

Summary
Although lineage-specific genes have been identified in the mammary gland, little is known about the contribution of the 3D genome organization to gene regulation in the epithelium. Here, we describe the chromatin landscape of the three major epithelial subsets through integration of long- and short-range chromatin interactions, accessibility, histone modifications, and gene expression. While basal genes display exquisite lineage specificity via distal enhancers, luminal-specific genes show widespread promoter priming in basal cells. Cell specificity in luminal progenitors is largely mediated through extensive chromatin interactions with super-enhancers in gene-body regions in addition to interactions with polycomb silencer elements. Moreover, lineage-specific transcription factors appear to be controlled through cell-specific chromatin interactivity. Finally, chromatin accessibility rather than interactivity emerged as a defining feature of the activation of quiescent basal stem cells. This work provides a comprehensive resource for understanding the role of higher-order chromatin interactions in cell-fate specification and differentiation in the adult mouse mammary gland.
Keywords: mammary gland, epigenetics, enhancer, chromatin looping, stem cells, transcription factors, polycomb
Graphical abstract
Highlights
-
•
Luminal progenitor genes are primed in basal cells but basal genes are “off” in luminal cells
-
•
Progenitors display extensive chromatin interactivity and super-enhancer involvement
-
•
Polycomb site interactions are enriched in luminal cells and influence gene expression
-
•
Chromatin accessibility (and not interactivity) defines the activation of stem cells
Milevskiy et al. investigate the epigenetic states of three epithelial lineages and two stem cell populations in the mouse mammary gland. Each epithelial cell type utilizes distinct epigenetic mechanisms to govern cell-specific expression: basal promoters display lineage activation; luminal promoters are promiscuous and rely on intricate chromatin looping patterns to achieve lineage specification.
Introduction
Chromatin structure is fundamental to the transcriptional control of genes that dictate lineage commitment and differentiation, enabling specific interactions between promoters, enhancers, and other cis-regulatory elements (CREs) to regulate transcription.1,2 Genome structure exhibits a hierarchical organization of multiscale structural units ranging from chromosome territories to active (A) and inactive (B) compartments to topological-associated domains (TADs) to enhancer-promoter chromatin loops.3,4,5 Gene promoter and CRE activities are coordinately regulated at the epigenetic level through methylation, acetylation, and/or ubiquitination of histones at varying lysine residues.6,7 Silencer elements have only recently come into focus, with polycomb complexes implicated in controlling their activity via chromatin looping.8,9 Both enhancers and silencers play key roles in development, and alterations in either element can lead to aberrant reprogramming of cells and pathogenic states.10,11
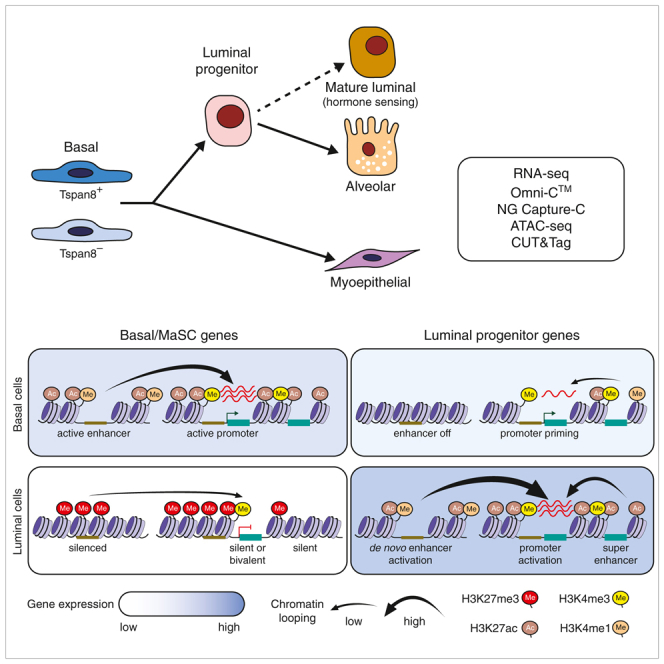
The mammary gland is composed of an epithelial ductal tree that undergoes dramatic morphogenesis across the different developmental phases.12 Structurally, each duct comprises an inner layer of luminal cells and an outer layer of elongated, contractile myoepithelial (basal) cells. In the steady-state adult gland, there are three major cell types: basal, luminal progenitor (LP), and hormone-sensing (HS)/mature luminal (ML) cells. The basal compartment is enriched for cells with mammary repopulating capacity defined through in vivo transplantation studies, including a small subset of deeply quiescent mammary stem cells (MaSCs).13,14 The luminal compartment is composed of two lineages, which are primarily distinguished by their hormone receptor (HR) status. The LP pool predominantly contains HR-negative progenitors for the alveolar lineage but also a small population of HR-positive cells.15,16 HS/ML cells constitute the dominant luminal cell type in the homeostatic mammary gland and are enriched for HR-positive cells.
A number of studies have investigated the gene expression profiles, DNA methylation, and histone epigenomes of the different mouse mammary epithelial subsets, leading to the definition of lineage-specific genes.17,18,19,20,21 Chromatin accessibility assays have uncovered mixed-lineage chromatin accessibility in fetal and adult basal cells.19,22,23,24,25 In human breast epithelial cells, luminal enhancer elements can be pre-marked in basal cells.26,27 The most well-characterized mammary CRE is the super-enhancer associated with the mouse whey acidic protein (Wap) gene, perturbation of which results in reduced interactions and decreased Wap expression during pregnancy.28,29 Fundamental questions remain about the nature and repertoire of promoter and enhancer states that operate in the adult mammary gland and the role of chromatin interactivity in instructing lineage commitment. In this report, we systematically characterize the epigenetic states of three epithelial lineages in the mouse mammary gland as well as the quiescent and activated basal populations through the integration of genome-wide chromatin interaction, chromatin accessibility, histone modification, and gene expression data. Remarkably, each epithelial lineage was found to utilize distinct epigenetic mechanisms to govern cell-specific gene expression. This epigenomic atlas provides a resource for understanding the role of higher-order chromatin interactions in dictating cell-fate specification and differentiation in the mammary gland and for uncovering novel transcription factors (TFs) implicated in these processes.
Results
Generation of an epigenetic atlas of murine mammary epithelial cells
To further explore gene regulation in the mammary gland, we have generated an extensive epigenetic atlas for mammary epithelial cells (MECs) that includes lineage-negative basal (CD29hiCD24+), LP (CD61+CD29loCD24+), and ML/HS (hereafter termed ML; CD61−CD29loCD24+) cellular populations sorted from 9-week adult female mice, in addition to quiescent (Tspan8+CD29hiCD24+) and activated “stem cells” (Tspan8−CD29hiCD24+) in the basal compartment14 (Figures 1A and S1A). The atlas encompasses chromatin interaction analysis for both short- (NG Capture-C)30 and long-range interactions (Omni-C), chromatin accessibility analysis via assay with transposase-accessible chromatin with sequencing (ATAC-seq),31 and RNA sequencing (RNA-seq; Figure 1B), together with the mapping of nine histone marks and RNA polymerase II (Pol II) via cleavage under targets and tagmentation (CUT&Tag)32 (Figure S1B; Table S1). The following chromatin modifications were determined by CUT&Tag sequencing: H2AK119ub (polycomb repressive complex 1 mark; PRC1), H3K4me1 (enhancer marking), H3K4me3 (promoter marking), H3K9ac (active promoters), H3K9me2 (heterochromatin), H3K9me3 (repeat elements and heterochromatin), H3K27ac (active regulatory elements), H3K27me3 (PRC2), and H3K36me3 (polymerase elongation). Different datasets were subjected to a range of differential analyses with pairwise comparisons and then integrated for chromatin-state modeling and TF networking.
Figure 1.

Chromatin modifications display lineage-specific associations with gene expression
(A) Experimental design for the mammary epithelial epigenetic atlas encompassing basal, LP, and ML cells as well as Tspan8+ (quiescent) and Tspan8− (activated) basal cells.
(B) Multidimensional scaling (MDS) plot of RNA-seq data.
(C) Pearson correlation coefficients (p) for each epigenetic mark measured by CUT&Tag and ATAC-seq versus RNA expression.
(D) Heatmap of chromatin changes for DEGs between cell types for ATAC-seq and CUT&Tag (reads per kilobase per million [RPKM], log2) data.
(E) Coverage track plots for ATAC-seq and CUT&Tag at gene promoters.
(F) Heatmap showing the percentage of regions that display significant differential logFC (>0 up/<0 down) for ATAC-seq and CUT&Tag marks between the three cell types (FDR < 0.05).
(G) Bar plot showing TSSs considered to be primed or active across DEGs. See also Figures S1 and S2; Tables S1, S2, and S3.
Promoter regions display unique chromatin patterns in the three major mammary epithelial cell types
Correlation of gene expression, accessibility, and histone modification data was performed for each cell population (Figures 1C and S1C). For most epigenetic marks, except for H3K9me2/3, there was a clear increase in correlation with gene expression in both luminal populations, and as anticipated, the PRC1/2 histone modifications displayed an increased negative correlation with expression, consistent with previous findings.17,21,26 The associations between promoter epigenetic modifications and gene expression in luminal cells were particularly evident when examining differentially expressed genes (DEGs) between pairwise comparisons (Figures 1D and S1D; Table S2). For DEGs enriched in basal cells, there were increases in active marks, including RNA Pol II, H3K27ac, H3K4me3, and H3K9ac, compared with luminal cells, as well as increased chromatin accessibility, and a decrease in the repressive marks H2AK119ub and H3K27me3 (Figure 1E). For genes enriched in LP compared with basal cells, there was no increase in chromatin accessibility at the transcription start site (TSS) in the LP population (Figure 1D); rather, nucleosome-free regions (NFRs) were decreased across the LP epigenome (Figures S1E and S1F). The LP gene NFRs appear to be established in basal cells prior to increased gene expression in the LP population (Figure S1G). Active marks (RNA Pol II, H3K4me3, H3K9ac, and H3K27ac) increased across some luminal gene promoters in both luminal subsets, such as for Cd14, Foxi1, and Wnt4; however, a number of genes exhibited similar activity at their promoter across all three cell types, e.g., Hey1 and Prom1 (Figure 1E). Interestingly, H3K27me3 associated more with gene repression than H2AK119ub (Figure S1D); however, many luminal genes exhibited minimal changes in both marks upon comparison of basal and luminal populations (e.g., Cd14, Foxi1, Hey1, Prom1, and Wnt4) (Figure 1E).
The genome-wide changes in chromatin modifications, transcriptional activity, and accessibility were next examined by de novo detection of differential regions (Figure 1F; Table S3),33 confirming that highly dynamic changes primarily occur at basal gene promoters (Figures S1H and S2A). Chromatin accessibility was found to be dynamic between the basal and the luminal lineages across a large portion of the genome, as were H3K27me3, H3K36me3, H3K4me1, and H3K9me3 marks. Not surprisingly, H3K36me3 displayed large differences between luminal populations, as this “elongation mark” was associated with the gene bodies of most DEGs (71% of “up” genes in LP and 65% of up genes in ML cells) (e.g., Esr1, Fxyd2, Prom1, Pgr, Pinc, and Hey1). H2AK119ub was found at most sites marked by H3K27me3; however, H3K27me3 displayed a greater number of differential regions particularly between the basal and the luminal populations (Figure 1F). Recent reports suggest that PRC1 has functions independent of PRC2-mediated gene repression.34,35 We found several regions of discordance between these polycomb marks exemplified by Acaa1b, Ankrd53, Cdk6, Fgfr3, and Mmp17, suggesting that differential regulation by PRC1 versus 2 occurs in a subset of genes in MECs (Figures 1F and S2B; Table S3). Collectively, these data indicate that the promoter regions of genes highly expressed in luminal cells are often active and primed in the basal population (34% up genes in LP and 32% up genes in ML). Conversely, most genes enriched in the basal population have repressed or silent promoter regions in luminal cells (∼83% up genes in basal cells) (Figure 1G).
Chromatin looping and compartmental changes influence lineage-restricted gene expression
To explore 3D genome architecture in the different lineages, we performed Omni-C (Figures S2C and S2D), which measures the interaction frequency of all loci with all other loci in the genome. Differential interaction (DI) analysis based on the diffHic36 pipeline was used to partition the genome into 50-kb bins and to count the number of read pairs mapping to each pair of bins (an interaction) (Figure 2A). We found more than 30,000 significant DIs between basal, LP, and ML cells (false discovery rate [FDR] < 0.1) (Figure 2B; Table S4) that positively correlated with DEGs (Figure 2C). Evaluation of the top DIs based on significance revealed several loci that displayed marked differences in interactivity, including a cluster of collagen genes (Col6a1, Col6a2, and Col18a1) in basal cells, Dock8 in LP cells, and Plac8, Kcnj11, and Abcc8 in ML cells (Figures 2D and S2E).
Figure 2.

Chromatin looping dictates lineage-specific gene expression in the adult mammary gland
(A) MDS plot of the Omni-C data for the basal, LP, and ML cells.
(B) Number of DIs for pairwise comparisons.
(C) Fold change (log2) of DIs overlapping DEGs for pairwise comparisons.
(D) Normalized contact matrices of the top clustered DIs between basal, LP, and ML cell comparisons. The Omni-C bin pairs are plotted at 20-kb resolution; arcs are DI Z scores (−log10 p values). Triangles indicate loci of significant change.
(E) Percentage of genome undergoing a compartment strength change between cell populations.
(F) Normalized contact matrices of Omni-C data at 20 kb resolution of the Snai2 and Kit loci in basal and LP cells with corresponding Capture-C reads (CPM) for the Snai2/Kit TSSs. Arcs are as in (E). Red, increasing logFC in basal cells; blue, increasing logFC in LP cells with A/B compartments indicated.
(G) Compartment strength (PC1 eigenvalues) and gene expression across the Snai2 and Kit loci. The mean and standard deviation are plotted.
(H) Expression of genes located within A and B compartments.
(I) Expression of DEGs within compartments that are altered in strength for pairwise comparisons of basal, LP, and ML cells. The differential expression of genes that go from A to weak-A/B is shown, demonstrating positive enrichment of expression for genes in the A compartments in each comparison. The p values were determined using two-tailed t tests with Welch’s corrections: ∗p < 0.05, ∗∗∗∗p < 0.0001. See also Figure S2;Tables S4 and S5.
We next determined the percentage of the genome classified into active (A) or inactive (B) chromatin compartments (Figure S2F). Compartments were altered in ∼6% of the genome between basal and luminal populations, while remaining stable between LP and ML cells (<0.7% altered) (Figure 2E; Table S5). Two regions of significant compartment switching encompassed the Snai2 (basal specific) and Kit genes (LP specific) (Figure 2F). The Kit locus displayed increased chromatin looping in LP cells (indicative of an A compartment) compared with basal cells, which had significantly reduced chromatin contacts, characteristic of B compartments. Snai2 displayed the reverse pattern compared with Kit, with enrichment of interactions in basal cells relative to LP cells. Analysis of genes located within compartments that differed between cell populations demonstrated that >75% show increased expression when their chromatin region changed from a B to an A compartment or when the A compartment showed an increase in interactivity (Figure S2G), as apparent for the Snai2 and Kit loci (Figure 2G). As expected, gene expression was higher in A versus B compartments (Figure 2H). Interestingly, gene expression was significantly higher for genes in B compartments in basal cells compared with those in B compartments for luminal cells (Figure 2H). In altered compartments, there was stronger gene repression from the basal to the luminal populations (basal A to luminal weak-A or B compartment) (Figure 2I). While B compartments exhibited reduced gene expression, these data raise the possibility that gene expression in basal cells is less reliant on chromatin interactions and compartment strength compared with the luminal lineages.
As expected, all active chromatin marks were higher in A than B compartments, whereas the heterochromatin mark H3K9me2 was enriched in B compartments (Figure S2H). Interestingly, H2AK119ub, but not H3K27me3, was higher in A compartments. H3K4me1, H3K36me3, H3K4me3, and H3K27me3 correlated with changes in compartment strength between cell comparisons (Figure S2I). Surprisingly, we saw little correlation between compartment switching, chromatin accessibility, and H3K9ac, suggesting that these epigenetic features may be regulated independent of chromatin interactivity. Collectively, these data indicate that major changes in genome architecture and epigenetic modifications distinguish the basal versus luminal lineages, accompanied by alterations in gene expression. Genome architecture, however, was largely unchanged between LP and ML cells despite dynamic changes in epigenetic marks.
