 | T | stop_gained | ENSMUSP00000100116.2:p.Arg82Ter |
| Igkv17-121 | 6_68037297_T/A | 6:68037297 | A | stop_gained | ENSMUSP00000100116.2:p.Leu110Ter |
| Igkv17-121 | 6_68037299_C/T | 6:68037299 | T | stop_gained | ENSMUSP00000100116.2:p.Gln111Ter |
| Igkv6-25 | 6_70215666_ACGGAGA/- | 6:70215666-70215672 | - | frameshift_variant | ENSMUSP00000100184.1:p.Asp19AlafsTer3 |
| Suclg1 | 6_73275183_CTCTCTCTCTCTCTC/CTCTCTCTCTCTCTCTC | 6:73275183-73275197 | CTCTCTCTCTCTCTCTC | splice_acceptor_variant,intron_variant | - |
| Crym | 7_120195337_CCCCC/CCC | 7:120195337-120195341 | CCC | frameshift_variant | ENSMUSP00000033198.5:p.Ala220SerfsTer11 |
| Zfp629 | 7_127612556_C/T | 7:127612556 | T | stop_gained | ENSMUSP00000140505.1:p.Trp33Ter |
| 4930543N07Rik | 7_138563954_G/A | 7:138563954 | A | splice_donor_variant,non_coding_transcript_variant | - |
| Vmn1r101 | 7_20442350_TTTT/TTTTT | 7:20442350-20442353 | TTTTT | frameshift_variant | ENSMUSP00000129819.1:p.Thr82AsnfsTer5 |
| Vmn1r117 | 7_20883467_G/A | 7:20883467 | A | stop_gained | ENSMUSP00000131580.1:p.Arg219Ter |
| Tyrobp | 7_30417327_TTTTTTTTTTTTTT/TTTTTTTTTTTTTTTTTTTTTT | 7:30417327-30417340 | TTTTTTTTTTTTTTTTTTTTTT | splice_acceptor_variant,intron_variant | - |
| Cldn18 | 9_99696053_C/T | 9:99696053 | T | splice_donor_variant | - |
| Cldn18 | 9_99696053_C/T | 9:99696053 | T | splice_donor_variant | - |
Supplementary Material
Acknowledgments
This work was supported by NIH awards (1R01DC021133 and 1 R24 OD035408) to GP. TT, VN, and AP were funded by Alphabet Inc. and/or a subsidiary thereof (Alphabet). We thank Pi-Chuan Chang and Greg Corrado for their valuable insights and feedback during our research.
Footnotes
Competing interests
The Stanford University Medical School authors have no competing interests. T.T., V.N., and A.P. are employees of Alphabet and may own stock as part of their standard compensation package.
Data Availability
SNP allele data is available at the Mouse Phenome Database (GenomeMUSter https://mpd.jax.org/genotypes).
Code Availability
Med-PaLM 2 is an LLM for the biomedical domain. We are not open-sourcing model code and weights owing to the safety implications of unmonitored use of such a model in medical settings. In the interest of responsible innovation, we will be working with academic and industry research partners, providers, regulators, and policy stakeholders to validate and explore safe onward uses of Med-PaLM 2. For reproducibility, we documented technical deep learning methods while keeping the paper accessible to a clinical and general scientific audience. Our work builds upon PaLM, for which technical details have been described extensively, and our institution has open-sourced several related LLMs to further the development of research methods in the field (https://huggingface.co/google/flan-t5-xl).
References
- 1.Fang Z. & Peltz G. An automated multi-modal graph-based pipeline for mouse genetic discovery. Bioinformatics 38, 3385–3394 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A. N., Kaiser Ł. & Polosukhin I. Attention is all you need. Advances in neural information processing systems 30 (2017). [Google Scholar]
- 3.Chowdhery A., Narang S., Devlin J., Bosma M., Mishra G., Roberts A., Barham P., Chung H. W., Sutton C., Gehrmann S., et al. Palm: Scaling language modeling with pathways. arXiv preprint arXiv:2204.02311 (2022). [Google Scholar]
- 4.Singhal K., Azizi S., Tu T., Mahdavi S. S., Wei J., Chung H. W., Scales N., Tanwani A., Cole-Lewis H., Pfohl S., et al. Large language models encode clinical knowledge. Nature 620, 172–180 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singhal K., Tu T., Gottweis J., Sayres R., Wulczyn E., Hou L., Clark K., Pfohl S., Cole-Lewis H., Neal D., et al. Towards expert-level medical question answering with large language models. arXiv preprint arXiv:2305.09617 (2023). [Google Scholar]
- 6.Liao G., Wang J., Guo J., Allard J., Cheng J., Ng A., Shafer S., Puech A., McPherson J. D., Foernzler D., et al. In silico genetics: identification of a functional element regulating H2-E α gene expression. Science 306, 690–695 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Guo Y., Weller P., Farrell E., Cheung P., Fitch B., Clark D., Wu S.-y., Wang J., Liao G., Zhang Z., et al. In silico pharmacogenetics of warfarin metabolism. Nature biotechnology 24, 531–536 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorge R. E., Trang T., Dorfman R., Smith S. B., Beggs S., Ritchie J., Austin J.-S., Zaykin D. V., Meulen H. V., Costigan M., et al. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nature medicine 18, 595–599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith P. A. K+ channels in primary afferents and their role in nerve injury-induced pain. Frontiers in Cellular Neuroscience 14, 566418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cregg R., Momin A., Rugiero F., Wood J. N. & Zhao J. Pain channelopathies. The Journal of physiology 588, 1897–1904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alloui A., Zimmermann K., Mamet J., Duprat F., Noël J., Chemin J., Guy N., Blondeau N., Voilley N., Rubat-Coudert C., et al. TREK-1, a K+ channel involved in polymodal pain perception. The EMBO journal 25, 2368–2376 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi J. S. Transcriptional architecture of the mammalian circadian clock. Nature Reviews Genetics 18, 164–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bumgarner J. R., Walker W. H. II & Nelson R. J. Circadian rhythms and pain. Neuroscience & Biobehavioral Reviews 129, 296–306 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Q. Y., Johnson K. R. & Erway L. C. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hearing research 130, 94–107 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noben-Trauth K., Zheng Q. Y. & Johnson K. R. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nature genetics 35, 21–23 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boëda B., El-Amraoui A., Bahloul A., Goodyear R., Daviet L., Blanchard S., Perfettini I., Fath K. R., Shorte S., Reiners J., et al. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. The EMBO journal 21, 6689–6699 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siemens J., Kazmierczak P., Reynolds A., Sticker M., Littlewood-Evans A. & Müller U. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proceedings of the National Academy of Sciences 99, 14946–14951 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arslan A., Guan Y., Fang Z., Chen X., Donaldson R., Zhu W., Ford M., Wu M., Zheng M., Dill D. L., et al. High throughput computational mouse genetic analysis. BioRxiv, 2020–September (2020). [Google Scholar]
- 19.Arslan A., Fang Z., Wang M., Cheng Z., Yoo B., Bejerano G. & Peltz G. Analysis of structural variation among inbred mouse strains identifies genetic factors for autism-related traits. BioRxiv, 2021–February (2021). [Google Scholar]
- 20.Arslan A., Fang Z., Wang M., Tan Y., Cheng Z., Chen X., Guan Y., Pisani J., L., Yoo B., Bejerano G., et al. Analysis of structural variation among inbred mouse strains. BMC genomics 24, 97 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prochazka M., Serreze D. V., Frankel W. N. & Leiter E. H. NOR/Lt mice: MHC-matched diabetes-resistant control strain for NOD mice. Diabetes 41, 98–106 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Yoshimura H., Takumi Y., Nishio S.-y., Suzuki N., Iwasa Y.-i. & Usami S.-i. Deafness gene expression patterns in the mouse cochlea found by microarray analysis. PLoS One 9, e92547 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abe S., Katagiri T., Saito-Hisaminato A., Usami S.-i., Inoue Y., Tsunoda T. & Nakamura Y. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. The American Journal of Human Genetics 72, 73–82 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oshima A., Suzuki S., Takumi Y., Hashizume K., Abe S. & Usami S. CRYM mutations cause deafness through thyroid hormone binding properties in the fibrocytes of the cochlea. Journal of medical genetics 43, e25–e25 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinney C. J. & Bloch R. J. μ-Crystallin: A thyroid hormone binding protein. Endocrine regulations 55, 89–102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aksoy O., Hantusch B. & Kenner L. Emerging role of T3-binding protein μ-crystallin (CRYM) in health and disease. Trends in Endocrinology & Metabolism (2022). [DOI] [PubMed] [Google Scholar]
- 27.Borel F., Hachi I., Palencia A., Gaillard M.-C. & Ferrer J.-L. Crystal structure of mouse mu-crystallin complexed with NADPH and the T3 thyroid hormone. The FEBS journal 281, 1598–1612 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Hou X., Yasuda S. P., Yamaguchi M., Suzuki S., Seki Y., Ouchi T., Mao T., Prakhongcheep O., Shitara H. & Kikkawa Y. Impacts of an age-related hearing loss allele of cadherin 23 on severity of hearing loss in ICR and NOD/Shi mice. Biochemical and Biophysical Research Communications 674, 147–153 (2023). [DOI] [PubMed] [Google Scholar]
- 29.Suzuki S., Suzuki N., Mori J.-i., Oshima A., Usami S. & Hashizume K. μ-Crystallin as an intracellular 3, 5, 3-triiodothyronine holder in vivo. Molecular endocrinology 21, 885–894 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Church G. Compelling reasons for repairing human germlines. N Engl J Med 377, 1909–1911 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Yang Y., Muzny D. M., Reid J. G., Bainbridge M. N., Willis A., Ward P. A., Braxton A., Beuten J., Xia F., Niu Z., et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. New England Journal of Medicine 369, 1502–1511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee H., Deignan J. L., Dorrani N., Strom S. P., Kantarci S., Quintero-Rivera F., Das K., Toy T., Harry B., Yourshaw M., et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama 312, 1880–1887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng S. B., Buckingham K. J., Lee C., Bigham A. W., Tabor H. K., Dent K. M., Huff C. D., Shannon P. T., Jabs E. W., Nickerson D. A., et al. Exome sequencing identifies the cause of a mendelian disorder. Nature genetics 42, 30–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng S. B., Turner E. H., Robertson P. D., Flygare S. D., Bigham A. W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E. E., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 461, 272–276 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dragojlovic N., Elliott A. M., Adam S., van Karnebeek C., Lehman A., Mwenifumbo J. C., Nelson T. N., du Souich C., Friedman J. M. & Lynd L. D. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genetics in Medicine 20, 1–9 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Wang K., Li M. & Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164–e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jagadeesh K. A., Wenger A. M., Berger M. J., Guturu H., Stenson P. D., Cooper D. N., Bernstein J. A. & Bejerano G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nature genetics 48, 1581–1586 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Jagadeesh K. A., Paggi J. M., Ye J. S., Stenson P. D., Cooper D. N., Bernstein J. A. & Bejerano G. S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nature genetics 51, 755–763 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Singleton M. V., Guthery S. L., Voelkerding K. V., Chen K., Kennedy B., Margraf R. L., Durtschi J., Eilbeck K., Reese M. G., Jorde L. B., et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. The American Journal of Human Genetics 94, 599–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smedley D., Jacobsen J. O., Jäger M., Köhler S., Holtgrewe M., Schubach M., Siragusa E., Zemojtel T., Buske O. J., Washington N. L., et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nature protocols 10, 2004–2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jagadeesh K. A., Birgmeier J., Guturu H., Deisseroth C. A., Wenger A. M., Bernstein J. A. & Bejerano G. Phrank measures phenotype sets similarity to greatly improve Mendelian diagnostic disease prioritization. Genetics in Medicine 21, 464–470 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Birgmeier J., Haeussler M., Deisseroth C. A., Steinberg E. H., Jagadeesh K. A., Ratner A. J., Guturu H., Wenger A. M., Diekhans M. E., Stenson P. D., et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Science translational medicine 12, eaau9113 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Caro-Llopis A., Rosello M., Orellana C., Oltra S., Monfort S., Mayo S. & Martinez F. De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy? Pediatric research 80, 809–815 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Rohowetz L. J., Mardelli M. E., Duncan R. S., Riordan S. M. & Koulen P. The contribution of anterior segment abnormalities to changes in intraocular pressure in the DBA/2J mouse model of glaucoma: DBA/2J-Gpnmb+/SjJ mice as critical controls. Frontiers in Neuroscience 15, 801184 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng J., Novati G., Pan J., Bycroft C., Žemgulytė A., Applebaum T., Pritzel A., Wong L. H., Zielinski M., Sargeant T., et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381, eadg7492 (2023). [DOI] [PubMed] [Google Scholar]
- 46.Kilkenny C., Browne W. J., Cuthill I. C., Emerson M. & Altman D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. Journal of Pharmacology and Pharmacotherapeutics 1, 94–99 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seist R., Landegger L. D., Robertson N. G., Vasilijic S., Morton C. C. & Stankovic K. M. Cochlin deficiency protects against noise-induced hearing loss. Frontiers in Molecular Neuroscience 14, 670013 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Early S., Saad M. A., Mallidi S., Mansour A., Seist R., Hasan T. & Stankovic K. M. A fluorescent photoimmunoconjugate for imaging of cholesteatoma. Scientific Reports 12, 19905 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J., Landegger L. D., Sun Y., Ren J., Maimon N., Wu L., Ng M. R., Chen J. W., Zhang N., Zhao Y., et al. A cerebellopontine angle mouse model for the investigation of tumor biology, hearing, and neurological function in NF2-related vestibular schwannoma. Nature protocols 14, 541–555 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H. & Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. & Subgroup, 1. G. P. D. P. The sequence alignment/map format and SAMtools. bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLaren W., Gil L., Hunt S. E., Riat H. S., Ritchie G. R., Thormann A., Flicek P. & Cunningham F. The ensembl variant effect predictor. Genome biology 17, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SNP allele data is available at the Mouse Phenome Database (GenomeMUSter https://mpd.jax.org/genotypes).
Med-PaLM 2 is an LLM for the biomedical domain. We are not open-sourcing model code and weights owing to the safety implications of unmonitored use of such a model in medical settings. In the interest of responsible innovation, we will be working with academic and industry research partners, providers, regulators, and policy stakeholders to validate and explore safe onward uses of Med-PaLM 2. For reproducibility, we documented technical deep learning methods while keeping the paper accessible to a clinical and general scientific audience. Our work builds upon PaLM, for which technical details have been described extensively, and our institution has open-sourced several related LLMs to further the development of research methods in the field (https://huggingface.co/google/flan-t5-xl).