Super-enhancers frequently interact with luminal progenitor genes
To further examine lineage-restricted differences in chromatin interactions at high resolution, we employed Capture-C.30 This enabled mapping of short-range interactions involving the TSSs of 18 genes, which were mainly lineage-restricted TFs (Figures S3A and S3B). Capture-C and Omni-C data displayed high concordance, as illustrated by Cited1 and Foxa1, which show increased interactivity between their promoter regions and distal sites in ML cells (Figures 3A and S3C). A number of genes displayed increases in TSS connectivity between cell types that positively correlated with expression (ΔNp63, Snai2, Kit, and Msx2) (Figure 3B). LP gene TSSs were the most interactive with their surrounding chromatin (Figures 3B and S3D). This increased TSS interactivity suggests the involvement of super-enhancers (SEs), as they have previously been shown to interact more frequently than typical enhancers (TEs).37,38
Figure 3.

LP genes are highly interconnected to intragenic super-enhancers
(A) Normalized contact matrices of the Omni-C data at 20 kb resolution and Capture-C reads (CPM) for the Cited1 and Foxa1 loci. Arrows indicate sites connected to the TSSs in ML cells.
(B) Normalized Capture-C reads of the 18 selected genes.
(C) Identification of SEs and TEs in MECs. Examples of SEs associated with lineage-enriched genes are highlighted.
(D) Expression of genes associated with either TE or SEs. Fold change expression between TE and SE is indicated.
(E) The number of interactions, as determined by FitHiChIP, connecting genes to a TE or SE.
(F) SE acetylation, shown as the percentage of the pooled biological replicate counts.
(G) The distance of cDI regions for pairwise comparison between basal, LP, and ML cells.
(H) Coverage track plots of gene bodies showing Capture-C (CPM) (500-bp sliding windows), CUT&Tag (RPKM), and SEs. Arrows highlight significant regions of TSS to gene body interactivity.
(I) In silico 5C analysis of Omni-C data of DEGs.
(J) In silico 4C analysis, measuring TSS-to-intergenic regions (left) and TSS-to-gene-body interactions (right). The p values were determined using two-tailed t tests with Welch’s correction (D, E, F, and G) or paired two-tailed t tests (I and J): n.s., not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Boxes show median, quartiles, and 5th and 95th percentiles. See also Figure S3; Table S6.
To identify potential SEs, we applied the ROSE algorithm39 to the H3K27ac data and ranked regions based on read density (Figure 3C). SEs were identified for a number of lineage-specific genes derived from signature analysis (35% of basal genes, e.g., Trp63, Col16a1, and Myh11; 42% of LP genes, e.g., Notch1, Ehf, Kit, and Elf5; 33% of ML genes, e.g., Esr1, Foxa1, and Prlr). Overall, 444, 174, and 170 SEs were unique to the basal, LP, and ML populations, respectively. Basal cells comprised the most enhancer regions (either TEs or SEs), and surprisingly, LP cells displayed ∼40% fewer TEs and ∼20% fewer SEs. However, an increased proportion of SEs (11%) were unique to LP cells compared with TEs (6%) (Figure S3E). Relative to TEs, SEs were associated with genes showing increased expression (Figure 3D) and TSS-SE interactions, with SEs in LP cells showing the highest TSS-SE interactivity (Figures 3E and S3F). In conjunction, SEs in LP cells showed increased H3K27ac compared with basal SEs (Figures 3F and S3G) and spanned shorter distances compared with SEs in either basal or ML cells (Figure S2H).
To determine whether increased interactivity in LP cells associated with the span of TSS-distal regions, we performed a differential analysis based on the Capture-C data (capture DIs [cDIs]) (Table S6). Both luminal populations displayed an enrichment for cDIs with a smaller span compared with basal cells (cDIs spanning <200 kb: ∼27% of basal up genes, ∼48% of LP up genes, and ∼35% of ML up genes) (Figure 3G). Indeed, the genome-wide Omni-C data showed a similar pattern, with enrichment of interactions spanning 5–100 kb in luminal cells, while those spanning >500 kb were highest in the basal population (Table S1). When examining the Capture-C signals for genes >30 kb in length (9/18 capture genes), we observed highly significant (FDR < 0.05) intragenic cDIs for five genes, Trp63 and Tcf7 in basal cells, and Elf5, Ehf, and Kit in LP cells, with overlapping intragenic SE sites (Figure 3H; Table S6). Interestingly, when extended to all lineage-specific TFs, we observed that 45% of LP TFs contained SEs across their gene bodies compared with 25% of basal and 17% of ML TFs, suggesting that high TSS interactivity relates to short-range interactions with SEs within gene bodies.
To examine TSS interactivity on a genome-wide scale, we performed in silico 4C and 5C analysis on the unbiased Omni-C libraries (Figure S3I). These global and unbiased analyses revealed that LP cells are more enriched for TSS-intragenic and TSS-intergenic interactions compared with basal and ML cells (Figures S3J). When assessing DEGs, we observed an association between expression, gene-body connections (Figure 3I), and intragenic and intergenic connectivity in all comparisons performed (Figure 3J), and these correlated with differences in the number of DE genes connected to an SE (Figure S3K). These genome-wide data confirm findings from Capture-C experiments that LP TSSs are more interactive than basal or ML promoter regions. Moreover, the data suggest that short-range interactions, especially those from TSSs to gene bodies, are more frequent in LP cells despite widespread SE involvement across the different cell types.
Chromatin looping determines transcriptional start-site usage
Alternative TSS usage from the canonical TSS provides an additional regulatory mechanism for cell-specific expression. We therefore investigated whether alternative promoter usage displays lineage specificity and associates with differential enhancer activation in MECs. To explore “on” or “off” TSSs, we identified TSSs that have little or no RNA expressed in any epithelial subset (e.g., TAp63, “off” 12,817 TSSs) and those where the TSS was expressed in at least one cell type (e.g., ΔNp63, “on” 16,945 TSSs) (Figure 4A). Trp63, encoding a key basal-restricted TF required for mammary gland development,40,41 has two known TSSs that produce the longer TAp63 (not expressed in MECs) and shorter ΔNp63 (basal-enriched) isoforms (Figures 4B and S4A). The ΔNp63 isoform interacts extensively with a basal SE within its gene body and another within the Tprg gene, which is a transcriptional target of p63.42 Many genes showed a similar pattern to Trp63 with one “off” TSS and one or more “on” TSSs, including Elf5, Grhl1, Runx1, and Cd36. TSS to gene-body chromatin interactions were higher for “on” TSSs, as were typical promoter activation marks (Figures 4A and S4B). Interestingly, intragenic interactions appeared to be dependent on the position of the alternative TSS within the gene body, with those at the 5′ end being the least interactive (Figure 4C). These data indicate that the “on” state of a TSS is associated with increased chromatin interactivity in conjunction with increased active histone marks, compared with those TSSs switched “off.”
Figure 4.

Super-enhancer interactivity determines Trp63 and Foxp1 isoform expression
(A) Expression and connectivity of TSSs identified as “off” or “on.” t test with Welch’s correction for “off” versus “on” TSSs. ∗∗∗∗p < 0.0001. Boxes shows median, quartiles, and 5th and 95th percentiles.
(B) Coverage track plot of the Trp63 locus. Shown are the isoforms, alternative TSSs, in silico 4C for ΔNp63 and TAp63 TSSs (CCPM), CUT&Tag (RPKM), SEs, and interactions between the TSSs and the distal regions.
(C) Heatmaps of expression and chromatin connectivity for TSSs based on their position within the gene.
(D) Heatmap of DEATSS expression with examples indicated.
(E) Western blot for Foxp1 expression. Histone 3 (H3) was used as a loading control.
(F) Foxp1 TSS expression, annotated as genomic coordinates.
(G) Chromatin state of the Foxp1 locus. Shown are the isoforms, SEs, in silico 4C (CCPM) for Foxp1-A and Foxp1-D TSSs, coverage (RPKM) for CUT&Tag, and Omni-C interactions called by FitHiChIP.
(H) Foxp1 gene-body (GB) connectivity determined by in silico 5C analysis.
(I) Foxp1 chromatin interactivity between the alternative TSSs and the GB determined by in silico 4C analysis. Genome coordinates are from Mus musculus mm10 (GRCm38). The p values were determined using one-way ANOVA: ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. See also Figure S4; Table S7.
To identify differentially expressed alternative TSSs (DEATSSs), we determined differentially expressed TSSs (DETSSs) across lineages and then measured intragene variation between alternative TSSs (DEATSSs) (Figure S4C). We identified 196 DEATSSs between basal and LP cells: 237 for basal versus ML cells, and 102 for LP versus ML cells (Figure 4D; Table S7). For example, Eya2 and Lrrfip1 displayed DEATSS usage between the basal and the luminal lineages (Figures S4A and S4D).
The TF Foxp1, which is critical for the activation of MaSCs,43 is translated as two major isoforms (A and D) that originate from DEATSSs. Foxp1-D is strongly enriched in basal cells, while Foxp1-A occurs at similar levels across the three populations (Figure 4E). Expression of the Foxp1-A and -D mRNAs parallels these findings (Figure 4F). Interestingly, the Foxp1-D TSS interacted more frequently with three intragenic SE regions and the Foxp1-A TSS in basal relative to luminal cells (Figure 4G). Notably, interactions within the gene body of Foxp1 and those specifically involving Foxp1 TSSs were higher in basal than in luminal cells (Figures 4H and 4I). Given these lineage-specific chromatin interactions with SEs and the critical function of Foxp1, the role of Foxp1-D in mammary gland development warrants further attention.
Chromatin modeling reveals dynamic changes in enhancer and promoter states in mammary epithelial cells
To decipher the contributions of different histone modifications to chromatin states, we integrated the CUT&Tag and ATAC-seq datasets and utilized ChromHMM44 to model genome-wide chromatin states (Figure 5A). With 12 input datasets, we produced a 24-state model comprising five promoter (TSS), six enhancer (Enh), two transcription (Tx), three polycomb (PRC), three quiescent/silent (Quies), and five heterochromatin and repeat element states (Het) (Figures S5A and S5B). An increased percentage of the genome in basal cells was covered by active promoter states 1 and 3 (Figure 5B), concordant with widespread promoter priming evident in these cells. Consistent with the Capture-C analysis, intragenic enhancers (Enh_A_G, state 6) were most abundant in LP cells. Regions covered by H2AK119ub (primed and PRC1/2 states 10, 14, and 16) also appeared more abundant in LP cells, while bivalent promoters (state 5) increased from basal to LP to ML cells. Moreover, the five promoter (1–5) and three polycomb states (14–16) differentially associated with transcriptional activity, with state 1 reflecting the most highly expressed genes and states 14 and 15 (promoters covered by PRC1/2 and PRC2) associated with the lowest expressed genes (Figure 5C). Interestingly, genes marked by bivalent or polycomb-repressed chromatin appeared to have higher expression in basal cells compared with those similarly marked in LP or ML cells. To explore this further, we devised a summary score of TSS and CRE activity based on our ChromHMM modeling and chromatin looping, enabling a hierarchical clustering of DEGs by their scores and gene expression (Figure 5D). Cluster C2 highlights a number of genes (e.g., Tbx21, Tcf24) that are marked with bivalent or polycomb-enriched chromatin across all cell types but are significantly upregulated in basal cells (Figure S5C).
Figure 5.

Chromatin state modeling reveals differential usage of enhancers and polycomb regions in the different mammary epithelial lineages
(A) Epigenomes of epithelial subsets modeled as 24 chromatin states with ChromHMM using ATAC-seq and CUT&Tag data. Heatmap displaying frequencies of chromatin marks and IgG (control) across each state, with annotations listed on the left and terms on the right. A1/2, active; Biv, bivalent; Flk, flanking; G, genic; Prm, primed; Wk, weak.
(B) Genomic overlap of ChromHMM states across MECs.
(C) Expression of genes with a defined TSS, states 1–5, and polycomb states 14–16. The p values were determined using two-tailed t test with Welch’s correction: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(D) Hierarchical clustering of DEGs by expression, TSS, and enhancer score. Shown from left to right is the cluster dendrogram, DEGs for pairwise comparisons, differential expression log2 FC (DE), TSS score (TS), CRE score (CS), expression (RNA-seq, RPKM, log2), TSS ChromHMM states, number of significant interactions as determined by FitHiChIP, CRE ChromHMM states, cluster numbers, and highlighted genes.
(E) State similarity (Jaccard values) for pairwise comparisons of basal, LP, and ML cells.
(F) Active enhancer regions (states 6 and 7) displayed as altered state for each pairwise comparison. Shown is the percentage of active enhancer regions in one subset and the percentage of those same regions that are weak/marked (state 8) or primed (state 10), polycomb only (states 14, 15, and 16), or silent/heterochromatin (states 17 to 24) within the comparator subset.
(G) Hierarchical clustering of LP-alveolar genes, as identified in Pal et al.25 Shown from the left to right: cluster dendrogram, single-cell RNA expression (Z scores), TSS score, TSS ChromHMM states contributing to the TSS score, CRE score, number of significant interactions as called by FitHiChIP, CRE ChromHMM states contributing to the CRE score, subclusters, and highlighted genes.
(H) Chromatin state for TSS regions of DEGs.
(I) The number of interactions detected by FitHiChIP for genes categorized based on their TSS chromatin state.
(J) The median genomic distance between TSSs and CREs. See also Figures S5, S6, and S7; Table S8.
To explore chromatin state similarities between MECs, we examined Jaccard values and Sankey transition plots (Figures 5E and S5D). State 1 active promoters and intragenic enhancers (state 6) were the most stable between cell types, while the weaker promoters (states 2 and 3), weaker enhancers (state 8), and active intergenic enhancers (state 7) were the most dynamic regulatory elements (Figure 5E). Most active enhancers (states 6 and 7) exhibited de novo formation from silent to active chromatin (Figure 5F). Active enhancers in luminal cells were more likely to be weak/marked enhancers (state 8, 20%) in basal cells, whereas active enhancers in basal cells were more often marked by polycomb in luminal cells (16% state 10 and 21% states 14–16). Enhancer marking in LP cells for activation in ML cells (e.g., enhancers of Cldn6, Fxyd2, Wnt4, Mgat5, and Fzd10) was slightly less prevalent (25%–34%) than de novo formation of active enhancers in ML cells (38%–40%) (e.g., Foxa1, Prom1, Syt17, Fxyd2). These observations indicate that basal cells are more likely to mark enhancers without repressive polycomb modifications, while luminal cells utilize polycomb-marked chromatin to either repress enhancers or prime them for activation.
Little concordance was observed between PRC1- and PRC2-marked chromatin, as PRC1 states (states 10 and 16) were more dynamic between cell populations compared with those defined by PRC2 (states 5, 14, and 15) (Figures 5E and S5D). Despite the strong genome-wide correlation between H3K27me3 and H2AK119ub, we observed a reduced correlation in basal cells at regions where PRC2 was highest (state 5, bivalent promoters, and state 14, PRC1/2) (Figure S5E). Thus, PRC1 may not be required for all bivalent and polycomb-repressed regions in basal cells but may be more critical for repression in luminal cells (Figure 1C). Furthermore, we observed more polycomb-marked enhancers in luminal cells that were active in basal cells, compared with the reverse (Figure 5F).
State 10, which exhibits increased H2AK119ub over H3K27me3, appears to represent a “primed” enhancer state, and active basal enhancers were more likely to be primed rather than weak/marked in luminal cells (Figure 5F). Interestingly, these primed regions were more enriched for CpG islands than active enhancers, consistent with the known link between DNA methylation and polycomb repression (Figures S5A and S5F). We speculated that these regions may become active during pregnancy, as LP cells comprise precursors for the alveolar lineage. To assess gene changes in LP cells during pregnancy, we assessed the expression of marker genes for the LP-alveolar lineage cluster from our single-cell RNA-seq dataset spanning adult, early and late pregnancy, lactation, and involution (cluster 1 in the pregnancy cycle).25 We reclustered the marker genes and overlaid the ChromHMM TSS and CRE states onto the four subclusters formed (Figure 5G). Genes that were upregulated in pregnancy, such as Wap, Wfdc18, and Litaf (sub-clusters C1, C2, and C3), displayed increased chromatin contacts with primed enhancers and polycomb-repressed regions (Figure S5G). The previously identified Wap SE region28 was found to be marked by H2AK119ub in luminal cells, with enhancer 1 (proximal to the Wap TSS and the first to be activated during pregnancy) exhibiting state 10 chromatin marked by H3K4me1, low-H3K27ac, low-H2AK119ub, and low chromatin accessibility, especially evident in the LP population (Figure S6A). While Wfdc18 exhibited an active promoter region, the TSS interacted with distant primed and polycomb regions exclusively in luminal cells (Figure S6B). Hence, through the inclusion of the PRC1 mark (H2AK119ub) combined with the enhancer mark (H3K4me1) in our ChromHMM analysis, “primed” regions were shown to harbor low levels of H2AK119ub without H3K27me3, suggesting that these regions may serve to prime enhancers for activation during pregnancy, such as in the case of Wap.
Previous reports have shown that bivalent domains can poise genes for activation during development.45,46 We therefore determined genes that displayed bivalent promoters unique to each lineage (248 basal, 123 LP, and 209 ML genes) and performed a Gene Ontology (GO) analysis (Table S8). Interestingly, we found that bivalent genes in LPs were enriched for GOs associated with pregnancy, consistent with these cells serving as progenitors for alveolar cells. Bivalent genes within the basal population were enriched for membranal proteins and signaling genes, while those in ML cells were enriched for cell adhesion and mammary morphogenesis and, surprisingly, included genes enriched in the ML population (Figures S6C and S6D). These data suggest that bivalent chromatin may poise gene expression in the LP population but associate with expressed genes in ML cells.
Promoter states exhibit differential interactivity with enhancer and silencer regions
We next analyzed the impact of chromatin states on the expression of cell-specific genes (Table S2; Figures 5H and S6C). Promoter regions of genes highly expressed in basal cells (e.g., Tbx3, Mycn, and Snai2) were mostly active (states 1 to 3) in basal cells and weak, bivalent, or repressed (states 2, 5, 14, 15, and 16) in the luminal populations (clusters C4, C9–C11) (Figures 5D and S6C). By contrast, for genes highly expressed in LP cells (e.g., Rora, Hey1, and Tcf7l2) (Figure 5D), many promoters showed similar activity in basal (70% with state 1, 2, or 3) compared with LP cells (88%) and minimal repressive or bivalent states. Many genes enriched in ML cells, 58% of those with lower expression in LP cells (e.g., Myb, Esr1, Esrrb, Pgr, Cited1, Bmp3), exhibited bivalent and polycomb-enriched chromatin at their promoter regions (cluster C6, states 5 and 14) (Figures 5D, S6C, and S6D), suggesting either heterogeneity within the ML population or a more complex mechanism of promoter-mediated transcription such as allele-specific repression.
To identify significant chromatin interactions (FDR < 0.1) within each cell type, the FitHiChIP pipeline was applied to link distal CREs (>20 kb up- or downstream of the TSS) and TSSs genome wide. Examination of the average number of significant interactions for each promoter and polycomb state (H3K27me3 and H2AK119ub states overlapping TSSs) revealed that state 1 promoters showed the highest level of interactivity with distal regions (Figure 5I). Similar to the Capture-C data, LP gene promoters (states 1–4) were more interactive, interacting with ∼2.4- and ∼1.3-fold more chromatin than basal and ML promoters, respectively (clusters C7 and C8) (Figure 5D). We then examined the expression of genes that have no interacting CREs or increasing numbers (up to >9) (Figure S6E). Enhancer regions (state 7) had the greatest positive influence on expression, while bivalent promoters (state 5) and polycomb regions containing PRC2 (states 14 and 15) were negatively associated with expression, serving as putative silencer elements. State 10 CREs had no impact on expression, consistent with these regions being “primed” and not yet influencing transcription. We also observed an association between CREs and the genomic distance they span to interact with their target TSS, with enhancers generally located closer to TSSs than to the more distal polycomb-marked sites (Figure 5J).
We next investigated the type of distal CRE that interacted with promoter elements. Active promoters (state 1) interacted more frequently with active enhancers and transcribed regions, while repressed promoters (states 5, 14, and 15) were more often linked with polycomb-silenced distal regions (Figures 5D and S6F). There was a clear positive association between expression and active promoters, enhancers, and transcribed regions (states 1–4, 6–8, and 11–13) and a negative association with polycomb regions (states 14–16) for each population (Figure S6G). Genes enriched in ML compared with LP cells displayed a different pattern, with a lack of interacting active enhancers (states 6 and 7) but an increased involvement of weaker enhancers and interacting active or weak promoters (states 1–3 and 8). To deconvolute chromatin interaction complexity, we turned to UpSet plot analysis (Figures S7A and S7B). Gene interactions involving polycomb silencer regions displayed decreased expression in the luminal populations but minimal impact on basal cell expression (clusters C9, C10, and C11) (Figure 5D). This was particularly evident for the basal-specific gene Cxcl14, where an intergenic region comprising multiple enhancers displayed polycomb-marked chromatin in both luminal populations but maintained chromatin interactivity (Figure S7C). Similarly, the basal-restricted TF Dzip1 demonstrated the highest level of interactivity in LP cells where the promoter appears to interact with distant PRC1 and primed enhancer sites. Overall, these data indicate that basal lineage specification relies on typical promoter and enhancer epigenetic profiles. By contrast, promoters of the luminal lineages appear promiscuous, with many LP TSSs exhibiting a weak/active state and bivalent modifications occurring across ML TSSs in all cell types. Notably, luminal enhancer and silencer elements displayed exquisite cell specificity, likely accounting for lineage determination.
Lineage-specific transcription factors are defined by their chromatin binding profiles
To test whether gene activation could be achieved through distal CRE involvement, three lineage-specific TF genes representing the primary lineages were investigated using CRISPRa-dCas9 editing47 in COMMA-DβGeo cells: Snai2, Elf5, and Msx2 (Figures 6A and 6B). The Snai2 promoter interacts with at least three distal enhancer loci that display basal cell specificity. Targeting of an 18-kb region within an Efcab1 intron (221/239 kb downstream) had the strongest impact on Snai2 expression (Figures 6C and S8A). Interestingly, the effect of enhancers on Snai2 transcription appeared to be independent of distance from the TSS, with similar increases observed for the closest (94/95 kb) and furthest enhancers (405/409 kb downstream). The Elf5 promoter region interacts with distal and intragenic SEs that display de novo activation in LP cells and retain activity in the ML population (Figures 6A and S8A). Targeting the intragenic enhancer or the upstream enhancer (58/59 kb) led to increased Elf5 expression (Figure 6D). Msx2 harbors a promoter region that displays bivalent chromatin across each cell type but paradoxically interacts with enhancers that are active in ML cells (Figures 6A and S8A). Targeting the distal upstream enhancer region (184/186 kb) led to a robust increase in Msx2 (Figure 6D). Thus, enhancers can be specifically targeted in MECs to augment target gene expression.
Figure 6.

Long-range regulation of transcription factor genes defines the mammary epithelial lineages
(A) Coverage track plots. Capture-C is shown as CPM, 500-bp sliding windows, Omni-C (CCPM) for interactions anchored at TSS regions (10 kb resolution).
(B) Experimental design of sgRNA targeting of lineage-restricted enhancers using CRISPRa-dCas9.
(C) Western blot analysis of Snai2 expression following sgRNA-mediated enhancer activation versus non-targeting control (NTC). Hsp70, loading control.
(D) qRT-PCR of Elf5 and Msx2 expression, following enhancer activation (n = 2). The mean and standard deviation are shown.
(E) TF networks for lineage-specific TFs. Node color represents gene expression (RNA-seq, RPKM, log2), node size shows network connectivity, lines represent footprinted motifs with thickness as relative enrichment. See also Figure S8.
To identify potentially novel regulators of cell fate and TF binding dynamics across the genome, we performed footprinting analysis with TOBIAS48 on the ATAC-seq data. Networks were constructed for the cell-specific TF signatures (Figures S8B and S8C) by mapping footprinted binding sites to gene promoters and interacting regions (Figure 6E).49 This analysis revealed highly interconnected TFs in both basal and LP cells, but less so in ML cells, due to a reduced number of interactions (Figures 5I and 6E). In basal cells, Snai2, Trp63, and Foxp1 were highly interconnected, but several other regulators dominated the network, including Egr2, Egr3, Prdm1, Runx1, Tead1, Nfatc1, and Nfyb. The Egr, Nfat, Runx, Tcf, and Tead motifs were also found to be enriched in human breast basal TF networks.26,50 Using ChromHMM-defined states to map the DNA-binding elements bound by these TFs, we predicted that Egr2, Nfyb, and Egr3 primarily bound to active TSSs, while Prdm1, Snai2, Trp63, Tead1, Nfatc1, and Runx1 were likely enhancer-bound factors (Figure S8D). In LP cells, the known alveolar lineage regulator Elf551 and the Notch pathway effector Hey141,52 emerged as key TFs together with potentially novel regulators Ror⍺, Rorγ, and Crem, which demonstrated high expression and interconnectivity, suggesting central roles in LP maintenance or differentiation (Figure 6E). Hey1 displayed strong enrichment at active TSSs, while Elf5 and Crem were predicted to bind promoter and enhancer regions and Ror⍺ and Rorγ to serve as strong enhancer-bound factors (Figure S8E). Many ML-enriched TFs have been previously studied (e.g., PR, ER, and Foxa1), but the developmental roles of other well-connected TFs such as Smad3, Myb, and Meis1 are yet to be determined (Figure 6E). The binding of these TFs was enriched at active enhancers, suggesting that distal elements are a central point of gene regulation in ML cells, such as the case for Msx2 (Figure S8F).
Chromatin accessibility dictates cellular states within the basal compartment
To interrogate chromatin structure in the Tspan8+ (quiescent basal; QBa) and Tspan8− (active basal; ABa) sub-populations,14 sorted cells were subjected to ATAC-seq, low-cell CUT&Tag, and Omni-C sequencing (Figure 1A; Table S1). Differential gene expression and differential accessibility (ATAC-seq data) analysis identified 8.1 × 106 100-bp bins with increased accessibility in Tspan8− cells and 5 × 106 in Tspan8+ cells (Figure 7A; Tables S2 and S3), revealing that accessibility at promoter and distal CRE sites increased with expression in most cases (Figure 7B). Notably, we observed increased chromatin accessibility in Tspan8+ cells throughout the Slc14a1 locus (highest DEG) and the adjacent Lgr5 and Tspan8 loci, which together define the most quiescent state14 (Figure 7C). Broad changes to luminal gene accessibility were not observed between the two basal populations, consistent with the CUT&Tag data. Thus, luminal gene priming appears ubiquitous within the basal compartment (Figure S9A).
Figure 7.

Chromatin accessibility remodels the basal genome upon exit from quiescence
(A) Differentially accessible (DA) regions between basal Tspan8+ (quiescent; QBa) and Tspan8− cells (activated; ABa).
(B) DEGs between QBa and ABa cells with a DA region overlapping the promoter or DA region overlapping ≥1 CREs interacting with their promoter.
(C) Coverage track plots for QBa and ABa loci.
(D) Genomic enrichment (observed/expected) of chromatin states overlapping DA regions connected to DEGs.
(E) Differential TF network comparing QBa versus ABa. Node and line colors depict differential expression and binding, respectively; line width shows motif enrichment and node size depicts network connectivity.
(F) Chromatin states for TF loci, with footprinted TF motifs indicated. See also Figure S9; Tables S1, S2, and S3.
Widespread changes in chromatin interactions were not evident between the two basal subsets, as no DIs were found. Through ChromHMM-modeling, however, differentially accessible (DA) sites were shown to be enriched for intergenic enhancer states in Tspan8+ cells, while both intragenic and intergenic enhancers were enriched in more accessible regions in Tspan8− cells (Figure 7D). Alterations in accessibility were shown to associate with changes in enhancer states (i.e., active to weak enhancer: Lama1, Col4a2, Ptx3, Col6a1), enhancer repression (i.e., active to primed or repressed region: Ccnd1, Col4a2, Col6a1), and enhancers whose activity did not change (i.e., active to active or weak to weak: Col4a2, Plet1) (Figures S9B and S9C). These findings indicate that active enhancer states (states 6 and 7) are stable within the basal population, whereas de novo enhancer activation or repression tends to occur for weaker enhancers (states 8–11) and is likely the result of changing chromatin accessibility and not interactions (Figure S9D).
A differential TF network was next created for the enrichment of footprinted binding sites mapped to differentially expressed promoters and interacting regions in the two basal subsets (Figure 7E). This analysis identified a number of highly interconnected factors within the network: Fli1, Rara, Erg, Foxc1, Gli1, and Batf were enriched in Tspan8+ cells and E2f8, Dlx3, Egr4, Tbx2, Etv4, Etv5, Ahr, Zfhx4, and E2f1 in Tspan8− cells. The most differentially expressed TFs included Batf, a member of the AP-1/ATF superfamily of TFs,53 and Tbx2, which has been implicated in mammary development along with its family member Tbx3.54 Batf expression was higher in Tspan8+ cells in conjunction with enrichment of motifs for the putative regulators Egr2, Gata3, and Elf1, which are predicted to bind the promoter and enhancer regions of Batf (Figures 7E and 7F). The early-growth-responsive TF Egr2 was also implicated in the regulation of Tbx2, alongside Egr4 and E2f8, through binding at promoter and enhancer regions in basal Tspan8− cells. The roles of Batf, Egr, and Ets family members (with the exception of Elf5) in the mammary gland have yet to be elucidated.
Discussion
Epigenetic regulation is central to tissue-specific function, where higher-order chromatin structure allows cell-specific TFs to govern specification and differentiation.2,55 Through a multimodal analysis of chromatin structure, we provide evidence that gene regulation in the different epithelial cell types is underpinned by distinct epigenetic mechanisms. Basal-specific genes are characterized by typical interactions between promoters and distal enhancers and increased involvement of silencer elements in luminal cells. However, the promoter regions of many luminal-specific genes were active in basal cells, with lineage-specific expression achieved through extensive chromatin interactivity with enhancer regions. LP-specific genes were characterized by the highest level of chromatin looping between lineage-specific intragenic SEs and the promoters of TF genes. In ML cells, many genes exhibited bivalent chromatin domains over their promoters, despite interacting with active and/or weak enhancers. Together these data highlight the plastic nature of chromatin in basal cells, where the luminal gene program is primed for activation, while in the more differentiated luminal compartment, the basal program is repressed, consistent with the inability of these cells to reconstitute a functional gland upon transplantation.56,57
Investigation of chromatin interactivity on a global scale revealed extensive enhancer marking and de novo enhancer activation across the different MEC types. Enhancer marking of luminal genes (20% of enhancers), including Notch1, Foxi1, Fhad1, and Rora, was evident in basal cells, consistent with a previous report for human breast,26 while de novo activation in LP cells (60% of enhancers) was seen for enhancers of other LP genes, such as Cd14, Krt8/18, Klf6, and Gata3. Similarly, enhancers of Prlr, Meis1, Msx2, Myb, and Smad3 were marked in LP cells (25% of enhancers), presumably for activation in ML cells, but enhancer priming (state 10) in luminal cells was more frequent than in basal cells (24%–30% versus 8%–16%). The enhancer marking together with promoter priming and bivalency seen for ML genes in LP cells is consistent with the LP pool contributing to both the HR− and the HR+ lineages.15,16 Thus, luminal lineage priming predominantly occurs in basal cells at the promoter level, while lineage priming of ML genes in the LP population occurs extensively at both promoter and enhancer regions. These data also suggest that de novo activation of enhancers is at least as prevalent as enhancer marking.
Substantial evidence for enhancer priming was seen in the LP pool, which may be PRC mediated through H2AK119ub. Recent reports have shown that PRC1 is critical for chromatin structure and that its loss diminishes chromatin looping irrespective of whether the target gene transcription increases or decreases.58 Potential chromatin remodeling by PRC1 and enhancer activation in the mammary gland during periods of expansion would support earlier findings that H3K27ac permanently increases across the epigenome in post-pregnancy glands.59 While a positive association between enhancer marking (low H3K4me1, H3K27ac, chromatin accessibility, states 8/9) and expression was observed, primed enhancers (H3K4me1 and low H2AK119ub, state 10) interacted minimally with target genes, with no effect on expression, indicative of distinct enhancer states. Thus, PRC1 may play a novel role in hormone-driven “epigenetic priming” of mammary epithelium.17,18,21,60 Furthermore, we identified lineage-specific SEs that overlapped crucial mammary regulatory genes, including Trp63, Foxp1, Foxa1, Gata3, Esr1, and Notch1. In the case of Foxp1, we identified differential SE interactions with TSSs in basal cells that likely control alternative isoform expression, implicating SEs in influencing promoter usage.
Through a multifaceted integrative analysis, we identified candidate TFs likely to execute roles in lineage restriction along the mammary epithelial hierarchy. In basal cells, these include known regulators (e.g., Trp63, Foxp1, Snai2, and Id4) and novel factors (e.g., Egr and Batf) that were linked through chromatin interactivity studies. Our data also highlight the emergence of the retinoic acid-related orphan receptors Ror⍺ and Rorγ as putative regulators of TFs in LP cells. The majority of ML TFs preferentially bound to enhancer sites; in combination with active genes exhibiting promoter bivalency, these findings suggest that distal enhancers are critical for specification in ML cells. It is likely that enhancer-binding TFs such as Trp63, Foxp1, Rorγ, Foxa1, and Pgr contribute to the maintenance of chromatin interactions or control loop formation, analogous to the roles of TFs in driving changes in chromatin architecture in other systems.61,62 Future work will entail dissecting the novel TF networks in refined mammary epithelial subsets at a functional level, including their physiological roles during development.
Limitations of the study
First, based on population-level sequencing of RNA and epigenetic features, we cannot exclude the possibility that bivalent domains and weak promoters/enhancers represent heterogeneity within the basal, LP, or ML/HS populations. For example, the small HR+ LP subset (5%–10%) in the luminal compartment could display different epigenetic patterns across regulatory elements. Due to the input required for interactivity studies, it was not possible to isolate sufficient cells from this subset. Second, we utilized ChromHMM to model chromatin states based on nine histone marks, RNA Pol II, and chromatin accessibility. Modeling is predictive by nature and warrants confirmation by functional studies based on in vivo perturbation of key regulators coupled with chromatin state analyses. For example, ablation of PRC1 within the mammary gland could assess the importance of this mark in establishing primed enhancers (state 10). Third, although validation of enhancers was performed in a relevant cell line, chromatin interactions and enhancer activity may differ in primary cells. Finally, despite the rapid growth of single-cell technologies, it is not yet possible to assess the plethora of histone marks and RNA and chromatin interactions within a cell at the same level of precision as for bulk populations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rat FITC anti-mouse CD29 (clone HMβ1-1) | Biolegend | Cat# 102206; RRID: AB_312882 |
| armenian hamster Pacific blue anti-mouse CD24 (clone M1/69) | Biolegend | Cat# 101820; RRID: AB_572010 |
| armenian hamster PE anti-mouse CD61 (clone HMβ3-1) | Biolegend | Cat# 104308; RRID: AB_313084 |
| rat APC anti-mouse CD31 (clone 390) | Biolegend | Cat# 102410; RRID: AB_312905 |
| rat APC anti-mouse CD45 (clone 30-F11) | Biolegend | Cat #103112; RRID: AB_312977 |
| rat APC anti-mouse TER-119 (clone TER-119) | Biolegend | Cat #116212; RRID: AB_313713 |
| anti-Foxp1 | Cell Signaling Technologies | Cat# 2005; RRID: AB_2106979 |
| anti-total Histone 3 | Merck Millipore | Cat# 07-690; RRID: AB_417398 |
| anti-α-tubulin | Sigma Aldrich | Cat# T6199; RRID: AB_477583 |
| anti-Snai2 | Cell Signaling Technologies | Cat# C19G7; RRID: AB_2239535 |
| anti-HSP70 (clone N6) | In house | N/A |
| mouse IgG (Western blot) | Southern Biotech | Cat# 1010-05 |
| rabbit IgG (Western blot) | Southern Biotech | Cat# 4010-05 |
| Guinea pig anti-Rabbit IgG (H+L) | Sigma Aldrich | Cat# SAB3700890 |
| anti-H2AK119ub | Cell Signaling Technologies | Cat# 8240; RRID: AB_10891618 |
| anti-H3K4me1 | Abcam | Cat# 8895; RRID: AB_306847 |
| anti-H3K4me3 | Merck Millipore | Cat# 07-473; RRID: AB_1977252 |
| anti-H3K27me3 | Merck Millipore | Cat# 07-449; RRID: AB_310624 |
| anti-H3K27ac | Abcam | Cat# 4729; RRID: AB_2118291 |
| anti-H3K9me3 | Abcam | Cat# 8898; RRID: AB_306848 |
| anti-H3K9ac | Abcam | Cat# 4441; RRID: AB_2118292 |
| anti-H3K9me2 | Abcam | Cat# 1220; RRID: AB_449854 |
| anti-H3K36me3 | Active Motif | Cat# 61021; RRID: AB_2614986 |
| anti-RNA Polymerase II | Merck Millipore | Cat# 05-623; RRID: AB_309852 |
| anti-Mouse IgG (CUT&Tag) | Sigma Aldrich | Cat# SAB3701102 |
| anti-Rabbit IgG (CUT&Tag) | Sigma Aldrich | Cat# SAB3700926 |
| Bacterial and virus strains | ||
| One Shot Stbl3 Chemically Competent E. coli | Thermo Fisher Scientific | Cat# C737303 |
| Chemicals, peptides, and recombinant proteins | ||
| Trypsin (2.5%) | Thermo Fisher Scientific | Cat# 15090-046 |
| Dispase II (neutral protease, grade II) | Sigma Aldrich | Cat# 4942078001 |
| Gibco™ DMEM/F12, GlutaMAX™ Supplement | Thermo Fisher Scientific | Cat# 10565018 |
| Gibco™ Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| Insulin (Roche) | Sigma Aldrich | Cat# 11376497001 |
| Cyclodextrin-encapsulated hydrocortisone | Sigma Aldrich | H0396 PubChem Substance ID: 24895401 |
| Epidermal growth factor (EGF) | Sigma Aldrich | Cat# E9644 |
| Cholera Toxin | Sigma Aldrich | Cat# C-8052 |
| Trypan Blue 0.4% | Thermo Fisher Scientific | Cat# T10282 |
| Deoxyribonuclease I (DNAse I) | Worthington Biochemical Corp | Cat# LS002140 |
| Clostridiopeptidase A (Collagenase) | Sigma Aldrich | Cat# C9891 |
| Hyaluronate 4-glycanohydrolase (Hyaluronidase) | Sigma Aldrich | Cat# H3506 |
| EGTA | Sigma Aldrich | Cat# E0396 |
| 7-Aminoactinomycin D (7AAD) | Sigma Aldrich | Cat#A9400 |
| Clarity electrochemiluminescence substrate | Biorad | Cat# 1705060 |
| blot via NuPAGE 4–12% Bis-Tris 1.5 mm gel | Life Technologies | Cat# NP0335BOX |
| DPBS, no calcium, no magnesium | Gibco | Cat# 14190144 |
| Tris-HCl | Fisher | Cat# BP1521 |
| Sodium chloride | Sigma Aldrich | Cat# S5150-1L |
| Magnesium chloride | Sigma Aldrich | Cat# #M8266-100G |
| IGEPAL CA-630 | Sigma-Aldrich | Cat# I8896 |
| UltraPure™ DNase/RNase-Free Distilled Water | Invitrogen | Cat# 10977015 |
| NEBNext High-Fidelity 2× PCR Master Mix | New England Biolabs | Cat# M0541 |
| 100x SYBR Green I | Invitrogen | Cat# S-7563 |
| Bovine Serum Albumin | Sigma Aldrich | Cat# A80577 |
| Calcium chloride | Sigma Aldrich | Cat# C4901 |
| Digitonin | Millipore | Cat #300410 |
| Dimethyl sulfoxide | Sigma Aldrich | Cat# D4540 |
| Ethanol 100% | VWR | Cat# 20821.330 |
| HEPES pH 7.5 | Sigma Aldrich | Cat# H3375 |
| Manganese chloride | Sigma Aldrich | Cat# 203734 |
| Potassium chloride | Sigma Aldrich | Cat# P3911 |
| SDS | Invitrogen | Cat# AM9820 |
| Sodium acetate | Invitrogen | Cat# AM9740 |
| Spermidine | Sigma Aldrich | Cat# S2501 |
| cOmplete™, Mini Protease Inhibitor Cocktail | Roche | Cat# 4693159001 |
| Concanavalin A-coated magnetic beads | Bangs Laboratories | Cat# BP531 |
| Protein A-Tn5 (pA-Tn5) fusion protein | Fred Hutchinson Cancer Center | N/A |
| Proteinase K (thermolabile) | New England Biolabs | Cat# P811 |
| RNase | Roche | Cat# 10109169001 |
| AMPure® XP Beads | Beckman Coulter | Cat# A63881 |
| UltraPure™ 0.5M EDTA, pH 8.0 | Invitrogen | Cat# 15575020 |
| DpnII | New England Biolabs | Cat# R0543M |
| Glycine | Sigma Aldrich | Cat# G7126 |
| Formaldehyde (37%) | Sigma Aldrich | Cat# 252549 |
| T4 DNA ligase (30 U/μL) | Thermo Fisher Scientific | Cat# EL0013 |
| Proteinase K | Thermo Scientific | Cat# EO0491 |
| Phenol-chloroform-isoamyl alcohol (PCI, 25:24:1) | Sigma Aldrich | Cat# 77617 |
| 1 μg/μl COT DNA of relevant species (mouse) | Thermo Fisher Scientific | Cat# 18440016 |
| M-270 Streptavidin Dynabeads | Thermo Fisher Scientific | Cat# 65305 |
| Disuccinimidyl Glutarate (DSG) | Thermo Fisher Scientific | Cat# A35392 |
| Triton-X-100 | Biorad | Cat# 1610407 |
| BbsI-HF | New England Biolabs | Cat# R3539 |
| T4 DNA ligase (plasmid cloning) | Promega | Cat #M1801 |
| Critical commercial assays | ||
| miRNeasy™ Micro Kit | Qiagen | Cat# 217084 |
| TruSeq RNA Library Preparation Kit v2 | Illumina | Cat# RS-122-2001 |
| On-column Dnase | Qiagen | Cat# 79256 |
| ATAC | Illumina | Cat# FC-121-1030 |
| MinElute | Qiagen | Cat# 28204 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32854 |
| Qubit dsDNA BR Assay Kit | Thermo Fisher Scientific | Cat# Q32853 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7370S |
| NEBNext Multiplex Oligos for Illumina Primer set 1 | New England Biolabs | Cat# E7335S |
| NEBNext Multiplex Oligos for Illumina Primer set 2 | New England Biolabs | Cat# E7500S |
| Herculase II Fusion Polymerase Kit | Agilent Technologies | Cat# 600675 |
| Nimblegen SeqCap EZ Hybridization and wash kit | Roche | Cat# 05634261001 |
| Nimblegen SeqCap EZ Accessory kit v2 | Roche | Cat# 07145594001 |
| Nimblegen SeqCap EZ HE-oligo kit A | Roche | Cat# 06777287001 |
| Nimblegen SeqCap EZ HE-oligo kit B | Roche | Cat# 06777317001 |
| Omni-C™ Kit | Dovetail | Cat# 21005 |
| Omni-C™ Library Module for Illumina 8Rx | Dovetail | Cat# 25004 |
| Omni-C™ Library Module for Illumina 8Rx | Dovetail | Cat# 25010 |
| Omni-C™ Filter Set | Dovetail | Cat# 25003 |
| Zymo DNA Clean & Concentrator-5 | Zymo Research | Cat# D4013 |
| Plasmid Max Kit | Qiagen | Cat# 12162 |
| Deposited data | ||
| RNA-seq, ATAC-seq, Omni-C™ and CUT&Tag | This paper | NCBI GEO accession number: GSE227750 |
| scRNA-seq mouse atlas | Pal et al.25 | NCBI GEO accession number: GSE164017 |
| Experimental models: Organisms/strains | ||
| Mouse: FVB/NJ | The Jackson Laboratory | Cat# JAX:001800: RRID: IMSR_JAX:001800 |
| Experimental models: Cell lines | ||
| Mouse: COMMA-DβGeo | Dr D. Medina | RRID: CVCL_5733 |
| Others | ||
| BD FACSAria™ C | BD Biosciences | Flow Cytometry, Walter and Eliza Hall Institute of Medical Research |
| BD FACSAria™ Fusion | BD Biosciences | Flow Cytometry, Walter and Eliza Hall Institute of Medical Research |
| LowBind DNA tubes | Eppendorf | Cat# 0030108051 |
| Tapestation loading tips | Agilent Technologies | Cat# 5067-5153 |
| Genomic DNA ScreenTape | Agilent Technologies | Cat# 5067-5365 |
| Genomic DNA Reagents | Agilent Technologies | Cat# 5067-5366 |
| D1000 DNA ScreenTape | Agilent Technologies | Cat# 5067- 5582 |
| D1000 DNA Reagents | Agilent Technologies | Cat# 5067- 5583 |
| D5000 DNA ScreenTape | Agilent Technologies | Cat# 5067- 5588 |
| D5000 DNA Reagents | Agilent Technologies | Cat# 5067- 5589 |
| Covaris microTUBE AFA Fiber pre-split snap-cap 6 x 16 mm | Covaris | Cat# 520045 |
| Recombinant DNA | ||
| CRISPRa plasmid: pLH-gRNA-MSx2-sfGFP | addgene | Cat# 75389; RRID: Addgene_75389 |
| CRISPRa plasmid: pLH-MS2-p65-HSF1-P2A-BFP | addgene | Cat# 61423; RRID: Addgene_61423 |
| CRISPRa plasmid: pLH-dCas9-VP64-T2A-mCherry | addgene | Cat# 61422; RRID: Addgene_61422 |
| Software | ||
| R | R Project for Statistical Computing | RRID: SCR_001905 |
| R packages Rsubread, edgeR, limma, Glimma, csaw, diffHic, HiCRep, Sushi, plyranges, Iranges, rtracklayer, BSgenome.Mmusculus.UCSC.mm10 | Bioconductor | RRID: SCR_006442 |
| R package gplots, ComplexUpset, Pheatmap, viridisLite | CRAN | RRID: SCR_003005 |
| Mus musculus gene information | NCBI | ftp://ftp.ncbi.nlm.nih.gov |
| Picard-tools | The Broad Institute | RRID:SCR_006525 |
| BEDtools | Quinlan et al.63 Bioinformatics | RRID: SCR_006646 |
| Trim Galore | The Babraham Institute | RRID: SCR_011847 |
| Bowtie2 | Langmead et al.64 | RRID: SCR_016368 |
| CCanalyser3 | Davies et al.65 | https://github.com/Hughes-Genome-Group/Capture-C/releases |
| Deeptools | Ramirez et al.66 | RRID: SCR_016366 |
| Samtools | Danecek et al.67 | RRID: SCR_002105 |
| HOMER | Heinz et al.68, Heinz et al.69 | RRID: SCR_010881 |
| SEACR | Meers et al.70 | https://github.com/FredHutch/SEACR |
| HiC-Pro | Servant et al.71 | RRID: SCR_017643 |
| FitHiChIP | Bhattacharyya et al.72 | https://github.com/ay-lab/FitHiChIP |
| ChromHMM | Ernst et al.73 | RRID: SCR_018141 |
| TOBIAS | Bentsen et al.48 | https://github.com/loosolab/TOBIAS |
| Cytoscape | Shannon et al.49 | RRID: SCR_003032 |
| JASPAR2018_CORE_vertebrates_non-redundant | Castro-Mondragon et al.74 | RRID: SCR_003030 |
| HOCOMOCOv11_core_MOUSE_mono | Kulakovskiy et al.75 | RRID: SCR_005409 |
| Python | Python Programming Language | RRID: SCR_008394 |
Resource availability
Lead contact
Further information regarding the resources and reagents used in this study should be directed to Jane Visvader (visvader@wehi.edu.au).
Materials availability
The study did not generate any new unique reagents.
Experimental model and study participant details
FVB/N mice were provided by the WEHI animal facility. All mice were bred and maintained in the WEHI animal facility according to institutional guidelines and all experiments approved by the WEHI Animal Ethics Committee. COMMA-DβGeo cells were kindly provided by Dr. Daniel Medina.
Method details
Isolation of mammary epithelial subsets
Mammary epithelial cells were isolated as previously described (Shackleton et al 2006).56 Briefly, mammary glands from six adult (9-10 week-old) female FVB/N mice were pooled and cell suspensions were generated via tissue chopping and digestion with collagenase/hyaluronidase/DNase, TEG, Dispase/TEG and a final red-cell lysis performed. Cell suspensions were stained for flow cytometry using the following antibodies: FITC anti-mouse CD29 (rat, clone HMβ1-1, BioLegend Cat#102206, 1/200 dilution), Pacific Blue anti- mouse CD24 (Armenian Hamster, clone M1/69, BioLegend Cat#101820, 1/200), PE anti-mouse CD61 (Armenian Hamster, clone HMβ3-1, BioLegend Cat#104308, 1/ 100), APC anti-mouse CD31 (rat, clone 390, BioLegend Cat#102410, 1/40 dilution), APC anti-mouse CD45 (rat, clone 30-F11, BioLegend, Cat#103112, 1/100 dilution), and APC anti-mouse TER-119/erythroid cell (rat, clone TER-119, BioLegend Cat#116212, 1/80 dilution). Stained cells were resuspended in 2% FCS/PBS with 7-AAD to exclude dead cells.
RNA-sequencing and differential gene expression
Sorted cell pellets were washed in PBS and snap-frozen on dry ice. Pellets were then resuspended in QIAzol™ and RNA prepared following Qiagen’s recommended protocol for the miRNeasy™ micro kit (Qiagen Cat#217084), including the on-column DNase digestion (Qiagen Cat#79256). 100 ng total RNA was used to prepare sequencing libraries following Illumina’s TruSeq RNA v2 protocol (Illumina #RS-122-2001, RS-122-2002). RNA libraries were sequenced on the Illumina NextSeq 500, aiming for >30M 80 bp paired-end reads. Reads were mapped to the mouse genome (mm10) and using Rsubread counted with the inbuilt mm10 RefSeq gene annotation.76 Differential gene expression analysis was performed using the edgeR package, with lowly expressed genes removed via filterByExpr and library normalization with the TMM method.77 To select genes with large differences in expression we applied a fold-change cut-off using glmTreat (fc=2.5) between basal, LP and ML comparisons and glmTreat (fc=1.2) between Tspan8+ and Tspan8– comparisons. Signature genes of basal, LP and ML were obtained by intersecting the up-regulated DE genes between the pairwise comparisons, where the glmTreat fold-change cut-off was 2 for basal signatures and 1.2 for LP and ML signatures.
ATAC-sequencing
Cells (50,000) from sorted mammary epithelium were reserved for bulk ATAC-sequencing. ATAC-sequencing was performed as previously described78,79: cells were lysed (50 μl lysis = 10 mM Tris-Cl (Tris-Cl pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% NP-40 in nuclease-free H2O) and transposed with Illumina’s Tn5 enzyme (50 μl trans-position = 25 μl 2X TD Buffer (Illumina Cat#FC-121-1030), 2.5 μl TDE1 (Illumina Cat#FC-121-1030), 22.5 μl nuclease-free H2O). Cells were incubated for 20 min at 22°C then 30 min at 37°C and purified with the MinElute kit from Qiagen (Cat#28204). ATAC libraries were sequenced on Illumina’s NextSeq 500, aiming for >100M 80 bp paired-end reads per biological replicate. Reads were processed as previously described.25
CUT&Tag sequencing
CUT&Tag sequencing was performed as described.32 100,000 FACS purified cells were bound to Concanavalin A-coated magnetic beads and incubated overnight at 4°C in 50 μl of primary antibody buffer: 20 mM HEPES pH 5.5, 150 mM NaCl, 0.5 mM spermidine (Sigma-Aldrich, Cat#S2501), 1x cOmplete™ Mini Protease Inhibitors (Roche Cat#4693159001), 0.05% Digitonin (Millipore Cat#300410), 0.1% BSA (Sigma-Aldrich Cat#A80577), 2 mM EDTA and MilliQ H2O. Primary antibodies used for CUT&Tag: H2AK119ub (CST Cat#8240, 1:50), H3K4me1 (abcam Cat#8895, 1:50), H3K4me3 (Merck Cat#07-473, 1:50), H3K9ac (abcam Cat#4441, 1:50), H3K9me2 (abcam Cat#1220, 1:50), H3K9me3 (abcam Cat#8898, 1:50), H3K27ac (abcam Cat#4729, 1:50), H3K27me3 (Merck Cat#07-449, 1:50), H3K36me3 (Active Motif Cat#61021, 1:50), RNA Pol II (Merck Cat#05-623, 1:50) and mouse IgG (Merck Cat#12-371, 1:50). Cells were then incubated in secondary antibody, either anti-mouse IgG (Sigma Cat#SAB3701102) or anti-rabbit IgG (Sigma Cat#SAB3700926) before addition of pA-Tn5 (kind gift from the Henikoff laboratory, 1:250) and tagmentation. Tagmented DNA was solubilized with Thermolabile Proteinase K (NEB Cat#P8111S) for 1-2 hrs at 37oC to 300 rpm, then purified with elution buffer (10 mM Tris-HCl pH 8, 1 mM EDTA, 1/400 RNase A (Roche Cat#10109169001) and DNase-free H2O) and PCR amplified (17 cycles). Libraries were subjected to SPRI bead purification and sequenced on an Illumina NextSeq 500, aiming for >2M 80 bp paired-end reads per antibody. Small aliquots from libraries of untested antibodies were sequenced as pilot experiments to ensure appropriate enrichment for histone modifications and RNA Pol II (Table S1). Trim Galore (The Babraham Institute) was used to trim adapter sequences before mapping with Bowtie264 as previously described. PCR duplicates were removed with picard-tools and files converted to bam format for counting with bedtools, peak calling and differential analysis. For CUT&Tag of the Tspan8-defined cell populations, adaptations were made for low-cell inputs of 10,000-20,000 cells. Cells were incubated with in 10 μl primary antibody buffer, 20 μl secondary antibody buffer, 20 μl pA-Tn5 mix and PCR amplified for 19 cycles.
Omni-C™ library preparation and sequencing
Chromatin interaction libraries were generated using the Omni-C™ protocol from Dovetail™ genomics (Cat#21005). Purified MECs (300,000-500,000 cells) were pelleted and snap-frozen on dry ice before fixation with formaldehyde and in situ DNase digestion (1/8 dilution of DNase following optimization). Fixed chromatin (1 μg) was bound to chromatin capture beads before end-polishing, bridge ligation to insert biotin labels on DNA-ends, intra-aggregate ligation and crosslink reversal followed by a SPRI bead clean-up and size selection. End-repair was performed on 150 ng of ligated DNA, followed by Illumina adapter ligation. Ligation products were bound to streptavidin beads, and final products were indexed and sequenced on an Illumina NextSeq 500, aiming for >100M 80 bp paired-end reads per biological replicate.
NG Capture-C library sequencing and processing
Capture-C was performed as described80 on 300,000 sorted cells. Briefly, 3C libraries were generated on fixed cells and digested with DpnII (NEB Cat#R0543M) overnight. Following heat-inactivation, DNA was ligated and chromatin de-crosslinked. Up to 1 μg of 3C library was sonicated on a Covaris S220 Focused Ultrasonicator using the following settings, 10% duty cycle, 5 intensity, 200 cycles/burst for 360 seconds before adding SPRI beads. End repair and adapter ligation were performed using the recommended conditions for NEBNext Ultra DNA Library Prep Kit for Illumina (NEB Cat#E7370S) and NEDNext Multiple Oligos for Illumina Primer set 1 and 2 (NEB Cat#E7335S and E7500S). Biotinylated capture oligonucleotides (probes) were 5’ biotin-tagged 120 bp oligonucleotides. Capture probe design was adapted from80, targeted at the TSS of ΔNp63, Foxi1, Gli3, Esr1, Lgr5, Tcf7, Tbx2, Foxa1, Msx2, Snai2, Ehf, Elf5, Hey1, Kit, Tbx3, Pgr, Cited1 and Maf see below. Enrichment of capture sites was adapted from the protocols for Nimblegen SeqCap EZ Hybridization (Roche Cat#05634261001, 07145594001, 06777287001 and 0677317001). Capture probes were pooled to a concentration of 2.9 nM and hybridization was performed over three days before streptavidin bead pulldown, SPRI bead clean-up and size selection. Capture probe hybridization was repeated on the library. Following the second hybridization, libraries were sequenced on an Illumina NextSeq 500 aiming for >2M 150 bp paired-end reads per biological replicate. Reads were mapped to mm10 and processed using the recommended pipeline from CCanalyser3 (https://github.com/Hughes-Genome-Group/Capture-C/releases) aiming for >10,000 capture fragments per TSS. Analysis of capture differential interactions (cDIs) was performed using Glimma and edgeR. Briefly, capture counts were counted across each DpnII restriction fragment (∼500 bp) on the same chromosome ± 2.5 Mb of each gene TSS (TSS based off RefSeq refGene.gtf, downloaded 8/2/2017) (except Esr1 which lies close to the telomere). Counts were then TMM normalized and differential interactivity determined from a linear model (lmFit) with an empirical Bayes moderation and FDR < 0.05 calculated by the Benjamini-Hochberg method.
NG Capture-C biotinylated probes for 18 genes with lineage-restricted expression. 5’ Biotin is denoted by the IDT code /5Biosg/
| Oligo_Name | Sequence with mod |
|---|---|
| Trp63_CapC_5p_A | /5Biosg/GATCTAGATTTTTTTCCAGAATTTTAATCCCCTAAATTTTAGAAGAAATTTAACATTCACTCTTACTAGT CTTAGGCAGTTAGAATCTTAGGTACATTAGAGAAAACTGTGCTGCGGATT |
| Trp63_CapC_3p_A | /5Biosg/CAGAAAAGAGGAGAGCAGCCTTGACCAGTCTCACTGCTAACATGTTGTACCTGGAAAACAATGCCC AGACTCAATTTAGTGAGGTAAGGCTTTAAGATTTTAGCCCTCTGCATAGAGATC |
| Trp63_CapC_5p_B | /5Biosg/GATCCTTTTCTTCGCTTCTTCTTTTAAATAAAAACATACTTAGCTGTGGTTCTAGTGACCCAATGTAAAT GTGTTTGCAAGTCGCATGTAGGAAACTGCTTTTCTGAGTGAATGTTTTCT |
| Trp63_CapC_3p_B | /5Biosg/AGTGAGAAGACCCTGCTGGGAATATTGGTGGGTGTGAATGTACCAGTGATAGAAACGAATTGATTGT GTATTAACTGTATAGTAAAGTTCTCCAGGCTTCATACTAAAAGGAAATGGATC |
| Foxi1_CapC_5p | /5Biosg/GATCTGGCTCAGTGTGAGGCGCTGGTCGGGCGCACCGTGGATTGCCATGGCAATGAGGGCTGAAT AGGAATATGGTGGGCGTACCAGCTTCATGAGCTCTTCCTGAGAGGGGATGGGCAG |
| Foxi1_CapC_3p | /5Biosg/GGCTCTTAAAAAGAACATCAAAAGATAGAGGATGGGAACTGGATTTTAGGTAGAAATGAGAAAGAAC ACCCACGATTCAAGTTTCTTTCTAGCTTTAAGGCACAGCAGTCTCCCCTGATC |
| Gli3_CapC_5p | /5Biosg/GATCCGCGCGCGCGGAGCGGGACCCCGCCGGGCGCGGCCTGGAAAGGAGCGGGAAAGCAAAGT AAGGCGAGCAGTCTTCCCAAGTTTTTAAACCAACTTCGCCCCCTGCGGCGGCGGCGG |
| Gli3_CapC_3p | /5Biosg/GCCCTTAGTCGTTGAAAAGTCGACTTGACGGTCGGGGGGCTCCTTTCTTTCTCTTTCTCCTCCCACC CGCGGTTGAACCAAATCAGAACTGTCACTCAGGGCCGGTGACTCAGAGGGATC |
| Esr1_CapC_Prom1_5p | /5Biosg/GATCACTTTTTTTTTTTTTTTTCAGCCAGAGGCTTGTCCTTCTGCAAACATCTATCTGTAGGCAAACGC TGAGAAAGTTGGAAGCAGACCTGGCTGGAGAGAGAGAGGAAGGCCACCCCC |
| Esr1_CapC_Prom1_3p | /5Biosg/TTAAAGCTGACCTTGGTCCCATGGAAATTCACTCCGGGGAAGCTACATTCAGCTGAGTCAGACACAG TTATCTGTTCTTGGTGCAGCACTGGCAGGCTCTCTCTCTGCCTGCCTCAGATC |
| Esr1_CapC_5p_Up | /5Biosg/GATCAGAGGTAATGTGTTTGCTTAGCACATCAAAGTTTTGGGTTTATCCCTAGTACCAAAAAGGAAAA TAAATTTAAAATGCTTTTTATCTCATATTATTGCTTCAGCGTGTTTGTCTCT |
| Lgr5_CapC_5p | /5Biosg/GATCGGGTTACCTCAAGGTGGCCTGACCCTGGTCTCTGGGCCCCAAGGAACCGAGCTTCAGATTTG AAGGAGAGTCGGGAGGAGTACTTATCTCGGTCCCACAGAGATGAGTCGCTTCTG |
| Lgr5_CapC_3p | /5Biosg/GCTTCAAAACAACCTAAGGGGACACTTAGGGACTGAAAGATAACACAGGCTTTCTCTGGTCCATGTC AGATAACCTAGGGGGATGCATGCAGTGCTTGTTAAAAGGACCAAAATCTGATC |
| Tcf7_CapC_5p | /5Biosg/GATCTCCTTTTGGGTCAGACTCCTCTGGATGTTAACTGGGCAAGAGCATCTAGGAATTGGGCAGAGA AGTCCTTTTTTGTCCAAGCATCACCTGTGTGGTCAAGTAGGTAGCCAGCCCCA |
| Tcf7_CapC_3p | /5Biosg/ACAGCGTCCTTTGCTCAATCTGGAGGCTTCTTACGTCCCCGGGATACTAGATGGACCCTGacacacac acacacacacacacacacacacacacacacacaaacacCTGCCCTTTTGATC |
| Tbx2_CapC_Prom2_5p | /5Biosg/GATCGGTCCTCTGCGCTTTCCAGCCCTCGCCCAGGCAGCGGCGGGCGCGGGCGGCGAGGTGGG GGCCAGGCCAGGGGGAGGGGTCTCGGGGCCCGCTGGCCCGTCATTGGTTAATATTTT |
| Tbx2_CapC_Prom2_3p | /5Biosg/TCCCCTCCCGTCCAGAGCTTGGCCTGAGCTGTCAAAACCCCGCCCCCGGAGACCCACAATTGGTC CAAAAAGCGTAAAATCAGCAATCAAGGGGGGCCTGGCTCGTTAGCGCAGGGGATC |
| Foxa1_CapC_5p | /5Biosg/GATCTTACGTCGCCGGAGTGCCCACCTCCTCGTCCTCTCCCCATTTGTCCGCCGCACAAAGACGCT CGCACCTACAAAGCCCGAGGTGCACCTGTGAGGCGGCCGCCCGCCAGTCCAgcc |
| Foxa1_CapC_3p | /5Biosg/aaaaagaagaaaaggaaaaATAGGCGGTGCCTTGGAGGACAGGCCAGGGCTCTGGACCCAAAGAGCTGT GCTGCGGGAGAAGGACCTGGGTGGTGGCATCAGAGCTACAGCGCAAGGATC |
| Msx2_CapC_5p | /5Biosg/GATCCCGTCCCAGACGCGCACTCACTGGGCGGCGGGGAGTATCTGCCGGGCTCCTGTATCCACG GTGCTCCGTCTTCGGAATTTTCCGACTTGACCGAGGCGGTCTCGAAGGGCTTGACG |
| Msx2_CapC_3p | /5Biosg/CACGGACGCTCGCCACCGATTGGCTCTCCCTGGAGAGGCTTGGGGCCCTTCCCCCGCCCCGTTT GAAATAAATTAGGAGTTAATTACAGGAGCAGTCAGCAGAGTTGTTATTAGGCGATC |
| Snai2_CapC_5p | /5Biosg/GATCAGCAAGTTAACTTTCTGGCACGCCGCCCTAGACCTGCTGTGGCAGCTGCGGGGAGCCTTTA CCTTCCTTTCCCAAAAGCCAGAGCCTACAGCTGCTTGTGTGCAATAACCCCCCTC |
| Snai2_CapC_3p | /5Biosg/CGGTTCTATTGCTTGACTCAGAGAACACACCGGGCCGCTTTCCTTTTAATGCTGTGCCAAAACTGTC CTTGCAGTCTCTTGATTACTTAGGTTAAGTTTTAATTCAAAACCTTTGAGATC |
| Ehf_CapC_5p | /5Biosg/GATCATTTTTAAGGGTACCACCTGCCTAACCTTGACCACCTGACTCCTGCCGCCTTTGTGAATATAG ATTCTTTTACCTATCTACACCTATTTCCTCAAACTGGAATACACTAAAGATGA |
| Ehf_CapC_3p | /5Biosg/GAATAGAAAAGCCAAGTCCAAGTCCCTGTCAAGCAAATGAAGGAAGAGGGCCTGAGGTGCTCTTAA ACATCCGGCTCATTTTCTAACCCGTATTTAGTCCTCTGCTATGTCATCAAGATC |
| Elf5_CapC_5p | /5Biosg/GATCCCTGAAGCACCTTTATTCTTTACCACTTCTTGGCACTGCTCTTCTTTCCTAACACGCACAGAAT AGGGGATAACACTACATACAGAGGTTCGGGACTTGGCCAGCCCAGGCAAAGG |
| Elf5_CapC_3p | /5Biosg/GTCTGTATGCGTTCAGGGGTTTGTGCGCATTTGCCCCCTACTGGCAGCAACTGGAAACACATGCTC TCCCGCATCTGCGTTAAGGTAGGagaatttaagacagaataggggtcctagatc |
| Hey1_CapC_5p | /5Biosg/GATCCCACTCACGCTCAGTCTCCGGTTAAAACTCAACCATCCCTTTCCCACGCTGCGCCCCTTCCC ATGGATAGGGGGAGGGCGGGGAAGGCGGAGAGGTTGGGGCAGGGGCGGGGCCAC |
| Hey1_CapC_3p | /5Biosg/TTTTCTGGGTTAGCTTGAGGGAGGAGTAGCTATCCCCCGAGACATTCATTATGTTGGGATTTTTGCT GTTGTTTTGTTTTGTTTTTGTCCTCCCTTCCCCCTAGTGTTGGCGGCCAGATC |
| cKit_CapC_5p | /5Biosg/GATCTGCTCTGCGTCCTGTTGGTCCTGCTCCGTGGCCAGACAGGTGGGAAAGAGCGGCAGACAAG AGGACTGCACCCTCTGTGGGCGCAGCCCGGGTCCGGGAGGGGTGCCACCTGGGTG |
| cKit_CapC_3p | /5Biosg/ACCAGGACCGAGTCAGTTCTCCCCAGCTTTGGAAACCTCTGGTTTCAGTGTATGCGACTTGTAATCG CAGGTGGCCGttttttttttttttttttttttttGGAGGGGAATCCCGAGATC |
| Tbx3_CapC_5p_Up | /5Biosg/GATCTCGCTGCCTGTGAAAGCCAGAGTCTAGCTCAACTAAGACGCCTCCTGCGAGAAAGCCAGAG AAGAGCTAGGGGGCGGGGGAAGGAGTCGAAAAAAAGGTTAAAAAAAAAAGTCTCC |
| Tbx3_CapC_3p_Up | /5Biosg/CTTGGGCGCCAGTCGAGCCCCTGCTTGCTGCTTGCCCTACTGAAACCGACTTCCAGGAGCGGCTT TTCCAACACACTCCACGCACCAGGACAGCCCCTGCAGCGGCTATGTCTCCAGATC |
| Pgr_CapC_PromB_5p | /5Biosg/GATCTAGCCAGTGATTGGCTAGGGAGGGGCTTTGGGCGGGCCTTCCTAGAGCGCCAACGCTTGCT AGAAAGCTATGGAGCCAGTCTAGACTGTCACTATCAGTCTTTGTAGTATTTACGG |
| Pgr_CapC_PromB_3p | /5Biosg/TTCCTGTCCTCACCCCACCGCGACCGGGACAGCGCGACTACCACCCTTCCTCTGCGTCTGGGTGG AGGGTAAGGACAGGAGCTGACCAAGACCGCCCCTCCCAACCAGGAGGTGGAGATC |
| Pgr_CapC_Exon_5p | /5Biosg/GATCAAGGAGGAGGAGGAGGGCGCGGATGCTGCTGTGCGCTCGCCGCGCCCCTACCTGTCGGCT GGAGCCAGCTCCTCCACCTTCCCAGACTTCCCGCTGGCACCCGCGCCGCAGCGAGC |
| Pgr_CapC_Exon_3p | /5Biosg/CGGGCATAAAAGGGAGTGCAGATATACCATTTTATTGTGTACCATTCTCCCAAACATTGCCTGCAAA CTTCCTGAAGCCTGAGCACCCAGGTTCAAACCCTGAGGCCTCGCTCTAAGATC |
| Cited1_CapC_5p | /5Biosg/GATCACAGCCACCACGCTTGGTTCTCGGCGCGGGGACCGGGCTCTTAAGCCCCCACTGCCTCCC GTAGAGCCCTGGCGCACAAGCACACAGCAGCAGGCTGGCCTCGGCTAGGAATCCCA |
| Cited1_CapC_3p | /5Biosg/CTTCCTCACCGCAGCTCGGCAGCGCACTTCTGCAGCTGTGCCGCCGGGAACATTTTATAGCGGCG GGCTGGCGTGTGTGGCCCTTTAAAGGCGCTCGAGCTGGTGCAGTCACCGTGGATC |
| Maf_CapC_Up_5p | /5Biosg/GATCCCGGGCGCTTCAGGCTCGGGAAGGTCCTCCGCGGCTGGCGGTGACGGTGGTGATGACAGC GGCAGAGAGGATGCCGGAGGAGCACGGCCCGGTGCGCGGCGTCCCCGGCTCGCCGC |
| Maf_CapC_Up_3p | /5Biosg/GCGGGTGGCTGTCCCGGAGGCGCCGGCCTCCACACCGGAGTGGTTAACACTTCACGCTTCTCTCC TCTTCTGCCTGGCTCTTATGGTTACTATTATTATTTTCTTTTCTCTCTCCGGATC |
Chromatin accessibility and histone mark analysis
For analysis of chromatin accessibility and histone marks associated with genes, we mapped the merged biological replicate bam files using bedtools to three separate regions. Using genes from RefSeq, we annotated 500 bp up and downstream of the start of the gene as the TSS, the next ± 2 kb (excluding 1 kb TSS) as the TSS-proximal region and finally, we took the entire gene-body and subtracted counts within the TSS and TSS-proximal region. We then used the TSS with the highest number of CUT&Tag and ATAC counts when multiple TSSs were present. Data were normalized by subtracting IgG counts from each region (except ATAC, which does not have a control) and log2 RPKM determined using edgeR’s rpkm function. For coverage track-plots, bedgraphs were produced from bam files using the RPKM setting from DeepTools bamCoverage.81 To ascertain TSSs that were primed or active, we determined the percentile ranks across all three cell types for the CUT&Tag and ATAC-seq datasets and considered as TSS primed or active if the ATAC, H3K27ac and H3K4me3 percentile ranks were all >0.25.
CRISPRa/dCas9 guide and plasmid generation
Guide RNAs (gRNAs) for enhancers were designed to target peaks from the ATAC and CUT&Tag sequencing , see below for primer sequences. Guides that showed the largest increase in gene expression tended to be adjacent to the central ATAC peak at an enhancer and not overlapping conserved motif sites. gRNAs were designed with Benchling as 20 bp in length, targeting the mm10 genome and using the NGG PAM site design. Guides were cloned into pLH-gRNA1-MS2x2-sfGFP plasmid via the BbsI restriction site. In brief, pLH-gRNA-MS2x2-sfGFP was modified from pLH-sgRNA1-2XMS2 (Addgene #75389) by replacing the ccdB fragment with the BbsI cloning cassette and replacing the hygromycin resistance with a superfolder GFP with the BbsI site removed , see below for cloning sequences. The transactivator and deactivated-Cas9 plasmids were constructed from Addgene plasmids #61423 and #61422, respectively, as previously reported.82 sgRNAs were ordered from IDT Signapore and were cloned as described.82 Annealed oligos were phosphorylated by T4 polynucleotide kinase (PNK) (Promega #M4101): pLH-sgRNA1-2XMS2 was digested with BbsI-HF ® (New England Biolabs, #R3539) and ligated with the phosphorylated oligonucleotides. Plasmids were cloned using the Stbl3 Escherichia coli strain.
CRISPRa/dCas9 sgRNA target sequences used for enhancer activation
| sgRNA name | 20 bp sgRNA target sequence |
|---|---|
| Snai2 -94/95 sgRNA #1 | GACACCGTTAGTCATCCTAG |
| Snai2 -94/95 sgRNA #2 | AGCATCCTAGACACTGCACA |
| Snai2 -221/239 sgRNA #1 | TGTACACAGAGATATCTGAG |
| Snai2 -221/239 sgRNA #2 | AAGAATGGTGATGATTTGTG |
| Snai2 -221/239 sgRNA #3 | GTGCCCTGCTAACAGCCTAG |
| Snai2 -221/239 sgRNA #4 | AATCTTGCAGACAGACAATG |
| Snai2 -405/409 sgRNA #1 | ACACCTGTTCTGCCCAGGCA |
| Snai2 -405/409 sgRNA #2 | AGCTAGTAAGAAACCCCCAG |
| Elf5 +278/280 sgRNA #1 | ATCAGAAGGAATCCACCAGG |
| Elf5 +278/280 sgRNA #2 | TCATGGGCTCAGGAGCGGGG |
| Elf5 +58/59 sgRNA #1 | GGCAAGAACTCACTCCCCAA |
| Elf5 +58/59 sgRNA #2 | TGGCCTTTGAGAAGACGTCA |
| Elf5 -17/18 sgRNA #1 | CAACCAAGGGAGGTGCTAGG |
| Elf5 -17/18 sgRNA #2 | GGAGTTGGTATTGGCACTCA |
| Msx2 +184/186 sgRNA #1 | GAACTAGTGTCTCCTGAGCA |
| Msx2 +184/186 sgRNA #2 | ACCTAGTCAAGTCTAGCCTG |
| Msx2 +184/186 sgRNA #3 | TTGCTTCTCCCATCGCACTG |
| Non-targeting control (NTC) | GCCGTAAGCGGGCCGGTTGA |
Cloning sequences for pLH-gRNA-MS2x2-sfGFP
| Primer Sequence 5’ – 3’ | |
|---|---|
| BbsI cassette rowhead | ACCGGTGTCTTCGAGGCTTACAGGACGAAGACCC |
| AAACGGGTCTTCGTCCTGTAAGCCTCGAAGACAC | |
| Superfolder GFP rowhead | ATATGGATCCTTAATGCCGCCACCATGCGTAAAGGCGAAGAGCTGTTCAC |
| CAGGATATTGCCGTCCTCTTTAAAGTCAATG | |
| CATTGACTTTAAAGAGGACGGCAATATCCTG | |
| AATTGTCGACGTCGGCATCTACTTTATTTGTACAGTTCATCCATACCATGCGTGATG |
CRISPR-activation of target gene enhancers
COMMA-DβGeo cells were maintained in DMEM/F-12 GlutaMAX™ (Gibco, #10565018), supplemented with 2% FCS (Gibco, #10099-141), 10 μg/ml insulin and 5 ng/ml hEGF (Sigma Aldrich, #E9644). Cells were transduced with lentivirus by spin infection for 90 mins at 32°C. mCherryhighBFPhighGFP+ cells were sorted on the BD FACSAria™ Fusion one or two passages following transduction and frozen for protein and RNA analysis.
Quantitative real-time PCR
RNA was purified from sorted, transduced COMMA-DβGeo cell pellets using the RNeasy kit from Qiagen (Cat#74004) with on-column DNase digestion (Qiagen Cat#79256) performed. Purified RNA was used to produce cDNA with Invitrogen’s SuperScript™ IV and Oligo dTs (ThermoFisher Cat#18090010). qRT-PCR was then carried out with Bioline’s SensiMix™ Hi-Rox (Cat#QT605-05) on a Corbett Rotor-Gene 3000. qRT-PCR primers for Rplp0: forward, GACAACGGCAGCATTTATAACC and reverse, ACTCAGTCTCCACAGACAATGC; Elf5: forward, ACAGGATGACGTACGAGAAGC and reverse, ATCAAATGAGCCTGGTGTCC; Msx2: forward, CGGAAAATTCCGAAGACG and reverse, TTCAGAGAGCTGGAGAACTCG.
Western blot analysis
FACS purified cells were resuspended in RIPA buffer and sonicated, then lysates were clarified by centrifugation. Lysate was incubated at 95°C for 5 min with loading dye and reducing agent before being resolved by western blot via NuPAGE 4–12% Bis-Tris 1.5 mm gels (Life Technologies, #NP0335BOX). Proteins were transferred to polyvinylidene fluoride membrane using the iBlot 2 Dry Blotting System (Thermo Fisher Scientific) according to the manufacturer’s instructions. Membranes were probed with anti-FoxP1 (Cell Signaling Technology #2005, 1:1000), anti-total Histone 3 (Millipore #07-690, 1:5000), anti-α-tubulin (Sigma Aldrich #T6199, 1:5000), anti-Snai2 (Cell Signaling Technologies, #C19G7, 1:1000) or anti-HSP70 (clone N6, WEHI, 1:10,000) primary antibodies, followed by mouse or rabbit IgG secondary antibodies (1:10,000) conjugated to horseradish peroxidase. For Foxp1 blots, 22,500 sorted cells were probed per lane, and 100,000 cells per lane for Snai2. Membranes were developed using Clarity electrochemiluminescence substrate (Biorad #1705060) and imaged on ChemiDoc Touch Imaging System (Biorad).
Differential analysis of CUT&Tag and ATAC-seq data
Differential binding between libraries of each data type was assessed using the csaw package33 v1.30.1 with basal, LP and ML libraries in a separate analysis to the Tspan8+ and Tspan8– basal libraries. Biological replicates were kept separate for the analysis. For each data type, read parameters were defined with readParam with pe=”both”, minq=20 and reads in blacked regions were discarded. Max.frag was set for each data type depending on the fragment distribution and is shown in the “Bin sizes for csaw analysis” table below. Sliding windows of a width (depending on the library) were tiled across the genome of interval width/2. Windows were filtered with a global enrichment approach with the filterWindowsGlobal function. Data were binned with a larger window size (see below table) and required to be higher than a threshold plus the background. Loess-based normalization was performed with the normOffsets function. Differential binding was assessed for using the quasi-likelihood (QL) framework in the edgeR package v3.38.483 with robust=TRUE for the glmQLFit. The design matrix was constructed using a layout that specified the cell type and the mouse group (for basal Tspan8+ and Tspan8– cells and analysis with H3K27me3, H2AK119ub, H3K9me2, K3K9me3 CUT&Tag libraries, the mouse group was not included in the design matrix). Proximal tested windows were merged into regions by clusterWindows with a cluster level FDR target of 0.05 and where the maximum merged windows size and allowed distance between windows is given in the table below. DA regions between basal Tspan8+ and Tspan8– cells were allocated to a gene if they overlapped a TSS or an interacting region as determined by FitHiChIP (FDR < 0.1).
Multidimensional-scaling plots were constructed with the plotMDS function in the limma package applied to all the filtered and normalized logCPM values of windows. When required, logCPM values were corrected for experimental batch with the removeBatchEffect function of the limma package.
Bin sizes for csaw analysis
| Data type | Max.frag | Width (bp) | Binned size | Threshold Tspan8± | Threshold MEC | Distance between | Max merged width |
|---|---|---|---|---|---|---|---|
| ATAC | 500 | 100 | 2 kb | 8 | 6 | 500 | 10000 |
| RNA PolII | 700 | 1000 | 50 kb | 2 | 2 | 1000 | 50000 |
| H3K36me3 | 600 | 1000 | 50 kb | 1.8 | 1.8 | 1000 | 50000 |
| H3K4me1 | 1000 | 1000 | 50 kb | 1.8 | 1.8 | 1000 | 50000 |
| H3K4me3 | 1000 | 1000 | 50 kb | 1.8 | 1.8 | 1000 | 50000 |
| H3K9ac1 | 600 | 1000 | 50 kb | 1.8 | 1.8 | 1000 | 50000 |
| H3K27ac | 1000 | 1000 | 50 kb | 1.8 | 1.8 | 1000 | 50000 |
| H3K27me3 | 1000 | 10000 | 1 Mb | 1.8 | 1.8 | 10000 | 1000000 |
| H2AK119ub | 800 | 10000 | 1 Mb | 2 | 1.8 | 10000 | 1000000 |
| H3K9me2 | 600 | 100000 | 5 Mb | 2 | 1.5 | 100000 | 5000000 |
| H3K9me3 | 600 | 100000 | 5 Mb | 1.2 | 1.2 | 100000 | 5000000 |
Omni-C™ data processing with diffHic pipeline
Fastq libraries were aligned to the mm10 genome with the iter_map.py script in the diffHic package36 which implements iterative mapping84 and utilizes bowtie2 for alignment. The BAM files for the sequencing runs for libraries were merged with samtools then the FixMateInformation command from the Picard suite v1.117 (https://broadinstitute.github.io/picard/) was applied, duplicate reads were marked with MarkDuplicates and then re-sorted by name. Each BAM file was further processed to identify valid read pairs with the preparePairs function in diffHic. A param object was created from the genomic ranges object created by the emptyGenome function with the BSgenome.Mmusculus.UCSC.mm10 object. Read pairs were discarded if one read was unmapped, marked as duplicates or had a mapping quality score below 5. Read pairs were determined to be dangling ends or self-circling reads and removed if the pairs of inward-facing reads or outward-facing reads on the same chromosome were separated by less than 1000 bp.
Differential interactions analysis with diffHic
Differential interactions (DIs) were detected using the diffHic package. Two separate analyses were performed: for basal, LP and ML cells and for basal Tspan8+ and Tspan8– cells. Read pairs were counted into 50 kb bin pairs for all autosomes and sex chromosomes. Bins were discarded if they had a count of less than 5 or contained blacklisted genomic regions as defined by ENCODE for mm10.85 Filtering of bin-pairs was performed using the filterDirect function, where bin pairs were only retained if they had average interaction intensities more than 4-fold higher than the background ligation frequency. The ligation frequency was estimated from the inter-chromosomal bin pairs from a 2 Mb bin-pair count matrix. Diagonal bin pairs were also removed. The counts were normalized between libraries using a loess-based approach. Tests for DIs were performed using the quasi-likelihood (QL) framework86 of the edgeR package with a generalized linear model87 and empirical Bayes strategy88 as described previously.61 The design matrix was constructed using a layout that specified the cell type and the mouse group. A DI was defined as a bin pair with a FDR < 0.01. The DIs and genomic ranges were overlapped with the overlapsAny function from the IRanges package.
Multidimensional-scaling plots were constructed with the plotMDS function in the limma package applied to the filtered, normalized and mouse group corrected logCPM values of each bin pair. The logCPM values were corrected for mouse group with the removeBatchEffect function of the limma package. The distance between each pair of samples was the ‘leading log fold change’, defined as the root-mean-square average of the 10,000 largest log2-fold changes between that pair of samples. Upset plots were created using the ComplexUpset package.89
A/B compartment analysis
The HOMER HiC pipeline68,69 was used to identify A/B compartments, create contact matrices and decay curves. After preprocessing with the diffHic pipeline, libraries in HDF5 format were converted to the HiC summary format with R. Then input-tag directions were created for each library with the makeTagDirectory function, with the genome (mm10) specified. Summed biological-replicate tag directories for each cell type were also created. A/B compartments were identified at a resolution of 100 kb as described.90 With the summed biological-replicate tag directories, the runHiCpca.pl function was used on each library with -res/window 100,000 and the genome (mm10) specified. To identify changes in A/B compartments between libraries, the getHiCcorrDiff.pl function was used to directly calculate the difference in correlation profiles. Compartments were considered flipped if the difference in correlation was less than 0 and were considered melting if the difference was >0 and <0.4.
Omni-C™
Library reproducibility
To determine the reproducibility of the libraries the HiCRep R package was utilized to quantify the similarity between all libraries with the stratum adjusted correlation coefficient (SCC).91 For every combination of libraries, the raw contact matrices (50 kb resolution) of individual chromosomes for each replicate were used to compute the SCC with smoothing parameter 4 and maximum distance considered 5 Mb. For each pairwise comparison, a median SCC was calculated across all chromosomes. Values were plotted with pheatmap package.
Visualization of Omni-C™
Normalized contact matrices at 20 kb resolution were produced with the HOMER HiC pipeline for visualization with the summed biological-replicate tag directories. The analyzeHiC function was used with the -balance option. Contact matrix plots were constructed using the plotHic function from the Sushi R package v1.34.0.92 The color palette was reversed inferno from the viridisLite package v0.4.1.93 DI arcs were plotted with the plotBedpe function of the Sushi package. The Z-score shown on the vertical axis was calculated as –log10(P value). ChromHMM tracks and A/B compartments were plotted with the plotBed function. Capture-C data were plotted with the plotBedgraph function. Gene annotation was plotted with the plotGenes function.
TSS/genebody interactivity (in silico 4C/5C)
Using the diffHic processed data, for genes >100 kb, interactivity within the genebody, TSS and gene-body (intragenic) and with the TSS and the regions 2.5 Mb upstream and downstream of the TSS excluding the genebody (intergenic) was calculated. The TSS region was defined at 2.5 kb ± the TSS and included alternative TSSs. Any read pairs overlapping blacklisted regions were discarded. The genome was partitioned into 5 kb bins. For interactivity within the genebody, the connectCounts function was used with the bins across the genebody as the regions and second.regions arguments. For each gene the counts were summed. For the interactivity between the TSS and the genebody, the connectCounts function was used with TSS regions and the bins across the corresponding genebody excluding the TSS region as the regions and second.regions arguments. For each TSS region the counts were summed. For the interactivity between the TSS and the regions outside the genebody, the connectCounts function was used with the TSS regions as the regions argument and the bins in the 2.5 Mb regions upstream and downstream excluding the genebody and TSS regions as the second.regions argument. All gene/TSS counts were normalized with TMM normalization. Reads per interaction length (TSS) or area (within gene-body) per million were calculated with the RPKM from the edgeR package where the gene.length argument was set to: the gene length square for within gene-body interactions, the gene length for intragenic and the 2.5 Mb upstream and downstream of TSS excluding the gene-body for intergenic.
Identification of super-enhancers
H3K27ac CUT&Tag sequencing data were used to map super-enhancers with the ROSE39 (Rank ordering of super-enhancers) algorithm. Biological replicates were merged and down-sampled to 13.7M for H3K27ac and 1.9M reads for IgG from basal, LP and ML cells, while basal Tspan8+ and Tspan8– cells were down-sampled to 6.5M reads for H3K27ac and 2.5M for IgG. Peaks were called by SEACR70 (Sparse Enrichment Analysis for CUT&RUN) with normalization to IgG and a stringent peak curve cut-off. ROSE was performed using the H3K27ac peaks and merged read file from each cell population. SEs were linked to a gene if they overlapped their TSS proximal region (± 2.5 kb), gene-body regions (intragenic) or interacted with TSSs as determined using Omni-C™ data.
FitHiChIP significant interactions
To determine significant interactions within each cell type, Omni-C™ reads were mapped with HiC-Pro using Dovetail’s recommended pipeline. Interactions were centred around TSS as determined by RefSeq (refGene.gtf, downloaded 8/2/2017) and interactions were then called using HiC-Pro allValidPairs files with FitHiChIP72 using the following settings, “peak to all” interactions over 10 kb genomic bins, between 2 Mb and 20 kb from the TSSs with coverage bias regression and Q-value of <0.1.
Analysis of alternative TSSs
Read counts within ± 500 bp of the TSS regions were summarized using featureCounts for each transcript in the RefSeq annotation (refGene.gtf, downloaded 8/2/2017). Differential alternative TSSs analysis was performed using the edgeR quasi-likelihood pipeline, generating DETSSs. Regions with low read coverage were removed using filterByExpr, and the TMM normalization was applied. The testing was done by glmTreat. The fold-change cutoff was set at 2 for basal vs LP and basal vs ML, and 1.2 for LP vs ML comparisons. To distinguish differentially expressed TSSs from alternative and differentially expressed TSSs (DEATSSs), we applied a percent relative range calculation, determining the variability in all TSSs for a single gene across the three MEC populations. A final list of DEATSS was determined with the following parameters: 1- gene passes filtering using edgeR’s function filterByExpr, 2- a minimum FDR < 0.05 for at least one TSS within the gene, 3- gene contains ≥ 2 TSSs, 4- remove bi-directional promoters, 5- percent relative range of TSS CPM within a gene is ≥ 0.75.
Chromatin state modeling
Chromatin states of MECs were modeling using a hidden Markov model (ChromHMM).73 ChromHMM parameters were adjusted for CUT&Tag sequencing data, as there is very little background compared to ChIP-Seq and antibody-specific differences are larger. Genomic enrichment was first determined using DeepTools.81 plotFingerprint tool and calculated as “1-Elbow Point.” Each antibody and ATAC sample was down-sampled to match the number of reads from the sample with the minimum reads and maximum genomic enrichment. ChromHMM was then run on these down-sampled and genomic-enrichment normalized bam files with the following parameters: 24 states, bin size of 400 bp, information smoothing of 0.01, initialization method “information” and a pseudo count employed for 0 count regions. States were then reordered as TSSs, enhancers, transcription, polycomb, silent and heterochromatin and repeat regions. States are described as follows: (1) TSS_A1=active transcriptional start site #1, (2) TSS_Wk=weak TSS, (3) TSS_A2, active TSS #2, (4) TSS_Flk, TSS flanking region, (5) TSS_Biv=bivalent TSS, (6) Enh_G_A=active genic enhancer, (7) Enh_A=active enhancer, (8) Enh_W=weak enhancer, (9) Enh_Flk=enhancer flanking region, (10) Enh_Prm=primed enhancer, (11) Enh_G=genic enhancer region, (12) Tx_1=transcription #2, (13) Tx_2=transcription #2, (14) PRC1/2=polycomb repressive complex 1 and 2, (15) PRC2=PRC2 enriched, (16) PRC1=PRC1 enriched, (17-19) Quies_#=quiescent/silent chromatin, (20-24) Het_#=heterochromatin and repeat regions.
TSS and CRE scores
ChromHMM states were used to derive TSS and CRE scores. When multiple refseq promoters were available for a gene, the promoter with the highest read count was used. Promoter states were assigned a score based on their association with gene expression: state 1 = 2, states 2 and 3 = 1, states 5 and 16 = -1 and states 14 and 15 = -2. TSS scores were calculated as: maxscore(states 1:3) + minscore(states 5, 14:16). CREs were assigned to a gene using FitHiChIP analysis of the merged biological replicate Omni-C™ data at 10 kb resolution with a FDR < 0.1. CRE states were assigned scores based on their association with expression: states 1:3 = 1, states 6 and 7 = 2, states 5, 10 and 16 = 1, states 14 and 15 = 2. The number of interactions to the target TSS and the distance spanned are critical for CRE activity.94 Taking these data into account, the CRE score was calculated as: zscore((sumscores(state 1:3, 6 and 7) - sumscores(state 5, 10, 14:16)) x enriched CCPM / squareroot(span of interaction from TSS to CRE)).
Hierarchical clustering
DEGs were clustered using the mean-centered difference for the log2 RPKM gene expression and the TSS and CRE scores using the r package pheatmap. Expression (log2 RPKM), TSS and CRE ChromHMM states and interactions were then mapped based on the clustered gene order. Genes from the LP-alveolar single-cell cluster (Pal et al 2021,25 cluster C1) were clustered based on their scRNA-seq expression (NCBI GEO accession number: GSE164017), then ChromHMM states, scores and interactions were mapped based on the clustered gene order. Clustered dendrograms were used to determine the clusters marked on the heatmaps.
Transcription factor motif analysis
To predict transcription factor occupancy, we utilized the TOBIAS48 tools (Transcription factor Occupancy prediction By Investigation of ATAC-seq Signal). We merged JASPAR74 (JASPAR2018_CORE_vertebrates_non-redundant) and HOCOMOCO75 (HOCOMOCOv11_core_MOUSE_mono) motif databases and ran TOBIAS with default settings on narrow peaks called by MACS295 (parameters: callpeak --nomodel –shift -75 --extsize 150 – qvalue 0.05) on merged biological ATAC replicates.
To create networks and heatmaps of motif enrichment the bound sites for individual motifs from the TOBIAS analysis were imported into R and converted to Granges objects. ChromHMM states were imported into R as GRanges objects and split into 400 bp-width bins with tile ranges function from the plyranges package. The motifs were overlapped (connected) with the ChromHMM states, genes or DIs with the overlapsAny function from the IRanges package. Networks were constructed for each lineage (basal, LP and ML) for the TFs identified in the RNA-seq signature analysis that also had binding sites identified by the TOBIAS analysis and had expression more than 1 RPKM for LP and ML and 10 RPKM for basal cells. Footprinted motif sites mapped were to TSSs, proximal regions and distal interacting regions as determined by FitHiChIP. The distal region of an interaction was defined as the anchor not containing a gene; if an interaction contained a gene in both anchors an interaction was duplicated to include both overlapping events. Motif enrichment was the number of motifs in a region per bp. Networks were created Cytoscape49 using the gene as the target and the TF binding site as the source. Interconnectivity (betweenessCentrality) was calculated with the NetworkAnalyzer and treating the network as directed. For the differential transcriptional network, the network was created using the difference in motif enrichment between the basal Tspan8+ and Tspan8– populations. TFs were expressed at >1 RPKM and genes were DEGs.
Quantification and statistical analysis
Statistical tests between two groups used a two-tailed T-test with Welch’s corrections, unless otherwise specified. Statistical analysis was performed in R v4.2.0 and GraphPad Prism 9. P-values of statistical tests were as follows: n.s. = not significant, ∗ P<0.05, ∗∗ P<0.01, ∗∗∗ P<0.001 and ∗∗∗∗ P<0.0001.
Acknowledgments
We are grateful to B. Pal, H. Huckstep, and L. Prokopuk for expert advice and to the WEHI Bioservices, FACS, Research Computing, and Genomics facilities. This work was supported by the NBCF (IIRS-20-022), NHMRC (1054618, 1100807, 1113133, 1153049), NHMRC IRIISS, Victorian State Government Operational Infrastructure Support, and BCRF (USA). M.J.G.M. was supported by a VCA fellowship (ECRF19011), H.D.C. was supported by the Marian and E.H. Flack Fellowship, Y.C. was supported by an MRFF Investigator grant (1176199), and G.J.L., G.K.S., and J.E.V. were supported by NHMRC fellowships (G.J.L. 1078730, 1175960; G.K.S. 1058892; J.E.V. 1037230, 1102742).
Author contributions
M.J.G.M., H.D.C., and J.E.V. designed the study; M.J.G.M., S.K., T.M.J., S.R.K., W.F.C., M.T., and E.S. performed experiments; M.J.G.M., H.D.C., Y.C., and G.K.S. performed bioinformatic analyses; S.W. provided technique optimization; M.J.G.M., H.D.C., T.M.J., R.S.A., G.J.L. and J.E.V. interpreted data; M.J.G.M., H.D.C., and J.E.V. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100424.
Supplemental information
Data and code availability
Sequencing data for ATAC-seq, CUT&Tag, Omni-C and RNA-seq have been deposited at GEO under the superseries accession number GSE227750. Data will be publicly available on the date of publication.
References
- 1.Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K., Young R.A. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014;159:374–387. doi: 10.1016/j.cell.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng H., Xie W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019;20:535–550. doi: 10.1038/s41580-019-0132-4. [DOI] [PubMed] [Google Scholar]
- 3.Gibcus J.H., Dekker J. The hierarchy of the 3D genome. Mol. Cell. 2013;49:773–782. doi: 10.1016/j.molcel.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowley M.J., Corces V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018;19:789–800. doi: 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon J.R., Gorkin D.U., Ren B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell. 2016;62:668–680. doi: 10.1016/j.molcel.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guenther M.G., Levine S.S., Boyer L.A., Jaenisch R., Young R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. S0092-8674(07)00681-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Doni Jayavelu N., Jajodia A., Mishra A., Hawkins R.D. Candidate silencer elements for the human and mouse genomes. Nat. Commun. 2020;11:1061. doi: 10.1038/s41467-020-14853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ngan C.Y., Wong C.H., Tjong H., Wang W., Goldfeder R.L., Choi C., He H., Gong L., Lin J., Urban B., et al. Chromatin interaction analyses elucidate the roles of PRC2-bound silencers in mouse development. Nat. Genet. 2020;52:264–272. doi: 10.1038/s41588-020-0581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonen N., Futtner C.R., Wood S., Garcia-Moreno S.A., Salamone I.M., Samson S.C., Sekido R., Poulat F., Maatouk D.M., Lovell-Badge R. Sex reversal following deletion of a single distal enhancer of Sox9. Science. 2018;360:1469–1473. doi: 10.1126/science.aas9408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gisselbrecht S.S., Palagi A., Kurland J.V., Rogers J.M., Ozadam H., Zhan Y., Dekker J., Bulyk M.L. Transcriptional Silencers in Drosophila Serve a Dual Role as Transcriptional Enhancers in Alternate Cellular Contexts. Mol. Cell. 2020;77:324–337.e8. doi: 10.1016/j.molcel.2019.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu N.Y., Nolan E., Lindeman G.J., Visvader J.E. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol. Rev. 2020;100:489–523. doi: 10.1152/physrev.00040.2018. [DOI] [PubMed] [Google Scholar]
- 13.Cai S., Kalisky T., Sahoo D., Dalerba P., Feng W., Lin Y., Qian D., Kong A., Yu J., Wang F., et al. A Quiescent Bcl11b High Stem Cell Population Is Required for Maintenance of the Mammary Gland. Cell Stem Cell. 2017;20:247–260.e5. doi: 10.1016/j.stem.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu N.Y., Rios A.C., Pal B., Law C.W., Jamieson P., Liu R., Vaillant F., Jackling F., Liu K.H., Smyth G.K., et al. Identification of quiescent and spatially restricted mammary stem cells that are hormone responsive. Nat. Cell Biol. 2017;19:164–176. doi: 10.1038/ncb3471. [DOI] [PubMed] [Google Scholar]
- 15.Asselin-Labat M.L., Sutherland K.D., Barker H., Thomas R., Shackleton M., Forrest N.C., Hartley L., Robb L., Grosveld F.G., van der Wees J., et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 16.Shehata M., Teschendorff A., Sharp G., Novcic N., Russell I.A., Avril S., Prater M., Eirew P., Caldas C., Watson C.J., Stingl J. Phenotypic and functional characterisation of the luminal cell hierarchy of the mammary gland. Breast Cancer Res. 2012;14:R134. doi: 10.1186/bcr3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pal B., Bouras T., Shi W., Vaillant F., Sheridan J.M., Fu N., Breslin K., Jiang K., Ritchie M.E., Young M., et al. Global changes in the mammary epigenome are induced by hormonal cues and coordinated by Ezh2. Cell Rep. 2013;3:411–426. doi: 10.1016/j.celrep.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Huh S.J., Clement K., Jee D., Merlini A., Choudhury S., Maruyama R., Yoo R., Chytil A., Boyle P., Ran F.A., et al. Age- and pregnancy-associated DNA methylation changes in mammary epithelial cells. Stem Cell Rep. 2015;4:297–311. doi: 10.1016/j.stemcr.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dravis C., Chung C.Y., Lytle N.K., Herrera-Valdez J., Luna G., Trejo C.L., Reya T., Wahl G.M. Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell. 2018;34:466–482.e6. doi: 10.1016/j.ccell.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casey A.E., Sinha A., Singhania R., Livingstone J., Waterhouse P., Tharmapalan P., Cruickshank J., Shehata M., Drysdale E., Fang H., et al. Mammary molecular portraits reveal lineage-specific features and progenitor cell vulnerabilities. J. Cell Biol. 2018;217:2951–2974. doi: 10.1083/jcb.201804042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dos Santos C.O., Dolzhenko E., Hodges E., Smith A.D., Hannon G.J. An epigenetic memory of pregnancy in the mouse mammary gland. Cell Rep. 2015;11:1102–1109. doi: 10.1016/j.celrep.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung C.Y., Ma Z., Dravis C., Preissl S., Poirion O., Luna G., Hou X., Giraddi R.R., Ren B., Wahl G.M. Single-Cell Chromatin Analysis of Mammary Gland Development Reveals Cell-State Transcriptional Regulators and Lineage Relationships. Cell Rep. 2019;29:495–510.e6. doi: 10.1016/j.celrep.2019.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giraddi R.R., Chung C.Y., Heinz R.E., Balcioglu O., Novotny M., Trejo C.L., Dravis C., Hagos B.M., Mehrabad E.M., Rodewald L.W., et al. Single-Cell Transcriptomes Distinguish Stem Cell State Changes and Lineage Specification Programs in Early Mammary Gland Development. Cell Rep. 2018;24:1653–1666.e7. doi: 10.1016/j.celrep.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pervolarakis N., Nguyen Q.H., Williams J., Gong Y., Gutierrez G., Sun P., Jhutty D., Zheng G.X.Y., Nemec C.M., Dai X., et al. Integrated Single-Cell Transcriptomics and Chromatin Accessibility Analysis Reveals Regulators of Mammary Epithelial Cell Identity. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pal B., Chen Y., Milevskiy M.J.G., Vaillant F., Prokopuk L., Dawson C.A., Capaldo B.D., Song X., Jackling F., Timpson P., et al. Single cell transcriptome atlas of mouse mammary epithelial cells across development. Breast Cancer Res. 2021;23:69. doi: 10.1186/s13058-021-01445-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pellacani D., Bilenky M., Kannan N., Heravi-Moussavi A., Knapp D.J.H.F., Gakkhar S., Moksa M., Carles A., Moore R., Mungall A.J., et al. Analysis of Normal Human Mammary Epigenomes Reveals Cell-Specific Active Enhancer States and Associated Transcription Factor Networks. Cell Rep. 2016;17:2060–2074. doi: 10.1016/j.celrep.2016.10.058. [DOI] [PubMed] [Google Scholar]
- 27.Gascard P., Bilenky M., Sigaroudinia M., Zhao J., Li L., Carles A., Delaney A., Tam A., Kamoh B., Cho S., et al. Epigenetic and transcriptional determinants of the human breast. Nat. Commun. 2015;6:6351. doi: 10.1038/ncomms7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin H.Y., Willi M., HyunYoo K., Zeng X., Wang C., Metser G., Hennighausen L. Hierarchy within the mammary STAT5-driven Wap super-enhancer. Nat. Genet. 2016;48:904–911. doi: 10.1038/ng.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Willi M., Yoo K.H., Reinisch F., Kuhns T.M., Lee H.K., Wang C., Hennighausen L. Facultative CTCF sites moderate mammary super-enhancer activity and regulate juxtaposed gene in non-mammary cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies J.O.J., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016;13:74–80. doi: 10.1038/nmeth.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019;10:1930. doi: 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lun A.T.L., Smyth G.K. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016;44:e45. doi: 10.1093/nar/gkv1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobrinić P., Szczurek A.T., Klose R.J. PRC1 drives Polycomb-mediated gene repression by controlling transcription initiation and burst frequency. Nat. Struct. Mol. Biol. 2021;28:811–824. doi: 10.1038/s41594-021-00661-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen I., Bar C., Liu H., Valdes V.J., Zhao D., Galbo P.M., Jr., Silva J.M., Koseki H., Zheng D., Ezhkova E. Polycomb complexes redundantly maintain epidermal stem cell identity during development. Genes Dev. 2021;35:354–366. doi: 10.1101/gad.345363.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lun A.T.L., Smyth G.K. diffHic: a Bioconductor package to detect differential genomic interactions in Hi-C data. BMC Bioinf. 2015;16:258. doi: 10.1186/s12859-015-0683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.See Y.X., Chen K., Fullwood M.J. MYC overexpression leads to increased chromatin interactions at super-enhancers and MYC binding sites. Genome Res. 2022;32:629–642. doi: 10.1101/gr.276313.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao F., Fang Y., Tan H.K., Goh Y., Choy J.Y.H., Koh B.T.H., Hao Tan J., Bertin N., Ramadass A., Hunter E., et al. Super-Enhancers and Broad H3K4me3 Domains Form Complex Gene Regulatory Circuits Involving Chromatin Interactions. Sci. Rep. 2017;7:2186. doi: 10.1038/s41598-017-02257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H., Rahl P.B., Lee T.I., Young R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R.T., Tabin C., Sharpe A., Caput D., Crum C., McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 41.Yalcin-Ozuysal O., Fiche M., Guitierrez M., Wagner K.U., Raffoul W., Brisken C. Antagonistic roles of Notch and p63 in controlling mammary epithelial cell fates. Cell Death Differ. 2010;17:1600–1612. doi: 10.1038/cdd.2010.37. [DOI] [PubMed] [Google Scholar]
- 42.Antonini D., Dentice M., Mahtani P., De Rosa L., Della Gatta G., Mandinova A., Salvatore D., Stupka E., Missero C. Tprg, a gene predominantly expressed in skin, is a direct target of the transcription factor p63. J. Invest. Dermatol. 2008;128:1676–1685. doi: 10.1038/jid.2008.12. [DOI] [PubMed] [Google Scholar]
- 43.Fu N.Y., Pal B., Chen Y., Jackling F.C., Milevskiy M., Vaillant F., Capaldo B.D., Guo F., Liu K.H., Rios A.C., et al. Foxp1 Is Indispensable for Ductal Morphogenesis and Controls the Exit of Mammary Stem Cells from Quiescence. Dev. Cell. 2018;47:629–644.e8. doi: 10.1016/j.devcel.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsumura Y., Nakaki R., Inagaki T., Yoshida A., Kano Y., Kimura H., Tanaka T., Tsutsumi S., Nakao M., Doi T., et al. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell. 2015;60:584–596. doi: 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 46.Liu L., Cheung T.H., Charville G.W., Hurgo B.M.C., Leavitt T., Shih J., Brunet A., Rando T.A. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H., et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bentsen M., Goymann P., Schultheis H., Klee K., Petrova A., Wiegandt R., Fust A., Preussner J., Kuenne C., Braun T., et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020;11:4267. doi: 10.1038/s41467-020-18035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Raths F., Karimzadeh M., Ing N., Martinez A., Yang Y., Qu Y., Lee T.Y., Mulligan B., Devkota S., Tilley W.T., et al. The molecular consequences of androgen activity in the human breast. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oakes S.R., Naylor M.J., Asselin-Labat M.L., Blazek K.D., Gardiner-Garden M., Hilton H.N., Kazlauskas M., Pritchard M.A., Chodosh L.A., Pfeffer P.L., et al. The Ets transcription factor Elf5 specifies mammary alveolar cell fate. Genes Dev. 2008;22:581–586. doi: 10.1101/gad.1614608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bouras T., Pal B., Vaillant F., Harburg G., Asselin-Labat M.L., Oakes S.R., Lindeman G.J., Visvader J.E. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell. 2008;3:429–441. doi: 10.1016/j.stem.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 53.Wu Z., Nicoll M., Ingham R.J. AP-1 family transcription factors: a diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL. Exp. Hematol. Oncol. 2021;10:4. doi: 10.1186/s40164-020-00197-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Douglas N.C., Papaioannou V.E. The T-box transcription factors TBX2 and TBX3 in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia. 2013;18:143–147. doi: 10.1007/s10911-013-9282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stadhouders R., Filion G.J., Graf T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature. 2019;569:345–354. doi: 10.1038/s41586-019-1182-7. [DOI] [PubMed] [Google Scholar]
- 56.Shackleton M., Vaillant F., Simpson K.J., Stingl J., Smyth G.K., Asselin-Labat M.L., Wu L., Lindeman G.J., Visvader J.E. Generation of a functional mammary gland from a single stem cell. Nature. 2006;439:84–88. doi: 10.1038/nature04372. [DOI] [PubMed] [Google Scholar]
- 57.Stingl J., Eirew P., Ricketson I., Shackleton M., Vaillant F., Choi D., Li H.I., Eaves C.J. Purification and unique properties of mammary epithelial stem cells. Nature. 2006;439:993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- 58.Boyle S., Flyamer I.M., Williamson I., Sengupta D., Bickmore W.A., Illingworth R.S. A central role for canonical PRC1 in shaping the 3D nuclear landscape. Genes Dev. 2020;34:931–949. doi: 10.1101/gad.336487.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feigman M.J., Moss M.A., Chen C., Cyrill S.L., Ciccone M.F., Trousdell M.C., Yang S.T., Frey W.D., Wilkinson J.E., Dos Santos C.O. Pregnancy reprograms the epigenome of mammary epithelial cells and blocks the development of premalignant lesions. Nat. Commun. 2020;11:2649. doi: 10.1038/s41467-020-16479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Asselin-Labat M.L., Vaillant F., Sheridan J.M., Pal B., Wu D., Simpson E.R., Yasuda H., Smyth G.K., Martin T.J., Lindeman G.J., Visvader J.E. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- 61.Johanson T.M., Lun A.T.L., Coughlan H.D., Tan T., Smyth G.K., Nutt S.L., Allan R.S. Transcription-factor-mediated supervision of global genome architecture maintains B cell identity. Nat. Immunol. 2018;19:1257–1264. doi: 10.1038/s41590-018-0234-8. [DOI] [PubMed] [Google Scholar]
- 62.Dall'Agnese A., Caputo L., Nicoletti C., di Iulio J., Schmitt A., Gatto S., Diao Y., Ye Z., Forcato M., Perera R., et al. Transcription Factor-Directed Re-wiring of Chromatin Architecture for Somatic Cell Nuclear Reprogramming toward trans-Differentiation. Mol. Cell. 2019;76:453–472.e8. doi: 10.1016/j.molcel.2019.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Davies J.O., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016;13:74–80. doi: 10.1038/nmeth.3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dundar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. Gigascience. 2021 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heinz S., Texari L., Hayes M.G.B., Urbanowski M., Chang M.W., Givarkes N., Rialdi A., White K.M., Albrecht R.A., Pache L., et al. Transcription Elongation Can Affect Genome 3D Structure. Cell. 2018;174:1522–1536.e22. doi: 10.1016/j.cell.2018.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meers M.P., Tenenbaum D., Henikoff S. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenet. Chromatin. 2019;12:42. doi: 10.1186/s13072-019-0287-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.J., Vert J.P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Bio. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhattacharyya S., Chandra V., Vijayanand P., Ay F. Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 2019;10:4221. doi: 10.1038/s41467-019-11950-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ernst J., Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012;9:215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castro-Mondragon J.A., Riudavets-Puig R., Rauluseviciute I., Lemma R.B., Turchi L., Blanc-Mathieu R., Lucas J., Boddie P., Khan A., Manosalva Pérez N., et al. JASPAR 2022: the 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022;50:D165–D173. doi: 10.1093/nar/gkab1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A., et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018;46:D252–D259. doi: 10.1093/nar/gkx1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liao Y., Smyth G.K., Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. doi: 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen Y., Lun A.T.L., Smyth G.K. From reads to genes to pathways: differential expression analysis of RNA-Seq experiments using Rsubread and the edgeR quasi-likelihood pipeline. F1000Res. 2016;5:1438. doi: 10.12688/f1000research.8987.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015;109 doi: 10.1002/0471142727.mb2129s109. 21.29.1 29 21.29.9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Michalak E.M., Milevskiy M.J.G., Joyce R.M., Dekkers J.F., Jamieson P.R., Pal B., Dawson C.A., Hu Y., Orkin S.H., Alexander W.S., et al. Canonical PRC2 function is essential for mammary gland development and affects chromatin compaction in mammary organoids. PLoS Biol. 2018;16 doi: 10.1371/journal.pbio.2004986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oudelaar A.M., Davies J.O.J., Downes D.J., Higgs D.R., Hughes J.R. Robust detection of chromosomal interactions from small numbers of cells using low-input Capture-C. Nucleic Acids Res. 2017;45:e184. doi: 10.1093/nar/gkx1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan W.F., Coughlan H.D., Chen Y., Keenan C.R., Smyth G.K., Perkins A.C., Johanson T.M., Allan R.S. Activation of stably silenced genes by recruitment of a synthetic de-methylating module. Nat. Commun. 2022;13:5582. doi: 10.1038/s41467-022-33181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lun A.T.L., Chen Y., Smyth G.K. It's DE-licious: A Recipe for Differential Expression Analyses of RNA-seq Experiments Using Quasi-Likelihood Methods in edgeR. Methods Mol. Biol. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- 84.Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lund S.P., Nettleton D., McCarthy D.J., Smyth G.K. Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Stat. Appl. Genet. Mol. Biol. 2012;11 doi: 10.1515/1544-6115.1826. [DOI] [PubMed] [Google Scholar]
- 87.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Phipson B., Lee S., Majewski I.J., Alexander W.S., Smyth G.K. Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann. Appl. Stat. 2016;10:946–963. doi: 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014;20:1983–1992. doi: 10.1109/TVCG.2014.2346248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O., et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang T., Zhang F., Yardımcı G.G., Song F., Hardison R.C., Noble W.S., Yue F., Li Q. HiCRep: assessing the reproducibility of Hi-C data using a stratum-adjusted correlation coefficient. Genome Res. 2017;27:1939–1949. doi: 10.1101/gr.220640.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Phanstiel D.H., Boyle A.P., Araya C.L., Snyder M.P. Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics. 2014;30:2808–2810. doi: 10.1093/bioinformatics/btu379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garnier S., Noam R., Rudis B., Filipovic-Pierucci A., Galili T., Greenwell B., Sievert C., Harris D.J., Chen J.J. 2021. Viridis - Colorblind-Friendly Color Maps for R. [DOI] [Google Scholar]
- 94.Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A., et al. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019;51:1664–1669. doi: 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Feng J., Liu T., Qin B., Zhang Y., Liu X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data for ATAC-seq, CUT&Tag, Omni-C and RNA-seq have been deposited at GEO under the superseries accession number GSE227750. Data will be publicly available on the date of publication.